 Mai A. Amer
Mai A. Amer Manal M. Darwish
Manal M. Darwish Noha S. Soliman3
Noha S. Soliman3 Heba M. Amin
Heba M. Amin
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Cell. Infect. Microbiol. , 02 February 2024
Sec. Molecular Bacterial Pathogenesis
Volume 14 - 2024 | https://doi.org/10.3389/fcimb.2024.1328390
This article is part of the Research Topic Epidemiology of Antimicrobial Resistance and Virulence Factors of Emerging and Re-emerging bacteria View all 11 articles
Coagulase-negative staphylococci (CoNS) are a group of gram-positive staphylococcal species that naturally inhabit the healthy human skin and mucosa. The clinical impact of CoNS-associated infections has recently been regarded as a challenge for diagnosis and therapeutic options. CoNS-associated infections are primarily caused by bacterial resistance to antibiotics and biofilm formation. As antibiotics are still the most used treatment, this problem will likely persist in the future. The present study aimed to investigate the resistance and virulence of CoNS recovered from various acne lesions and explore their genetic basis. Skin swab samples were collected from participants with acne and healthy skin. All samples underwent conventional culture for the isolation of CoNS, MALDI-TOF confirmation, antibiotic susceptibility, and biofilm formation testing. A total of 85 CoNS isolates were recovered from the samples and preliminarily identified as Staphylococcus epidermidis. Isolates from the acne group (n = 60) showed the highest rates of resistance to penicillin (73%), cefoxitin (63%), clindamycin (53.3%), and erythromycin (48%), followed by levofloxacin (36.7%) and gentamycin (31.7%). The lowest rates of resistance were observed against tetracycline (28.3%), doxycycline (11.7%), and minocycline (8.3%). CoNS isolated from mild, moderate acne and healthy isolates did not show strong biofilm formation, whereas the isolates from the severe cases of the acne group showed strong biofilm formation (76.6%). Four extensively drug-resistant and strong biofilm-forming staphylococcal isolates recovered from patients with severe acne were selected for whole-genome sequencing (WGS), and their genomes were investigated using bioinformatics tools. Three of the sequenced genomes were identified as S. epidermidis; however, isolate 29AM was identified as Staphylococcus warneri, which is a newly emerging pathogen that is not commonly associated with acne and was not detected by MALDI-TOF. All the sequenced strains were multidrug-resistant and carried multiple resistance genes, including blaZ, mecA, tet(K), erm(C), lnuA, vgaA, dfrC, fusB, fosBx1, norA, and vanT, which were found to be located on plasmids and chromosomes. Virulence features were detected in all genomes in the presence of genes involved in adherence and biofilm formation (icaA, icaB, icaC, sdrG, sdrH, atl, ebh, and ebp). Only the S. warneri isolate 29AM contained immune evasion genes (capB, capC, acpXL, and manA), an anti-phagocytosis gene (cdsA), and other unique features. As a result of their potential pathogenicity and antibiotic resistance, CoNS must be monitored as an emerging pathogen associated with acne infections. To the best of our knowledge, this is the first report to isolate, identify, and correlate S. warneri with severe acne infections among Egyptian patients using WGS and bioinformatic analysis.
Staphylococcus species frequently colonize the skin of birds and mammals. Staphylococcus species are distinguished by their ability to coagulate blood into two main groups: coagulase-positive staphylococci, Staphylococcus aureus, and coagulase-negative staphylococci (CoNS), which include most species, such as Staphylococcus epidermidis (Otto, 2010). CoNS is a common skin microbiome organism and can inhibit the adhesion of virulent S. aureus and other pathogens (Christensen and Brüggemann, 2014). S. epidermidis is the most commonly isolated staphylococcal species from the human skin (Becker et al., 2014). It primarily colonizes the head, nose, and axilla. Staphylococcus hominis and Staphylococcus capitis are two additional common human skin colonizers. The latter is more commonly detected in the head and is more prevalent in adolescence. Staphylococcus haemolyticus and Staphylococcus warneri are less frequently observed in the human skin. Additionally, species that typically reside on farms or domestic animals, such as Staphylococcus sciuri or Staphylococcus intermedius, may transiently colonize humans (Otto, 2010). CoNS frequently exhibit multiple drug resistance, have few effective therapeutic choices, result in incurable diseases, and accumulate resistant strains in communities and hospitals. Antimicrobial resistance has various major causes, ranging from a lack of infection control to the inappropriate use of antibiotics. Recent investigations have shown that CoNS are highly resistant to erythromycin, vancomycin, oxacillin, methicillin, and penicillin (França, 2023). Scientists are paying attention to the emergence of multidrug-resistant (MDR) strains among CoNS and S. warneri, which is considered an emerging pathogen that can cause serious infections (Alawad et al., 2022).
S. epidermidis is commonly regarded as a commensal microorganism because it is beneficial to the skin in healthy environments (Cogen et al., 2008; Yang et al., 2022). Commensal S. epidermidis undergoes mutualistic and symbiotic interaction with the cutaneous system (Brüggemann, 2010). The skin hosts and supplements S. epidermidis with nutrients; in exchange, bacteria participate in host defense, innate immunity, and skin homeostasis. Through their microbial surface components, these bacteria interact with extracellular matrix proteins in the human skin (Arrecubieta et al., 2007; Brüggemann, 2010), which subsequently permits the interaction pathways between skin cells and bacteria (Yang et al., 2022). S. epidermidis can prevent biofilm formation of pathogenic strains via the secretion of bacteriocins (Paluch et al., 2020). Additionally, during the healing process of wounds or skin diseases, S. epidermidis lipoteichoic acid (LTA) can reduce skin inflammation (Fournière et al., 2020).
Although S. epidermidis plays a physiological role in maintaining skin homeostasis, it can be linked to some skin pathologies such as acne vulgaris, a prevalent chronic inflammatory skin condition affecting the pilosebaceous unit, where S. epidermidis was found to be overrepresented (Fitz-Gibbon et al., 2013; O’Neill and Gallo, 2018). Acne is primarily caused by three factors: (1) bacterial strains (Downing et al., 1986; Aydemir, 2014; Kutlu et al., 2023), C. acnes and S. epidermidis are both present, with C. acnes making up less than 2% of the skin surface bacteria and S. epidermidis being overrepresented (Fitz-Gibbon et al., 2013; O’Neill and Gallo, 2018); (2) qualitative and quantitative excessive seborrhea in sebaceous glands in acne lesions; and (3) keratinocytes with hyperkeratinization of the pilosebaceous unit, which causes the production of comedones, papules, and pustules (Jahns et al., 2012). The role of biofilms in acne development is an active area of research. Biofilms are complex communities of microorganisms that adhere to surfaces and produce a protective extracellular matrix. The formation of microbial communities, notably Propionibacterium acnes (P. acnes), as biofilms, contributes to the obstruction of follicles and the accumulation of sebum, dead skin cells, and bacteria. In this structured matrix, P. acnes exhibits increased resistance to antimicrobial agents and host immune responses, fostering inflammation and contributing to the characteristic inflammatory lesions seen in acne. The resilience of biofilms poses challenges in treatment because their protective nature makes bacteria less susceptible to conventional therapies, potentially leading to treatment resistance and recurrent lesions. The chronic nature of acne, characterized by periods of exacerbation and remission, may be linked to the persistence of biofilms, emphasizing the need for research on disrupting biofilm formation as a potential target for innovative acne management strategies (Chen et al., 2017; Coenye et al., 2022).
Notably, some phylogenetic sequence types (STs) of S. epidermidis (e.g., ST2, ST5, ST23, and ST215) are associated with nosocomial infections, raising the possibility of their pathogenicity owing to their virulence and multidrug resistance properties (Månsson et al., 2015; Lee et al., 2018). Several virulence genes play an essential role in adhesion, biofilm development, and phenol-soluble modulation, in addition to the presence of mobile genetic elements (MGEs) that are involved in the acquisition and transmission of virulence and resistance features that enhance the pathogenicity of S. epidermidis (Bouchami et al., 2016; Rolo et al., 2017). One of the most important virulence factors of S. epidermidis is biofilm formation, which is mediated by intercellular adhesin (ica) and accumulation-associated protein (aap) genetic determinants (Arciola et al., 2015; Schaeffer et al., 2016). Biofilms are recognized as a common form of microbial growth that confer protection against the host immune system and antibiotic challenges (França, 2023). In CoNS biofilms, especially in S. epidermidis, the polysaccharide poly-N-acetylglucosamine (PNAG) is one of the most dominant metabolites, and up to 60% of the recovered clinical isolates produce the proteins encoded by the icaADBC genes (Ahmed et al., 2019). The first staphylococcal component with a key role in biofilm accumulation was identified as polysaccharide intercellular adhesin (PIA). However, not all strains of S. epidermidis strains possess the icaADBC operon and are formed by the protein products of this gene (Formosa-Dague et al., 2016; Schaeffer et al., 2016). Horizontal gene transfer (HGT) is one of the most significant methods for CoNS to acquire exogenous DNA and, as a result, antibiotic resistance genes in biofilms (Águila-Arcos et al., 2017). In recent years, attention has been focused on the arginine catabolic mobile element (ACME) system, a pathogenicity island hypothesized to promote host colonization and immune evasion (O’Connor et al., 2018). ACME likely descends from S. epidermidis and spreads horizontally to S. aureus (Onishi et al., 2013; Planet et al., 2013).
Considering emerging evidence for the pathogenicity of some specific strains of CoNS, assuming that not all staphylococcal isolates behave similarly, it has been challenging to identify clinically significant strains. To this end, we aimed to characterize CoNS isolated from healthy skin and acne lesions phenotypically by testing antimicrobial susceptibility and biofilm production, and genetically characterized CoNS isolated from severe acne infections using whole genome sequence (WGS) technology in order to understand the genetic basis of bacterial virulence, antibiotic resistance, and phylogenetic background.
The present study was conducted with a total of 140 participants, divided into two groups: i) the acne group (n = 100) with various levels of severity, including mild (n = 25), moderate (n = 45), and severe acne (n = 30), and ii) the healthy group with healthy skin (n = 40). All participants were university students in Egypt aged 18–24 years. The patients recruited in this study were all non-diabetic, not previously treated with topical antibiotics, did not previously receive acne treatment, and had no history of underlying medical illnesses. For the female participants, skin samples were not collected during the menstrual period.
The participant clinician classified the degree of acne based on the clinical features into mild, moderate, and severe acne. The mild level was Acne comedonica (comedones and congestion), moderate level was papulopustular acne (mild papules and pustules), and severe form was pustule-nodular acne (severe pustules and nodules).
All participants provided written informed consent, and the study was approved by the research ethics committee of the pharmacy faculty of the October University for Modern Sciences and Arts (MSA) (approval number (M1/Ec1/2022PD).
Skin swabs of acne lesions were collected from the participants. For the growth of bacterial colonies, skin swabs were cultured in tryptic soy broth (TSB) (Oxoid, UK) at 37°C for 3 days (Huang et al., 2022). Purification was performed by serial subcultivation on 5% sheep blood agar (SBA) (Oxoid, UK) and mannitol salt agar (MSA) (Oxoid, UK) at 37°C for 24 h to obtain single colonies in pure culture. Isolates obtained from the blood and MSA were preliminarily identified as CoNS based on colony morphology and conventional biochemical reactions. Microscopic characteristics were studied using Gram staining. Catalase, DNase, slide coagulase activity, motility, and blood agar hemolysis tests were conducted according to the method by Freney et al. (1999). Isolate identity was subsequently confirmed using Matrix-Assisted Laser Desorption/Ionization Time-of-Flight Mass Spectrometry (MALDI-TOF MS) (Dubois et al., 2010).
The Kirby–Bauer disc diffusion method was used to screen CoNS isolates for antibiotic susceptibility. The adjusted bacterial culture, equivalent to 0.5 McFarland standard, was inoculated onto Muller Hinton agar (Lab M, UK) and incubated at 37°C for 24 h. Commercial antibiotic discs (Oxoid, England) were tested: penicillin (10 U), cefoxitin (30 μg), erythromycin (15 μg), vancomycin (30 μg), clindamycin (2 μg), tetracycline (30 μg), trimethoprim/sulfamethoxazole (1.25/23.75 μg), gentamicin (10 μg), levofloxacin (5 μg), tetracycline (30 μg), rifampin (5 μg), minocycline (30 μg), and doxycycline (30 μg). The interpretative results matched the Clinical and Laboratory Standards Institute standards (CLSI) (CLSI, 2022).
The crystal violet assay was used to evaluate the ability of the staphylococcal isolates to form biofilms (Amer et al., 2021). Briefly, overnight cultures were adjusted to 105 CFU/ml and inoculated in 200 μl of TSB supplemented with 1% (v/v) glucose (LOBA Chemie, India), in 96-flat bottom polystyrene microtiter plate (Greiner Bio-one®, Germany). The microtiter plates were incubated at 37°C without shaking for 24 h. After incubation, microbial growth was determined by measuring the turbidity at 600 nm, after which the cells were removed, washed twice with sterile phosphate buffered saline (PBS), and left to air dry. The dried biofilms were then stained with 0.1% crystal violet (CV) (LOBA Chemie, India) for 15 min, excess stain was removed, the wells were washed with 200 μl sterile distilled water, excess water was removed, and the plates were dried. Finally, the stain was solubilized in 33% glacial acetic acid (LOBA Chemie, India). Colorimetric analysis of the biofilm biomass was performed at 545 nm. Biofilm formation was evaluated using the biofilm formation index, [BFI]: (AB − CW)/(GB − GW), where AB is the OD545 of the CV-stained microorganisms, CW is the OD545 nm of the stained blank wells containing only media, GB is the OD600 of the cell culture, and GW is the OD600 of the blank well. Isolates were classified into four groups: nonadherent (0.35), mild (0.35 to 0.69), moderate (0.70 to 1.09), and strong (>1.10) biofilm forming according to the specified semi-quantitative biofilm production classification described by Lucero-Mejía et al. (2020).
Bacterial pellets of CoNS strains were pretreated with 180 µl lysis buffer formulated with 20 mM Tris–HCl, pH 8.0, 2 mM EDTA (ADWIC, Egypt), 1.2% Triton X-100, and lysozyme that was added immediately before use, at a final concentration of 20 mg/ml. After pretreatment, genomic DNA was extracted using the QIAamp® DNA Mini Kit (QIAGEN, Germany) according to the manufacturer’s recommendations for gram-positive bacteria.
The presence of icaA, icaD, and mecA in the extracted DNA was detected by polymerase chain reaction using forward and reverse primers, as shown in Table 1. The amplification reaction was performed according to the method described by Petrelli et al. (2006), using a Biometra DNA thermal cycler (Hamburg, Germany). Amplicons were analyzed by electrophoresis on 1% agarose gels (Lonza, USA), stained with ethidium bromide dye (Sigma-Aldrich, USA), and visualized under a UV light transilluminator (LTF Labortechnik, Germany). Ruler Gene 100 bp A DNA ladder served as a DNA size indicator (Zhou et al., 2013).
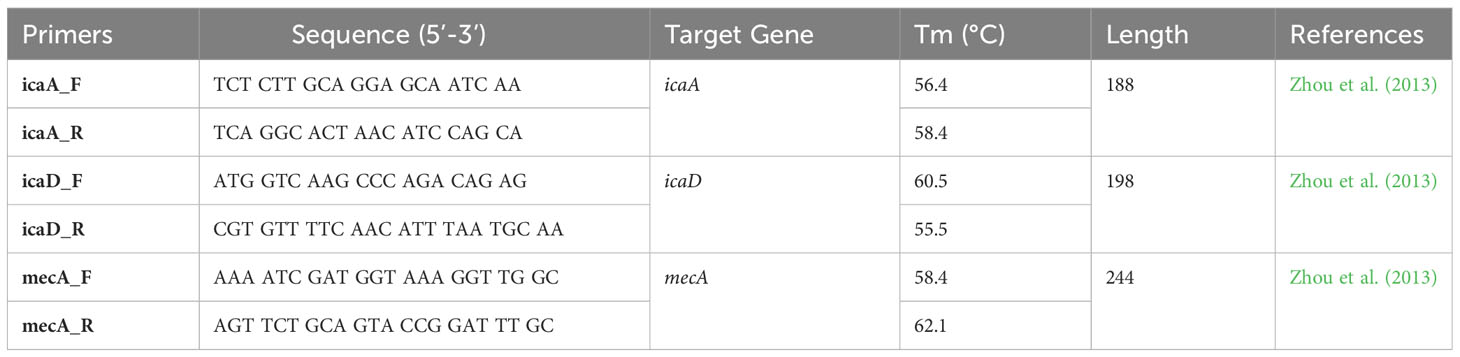
Table 1 List of primers used in PCR.
Following DNA extraction using the QIAamp® DNA Mini Kit (QIAGEN, Germany), sequencing libraries were prepared using the Nextera XT DNA Library Preparation Kit (Illumina, USA) according to the manufacturer’s instructions. Quality control was performed using an Agilent DNA 1000 chip, prior to shotgun sequencing using MiSeq (Illumina, USA). Preassembly processing of the generated reads was performed using FastQC. Trimmomatic v0.32 was used to remove low-quality (mean quality less than 25) or low-complexity reads, reads mapping to the human genome or large and small ribosomal units of bacteria, fungi, and humans, and known contaminants (e.g., phiX174, Illumina spike-in) (Bolger et al., 2014). All genomes are available in the NCBI genome (BioProject accession number PRJNA993660).
Pre-processed reads were assembled de novo using SPAdes version 3.6.1, with contigs shorter than 1,000 nucleotides discarded (Bankevich et al., 2012). The analysis was only selected for reconstructed genomes with an N50 > 50,000. The assembled contiguous sequences were submitted via the National Center for Biotechnology Information (NCBI) Prokaryotic Genome Annotation Pipeline to GenBank for gene annotation. The generated contigs were further analyzed to investigate the genetic elements of interest. After that, the most similar sequences were identified and a phylogenetic tree was constructed using the Bacterial and Viral Bioinformatics Resource Center (BV-BRC) (https://www.bv-brc.org/) (Olson et al., 2023).
PathogenFinder was used to predict the pathogenicity of the isolates for human hosts (https://cge.cbs.dtu.dk/services/PathogenFinder/). The assembled genomes obtained from the WGS data were annotated to identify the predicted resistome using ResFinder 4.1 (with a minimum length and threshold of 60% and 90%, respectively) (https://cge.cbs.dtu.dk/services/ResFinder/), and the Comprehensive Antibiotic Resistance Database (CARD) available at (https://card.mcmaster.ca/analyze/rgi) (Alcock et al., 2020), using the default selection criteria “perfect and strict hits only.” These platforms were employed in combination to avoid the drawbacks of each platform.
The genetic basis (chromosomal single-nucleotide polymorphism [SNP]) for fluoroquinolone and rifampicin resistance genes from the assembled genomes was studied (Altschul et al., 1990). Alignment of gyrA, gyrB, parC, parE, and rpoB genes to the reference strains of S. epidermidis (ATCC®12228 and ATCC®35984) with their corresponding genes from the assembled isolates in this study. Mutations were analyzed using multiple sequence alignment (MSA) and SNP analysis tool provided by the BV-BRC (https://www.bv-brc.org/). Clustal Omega (1.2.4) provided by EMBL’s European Bioinformatics Institute (EMBL-EBI) (https://www.ebi.ac.uk/Tools/msa/clustalo/) was used to perform MSA alignments of the predicted amino acid sequences. Colored alignments were visualized using the multiple alignment viewer tool MView 1.63, available at (https://www.ebi.ac.uk/Tools/msa/mview/) and hosted by EMBL-EBI.
VirulenceFinder 2.0 (using a minimum length of 60% and a threshold of 90%) (https://cge.cbs.dtu.dk/services/VirulenceFinder/) (Joensen et al., 2014), virulence factor database (VFDB) (http://www.mgc.ac.cn/cgi-bin/VFs/v5/main.cgi?func=VFanalyzer) and BacWGSTdb (http://bacdb.cn/BacWGSTdb) were used for screening the presence of virulence genes. The virulence determinants related to S. epidermidis were examined including adhesion, enzymes, biofilm formation, secretion, immune evasion, toxins, anti-phagocytosis, and intracellular survival.
Multilocus sequence typing (MLST) was performed in silico using MLST 2.0, which is available on the website of the Center for Genomic Epidemiology (https://cge.cbs.dtu.dk/services/MLST/) (Larsen et al., 2012) and the public molecular typing database PubMLST (https://pubmlst.org/).
Sequence types were assigned comparing the internal fragments of the seven housekeeping genes (arcC, aroE, gtr, mutS, pyrR, tpiA, and yqiL) from S. epidermidis to determine the MLST sequence types (STs) (Thomas et al., 2007).
Mobile genetic elements (MGEs) related to ARGs and their genomic context were investigated using NCBI annotations. The web-based typing tool SCCmecFinder was used to determine the SCCmec types and their structural position in the S. epidermidis isolates in silico (https://cge.cbs.dtu.dk/services/SCCmecFinder/). Plasmid replicon types were detected in silico using PlasmidFinder 2.1, available at (https://cge.cbs.dtu.dk/services/PlasmidFinder/) (Carattoli et al., 2014).
Prophage sequences within the assembled genomes were detected and annotated using PHASTER tool (https://phaster.ca/) (Arndt et al., 2016). Only “intact” prophage regions discovered by PHASTER were considered. The sites of the prophage regions were BLASTED against CARD to determine whether they included resistance genes. MobileElementFinder v1.0.3 was used to identify ISs and transposons flanking the resistance genes (Johansson et al., 2021), available at https://cge.cbs.dtu.dk/services/MGE/. NCBI annotations were used to investigate the support environment for resistance genes. Insightful Science’s SnapGene viewer software v5.1.3.1, was used to examine the context of resistance genes by visualizing annotated contigs software (http://www.snapgene.com). Insertion sequences (ISs) were identified using BLAST analysis against the NCBI nucleotide database. Resistance islands were predicted using the IslandViewer4 web tool (http://www.pathogenomics.sfu.ca/islandviewer/) (Li and Durbin, 2010).
CRISPRCasFinder tool available at (https://crisprcas.i2bc.paris-saclay.fr/CrisprCasFinder/Index) utilizes the default advanced parameters for CRISPR and the clustering model “SubTyping” for Cas to search the genomes for clustered regularly interspaced short palindromic repeats (CRISPR) and cas genes.
The restriction–modification system (R–M system) was detected using a minimum length of 60% and %ID threshold of 95% using Restriction–ModificationFinder 1.1, available at (https://cge.cbs.dtu.dk/services/Restriction-ModificationFinder/) (Camacho et al., 2009). ACME genes were identified and mapped within the genomes. The arc, opp3, and kdp operons were used to align the ACME components, which were then classified as arc and opp3 operons (type I), arc operons alone (type II), opp3 operons alone (type III), arc and kdp operons (type IV), and arc, opp, and kdp operons (type V).
CSIPhylogeny (https://cge.cbs.dtu.dk/services/CSIPhylogeny/) was used to construct phylogenetic trees based on the maximum likelihood method of concatenated alignment of high-quality SNPs, which uses assembled contigs to perform SNP calling, SNP filtering, and phylogeny inference (Kaas et al., 2014). The analysis was performed on a platform using default parameters. The assembled genomes have been uploaded for comparison. To compare our isolates to S. epidermidis and S. warneri genomes available on the BV-BRC website, we searched and downloaded the S. epidermidis and S. warneri genomes and included them in the analysis. Figtree program was used to edit and visualize the phylogenetic tree (http://tree.bio.ed.ac.uk/software/figtree/).
The core genome single nucleotide polymorphism (cgSNP)-based phylogenetic tree was visualized using the interactive tree of life (iTOL) web tool v6.7 (https://itol.embl.de/itol.cgi) associated with the isolates, and other genomic information, and antibiotic resistance determinants. A phylogenetic tree was also constructed using the isolates and closely related genomes derived from BV-BRC (https://www.bv-brc.org/).
The assembled draft genomes from the Whole Genome Shotgun project were uploaded to the GenBank database under BioProject number PRJNA993660.
GraphPad Prism 8.0.0 for Windows (GraphPad Software Inc., CA, USA) was used for the statistical analysis. The independent samples t-test and two-way analysis of variance (ANOVA) were used to compare the antibiotic resistance determinants, biofilm-forming ability, and prevalence of biofilm-associated genes among CoNS isolates, with a P-value of 0.05 regarded as statistically significant.
A total of 140 participants were enrolled in this study and divided into two groups: i) the acne group (n = 100) with various levels of severity, including mild (n = 25), moderate (n = 45), and severe acne (n = 30), and ii) the healthy group with healthy skin (n = 40). Sex distribution was represented in the form of 60 (60%) males and 40 (40%) females in the acne group, and 25 (62.5%) males and 15 (37.5%) females in the healthy group.
The culture of skin swabs yielded a total of 85 staphyloccal isolates that were preliminarily identified based on conventional biochemical reactions, all of which were gram-positive arranged in grape-like clusters, catalase-positive, DNase-negative, coagulase-negative, non-motile, and non-hemolytic on blood agar. All isolates were identified as S. epidermidis by MALDI-TOF MS. Sixty isolates were obtained from the acne group (60%), and 25 isolates (62.5%) were obtained from the healthy group.
The results of antimicrobial susceptibility testing of CoNS isolates from the acne and healthy groups are displayed in Supplementary Table 1, where isolates from the acne group (N = 60) showed the highest rates of resistance to penicillin (73%), cefoxitin (63%), clindamycin (53.3%), and erythromycin (48%), followed by levofloxacin (36.7%) and gentamycin (31.7%), while least rates of resistance were observed for tetracycline (28.3%), doxycycline (11.7%), and minocycline (8.3%). The antimicrobials with the highest rates of resistance among the healthy group isolates were clindamycin (56%), erythromycin (44%), penicillin (40%), and cefoxitin (40%), followed by levofloxacin (32%) and tetracycline (32%), whereas those with the lowest rates of resistance were gentamycin (20%), doxycycline (8%), and minocycline (0%). Comparing the resistance prevalence between the two groups of isolates showed a statistically significant difference (P-value <0.05) for penicillin and cefoxitin, however, no significant difference was observed for the other antimicrobials (Supplementary Table 1) (Figure 1). Out of the acne group, 15 isolates (25%) demonstrated multidrug resistance (MDR) to antibiotics, four of which were extensively drug resistant (XDR), whereas in the control group, two isolates exhibited resistance to multiple drugs (8%), and none were XDR. Statistical analysis revealed a significant difference between the two groups (P-value <0.05).
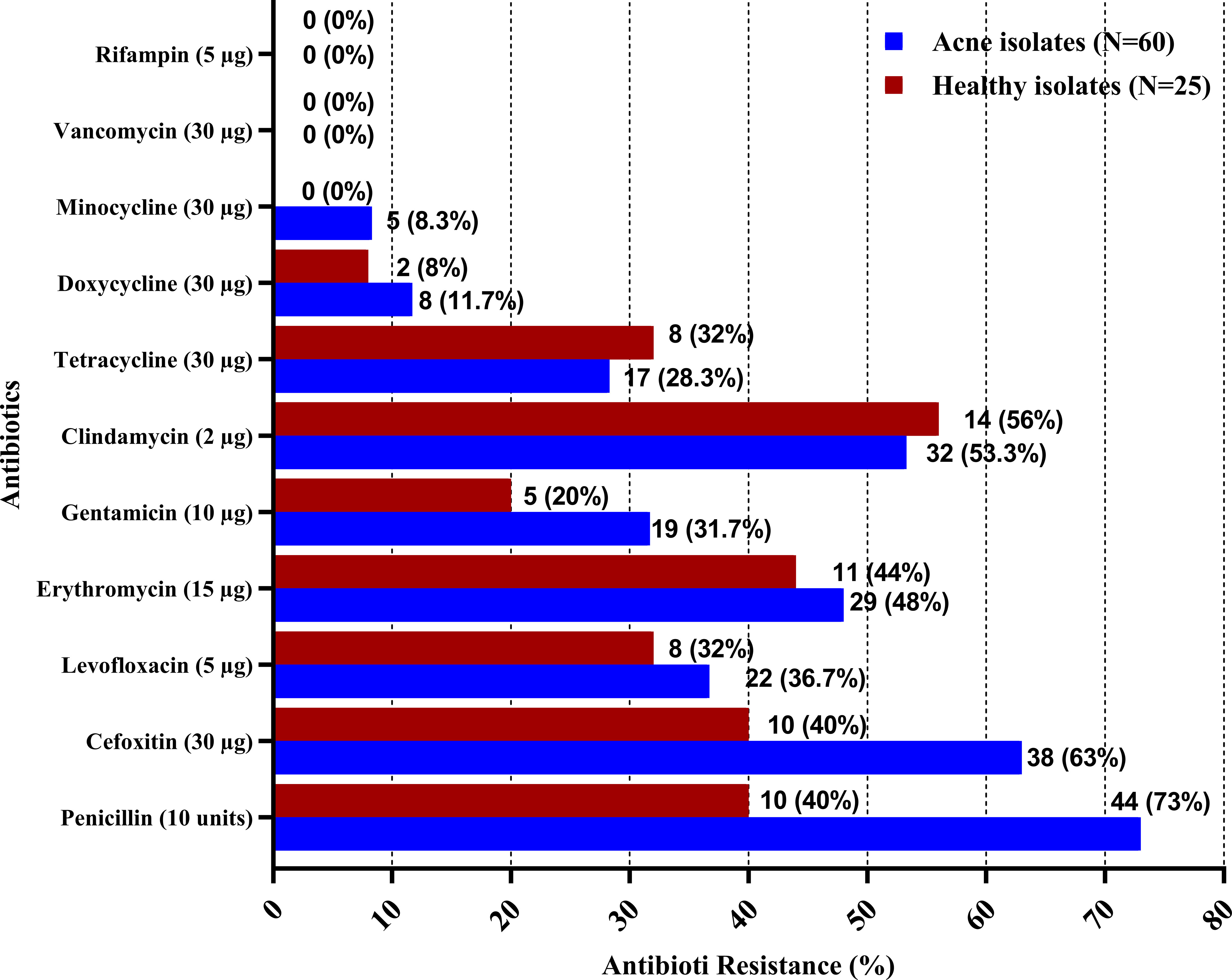
Figure 1 Antibiotic resistance among CoNS isolates from acne and healthy groups. Antibiotic susceptibility testing was performed for CoNS isolates using the Kirby–Bauer disk diffusion method, in accordance with the Clinical and Laboratory Standards Institute (CLSI).
Biofilm formation ability of CoNS isolates was tested using crystal violet biofilm assay method. Forty-four isolates recovered from the acne group (73.3%) and 11 isolates recovered from the healthy group (44%) were able to form biofilm. Biofilm-producers demonstrated strong, moderate, and weak biofilm production at rates of 38.3%, 28.3%, and 6.6% among CoNS isolates of the acne group (n=60), while rates of 0%, 8%, and 36% among CoNS isolates of healthy group (n=25), respectively (Supplementary Table 2). The utilization of an unpaired t-test in statistical analysis indicated a significant contrast between the acne and healthy groups. Notably, only isolates retrieved from the severe acne group exhibited the ability to form strong biofilms (76.6%). All MDR isolates recovered from the acne group formed strong biofilms. (Supplementary Table 2) (Figure 2).
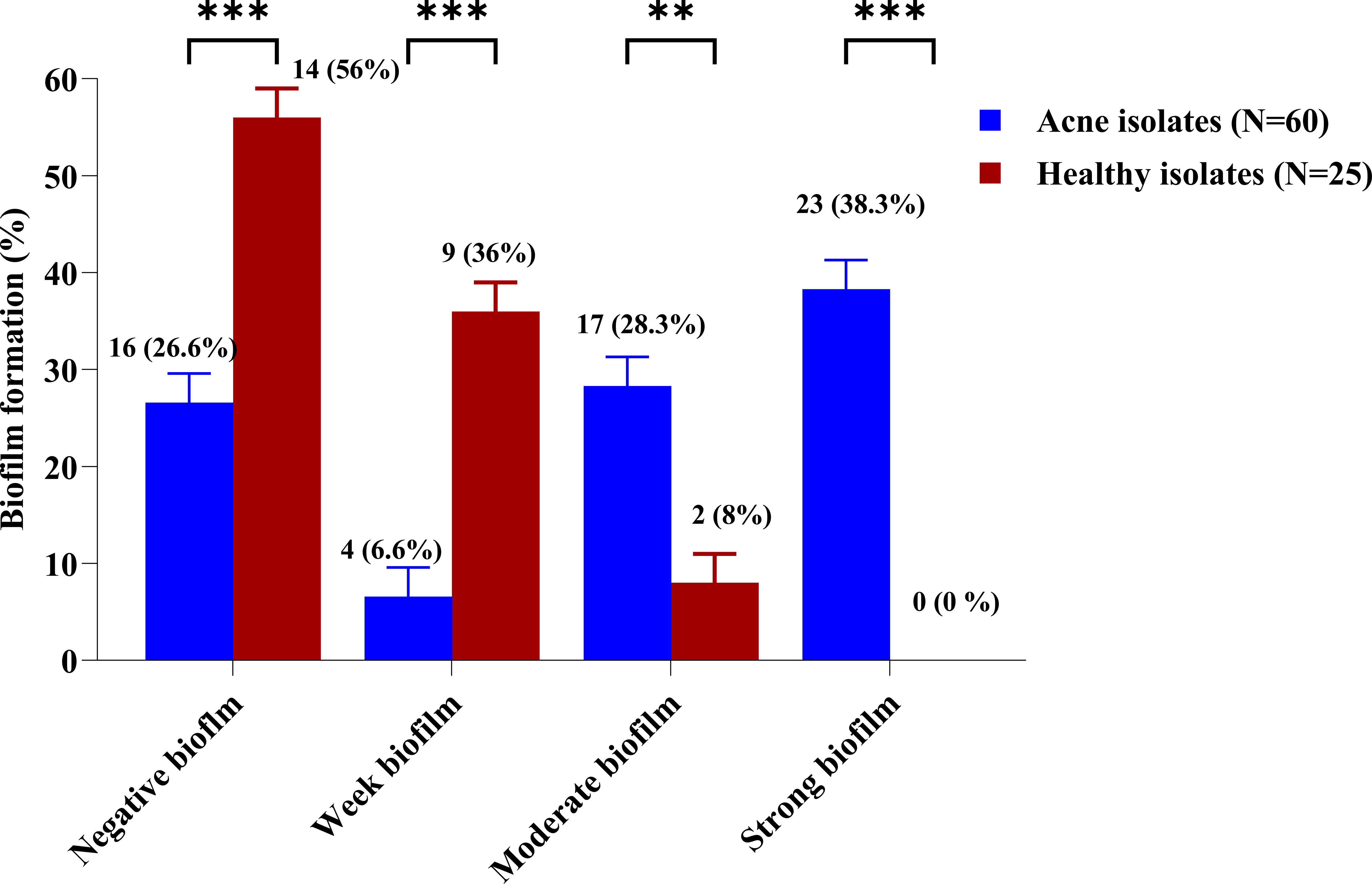
Figure 2 Biofilm formation percentage among CoNS isolates from the acne and healthy groups. The biofilm-forming ability of the CoNS isolates was assessed using a crystal violet assay. Adjusted overnight cultures were inoculated in 200 μl of TSB supplemented with 1% (v/v) glucose, in a 96-flat bottom polystyrene microtiter plate. The biofilms were then washed and stained with 0.1% crystal violet. The biofilm biomass was measured colorimetrically at 545 nm and evaluated using the biofilm formation index. Statistical analysis using two-way ANOVA, which was followed by multiple comparisons test with a significance level at ** p < 0.01; *** p < 0.001.
Biofilm-associated gene carriage was assessed in the isolates recovered from acne and healthy samples. The distribution of the biofilm-associated genes is shown (Figure 3). Two-way ANOVA in statistical analysis revealed a significant difference in the prevalence of biofilm-associated genes among CoNS isolates obtained from individuals with moderate and severe acne compared to those from mild acne and healthy individuals (P-value <0.05). Four XDR and strong biofilm-forming CoNS isolates were coded as 29AM, 36AM, 48AF, and 54AF were selected for further study using WGS. The antibiotic resistance characteristics of the four isolates are presented in Table 2.
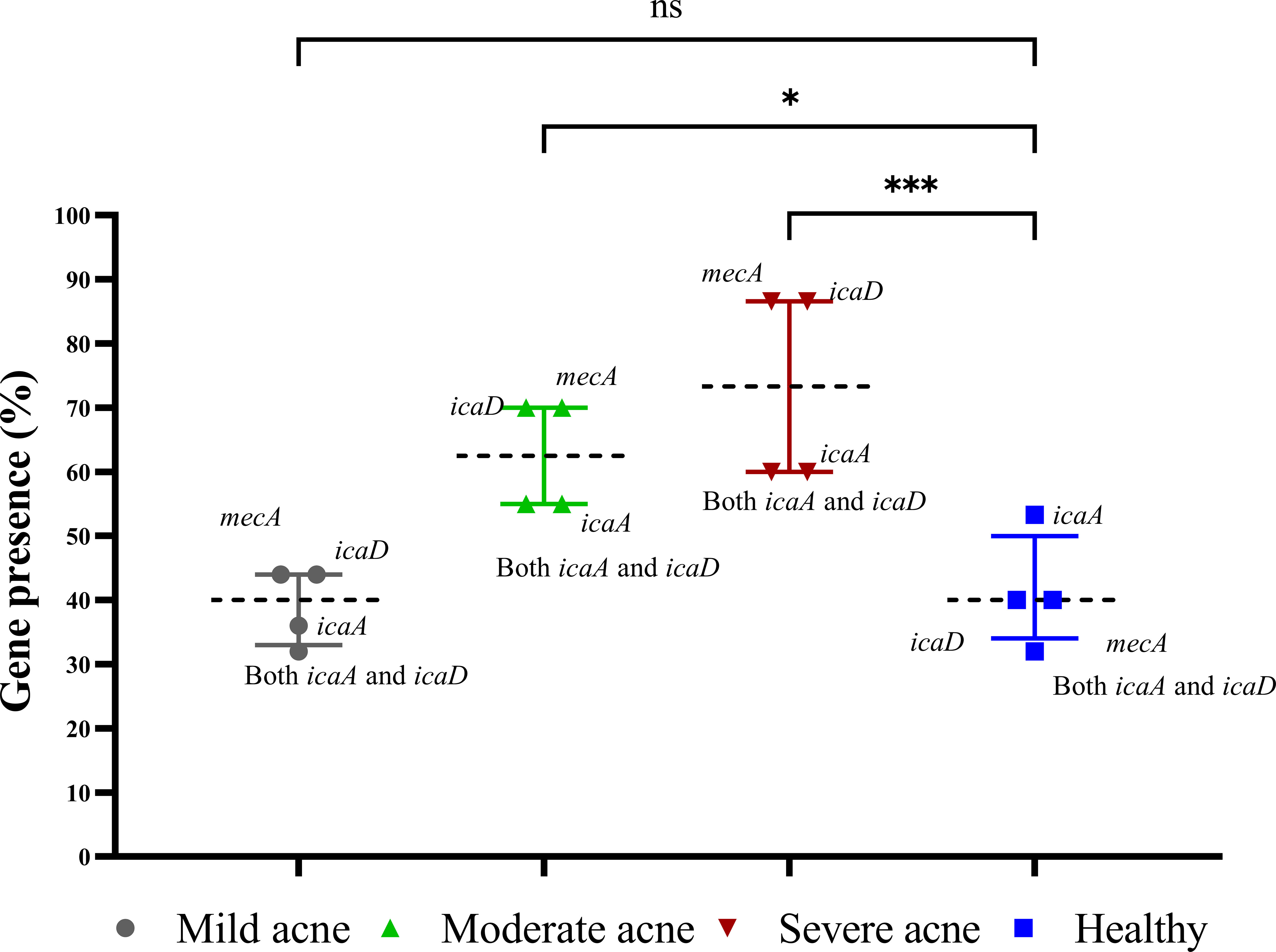
Figure 3 Prevalence of biofilm genes among CoNS isolates. The presence of icaA, icaD, and mecA in the extracted DNA was detected by polymerase chain reaction. The presence of the gene was represented as a percentage of the different acne and healthy groups, and the results are shown as medians with interquartile ranges. Statistical analysis using two-way ANOVA, which was followed by multiple comparisons test with a significance level at * p < 0.05; *** p < 0.001. ns, non-significant.
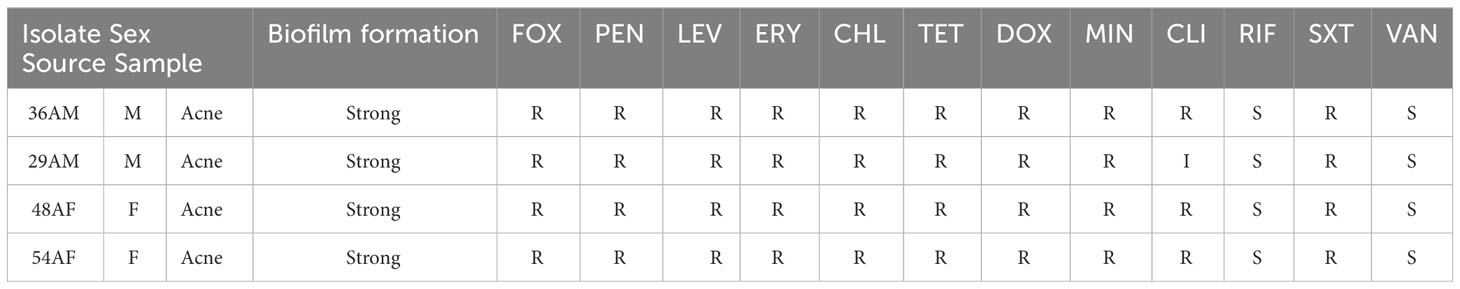
Table 2 The antibiotic susceptibility profile and accompanying metadata for the four staphylococcal isolates.
The CLSI breakpoints for CoNS were used to interpret antibiotic susceptibility tests. R, resistant; I, intermediate; S, susceptible; M, male; F, female. FOX, cefoxitin; PEN, penicillin; LEV, levofloxacin; ERY, erythromycin; CHL, chloramphenicol; TET, tetracycline; DOX, doxycycline; MIN minocycline; CLI, clindamycin; RIF, rifampin; SXT, sulfamethoxazole/trimethoprim; VAN, vancomycin.
Based on the short-read sequences, draft genomes comprising 65–294 contigs were assembled with a mean N50 of 80,333 bp, covering approximately 81%–86% of the reference genome. Supplementary Table 3 displays the genome sequences and assembly parameters, such as size, number of contigs, number of RNAs, guanine–cytosine (GC) content (%), number of coding sequences, N50, and L50.
The isolates’ draft genome size ranged from 2.3 Mb to 2.6 Mb, with a GC content of 31.88% to 32.44%. Three isolates coded as 36AM, 48AF, and 54AF were confirmed to be S. epidermidis using WGS, while the isolate coded as 29AM was identified as S. warneri, another multidrug-resistant CoNS.
Antibiotic resistance genes (ARGs) conferring resistance to β-lactams (blaZ), methicillin/oxacillin (mecA), glycopeptides (vanT, vanY), fluoroquinolone [norA, norC, sdrM], tetracyclines [tet(K)], macrolide–lincosamide–streptogramin B antibiotic (MLSB) [erm(C), vgaA, and InuA], trimethoprim-sulfamethoxazole (dfrC), aminocoumarin (gyrB), fosfomycin (fosBx1), and fusidic acid (fusB) were detected in the isolated genomes (Table 3). All staphylococcal isolates except isolate 54AF possessed the blaZ gene, while all isolates except S. warneri 29AM possessed the mecA gene. ARGs related to trimethoprim-sulfamethoxazole-, tetracycline-, doxycycline-, and erythromycin-resistance followed resistance phenotypes. Isolates phenotypically resistant to these antibiotics and their corresponding ARGs were detected in the genomic context.
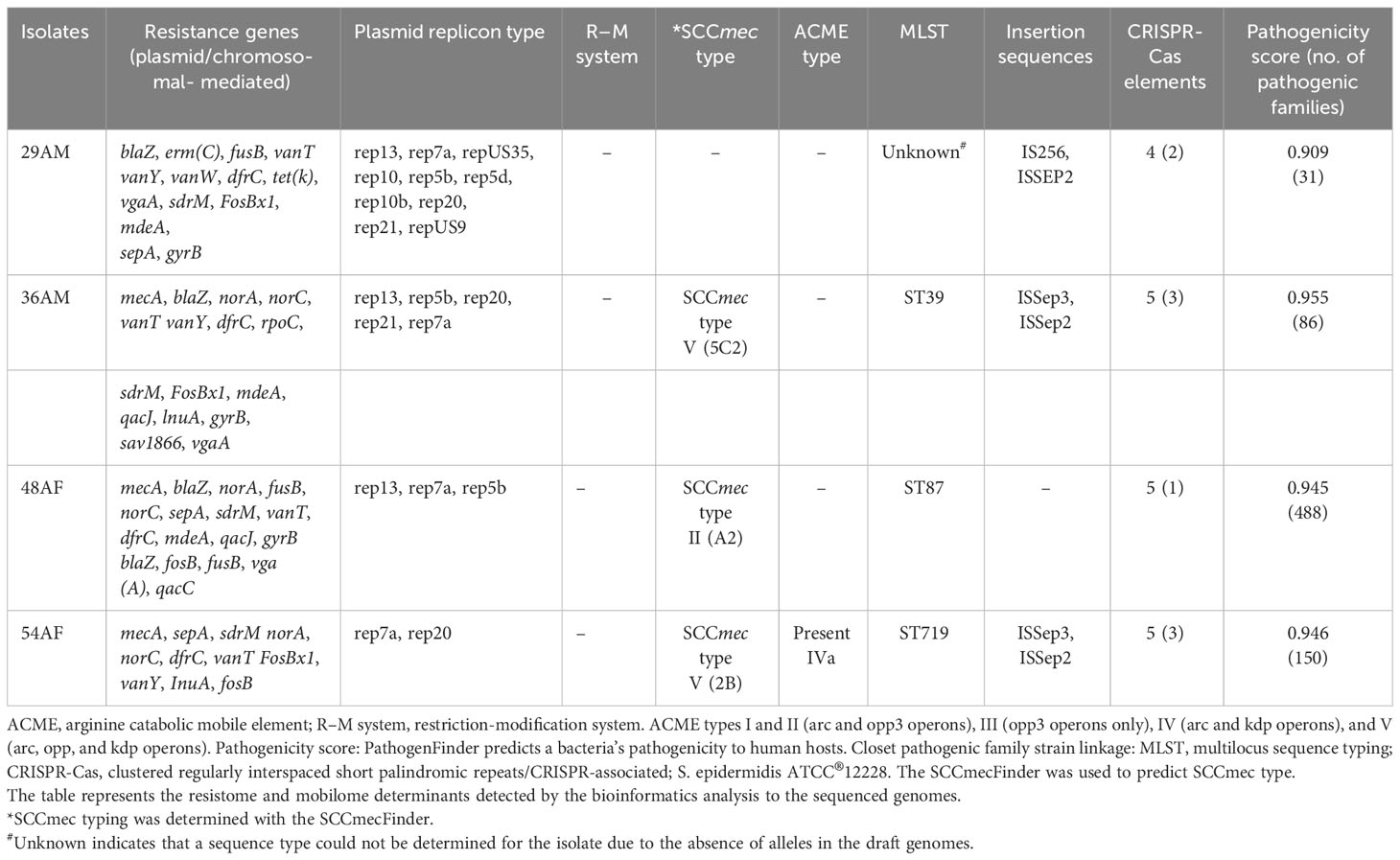
Table 3 Genotypic characteristics of Staphylococcus isolates.
The four isolates showed agreement between the cefoxitin-resistant phenotype and either mecA or blaZ gene. The tetracycline-resistance gene tet(K) was only found in isolate 29AM, although all the tested isolates were phenotypically resistant to tetracycline.
Aminoglycoside resistance mechanisms were absent in all isolates, and none of the isolates were phenotypically resistant to gentamicin or amikacin. Furthermore, the MLSB resistance mechanism erm (C) was only detected in isolate 29AM, vga (A) was detected in all isolates except 54AF, lnu (A) was also detected in 36AM and 54AF isolates, and dfrC was detected in all isolates except 36AM. Antibiotic efflux pump genes (norA, norC, sdrM, mdeA, and qacC) were detected, which can also provide fluoroquinolone and macrolide resistance.
Mutations in gyrA, gyrB, parC, pare, and rpoB in S. epidermidis isolates compared to S. epidermidis ATCC®12228 were manually curated (Table 4). MSA of the predicted amino acid sequences of the genes carried by S. epidermidis isolates and close genomes retrieved from the BV-BRC database (Supplementary Table 5) compared to that of S. epidermidis strain ATCC®12228 was performed and visualized using M.View (Supplementary Figures 1–5). Moreover, MSA of S. warneri 29AM with the closely related S. warneri strains retrieved from the BV-BRC database (Supplementary Table 6) was also performed (Supplementary Figures 6–10), and there was no standard strain of S. warneri, so the gene mutation could not be detected. The genome of isolate 36AM possessed eight mutations in gyrA, two mutations in gyrB, seven mutations in parC, and two mutations in pare, but no mutations were detected in rpoB. The gemome of isolate 48AF did not harbor any mutations. Isolate 54AF contained eight mutations in gyrA, one in gyrB, seven in parC, four in parE, and one in rpoB.
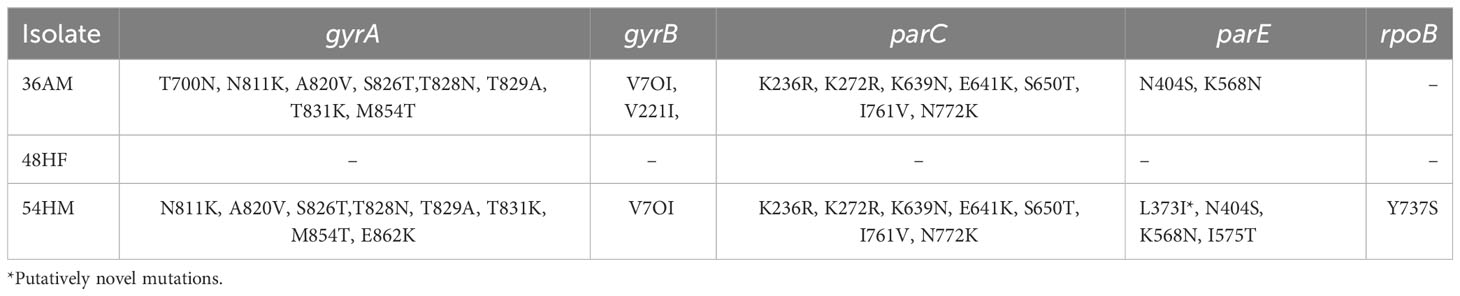
Table 4 S. epidermidis isolates mutations in gyrA, gyrB, parC, parE, and rpoB genes.
PathogenFinder estimated that the mean chance of isolates being pathogenic to humans ranged from 0.909 to 0.955 and matched numerous pathogenic families. Virulome analysis showed probable virulence genes encoding proteins from numerous S. epidermidis virulence categories, including adherence/biofilm production, enzymes, immune evasion, invasion, toxin, anti-phagocytosis, intracellular survival, and stress adaptability (Figure 4). Interestingly, the S. warneri isolate 29AM exclusively exhibited a higher number of virulence genes associated with acid resistance (ureB, ureG), anaerobic respiration (narH), antiphagocytosis (cspA), immune evasion (capB, capC, acpXL, manA), intracellular survival (lplA1), invasion (lpeA), iron uptake (fagC, hemL, vctC), lipid and fatty acid metabolism (panD), phagosome arresting (ndK), regulation (lisR, sigA/rpoV), stress adaptation (katA), and surface protein anchoring (lgt).
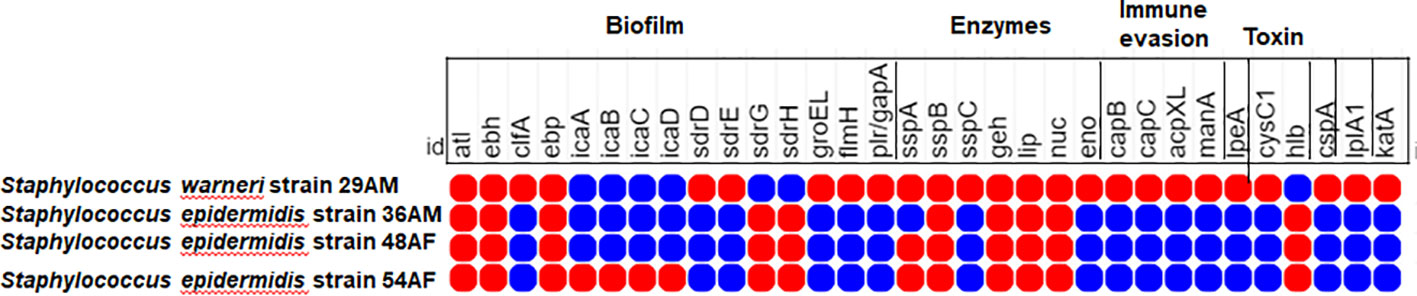
Figure 4 Heatmap showing the distribution of different virulence determinants among the sequenced isolates in the current study. The red circles denote the presence of virulence genes, whereas the lack of virulence genes is denoted by blue circles. The figure was created using the MORPHEUS online tool (https://software.broadinstitute.org/morpheus).
In silico MLST analyses identified three different MLST types: sequence type (ST) isolate 36AM: ST39, isolate 48AF: ST87, and isolate 54AF: ST719. Isolate 29AM was not identified by MLST. The most resistant isolate belonged to ST87 and contained 16 ARGs encoding for resistance to multiple antibiotic drug classes (Table 3).
In silico SCCmec typing/subtyping revealed three SCCmec types/subtypes, isolate 36AM belongs to SCCmec type V(5C2), isolate 48AF belongs to SCCmec type II (A2), and isolate 54AF belongs to SCCmec type V(2B),
PlasmidFinder and BacWGSTdb plasmid analysis showed ten distinct plasmid replicon types. Rep13 (3), rep7a (4), rep5a (3), and rep20 (3) were the most common types of plasmid replicons. Replicon types repUS35, rep10, rep5d, rep10b, rep21, repUS9 were only found in S. warneri 29AM.
ISs were detected in all tested isolates, except for isolate 48HF. Three distinct IS types from three distinct IS families were identified. IS families were IS256, 1S110, and IS200/IS605. IS256, family is well known to be associated with biofilm formation and virulence. IS256 family was found in S. warneri 29AM only. Furthermore, the resistance gene erm(C) was discovered exclusively in conjunction with transposon Tn554 in one isolate (29AM). The NCBI annotation of isolates 29AM, 36AM, and 48AF identified the presence of the blaZ gene flanked by the regulatory genes blaR1 and blaI; however, this configuration was absent in isolate 54AF. The genetic context of the specific resistance genes investigated in this study was analyzed with a focus on illustrating the interplay between Mobile Genetic Elements (MGEs), ARGs, and virulence genes (Table 5).
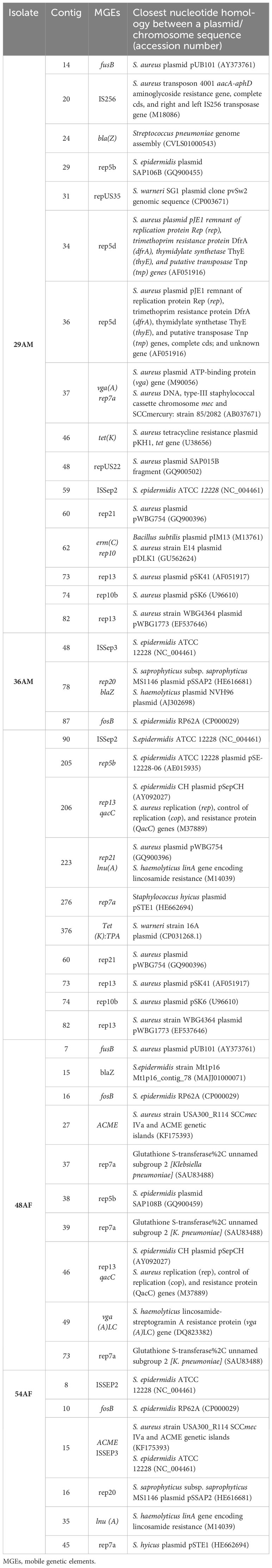
Table 5 MGEs in Staphylococcus strains linked to antibiotic resistance genes.
The PHASTER tool discovered intact prophages incorporated into the genomes of isolates 29AM and 54AF; no phages were detected in isolates 36AM and 48AF. The most common prophage was PHAGE_Staphy_StB12 (n = 4). No resistance genes were found in any of the prophages. The characteristics of the prophages, such as GC content and the number of coding sequences, are shown in Supplementary Table 4.
Repeats/CRISPR-Associated Elements, Arginine Catabolic Mobile Element (ACME), and Restriction–Modification (RM) systems. CRISPRCasFinder detects CRISPR sequences. Each isolate contained at least one CRISPR sequence. CRISPR-associated (Cas) genes were found in all isolates. None of the isolates contained an R–M system. ACME was identified in S. epidermidis isolates 48AF and 54AF and was classified as type IV (Table 3).
The phylogenetic relationships between the study isolates and their closely related genomes were determined for S. epidermidis isolates 36AM, 48AF, and 54AF core genomes and for S. warneri 29AM and were compared to the similar genomes detected by the BV-BRC database (Supplementary Tables 5, 6). A phylogenetic tree illustrating their genomic relatedness is shown in (Figures 5, 6). An SNP-based phylogenetic tree was generated using the tested genomes and S. epidermidis ATCC®12228, S. epidermidis ATCC®35984, S. epidermidis strain 785SEPI, and S. warneri strain SG1 (Figure 7).
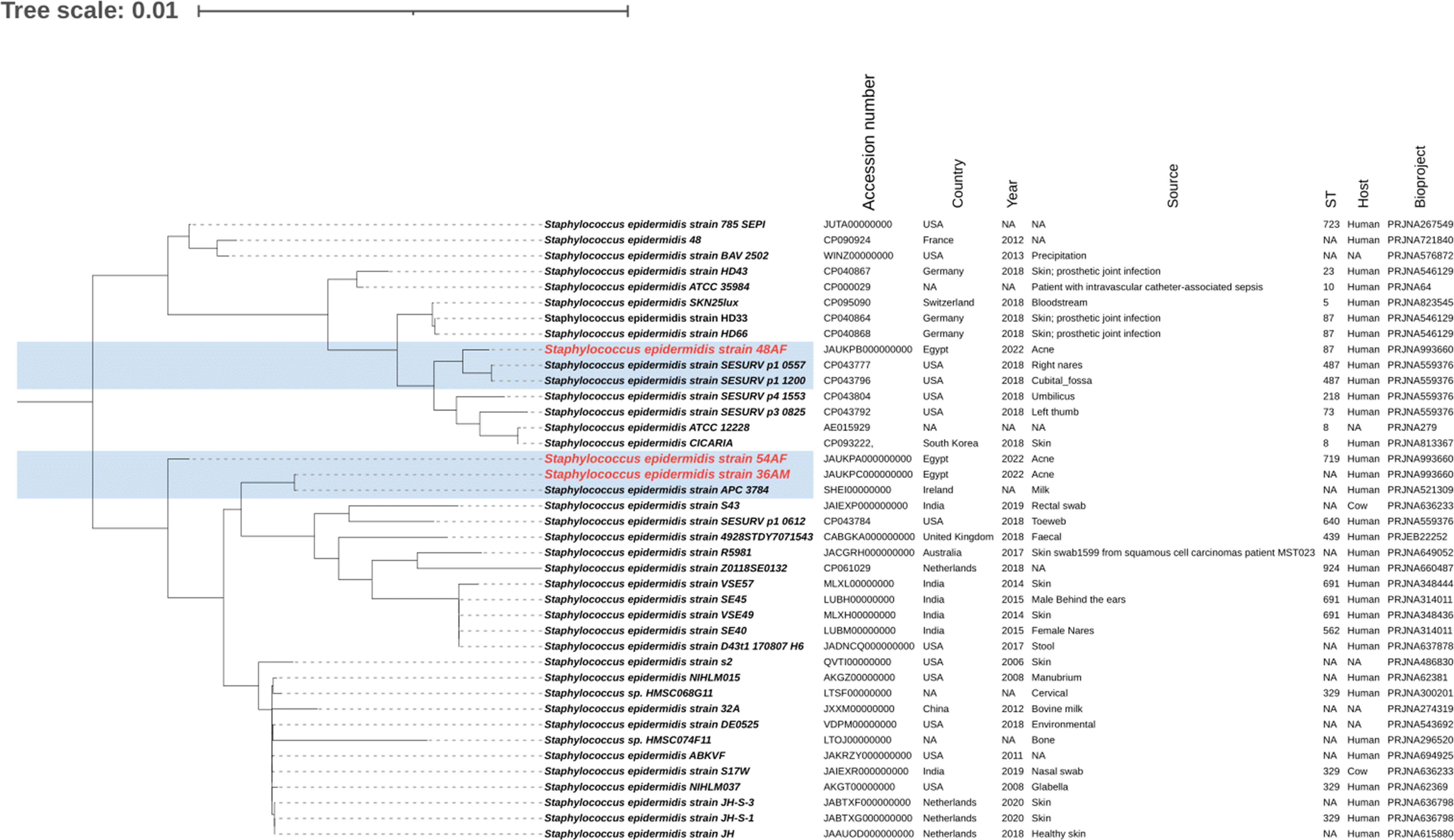
Figure 5 Whole genome-based phylogenetic tree of S. epidermidis isolates. The S. epidermidis genome sequenced in the current study was compared to the closely related genomes, highlighted in blue. The labels of the S. epidermidis strains sequenced in the current study are shown in red. The figure was created using the iTOL online tool v6.7 (https://itol.embl.de/).

Figure 6 Whole genome-based phylogenetic tree of the S. warneri isolate 29AM. The S. warneri 29AM genome sequenced in the current study was compared to the closely related genomes, highlighted in blue. The label of the S. warneri strain sequenced in this study is shown in red. The figure was created using the iTOL online tool v6.7 (https://itol.embl.de/).
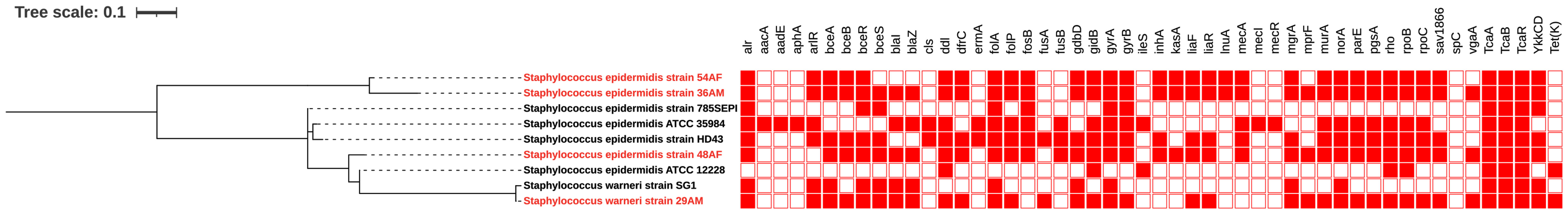
Figure 7 SNP-based phylogenetic tree of Staphylococcus isolates. SNP base phylogeny and heat map displaying the antimicrobial resistance genetic determinants among the sequenced isolates generated and selected genomes: S. epidermidis ATCC®12228, S. epidermidis ATCC®35984, S. epidermidis strain 785SEPI, and S. warneri strain SG1. Red and white indicate the presence and absence of ARGs, respectively. The labels of the strains sequenced in this study is shown in red. The figure was created using the iTOL online tool v6.7 (https://itol.embl.de/).
Acne is a common inflammatory skin disorder that affects the sebaceous glands. Although acne develops through the interaction of several factors, the exact cause of acne remains unknown. The interaction between skin bacteria and host immunity is increasingly thought to play an essential role in this condition, with variable microbial composition and activity detected in patients with acne (Lee et al., 2018).
Our research explored the microbial dynamics within acne, studied the CoNS recovered from acne, and compared them to healthy skins. While the recovery rates of CoNS isolates from acne-diseased individuals closely paralleled those from their healthy skin counterparts, a significant contrast emerged in the phenotypic characterization of antibiotic resistance and biofilm production. Antibiotic susceptibility testing revealed a landscape of antibiotic resistance patterns. Although the antibiotic resistance profiles showed no substantial differences between the two groups, except for penicillin and cefoxitin, it is noteworthy that isolates from individuals with acne displayed significantly increased biofilm production compared to those from individuals with healthy skin. In the acne group, isolates exhibited the ability to form moderate (28.3%) and strong biofilms (38.3%), in marked contrast to the healthy isolates, where only 8% demonstrated moderate biofilm formation, and none exhibited strong biofilm production. This highlights the significance of going beyond the bacterial existence to comprehend the functional distinctions that contribute to the development and pathogenesis of acne. The present study explored the biofilm-associated genes (icaA, icaD, and mecA), which were particularly prevalent in clinical isolates from participants with moderate to severe acne. Co-expression of icaA and icaD significantly augmented biofilm formation, with icaD exhibiting a higher positive detection rate. The correlation between drug resistance and biofilm formation was significant. Bacteria in biofilms exhibit increased resistance to antibiotics because of the surrounding protective matrix. This enhanced resistance may contribute to the development of MDR and XDR strains. In our study, all the strong biofilm-forming CoNS were MDR. Strong biofilm formation may serve as a mechanism by which bacteria evade the effects of antimicrobial agents, allowing them to persist and proliferate. A protective biofilm matrix can shield bacteria from the action of antibiotics, making it more difficult to eradicate the infection. Additionally, the coexistence of drug resistance and strong biofilm formation may contribute to the persistence of bacterial infections leading to chronic and recurrent conditions (Coenye et al., 2022).
Four XDR and strong biofilm-forming CoNS isolates, recovered from severe acne infections, were selected for genomic analysis. We investigated the genomic features using WGS, including MGEs, genetic contexts of the identified resistance genes, and virulence factors. Upon analyzing the genomes of the four strains, three isolates (36AM, 48AF, and 54AF) were confirmed to be S. epidermidis, whereas isolate 29AM was identified as S. warneri, a common CoNS that is regularly found in human mucous membranes and various sites, including the eye, peritoneum, wounds, and urethra. While constituting approximately 1% of skin staphylococci and prevalent in 50% of healthy adults, S. warneri is typically non-pathogenic. However, recent studies have highlighted its emergence as a potential pathogen, particularly associated with implanted materials, even in the absence of foreign bodies, and in immunocompromised individuals. This shift challenged previous perceptions of its limited pathogenicity (Otto, 2010; França, 2023). The pathophysiology and epidemiology of this species are not well understood. Furthermore, methods used to distinguish S. warneri from S. epidermidis, such as conventional culturing techniques and MALDI-TOF MS, are not sufficiently sensitive (Dubois et al., 2010; Rosa et al., 2022). S. epidermidis, a common commensal on human epithelia, displays significant genetic diversity with over 400 recognized sequence types (STs), primarily clustering in CC2. The prevalent ST2, associated with invasiveness due to IS256 insertion sequences and ica genes, dominates in clinical isolates (Méric et al., 2015). The tested genomes were investigated for their ST types, and isolates 36AM, 48AF, and 54AF were identified as ST39, ST87, and ST719, respectively. Understanding the distribution of these STs in acne isolates could be relevant for investigating the genetic diversity and potential virulence factors associated with S. epidermidis in individuals with acne. Further analysis and comparison with STs from other contexts may provide insights into the role of specific genetic variants in the pathogenesis of acne or in the skin microbiome of patients with acne.
Genomic analysis of the isolates revealed a diverse and extensive drug resistance pattern involving various mechanisms, such as enzyme inactivation, target modification, and efflux pumps. Specific resistance genes (dfrC, vanT, erm©, FOSBx1, blaZ, and norA) and mutations in key genes (gyrA, gyrB, parC, parE, and rpoB) were identified. These findings align with those of previous studies and emphasize the adaptability of CoNS in acquiring resistance. The observed genetic diversity underscores the complexity of antibiotic resistance profiles (Xu et al., 2018; Cabrera-Contreras et al., 2019; Raue et al., 2020). The detection of blaZ in the isolates, along with their regulatory genes blaR and blaI in addition to the presence of the mecA gene, may explain the resistance to penicillin and cephalosporins observed in phenotypic antibiotic susceptibility testing, as reported by a previous study by Cabrera-Contreras et al. (2019). Despite the correlation between genotypic and phenotypic resistance to penicillin and cephalosporins, potential inconsistencies highlight the importance of considering factors such as sequencing quality.
Both pathogenic and commensal S. epidermidis strains share many virulence genes (Otto, 2009). Adherence/biofilm-forming genes and multidrug resistance were found in the sequenced isolates and confirmed by the identification of adherence-related genes. The ica operon and IS256, both of which have been linked to the pathogenicity of S. epidermidis, were not detected in any of the isolates (Murugesan et al., 2018). This is comparable to the results of a study in Mexico, where 50% of S. epidermidis isolates were negative for the ica operon (Cabrera-Contreras et al., 2019).
All sequenced genomes contained several virulence genes encoding proteins associated with the pathogenesis of acne. Several genes such as atl, ebh, epb, sdrG, and sdrH are involved in adhesion and biofilm formation. Enzymes implicated in acne production, including proteases (sspA, sspB) and lipases (geh, lip, nuc), were produced by all isolates. The presence of the hlb gene encoding β-hemolysin toxin was supported by Kumar et al. (2016), suggesting a role in tissue damage and pathogen spread into deeper tissues (O’Neill et al., 2020). Similar virulence determinants were detected in many studies on S. epidermidis, including the elastin binding protein gene ebp, the serine protease gene sspA, the autolysin gene atlE, the lipase gene geh, the cell wall-associated fibronectin-binding protein gene ebh, the nuclease gene nuc, and the ica genes (Salgueiro et al., 2017; Xu et al., 2018; Raue et al., 2020).
Compared to S. epidermidis, S. warneri strain 29AM exhibited additional virulence factors related to the production of toxins, resistance to acidic conditions, evasion of the immune system, intracellular survival capabilities, invasion mechanisms, iron uptake processes, lipid metabolism modulation, phagosome arresting ability, stress adaptation strategies, and surface protein anchoring. All of these virulence factors play integral roles in shaping the course of acne development. These findings provide insights into the diverse virulence profiles contributing to acne pathophysiology, with S. warneri strain 29AM displaying enhanced virulence factors that are not commonly associated with acne pathogens.
These antibiotic resistance and virulence genes are part of the accessory genome, which is organized both within and between species. The prediction of pathogenicity of isolates towards human hosts produced a high average likelihood score (Pscore 0.937), close to 1.00. This pathogenicity score is attributed to the presence of numerous virulence genes in the isolates, which supports their pathogenic potential in humans.
MGEs play a significant role in acne pathophysiology, contributing to the genetic diversity and adaptability of bacteria involved in acne, such as S. epidermidis and S. warneri. MGEs, including SCCmec and ACME, serve as repositories for both resistance and virulence genes. Their mobility allows the transfer of genetic material within and across bacterial species through horizontal gene transfer, contributing to the exchange of traits within the bacterial population (Sheppard et al., 2016; Foster, 2017).
In the context of acne, the presence of MGEs is associated with the acquisition of virulence and resistance genes. For instance, the study identified IS256, an insertion sequence linked to virulence and biofilm development, in a strong biofilm-forming S. warneri 29AM isolate. This suggests that MGEs may enhance the genomic organization of bacteria involved in acne, potentially influencing their pathogenicity and ability to form biofilms, which is a key factor in acne development (Murugesan et al., 2018). IS256 has been shown in previous investigations to enhance the genomic organization of pathogenic S. epidermidis isolates (Cabrera-Contreras et al., 2019). The discovery of the IS families IS110 and IS200/IS605 coincides with the findings of Raue et al. (2020). Moreover, this study identified resistance genes frequently carried on plasmids, demonstrating how MGEs can facilitate the spread of antibiotic resistance. Plasmid-borne genes, such as erm©, lnu(A), tet(K), qacC, and blaZ, can be easily transferred between bacterial cells through conjugation, contributing to the dissemination of resistance traits within the bacterial population associated with acne (Cabrera-Contreras et al., 2019).
In this study, the prophages were found to be incapable of carrying resistance genes through transduction. However, transposons identified in two isolates, 36AM and 48AF, including Tn554 flanking erm (A), may facilitate the movement of beneficial genes, particularly those linked to antibiotic resistance, in the context of acne-associated bacteria (Mbelle et al., 2019).
The CRISPR-Cas system, a bacterial defense mechanism against phage infection, was identified in all isolates, providing a memory of the viral genetic code to prevent future infections. Each isolate also contained at least one intact prophage (Makarova et al., 2020). In a recent study by Raue et al. (2020), four CRISPR elements were discovered in the genome of biofilm-positive and methicillin-susceptible S. epidermidis O47 strain. The restriction–modification (R–M) system was not detected in any of the isolates in this study. The R–M system, similar to the CRISPR-Cas system, is a bacterial defense system against bacteriophage invasion (Vasu and Nagaraja, 2013). These defense systems are crucial for protecting bacteria from viral infections, contributing to our understanding of their role in the complex pathogenesis of acne.
The ACME system, recognized as a pathogenicity island, was found in two isolates (48AF and 54AF), raising interest because of its potential role in host colonization, immune evasion, and the transfer of virulence or survival genes (O’Connor et al., 2018). Studies have evaluated the resistance and virulence profiles of S. epidermidis isolates from bloodstream infections and neonatal nares, and an ACME carriage rate of 16% was found (Salgueiro et al., 2017). ACME have a greater prevalence and variety in S. epidermidis than in S. aureus. ACME is present in bacterial chromosomes adjacent to the SCCmec type IV element in S. aureus (Diep et al., 2006; Ellington et al., 2008). The present study demonstrated an association between ACME and SCCmec type IV, which is consistent with the findings of Du et al. (2013). Most resistance genes are flanked by transposases, ISs, or both, and these can transmit resistance genes within and between plasmids and chromosomes, potentially within and between bacterial strains (Mbelle et al., 2019).
These findings provide insights into the genetic factors influencing acne pathogenesis, particularly in the context of bacterial defense mechanisms and the interplay of virulence and resistance genes. The diversity of MGEs observed in the Staphylococcus genomes indicates an active process of gene exchange, suggesting that MGEs contribute to the evolution and adaptation of bacteria involved in acne pathophysiology.
This study diverges from the previous focus on pathogenic S. aureus by emphasizing the potential pathogenicity of CoNS, which are typically considered commensals. To the best of our knowledge, this is the first report in Egypt that offers insights into the diversity, distribution, virulence, and resistance profiles of S. epidermidis and S. warneri isolates recovered from acne, establishing their relatedness to global strains through WGS. Despite the limited number of isolates, our research establishes a foundational understanding for future investigations, contributing to the comprehension of the potential role of CoNS in acne vulgaris. In conclusion, this study highlights the complex nature of acne, which explains its microbial interactions, biofilm formation, and virulence factors. This comprehensive insight is vital for developing targeted and personalized therapeutic strategies and advancing the understanding of acne pathophysiology for more effective treatment approaches.
CoNS is as a significant contributor to acne development and pathogenesis by releasing virulence factors, which are more abundant in acne patients than in normal skin. The diverse combinations of ARGs, virulence genes, and MGEs found in S. epidermidis and S. warneri isolates recovered from acne highlight the enriched content of mobilized antibiotic resistance and pathogenic features. This study emphasizes the importance of tracking CoNS genomes as emerging infectious agents and shedding light on their pathogenicity. Notably, our findings suggest that S. warneri is an emerging pathogen implicated in acne pathogenesis, urging reconsideration of its role in infectious processes. This study sheds light on the complex interplay between microbial elements and acne and offers valuable insights for future research and targeted interventions in acne management.
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/Supplementary Material.
The studies involving humans were approved by MSA University-Ethics Committee. The studies were conducted in accordance with the local legislation and institutional requirements. The participants provided their written informed consent to participate in this study.
MA: Data curation, Formal analysis, Methodology, Resources, Software, Validation, Visualization, Writing – original draft, Writing – review & editing. MD: Data curation, Formal analysis, Methodology, Writing – original draft. NS: Investigation, Methodology, Project administration, Resources, Writing – original draft. HA: Conceptualization, Formal analysis, Investigation, Methodology, Project administration, Supervision, Validation, Writing – original draft, Writing – review & editing.
The author(s) declare that no financial support was received for the research, authorship, and/or publication of this article.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fcimb.2024.1328390/full#supplementary-material
Águila-Arcos, S., Álvarez-Rodríguez, I., Garaiyurrebaso, O., Garbisu, C., Grohmann, E., Alkorta, I. (2017). Biofilm-forming clinical staphylococcus isolates harbor horizontal transfer and antibiotic resistance genes. Front. Microbiol. 8. doi: 10.3389/fmicb.2017.02018
Ahmed, D. M., Messih, M., Ibrahim, N. H., Meabed, M. H., Abdel-Salam, S. M. (2019). Frequency of icaA and icaD determinants and biofilm formation among coagulase-negative staphylococci associated with nasal carriage in neonatal intensive care units. Germs 9 (2), 61–70. doi: 10.18683/germs.2019.1159
Alawad, M. J., Ali, G. A., Goravey, W. (2022). Underrecognized pathogen; Staphylococcus warneri-associated native mitral valve endocarditis in an immunocompetent host: A case report and literature review. Clin. Case Rep. 10 (4), e05591. doi: 10.1002/ccr3.5591
Alcock, B. P., Raphenya, A. R., Lau, T. T. Y., Tsang, K. K., Bouchard, M., Edalatmand, A., et al. (2020). CARD 2020: antibiotic resistome surveillance with the comprehensive antibiotic resistance database. Nucleic Acids Res. 48 (D1), D517–d525. doi: 10.1093/nar/gkz935
Altschul, S. F., Gish, W., Miller, W., Myers, E. W., Lipman, D. J. (1990). Basic local alignment search tool. J. Mol. Biol. 215 (3), 403–410. doi: 10.1016/S0022-2836(05)80360-2
Amer, M. A., Wasfi, R., Attia, A. S., Ramadan, M. A. (2021). Indole derivatives obtained from Egyptian enterobacter sp. soil isolates exhibit antivirulence activities against Uropathogen. Proteus mirabilis. Antibiotics (Basel) 10 (4), 363. doi: 10.3390/antibiotics10040363.
Arciola, C. R., Campoccia, D., Ravaioli, S., Montanaro, L. (2015). Polysaccharide intercellular adhesin in biofilm: structural and regulatory aspects. Front. Cell Infect. Microbiol. 5, 7. doi: 10.3389/fcimb.2015.00007
Arndt, D., Grant, J. R., Marcu, A., Sajed, T., Pon, A., Liang, Y., et al. (2016). PHASTER: a better, faster version of the PHAST phage search tool. Nucleic Acids Res. 44 (W1), W16–W21. doi: 10.1093/nar/gkw387
Arrecubieta, C., Lee, M. H., Macey, A., Foster, T. J., Lowy, F. D. (2007). SdrF, a Staphylococcus epidermidis surface protein, binds type I collagen. J. Biol. Chem. 282 (26), 18767–18776. doi: 10.1074/jbc.M610940200
Aydemir, E. H. (2014). Acne vulgaris. Turk. Pediatri. Ars. 49 (1), 13–16. doi: 10.5152/tpa.2014.1943
Bankevich, A., Nurk, S., Antipov, D., Gurevich, A. A., Dvorkin, M., Kulikov, A. S., et al. (2012). SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 19 (5), 455–477. doi: 10.1089/cmb.2012.0021
Becker, K., Heilmann, C., Peters, G. (2014). Coagulase-negative staphylococci. Clin. Microbiol. Rev. 27 (4), 870–926. doi: 10.1128/CMR.00109-13
Bolger, A. M., Lohse, M., Usadel, B. (2014). Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30 (15), 2114–2120. doi: 10.1093/bioinformatics/btu170
Bouchami, O., De Lencastre, H., Miragaia, M. (2016). Impact of insertion sequences and recombination on the population structure of staphylococcus haemolyticus. PLoS One 11 (6), e0156653. doi: 10.1371/journal.pone.0156653
Brüggemann, H. (2010). "Skin: Acne and Propionibacterium acnes Genomics", in Handbook of Hydrocarbon and Lipid Microbiolog. Eds. Timmis, K. N. (Berlin, Heidelberg: Springer). doi: 10.1007/978-3-540-77587-4_244
Cabrera-Contreras, R., Santamaría, R. I., Bustos, P., Martínez-Flores, I., Meléndez-Herrada, E., Morelos-Ramírez, R., et al. (2019). Genomic diversity of prevalent Staphylococcus epidermidis multidrug-resistant strains isolated from a Children’s Hospital in México City in an eight-years survey. PeerJ 7, e8068. doi: 10.7717/peerj.8068
Camacho, C., Coulouris, G., Avagyan, V., Ma, N., Papadopoulos, J., Bealer, K., et al. (2009). BLAST+: architecture and applications. BMC Bioinf. 10, 421. doi: 10.1186/1471-2105-10-421
Carattoli, A., Zankari, E., García-Fernández, A., Voldby Larsen, M., Lund, O., Villa, L., et al. (2014). In silico detection and typing of plasmids using PlasmidFinder and plasmid multilocus sequence typing. Antimicrob. Agents Chemother. 58 (7), 3895–3903. doi: 10.1128/AAC.02412-14
Chen, X.-P., Li, W.-G., Zheng, H., Du, H.-Y., Zhang, L., Zhang, L., et al. (2017). Extreme diversity and multiple SCCmec elements in coagulase-negative Staphylococcus found in the Clinic and Community in Beijing, China. Ann. Clin. Microbiol. Antimicrobials 16 (1), 57. doi: 10.1186/s12941-017-0231-z
Christensen, G. J., Brüggemann, H. (2014). Bacterial skin commensals and their role as host guardians. Benef Microbes 5 (2), 201–215. doi: 10.3920/BM2012.0062
CLSI (2022). Performance Standards for Antimicrobial Susceptibility Testing; Twenty-First Informational Supplement. CLSI Document M100-S21 (Wayne, PA: Clinical and Laboratory Standards Institute).
Coenye, T., Spittaels, K.-J., Achermann, Y. (2022). The role of biofilm formation in the pathogenesis and antimicrobial susceptibility of Cutibacterium acnes. Biofilm 4, 100063. doi: 10.1016/j.bioflm.2021.100063
Cogen, A. L., Nizet, V., Gallo, R. L. (2008). Skin microbiota: a source of disease or defence? Br. J. Dermatol. 158 (3), 442–455. doi: 10.1111/j.1365-2133.2008.08437.x
Diep, B. A., Gill, S. R., Chang, R. F., Phan, T. H., Chen, J. H., Davidson, M. G., et al. (2006). Complete genome sequence of USA300, an epidemic clone of community-acquired meticillin-resistant Staphylococcus aureus. Lancet 367 (9512), 731–739. doi: 10.1016/S0140-6736(06)68231-7
Downing, D. T., Stewart, M. E., Wertz, P. W., Strauss, J. S. (1986). Essential fatty acids and acne. J. Am. Acad. Dermatol. 14 (2 Pt 1), 221–225. doi: 10.1016/S0190-9622(86)70025-X
Du, X., Zhu, Y., Song, Y., Li, T., Luo, T., Sun, G., et al. (2013). Molecular analysis of Staphylococcus epidermidis strains isolated from community and hospital environments in China. PLoS One 8 (5), e62742. doi: 10.1371/journal.pone.0062742
Dubois, D., Leyssene, D., Chacornac, J. P., Kostrzewa, M., Schmit, P. O., Talon, R., et al. (2010). Identification of a variety of Staphylococcus species by matrix-assisted laser desorption ionization-time of flight mass spectrometry. J. Clin. Microbiol. 48 (3), 941–945. doi: 10.1128/JCM.00413-09
Ellington, M. J., Yearwood, L., Ganner, M., East, C., Kearns, A. M. (2008). Distribution of the ACME-arcA gene among methicillin-resistant Staphylococcus aureus from England and Wales. J. Antimicrob. Chemother. 61 (1), 73–77. doi: 10.1093/jac/dkm422
Fitz-Gibbon, S., Tomida, S., Chiu, B. H., Nguyen, L., Du, C., Liu, M., et al. (2013). Propionibacterium acnes strain populations in the human skin microbiome associated with acne. J. Invest. Dermatol. 133 (9), 2152–2160. doi: 10.1038/jid.2013.21
Formosa-Dague, C., Feuillie, C., Beaussart, A., Derclaye, S., Kucharíková, S., Lasa, I., et al. (2016). Sticky matrix: adhesion mechanism of the staphylococcal polysaccharide intercellular adhesin. ACS Nano 10 (3), 3443–3452. doi: 10.1021/acsnano.5b07515
Foster, T. J. (2017). Antibiotic resistance in Staphylococcus aureus. Current status and future prospects. FEMS Microbiol. Rev. 41 (3), 430–449. doi: 10.1093/femsre/fux007
Fournière, M., Latire, T., Souak, D., Feuilloley, M. G. J., Bedoux, G. (2020). Staphylococcus epidermidis and Cutibacterium acnes: Two Major Sentinels of Skin Microbiota and the Influence of Cosmetics. Microorganisms 8 (11), 1752. doi: 10.3390/microorganisms8111752
França, A. (2023). The role of coagulase-negative staphylococci biofilms on late-onset sepsis: current challenges and emerging diagnostics and therapies. Antibiotics 12 (3), 554. doi: 10.3390/antibiotics12030554
Freney, J., Kloos, W. E., Hajek, V., Webster, J. A., Bes, M., Brun, Y., et al. (1999). Recommended minimal standards for description of new staphylococcal species. Subcommittee on the taxonomy of staphylococci and streptococci of the International Committee on Systematic Bacteriology. Int. J. Syst. Bacteriol. 49 Pt 2, 489–502. doi: 10.1099/00207713-49-2-489
Huang, T. Y., Jiang, Y. E., Scott, D. A. (2022). Culturable bacteria in the entire acne lesion and short-chain fatty acid metabolites of Cutibacterium acnes and Staphylococcus epidermidis isolates. Biochem. Biophys. Res. Commun. 622, 45–49. doi: 10.1016/j.bbrc.2022.06.068
Jahns, A. C., Lundskog, B., Ganceviciene, R., Palmer, R. H., Golovleva, I., Zouboulis, C. C., et al. (2012). An increased incidence of Propionibacterium acnes biofilms in acne vulgaris: a case-control study. Br. J. Dermatol. 167 (1), 50–58. doi: 10.1111/j.1365-2133.2012.10897.x
Joensen, K. G., Scheutz, F., Lund, O., Hasman, H., Kaas, R. S., Nielsen, E. M., et al. (2014). Real-time whole-genome sequencing for routine typing, surveillance, and outbreak detection of verotoxigenic Escherichia coli. J. Clin. Microbiol. 52 (5), 1501–1510. doi: 10.1128/JCM.03617-13
Johansson, M. H. K., Bortolaia, V., Tansirichaiya, S., Aarestrup, F. M., Roberts, A. P., Petersen, T. N. (2021). Detection of mobile genetic elements associated with antibiotic resistance in Salmonella enterica using a newly developed web tool: MobileElementFinder. J. Antimicrob. Chemother. 76 (1), 101–109. doi: 10.1093/jac/dkaa390
Kaas, R. S., Leekitcharoenphon, P., Aarestrup, F. M., Lund, O. (2014). Solving the problem of comparing whole bacterial genomes across different sequencing platforms. PLoS One 9 (8), e104984. doi: 10.1371/journal.pone.0104984
Kumar, B., Pathak, R., Mary, P. B., Jha, D., Sardana, K., Gautam, H. K. (2016). New insights into acne pathogenesis: Exploring the role of acne-associated microbial populations. Dermatol. Sin. 34 (2), 67–73. doi: 10.1016/j.dsi.2015.12.004
Kutlu, Ö., Karadağ, A. S., Wollina, U. (2023). Adult acne versus adolescent acne: a narrative review with a focus on epidemiology to treatment. Anais Brasileiros Dermatol. 98 (1), 75–83. doi: 10.1016/j.abd.2022.01.006
Larsen, M. V., Cosentino, S., Rasmussen, S., Friis, C., Hasman, H., Marvig, R. L., et al. (2012). Multilocus sequence typing of total-genome-sequenced bacteria. J. Clin. Microbiol. 50 (4), 1355–1361. doi: 10.1128/JCM.06094-11
Lee, J. Y. H., Monk, I. R., Gonçalves Da Silva, A., Seemann, T., Chua, K. Y. L., Kearns, A., et al. (2018). Global spread of three multidrug-resistant lineages of Staphylococcus epidermidis. Nat. Microbiol. 3 (10), 1175–1185. doi: 10.1038/s41564-018-0230-7
Li, H., Durbin, R. (2010). Fast and accurate long-read alignment with Burrows-Wheeler transform. Bioinformatics 26 (5), 589–595. doi: 10.1093/bioinformatics/btp698
Lucero-Mejía, J. E., Romero-Gómez, S. D. J., Hernández-Iturriaga, M. (2020). A new classification criterion for the biofilm formation index: A study of the biofilm dynamics of pathogenic Vibrio species isolated from seafood and food contact surfaces. J. Food Sci. 85 (8), 2491–2497. doi: 10.1111/1750-3841.15325
Makarova, K. S., Wolf, Y. I., Iranzo, J., Shmakov, S. A., Alkhnbashi, O. S., Brouns, S. J. J., et al. (2020). Evolutionary classification of CRISPR-Cas systems: a burst of class 2 and derived variants. Nat. Rev. Microbiol. 18 (2), 67–83. doi: 10.1038/s41579-019-0299-x
Månsson, E., Hellmark, B., Sundqvist, M., Söderquist, B. (2015). Sequence types of Staphylococcus epidermidis associated with prosthetic joint infections are not present in the laminar airflow during prosthetic joint surgery. Apmis 123 (7), 589–595. doi: 10.1111/apm.12392
Mbelle, N. M., Feldman, C., Osei Sekyere, J., Maningi, N. E., Modipane, L., Essack, S. Y. (2019). The resistome, mobilome, virulome and phylogenomics of multidrug-resistant escherichia coli clinical isolates from pretoria, South Africa. Sci. Rep. 9 (1), 16457. doi: 10.1038/s41598-019-52859-2
Méric, G., Miragaia, M., De Been, M., Yahara, K., Pascoe, B., Mageiros, L., et al. (2015). Ecological Overlap and Horizontal Gene Transfer in Staphylococcus aureus and Staphylococcus epidermidis. Genome Biol. Evol. 7 (5), 1313–1328. doi: 10.1093/gbe/evv066
Murugesan, S., Mani, S., Kuppusamy, I., Krishnan, P. (2018). Role of insertion sequence element is256 as a virulence marker and its association with biofilm formation among methicillin-resistant Staphylococcus epidermidis from hospital and community settings in Chennai, South India. Indian J. Med. Microbiol. 36 (1), 124–126. doi: 10.4103/ijmm.IJMM_17_276
O’Connor, A. M., Mcmanus, B. A., Kinnevey, P. M., Brennan, G. I., Fleming, T. E., Cashin, P. J., et al. (2018). Significant enrichment and diversity of the staphylococcal arginine catabolic mobile element ACME in staphylococcus epidermidis isolates from subgingival peri-implantitis sites and periodontal pockets. Front. Microbiol. 9, 1558. doi: 10.3389/fmicb.2018.01558
Olson, R. D., Assaf, R., Brettin, T., Conrad, N., Cucinell, C., Davis, J. J., et al. (2023). Introducing the Bacterial and Viral Bioinformatics Resource Center (BV-BRC): a resource combining PATRIC, IRD and ViPR. Nucleic Acids Res 51 (D1), D678–D689. doi: 10.1093/nar/gkac1003
O’Neill, A., Gallo, R. (2018). Host-microbiome interactions and recent progress into understanding the biology of acne vulgaris. Microbiome 6 (1), 177. doi: 10.1186/s40168-018-0558-5
O’Neill, A. M., Nakatsuji, T., Hayachi, A., Williams, M. R., Mills, R. H., Gonzalez, D. J., et al. (2020). Identification of a human skin commensal bacterium that selectively kills cutibacterium acnes. J. Invest. Dermatol. 140 (8), 1619–1628.e1612. doi: 10.1016/j.jid.2019.12.026
Onishi, M., Urushibara, N., Kawaguchiya, M., Ghosh, S., Shinagawa, M., Watanabe, N., et al. (2013). Prevalence and genetic diversity of arginine catabolic mobile element (ACME) in clinical isolates of coagulase-negative staphylococci: identification of ACME type I variants in Staphylococcus epidermidis. Infect. Genet. Evol. 20, 381–388. doi: 10.1016/j.meegid.2013.09.018
Otto, M. (2009). Staphylococcus epidermidis–the A’ccidental’ pathogen. Nat. Rev. Microbiol. 7 (8), 555–567. doi: 10.1038/nrmicro2182
Otto, M. (2010). Staphylococcus colonization of the skin and antimicrobial peptides. Expert Rev. Dermatol. 5 (2), 183–195. doi: 10.1586/edm.10.6
Paluch, E., Rewak-Soroczyńska, J., Jędrusik, I., Mazurkiewicz, E., Jermakow, K. (2020). Prevention of biofilm formation by quorum quenching. Appl. Microbiol. Biotechnol. 104 (5), 1871–1881. doi: 10.1007/s00253-020-10349-w
Petrelli, D., Zampaloni, C., D’ercole, S., Prenna, M., Ballarini, P., Ripa, S., et al. (2006). Analysis of different genetic traits and their association with biofilm formation in Staphylococcus epidermidis isolates from central venous catheter infections. Eur. J. Clin. Microbiol. Infect. Dis. 25 (12), 773–781. doi: 10.1007/s10096-006-0226-8
Planet, P. J., Larussa, S. J., Dana, A., Smith, H., Xu, A., Ryan, C., et al. (2013). Emergence of the epidemic methicillin-resistant Staphylococcus aureus strain USA300 coincides with horizontal transfer of the arginine catabolic mobile element and speG-mediated adaptations for survival on skin. mBio 4 (6), e00889–e00813. doi: 10.1128/mBio.00889-13
Raue, S., Fan, S. H., Rosenstein, R., Zabel, S., Luqman, A., Nieselt, K., et al. (2020). The genome of staphylococcus epidermidis O47. Front. Microbiol. 11, 2061. doi: 10.3389/fmicb.2020.02061
Rolo, J., Worning, P., Nielsen, J. B., Bowden, R., Bouchami, O., Damborg, P., et al. (2017). Evolutionary origin of the staphylococcal cassette chromosome mec (SCCmec). Antimicrob. Agents Chemother. 61 (6), e02302-16. doi: 10.1128/AAC.02302-16
Rosa, N. M., Penati, M., Fusar-Poli, S., Addis, M. F., Tola, S. (2022). Species identification by MALDI-TOF MS and gap PCR-RFLP of non-aureus Staphylococcus, Mammaliicoccus, and Streptococcus spp. associated with sheep and goat mastitis. Vet. Res. 53 (1), 84. doi: 10.1186/s13567-022-01102-4
Salgueiro, V. C., Iorio, N. L., Ferreira, M. C., Chamon, R. C., Dos Santos, K. R. (2017). Methicillin resistance and virulence genes in invasive and nasal Staphylococcus epidermidis isolates from neonates. BMC Microbiol. 17 (1), 15. doi: 10.1186/s12866-017-0930-9
Schaeffer, C. R., Hoang, T.-M. N., Sudbeck, C. M., Alawi, M., Tolo, I. E., Robinson, D. A., et al. (2016). Versatility of Biofilm Matrix Molecules in Staphylococcus epidermidis Clinical Isolates and Importance of Polysaccharide Intercellular Adhesin Expression during High Shear Stress. mSphere 1 (5), e00165–e00116. doi: 10.1128/mSphere.00165-16
Sheppard, A. E., Stoesser, N., Wilson, D. J., Sebra, R., Kasarskis, A., Anson, L. W., et al. (2016). Nested Russian doll-like genetic mobility drives rapid dissemination of the carbapenem resistance gene blaKPC. Antimicrob. Agents Chemother. 60 (6), 3767–3778. doi: 10.1128/AAC.00464-16
Thomas, J. C., Vargas, M. R., Miragaia, M., Peacock, S. J., Archer, G. L., Enright, M. C. (2007). Improved multilocus sequence typing scheme for Staphylococcus epidermidis. J. Clin. Microbiol. 45 (2), 616–619. doi: 10.1128/JCM.01934-06
Vasu, K., Nagaraja, V. (2013). Diverse functions of restriction-modification systems in addition to cellular defense. Microbiol. Mol. Biol. Rev. 77 (1), 53–72. doi: 10.1128/MMBR.00044-12
Xu, Z., Misra, R., Jamrozy, D., Paterson, G. K., Cutler, R. R., Holmes, M. A., et al. (2018). Whole genome sequence and comparative genomics analysis of multi-drug resistant environmental staphylococcus epidermidis ST59. G3 (Bethesda) 8 (7), 2225–2230. doi: 10.1534/g3.118.200314
Yang, Y., Qu, L., Mijakovic, I., Wei, Y. (2022). Advances in the human skin microbiota and its roles in cutaneous diseases. Microbial. Cell Factories 21 (1), 176. doi: 10.1186/s12934-022-01901-6
Keywords: CoNS, acne, antibiotic resistance, virulence, genome analysis, mobilizable genetic elements, Staphylococcus epidermidis, Staphylococcus warneri
Citation: Amer MA, Darwish MM, Soliman NS and Amin HM (2024) Resistome, mobilome, and virulome explored in clinical isolates derived from acne patients in Egypt: unveiling unique traits of an emerging coagulase-negative Staphylococcus pathogen. Front. Cell. Infect. Microbiol. 14:1328390. doi: 10.3389/fcimb.2024.1328390
Received: 26 October 2023; Accepted: 10 January 2024;
Published: 02 February 2024.
Edited by:
Manuel Gerardo Ballesteros Monrreal, University of Sonora, MexicoReviewed by:
Yosainix Gaerste-Diaz, Cuauhtémoc University, MexicoCopyright © 2024 Amer, Darwish, Soliman and Amin. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Heba M. Amin, aHRtYWdkeUBtc2EuZWR1LmVn
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.