 Deborah Cook
Deborah Cook Mollee D. Flannigan1
Mollee D. Flannigan1 Julia H. Chariker
Julia H. Chariker Janelle M. Hare
Janelle M. Hare
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Cell. Infect. Microbiol. , 11 January 2024
Sec. Molecular Bacterial Pathogenesis
Volume 13 - 2023 | https://doi.org/10.3389/fcimb.2023.1324091
Introduction: Acinetobacter baumannii strain 17978 is an opportunistic pathogen possessing a DNA damage response (DDR) in which multiple error-prone polymerase genes are co-repressed by a UmuD homolog, UmuDAb, and the small Acinetobacter-specific protein DdrR. Additionally, these regulators coactivate nine other genes. We identified the DNA damage-inducible transcriptome for wildtype, umuDAb, and recA strains, and later established the ddrR DDR transcriptome. However, the ATCC 17978 reference genome had several assembly errors and lacked the 44 kb virulence locus, AbaAL44, that is present in the strain 17978 UN.
Methods: For this project, we combined our earlier single-end read RNAseq data with the ddrR paired-end reads and aligned these data to the improved 17978 UN genome assembly that resembled our laboratory strain, 17978 JH.
Results: New DESeq2 analyses verified previous differentially expressed genes (DEGs) but also found 339 genes in 17978 JH that were not annotated or physically present in the older genome assembly. Sixty-three were differentially expressed after DNA damage, and 182 had differential basal expression when comparing umuDAb, ddrR, or recA strains to wildtype, with 94 genes’ expression unchanged. This work identified and characterized the 55 gene DNA damage-repressible transcriptome, 98% of which required either umuDAb or ddrR for repression. Two-thirds of these DEGs required both regulators. We also identified 110 genes repressed only in the ddrR strain, ~50% of which were due to increased basal expression levels. Basal gene expression in the ddrR mutant was further dysregulated independent of the DDR. Over 800 genes were upregulated, and over 1200 genes were downregulated compared to wildtype expression. Half of A. baumannii’s essential genes were upregulated in the ddrR strain, including cell division genes, and two-thirds of these were downregulated in the umuDAb strain.
Discussion: The ddrR mutant upregulated genes enriched in translation, RNA metabolism, protein metabolism, AA/FA/cell-structure synthesis, and transport, while downregulating genes enriched in quorum sensing, biofilm production, secretion systems, pilus production, cell adhesion, and aromatics and chlorine degradation. Our data underscore the need for accurate and appropriately matched genome assemblies and indicate that ddrR affects approximately 60% of the genome, rendering it a potential target for Acinetobacter baumannii infection treatment.
Acinetobacter baumannii is a non-enteric, gram-negative opportunistic pathogen that commonly causes nosocomial infections and is challenging to treat due to many strains’ multiple antibiotic resistances (Peleg et al., 2008; Antunes et al., 2011; Visca et al., 2011). The emergence of carbapenem-resistant strains has prompted the World Health Organization to name A. baumannii as a top-priority critical pathogen (Tacconelli et al., 2018).
Several Acinetobacter species and strains are models for studying virulence-related processes such as antibiotic resistance, the DNA damage response (DDR), desiccation tolerance, and metabolism. Among the hundreds of A. baumannii strains that are currently studied, one of the oldest and most commonly used is A. baumannii ATCC 17978, isolated in 1951 from an infant with fatal meningitis and susceptible to most antibiotics (Smith et al., 2007). Antibiotic resistance in this pathogen is usually due to the gain or loss of genetic material, via plasmids or horizontal gene transfer (Dobrindt and Hacker, 2001; Antunes et al., 2014), but the DDR is mutagenic and can cause antibiotic resistance (Norton et al., 2013; Aranda et al., 2014; Durante-Mangoni et al., 2015; Zhu et al., 2019). The A. baumannii DDR that induces ~150 genes after DNA damage (Aranda et al., 2013; Hare et al., 2014) is unusual in not being regulated by LexA, as the genus does not encode a LexA homolog (Hare et al., 2012). Several features of this system and its coregulators UmuDAb and DdrR have been investigated in the non-pathogenic A. baylyi ADP1 and A. baumannii (Robinson et al., 2010; Hare et al., 2012; Aranda et al., 2013; Norton et al., 2013). Both species induce umuDAb and ddrR as part of their DDR (Hare et al., 2014). The products of this divergently transcribed gene cluster (Peterson et al., 2020) jointly repress the six plasmid- or prophage-based umuD and umuC genes (Hare et al., 2014) that have been acquired through horizontal gene transfer (Di Nocera et al., 2011; Hare et al., 2012). These umuDC genes encode subunits of the error-prone polymerase V (UmuD’2C) that conducts error-prone DNA repair and thus are under tight regulation. Polymerase V repair can lead to mutations in genes whose gain of function could potentially increase antibiotic resistance (Norton et al., 2013).
UmuDAb and DdrR constitute an unusual coregulatory system for DDR genes. ddrR encodes a ~9 kD protein that is unique to the Acinetobacter genus. When we investigated whether umuDAb and ddrR regulated all DNA damage-inducible genes in Acinetobacter, we conducted RNAseq analyses to identify the DDR-induced UmuDAb transcriptome in A. baylyi and A. baumannii (Hare et al., 2014), later extending the analysis to the DdrR transcriptome (Peterson et al., 2020). RNAseq is a cost-effective way to study the transcriptome of organisms, supplementing and often replacing microarray analysis. However, this powerful technique relies on an accurately sequenced and assembled genome to align cDNA reads. Unfortunately, the first and most used reference genome for ATCC 17978 (GCA_000015425.1) contains several errors introduced through bases lost during sequencing or by incorrectly combining contigs, notably the chromosome and one of the three plasmids, pAB3 (Smith et al., 2007; Weber et al., 2015). For example, the lon gene was annotated as two open reading frames (A1S_1031 and A1S_1030), because a base was missed in a poly A or T region during sequencing (Ching et al., 2018). After sequencing our lab strain 17978 JH, we observed that the gene transcribed convergently with ddrR (A1S_3662; dtpA (Green et al., 2022) contained a 400 bp high-GC repeat region that was missing from the ATCC 17978 assembly (D. Cook, personal communication). That apparent assembly mistake caused a frameshift, so dtpA appears to extend through both genes’ observed transcription termination sites (Kröger et al., 2018). A recent A. baumannii study confirms that this region with the full-length dtpA gene, is conserved among A. baumannii strains AB5075, 19606, and 17978-mff (Green et al., 2022).
Several documented genomic differences between different laboratories’ ATCC 17978 strains have also complicated past studies. In 2018, Kröger et al. investigated the transcriptome of 17978 using the University of Alberta’s A. baumannii 17978-mff assembly, which contained the pAB3 plasmid, as their reference genome. However, the Kröger lab strain had lost the large plasmid pAB3 during transit and their transcriptome data only represented the chromosome. In another example, a 44 kb virulence locus, AbaAL44 (Acinetobacter baumannii accessory locus 44 kb), was identified as both present and absent in an admixture found in two different laboratory A. baumannii 17978 strains purchased from the American Type Culture Collection (ATCC) (Wijers et al., 2021). The AbaAL44 locus is present in sequenced strains 17978-mff, 17978 UN, and our strain 17978 JH but is absent from the original ATCC 17978 and 17978 VU strains, probably due to the locus’ loss during laboratory propagation (Wijers et al., 2021). Similar genomic variations between different laboratories’ cultures have been seen in A. baumannii 19606 (Artuso et al., 2022). These examples show how variable and constantly changing the A. baumannii genome is: only 14.8% to 16.5% of the genome is shared among 12 (Imperi et al., 2011) or 47 (Philippe et al., 2022) isolates, respectively.
Our RNAseq study of the 17978 JH DNA damage transcriptome used the ATCC 17978 reference genome for read alignments and CuffDiff analysis to find differentially expressed genes (DEGs) (Hare et al., 2014; Peterson et al., 2020). However, due to assembly errors and the absence of AbaAL44 from the reference genome, our analysis was incomplete and less accurate than desired. Fortunately, technological advancements have improved the quality and accuracy of genome assembly. This project’s goals were to: (i) extend our earlier investigation of the induced DDR to obtain the repressed DDR and its regulation by ddrR and umuDAb, and (ii) improve our gene discovery methods. We therefore aligned the RNAseq data to the more accurate 17978 UN genome assembly that matches our lab strain 17978 JH and updated our analysis pipeline to use DESeq2. That Bioconductor package uses a statistical model to estimate the mean and variance of the count data, which controls differences in sequencing parameters between samples (such as single or paired-end reads). This allowed the single-read data for 17978 JH, the umuDAb mutant, and the recA mutant (Hare et al., 2014) to be combined with and directly compared to paired-read data from the ddrR mutant (Peterson et al., 2020).
With these improved methods, we verified previously reported DDR genes and found new ones that had not been annotated or were absent from the ATCC 17978 genome assembly. We also compared basal expression levels and found genes and pathways whose expression was affected by mutating the regulators umuDAb, ddrR, and recA, independent of DNA damage treatment. This analysis showed that ddrR plays a much more significant role than simply co-regulating error-prone polymerases and the ddrR-umuDAb overlapping promoters. ddrR affects the expression of genes for quorum sensing, efflux pumps, pilus production, the TCA cycle, RNA transcription and translation, and other metabolic processes. This means it could be an effective therapeutic target to aid in treating Acinetobacter baumannii infections.
RNAseq reads from previously published work were used (Hare et al., 2014, Peterson et al., 2020). In those projects, the RNAseq analysis was to find DEGs after the cells experienced DNA damage caused by mitomycin C (MMC). The first RNAseq dataset included samples from three Acinetobacter strains: wildtype ATCC 17978 JH, and two single-mutant strains 17978 JH recA::Km (Aranda et al., 2011), and 17978 JH ΔumuDAb::Km (JH1600; (Hare et al., 2014)).
Sequencing reads were collected as 160 bp unpaired cDNA reads (Hare et al., 2014). The read clusters were normalized by multiplying each sample’s coverage by the total reads of the lower read-count sample divided by the respective sample’s total reads, and the induction ratio of reads was calculated between MMC-treated and untreated samples. The gene was induced if the induction ratio was greater than or equal to 2.0 and repressed if it was less than 0.5. No p-values or false discovery rate values were used (Hare et al., 2014). The second study consisted of a new RNAseq data set for a 17978 ddrR::TnLK mutant strain (JH1700; (Peterson et al., 2020), with RNA processed as 75 bp paired-end cDNA reads. These data were normalized and analyzed separately from the data from the earlier study (Hare et al., 2014). Gene expression levels were calculated by FPKM (fragments per kilobase of transcript per million fragments mapped reads), then tested for differential gene expression with CuffDiff with a false discovery rate (FDR) threshold of q < 0.01 for genes that were repressed or induced greater than 2-fold according to FPKM (Peterson et al., 2020). Samples for both studies were collected by the same person, in the same make of test tubes, in the same shaking incubator in the same lab, in the same medium and volume at the same time, temperature, and aeration conditions, and RNA prepared with the same purification kit.
For this project, we first compared DEGs repressed after MMC treatment in the wildtype 17978 JH strain (WT) using data aligned to both the newer 17978 UN assembly GCA_019356215.1 (Wijers et al., 2021) and the earlier ATCC 17978 reference genome GCA_000015425.1 (for validation purposes). We used the final counts files, prepared by the KY INBRE Bioinformatics core, with the reads aligned to each genome using STAR aligner (Dobin et al., 2013). DESeq2 analysis was performed from the raw read counts. DESeq2 normalizes RNAseq experiment count data and finds DEGs between samples based on the negative binomial distribution (Love et al., 2014). For both MMC-treated and basal expression analyses, genes were considered differentially expressed if they had an adjusted p-value (q value) of < 0.05. The log2 fold change filter for values 1 < FC < -1 was not used in this analysis, to identify more biologically relevant differences in genes with low expression. Outliers were calculated in DESeq2 using Cook’s cutoff. Expression differences between WT and mutant strains were verified with RT-qPCR for several genes as performed previously (Hare et al., 2014; Peterson et al., 2020) and with new primers for additional genes (See Supplementary Table S1).
Pathway and gene enrichment analyses and images were created using the 17978 UN genome on the BioCyc website with our DESeq2 results overlaid in Cellular Overview (Paley and Karp, 2006) and enrichment scores calculated from the same data in the Omics Dashboard using padj < 0.05 as the threshold (Paley et al., 2017).
We previously analyzed and published the ddrR transcriptome separately from the WT, umuDAb mutant, and recA mutant transcriptomes (Hare et al., 2014; Peterson et al., 2020). In this project, we tested whether combining the ddrR paired-end reads with the single reads of the other three strains into a single data set would alter the DESeq2 results for induced WT genes after MMC treatment. DESeq2 was applied both to the original counts file that contained only the reads for WT and the umuDAb and recA mutants, and to a counts file containing these strains’ data and those of the ddrR mutant. The combined mean for each gene was different due to the broader range of values in the ddrR data, but the overall results were very similar. Virtually all induced DEGs were the same in both analyses (170/177), with only two genes unique to the unmerged analysis and five genes that only appeared in the merged data set (Supplementary Table S2). All the results presented in this manuscript are from DESeq2 analysis of the merged data counts files.
After alignment and DESeq2 analysis of the entire data set, induced or repressed DEGs were identified for each strain after MMC treatment. The DESeq2 summary results from each genome alignment and strain are in Table 1, and the complete data set is in Supplementary Table S3. The new (17978 UN) genome alignment found 339 genes in 17978 JH that were not annotated or physically present in the older (ATCC 17978) reference genome, 43 of which were differentially expressed after MMC treatment and 182 (181 in the ddrR mutant alone) that had differential basal expression compared untreated WT. Supplementary Table S3 only includes the WT 17978 JH strain and the umuDAb and ddrR mutants because no genes were induced or repressed after MMC treatment in the recA mutant.

Table 1 Summary data for genes differentially expressed after MMC treatment.
The WT DNA damage response resulted in 175 induced genes and 55 repressed genes (Figures 1A, 2B). Interestingly, there were even more genes induced (238) and repressed (131) in the ddrR mutant (Figure 1B), including many of the same genes induced as WT (Figure 2). Lastly, in the umuDAb mutant, DNA damage resulted in 223 genes induced and 10 repressed (Figures 1C, 2). Most of the induced genes reported here were previously published but used different analysis pipelines, fold change threshold, and genome assembly (Hare et al., 2014; Peterson et al., 2020) than this project. Additional findings from this study’s analysis are discussed in each pertinent section below.
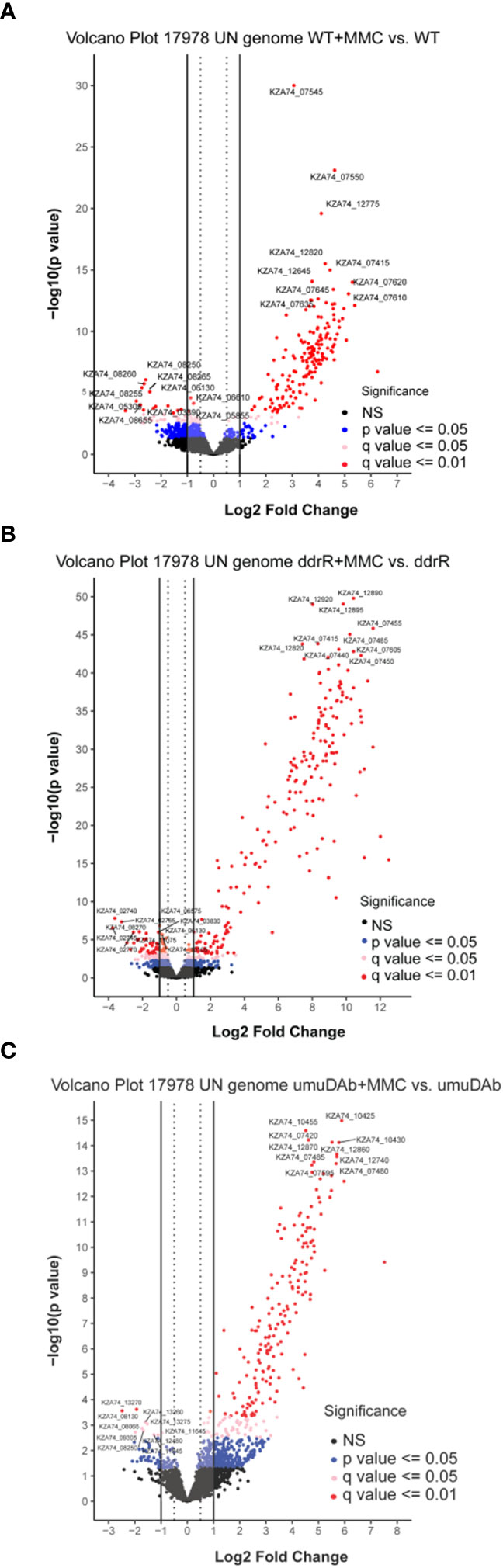
Figure 1 Global transcriptomic analysis of differentially expressed genes after MMC treatment for (A) wildtype (WT), (B) ddrR, and (C) umuDAb mutant strains of A. baumannii 17978 JH. Volcano plots show the log2 fold changes in gene expression where each dot represents one gene. Red and pink dots indicate significance at the q < 0.01 or q < 0.05 level. The log2 fold change is plotted on the x-axis, and the y-axis shows the log10 of the p value. The 10 genes with the greatest induction or repression are labelled.
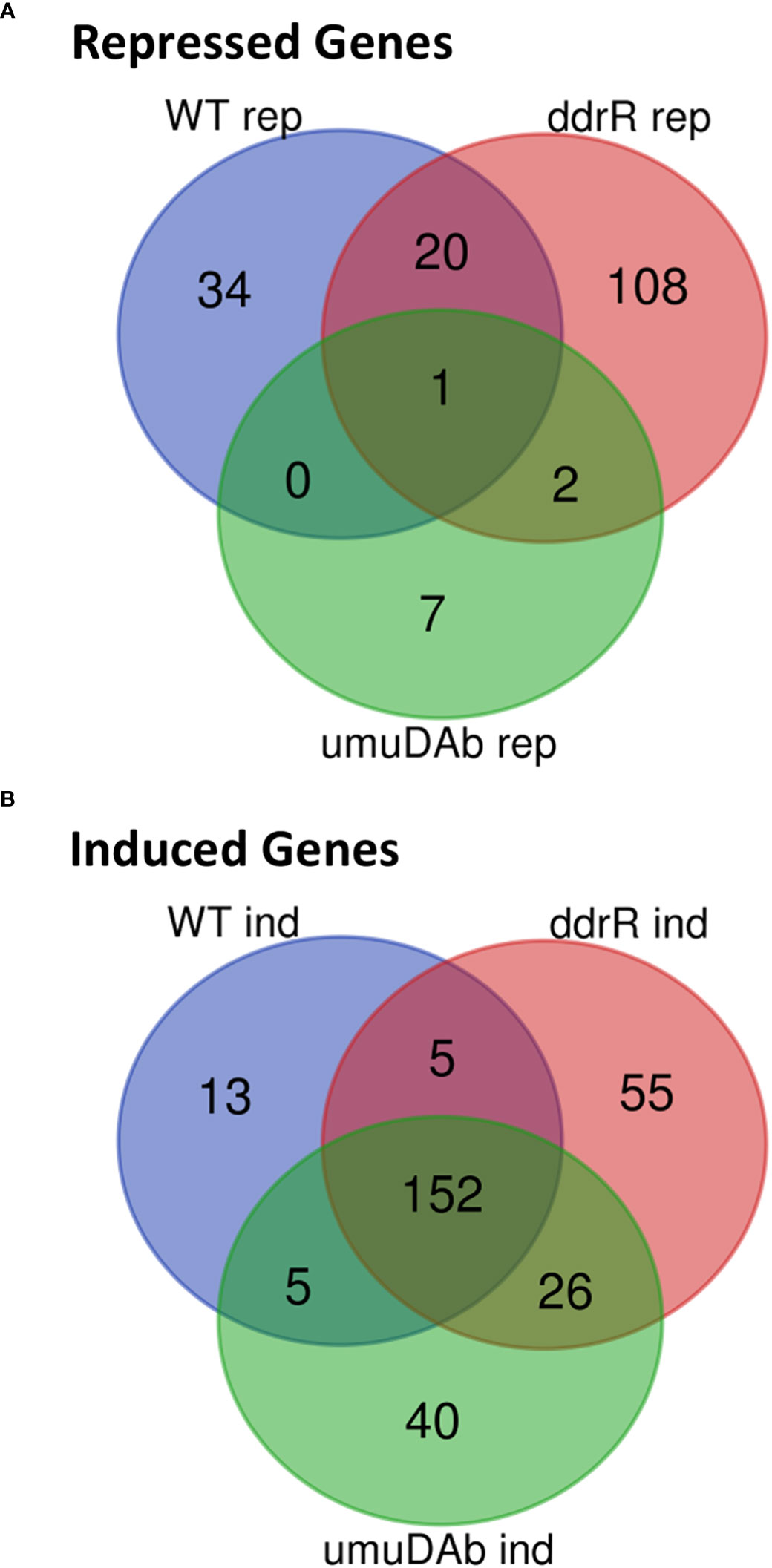
Figure 2 Venn diagram of differential gene expression, showing genes that were (A) repressed or (B) induced after MMC treatment (q < 0.05 for each) in wildtype (WT), ddrR, and umuDAb mutant strains of A. baumannii 17978 JH.
To complement our understanding of genes induced by DNA damage treatment, we investigated genes that were repressed in WT cells after MMC treatment. The results for both the older and the new reference genomes were combined in Table 2 to highlight DEG differences that directly resulted from the reference assembly used. Both genomes agreed for 43 genes of the 53 or 55 (using ATCC 17978 or 17978 UN, respectively) genes repressed in WT cells (Table 2). The categorization as being regulated by UmuDAb and DdrR, or only UmuDAb, differed for a minority (18) of the 43 genes repressed in both alignments. This could be due to the improved method of using q-values to assess significance rather than the simpler two-fold induction or repression ratio used previously, as well as improved annotations of entire genes rather than fragments of genes and newly annotated genes, including those in AbaAL44, causing changes in read depth for other genes. In the rest of our analysis of MMC-repressed genes, we considered only results from alignment to the 17978 UN genome.
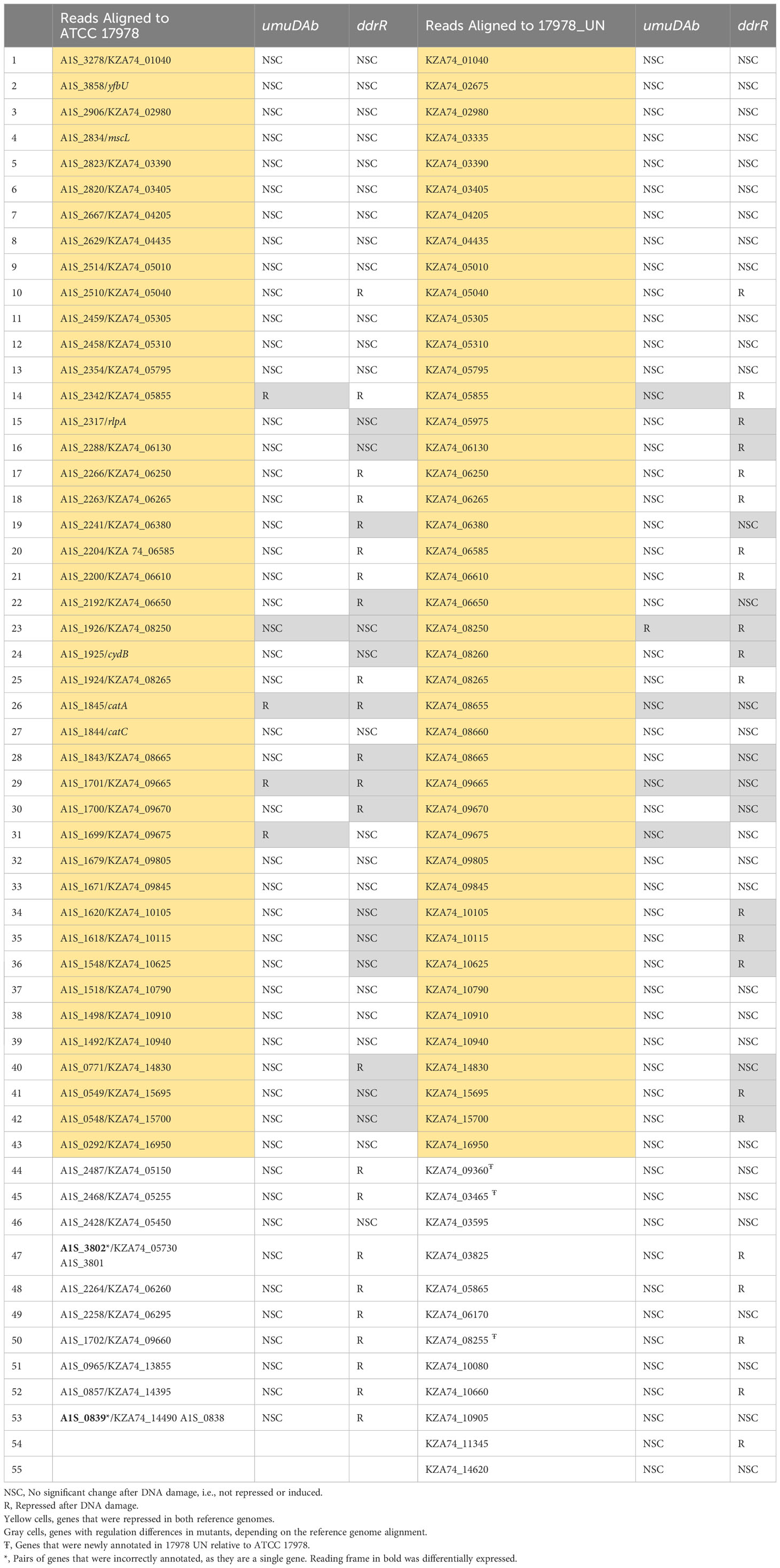
Table 2 Genes repressed after DNA damage that are regulated by ddrR or umuDAb.
Of the 55 genes that were repressed by MMC in the WT strain, 34 were no longer repressed in either the ddrR or umuDAb mutant (i.e., required each for repression) and 20 of the remaining 21 genes required only umuDAb for repression (Figures 1, 2A). No genes required only ddrR for repression, and only one gene (ybgE, KZA74_08250) was repressed in WT and not regulated by either ddrR or umuDAb after MMC treatment (Figure 3A). Six normally repressed genes were not repressed in the ddrR mutant after DNA damage because they were already downregulated more than two-fold (repressed with no treatment) before MMC treatment (catABC, acoAB, and aceF; involved in catechol metabolism and acetoin catabolism). Seven other genes normally repressed after DNA damage were dysregulated in the absence of ddrR. They were expressed at levels more than two-fold higher than untreated WT and were not repressed after DNA damage (Figure 3A).
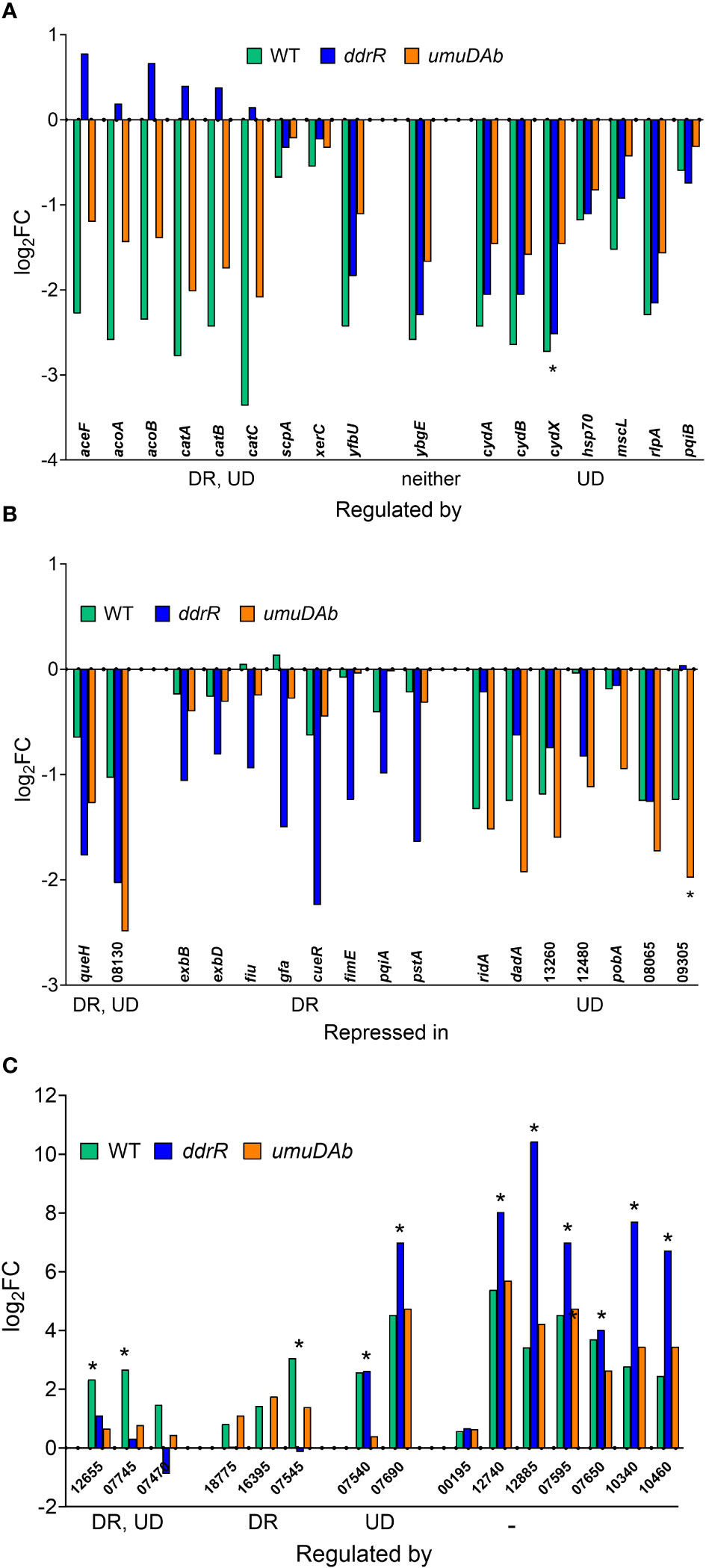
Figure 3 Genes responsive to MMC treatment in WT, ddrR and umuDAb mutant strains. RNASeq data of log2 fold changes is shown and categorized by response to MMC treatment, either repressed after treatment (panels A and B) or induced after treatment (panel C). (A) Genes that were repressed in the WT strain and whose repression was regulated by both ddrR and umuDAb (DR, UD), neither gene (-), or umuDAb (UD). (B) Genes that were not repressed in the WT strain but were repressed in the ddrR and/or umuDAb mutant strains. (C) Genes newly identified as induced after MMC treatment in this study. Shown are all eight induced genes regulated by either ddrR or umuDAb, or both genes, or neither gene (-). Unregulated genes include chromosomal gene KZA74_00195 and several CP genes (KZA74_12740 and 12885 from CP5, 07595 and 07650 from CP9, and 10340 and 10460 from CP14). Asterisks denote genes that had not been previously annotated in A. baumannii ATCC 17978. Numbers for gene names represent the KZA74 gene annotations.
The AbaAL44 locus pathogenicity island possesses 44 genes (Wijers et al., 2021). After aligning our cDNA reads to the 17978 UN genome, six of these genes, KZA74_9200 - 9210 and KZA74_09245 - 09255 had no read coverage from our strains. Of the 38 genes for which we had read coverage, three were repressed after MMC treatment, each in a different strain: ppk1 (KZA74_09360) in WT, clsC (KZA74_09315) in the ddrR mutant, and hypothetical KZA74_09305 in the umuDAb mutant (Supplementary Table S4).
Surprisingly, multiple genes not repressed in WT cells were repressed in either the ddrR or umuDAb mutant strains in response to DNA damage. Two genes were repressed in both umuDAb and ddrR mutant strains after MMC treatment: a hypothetical gene KZA74_08130 that was not annotated in the ATCC 17978 genome, and KZA74_11845 queH, epoxyqueuosine reductase (Figure 3B). Seven genes were repressed only in the umuDAb mutant: pobA, dadA homolog KZA74_13270, ridA homolog KZA74_13275, and four hypothetical genes (Figure 3B).
However, most (108) of the 117 genes were repressed solely in the ddrR mutant (Supplementary Table S5). Nearly half (49) of these genes repressed only in the ddrR mutant were repressed from dysregulated basal levels much higher than in WT. These included copper resistance genes cueR, copA, and copB (Williams et al., 2016; Williams et al., 2020). Interestingly, we found a highly upregulated 195 bp coding sequence that appeared to be a 5’ UTR of the cydA gene. Both cydA and cydB in the cydABX operon were modestly but not significantly upregulated and the cydX gene was severely downregulated in the ddrR mutant. After DNA damage, the 195 bp 5’ UTR region was repressed and the cydB gene was induced. This implies a possible regulatory effect of ddrR and the 5’ UTR region of cydA. The following pathways were enriched for these genes: amino acid, fatty acid/lipid, and cell structure synthesis, amino acid and amine degradation, translation, RNA metabolism, outer and plasma membrane components, and periplasm components.
In contrast, only ten of the genes repressed in the ddrR mutant had basal expression levels that were significantly lower than WT. Pathways enriched for these genes were involved in aromatic and chlorine degradation, aerobic respiration, adhesion, and pilus production, including the genes exbB and exbD, components of TonB dependent siderophore and vitamin B12 transport (Moeck and Coulton, 1998) and the fimbrial biogenesis gene, fimE, regulator of type I fimbriation in Escherichia coli (Blomfield et al., 1991). The remaining fifty genes were repressed from basal levels not significantly different from WT. None of these 108 genes were in a cryptic prophage (CP) and only one was on a plasmid, hypothetical (KZA74_19035) on pAB3.
The coregulators UmuDAb and DdrR that jointly repress the umuD-umuC error-prone polymerases before DNA damage, also are required to induce nine DDR genes (Peterson et al., 2020). We tested whether the new annotations in 17978 UN would reveal additional co-regulated genes that had either escaped annotation previously or were in the AbaAL44 region. Of the 175 genes induced in WT cells after MMC treatment (Supplementary Table S2), 50 were newly identified as induced, 40 were not annotated in the earlier reference genome and none were in AbaAL44 (Figure 2A, Table 3, Supplementary Table S4). All but three of these 50 DEGs were in cryptic prophages CP5, CP9, and CP14 (Di Nocera et al., 2011). One was in pAB3 (hypothetical gene KZA74_18775) and the other two were chromosomally encoded (hypothetical genes KZA74_00195 and KZA74_16395, which was transcribed convergently to the induced gst A1S_0408 (Figure 3C).
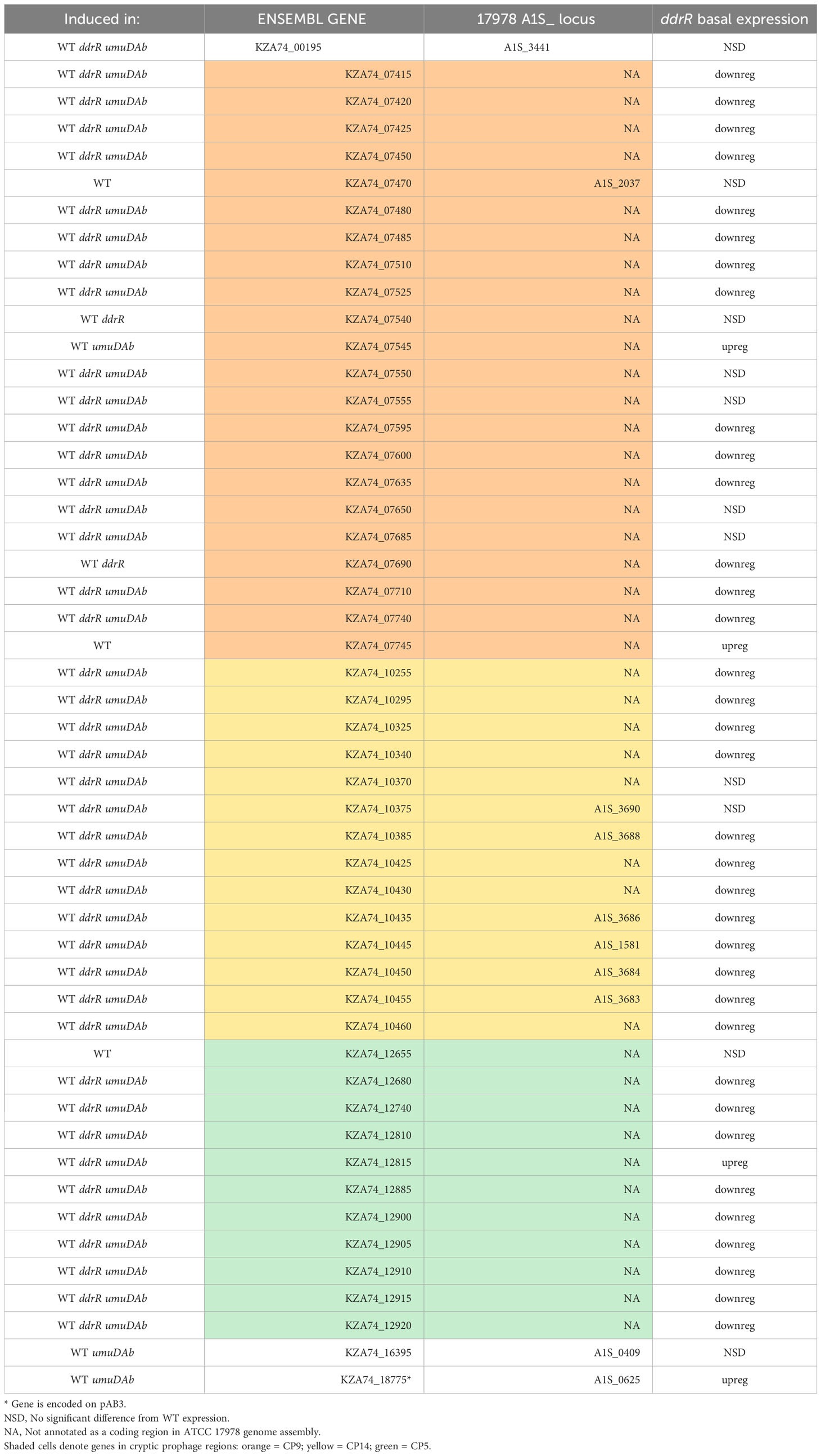
Table 3 Newly identified genes induced in WT after DNA damage.
Notably, we newly identified eight genes as being regulated by ddrR or umuDAb, five of which were not annotated in the ATCC 17978 genome assembly (Table 3). This brought the total number of induced genes controlled by these regulators to 23. Two coregulated genes (KZA74_12655 and KZA74_07745) are potentially part of the umuDC operons they follow, and the other coregulated gene was the CP9-encoded putative c1 prophage repressor esvI (KZA74_07470) (Figure 3C). Three ddrR-regulated genes were new DEGs: KZA74_18755, KZA74_16395 (located next to highly induced and conserved gst as described above), and KZA74_07545 (the latter two had increased basal expression in the ddrR mutant). Two umuDAb-regulated genes were new DEGs in CP9: KZA74_07540 and KZA74_07690 (Figure 3C).
We also identified 121 genes induced after DNA damage in the ddrR and umuDAb mutants that were not induced in WT cells (Supplementary Table S6). Fifty-five of these genes were only induced in the ddrR strain (Figure 2B), 33 of which were new to this study. Twenty-six genes were induced in both umuDAb and ddrR mutant strains, most of which (21) were not previously reported as induced (Table 4). These ddrR-regulated genes included five canonical DDR genes that were reported in a previous study (Hare et al., 2014) not to be induced: uvrA and ruvA (regulated by both umuDAb and ddrR), and ruvB, uvrC, and recX, (regulated by ddrR). Forty genes were induced only in the umuDAb strain, including 11 ribosomal proteins and translation-related factors (Table 5).
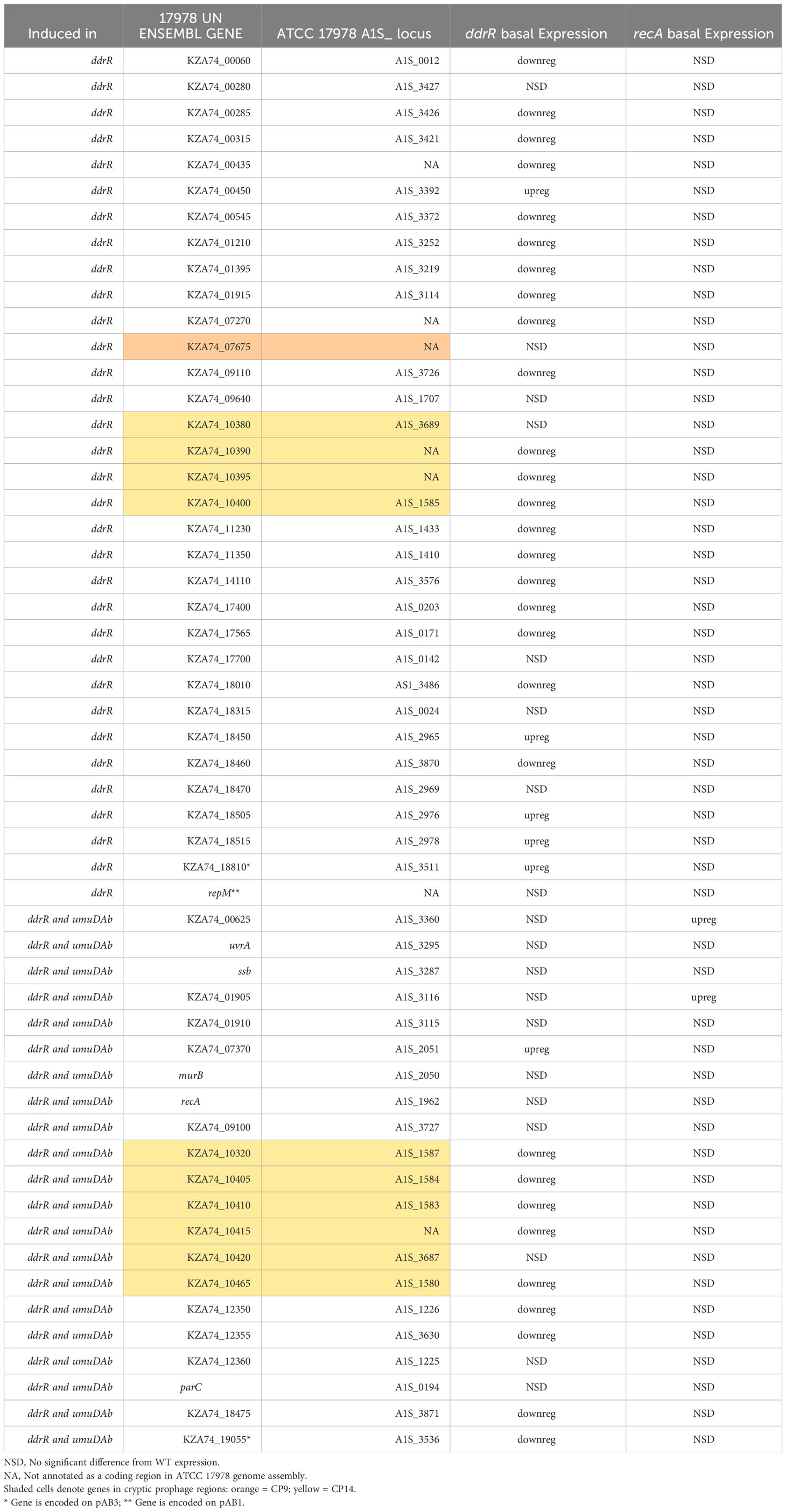
Table 4 Newly identified genes only induced in ddrR and umuDAb mutants.
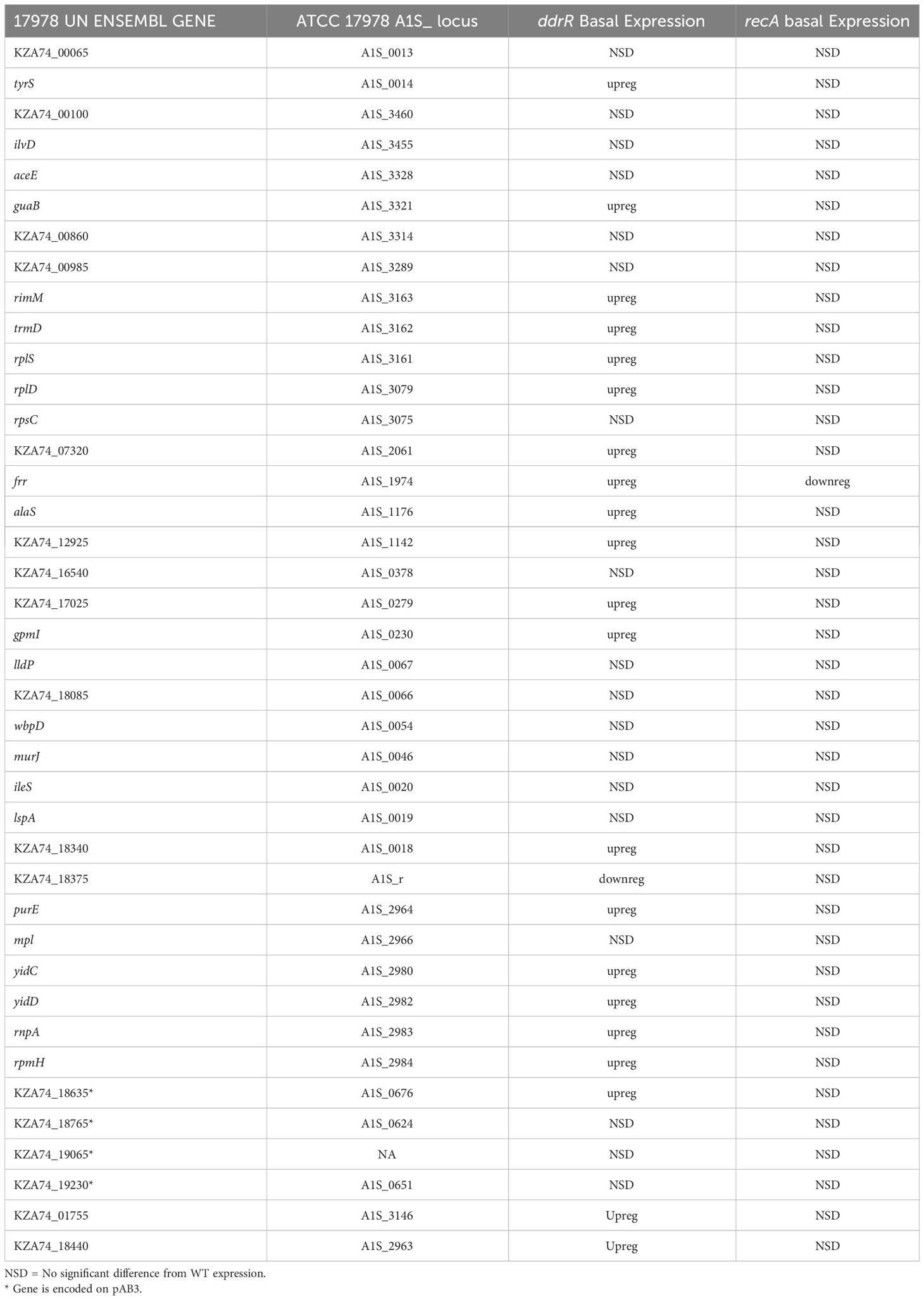
Table 5 Newly identified genes only induced in umuDAb mutant.
More than half of the induced genes showed altered basal expression levels in the ddrR or umuDAb mutants. Twenty-nine genes induced in ddrR cells showed induction from basal levels significantly lower in ddrR than in WT, including adeFGH, mgtA, mgtC, benE, czcC, sadH, gpi, and ltrA. Seven other genes were induced from levels significantly higher in ddrR than in WT, including pgpA (KZA74_00450), dgt (KZA74_04655), a low molecular weight phosphotyrosine protein phosphatase (KZA74_07370), a metal/formaldehyde-sensitive transcriptional repressor (KZA74_18515), a DsbC family protein (KZA74_18505), a DMT family protein (KZA74_18450), and a hypothetical (KZA74_18810). Half of the forty genes induced only in the umuDAb mutant had basal expression levels that trended lower than WT, but this trend was not statistically significant. These mostly transcription/translation genes were, conversely, basally upregulated in the ddrR mutant.
For basal expression comparisons we merged the untreated sample data for every strain’s growth in minimal media into a separate counts file to reduce any variance due to MMC treatment. A heatmap of the normalized read values for the top 200 differentially expressed genes was used to look for different expression patterns by strain. (Figure 4). The genes were classified as upregulated or downregulated for each untreated strain compared to the WT 17978 JH strain. The basal expression results for the mutant strains compared to the WT strain are in Supplementary Table S7.
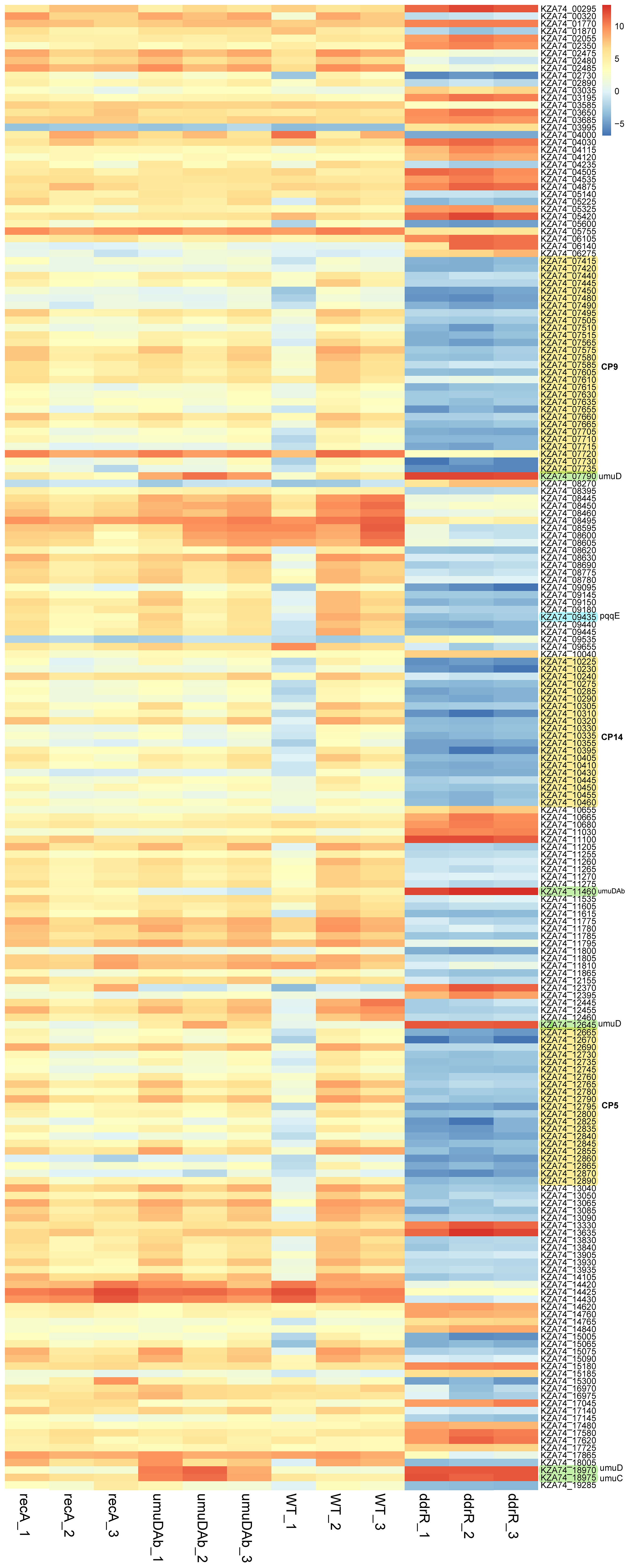
Figure 4 Heatmap of normalized read counts for the top 200 statistically significant DEGs (basal expression). Each column represents a biological replicate sample; each row represents a gene. Note the distinct and large-scale gene expression changes in the three clustered ddrR samples in the last three columns. The three cryptic prophages are highlighted in yellow and labelled, as are the umuDC and umuDAb loci. The log2 relative gene expression scale is depicted on the top right, with red representing upregulated and blue downregulated.
Mutation of ddrR tremendously impacted basal gene expression: 888 genes (23% of the genome) were significantly upregulated compared to WT (Figures 5A, 6A, SupplementaryTable S8) and 1241 genes (32.1%) were downregulated compared to WT expression levels (Figures 5A, 6B; Supplementary Table S9). Among the pathways enriched for upregulation were genes involved in cell division, DNA replication, protein transcription/translation/modification, phosphorylation, and non-acinetobactin iron acquisition. Also upregulated were three glutathione S-transferases (gst) and many genes involved in lipid metabolism, LPS biosynthesis, lysine biosynthesis, methionine import, pH regulation, phosphate import, phosphorylation, protein biosynthesis, OMP folding and transport, purine synthesis and metabolism, signal transduction, and sulfur metabolism. Notably, at least eleven transcriptional repressors and 72 transcription/translation genes (from ribosome proteins and assembly to extension factors and terminators) were also upregulated.
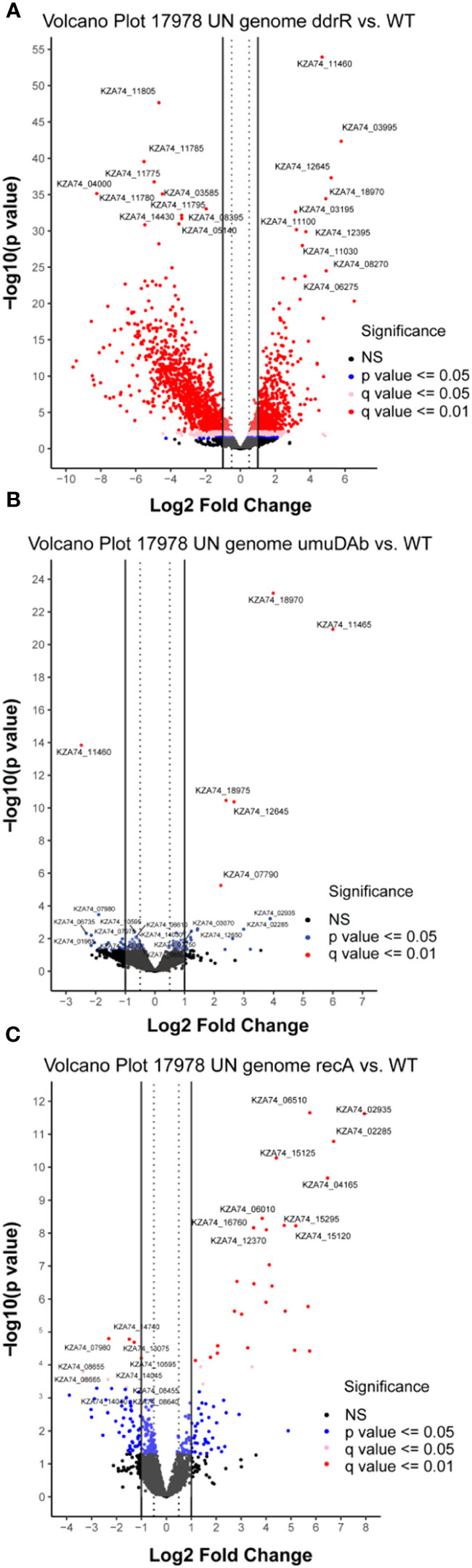
Figure 5 Global transcriptomic analysis of genes that were differentially expressed in WT A. baumannii 17978 JH cells vs mutants in (A) ddrR, (B) umuDAb, or (C) recA. Only untreated cells’ RNASeq data was combined and used in this analysis. Volcano plots show the log2 fold changes in gene expression where each dot represents one gene. Red and pink dots indicate significance at the q < 0.01 or q < 0.05 level. The log2 fold change is plotted on the x-axis, and the y-axis shows the log10 of the p value. The 10 genes with the highest induction or expression are labelled.
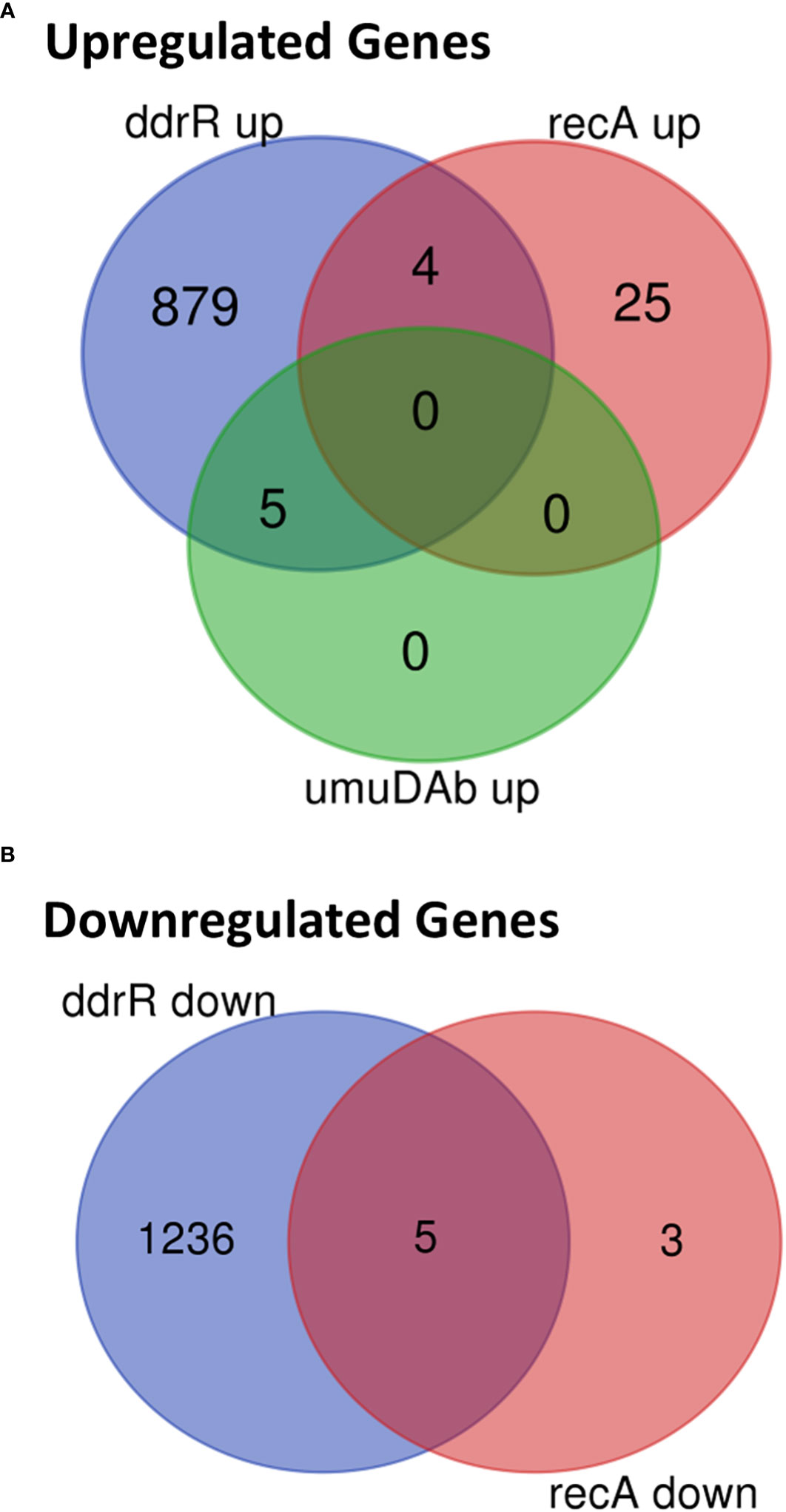
Figure 6 Venn diagram of differential gene expression, showing genes whose basal expression was (A) upregulated or (B) downregulated in WT A. baumannii 17978 JH cells vs ddrR and umuDAb mutants (q < 0.05 for each).
Two AbaAL44 genes, encoding a type 1 fimbrial protein (smf-1) and chaperone yadV were upregulated (Supplementary Table S4; Figure 7). Other chromosomal upregulated genes were involved in cell division processes that facilitate cellular elongation, septal formation, and daughter cell separation as part of the normal cell cycle. For example, the tol-pal system is a required part of cell division (Yakhnina and Bernhardt, 2020; Hale et al., 2022; Park and Cho, 2022). In the ddrR mutant, the tol-pal operon had a higher basal expression when compared to WT. Also upregulated was the newly characterized gene advA involved in chromosome segregation and cell division in A. baumannii (Geisinger et al., 2020) and t he lytic transglycosylase rlpA which facilitates daughter cell separation in Pseudomonas aeruginosa (Jorgenson et al., 2014) and Vibrio cholerae (Weaver et al., 2019). This upregulation of rlpA was confirmed with RT-qPCR analyses (Figure 8). In the ddrR mutant, rlpA and other genes mentioned in those studies (mltB and mltG) were all upregulated while amidase, amiE, and a lytic transglycosylase, slt, were downregulated.
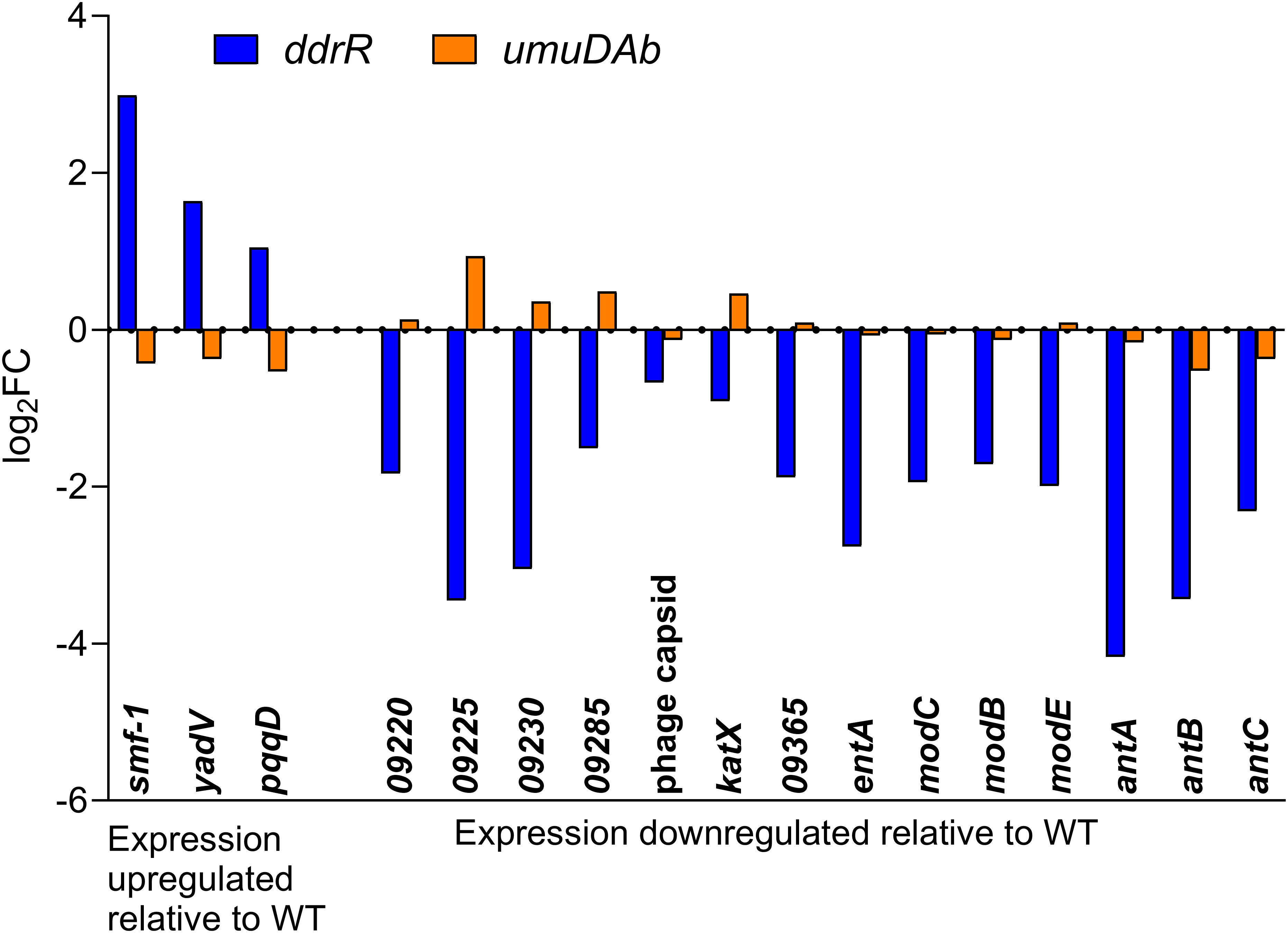
Figure 7 AbaAL44 genes displaying differential expression in the absence of MMC treatment in ddrR and umuDAb mutant strains. RNASeq data of log2 fold changes is shown for all 17 genes that were basally upregulated (n = 3) or downregulated (n = 14) in the ddrR strain.
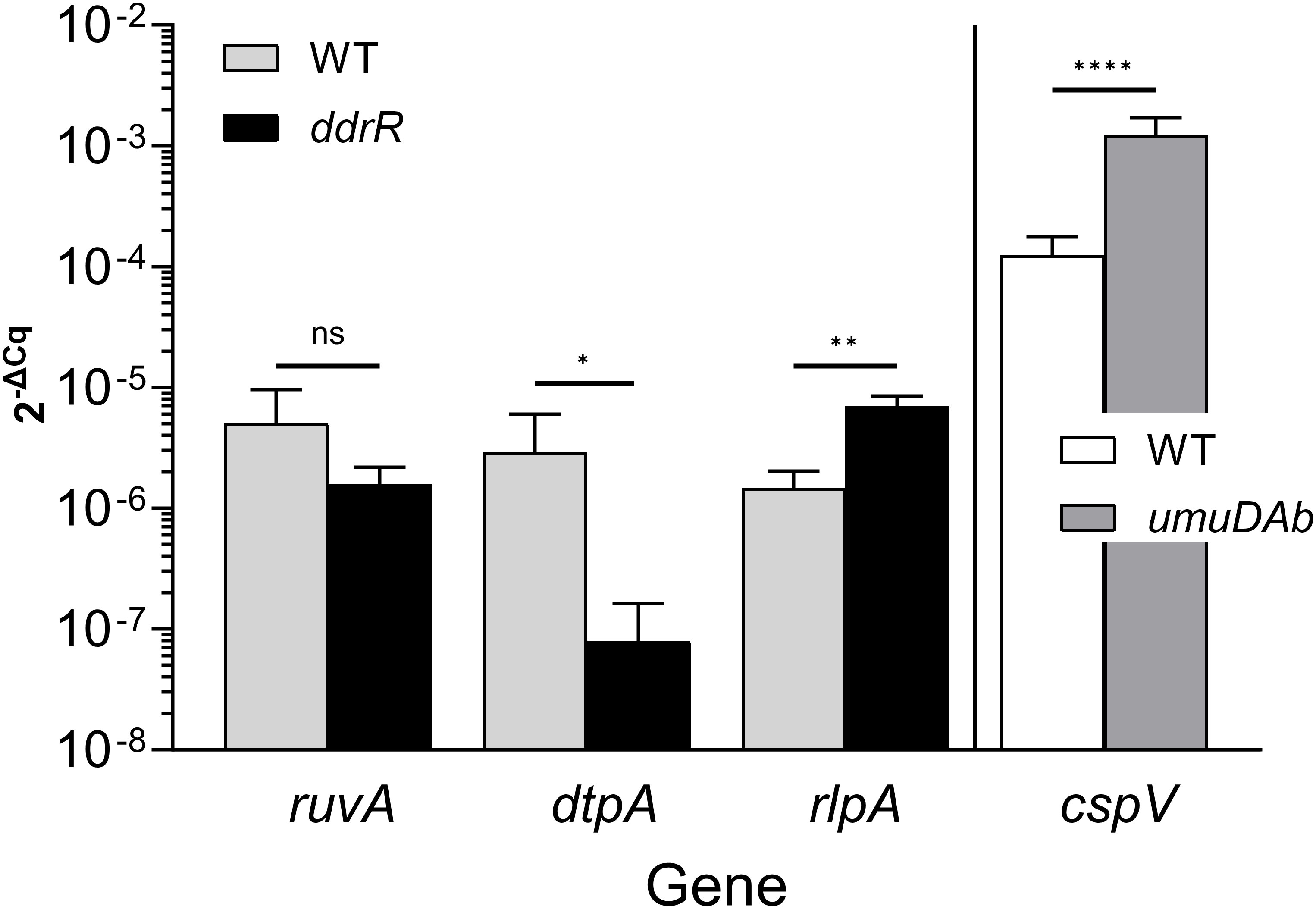
Figure 8 Representative RT-qPCR validation. This figure shows relative expression data for genes ruvA, dtpA, rlpA, and cspV in WT, ddrR, and/or umuDAb strains. RT-qPCR experiments measured gene expression (2-ΔCq) in the absence of DNA damage (basal expression). The expression of genes from different regulation categories is shown: genes whose expression was unchanged in ddrR and umuDAb mutant strains relative to WT (ruvA; KZA74_04650); those whose expression was decreased in the ddrR mutant relative to WT (dtpA; KZA74_11470); those whose expression was increased in the ddrR mutant relative to WT (rlpA; KZA74_05975); and those whose expression was increased in the umuDAb mutant relative to WT (cspV; KZA74_12340). The standard deviation of the mean from technical triplicates of biological triplicates is shown, where results from t-tests are shown: ****,P < 0.0001; ***,P < 0.001; **,P < 0.01; *,P < 0.05; ns = not significant.
The lack of ddrR also affected expression of stringent response genes. The stringent response can be triggered by production of the alarmones guanosine tetraphosphate (ppGpp) and guanosine pentaphosphate (pppGpp) in response to nutrient deprivation (Liu et al., 2017). The gene relA (KZA74_15545) is responsible for the ppGpp synthesis and accumulation of ppGpp, and spoT regulates ppGpp accumulation with hydrolytic activity and weak synthetase ability (Gaca et al., 2015). In A. baumannii, ppGpp deficiency can affect biofilm formation (Kim et al., 2021) and efflux pump expression (Jung et al., 2020). We observed spoT (KAZ74_01610) to be upregulated 3.4-fold in the ddrR mutant.
Genes downregulated in the ddrR mutant included 14 of the 38 genes in AbaAL44 (Figure 7, Supplementary Table S4), 80% of the prophage genes (146 out of 179 genes) and at least 71 transcriptional regulators/repressors. They also included at least 20 amino acid transporters and 23 ABC and MFS transporters. Pathway analysis showed that these downregulated genes are involved in acyclic terpene and leucine/isovalerate utilization, aromatic catabolism, amine metabolism, benzoate metabolism (20 genes), carnitine metabolism, catechol metabolism (for example analysis, see Figure 9), cell adhesion, fimbrial biogenesis, type II, IV, and VI secretion systems, cytolysis, fatty acid metabolism, glycerol metabolism, magnesium transport, nitrite metabolism, potassium transport, leucine catabolism, muconate metabolism, organosulfur metabolism, phenylalanine, tyrosine and tryptophan biosynthesis, purine metabolism, thiamine metabolism, isoleucine and valine catabolism, vanillin metabolism, acinetobactin-mediated iron acquisition, and copper and zinc regulation (Supplementary Table S9). Specific downregulated genes were the csuBABCDE biofilm genes, fimbrial genes; fimT and fimB, pilus genes; pilV, pilGHIJ, pilT, pilU, pilBC, pilR, pilNOPQ, and pilA; and efflux pump genes adeRS, adeFGH, and adeL. Of note are two separate clusters of oxidative and desiccation stress-related genes found downstream of ddrR in 17978 JH, all of which are downregulated in the ddrR mutant strain (Sun et al., 2021).
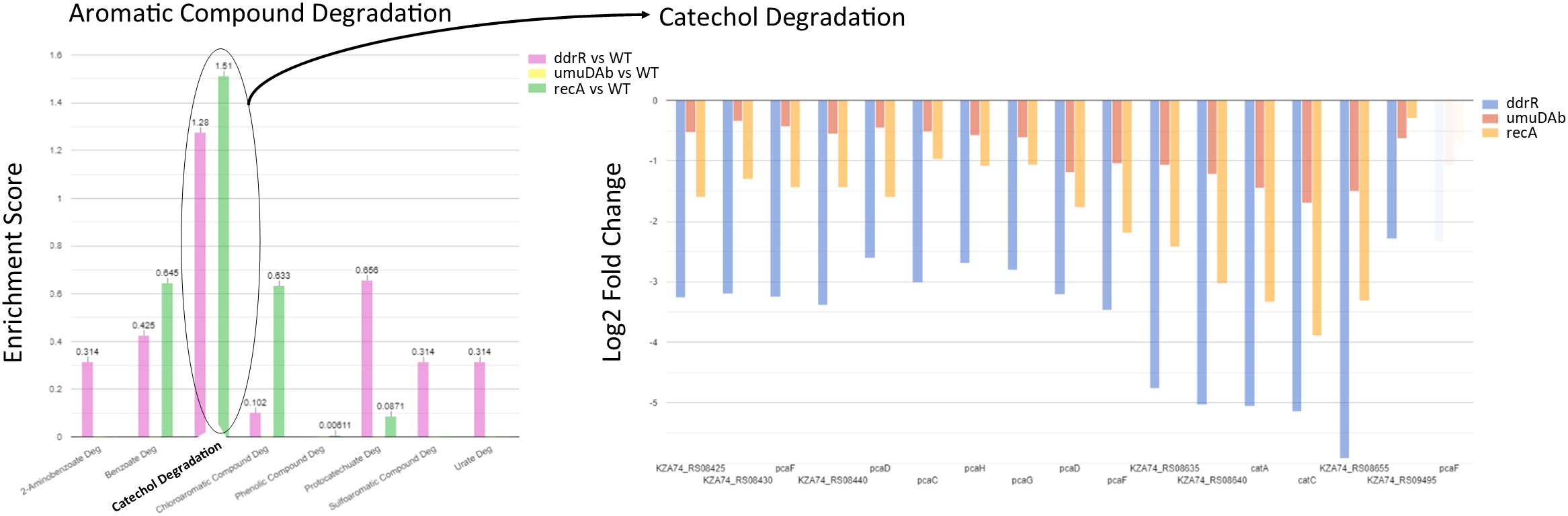
Figure 9 Example of pathway enrichment analysis. This figure shows a sample enrichment analysis for some genes whose basal expression was downregulated in the ddrR mutant. Multiple pathways were enriched for aromatic compounds. The ddrR cells were enriched for 16 of the 17 known genes involved in catechol degradation.
The significant amount of dysregulation seen in the ddrR mutant was not shared by the umuDAb or recA mutants. In the umuDAb mutant, basal expression was significantly higher than in WT cells for only five genes: the four error-prone polymerase V genes (three umuD and one umuC allele), and ddrR (Hare et al., 2014) (Figures 5B, 6A; Supplementary Table S8). These five genes are repressed by umuDAb and ddrR and induced after DNA damage (Hare et al., 2014; Peterson et al., 2020). No genes that showed decreased expression in the umuDAb mutant strain were statistically significant.
The recA mutant had a relatively small number of genes (37) differentially expressed. Eight were downregulated (Supplementary Table S9), and 29 were upregulated (Figures 5C, 6, Supplementary Table S8). Interestingly, several genes were similarly dysregulated in multiple mutants. Among upregulated genes, four were shared between the recA and ddrR mutants and five were shared between the umuDAb and ddrR mutants. The recA and ddrR mutants also shared five downregulated genes (Figure 6).
Several studies have determined which genes are essential in A. baumannii (Wang et al., 2014; Gallagher et al., 2015; Uddin et al., 2019; Bai et al., 2021). The most recent study found 373 genes considered essential (Bai et al., 2021). We considered a gene essential if it was on this list, as it compared their data to previous studies.
Many differences were seen in essential gene expression between our mutant strains and WT when grown under minimal media conditions. More than half (189) of these essential genes were expressed significantly higher in the ddrR mutant than in the WT strain (Supplementary Table S10). These essential genes are involved in amino acid (AA)-tRNA synthetases, ATP synthase and purine synthesis, capsular polysaccharide synthesis (CPS), DNA, replication, dNTP synthesis, fatty acid synthesis, Fe-S cluster synthesis, gluconeogenesis, heme synthesis, histidine synthesis, isoprenoid synthesis, lipopolysaccharide (LPS) synthesis, peptidoglycan (PG) synthesis, mRNA degradation, methionine synthesis, nicotinamide synthesis, phospholipid synthesis, protein secretion, purine synthesis, pyrimidine synthesis, ribosomal synthesis, ribosomal modification, ribosomal proteins, sodium efflux, transcription, transcription regulation, translation factors, tRNA modification, and tryptophan synthesis.
Second, many (85) of these 189 genes were inversely regulated in the ddrR and umuDAb mutant strains, with genes upregulated in the ddrR mutant being downregulated in the umuDAb mutant, often more than two-fold lower than WT levels, although the p-adjusted values were not significant. Several of the lower-expressed genes in the umuDAb mutant that code for ribosomal, transcription and translation proteins had expression levels increased more than 20-fold after DNA-damage, to the level of WT expression. Since these are essential genes, the reduced levels in the umuDAb mutant are likely biologically significant.
Conversely, only five essential genes were expressed significantly lower in the ddrR mutant than the WT strain. Two of these encode hypothetical proteins KZA74_09110 and KZA74_11475, which is transcribed divergently from dtpA and was not annotated in the ATCC 17978 genome. Both were induced after DNA damage in the ddrR mutant. The three other genes, prtC (KZA74_13285), hfq (KZA74_07030), and hemD (KZA74_17100) were downregulated in the ddrR mutant and unchanged after MMC treatment in all strains.
For this project we combined the use of the new reference genome, 17978 UN, that more closely matched our lab strain (Wijers et al., 2021), with an improved analysis pipeline that included DESeq2. The first widely used A. baumannii genome assembly and annotation (ATCC 17978) had all pAB3 genes combined with the chromosome. While it is possible that pAB3 may have integrated into the chromosome in the ATCC 17978 strain used for the genome assembly, others have observed no evidence of integration in their study of pAB3 (Weber et al., 2015). As the location and nature of gene acquisition matters for understanding the nature of any regulon, it is important to use an accurate genome assembly (with correct contig assembly and gene annotations).
Earlier analyses investigated genes controlled by the unusual UmuDAb and DdrR coregulators and focused on the genes induced after DNA damage (Aranda et al., 2013; Hare et al., 2014; Peterson et al., 2020). Those previous studies did not characterize genes repressed after DNA damage and genes that rely on ddrR or umuDAb for their repression or induction but are not part of the DDR. In this work, we showed how aligning our RNASeq data to the improved 17978 UN genome assembly for A. baumannii revealed 43 additional genes that were responsive to DNA damage, one-third of which were regulated by either ddrR or umuDAb. We expanded our knowledge of the control of the DDR, in observing that 100% of the 12 genes newly identified as repressed were regulated by either umuDAb or ddrR but only 17% of the 48 genes newly identified as induced after DNA damage were regulated by umuDAb or ddrR. Furthermore, we identified newly annotated genes such as the hypothetical genes KZA74_12655 and KZA74_07745 that appear to be part of the two umuDC operons possessed by 17978 JH. Their possible role in the unusual DDR of this genus is unexplored and highlights that ddrR and umuDAb control the repression of genes induced after DNA damage.
The induction of the five canonical SOS genes uvrA and ruvA, ruvB, uvrC, and recX in the mutants suggests that umuDAb and ddrR might prevent these damage response genes from being induced after MMC treatment, which could potentially allow more error-prone DNA repair to happen. This additional control is an example of distinctive features in A. baumannii strains that have the potential to increase genomic variation.
DEGs in the newly identified AbaAL44 accessory region (Wijers et al., 2021) revealed no genes that were induced after DNA damage in WT cells, but three AbaAL44 genes were repressed. One of these genes was not regulated by umuDAb or ddrR, another uniquely required ddrR for repression, and one uniquely required umuDAb. Almost half of the genes in AbaAL44 (17) were basally dysregulated in the absence of ddrR, indicating its importance in regulating genes in this pathogenicity island. These controlled genes encode a zonular occludens toxin domain protein (KZA74_09220), type 1 fimbrial protein and chaperone, and all three genes of the antABC operon (anthranilate dioxygenase) (Supplementary Table S4).
Furthermore, we reveal a large regulatory role for ddrR in controlling the expression of hundreds of genes in the absence of DNA damage. Quorum sensing (QS) is a form of cell-cell communication that allows pathogenic bacteria to coordinate virulence gene expression. QS is regulated in A. baumannii by the abaIMR operon (López-Martín et al., 2021; Sun et al., 2021), a gene cluster evolutionarily conserved among many Acinetobacter species The gene abaI encodes acyl homoserine lactone synthase, involved in signal transduction and potentially enhances A. baumannii virulence. AbaR is an AI synthase receptor. AbaM represses AHL biosynthesis and is a regulatory component of many other genes involved in QS and independent of QS. The genes abaI and abaR are upregulated in an abaM::TN26 mutant (López-Martín et al., 2021). In the ddrR mutant all three of these genes were downregulated.
Biofilms and fimbrial biogenesis are ways bacteria protect themselves from harsh living environments and biofilms have multiple avenues of regulation, such as cellular ppGpp levels. Many of the genes that are responsible for biofilm production, efflux pumps, motility, capsule formation, and siderophore production affected by ppGpp depletion (Kim et al., 2021), were downregulated in the ddrR mutant. Inversely, many essential genes are upregulated in the absence of ddrR and downregulated in the absence of umuDAb. None of the genes downregulated in the umuDAb mutant were statistically significant, but as essential genes, decreases in their expression are biologically significant as shown by CRISPR-based gene knockdown experiments by Bai et al., 2021. Without UmuDAb to regulate ddrR production, excess ddrR product could inhibit these genes.
The large number of genes dysregulated in the ddrR mutant was unexpected (1241 downregulated and 888 upregulated). One issue that can limit the confidence in comparisons of datasets is batch effects, due to the ddrR mutant samples being sequenced at a different time with paired-end reads rather than the single-end sequences used for all the other samples. Even though the same scientist used the same lab equipment and growth conditions to collect these samples, we examined our data in additional ways to address this concern. First, wondering if the adjusted p-value cutoff of 0.05 was too lenient, we reran the analysis with a more stringent cutoff of padj < 0.01 and adding a log2 fold change cutoff greater than 1 and less than -1. This method still yielded 82.8% (1028 genes) of the genes obtained with the higher p-value, and the number of basally unregulated genes only decreased to 59.9% (532 genes) of the number obtained with the higher p-value. Second, several genes were selected from those mentioned in this study for validation with RT-qPCR and several others had already been validated in previous work (see summary in Table 6). The results were not concordant for 0% of the DNA damage-responsive genes and only 20% of the genes whose basal expression was dysregulated. This was not surprising as the results for 15-20% of genes, when compared between RNAseq and RT-qPCR, can be non-concordant (not matching between methods) (Everaert et al., 2017; Coenye, 2021). However, it has been noted that in studies with large numbers of DEGs, if the RNAseq analyses pipeline is robust enough and the sample number is high enough there is not much value added with qPCR validation (Coenye, 2021). The DESeq2 pipeline used here is robust and can detect and reduce batch effect in its results (Love et al., 2014; Seyednasrollah et al., 2015). Even though all of the genes tested were not concordant, the validation of the majority of the genes selected for RT-qPCR lends credence to the conclusion that the product of ddrR affects many cellular systems and processes. Finally, we chose the DESeq2 algorithm that uses a statistical model to estimate the mean and variance of the count data, which controls for batch effects, such as our differences between sequencing methods (the single and paired-end reads). Crucially, this approach allowed us to compare the expression of DNA damage-responsive genes and other genes directly and comprehensively across WT and all the mutant strains without DNA damage. DESeq2 has good sensitivity for detecting low-expressed genes and is known for more consistent detections in cases with few biological replicates (Love et al., 2014; Seyednasrollah et al., 2015), as we had biological triplicates for each of our strains. Nevertheless, the effects of the DdrR and UmuDAb regulators on any particular operon, metabolic process or gene will require additional investigation to confirm.
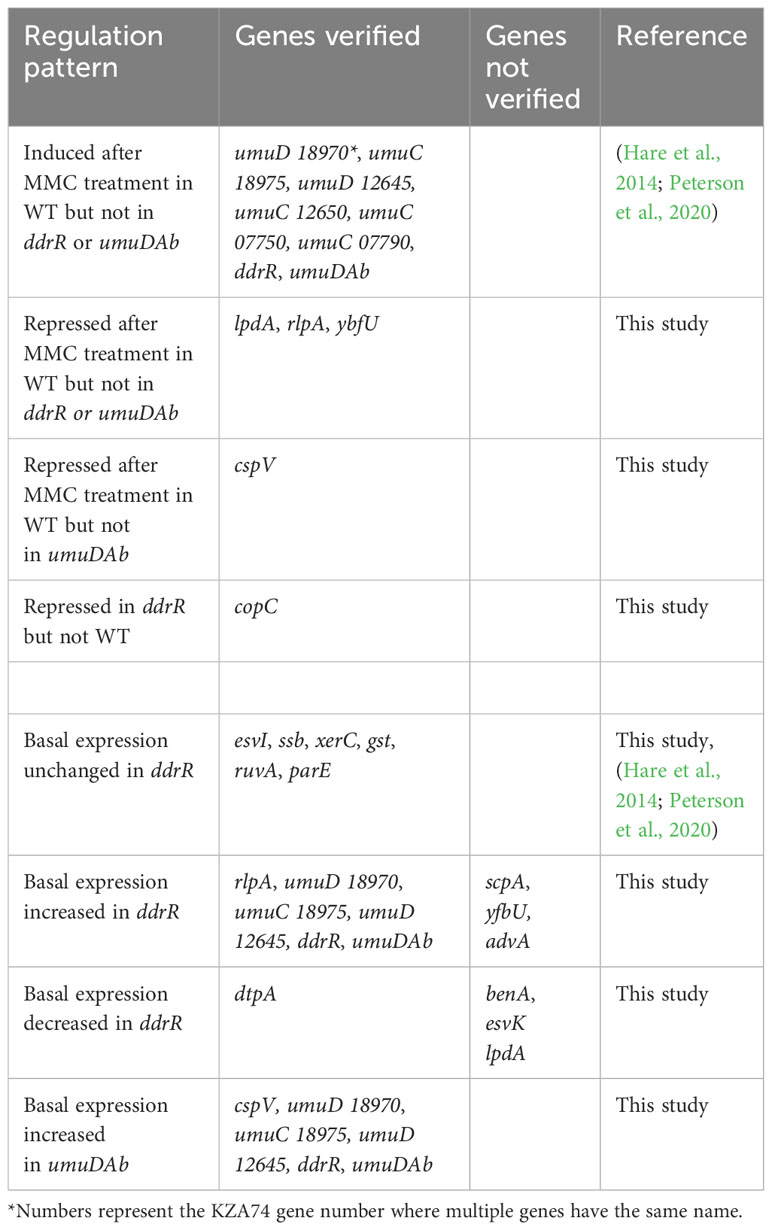
Table 6 Genes verified by RT-qPCR to match the pattern of regulation observed in RNASeq results.
Although it affects the expression of many genes, the mechanism of action of ddrR has not yet been elucidated. It co-regulates the error-prone polymerases, umuDAb and itself (Peterson et al., 2020) by enhancing the repression activity of the LexA-like repressor, UmuDAb (Cook et al., 2022), and DdrR has been shown to interact with UmuDAb protein in vitro (Pavlin et al., 2022). DdrR actions resemble in some ways those of the small bacteriophage protein, gp7, found in Bacillus thuringiensis, which binds to the bacterial LexA protein and increases its repression (Fornelos et al., 2015). However, DdrR is not prophage-encoded, shares no sequence similarity with gp7 (Cook et al., 2022; Pavlin et al., 2022), and interacts with the non-canonical UmuDAb repressor, not LexA. Nevertheless, its corepressor activities allow speculation that it may interact with other repressors or activators to modulate their regulatory activity. Alternately, DdrR is required for the induction of phage repressors KZA74_07470 (esvI) and KZA74_12875, which may control multiple phage genes. Further studies and testing are still needed to reveal how ddrR interacts and affects other gene expression in A. baumannii.
This study pinpointed canonical damage response genes induced after DNA damage solely in the absence of ddrR or umuDAb, suggesting their potential role in preventing the induction of those genes and influencing genomic variation in A. baumannii strains. Moreover, the lack of ddrR led to the upregulation of essential genes involved in DNA replication, transcription, translation, and membrane transport, alongside alterations in stringent response pathways indicating a broader impact on cellular functions beyond DDR. The findings highlighted intricate regulatory networks affected by ddrR, particularly in biofilm formation, quorum sensing, and cellular communication, with implications for A. baumannii virulence and survival strategies. Future investigations aim to elucidate ddrR’s functional mechanisms, potentially paving the way for therapeutic targeting in combating A. baumannii infections.
Publicly available datasets were analyzed in this study. This data can be found here: SRA run selector: SRR1165107 SRR1165108 SRR1165109 SRR1165110 SRR1165111 SRR1165112 SRR1165113 SRR1165114 SRR1165115 SRR1165116 SRR1165117 SRR1165118 SRR1165119 SRR1165120 SRR1165121 SRR1165122 SRR1165123 SRR1165124 SRR6150760 SRR6150759 SRR6150758 SRR6150757 SRR6150756 SRR6150755.
DC: Conceptualization, Data curation, Formal Analysis, Investigation, Methodology, Software, Validation, Visualization, Writing – original draft, Writing – review & editing. MF: Investigation, Visualization, Writing – review & editing. JC: Validation, Writing – review & editing, Formal Analysis, Methodology. JH: Conceptualization, Funding acquisition, Investigation, Supervision, Validation, Visualization, Writing – review & editing.
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This work was supported by NIH 2R15GM085722-04 to JH. and the KY IDeA Networks of Biomedical Research Excellence NIH grant 5P20GM103436-22. DC and MF were supported by R15GM085722 and 5P20GM103436-22.
We gratefully acknowledge the Kentucky INBRE Biostatistics core, particularly JC and Eric Rouchka, for their technical support.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fcimb.2023.1324091/full#supplementary-material
Antunes, L. C.S., Imperi, F., Carattoli, A., Visca, P. (2011). Deciphering the multifactorial nature of acinetobacter baumannii pathogenicity. PloS One 6 (8), e226745. doi: 10.1371/journal.pone.0022674
Antunes, L. C. S., Visca, P., Towner, K. J. (2014). Acinetobacter baumannii: evolution of a global pathogen. Pathog. Dis. 71 (3), 292–3015. doi: 10.1111/2049-632X.12125
Aranda, J., Bardina, C., Beceiro, A., Rumbo, S., Cabral, M. P., Barbe, J., et al. (2011). Acinetobacter Baumannii recA protein in repair of DNA damage, antimicrobial resistance, general stress response, and virulence. J. Bacteriol. 193 (15), 3740–3475. doi: 10.1128/JB.00389-11
Aranda, J., Lopez, M., Leiva, E., Magan, A., Adler, B., Bou, G., et al. (2014). Role of acinetobacter baumannii umuD homologs in antibiotic resistance acquired through DNA damage-induced mutagenesis. Antimicrob. Agents Chemother. 58 (3), 1771–1735. doi: 10.1128/AAC.02346-13
Aranda, J., Poza, M., Shingu-Vazquez, M., Cortes, P., Boyce, J. D., Adler, B., et al. (2013). Identification of a DNA-damage-inducible regulon in acinetobacter baumannii. J. Bacteriol. 195 (24), 5577–5825. doi: 10.1128/JB.00853-13
Artuso, I., Lucidi, M., Visaggio, D., Capecchi, G., Lugli, G. A., Ventura, M., et al. (2022). Genome diversity of domesticated acinetobacter baumannii ATCC 19606T strains. Microbial. Genomics 8 (1), 0007495. doi: 10.1099/mgen.0.000749
Bai, J., Dai, Y., Farinha, A., Tang, A. Y., Syal, S., Vargas-Cuebas, G., et al. (2021). Essential gene analysis in acinetobacter baumannii by high-density transposon mutagenesis and CRISPR interference. J. Bacteriol. e00565-20. doi: 10.1128/JB.00565-20
Blomfield, I. C., McClain, M. S., Princ, J. A., Calie, P. J., Eisenstein, B. I. (1991). Type 1 fimbriation and fimE mutants of escherichia coli K-12. J. Bacteriol. 173 (17), 5298–5307. doi: 10.1128/jb.173.17.5298-5307.1991
Ching, C., Yang, B., Onwubueke, C., Lazinski, D., Camilli, A., Godoy, V. G. (2018). Lon protease has multifaceted biological functions in. Acinetobacter. Baumannii.” J. Bacteriol. 201 (2), e00536–e00185. doi: 10.1128/JB.00536-18
Coenye, T. (2021). Do results obtained with RNA-sequencing require independent verification? Biofilm 3 (January), 100043. doi: 10.1016/j.bioflm.2021.100043
Cook, D., Flannigan, M. D., Candra, B. V., Compton, K. D., Hare, J. M. (2022). The ddrR coregulator of the acinetobacter baumannii mutagenic DNA damage response potentiates umuDAb repression of error-prone polymerases. J. Bacteriol. 204 (11), e00165–e00225. doi: 10.1128/jb.00165-22
Di Nocera, P. P., Rocco, F., Giannouli, M., Triassi, M., Zarrilli, R. (2011). Genome organization of epidemic Acinetobacter Baumannii strains. BMC Microbiol. 11, 224. doi: 10.1186/1471-2180-11-224
Dobin, A., Davis, C. A., Schlesinger, F., Drenkow, J., Zaleski, C., Jha, S., et al. (2013). STAR: ultrafast universal RNA-Seq aligner. Bioinf. (Oxford. England). 29 (1), 15-21. doi: 10.1093/bioinformatics/bts635
Dobrindt, U., Hacker, J. (2001). Whole genome plasticity in pathogenic bacteria. Curr. Opin. Microbiol. 4 (5), 550–557. doi: 10.1016/s1369-5274(00)00250-2
Durante-Mangoni, E., Del Franco, M., Andini, R., Bernardo, M., Giannouli, M., Zarrilli, R. (2015). Emergence of Colistin Resistance without Loss of Fitness and Virulence after Prolonged Colistin Administration in a Patient with Extensively Drug-Resistant Acinetobacter Baumannii. Diagn. Microbiol. Infect. Dis. 82 (3), 222–265. doi: 10.1016/j.diagmicrobio.2015.03.013
Everaert, C., Luypaert, M., Maag, J. L.V., Cheng, Q. X., Dinger, M. E., Hellemans, J., et al. (2017). Benchmarking of RNA-sequencing analysis workflows using whole-transcriptome RT-qPCR expression data. Sci. Rep. 7 (1), 15595. doi: 10.1038/s41598-017-01617-3
Fornelos, N., Butala, M., Hodnik, V., Anderluh, G., Bamford, J. K., Salas, M. (2015). Bacteriophage GIL01 gp7 interacts with host lexA repressor to enhance DNA binding and inhibit recA-mediated auto-cleavage. Nucleic Acids Res. 43 (15), 7315–7295. doi: 10.1093/nar/gkv634
Gaca, A. O., Colomer-Winter, C., Lemos, J. A. (2015). Many means to a common end: the intricacies of (p)ppGpp metabolism and its control of bacterial homeostasis. J. Bacteriol. 197 (7), 1146–1565. doi: 10.1128/JB.02577-14
Gallagher, L. A., Ramage, E., Weiss, E. J., Radey, M., Hayden, H. S., Held, K. G., et al. (2015). Resources for genetic and genomic analysis of emerging pathogen acinetobacter baumannii. J. Bacteriol. 197 (12), 2027–2355. doi: 10.1128/JB.00131-15
Geisinger, E., Mortman, N. J., Dai, Y., Cokol, M., Syal, S., Farinha, A., et al. (2020). Antibiotic susceptibility signatures identify potential antimicrobial targets in the acinetobacter baumannii cell envelope. Nat. Commun. 11 (1), 4522. doi: 10.1038/s41467-020-18301-2
Green, E. R., Fakhoury, J. N., Monteith, A. J., Pi, H., Giedroc, D. P., Skaar, E. P. (2022). Bacterial hydrophilins promote pathogen desiccation tolerance. Cell Host Microbe 30 (7), 975-987.e7. doi: 10.1016/j.chom.2022.03.019
Hale, C. A., Persons, L., de Boer, P. A.J. (2022). Recruitment of the tolA protein to cell constriction sites in escherichia coli via three separate mechanisms, and a critical role for FtsWI activity in recruitment of both TolA and TolQ. J. Bacteriol. 204 (1), e00464–e00421. doi: 10.1128/jb.00464-21
Hare, J. M., Bradley, J. A., Lin, C.-L., Elam, T. J. (2012). Diverse responses to UV light exposure in Acinetobacter include the capacity for DNA damage-induced mutagenesis in the opportunistic pathogens Acinetobacter Baumannii and Acinetobacter Ursingii. Microbiology 158, 601–611. doi: 10.1099/mic.0.054668-0
Hare, J. M., Ferrell, J. C., Witkowski, T. A., Grice, A. N. (2014). Prophage induction and differential recA and umuDAb transcriptome regulation in the DNA damage responses of Acinetobacter Baumannii and Acinetobacter Baylyi. PloS One 9 (4), e93861. doi: 10.1371/journal.pone.0093861
Imperi, F., Antunes, L. C. S., Blom, J., Villa, L., Iacono, M., Visca, P., et al. (2011). The genomics of acinetobacter baumannii: insights into genome plasticity, antimicrobial resistance and pathogenicity. IUBMB Life 63 (12), 1068–1745. doi: 10.1002/iub.531
Jorgenson, M. A., Chen, Y., Yahashiri, A., Popham, D. L., Weiss, D. S. (2014). The bacterial septal ring protein RlpA is a lytic transglycosylase that contributes to rod shape and daughter cell separation in pseudomonas aeruginosa. Mol. Microbiol. 93 (1), 113–285. doi: 10.1111/mmi.12643
Jung, H.-W., Kim, K., Maidul Islam, M., Lee, J. C., Shin, M. (2020). Role of ppGpp-regulated efflux genes in acinetobacter baumannii. J. Antimicrob. Chemother. 75 (5), 1130–1345. doi: 10.1093/jac/dkaa014
Kim, K., Islam, M., Jung, H.-w., Lim, D., Kim, K., Lee, S.-G., et al. (2021). ppGpp signaling plays a critical role in virulence of acinetobacter baumannii. Virulence 12 (1), 2122–2325. doi: 10.1080/21505594.2021.1961660
Kröger, C., MacKenzie, K. D., Alshabib, E. Y., Kirzinger, M. W.B., Suchan, D. M., Chao, T.-C., et al. (2018). The primary transcriptome, small RNAs and regulation of antimicrobial resistance in acinetobacter baumannii ATCC 17978. Nucleic Acids Res. 46 (18), 9684–9698. doi: 10.1093/nar/gky603
Liu, H., Xiao, Y., Nie, H., Huang, Q., Chen, W. (2017). Influence of (p)ppGpp on biofilm regulation in pseudomonas putida KT2440. Microbiol. Res. 204 (November), 1–8. doi: 10.1016/j.micres.2017.07.003
López-Martín, M., Dubern, J.-F., Alexander, M. R., Williams, P. (2021). AbaM regulates quorum sensing, biofilm formation, and virulence in acinetobacter baumannii. J. Bacteriol. 203 (8), e00635–e00205. doi: 10.1128/JB.00635-20
Love, M. I., Huber, W., Anders, S. (2014). Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15 (12), 5505. doi: 10.1186/s13059-014-0550-8
Moeck, G. S., Coulton, J. W. (1998). TonB-dependent iron acquisition: mechanisms of siderophore-mediated active transport†. Mol. Microbiol. 28 (4), 675–815. doi: 10.1046/j.1365-2958.1998.00817.x
Norton, M. D., Spilkia, A. J., Godoy, V. G. (2013). Antibiotic resistance acquired through a DNA damage-inducible response in acinetobacter baumannii. J. Bacteriol. 195 (6), 1335–1455. doi: 10.1128/JB.02176-12
Paley, S. M., Karp, P. D. (2006). The pathway tools cellular overview diagram and omics viewer. Nucleic Acids Res. 34 (13), 3771–3785. doi: 10.1093/nar/gkl334
Paley, S., Parker, K., Spaulding, A., Tomb, J.-F., O’Maille, P., Karp, P. D. (2017). The omics dashboard for interactive exploration of gene-expression data. Nucleic Acids Res. 45 (21), 12113–12245. doi: 10.1093/nar/gkx910
Park, S., Cho, H. (2022). The tol-pal system plays an important role in maintaining cell integrity during elongation in escherichia coli. Front. Microbiol. 13. doi: 10.3389/fmicb.2022.891926
Pavlin, A., Bajc, G., Fornelos, N., Browning, D. F., Butala, M. (2022). The small ddrR protein directly interacts with the umuDAb regulator of the mutagenic DNA damage response in acinetobacter baumannii. J. Bacteriol. 204 (3), e00601215. doi: 10.1128/jb.00601-21
Peleg, A. Y., Seifert, H., Paterson, D. L. (2008). Acinetobacter baumannii: emergence of a successful pathogen. Clin. Microbiol. Rev. 21 (3), 538–825. doi: 10.1128/CMR.00058-07
Peterson, M. A., Grice, A. N., Hare, J. M. (2020). A corepressor participates in lexA-independent regulation of error-prone polymerases in acinetobacter. Microbiology 166, 212–265. doi: 10.1099/mic.0.000866
Philippe, C., Valcek, A., Whiteway, C., Robino, E., Nesporova, K., Bové, M., et al. (2022). What are the reference strains of acinetobacter baumannii referring to? bioRxiv 1-36. doi: 10.1101/2022.02.27.482139
Robinson, A., Brzoska, A. J., Turner, K. M., Withers, R., Harry, E. J., Lewis, P. J., et al. (2010). Essential biological processes of an emerging pathogen: DNA replication, transcription, and cell division in acinetobacter spp. Microbiol. Mol. Biol. Reviews.: MMBR. 74 (2), 273–975. doi: 10.1128/MMBR.00048-09
Seyednasrollah, F., Laiho, A., Elo, L. L. (2015). Comparison of software packages for detecting differential expression in RNA-seq studies. Briefings Bioinf. 16 (1), 59–705. doi: 10.1093/bib/bbt086
Smith, M. G., Gianoulis, T. A., Pukatzki, S., Mekalanos, J. J., Ornston, L. N., Gerstein, M., et al. (2007). New insights into acinetobacter baumannii pathogenesis revealed by high-density pyrosequencing and transposon mutagenesis. Genes Dev. 21, 601–614. doi: 10.1101/gad.1510307
Sun, X., Ni, Z., Tang, J., Ding, Y., Wang, X., Li, F. (2021). The abaI/abaR quorum sensing system effects on pathogenicity in acinetobacter baumannii. Front. Microbiol. 12. doi: 10.3389/fmicb.2021.679241
Tacconelli, E., Carrara, E., Savoldi, A., Harbarth, S., Mendelson, M., Monnet, D. L., et al. (2018). Discovery, research, and development of new antibiotics: the WHO priority list of antibiotic-resistant bacteria and tuberculosis. Lancet. Infect. Dis. 18 (3), 318–327. doi: 10.1016/S1473-3099(17)30753-3
Uddin, R., Masood, F., Azam, S. S., Wadood, A. (2019). Identification of putative non-host essential genes and novel drug targets against acinetobacter baumannii by in silico comparative genome analysis. Microbial. Pathogenesis. 128, 28–35. doi: 10.1016/j.micpath.2018.12.015
Visca, P., Seifert, H., Towner, K. J. (2011). Acinetobacter infection – an emerging threat to human health. IUBMB Life 63 (12), 1048–1545. doi: 10.1002/iub.534
Wang, N., Ozer, E. A., Mandel, M. J., Hauser, A. R. (2014). Genome-wide identification of acinetobacter baumannii genes necessary for persistence in the lung. mBio 5 (3), 6033-41. doi: 10.1128/mBio.01163-14
Weaver, A. I., Jiménez-Ruiz, V., Tallavajhala, S. R., Ransegnola, B. P., Wong, K. Q., Dörr, T. (2019). Lytic transglycosylases rlpA and mltC assist in vibrio cholerae daughter cell separation. Mol. Microbiol. 112 (4), 1100–11155. doi: 10.1111/mmi.14349
Weber, B. S., Ly, P. M., Irwin, J. N., Pukatzki, S., Feldman, M. F. (2015). A multidrug resistance plasmid contains the molecular switch for type VI secretion in acinetobacter baumannii. Proc. Natl. Acad. Sci. 112 (30), 9442–9475. doi: 10.1073/pnas.1502966112
Wijers, C. D.M., Pham, L., Menon, S., Boyd, K. L., Noel, H. R., Skaar, E. P., et al. (2021). Identification of two variants of acinetobacter baumannii strain ATCC 17978 with distinct genotypes and phenotypes. Infection. Immun. 89 (12), e00454215. doi: 10.1128/IAI.00454-21
Williams, C. L., Neu, H. M., Alamneh, Y. A., Reddinger, R. M., Jacobs, A. C., Singh, S., et al. (2020). Characterization of acinetobacter baumannii copper resistance reveals a role in virulence. Front. Microbiol. 11. doi: 10.3389/fmicb.2020.00016
Williams, C. L., Neu, H. M., Gilbreath, J. J., Michel, S. L.J., Zurawski, D. V., Scott Merrell, D. (2016). Copper resistance of the emerging pathogen acinetobacter baumannii. Appl. Environ. Microbiol. 82 (20), 6174–6885. doi: 10.1128/AEM.01813-16
Yakhnina, A. A., Bernhardt, T. G. (2020). The tol-pal system is required for peptidoglycan-cleaving enzymes to complete bacterial cell division. Proc. Natl. Acad. Sci. 117 (12), 6777–6835. doi: 10.1073/pnas.1919267117
Keywords: DNA damage response, ddrR, Acinetobacter baumannii, RNAseq, pathway analysis
Citation: Cook D, Flannigan MD, Chariker JH and Hare JM (2024) DNA damage response coregulator ddrR affects many cellular pathways and processes in Acinetobacter baumannii 17978. Front. Cell. Infect. Microbiol. 13:1324091. doi: 10.3389/fcimb.2023.1324091
Received: 18 October 2023; Accepted: 20 December 2023;
Published: 11 January 2024.
Edited by:
Kaiying Cheng, Hangzhou Normal University, ChinaReviewed by:
Gisela Di Venanzio, Washington University in St. Louis, United StatesCopyright © 2024 Cook, Flannigan, Chariker and Hare. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Janelle M. Hare, am0uaGFyZUBtb3JlaGVhZHN0YXRlLmVkdQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.