 Liying Zhu
Liying Zhu Xuejiao Ji3†
Xuejiao Ji3† Feifei Wang
Feifei Wang- 1Technical Center for Animal, Plant and Food Inspection and Quarantine of Shanghai Customs, Shanghai, China
- 2Key Laboratory of Medical Molecular Virology (MOE/NHC/CAMS) and Shanghai Institute of Infectious Disease and Biosecurity, Shanghai Frontiers Science Center of Pathogenic Microorganisms and Infection, School of Basic Medical Sciences, Fudan University, Shanghai, China
- 3Shanghai Clinical Research Center for Infectious Disease (tuberculosis), Shanghai Pulmonary Hospital, Tongji University School of Medicine, Shanghai, China
Listeria monocytogenes (Lm) is associated with severe foodborne infections and ubiquitous in the nature. Identification of characteristics of Lm transmission through trading of food products is essential for rapidly tracking Lm sources and controlling dissemination of listeriosis. In this study, a total of 44 Lm strains were isolated from food products originating from 14 countries/regions during 2003-2018 at the Shanghai port. The genomes of these Lm strains were sequenced by high-throughput sequencing. Multilocus sequence typing (MLST) analysis showed that 43 isolates were divided into 17 sequence types (STs). The distribution of STs was decentralized, with the dominant ST2 accounting for only 18.18% of the strains. The LM63 strain did not match with any of the existing STs. Core-genome MLST (cgMLST) analysis based on 1748 core genes categorized the 44 strains into 30 cgMLST types (CTs), with CT10153 and CT7892 as the most predominant CTs. Notably, LM63 and LM67 shared the same CT in the cgMLST analysis. The phylogenetic analysis based on single-copy homologous genes revealed that the 44 Lm strains were primarily classified into two lineages. The SNP analysis also indicated that these strains were roughly divided into two clades, with strains in the first clade mainly collected earlier than those in the second clade, which were predominantly collected from 2010 onwards. The analysis using the virulence factor database (VFDB) indicated that the virulence gene inlJ was the most prevalent among these 44 strains. Notably, ddrA, msbA, and sugC were enriched in this dataset, requiring further clarification of their roles in Listeria through future studies. These results might provide a clue for understanding of the global epidemiology and surveillance of Lm and present insights for implementing effective measures to reduce or prevent Listeria contamination outbreaks in imported food products.
1 Introduction
Listeria monocytogenes (Lm), a facultative anaerobic Gram-positive bacterium, is a common foodborne pathogen ubiquitous in the nature (Bolívar and Pérez-Rodríguez, 2023). Upon infection of a susceptible host, Lm can cause a severe systemic infection, namely listeriosis, which can manifest as meningitis, sepsis, and even lead to abortion in pregnant women. Listeriosis is especially harmful to the elderly, pregnant women, children, newborns, and immunocompromised individuals(Koopmans et al., 2023). It is essential to recognize listeriosis outbreaks promptly and trace them to their food sources to prevent further infections.
Outbreak of listeriosis has been frequently reported in numerous countries worldwide (Moura et al., 2016; Halbedel et al., 2018; Shi et al., 2021), including China. For instance, listeriosis surveillance data collected from 2013 to 2017 revealed that 211 listeriosis cases were diagnosed in 64 sentinel hospitals of China (Li et al., 2019). Another study indicated that 155 Lm strains were isolated from different food products, such as ready-to-eat food, raw meat, raw poultry, and raw seafood in Shanghai (China) from 2009 to 2019 (Zhang et al., 2020). Recently, Lm contamination was identified in the ready-to-eat meat processing plants of two factories in Shanghai during 2019-2020 (Zhang et al., 2021a). These findings suggest that it is necessary to keep continuous monitoring and stringent surveillance of Lm strains due to the potential risk of listeriosis outbreak in Shanghai, which is a prestigious international trade center with a huge amount of ready-to-eat food import every year. However, the relationship between Lm carried by these imported foods and the outbreak of listeriosis in Shanghai has not been reported.
The pathogenicity of foodborne bacteria is closely associated with their antibiotic resistance and virulence (Banerji et al., 2021; Dubey et al., 2023). Strains that exhibit a strong antibiotic resistance can survive in unfavorable growth environments (Dharmasena et al., 2021). Besides, highly virulent strains can lead to more severe disease (Dubey et al., 2023). Accordingly, the antibiotic resistance and virulence of bacteria are mainly determined by the antibiotic resistance or pathogenic genes.
Multilocus sequence typing (MLST) analysis, one type of nucleotide sequence-based analysis methods concentrating on the single-nucleotide polymorphisms (SNPs) of Lm housekeeping genes, is widely utilized in the classification of Lm strains into different sequence types (STs) despite the low resolution (Maiden et al., 1998; Revazishvili et al., 2004). The results of epidemiological studies indicated that Lm strains with ST87 and ST8 were the most dominant types isolated from Chinese patients (Li et al., 2019), while Lm stain with ST9 was the most common type isolated from food (Wang et al., 2012; Zhang et al., 2020). Therefore, the MLST analysis of Lm strains may promote the identification of the relationship between Lm isolated from different sources, as well as determining sources of contamination. With the widespread extension of whole-genome sequencing (WGS), WGS-based high-resolution molecular subtyping technology has provided a significantly improved discrimination capability to identify the molecular characteristics of Lm. The core-genome MLST (cgMLST) and SNPs methods have been extensively applied in the investigation of listeriosis outbreak (Chen et al., 2016; Jackson et al., 2016; Moura et al., 2016). Consequently, the molecular typing of Lm isolated from food and clinical patients may promote exploration of crucial correlations between listeriosis cases and contaminated food sources. This, in turn, aids in the effective tracking of the origins of food contamination (Chen et al., 2017).
In the present study, food samples were collected from 14 different countries or regions over a span of 15 years, followed by isolation, and sequencing of 44 Lm genomes. Subsequently, the SNP mutation profiles of these strains were analyzed, a phylogenetic tree was constructed, and the tree was examined in the context of time and geographical location. The results revealed similarities among Lm strains from different regions at various time periods, suggesting transmission of these strains. These findings contribute to the understanding of the global epidemiology of listeriosis and underline the importance of monitoring of imported food products to prevent the outbreak of this disease.
2 Materials and methods
2.1 Collection of samples and bacterial DNA extraction
Among 49 isolates from 49 food samples collected from 14 countries or regions in East Asia (including China and Vietnam), Europe, North America, and South America during the period from 2003 to 2018, a total of 44 Lm strains were successfully identified at the Shanghai port of China. The remaining 5 samples were identified as contaminated with non-Listeria strains and were not used for further analysis. The isolated strains were obtained from samples of meat and chilled products that were imported from Shanghai Port. The demographic characteristics of the isolated strains are listed in Table 1.
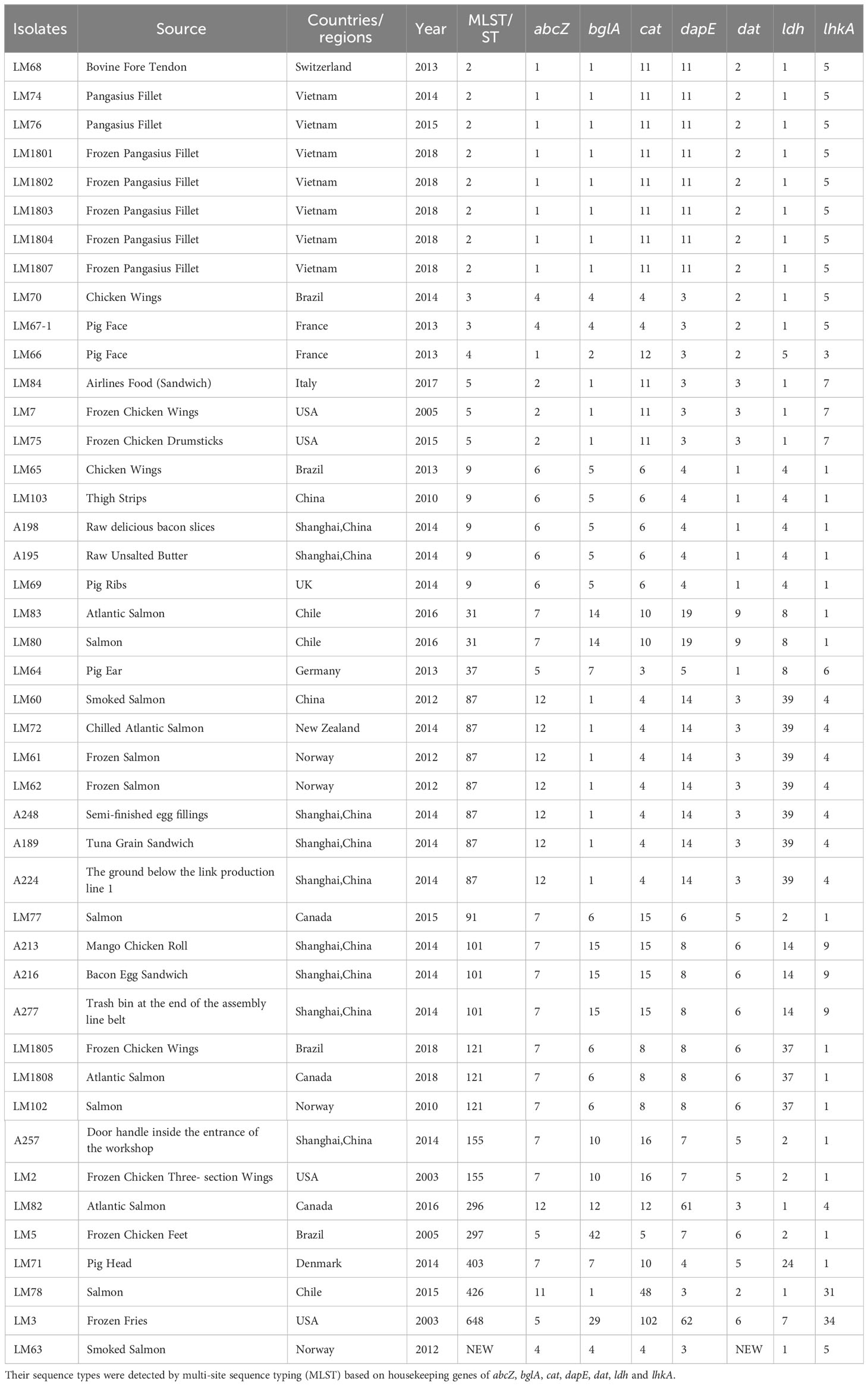
Table 1 The demographic characteristics of 44 L. monocytogenes isolated strains.
Before collecting samples, the surface of food bags was cleaned according to the aseptic operation protocols. About 25 g of each sample was collected and put in a homogenizing cup, containing 225 mL of LB1 enrichment solution under aseptic operation, and samples were homogenized at 8000~10000 r/min for 1~2 min, followed by incubation at 30°C for 24 h. Next, 0.1 mL suspension was transferred to 10 mL of LB2 enrichment solution, and incubated at 30°C for 24 h. Next, LB2 solution was seeded onto the Listeria chromogenic agar plate (Cat No: NCM1004; Neogen, New York, USA), and incubated at 36°C for 24~48 h. Subsequently, 3-5 colonies were collected from each plate and identified using Vitek Compact 2 kit (Meriere Biological Co., Ltd., Paris, France), which is an automatic biochemical analyzer. All isolated strains were stored at -80°C. DNA from each strain was extracted via a bacterial genome extraction kit according to the manufacturer’s instructions (Qiagen, Hilden, Germany), followed by storing at -20°C for further sequencing.
2.2 WGS, assembly and evaluation
To perform WGS, DNA quality and concentration were initially evaluated using the Qubit dsDNA HS Assay kit (Thermo Fisher Scientific, Waltham, MA, USA). Subsequently, the paired-end genomic libraries of each isolate were constructed, and sequencing was carried out on an Illumina instrument in PE250 mode. The raw reads were trimmed, draft genomes were then assembled using SOAPdenovo (Ver. 2.04) system (Van Der Zwan et al., 2018), and the gap-filling and base correction of the assembly genomes were carried out using GapCloser (ver. 1.12) system (Xu et al., 2020). The assembly results were comprehensively evaluated according to the overall length of the scaffolds (ranging from 2884308 to 3119043 bp), the number of scaffolds (ranging from 9 to 38), and the scaffold N50 (ranging from 215544 to 1551995 bp). Finally, the optimal genome assembly results were selected according to the K-mer values.
2.3 MLST and cgMLST analysis
The housekeeping genes for Lm (abcZ, bglA, cat, dapE, dat, ldh and lhkA) were utilized for MLST profiles of all isolated strains (Ragon et al., 2008; Carroll et al., 2017). The assembled genomes were uploaded to the MLST database of Lm (https://pubmlst.org/multilocus-sequence-typing) to determine MLST profiles (Maiden et al., 1998; Jolley et al., 2018). The alleles of the 7 loci of these strains were obtained, and the ST genotypes of these strains were identified according to the number of nucleotide differences between alleles.
In cgMLST analysis, Lm GCA_9001872251.1 (GenBank Accession No. LT906436.1) was utilized as a reference gene, and the best cgMLST values of each sample were matched according to the cgMLST1748 database (https://bigsdb.pasteur.fr/listeria/listeria.html) (Moura et al., 2016, Zhang et al., 2021b). Samples with a locus detection rate greater than 97% were included in the subsequent cgMLST typing analysis. Finally, these 44 Lm strains were divided into 30 CTs, including CT6150, CT3102, CT9387, CT5746, CT4677, CT7735, CT12277, CT10, CT3014, CT9470, CT7892, CT10153, CT6244, CT14053, CT2053, CT13475, CT96, CT11266, etc. A minimum spanning tree was generated using PHYLOViZ based on the results of cgMLST (Francisco et al., 2012).
2.4 Phylogenetic analysis
For the SNP-based phylogenetic tree construction, using GCA_9001872251.1 as the reference sequence, the trimmed reads were aligned by the Nucmer (Ver. 4.0.0) (Leekitcharoenphon et al., 2012). The SNPs, including DNA sequence polymorphisms caused by single-based conversion, transversion, and insertion/deletion, of each isolated strain were called with VarScan (Ver. 2.3) (Koboldt et al., 2009). Besides, SNPs with low sequencing depth and alignment quality values were filtered out. Subsequently, the matrix of 34,128 SNPs from all strains with reference alleles was generated, and the phylogenetic tree was constructed using the FastTree (Ver. 2.0.0) (Price et al., 2009).
For single-copy gene-based phylogenetic tree construction, single-copy homologous genes from homologous gene analysis were selected for multiple sequence alignment using MAFFT (http://mafft.cbrc.jp/alignment/software/) (Ver. 7.0) (Katoh and Standley, 2013), and alignment quality control with Gblocks (http://molevol.cmima.csic.es/castresana/Gblocks.html) (Ver. 0.91b) (Talavera and Castresana, 2007). Finally, the single-copy gene phylogenetic tree was inferred using RAxML (https://github.com/stamatak/standard-RAxML) (Ver. 8.0) (Stamatakis, 2014).
2.5 Analysis of mobile genetic elements
To identify MGEs in Listeria, it was attempted to analyze the number of prophages, clustered regularly interspaced short palindromic repeats (CRISPRs), and insertion sequences (ISs) in genome DNA sequence of isolated strains. Prophages were predicted in the genomes of 44 strains using the PHASTER database (Arndt et al., 2016). According to the predictive values, the prophages were classified into three categories: predictive value < 70, incomplete prophage; 70 ≤ predictive value < 90, questionable prophage; 90 ≤ predictive value, intact prophage. The numbers of all prophages and intact prophages are displayed in Appendix Table 1. CRISPRs were predicted using Minced (Ver. 0.2.0; https://sourceforge.net/projects/minced/) in isolated samples (Moller and Liang, 2017). The relevant statistics are presented in Appendix Table 2. ISs were predicted through Basic Local Alignment Search Tool (BLAST) (evaluate = 1-5) and ISfinder databases (Siguier et al., 2006). Furthermore, ISs were predicted in all 44 samples, while only 18 samples had complete ISs. The number of ISs is listed in Appendix Table 3.
2.6 Analysis of virulence genes
The virulence factor genes of 44 Lm strains were identified using the Virulence factors database (VFDB) (Chen et al., 2005), and confirmed by BLAST to determine the virulence of each strain. Heatmap and hierarchical clustering graph of gene distribution were drawn according to the abundance of virulence factor genes in each isolated strain, and samples and virulence factors were clustered using the pheatmap R package (Ver. 1.0.12). Samples illustrating similar distribution patterns of different virulence factors were grouped close to each other on the clustering tree. Moreover, the origin region of sample, isolation time, and cgMLST classification were marked on the map.
3 Results
3.1 Characteristics of bacterial strains
To analyze the genotype characteristics and virulence factors of bacteria isolated from 49 food samples collected from 2003 to 2018, the genomic DNA was sequenced using high-throughput sequencing. Among them, 44 isolates were identified as Lm, 2 isolates were identified as Enterococcus faecalis, and the other three were mixtures of various bacteria. Isolates identified as Enterococcus faecalis and those that were mixtures of various bacteria were excluded from subsequent analyses.
The demographic characteristics of 44 Lm strains are summarized in Table 1. Briefly, 11 strains were isolated from food products and food-processing environments in China, 7 strains were isolated from frozen food products imported from Vietnam, 4 strains from Brazil, the United States, and Norway, 3 strains from Canada and Chile, 2 strains from France, and the remaining 6 strains from Denmark, Germany, Switzerland, New Zealand, Italy, and the United Kingdom, respectively (Table 1).
To identify the genetic and evolutionary relationships between these Lm strains, MLST genotyping analysis was performed on the 44 isolated strains. The results showed that the 43 isolates were classified into 17 STs. The distribution of ST types was relatively dispersed. Specifically, ST2 was the most prevalent type, representing only 18.18% (8/44) of the strains. This was followed by ST87 (7/44, 15.91%), ST9 (5/44, 11.36%), with three occurrences each of ST5, ST101, and ST121, and two instances each of ST3, ST31, and ST155. The remaining ST types, including ST4, ST37, ST91, ST296, ST297, ST403, ST426, and ST648, each appeared only once (Table 1). Furthermore, the last strain, LM63 achieved from Norway in 2012, could not be classified as any of the existing STs because of an unknown genotype in the dat gene containing a new SNP, demonstrating that the mentioned strain might have a temporarily unknown activity or function.
3.2 cgMLST analysis of L. monocytogenes isolates
To achieve high-resolution species typing results, cgMLST analysis was carried out and the minimum spanning tree was drawn based on 1748 core gene loci (Figure 1). These 44 Lm strains were divided into 30 CTs. The CT10153 and CT7892 were the dominant types, while the CT10153, CT13475, and CT11266 had a relatively close genetic evolutionary distance, and the strains of CT10153 and CT13475 were all particularly isolated from imported food products from Vietnam at different time periods. The 10 strains collected from China between 2012 and 2014 exhibited distant genetic relationships, suggesting that the spread of Lm strains had also become more widespread over time. Moreover, LM63, belonging to none of existing STs in MLST analysis, was classified into the same type of CT9611 with LM67 strain in the cgMLST analysis.
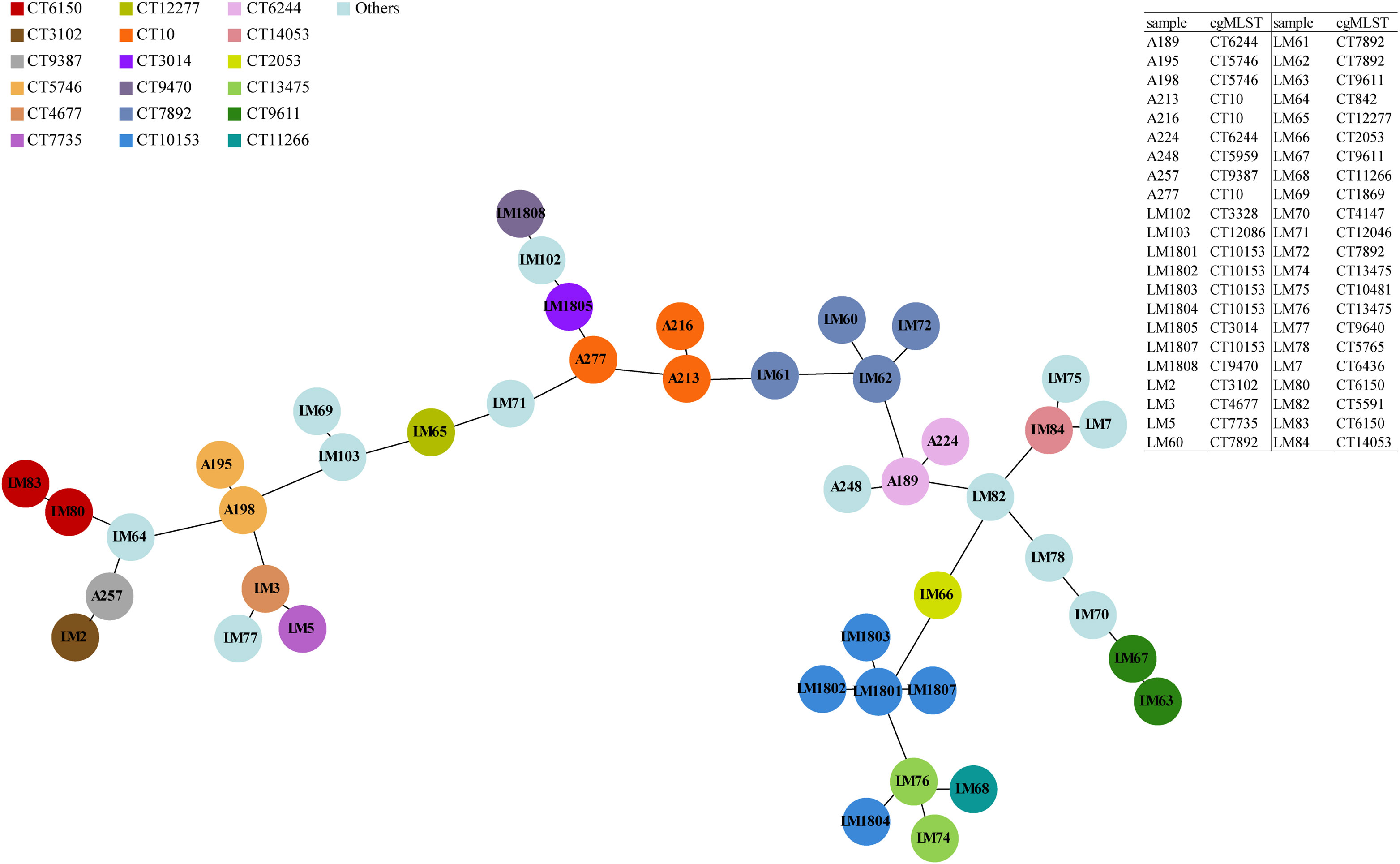
Figure 1 Phylogenetic relationships of L. monocytogenes strains as illustrated by a Minimum Spanning Tree. The minimum spanning tree is based on cgMLST and represents the relationships among 44 Lm strains with 30 distinct CTs. Each circle symbolizes an isolate, colored according to its specific CT (referenced in the key at the top left). Links between circles denote phylogenetic distances between isolates.
3.3 Genomic elements and phylogeny of 44 L. monocytogenes
Mobile genetic elements (MGEs) are potent drivers of genome evolution (Matereke and Okoh, 2020). To identify the impact of MGEs on Lm genome evolution, it was attempted to analyze the relationship of Lm evolution with the number of prophages, CRISPRs, and ISs in genome.
Prophages are a class of MGEs that are ubiquitous in bacterial genomes, and they also mediate transduction process causing the horizontal transfer of genetic materials between bacteria, including toxicity and antibiotic resistance genes (Vu et al., 2021). In the present study, prophages with the predictive values in the genomes of 44 Lm strains were predicted using the PHASTER database. Higher values indicated a greater likelihood of the fragment being incorporated into intact phages. The number of intact prophages in each sample is presented in Appendix Table 1. The majority of these prophages were Lm phages.
The CRISPR system is an acquired immune system that is widely existed in bacteria. It plays an important role in maintaining the stability of bacterial genome. The CRISPR system was predicted using MinCED (Ver. 0.2.0), and the CRISPR system was identified in 41 Lm samples. The relevant statistics are summarized in Appendix Table 2.
ISs are important genetic elements that drive the evolution of bacterial genome and lead to the instability of genome. In the present study, ISs were predicted through the BLAST (evaluate = 1-5) and ISfinder databases. Notably, ISs were predicted in all 44 Lm samples, while only 18 samples had complete ISs. The number of ISs is presented in Appendix Table 3. Most of these ISs were from Lm, and a limited number of them were from Leucanostoc mesenteroides. Among them, there was only one encoding gene of transposase, and no other functional elements were involved.
It was reported that MGEs are ideal loci for bacteria typing and evolutionary analysis (Frost et al., 2005; Botelho et al., 2019; Weisberg and Chang, 2023). In the present study, a phylogenetic tree was constructed by RAxML based on single-copy gene. The results revealed that 44 Lm strains were mainly classified into two lineages (Figure 2). One lineage consisted of 20 strains, spanning from A257 to LM83, while the remaining strains were in the other lineage.
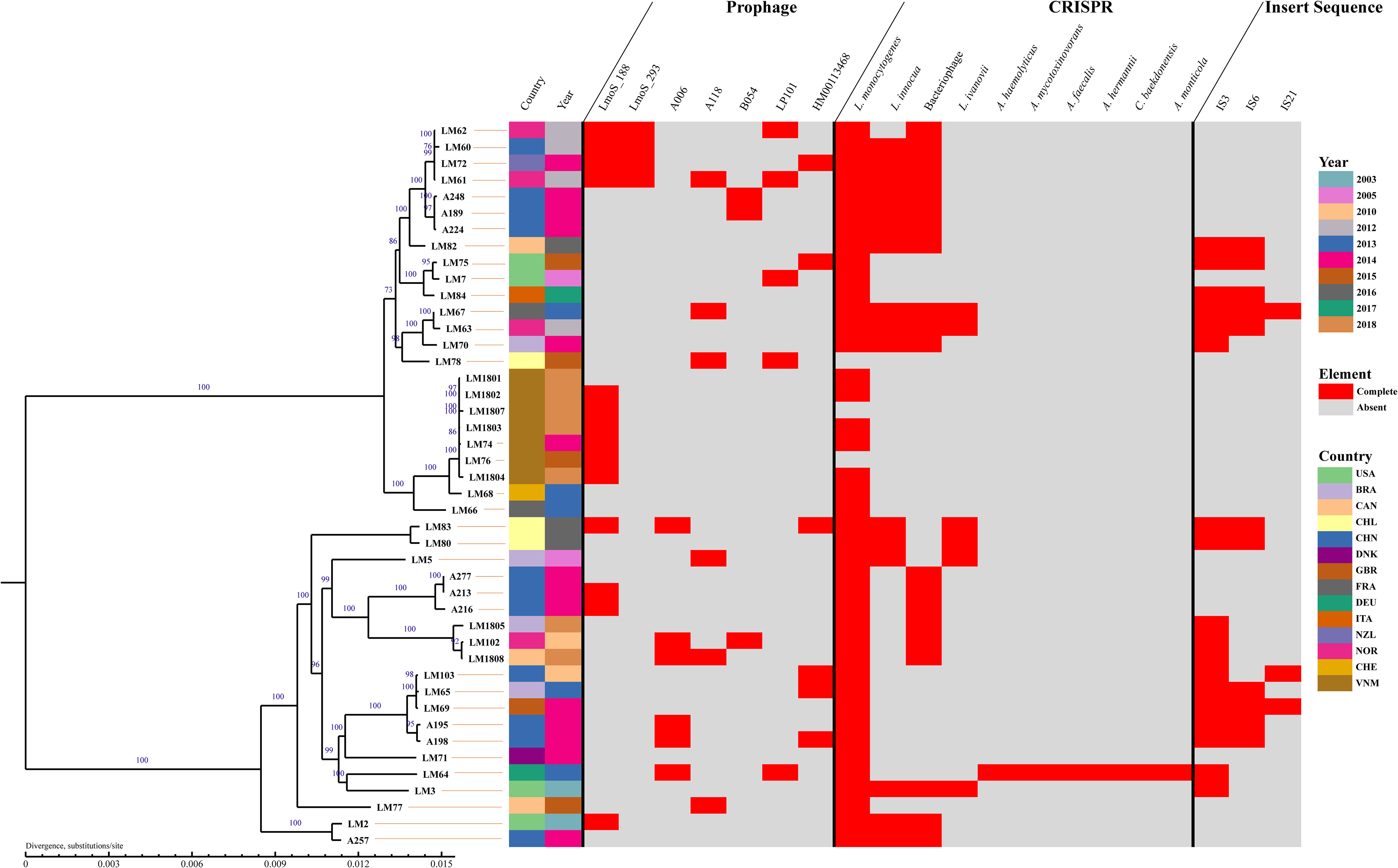
Figure 2 Phylogeny and mobile genomic elements of 44 L. monocytogenes strains. Using the RAxML software, a phylogenetic tree was constructed from gene sequences of identified homologous genes. Depicted within the tree are complete prophages, CRISPR, and IS identified in each strain. Country abbreviations: CHN-China, USA-the United States, BRA-Brazil, DNK-Denmark, DEU-Germany, FRA-France, CAN-Canada, NOR-Norway, CHE-Switzerland, NZL-New Zealand, ITA-Italy, GBR-the United Kingdom, VNM-Vietnam, CHL-Chile.
3.4 The whole-genome-based SNP phylogenetic analysis
Phylogeny is utilized to analyze the relationship among organisms, and it is widely applied in phylogenetic classification, epidemiology, and ecology (Lüth et al., 2020). To identify the epidemiological characteristics, the analysis was carried out based on SNP of these Lm strains at different time periods and from different countries. It was found that there were numerous SNP sites in 44 Lm strains.
Using Lm genome assembly GCA_900187225.1 published by the Sanger sequencing center in the UK in 2017 as the reference strain (Kremer et al., 2017), the 44 Lm strains were subsequently subjected to the whole-genome-based SNP phylogenetic analysis. These 44 strains could be roughly classified into two clades (Figure 3), in which the strains from LM5 to A195 belonged to the first clade, and the remaining strains were categorized as the second clade. The second clade had significantly more strains, whereas there were some differences between strains. In order to explore the reasons related to these differences, the evolutionary tree was annotated according to the time of collection and geographical regions of the strains.
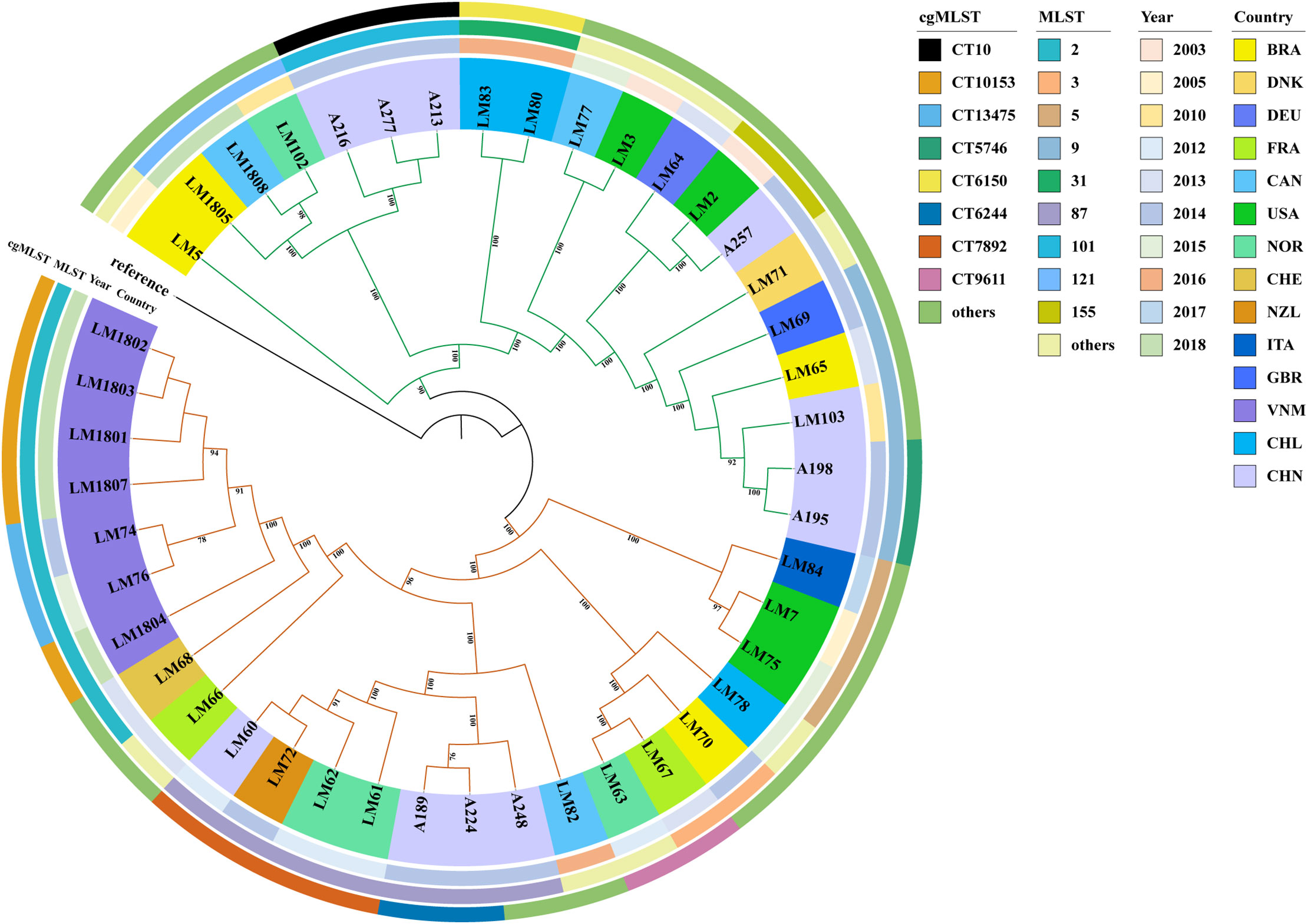
Figure 3 SNPs-based phylogenetic tree. The maximum-likelihood phylogenetic tree was established using34,128 SNPs called from 44 Lm strains. The reference strain was Lm GCA_900187225.1 published by Sanger sequencing center in the UK. All isolates were clustered into two distinct clades. The color strips in outer circle represent different cgMLST genotypes, those in middle circle represent different collecting time (year), and those in inner circle represent different collecting regions.
Significant differences were found in the time of Lm strain collection between the first and second clades. In the first clade, most of Lm strains were obtained in earlier. The Lm strains in the second clade were mainly collected from 2010 and later. The LM5 strain obtained in 2005 was the most similar to the reference.
Additionally, the characteristics of bacteria collected from different regions were analyzed in the phylogenetic tree. The relationship between Lm strains from Vietnam was relatively close, and there was also a certain relationship between Lm strains spanning 11 years (from 2003 to 2014) from other countries. In the first clade, the bacterial strains were collected over an approximately 11-year period, ranging from 2003 to 2014, and strains from the same regions exhibited similarity. This indicated that there was a mutual relationship between Lm strains from different time periods or regions.
3.5 Analysis of virulence genes
Virulence factors produced by pathogens play an crucial role in the occurrence of diseases (Wartha et al., 2023). To evaluate the virulence of these Lm strains, the virulence factors were identified through analyzing the genome sequences of these 44 strains in the VFDB (Chen et al., 2005; Matto et al., 2022). It was revealed that the majority of the known virulence factors associated with Lm were present in the 44 Listeria strains, inlJ was the richest one, and they were also rich in inlA and lap (Figure 4A). In Lm, inlJ and lap were associated with cell adhesion, and inlA was correlated with bacterial migration (Osek and Wieczorek, 2022). Meanwhile, 6 strains (LM3, LM5, LM64, LM71, LM80, and LM83) did not contain vip virulence gene. Moreover, it was found that some other common virulence factors did not exist in the 44 Lm strains, including the exoenzyme SMase, the pgdA gene related to immune evasion, the auto, inlP, and p60 genes in migration, the svpA gene in iron ion absorption, the peptidase Lsp, and the LLO and LLS encoding genes related to toxin.
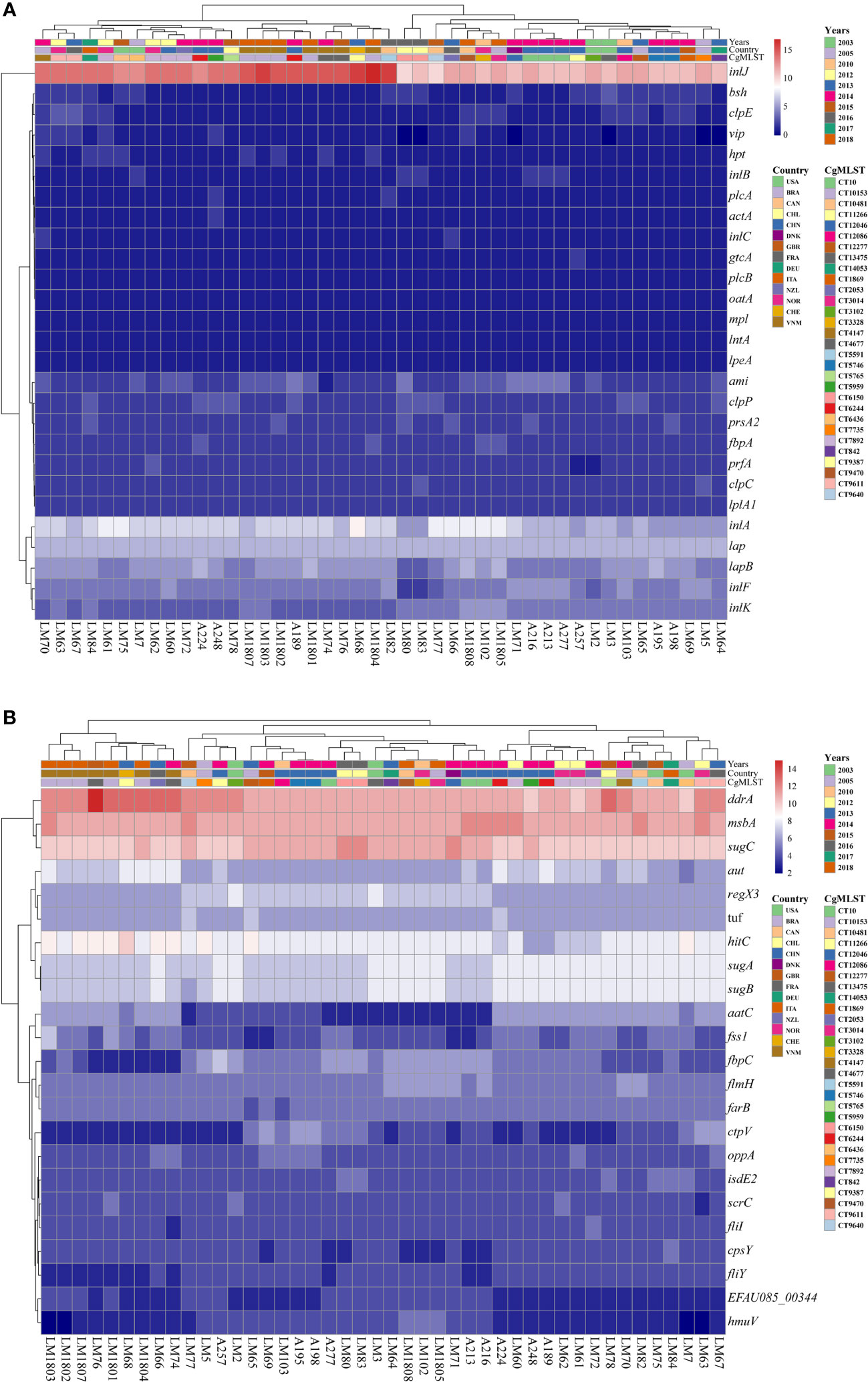
Figure 4 Virulence factor analysis results. Heatmap and hierarchical clustering was performed based on gene copies of virulence factors common across the Lm strains (A) or virulence factors with high abundance across our dataset (B) using Euclidean distance measurement method, and was visualized with the “pheatmap” package of R. The color key indicates the abundance of the virulence genes. Red: high abundance, Blue: low abundance.
In addition to the known virulence factors associated with Lm, the dataset exhibited an enrichment of other factors compared with VFDB. The heatmap showed that these 44 strains had the highest abundance of ddrA, msbA, and sugC genes, followed by hitC, sugA, and sugB genes (Figure 4B). Interestingly, the ddrA gene exhibited a higher copy number mainly in the strains from Vietnam, compared to strains derived from other countries/regions. However, the roles of these genes were not reported, and further studies are required to clarify their roles in Listeria.
4 Discussion
In the present study, 49 bacterial strains were collected from food samples originating from 14 countries/regions spanning 15 years. Their genomic DNA was sequenced using high-throughput sequencing technology. Among them, five isolated strains (A212, LM6, LM79, LM4, and LM1806) were found to be contaminated and were therefore excluded from further analysis. As a result, a total of 44 Lm strains were analyzed in terms of their genomic sequences to investigate the bacterial genotypes and SNPs. MLST is a widely utilized method for bacterial genome typing based on DNA sequences. It was initially developed in 1998 and has been used to discern natural genetic variations in N. meningitidis (Maiden et al., 1998). In contrast to traditional molecular biology genotyping methods, MLST possesses higher resolution and the ability to readily distinguish bacterial subtypes, highly facilitating the exploration of the relationship between bacterial subtypes and diseases. Currently, a huge amount of MLST data related to Listeria are stored in databases. In the present study, MLST typing analysis was conducted on the genomic sequence data of 44 Lm strains. As indicated in Table 1, it was found that the Lm strain LM63, isolated from Norway in 2012, had an unknown typing result due to the presence of new SNP information in the dat gene, a gene essential for D-alanine biosynthesis. D-alanine, not synthesized by vertebrates, is a crucial component required for bacterial cell wall synthesis and growth. While the deletion of the dat gene did not exhibit reversion or significant suppression of its auxotrophy under laboratory conditions, it alleviated the toxicity of Lm and could potentially serve as an attenuated vaccine (Zhao et al., 2005). This suggests that mutations in the dat gene may play a crucial role in regulation of Lm function. Although there were differences in the typing of other strains except for LM63, they were relatively centralized in genotype.
Subsequently, these genomic data were utilized to construct phylogenetic trees, and the evolutionary relationships between these isolated Lm strains from different regions and at different time periods were analyzed. Notably, SNPs, as polymorphisms in genomic sequences, are highly appropriate for survival analysis in the context of evolution (Hammarlof et al., 2018; Losic et al., 2020). In the present study, it was found that these 44 Lm strains had different SNP loci, and a phylogenetic tree was constructed based on these SNPs. Compared with the reference genome sequence, the 44 Lm strains were roughly divided into two clades. There was a difference in sampling time between strains in the first clade and those in the second clade. In addition, Lm strains obtained in earlier years were mainly concentrated in the first clade. Strains in the second clade were mostly collected from 2010 onwards. The strain LM5, obtained in 2005, displayed the highest similarity to the reference genome.
Research has indicated that the evolution of bacteria follows distinct biogeographic patterns (Martiny et al., 2006; Li and Hu, 2021; Gao et al., 2023). Huang et al. discovered the distinct evolutionary trajectories of the spread of E. coli ST1193 in China and abroad, and this was confirmed by the hybrid analysis of core genome and accessory genome phylogeny, virulence factors, antibiotic resistance genes, and plasmid replicon profiles (Huang et al., 2021). Similar results were obtained in the present study. It was found that Lm strains isolated from Vietnamese products clustered well together in the minimum spanning tree analysis, SNP-based evolutionary analysis, single-copy gene-based evolutionary analysis, mobile element analysis (including prophages, ISs, CRISPRs), and virulence gene analysis. Moreover, the results of evolutionary analysis indicated that these strains were different from others isolated from products of other countries/regions. Furthermore, Lm strains isolated from Chilean products were also relatively clustered together. These findings indicated that Lm had been evolved independently in specific geographical regions.
Additionally, it was found that the evolutionary relationships between Lm strains from Vietnam were relatively close. Furthermore, it was noted that the genetic relationships between Lm strains from different countries/regions were relatively close, which not only existed within the same continents, such as between the United States and Canada (LM77 and LM3), France and Norway (LM67-1 and LM63), but also found between intercontinental countries, including China and Brazil, China and the United States, China and New Zealand, Vietnam and Sweden, Vietnam and France, and the United States and Italy. The transmission of Lm strains between different regions might be achieved through direct vertical or horizontal gene transfer. According to the results of evolutionary analysis and the isolated time of Lm, it was hypothesized that strains existed in Vietnam might be spread to Vietnam through food trade after their transmission within Europe. This might be attributed to the biological characteristics of Lm, which is existed ubiquitously in the nature, it can survive and reproduce even at low temperatures, and it can easily spread into food processing environments and cause food contamination. Nonetheless, these findings are insufficient to rule out the effects of other traded products on the spreading of Lm. Moreover, it was revealed that there were certain similarities among strains from different regions spanning 11 years (from 2003 to 2014), suggesting that there might be possible transmission events of Lm strains between different sources and time periods.
Finally, the virulence genes involved in these 44 Lm strains were analyzed. Virulence is described as an ability of an organism to infect the host and cause a disease. Virulence factors, which are either secretory, membrane associated or cytosolic in the nature, comprise a group of molecules produced by pathogenic microorganisms that may cause disease in the host (Sharma et al., 2017). The VFDB database was established by the Institute of Pathogenic Microbiology, Chinese Academy of Medical Sciences, which is an integrated and comprehensive online database for curating information related to virulence factors of bacterial pathogens, and it is widely recognized due to its up-to-date status (Chen et al., 2005). In the VFDB database, there were 36 major virulence factors identified in Lm, including actin-based motility, adherence, bacterial cell wall modification, bile resistance, exoenzyme, immune evasion, immunomodulator, intracellular growth, invasion, iron uptake, metabolic adaptation, peptidase, regulation, stress protein, and toxin (Chen et al., 2005). In the present study, VFDB database was employed to annotate the virulence factors of the 44 Lm strains. The results showed that the 44 strains were all enriched in genes, such as inlJ, inlA, and lap. Besides, inlJ and lap genes were related to cell adhesion, and inlA gene was related to bacterial migration. These results demonstrated that there were similarities in virulence genes among different strains.
In the analysis of common virulence genes, it was indicated that the Lm strains contained the consistent compositions of virulence genes. This demonstrates that there might be no significant differences in human diseases caused by Lm infection. Among isolated strains, only 6 strains (LM3, LM5, LM64, LM71, LM80, and LM83) did not contain vip virulence gene, which might be attributed to random gene loss as there was no association between the time and region of their isolation. Prior researches have shown that vip gene related to migration did not exist in all nonpathogenic Listeria species as well as in particular in two rare Lm serovars (4a, 4c) (Doumith et al., 2004; Cabanes et al., 2005). Multiple studies have found that knocking out vip gene in Lm had no significant effect on the bacterial growth. These results indicated that vip gene may have other functions in the growth of Lm. Nevertheless, there were significant difference in the gene copy number of common virulence genes among these isolated Lm strains, especially in inlJ gene, which encoded internalin composed of 916 amino acids and presented only in Lm. Those inlJ gene knocking-out strains exhibited a lower invasive ability of Lm (Sabet et al., 2005), and this gene was missed in avirulent Listeria species (Liu et al., 2003; Doumith et al., 2004). Accordingly, the isolated strains could be classified into two distinct groups based on the copy number of the inlJ gene, suggesting that these strains might present significant differences in disease severity. However, there is no study concentrating on the relationship between the copy number of the inlJ gene and the disease severity, and this should be investigated in the future studies.
Notably, it was found that the copy number of virulence genes, such as inlJ and ddrA genes, in Lm strains isolated from Vietnam, and their closely related strains isolated from Sweden and France, was significantly higher than that from other regions. This not only indicated that there were specific biogeographical characteristics in Lm evolution and the possibility of Lm cross regional transmission, but also suggested the likelihood of cross-regional trade, leading to outbreak of foodborne diseases caused by Lm infection in other regions.
According to the proposed comprehensive monitoring system for imported products carried Lm, a reliable relationship was explored between foodborne diseases and potential sources. Given the feasibility of tracking imported food products, it becomes possible to promptly trace the origins of foodborne pathogenic strains responsible for disease outbreaks. By leveraging traceability results, appropriate measures can be taken, drawing from the treatment experience in the source areas. This approach allows for the rapid containment of foodborne diseases, thereby mitigating the associated social and economic losses. Therefore, the characterization of the foodborne pathogen Lm might play a crucial role in preventing further infections through rapid identification and precise allocation of contaminated food sources.
Data availability statement
The data presented in the study are deposited in the NCBI Bioproject repository, accession number PRJNA1036137..
Author contributions
LZ: Data curation, Formal analysis, Methodology, Software, Visualization, Writing – original draft. XJ: Data curation, Formal analysis, Writing – original draft. YW: Data curation, Formal analysis, Writing – review & editing. WX: Resources, Writing – review & editing. FW: Conceptualization, Funding acquisition, Methodology, Project administration, Supervision, Writing – review & editing. XH: Conceptualization, Funding acquisition, Project administration, Supervision, Writing – review & editing.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This work was supported by National Key Research and Development Program of China (2022YFF1103105). Shanghai Natural Science Foundation of Grant (19ZR1470400, 19ZR1470400). Shanghai Municipal Science and Technology Major Project (ZD2021CY001).
Acknowledgments
We gratefully thank Shanghai WinnerBio Technology Co., Ltd. (Shanghai, China) for the assistance with bioinformatics and statistical analyses.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fcimb.2023.1287564/full#supplementary-material
References
Arndt, D., Grant, J. R., Marcu, A., Sajed, T., Pon, A., Liang, Y., et al. (2016). PHASTER: a better, faster version of the PHAST phage search tool. Nucleic Acids Res. 44, W16–W21. doi: 10.1093/nar/gkw387
Banerji, R., Karkee, A., Kanojiya, P., Saroj, S. D. (2021). Pore-forming toxins of foodborne pathogens. Compr. Rev. Food Sci. Food Saf. 20, 2265–2285. doi: 10.1111/1541-4337.12737
Bolívar, A., Pérez-Rodríguez, F. (2023). Listeria in food: prevalence and control. Foods 12, 1378. doi: 10.3390/foods12071378
Botelho, J., Grosso, F., Peixe, L. (2019). Antibiotic resistance in Pseudomonas aeruginosa - Mechanisms, epidemiology and evolution. Drug Resist. Update 44, 100640. doi: 10.1016/j.drup.2019.07.002
Cabanes, D., Sousa, S., Cebria, A., Lecuit, M., Garcia-Del Portillo, F., Cossart, P. (2005). Gp96 is a receptor for a novel Listeria monocytogenes virulence factor, Vip, a surface protein. EMBO J. 24, 2827–2838. doi: 10.1038/sj.emboj.7600750
Carroll, L. M., Kovac, J., Miller, R. A., Wiedmann, M. (2017). Rapid, High-Throughput Identification of Anthrax-Causing and Emetic Bacillus cereus Group Genome Assemblies via BTyper, a Computational Tool for Virulence-Based Classification of Bacillus cereus Group Isolates by Using Nucleotide Sequencing Data. Appl. Environ. Microbiol. 83, e01096–17. doi: 10.1128/aem.01096-17
Chen, Y., Burall, L. S., Luo, Y., Timme, R., Melka, D., Muruvanda, T., et al. (2016). Listeria monocytogenes in stone fruits linked to a multistate outbreak: enumeration of cells and whole-genome sequencing. Appl. Environ. Microbiol. 82, 7030–7040. doi: 10.1128/AEM.01486-16
Chen, Y., Luo, Y., Carleton, H., Timme, R., Melka, D., Muruvanda, T., et al. (2017). Whole genome and core genome multilocus sequence typing and single nucleotide polymorphism analyses of listeria monocytogenes isolates associated with an outbreak linked to cheese, United States 2013. Appl. Environ. Microbiol. 83, e00633–17. doi: 10.1128/AEM.00633-17
Chen, L., Yang, J., Yu, J., Yao, Z., Sun, L., Shen, Y., et al. (2005). VFDB: a reference database for bacterial virulence factors. Nucleic Acids Res. 33, D325–D328. doi: 10.1093/nar/gki008
Dharmasena, M., Wang, H., Wei, T., Bridges, W. C., Jr., Jiang, X. (2021). Survival of Clostridioides difficile in finished dairy compost under controlled conditions. J. Appl. Microbiol. 131, 996–1006. doi: 10.1111/jam.15001
Doumith, M., Cazalet, C., Simoes, N., Frangeul, L., Jacquet, C., Kunst, F., et al. (2004). New aspects regarding evolution and virulence of Listeria monocytogenes revealed by comparative genomics and DNA arrays. Infect. Immun. 72, 1072–1083. doi: 10.1128/IAI.72.2.1072-1083.2004
Dubey, S., Ager-Wiick, E., Peng, B., Depaola, A., Sorum, H., Munang’andu, H. M. (2023). The mobile gene cassette carrying tetracycline resistance genes in Aeromonas veronii strain Ah5S-24 isolated from catfish pond sediments shows similarity with a cassette found in other environmental and foodborne bacteria. Front. Microbiol. 14, 1112941. doi: 10.3389/fmicb.2023.1112941
Francisco, A. P., Vaz, C., Monteiro, P. T., Melo-Cristino, J., Ramirez, M., Carriço, J. A. (2012). PHYLOViZ: phylogenetic inference and data visualization for sequence based typing methods. BMC Bioinf. 13, 87. doi: 10.1186/1471-2105-13-87
Frost, L. S., Leplae, R., Summers, A. O., Toussaint, A. (2005). Mobile genetic elements: the agents of open source evolution. Nat. Rev. Microbiol. 3, 722–732. doi: 10.1038/nrmicro1235
Gao, P., Wang, P., Ding, M., Zhang, H., Huang, G., Nie, M., et al. (2023). A meta-analysis reveals that geographical factors drive the bacterial community variation in Chinese lakes. Environ. Res. 224, 115561. doi: 10.1016/j.envres.2023.115561
Halbedel, S., Prager, R., Fuchs, S., Trost, E., Werner, G., Flieger, A. (2018). Whole-genome sequencing of recent listeria monocytogenes isolates from Germany reveals population structure and disease clusters. J. Clin. Microbiol. 56, e00119–18. doi: 10.1128/JCM.00119-18
Hammarlof, D. L., Kroger, C., Owen, S. V., Canals, R., Lacharme-Lora, L., Wenner, N., et al. (2018). Role of a single noncoding nucleotide in the evolution of an epidemic African clade of Salmonella. Proc. Natl. Acad. Sci. U.S.A. 115, E2614–E2623. doi: 10.1073/pnas.1714718115
Huang, J., Zhao, Z., Zhang, Q., Zhang, S., Zhang, S., Chen, M., et al. (2021). Phylogenetic analysis reveals distinct evolutionary trajectories of the fluoroquinolones-resistant escherichia coli ST1193 from Fuzhou, China. Front. Microbiol. 12, 746995. doi: 10.3389/fmicb.2021.746995
Jackson, B. R., Tarr, C., Strain, E., Jackson, K. A., Conrad, A., Carleton, H., et al. (2016). Implementation of nationwide real-time whole-genome sequencing to enhance listeriosis outbreak detection and investigation. Clin. Infect. Dis. 63, 380–386. doi: 10.1093/cid/ciw242
Jolley, K. A., Bray, J. E., Maiden, M. C. J. (2018). Open-access bacterial population genomics: BIGSdb software, the PubMLST.org website and their applications. Wellcome. Open Res. 3, 124. doi: 10.12688/wellcomeopenres.14826.1
Katoh, K., Standley, D. M. (2013). MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol. Biol. Evol. 30, 772–780. doi: 10.1093/molbev/mst010
Koboldt, D. C., Chen, K., Wylie, T., Larson, D. E., Mclellan, M. D., Mardis, E. R., et al. (2009). VarScan: variant detection in massively parallel sequencing of individual and pooled samples. Bioinformatics 25, 2283–2285. doi: 10.1093/bioinformatics/btp373
Koopmans, M. M., Brouwer, M. C., Vázquez-Boland, J. A., Beek, D. V. D. (2023). Human listeriosis. Clin. Microbiol. Rev. 36, e0006019. doi: 10.1128/cmr.00060-19
Kremer, P. H. C., Lees, J. A., Koopmans, M. M., Ferwerda, B., Arends, A. W. M., Feller, M. M., et al. (2017). Benzalkonium tolerance genes and outcome in Listeria monocytogenes meningitis. Clin. Microbiol. Infect. 23, 265.e1–265.e7. doi: 10.1016/j.cmi.2016.12.008
Leekitcharoenphon, P., Kaas, R. S., Thomsen, M. C. F., Friis, C., Rasmussen, S., Aarestrup, F. M. (2012). snpTree–a web-server to identify and construct SNP trees from whole genome sequence data. BMC Genomics Suppl 7, S6. doi: 10.1186/1471-2164-13-S7-S6
Li, W., Bai, L., Ma, X., Zhang, X., Li, X., Yang, X., et al. (2019). Sentinel listeriosis surveillance in selected hospitals, China 2013-2017. Emerg. Infect. Dis. 25, 2274–2277. doi: 10.3201/eid2512.180892
Li, Y., Hu, C. (2021). Biogeographical patterns and mechanisms of microbial community assembly that underlie successional biocrusts across northern China. NPJ Biofilms. Microbiomes. 7, 15. doi: 10.1038/s41522-021-00188-6
Liu, D., Ainsworth, A. J., Austin, F. W., Lawrence, M. L. (2003). Characterization of virulent and avirulent Listeria monocytogenes strains by PCR amplification of putative transcriptional regulator and internalin genes. J. Med. Microbiol. 52, 1065–1070. doi: 10.1099/jmm.0.05358-0
Losic, B., Craig, A. J., Villacorta-Martin, C., Martins-Filho, S. N., Akers, N., Chen, X., et al. (2020). Intratumoral heterogeneity and clonal evolution in liver cancer. Nat. Commun. 11, 291. doi: 10.1038/s41467-019-14050-z
Lüth, S., Halbedel, S., Rosner, B., Wilking, H., Holzer, A., Roedel, A., et al. (2020). Backtracking and forward checking of human listeriosis clusters identified a multiclonal outbreak linked to Listeria monocytogenes in meat products of a single producer. Emerg. Microbes Infect. 9, 1600–1608. doi: 10.1080/22221751.2020.1784044
Maiden, M. C., Bygraves, J. A., Feil, E., Morelli, G., Russell, J. E., Urwin, R., et al. (1998). Multilocus sequence typing: a portable approach to the identification of clones within populations of pathogenic microorganisms. Proc. Natl. Acad. Sci. U.S.A. 95, 3140–3145. doi: 10.1073/pnas.95.6.3140
Martiny, J. B., Bohannan, B. J., Brown, J. H., Colwell, R. K., Fuhrman, J. A., Green, J. L., et al. (2006). Microbial biogeography: putting microorganisms on the map. Nat. Rev. Microbiol. 4, 102–112. doi: 10.1038/nrmicro1341
Matereke, L. T., Okoh, A. I. (2020). Listeria monocytogenes virulence, antimicrobial resistance and environmental persistence: A review. Pathogens 9, 528. doi: 10.3390/pathogens9070528
Matto, C., D’alessandro, B., Mota, M. I., Braga, V., Buschiazzo, A., Gianneechini, E., et al. (2022). Listeria innocua isolated from diseased ruminants harbour minor virulence genes of L. monocytogenes. Vet. Med. Sci. 8, 735–740. doi: 10.1002/vms3.710
Moller, A. G., Liang, C. (2017). MetaCRAST: reference-guided extraction of CRISPR spacers from unassembled metagenomes. PeerJ 5, e3788. doi: 10.7717/peerj.3788
Moura, A., Criscuolo, A., Pouseele, H., Maury, M. M., Leclercq, A., Tarr, C., et al. (2016). Whole genome-based population biology and epidemiological surveillance of Listeria monocytogenes. Nat. Microbiol. 2, 16185. doi: 10.1038/nmicrobiol.2016.185
Osek, J., Wieczorek, K. (2022). Listeria monocytogenes-how this pathogen uses its virulence mechanisms to infect the hosts. Pathogens 11, 1491. doi: 10.3390/pathogens11121491
Price, M. N., Dehal, P. S., Arkin, A. P. (2009). FastTree: computing large minimum evolution trees with profiles instead of a distance matrix. Mol. Biol. Evol. 26, 1641–1650. doi: 10.1093/molbev/msp077
Ragon, M., Wirth, T., Hollandt, F., Lavenir, R., Lecuit, M., Le Monnier, A., et al. (2008). A new perspective on Listeria monocytogenes evolution. PloS Pathog. 4, e1000146. doi: 10.1371/journal.ppat.1000146
Revazishvili, T., Kotetishvili, M., Stine, O. C., Kreger, A. S., Morris, J. G., Jr., Sulakvelidze, A. (2004). Comparative analysis of multilocus sequence typing and pulsed-field gel electrophoresis for characterizing Listeria monocytogenes strains isolated from environmental and clinical sources. J. Clin. Microbiol. 42, 276–285. doi: 10.1128/JCM.42.1.276-285.2004
Sabet, C., Lecuit, M., Cabanes, D., Cossart, P., Bierne, H. (2005). LPXTG protein InlJ, a newly identified internalin involved in Listeria monocytogenes virulence. Infect. Immun. 73, 6912–6922. doi: 10.1128/IAI.73.10.6912-6922.2005
Sharma, A. K., Dhasmana, N., Dubey, N., Kumar, N., Gangwal, A., Gupta, M., et al. (2017). Bacterial virulence factors: secreted for survival. Indian J. Microbiol. 57, 1–10. doi: 10.1007/s12088-016-0625-1
Shi, D., Anwar, T. M., Pan, H., Chai, W., Xu, S., Yue, M. (2021). Genomic determinants of pathogenicity and antimicrobial resistance for 60 global listeria monocytogenes isolates responsible for invasive infections. Front. Cell Infect. Microbiol. 11, 718840. doi: 10.3389/fcimb.2021.718840
Siguier, P., Perochon, J., Lestrade, L., Mahillon, J., Chandler, M. (2006). ISfinder: the reference centre for bacterial insertion sequences. Nucleic Acids Res. 34, D32–D36. doi: 10.1093/nar/gkj014
Stamatakis, A. (2014). RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 30, 1312–1313. doi: 10.1093/bioinformatics/btu033
Talavera, G., Castresana, J. (2007). Improvement of phylogenies after removing divergent and ambiguously aligned blocks from protein sequence alignments. Syst. Biol. 56, 564–577. doi: 10.1080/10635150701472164
Van Der Zwan, H., van der Westhuizen, F., Visser, C., van der Sluis, R. (2018). Draft de novo genome sequence of agapornis roseicollis for application in avian breeding. Anim. Biotechnol. 29, 241–246. doi: 10.1080/10495398.2017.1367692
Vu, H. T. K., Stasiewicz, M. J., Benjakul, S., Vongkamjan, K. (2021). Genomic analysis of prophages recovered from listeria monocytogenes lysogens found in seafood and seafood-related environment. Microorganisms 9, 1354. doi: 10.3390/microorganisms9071354
Wang, Y., Zhao, A., Zhu, R., Lan, R., Jin, D., Cui, Z., et al. (2012). Genetic diversity and molecular typing of Listeria monocytogenes in China. BMC Microbiol. 12, 119. doi: 10.1186/1471-2180-12-119
Wartha, S., Bretschneider, N., Dangel, A., Hobmaier, B., Hörmansdorfer, S., Huber, I., et al. (2023). Genetic characterization of listeria from food of non-animal origin products and from producing and processing companies in bavaria, Germany. Foods 12, 1120. doi: 10.3390/foods12061120
Weisberg, A. J., Chang, J. H. (2023). Mobile genetic element flexibility as an underlying principle to bacterial evolution. Annu. Rev. Microbiol. 77, 603–624. doi: 10.1146/annurev-micro-032521-022006
Xu, M., Guo, L., Gu, S., Wang, O., Zhang, R., Peters, B. A., et al. (2020). TGS-GapCloser: A fast and accurate gap closer for large genomes with low coverage of error-prone long reads. Gigascience 9, giaa094. doi: 10.1093/gigascience/giaa094
Zhang, X., Liu, Y., Zhang, P., Niu, Y., Chen, Q., Ma, X. (2021b). Genomic Characterization of Clinical Listeria monocytogenes Isolates in Beijing, China. Front Microbiol 12, 751003. doi: 10.3389/fmicb.2021.751003
Zhang, H., Chen, W., Wang, J., Xu, B., Liu, H., Dong, Q., et al. (2020). 10-year molecular surveillance of listeria monocytogenes using whole-genome sequencing in shanghai, China 2009-2019. Front. Microbiol. 11, 551020. doi: 10.3389/fmicb.2020.551020
Zhang, H., Wang, J., Chang, Z., Liu, X., Chen, W., Yu, Y., et al. (2021a). Listeria monocytogenes contamination characteristics in two ready-to-eat meat plants from 2019 to 2020 in shanghai. Front. Microbiol. 12, 729114. doi: 10.3389/fmicb.2021.729114
Keywords: Listeria monocytogenes, foodborne pathogen, cgMLST, WGS, SNP, virulence genes
Citation: Zhu L, Ji X, Wu Y, Xu W, Wang F and Huang X (2023) Molecular characterization of Listeria monocytogenes strains isolated from imported food in China from 14 countries/regions, 2003-2018. Front. Cell. Infect. Microbiol. 13:1287564. doi: 10.3389/fcimb.2023.1287564
Received: 02 September 2023; Accepted: 04 December 2023;
Published: 20 December 2023.
Edited by:
Kokouvi Kassegne, Shanghai Jiao Tong University, ChinaReviewed by:
Alexandre Leclercq, Institut Pasteur, FranceDavid A. Montero, University of Concepcion, Chile
Irshad Sulaiman, United States Food and Drug Administration, United States
Copyright © 2023 Zhu, Ji, Wu, Xu, Wang and Huang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Xinxin Huang, aHh4MjAxOTA1MjlAMTI2LmNvbQ==; Feifei Wang, d2FuZ2ZlaWZlaUBmdWRhbi5lZHUuY24=
†These authors have contributed equally to this work