 Jacob H. Elnaggar1
Jacob H. Elnaggar1 Caleb M. Ardizzone1
Caleb M. Ardizzone1 Nuno Cerca2
Nuno Cerca2 Evelyn Toh3
Evelyn Toh3 Paweł Łaniewski4
Paweł Łaniewski4 Rebecca A. Lillis5
Rebecca A. Lillis5 Melissa M. Herbst-Kralovetz4
Melissa M. Herbst-Kralovetz4 Alison J. Quayle1
Alison J. Quayle1 Christina A. Muzny6
Christina A. Muzny6 Christopher M. Taylor1*
Christopher M. Taylor1*- 1Department of Microbiology, Immunology, and Parasitology, Louisiana State University Health Sciences Center, New Orleans, LA, United States
- 2Laboratory of Research in Biofilms Rosário Oliveira (LIBRO), Centre of Biological Engineering (CEB), University of Minho, Braga, Portugal
- 3Department of Microbiology & Immunology, Indiana University School of Medicine, Indianapolis, IN, United States
- 4Department of Basic Medical Sciences, College of Medicine-Phoenix, University of Arizona, Phoenix, AZ, United States
- 5Division of Infectious Diseases, Department of Medicine, Louisiana State University Health Sciences Center, New Orleans, LA, United States
- 6Division of Infectious Diseases, University of Alabama at Birmingham, Birmingham, AL, United States
Bacterial vaginosis (BV) is the most common vaginal dysbiosis. In this condition, a polymicrobial biofilm develops on vaginal epithelial cells. Accurately quantifying the bacterial burden of the BV biofilm is necessary to further our understanding of BV pathogenesis. Historically, the standard for calculating total bacterial burden of the BV biofilm has been based on quantifying Escherichia coli 16S rRNA gene copy number. However, E. coli is improper for measuring the bacterial burden of this unique micro-environment. Here, we propose a novel qPCR standard to quantify bacterial burden in vaginal microbial communities, from an optimal state to a mature BV biofilm. These standards consist of different combinations of vaginal bacteria including three common BV-associated bacteria (BVAB) Gardnerella spp. (G), Prevotella spp. (P), and Fannyhessea spp. (F) and commensal Lactobacillus spp. (L) using the 16S rRNA gene (G:P:F:L, G:P:F, G:P:L and 1G:9L). We compared these standards to the traditional E. coli (E) reference standard using known quantities of mock vaginal communities and 16 vaginal samples from women. The E standard significantly underestimated the copy numbers of the mock communities, and this underestimation was significantly greater at lower copy numbers of these communities. The G:P:L standard was the most accurate across all mock communities and when compared to other mixed vaginal standards. Mixed vaginal standards were further validated with vaginal samples. This new G:P:L standard can be used in BV pathogenesis research to enhance reproducibility and reliability in quantitative measurements of BVAB, spanning from the optimal to non-optimal (including BV) vaginal microbiota.
1 Introduction
Bacterial vaginosis (BV) is the most common vaginal infection, affecting more than 30% of women in the United States (Bautista et al., 2016). Although the exact etiology is yet to be identified, BV is known to be associated with loss of protective lactic acid- and hydrogen peroxide-producing Lactobacillus spp. and a dramatic increase in facultative and strict anaerobic bacteria (BV-associated bacteria; BVAB) (from 107 to 109 bacterial genomes per sample) including Gardnerella spp. (107), Prevotella spp. (108), and Fannyhessea vaginae (106) (Zozaya-Hinchliffe et al., 2010; Ravel et al., 2011). The current recommended treatment regimens for BV consist of oral and intra-vaginal metronidazole and clindamycin (“Bacterial Vaginosis - 2021 Sexually Transmitted Diseases Treatment Guidelines”, 2021), each with high initial success rates (80%). However, more than 60% of women will have an episode of recurrent BV (Bradshaw et al., 2006).
BV is associated with the formation of a polymicrobial biofilm on the surface of vaginal epithelial cells, which likely contributes to high recurrence rates after treatment (Patterson et al., 2007; Swidsinski et al., 2008). The sequence of events leading to BV biofilm formation is controversial and under active study (Machado et al., 2016; Sousa et al., 2023). One hypothesis is that a virulent strain of G. vaginalis, likely sexually transmitted, is the primary pathogen that displaces protective Lactobacillus spp. and adheres to the vaginal epithelium, initiating BV biofilm formation and allowing secondary colonizers to attach and multiply (Patterson et al., 2010; Santiago et al., 2011). In this hypothetical model, one additional early colonizer is thought to be P. bivia, which is recruited into the lower layers of the biofilm that is initiated by G. vaginalis (Verstraelen et al., 2009). Vaginal sialidase, produced by both G. vaginalis and P. bivia, promotes breakdown of the protective mucous layer on the vaginal epithelium (Wiggins et al., 2001). Loss of the protective mucous layer on the vaginal epithelium leads to increased adherence of other BVAB, including F. vaginae (Hardy et al., 2015), which join the BV biofilm in the upper layers, leading to the formation of a mature, polymicrobial entity (Castro et al., 2021). A better understanding of the development of the polymicrobial BV biofilm is crucial for improving BV diagnosis and developing more effective treatments.
Determining the burden of specific BVAB is important for better understanding the pathogenesis of incident BV (Gajer et al., 2012; Muzny et al., 2018). The 16S ribosomal ribonucleic acid (rRNA) gene, present in all bacterial genomes, is used to estimate bacterial burden (Woese et al., 1990). Polymerase chain reaction (PCR) amplification and sequencing of hypervariable regions of the 16S rRNA gene has been widely applied to characterize the vaginal microbiota and further investigate BV pathogenesis. However, this method only provides the relative abundance of each micro-organism, rather than absolute quantities (Poretsky et al., 2014). Quantitative PCR (qPCR) targeting the 16S rRNA gene is commonly used to measure the burden of bacterial species (Santiago et al., 2012; Ricchi et al., 2017; Galazzo et al., 2020). Typically, these assays use plasmid standards containing the Escherichia coli 16S rRNA gene. The E. coli (E) reference standard is a common tool for measuring bacterial burden in many types of communities (Zozaya-Hinchliffe et al., 2010; Jian et al., 2020; Tettamanti Boshier et al., 2020), however, this approach has not been evaluated for its accuracy in vaginal samples. Generally, universal primers amplify 16S rRNA genes from different genera with varying efficiencies due to differences in intervening sequences (e.g., guanine or cytosine [GC] content and length) (Aird et al., 2011), and E. coli is not a natural colonizer of the vaginal micro-environment (Acinas et al., 2004; Větrovský and Baldrian, 2013). Therefore, the use of an E standard may influence the results of the quantification of 16S rRNA gene copy number within the vaginal microbiota.
Accurate quantitation of bacterial burden in complex vaginal communities requires specifically designed standards. We generated standards specific for common vaginal micro-organisms to establish a method that allows for estimation of the bacterial burden of the vaginal micro-environment, from an optimal state to a mature, polymicrobial BV biofilm. These novel standards are composed of Gardnerella spp. (G), Prevotella spp. (P), Fannyhessea spp. (F), and Lactobacillus spp. (L) were compared to the traditional E standard using mock vaginal communities and patient-derived vaginal samples.
2 Materials and methods
2.1 Vaginal samples
We used stored isolated deoxyribonucleic acid (DNA) from vaginal swabs that were previously obtained in a study described in Mott et al., 2021 (Louisiana State University Health Sciences Center [LSUHSC] IRB protocol #1081). In brief, vaginal samples were collected from women seeking care at the LSUHSC CrescentCare Sexual Health Center Clinic in New Orleans, LA using a Copan Swab (Copan, Murrieta, CA, USA). DNA extraction and sequencing were performed by the LSUHSC Microbial Genomics Resource Group. Genomic DNA was extracted using the QIAamp DNA Stool Mini Kit (Qiagen, Hilden, Germany), modified to include bead beating. In addition to a swab being used for sequencing, a swab was also used for Gram stain and Nugent scoring to characterize the vaginal microbiome (Nugent et al., 1991). BV was common in this population of women (38% by Nugent score) and representative samples were selected from across the spectrum of the vaginal microbiota, characterized by Nugent score (n=6 normal vaginal microbiota, 5 intermediate vaginal microbiota, and 5 BV) and ethnicity (n=8 black, 7 white, and 1 other). Complete metadata is listed in Table S1B. Bacterial relative abundance can be observed in heatmaps (Figure S2) (Agile Toolkit for Incisive Microbial Analysis).
2.2 Primer design
We used published universal qPCR primers (UP) targeting the sixth hypervariable region (V6) of the bacterial 16S rRNA gene of multiple bacterial taxa (Bacchetti De Gregoris et al., 2011). We also designed taxa-specific primers (SP) outside the V6 amplicon of the UP for our bacterial taxa of interest, Gardnerella spp., Prevotella spp., Fannyhessea spp., Lactobacillus spp., and E. coli (Figure 1A). We developed python and bash programs to computationally nominate specific primers with set parameters (GC content: >50%, melting temperature: 55-65°C, and primer length: 18-22 bp) and validated them using BLAST queries (https://github.com/elnaggarj/16S-primer-design) (Altschul et al., 1990). Once chosen, these primers were synthesized (Integrated DNA Technologies, Coralville, IA) and sensitivity and specificity was tested via PCR using vaginal samples and American Type Culture Collection (ATCC) bacterial isolates when available; G. vaginalis ATCC 10287, L. crispatus ATCC 33197, and E. coli ATCC 25922.
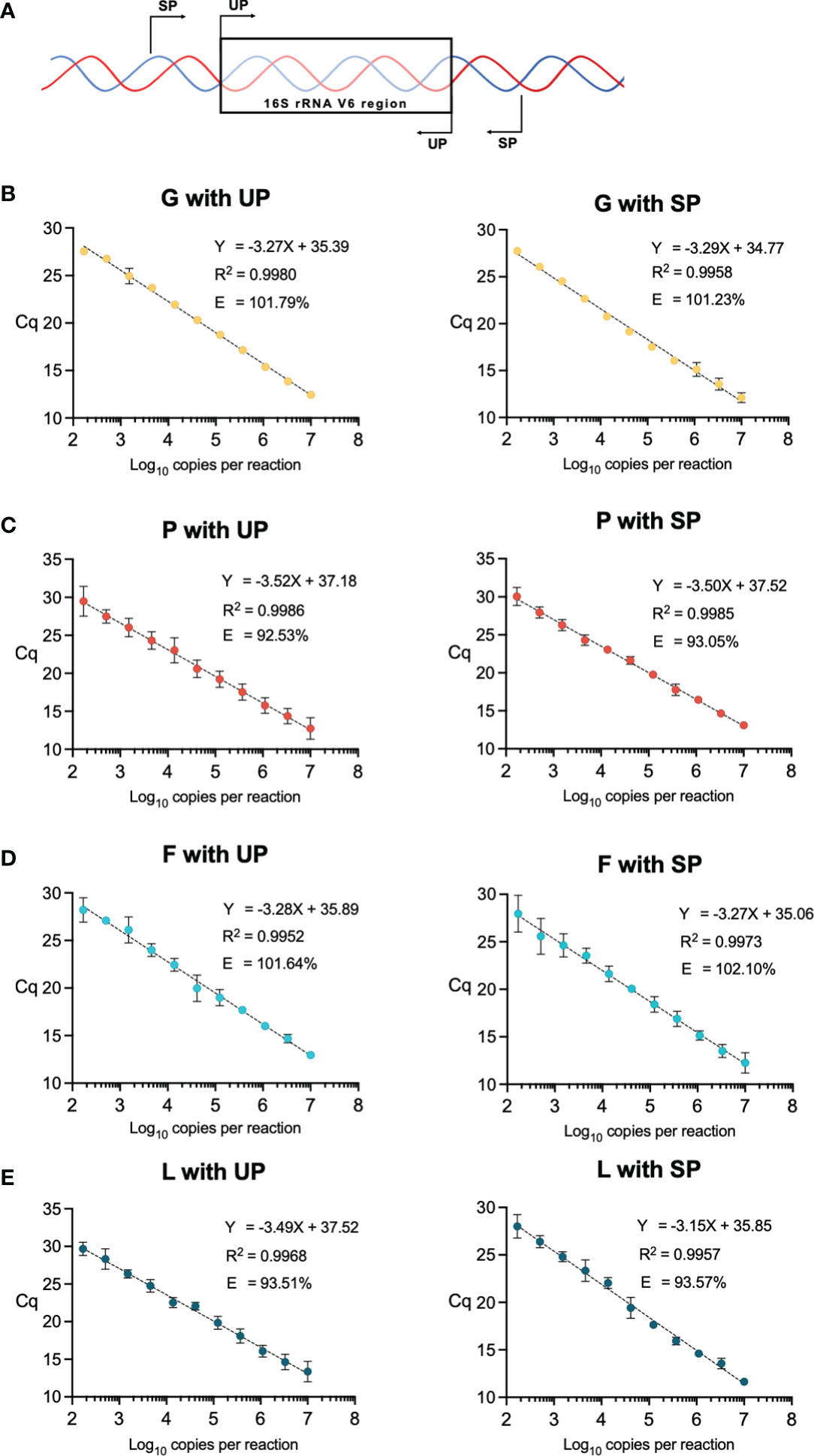
Figure 1 (A) Diagram of universal primer (UP) and specific primer (SP) locations in the V6 region of the 16S rRNA gene. (B–E) qPCR standards using the SP and UP for each vaginal bacteria of interest, Gardnerella spp. (G), Prevotella spp. (P), and Fannyhessea spp. (F), and Lactobacillus spp. (L). Data points are generated from three-fold dilutions of the purified plasmids starting at 107 copies to 167 copies per reaction. Each point represents an average of 3 replicate qPCR reactions with corresponding error bars. Efficiency (E) is calculated based on the slope of the linear regression. Axes are labeled by the quantification cycle (Cq) and the number of copies per reaction (log10).
2.3 qPCR conditions
Each qPCR reaction contained 0.5 nmol of primers, 1 µl of template, and 7 µl of SsoAdvanced Universal SYBR Green Supermix (Bio-Rad, Hercules, CA) in a total of 10 µl. The qPCR conditions included a premelt at 98°C for 3 minutes, and then 40 cycles of 98°C for 15 seconds and 60°C for 15 seconds, followed by a final melt curve where the temperature was incrementally increased 0.5°C for 5 seconds from 65°C to 98°C (Figure S1A). All qPCR reactions were performed in triplicate and values are listed in Table S1A.
2.4 Standard curve generation
Standard curves were generated for bacteria taxa of interest, Gardnerella spp., Prevotella spp., Lactobacillus spp., Fannyhessea spp., and E. coli. The rRNA gene was amplified for each bacterial micro-organism of interest from vaginal samples, and the E. coli rRNA gene was amplified from the ATCC strain using the SP. Specific V6 amplicons for each bacterial micro-organism was generated using OneTaq 2X Master Mix with Standard Buffer (New England Biolabs [NEB] Ipswich, MA). 16S rRNA gene amplicons for each micro-organism were sequenced and verified to be identical to the reference genome (Eurofins genomics, Louisville, KY, USA). For Gardnerella spp. we compared the amplicon to the G. vaginalis ATCC 10287, Prevotella spp. to P. bivia ATCC 29303, Fannyhessea spp. to F. vaginae ATCC BAA-55, Lactobacillus spp. to L. crispatus ATCC 33197, and E. coli to E. coli ATCC 25922. These amplicons were ligated into a pGEM-T Easy vector (Promega, Madison, WI) (Figure S1B) and transformed into chemically competent DH5α E. coli (Invitrogen Waltham, MA) to isolate single colonies (Bertani, 2004). We performed validation for correct inserts via PvuII-HF (NEB) digests on gel and Sanger sequencing (Eurofins, Luxembourg). Vectors/plasmids with specific amplicons for each vaginal bacterial organism were purified using HiSpeed Plasmid Maxi Kit (Qiagen). Concentrations of purified vectors were measured using a NanoDrop 2000 (Thermo Scientific, Waltham, MA). The copy number of each vector was calculated from concentrations derived from quantitative gel densitometry. qPCR standards for each bacterial taxa of interest were generated from ten three-fold dilutions of the purified plasmids starting at 107 copies to 102 copies per reaction. Taxa-specific standards were then amplified using SP and UP. Vaginal mix standards and E standard assays were amplified with UP (Bacchetti De Gregoris et al., 2011). The regression coefficients and reaction efficiencies, based on the slope of the regression (-1 + 10-1/slope), were compared for each standard. A 100% efficiency indicated that the PCR product of interest was accurately doubling with each cycle (Stolovitzky and Cecchi, 1996), and an appropriate efficiency is between 90-110%.
2.5 Mock vaginal communities
Gardnerella spp., Prevotella spp., Fannyhessea spp., and Lactobacillus spp. 16S rRNA genes in plasmids were combined in known quantities to simulate mock vaginal communities. Mock vaginal communities were generated at 106, 105, 104 total copy numbers, and quantified using both the E standard and the mixed vaginal standards. The calculated output from each standard was compared to the known input of each mock vaginal community. The output was divided by the input to generate a percentage. A 100% output/input indicated that identical input and output copy numbers were read from the standard.
2.6 Assessing standards using vaginal samples
Three ten-fold dilutions were made from the original isolated DNA from the vaginal samples. The 16S rRNA gene copy number per reaction was measured using the E and mixed vaginal standards to determine if there was a change in the difference between the two standards at different dilutions. Next, 16S rRNA gene copy number per sample was calculated and compared between the two standards and across Nugent categories (normal vaginal microbiota [0-3], intermediate vaginal microbiota [4-6], and BV [7-10]) (Nugent et al., 1991).
2.7 Statistical analysis and power
Analyses were performed using Prism (version 9.3.1; GraphPad Software, Inc., La Jolla, CA). Linear regressions were performed on standard curves. qPCR-derived quantification cycle (Cq) value was converted to 16S rRNA gene copies and compared using two-way analysis of variance (ANOVA). Test statistics and p-values are listed in Table S1C. Comparing 16 vaginal samples, we have over 95% power to detect differences between the E and vaginal standard using a two-tailed Type I error rate of 0.05.
3 Results
3.1 Taxa-specific primers to 16S rRNA gene
SP were generated for: Gardnerella spp. (G), Prevotella spp. (P), Fannyhessea spp. (F), Lactobacillus spp. (L), and E. coli (EC) (primer sequences listed in Table S1A), and quantitative standards for each species using SP and UP were generated from three-fold dilutions of the purified plasmids from 107 copies to 102 copies per reaction (Figures 1B–E). The length of the UP amplicon was similar across all tested bacterial species and the SP amplicon varied between 230 and 290 base pairs (Table 1). Although there was a difference in length between the UP and SP for a given micro-organism, the efficiency did not vary more than 1% (Table 1). Furthermore, there was no notable difference in the R2 of the standards and all R2 were greater than 0.99, meaning that the standards approximated a linear slope.
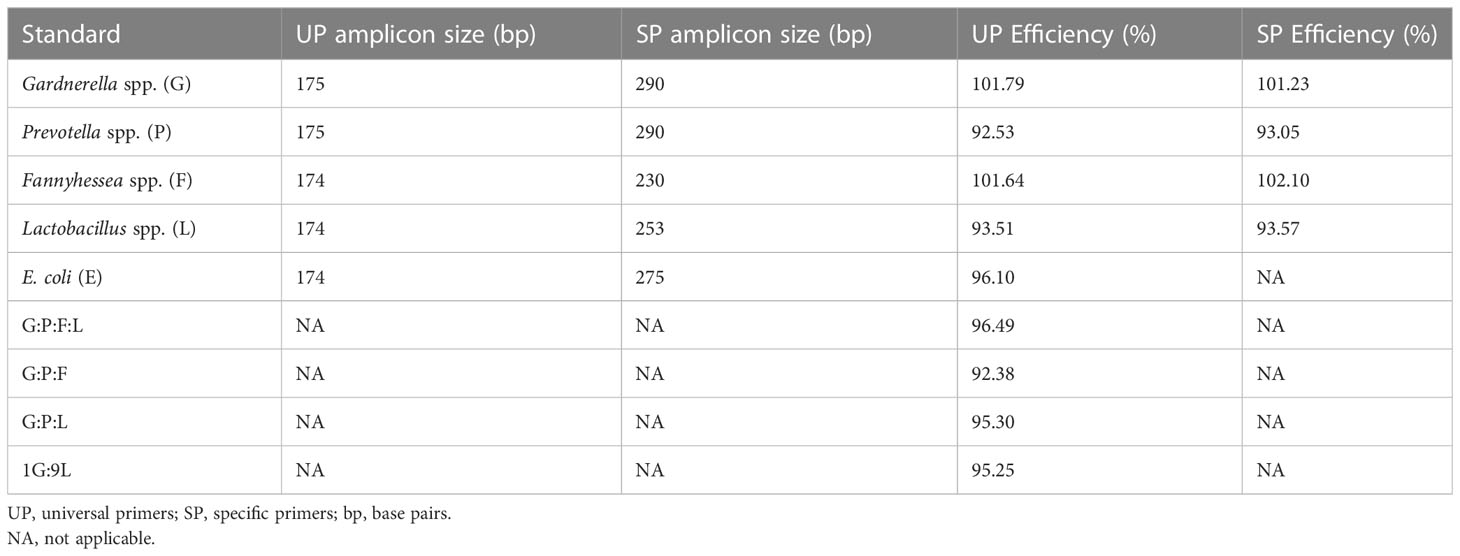
Table 1 Length and efficiencies of standards.
3.2 Vaginal specific standard for bacterial burden
As the taxa-specific standards performed well on their own, we next set out to generate a single vaginal standard to measure bacterial burden in the vaginal micro-environment. We compared standards that utilized common vaginal bacterial micro-organisms and generated standards consisting of different combinations of the G, P, F, and L 16S rRNA genes. This included a standard consisting of all four taxa in equal proportions (G:P:F:L), the BVAB (G:P:F), a standard representing an intermediate community (G:P:L), and an optimal community favoring lactobacilli (1G:9L).
Comparing these standards to each other and to the traditional E reference standard, we observed that vaginal mix standards and E standards were similar in efficiency (Table 1; Figures S3A–E). These standards were then compared using mock vaginal communities, where an optimal vaginal microbiota was represented by a mix of nine parts L to one-part G 16S rRNA gene plasmids, an intermediate community was represented by a mix of equal parts G, P, and L, and a BV-like microbiota consisting of G, P, F (Figure 2). Interestingly, the E standard significantly underestimated the mock vaginal communities, indicated by the measured output being less than the known input (all p < 0.05 when compared to 100%). Differences in other mixed vaginal standards in mock communities were observed. The measured output of the G:P:L mixed standard did not significantly differ from the known input in any of the three communities (all p > 0.05 when compared to 100%). Interestingly, this mixed vaginal standard never significantly differed regardless of the copy number of the vaginal community being measured. Whereas, in the E standard, the difference between measurements was greater at decreasing dilutions and was significant when comparing 106 to 104 copy numbers (Figure S4A), indicating that the E standard error varied based on the copy number of the 16S rRNA gene in the sample. Overall, these results indicate that the G:P:L mix standard was the most accurate for measuring all tested mock vaginal communities as it did not significantly differ in any dilution.
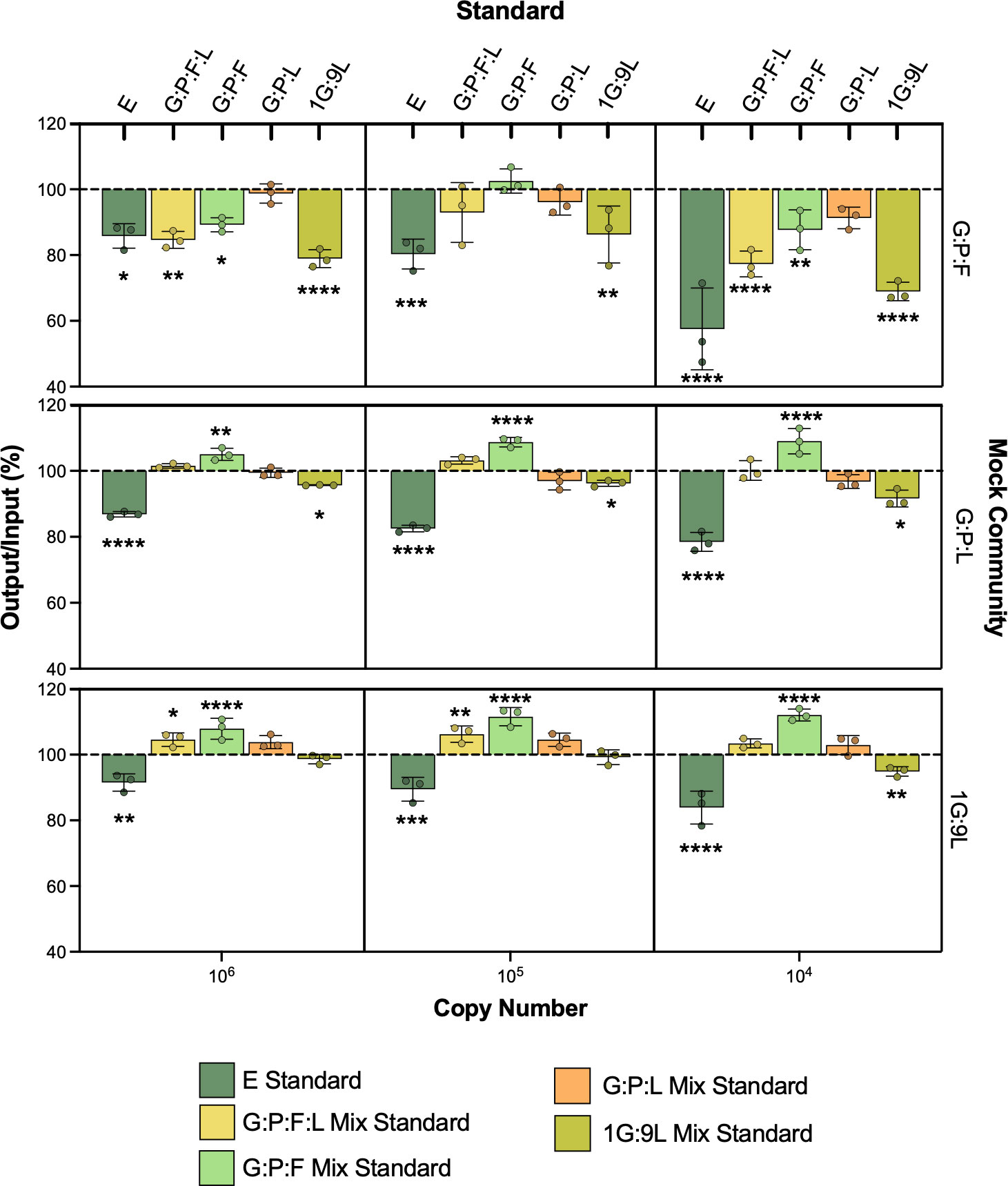
Figure 2 Mock vaginal communities using E and G:P:L mix standard. The equal parts G:P:L community is meant to represent a BV-like vaginal microbiome (top), G:P:L is representative of an intermediate microbiome (middle), and the 1G:9L is meant to represent an optimal vaginal microbiome (bottom). Known copy numbers are used as input, listed on the x-axis. The output is calculated using the corresponding standard and divided by the input to generate a percentage. A 100% output/input indicates the same copy number that was input was read as output from the standard. The resulting output/input was averaged and compared to 100% using two-way ANOVA, *P ≤ 0.05, **P ≤ 0.01, ***P ≤ 0.001, ****P ≤ 0.0001.
We continued to assess these mixed vaginal standards using vaginal samples collected from a diverse group of women enrolled in a previous study (Mott et al., 2021). First, we wanted to determine if this variation was based on the communities within these samples. 16 vaginal samples were stratified based on Nugent score category (6 normal, 4 intermediate, and 6 BV) (Nugent et al., 1991) We measured each sample with the different mixed standards and compared them to the E standard (Figures 3A–D). Interestingly, the difference between the G:P:F:L, G:P:L, and 1G:9L standards to the E standard significantly varied regardless of the Nugent category. Samples in all three Nugent categories varied consistently between the vaginal mix standard and the E standard. The G:P:L standard was the most significantly different (all p < 0.0001), followed by 1G:9L (all p < 0.003), and G:P:L:F (all p < 0.01). However, the G:P:F standard was the most similar to the E standard and was not significantly different at any Nugent score category. To determine if there was a variation in copy number similar to that observed in the mock vaginal communities, we compared the difference in the 16S rRNA gene copy number at three different dilutions (Figure S4B). The difference between the E and vaginal mix standards became significantly greater at increasing dilutions, supporting the results from the mock vaginal communities, indicating a similar variation based on dilution.
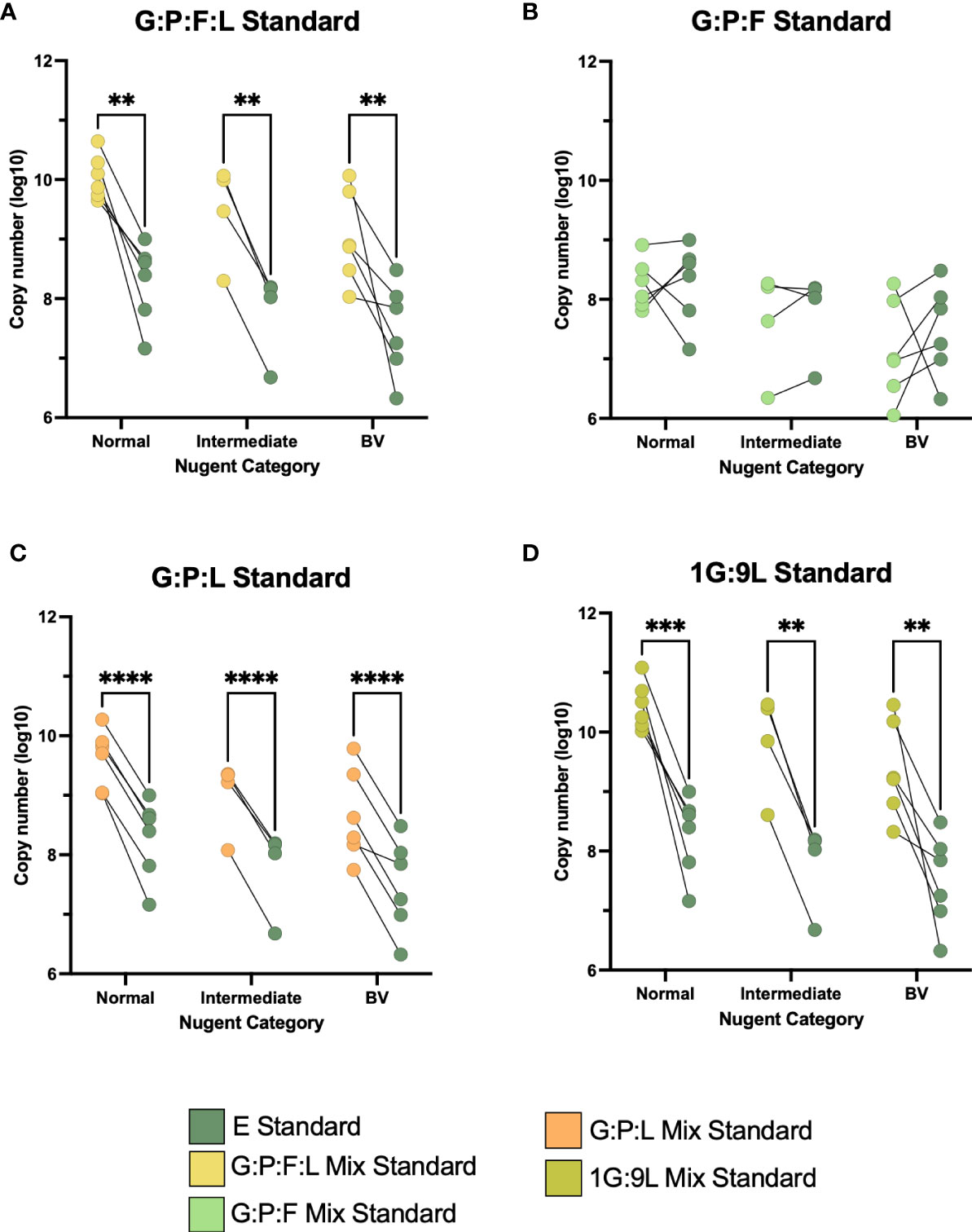
Figure 3 (A–D) Paired comparisons between the E and the four vaginal mixed standards across the three categories of Nugent score, normal, intermediate, and BV. The line between two points represents the same vaginal sample with each standard. The variability between these points was compared. All experiments were performed in triplicate and comparisons were made using two-way ANOVA, **P ≤ 0.01, ***P ≤ 0.001, ****P ≤ 0.0001.
4 Discussion
In this study, we generated novel qPCR standards for measuring the bacterial burden in the vaginal micro-environment. These standards were composed of 16S rRNA genes of common vaginal micro-organisms, Gardnerella spp., Prevotella spp., Fannyhessea spp., and Lactobacillus spp., and were compared to the traditional E standard. These new standards, specifically the one composed of G:P:L, were able to improve the accuracy of bacterial burden measurements within the vaginal microbial community and the complex BV biofilm.
Strengths of this study include the use of standards that are representative of bacteria that are common members of the vaginal microbiota (Salinas et al., 2020; Pacha-Herrera et al., 2022), rather than E. coli, which is not typically present in this micro-environment (Chen et al., 2021). An optimal vaginal microbiota is dominated by Lactobacillus spp. such as L. crispatus, L. jensenii, and L. gasseri, whereas in BV there is an increasing abundance of facultative and strict anaerobic bacteria including G. vaginalis, P. bivia, and F. vaginae. To generate taxa-specific primers, our SP amplicons varied in length. However, we observed that the efficiency between the UP and SP for a given micro-organism did not vary more than 1%. This suggests that the UP region contributes more to the efficiency of the standard rather than to the variation in the length of the SP region.
There are other BVAB present in the vaginal micro-environment and the BV biofilm; therefore, the absolute bacterial burden is difficult to quantify. To help account for this in our study, we created mock vaginal communities where the quantities of 16S rRNA gene copies are known. Since this is only a model of in vivo conditions, variables such as biofilm resistance and efficiency of DNA isolation cannot be actively replicated in these mock communities and require additional experimentation and validation (Lima et al., 2022). We tested our mock communities at varying copy numbers, from 106 to 104. This was designed to assess various points along the standard which ranges from 107 to 102 16S rRNA gene copy numbers. Interestingly, the different combinations of mixed vaginal bacteria performed variably across the different mock communities, and the G:P:L mix standard was observed to be the most accurate as it did not differ from 100% in any case. Perhaps a mixed standard that closely represents an intermediate vaginal community allows for a more robust measurement of bacterial burden across the spectrum of BV. We also observed Nugent-independent differences between the G:P:L and the E standard in a diverse range of vaginal samples.
A limitation of this study is that we chose to generate our mixed vaginal standard using a subset of bacteria found in the vaginal microbiome (Zozaya-Hinchliffe et al., 2010; Gajer et al., 2012). These organisms are most common in both optimal (Lactobacillus spp.) and BV (Gardnerella spp., Prevotella spp., and Fannyhessea spp.) vaginal microbiomes. Also, these specific micro-organisms have been found to change in relative abundance in the days leading up to incident BV (Muzny et al., 2018). Additional vaginal bacteria can be tested and combined in standards if there is particular interest moving forward. For example, Sneathia spp. can be included. There is growing evidence that Sneathia spp. are involved in preterm birth and vaginal inflammation (Anahtar et al., 2015; Fettweis et al., 2019; Łaniewski and Herbst-Kralovetz, 2021) and may be a secondary colonizer to the BV biofilm. Another limitation is that we only tested the V6 region of the 16S rRNA gene because we based our approach on previous work in this area (Bacchetti De Gregoris et al., 2011). We are unsure how standards constructed from other regions would compare to our findings, as primers for different variable regions amplify specific micro-organisms at varying efficiencies (Van Der Pol et al., 2019). The V6 region is similar between species within the same genus, for example different Lactobacillus spp. share the same 16S rRNA gene sequence. Thus, we were not able to develop species-specific standards for our micro-organisms of interest, Gardnerella spp., Prevotella spp., Fannyhessea spp., and Lactobacillus spp. (Janda and Abbott, 2007; Větrovský and Baldrian, 2013). However, since there are multiple different species of the same genus known to be found in the vaginal microbiota (e.g., L. crispatus and L. gasseri), genus-specific standards may perform better than ones generated using specific species. A future step would be to test other taxa as well as multiple variable regions of the 16S rRNA gene to identify a standard most optimal for the vaginal microbiota.
Overall, the ability to accurately measure the bacterial burden of the vaginal microbiota will improve our understanding of BV pathogenesis. For example, different Gardnerella species may be present in women who do not have BV, such as G. vaginalis, G. leopoldii, G. piotii, and G. swidsinskii (Castro et al., 2020), but, perhaps at a certain threshold or level of virulence, Gardnerella spp. can overcome the opposing forces in the vaginal micro-environment to initiate BV biofilm development (Roy et al., 1994). A better understanding of this sequence of events and changes in bacterial burden over time will help inform improvements in BV diagnosis and treatment. This method can also be synergized with complementary bioinformatics approaches such as calculating inferred absolute abundance (Tettamanti Boshier et al., 2020). In this method, the total bacterial burden is multiplied by the relative abundance through 16S rRNA gene sequencing to obtain a value comparable to the burden of the specific vaginal bacterial species. Improving the method to calculate the bacterial burden will result in improved overall accuracy of this calculation. Accurate bacterial burden measurements will also improve vaginal microbiome studies where vaginal sample collection takes place. Since this is a relatively cost-effective method, bacterial burden can be determined to guide selection and prioritization of samples for deeper sequencing. This method can also be adopted to better understand the etiology of incident BV. Dramatic bacterial shifts can occur in the vaginal micro-environment within short periods of time (Gajer et al., 2012), and high resolution of these changes is required to elucidate bacterial dynamics in the pathogenesis of BV. We plan to use our newly generated standard in longitudinal studies in combination with inferred absolute abundance to enhance our understanding of the changes in the burden of key vaginal bacteria prior to the onset of incident BV.
Data availability statement
The original contributions presented in the study are included in the article/Supplementary Material. Further inquiries can be directed to the corresponding author.
Ethics statement
The studies involving human participants were reviewed and approved by Louisiana State University Health Sciences Center [LSUHSC] IRB protocol #1081. The patients/participants provided their written informed consent to participate in this study.
Author contributions
JE and CA conceived this study. JE performed the experiments. JE and CA analyzed the data. RL clinically categorized patient samples and established the sample bank. NC, ET, PL, MH-K, AQ, CM, CT jointly supervised the study and provided oversight during the manuscript editing process All authors contributed to the article and approved the submitted version.
Funding
This work was supported by the National Institute of Allergy and Infectious Diseases (grants R01AI146065 to CM and R01AI118860 to AQ and CT), National Center for Advancing Translational Sciences (grant UL1TR003096 to the University of Alabama-Birmingham Center for Clinical and Translational Science), and an American Sexually Transmitted Diseases Association Small Project Assistance award (to JE).
Acknowledgments
The authors thank Julia Siren, CRNP and Emily Duffy, RN, for their assistance in enrolling patients in the study from which we used the stored vaginal samples. In addition, the authors thank David Nelson, PhD, for helpful discussions on study design for this manuscript.
Conflict of interest
Author CM is a consultant for Scynexis, Cepheid, and BioNTech. Author CM has received research funding support from NIH/NIAID, Lupin Pharmaceuticals, Abbott Molecular, Visby, and Gilead as well as honoraria from Visby Medical, Elsevier, Abbott Molecular, Scynexis, Roche, and Cepheid. Author RL is the clinical trial site PI for Cepheid, Hologic, Gilead, Visby, Roche, Becton Dickenson, OrthoQuidel, Merck, BioFire, CUE, CovarsaDx, and a consultant for Roche and Cepheid. Author MH-K is on the scientific advisory board for Freya Biosciences and a consultant for Vaginal Biome Sciences.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fcimb.2023.1198113/full#supplementary-material
Supplementary Figure 1 | (A) qPCR diagram. (B) pGEM-T Easy vector diagram with 16S rRNA gene.
Supplementary Figure 2 | Relative abundance heatmap for vaginal samples used in this study.
Supplementary Figure 3 | (A–E) qPCR standards using UP for Escherichia coli (E) and vaginal mixed standards composed of Gardnerella spp. (G): Prevotella spp. (P): Fannyhessea spp. (F), and Lactobacillus spp. (L). Data points are generated from three-fold dilutions of the purified plasmids starting at 107 copies to 102 copies per reaction. Each point represents an average of 3 replicate qPCR reactions with standard deviation error bars. Efficiency (E) is calculated based on the slope of the linear regression. Axes are labeled by the quantification cycle (Cq) and the number of copies per reaction (log10).
Supplementary Figure 4 | (A) E standard for the 3 mock communities showing a decrease in accuracy. (B) The E and G:P:L mix standard were performed on DNA isolated from vaginal swabs. These standards were performed at three ten-fold dilutions D1 (1/10), D2 (1/100), and D3 (1/1000). The difference in the resulting copy number from the E and G:P:L mix standard was calculated and compared across the three dilutions.
Supplementary Table 1 | (A) qPCR information. (B) Metadata for patients. (C) Statistical tests.
References
Acinas, S. G., Marcelino, L. A., Klepac-Ceraj, V., Polz, M. F. (2004). Divergence and redundancy of 16S rRNA sequences in genomes with multiple rrn operons. J. Bacteriol 186, 2629–2635. doi: 10.1128/JB.186.9.2629-2635.2004
Aird, D., Ross, M. G., Chen, W.-S., Danielsson, M., Fennell, T., Russ, C., et al. (2011). Analyzing and minimizing PCR amplification bias in illumina sequencing libraries. Genome Biol. 12, R18. doi: 10.1186/gb-2011-12-2-r18
Altschul, S. F., Gish, W., Miller, W., Myers, E. W., Lipman, D. J. (1990). Basic local alignment search tool. J. Mol. Biol. 215, 403–410. doi: 10.1016/S0022-2836(05)80360-2
Anahtar, M. N., Byrne, E. H., Doherty, K. E., Bowman, B. A., Yamamoto, H. S., Soumillon, M., et al. (2015). Cervicovaginal bacteria are a major modulator of host inflammatory responses in the female genital tract. Immunity 42, 965–976. doi: 10.1016/j.immuni.2015.04.019
Bacchetti De Gregoris, T., Aldred, N., Clare, A. S., Burgess, J. G. (2011). Improvement of phylum- and class-specific primers for real-time PCR quantification of bacterial taxa. J. Microbiol. Methods 86, 351–356. doi: 10.1016/j.mimet.2011.06.010
Bacterial Vaginosis - 2021 Sexually Transmitted Diseases Treatment Guidelines (2021) Centers for disease control and prevention. Available at: https://www.cdc.gov/std/treatment-guidelines/STI-Guidelines-2021.pdf (Accessed 7.22.21).
Bautista, C. T., Wurapa, E., Sateren, W. B., Morris, S., Hollingsworth, B., Sanchez, J. L. (2016). Bacterial vaginosis: a synthesis of the literature on etiology, prevalence, risk factors, and relationship with chlamydia and gonorrhea infections. Military Med. Res. 3, 4. doi: 10.1186/s40779-016-0074-5
Bertani, G. (2004). Lysogeny at mid-twentieth century: P1, P2, and other experimental systems. J. Bacteriol 186, 595–600. doi: 10.1128/JB.186.3.595-600.2004
Bradshaw, C. S., Morton, A. N., Hocking, J., Garland, S. M., Morris, M. B., Moss, L. M., et al. (2006). High recurrence rates of bacterial vaginosis over the course of 12 months after oral metronidazole therapy and factors associated with recurrence. J. Infect. Dis. 193, 1478–1486. doi: 10.1086/503780
Castro, J., Jefferson, K. K., Cerca, N. (2020). Genetic heterogeneity and taxonomic diversity among gardnerella species. Trends Microbiol. 28, 202–211. doi: 10.1016/j.tim.2019.10.002
Castro, J., Rosca, A. S., Muzny, C. A., Cerca, N. (2021). Atopobium vaginae and prevotella bivia are able to incorporate and influence gene expression in a pre-formed gardnerella vaginalis biofilm. Pathogens 10, 247. doi: 10.3390/pathogens10020247
Chen, X., Lu, Y., Chen, T., Li, R. (2021). The female vaginal microbiome in health and bacterial vaginosis. Front. Cell. Infect. Microbiol. 11. doi: 10.3389/fcimb.2021.631972
Fettweis, J. M., Serrano, M. G., Brooks, J. P., Edwards, D. J., Girerd, P. H., Parikh, H. I., et al. (2019). The vaginal microbiome and preterm birth. Nat. Med. 25, 1012–1021. doi: 10.1038/s41591-019-0450-2
Gajer, P., Brotman, R. M., Bai, G., Sakamoto, J., Schütte, U. M. E., Zhong, X., et al. (2012). Temporal dynamics of the human vaginal microbiota. Sci. Transl. Med. 4, 132ra52. doi: 10.1126/scitranslmed.3003605
Galazzo, G., van Best, N., Benedikter, B. J., Janssen, K., Bervoets, L., Driessen, C., et al. (2020). How to count our microbes? the effect of different quantitative microbiome profiling approaches. front. Cell. Infect. Microbiol. 10. doi: 10.3389/fcimb.2020.00403
Hardy, L., Jespers, V., Dahchour, N., Mwambarangwe, L., Musengamana, V., Vaneechoutte, M., et al. (2015). Unravelling the bacterial vaginosis-associated biofilm: a multiplex gardnerella vaginalis and atopobium vaginae fluorescence In situ hybridization assay using peptide nucleic acid probes. PloS One 10, e0136658. doi: 10.1371/journal.pone.0136658
Janda, J. M., Abbott, S. L. (2007). 16S rRNA gene sequencing for bacterial identification in the diagnostic laboratory: pluses, perils, and pitfalls. J. Clin. Microbiol. 45, 2761–2764. doi: 10.1128/JCM.01228-07
Jian, C., Luukkonen, P., Yki-Järvinen, H., Salonen, A., Korpela, K. (2020). Quantitative PCR provides a simple and accessible method for quantitative microbiota profiling. PloS One 15, e0227285. doi: 10.1371/journal.pone.0227285
Łaniewski, P., Herbst-Kralovetz, M. M. (2021). Bacterial vaginosis and health-associated bacteria modulate the immunometabolic landscape in 3D model of human cervix. NPJ Biofilms Microbiomes 7, 88. doi: 10.1038/s41522-021-00259-8
Lima, A., França, A., Muzny, C. A., Taylor, C. M., Cerca, N. (2022). DNA Extraction leads to bias in bacterial quantification by qPCR. Appl. Microbiol. Biotechnol. 106, 7993–8006. doi: 10.1007/s00253-022-12276-4
Machado, D., Castro, J., Palmeira-de-Oliveira, A., Martinez-de-Oliveira, J., Cerca, N. (2016). Bacterial vaginosis biofilms: challenges to current therapies and emerging solutions. Front. Microbiol. 6. doi: 10.3389/fmicb.2015.01528
Mott, P. D., Taylor, C. M., Lillis, R. A., Ardizzone, C. M., Albritton, H. L., Luo, M., et al. (2021). Differences in the genital microbiota in women who naturally clear chlamydia trachomatis infection compared to women who do not clear; a pilot study. Front. Cell Infect. Microbiol. 11. doi: 10.3389/fcimb.2021.615770
Muzny, C. A., Blanchard, E., Taylor, C. M., Aaron, K. J., Talluri, R., Griswold, M. E., et al. (2018). Identification of key bacteria involved in the induction of incident bacterial vaginosis: a prospective study. J. Infect. Dis. 218, 966–978. doi: 10.1093/infdis/jiy243
Nugent, R. P., Krohn, M. A., Hillier, S. L. (1991). Reliability of diagnosing bacterial vaginosis is improved by a standardized method of gram stain interpretation. J. Clin. Microbiol. 29, 297–301. doi: 10.1128/JCM.29.2.297-301.1991
Pacha-Herrera, D., Erazo-Garcia, M. P., Cueva, D. F., Orellana, M., Borja-Serrano, P., Arboleda, C., et al. (2022). Clustering analysis of the multi-microbial consortium by lactobacillus species against vaginal dysbiosis among Ecuadorian women. Front. Cell. Infect. Microbiol. 12. doi: 10.3389/fcimb.2022.863208
Patterson, J. L., Girerd, P. H., Karjane, N. W., Jefferson, K. K. (2007). Effect of biofilm phenotype on resistance of gardnerella vaginalis to hydrogen peroxide and lactic acid. Am. J. Obstet Gynecol 197, 170.e1–170.e7. doi: 10.1016/j.ajog.2007.02.027
Patterson, J. L., Stull-Lane, A., Girerd, P. H., Jefferson, K. K. (2010). Analysis of adherence, biofilm formation and cytotoxicity suggests a greater virulence potential of gardnerella vaginalis relative to other bacterial-vaginosis-associated anaerobes. Microbiol. (Reading) 156, 392–399. doi: 10.1099/mic.0.034280-0
Poretsky, R., Rodriguez-R, L. M., Luo, C., Tsementzi, D., Konstantinidis, K. T. (2014). Strengths and limitations of 16S rRNA gene amplicon sequencing in revealing temporal microbial community dynamics. PloS One 9, e93827. doi: 10.1371/journal.pone.0093827
Ravel, J., Gajer, P., Abdo, Z., Schneider, G. M., Koenig, S. S. K., McCulle, S. L., et al. (2011). Vaginal microbiome of reproductive-age women. Proc. Natl. Acad. Sci. U.S.A. 108 Suppl 1, 4680–4687. doi: 10.1073/pnas.1002611107
Ricchi, M., Bertasio, C., Boniotti, M. B., Vicari, N., Russo, S., Tilola, M., et al. (2017). Comparison among the quantification of bacterial pathogens by qPCR, dPCR, and cultural methods. Front. Microbiol. 8. doi: 10.3389/fmicb.2017.01174
Roy, S., Sharma, M., Ayyagari, A., Malhotra, S. (1994). A quantitative microbiological study of bacterial vaginosis. Indian J. Med. Res. 100, 172–176.
Salinas, A. M., Osorio, V. G., Pacha-Herrera, D., Vivanco, J. S., Trueba, A. F., Machado, A. (2020). Vaginal microbiota evaluation and prevalence of key pathogens in ecuadorian women: an epidemiologic analysis. Sci. Rep. 10, 18358. doi: 10.1038/s41598-020-74655-z
Santiago, G. L., dos, S., Deschaght, P., El Aila, N., Kiama, T. N., Verstraelen, H., et al. (2011). Gardnerella vaginalis comprises three distinct genotypes of which only two produce sialidase. Am. J. Obstet Gynecol 204, 450.e1–450.e7. doi: 10.1016/j.ajog.2010.12.061
Santiago, G. L., dos, S., Tency, I., Verstraelen, H., Verhelst, R., Trog, M., et al. (2012). Longitudinal qPCR study of the dynamics of l. crispatus, l. iners, a. vaginae, (sialidase positive) g. vaginalis and p. bivia in the vagina. PloS One 7, e45281. doi: 10.1371/journal.pone.0045281
Sousa, L. G. V., Pereira, S. A., Cerca, N. (2023). Fighting polymicrobial biofilms in bacterial vaginosis. Microbial Biotechnol 1751–7915.14261. doi: 10.1111/1751-7915.14261
Stolovitzky, G., Cecchi, G. (1996). Efficiency of DNA replication in the polymerase chain reaction. Proc. Natl. Acad. Sci. U.S.A. 93, 12947–12952. doi: 10.1073/pnas.93.23.12947
Swidsinski, A., Mendling, W., Loening-Baucke, V., Swidsinski, S., Dörffel, Y., Scholze, J., et al. (2008). An adherent gardnerella vaginalis biofilm persists on the vaginal epithelium after standard therapy with oral metronidazole. Am. J. Obstet Gynecol 198, 97.e1–97.e6. doi: 10.1016/j.ajog.2007.06.039
Tettamanti Boshier, F. A., Srinivasan, S., Lopez, A., Hoffman, N. G., Proll, S., Fredricks, D. N., et al. (2020). Complementing 16S rRNA gene amplicon sequencing with total bacterial load to infer absolute species concentrations in the vaginal microbiome. mSystems 5, e00777–e00719. doi: 10.1128/mSystems.00777-19
Van Der Pol, W. J., Kumar, R., Morrow, C. D., Blanchard, E. E., Taylor, C. M., Martin, D. H., et al. (2019). In silico and experimental evaluation of primer sets for species-level resolution of the vaginal microbiota using 16S ribosomal RNA gene sequencing. J. Infect. Dis. 219, 305–314. doi: 10.1093/infdis/jiy508
Verstraelen, H., Verhelst, R., Claeys, G., De Backer, E., Temmerman, M., Vaneechoutte, M. (2009). Longitudinal analysis of the vaginal microflora in pregnancy suggests that l. crispatus promotes the stability of the normal vaginal microflora and that l. gasseri and/or l. iners are more conducive to the occurrence of abnormal vaginal microflora. BMC Microbiol. 9, 116. doi: 10.1186/1471-2180-9-116
Větrovský, T., Baldrian, P. (2013). The variability of the 16S rRNA gene in bacterial genomes and its consequences for bacterial community analyses. PloS One 8, e57923. doi: 10.1371/journal.pone.0057923
Wiggins, R., Hicks, S. J., Soothill, P. W., Millar, M. R., Corfield, A. P. (2001). Mucinases and sialidases: their role in the pathogenesis of sexually transmitted infections in the female genital tract. Sex Transm Infect. 77, 402–408. doi: 10.1136/sti.77.6.402
Woese, C. R., Kandler, O., Wheelis, M. L. (1990). Towards a natural system of organisms: proposal for the domains archaea, bacteria, and eucarya. Proc. Natl. Acad. Sci. U.S.A. 87, 4576–4579. doi: 10.1073/pnas.87.12.4576
Keywords: bacterial burden, vaginal microbiome, bacterial vaginosis, biofilm, qPCR standard, Gardnerella, Prevotella, Lactobacillus
Citation: Elnaggar JH, Ardizzone CM, Cerca N, Toh E, Łaniewski P, Lillis RA, Herbst-Kralovetz MM, Quayle AJ, Muzny CA and Taylor CM (2023) A novel Gardnerella, Prevotella, and Lactobacillus standard that improves accuracy in quantifying bacterial burden in vaginal microbial communities. Front. Cell. Infect. Microbiol. 13:1198113. doi: 10.3389/fcimb.2023.1198113
Received: 31 March 2023; Accepted: 05 June 2023;
Published: 19 June 2023.
Edited by:
Daniel Pletzer, University of Otago, New ZealandReviewed by:
António Machado, Universidad San Francisco de Quito, EcuadorMathys Jacobus Redelinghuys, University of the Witwatersrand, South Africa
Copyright © 2023 Elnaggar, Ardizzone, Cerca, Toh, Łaniewski, Lillis, Herbst-Kralovetz, Quayle, Muzny and Taylor. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Christopher M. Taylor, Y3RheTE1QGxzdWhzYy5lZHU=