 Anne Vatland Krøvel1,2*†
Anne Vatland Krøvel1,2*† Marit A. K. Hetland1,3*†
Marit A. K. Hetland1,3*† Eva Bernhoff1,2Anna Steensen Bjørheim1,2Markus André Soma1
Eva Bernhoff1,2Anna Steensen Bjørheim1,2Markus André Soma1 Iren H. Löhr1,2,4
Iren H. Löhr1,2,4- 1Department of Medical Microbiology, Stavanger University Hospital, Stavanger, Norway
- 2National Reference Laboratory for Legionella, Stavanger University Hospital, Stavanger, Norway
- 3Department of Biological Sciences, Faculty of Mathematics and Natural Sciences, University of Bergen, Bergen, Norway
- 4Department of Clinical Science, Faculty of Medicine, University of Bergen, Bergen, Norway
Sequence-based typing (SBT) of Legionella pneumophila is a valuable tool in epidemiological studies and outbreak investigations of Legionnaires’ disease. In the L. pneumophila SBT scheme, mompS2 is one of seven genes that determine the sequence type (ST). The Legionella genome typically contains two copies of mompS (mompS1 and mompS2). When they are non-identical it can be challenging to determine the mompS2 allele, and subsequently the ST, from Illumina short-reads. In our collection of 233 L. pneumophila genomes, there were 62 STs, 18 of which carried non-identical mompS copies. Using short-reads, the mompS2 allele was misassembled or untypeable in several STs. Genomes belonging to ST154 and ST574, which carried mompS1 allele 7 and mompS2 allele 15, were assigned an incorrect mompS2 allele and/or mompS gene copy number when short-read assembled. For other isolates, mainly those carrying non-identical mompS copies, short-read assemblers occasionally failed to resolve the structure of the mompS-region, also resulting in untypeability from the short-read data. In this study, we wanted to understand the challenges we observed with calling the mompS2 allele from short-reads, assess if other short-read methods were able to resolve the mompS-region, and investigate the possibility of using long-reads to obtain the mompS alleles, and thereby perform L. pneumophila SBT from long-reads only. We found that the choice of short-read assembler had a major impact on resolving the mompS-region and thus SBT from short-reads, but no method consistently solved the mompS2 allele. By using Oxford Nanopore Technology (ONT) sequencing together with Trycycler and Medaka for long-read assembly and polishing we were able to resolve the mompS copies and correctly identify the mompS2 allele, in accordance with Sanger sequencing/EQA results for all tested isolates (n=35). The remaining six genes of the SBT profile could also be determined from the ONT-only reads. The STs called from ONT-only assemblies were also consistent with hybrid-assemblies of Illumina and ONT reads. We therefore propose ONT sequencing as an alternative method to perform L. pneumophila SBT to overcome the mompS challenge observed with short-reads. To facilitate this, we have developed ONTmompS (https://github.com/marithetland/ONTmompS), an in silico approach to determine L. pneumophila ST from long-read or hybrid assemblies.
1 Introduction
The Legionella-bacteria can cause a severe and potentially fatal form of pneumonia called Legionnaires’ disease (LD). When the bacteria colonize and multiply in man-made systems with favorable conditions for growth, it may pose a threat to human health, through inhalation of bacteria-contaminated aerosols (Bartram et al., 2007; Whiley et al., 2014). There are more than 60 known species of Legionella with varying pathogenicity (Parte et al., 2020). Legionella pneumophila is the species implicated in at least 90% of the reported LD cases worldwide (reviewed in Herwaldt and Marra, 2018; Chauhan and Shames, 2021). L. pneumophila can be subtyped into at least 15 serogroups based on surface molecules and also into sequence types (STs) determined by the seven genes flaA, pilE, asd, mip, mompS, proA and neuA (Gaia et al., 2005; Ratzow et al., 2007). The current number of defined STs is over 3100 (Legionella-SBT, 2023).
Sequence-based typing (SBT) is a valuable tool for source investigations and epidemiological studies of L. pneumophila that allows for rapid molecular typing and inter-laboratorial comparison (Gaia et al., 2005; Ratzow et al., 2007). Although L. pneumophila SBT performed by Sanger sequencing is still considered the gold standard, more recently, whole-genome sequencing (WGS) has become the method of choice, providing both ST and superior information about genetic relatedness, and is today indispensable in surveillance and outbreak investigations (Moran-Gilad et al., 2015; Khodr et al., 2016; Raphael et al., 2016; Krøvel et al., 2022; Ricci et al., 2022). Analyses like core or whole genome multi locus sequence typing (cgMLST/wgMLST) and single nucleotide polymorphism (SNP) analysis have higher discriminatory power than SBT and would provide more information to aid the identification of the source of an outbreak.
A challenge in the L. pneumophila SBT scheme is that the mompS gene, which is used to determine the ST, is usually present with two copies in the genome, mompS1 and mompS2. Only mompS2 contributes to the ST (Gaia et al., 2005; Gordon et al., 2017). The two mompS copies are closely positioned in the genome, resulting in a mompS-region of about 2100 bp (see Figure 1). L. pneumophila SBT by Sanger sequencing amplifies mompS2 using specific primers. Short-read WGS gives a maximum of 300 bp reads, which are too short to cover the mompS gene let alone the entire mompS-region. When the two mompS copies are non-identical, existing in silico approaches for L. pneumophila SBT have been shown to have limitations, related to erroneous calling of the mompS alleles or incorrect assembly of the mompS-region, which may result in incorrect ST determination and/or untypeabilty (Gordon et al., 2017).
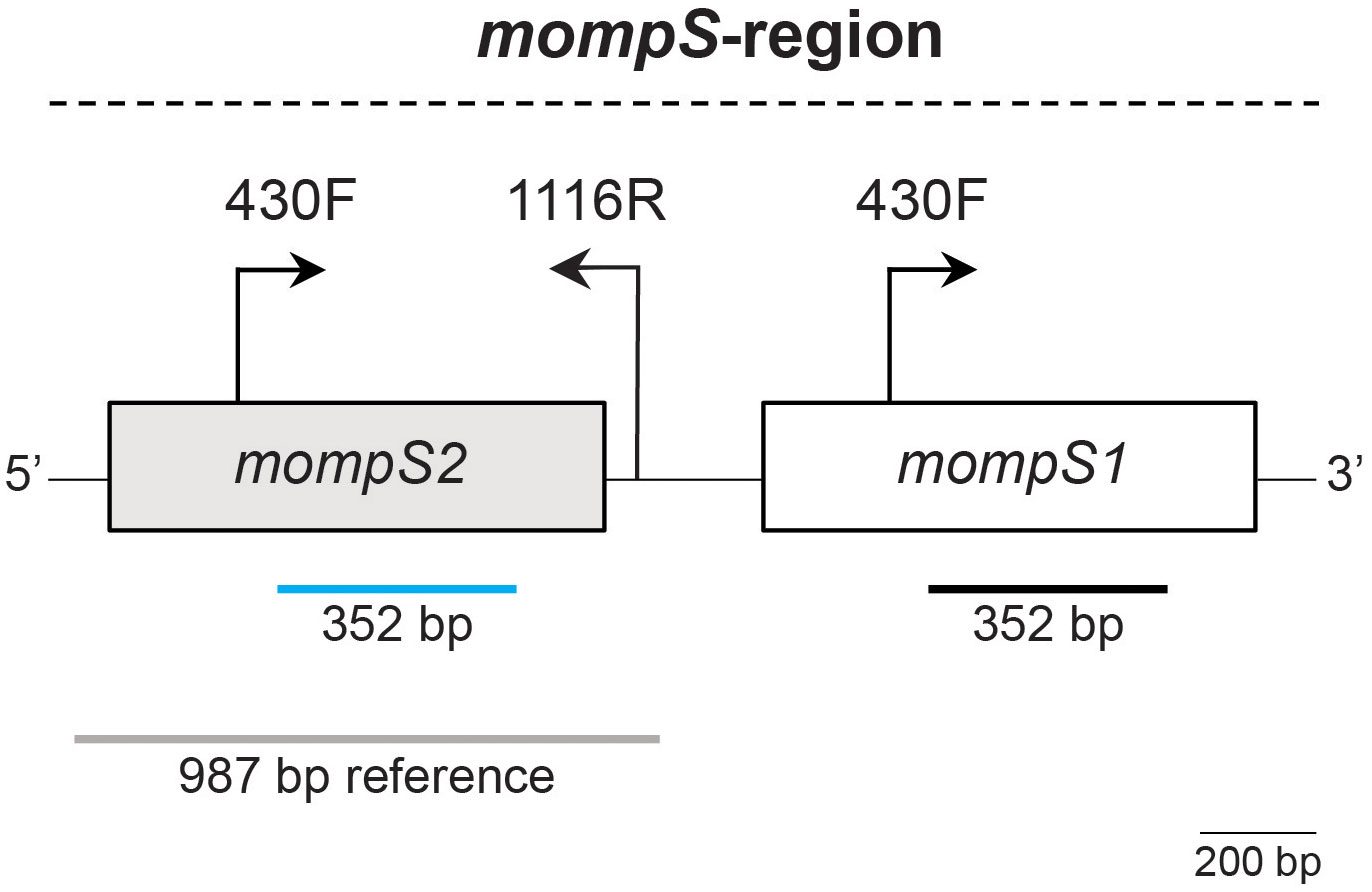
Figure 1 The mompS-region. The two mompS copies are closely positioned in the genome. The first step of Sanger sequencing uses primers 430F and 1116R to specifically amplify mompS2 (Gaia et al., 2005; Gordon et al., 2017). The 352 bp conserved region used for determination of mompS2 in L. pneumophila SBT is marked by a blue line. The mompS1 gene also contains the primer binding site for primer 430F and a 352 bp conserved region, marked by a black line. A reference sequence defined by Gordon et al. (2017) to distinguish mompS2 from mompS1, which is also used in our ONTmompS in silico approach, is indicated by a grey line. The 987 bp reference sequence covers the 5’ flanking region from position 157, the entire coding sequence of mompS2 (204-1070) and the 3’ flanking region including primer 1116R. The positions refer to those in the GenBank sequence AF078136, which was used to design the original primers for Sanger sequencing. A 200 bp scalebar is indicated.
Illumina short-read WGS has so far been the method of choice for genomic surveillance because of the higher basecalling accuracy compared to long-read sequencing technologies like Oxford Nanopore Technologies (ONT). However, challenges with untypeability and resolution of repetitive regions due to the short read length has led to exploration of long-read sequencing as an alternative (Ben Khedher et al., 2022). At the moment, hybrid-assemblies of short- and long-reads are considered the WGS gold standard over ONT-only assemblies with regards to accuracy (Wick et al., 2023).
In our laboratory, we have observed several challenges related to calling mompS from short-reads. In diagnostics and surveillance of Legionella, time and cost is of the essence and generation of hybrid assemblies for all samples is not likely to be standard procedure. Therefore, the possibility of using ONT-only data to overcome the shortcomings observed in L. pneumophila SBT from short-reads is highly relevant and has to our knowledge not previously been investigated. In this study, we wanted to understand the challenges we observed using short-reads, assess if other short-read methods were able to resolve the mompS-region and investigate the possibility of using long-reads to obtain the mompS allele, and thereby perform L. pneumophila SBT from long-reads only.
2 Materials and methods
2.1 Sample collection
At the National Reference Laboratory for Legionella, Stavanger University Hospital, Norway, we have a collection of Illumina sequenced clinical and environmental isolates, collected between 2001 and 2022 (n=233). Of these, 35 were selected for further analysis in this study, based on discrepancies between the mompS result from Illumina short-read and Sanger sequencing, presence of non-identical mompS copies or untypeability of mompS (n=27). Isolates with identical copies of mompS were included as controls (n=8).
A further 46 isolates were included later in the study to validate the ONTmompS in silico SBT approach that we developed for assigning L. pneumophila ST (see section 3.3).
2.2 Whole-genome sequencing
The 35 L. pneumophila isolates had previously been Illumina short-read sequenced and the fastq-files deposited in our in-house sequence database. In short, DNA had been extracted using MagNA Pure 96 with the Pathogen Universal 200 4.0 purification protocol (Roche Applied Science, Penzberg, Germany). Genomic libraries were prepared using Illumina DNA library prep and sequenced to ≥30X read depth using the Illumina MiSeq platform (see Table S1 for details and BioSample accessions).
The same 35 isolates were long-read sequenced in this study. DNA was extracted using the GenFind V3 kit (Beckman Coulter, Indianapolis, United States). Genomic libraries for long-read sequencing were prepared using the Kit12 chemistry and the ligation sequencing kit (SQK-LSK112) in combination with the R10.4 MinION flow cells (n=34), or the Kit9 ligation sequencing kit (SQK-LSK-109) in combination with the R9.4.1 flow cell (n=1) from ONT (Oxford, UK). All libraries were sequenced on the ONT GridION device (GRD-X5G003), aiming for ≥100X read depth. Guppy v6.4.2 was used to basecall and demultiplex the fast5-files with the super accurate basecalling model (see Table S1 for details).
The 46 isolates used for validation of the ONTmompS in silico L. pneumophila SBT approach were ONT sequenced using Kit9/R9.4.1 (n=39) or Kit12/R10.4 (n=7) and basecalled with Guppy v6.4.2 using the super accurate model (see Table S2 for source details and BioSample accessions).
2.3 Assembly and polishing
For the short-reads we used Asmbl v0.2.0 (https://github.com/marithetland/Asmbl) to perform read trimming, de novo assembly and quality control: TrimGalore v0.6.7 (https://github.com/FelixKrueger/TrimGalore) was used to remove adapter-contamination and low-quality reads, prior to assembly. We generated two sets of short-read assemblies (for all 35 genomes): The first with Unicycler v0.5.0 (Wick et al., 2017), which uses SPAdes v3.15.5 (Bankevich et al., 2012) for assembly, and the second with Unicycler v0.4.8, which uses SPAdes v3.13.0, includes read error correction and short-read polishes the assembly with Pilon v1.24. In addition, we created assemblies with the following tools and parameters: 1) Unicycler v0.4.8 with SPAdes v3.13.0 without read error correction, 2) without Pilon short-read polishing, and 3) without both, 4) Unicycler v0.5.0 with SPAdes v3.13.0, 5) SPAdes v3.15.5 on its own, 6) SPAdes v3.13.0, and 7) SKESA v2.4.0 (Souvorov et al., 2018).
For the long-reads, Filtlong v0.2.1 (https://github.com/rrwick/Filtlong) was used to discard the worst 5% of read bases and any reads shorter than 1 kbp prior to assembly. We then generated long-read assemblies with three different tools: Unicycler v0.5.0 (which uses a miniasm v0.3-r179 + racon v1.5.0 pipeline), Flye v2.9 (Kolmogorov et al., 2019), and Trycycler v0.5.3 (Wick et al., 2021). Trycycler was run according to the instructions at https://github.com/rrwick/Trycycler/wiki/How-to-run-Trycycler. In short, the reads were subsampled into 12 read-sets and assembled using Flye v2.9 (Kolmogorov et al., 2019), miniasm v0.3-r179 (Li, 2016) + minipolish v0.1.3 (Wick and Holt, 2019) and raven v1.8.1 (Vaser and Šikić, 2021), providing a total of 12 assemblies per isolate, four from each assembly method. The assemblies were then clustered and reconciled. Next, a multiple sequence alignment using MUSCLE v3.8.1551 (Edgar, 2004) was performed, followed by partitioning of the reads. Lastly, a single consensus assembly was generated for each isolate. To correct any small-scale errors, the final assemblies were polished with the long-reads using Medaka v1.7.2 with model r1041_e81_sup_g610 for R10.4 flow cells and model r941_min_sup_g507 for R9.4.1 flow cells, respectively (https://github.com/nanoporetech/medaka).
To assess if ONT-only sequencing with the Kit12/R10.4 chemistry/flow cells was as accurate as the current gold-standard of using ONT data in hybrid with Illumina reads, and thus sufficient for L. pneumophila SBT, we also generated long-read first hybrid assemblies. This was done by further polishing the Trycyler+Medaka assemblies with short-reads using Polypolish v0.5.0 and Polca v4.0.5.
2.4 Sequence-based typing and identification of mompS1 and mompS2
Sanger sequencing was the method used for SBT at the National Reference Laboratory for Legionella until August 2019, when it was replaced by Illumina short-read WGS. All isolates in this study (n=35) were analyzed using Sanger sequencing according to the standardised protocol of the ESCMID Study Group for Legionella Infections (ESGLI) (Gaia et al., 2005; Ratzow et al., 2007) or the ST of the isolate was previously confirmed as part of an External Quality Assessment (EQA) programme.
For the Illumina sequenced isolates, the STs were determined with legsta v0.5.1 (https://github.com/tseemann/legsta) and/or with BLASTn v2.12.0+ (Camacho et al., 2009). Presence of non-identical copies along with the confirmation of mompS2 were identified by examination of the assembly sequence. For investigations of the mompS alleles in STs 154 and 574, we created core genome alignments with RedDog v1b11 (https://github.com/katholt/RedDog) against hybrid assemblies of these STs and viewed the bam files in IGV v2.15.2 (Robinson et al., 2011).
To identify both mompS1/mompS2 and the L. pneumophila ST from ONT-only and hybrid assemblies, we have developed ONTmompS v2.0.0 (https://github.com/marithetland/ONTmompS), see section 3.3 for details.
3 Results
3.1 Challenges with identifying mompS2 for use in SBT from short-read sequencing data
In our in-house sequence database of L. pneumophila genomes (n=233), there were 62 unique STs. Of these, 18 STs (29%) carried non-identical copies of mompS. We included a set of 35 isolates in this study; 27 isolates representing each of the 18 STs with non-identical mompS copies and 8 isolates (7 STs) with identical copies. Currently, to identify L. pneumophila SBT at the National Reference Laboratory for Legionella, isolates are subjected to Illumina short-read sequencing, followed by Unicycler assembly (which uses SPAdes), and legsta, which is an in silico SBT tool, to determine the ST. We have encountered three main challenges with this method: 1) erroneous calling of the mompS1 allele instead of mompS2 by legsta, 2) misassembly of the mompS genes when using Unicycler v0.4.8, and 3) failure to resolve the structure of the mompS-region with Unicycler v0.4.8 and v0.5.0.
The first challenge occurs when an isolate contains non-identical copies of the mompS gene. Upon repeated runs of legsta v0.5.1 using the same input assembly-files, the mompS2 allele called varied between the two alleles found in mompS1 and mompS2. This was the case for all genomes with non-identical mompS copies where both alleles were defined in the database. This issue was solved by examining the assembly graph in Bandage (Wick et al., 2015) and using the mompS2 flanking sequence in BLASTn searches to identify the correct mompS2 allele for use in the SBT scheme.
The second challenge is more demanding as it is due to misassembly of the reads in the mompS region, i.e. some reads that belonged to mompS2 assembled into mompS1 and vice versa. This challenge applies to at least genomes belonging to ST154 and ST574 when using Unicycler v0.4.8 (Table 1). Both these STs carry mompS1 allele 7 and mompS2 allele 15, differing by 1 nucleotide at position 63, between A and G. For the isolates belonging to these STs (n=8), Sanger sequencing always determined the mompS2 gene with allele 15, while the short-read assembly often determined the mompS1 and mompS2 genes as identical, either with allele 7 or 15. Further, the identified mompS2 allele sometimes varied between 7 and 15 when the same isolate was re-sequenced. We investigated this with read mapping, which showed that the two nucleotides were mapped to both mompS1 and mompS2 at different frequencies. Further, for some of the ST154 and ST574 isolates, the short-read assemblies contained an additional copy of the mompS gene, resulting in three identical mompS copies per isolate. This misalignment and misassembly of reads occurs because of the repetitiveness of the mompS-region and that the Illumina read lengths are shorter (≤300 bp) than the 352 bp part of the mompS2 gene that is used for allele determination (Gaia et al., 2005).
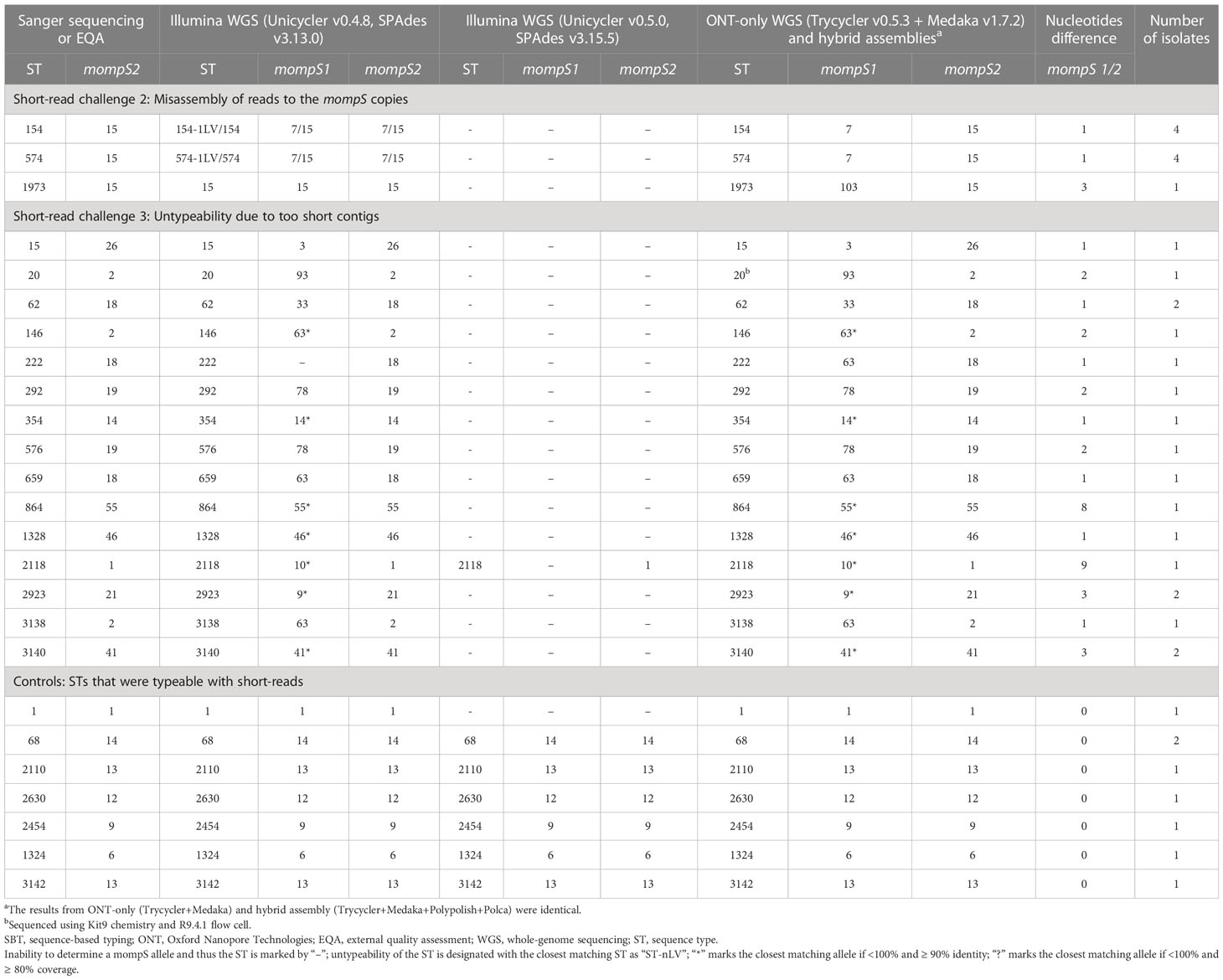
Table 1 Comparison of L. pneumophila SBT results for Sanger, Illumina short-read (assemblers with highest and lowest accuracy are shown), ONT-only and hybrid assemblies for genomes in the dataset.
The third challenge; failure to resolve the structure of the mompS-region, which resulted in untypeability of isolates, was seen for 27 isolates when using Unicycler v0.5.0, including the eight that were misassembled with Unicycler v0.4.8 (see Table 1). Of these, 26 genomes carried non-identical mompS copies and one genome (ST1) had identical mompS copies. Investigations of the assembly graphs showed that the mompS-region of these isolates consisted of several short contigs that did not span the mompS genes and the flanking regions, which are used to distinguish mompS2 from mompS1.
We investigated whether the read length or read depth affected the results we observed with the short-read assemblers. The isolates were sequenced with different Illumina kits leading to paired-end read lengths of 2x300 bp, 2x250 or 2x150 bp (see Table S1); we observed no pattern indicating that either kit was linked to typeability. Similarly, we saw no link between the read depth and typeability (the read depths ranged 32-212X). As there were differences in the short-read assemblies with the two Unicycler versions, we tested several methods of assembly with/without polishing and read error correction to see if any would consistently type the ST of all the genomes (see section 2.3). Unfortunately, none of the methods consistently typed all the isolates, however those that included short-read polishing did produce better results (for details see Table S3).
In sum, the observed misassembly or untypeability of mompS2, which varied with different assemblers or versions of the same tools, indicates major challenges with using short-read sequencing to identify the mompS2 allele, and thus the ST, for several L. pneumophila isolates.
3.2 Long-read sequencing resolves the mompS-region and provides the necessary accuracy for L. pneumophila SBT
The combination of long-reads spanning the entire mompS-region (Figure 1) with the recent improvements in accuracy to the ONT sequencing technology, led us to the hypothesis that ONT reads on their own would be sufficient for calling mompS and subsequently L. pneumophila SBT. We assessed three common methods of long-read assembly (Trycycler, Flye and Unicycler) to see if mompS2 could be reliably identified using ONT-only reads. The 35 isolates were ONT sequenced, assembled and polished to create seven sets of assemblies for each isolate (Figure 2).

Figure 2 Accuracy of Legionella pneumophila ST calls using ONT-only or hybrid assemblies. Three assemblers (Trycycler v0.5.3, Flye v2.9, Unicycler v0.5.0) were used for long-read only assembly, followed by long-read polishing with Medaka v1.7.2. The Trycycler+Medaka assemblies were further short-read polished with Polca v4.0.5 and Polypolish v0.5.0 to create hybrid assemblies. Genomes were sequenced with Kit12/R10.4 (n=34) and Kit9/R9.4 (n=1). The number (and percentage) in each box indicates the amount of isolates whose ST was accurately called when using these tools. See also Table S1 for more details.
The mompS-region of all 35 isolates was resolved using either of the methods, but only Trycycler+Medaka and hybrid assemblies with long and short-read polishing assigned the correct ST in accordance with Sanger/EQA results in all isolates (Figure 2). For the Trycycler-only assemblies, one genome had an incorrect proA allele, which was corrected with Medaka long-read polishing. For the Flye+Medaka and Unicycler+Medaka assemblies, three and two genomes had one incorrect allele call, respectively.
To perform in silico SBT from long-read assemblies we first ran legsta, but this tool gave inconsistent results similar to what we observed with short-reads: some alleles were not called, even though we could identify them with BLASTn searches (most commonly flaA allele 11 and neuA allele 11), and the mompS allele was sometimes incorrectly called in genomes with non-identical copies.
3.3 ONTmompS for in silico SBT of L. pneumophila from long-reads
To identify the mompS1 and mompS2 alleles and the L. pneumophila ST from ONT-only and hybrid assemblies, we have developed the open source tool ONTmompS v2.0.0 (https://github.com/marithetland/ONTmompS). For each input assembly, the tool first identifies the mompS2 allele by distinguishing it from mompS1: BLASTn v2.12.0+ is used to query the assembly against a 987 bp reference sequence (see Figure 1). This reference was originally defined for use in the mompS tool (https://github.com/bioinfo-core-BGU/mompS) developed by Gordon et al. (2017), and is a conserved sequence that covers the 352 nucleotides of the mompS2 gene that are used for allele definition, and flanking regions, including the 430F upstream primer and the downstream primer 1116R. Next a pairwise Smith-Waterman local sequence alignment (EMBOSS v6.6.0.0) (Smith and Waterman, 1981) is performed on each mompS copy against the 1116R primer sequence, which is the downstream primer traditionally used in Sanger sequencing and which can be used to distinguish mompS2 from mompS1 (Gaia et al., 2005; Gordon et al., 2017). The copy that aligns to the primer is assigned as mompS2 and the other copy as mompS1. Once the mompS2 copy has been identified, the alleles of all seven genes in the SBT scheme and the resulting ST are determined using the same logic as that developed for in silico multi-locus sequence typing (MLST) of Klebsiella pneumoniae in Kleborate v2.3.1 (https://github.com/katholt/Kleborate) (Lam et al., 2021). ONTmompS reports two main results: 1) the allele numbers of the mompS1 and mompS2 copies and 2) the ST together with the alleles of the seven genes in the scheme.
Using ONTmompS v2.0.0, we were able to identify mompS2 (and thus ST) for all Trycycler+Medaka and hybrid assemblies, in accordance with Sanger sequencing or EQA results for all 35 isolates.
This analysis showed that all the isolates that were misassembled or untypeable when using short-read assembly methods were typeable with ONT-only assemblies when using Trycycler for assembly together with Medaka long-read polishing. The controls, i.e. the isolates that had identical mompS copies and were typeable with short-read sequencing, were also typeable with this ONT-only workflow.
To further test and confirm that ONT-only reads are sufficient for L. pneumophila SBT, and potentially also from the Kit9/R9.4.1 chemistry/flow cells, we tested our workflow on an additional 46 ONT-sequenced isolates (Table S2). Of these isolates, 39 were sequenced using Kit9 chemistry/R9.4.1 flow cells, the remaining seven with Kit12/R10.4. All isolates were basecalled with the super accurate model and assembled with Trycycler+Medaka followed by analyses using ONTmompS. The mompS-region was resolved for all the isolates and STs were typeable and in accordance with Illumina and Sanger sequencing where that was available. This indicates that ONT-only reads (from at least Kit9/R9.4.1 and Kit12/R.10.4) are a reliable and effective solution for L. pneumophila SBT.
4 Discussion and conclusion
In this study, we encountered several challenges when calling mompS2 from Illumina short-read sequences and have shown that ONT long-read sequences are a reliable alternative for determining mompS2 and the ST of L. pneumophila. Today, epidemiological investigations of L. pneumophila and associated disease or outbreaks are mainly done by Sanger SBT and/or WGS analyses. The challenges with identifying mompS2 due to non-identical copies of mompS and the short length of Illumina reads is a hindrance for routine use of short-reads in typing and surveillance. In 2015, a WGS-based cgMLST typing scheme for L. pneumophila was proposed, which showed high resolution of strains within the same ST/clonal complex from short-reads, but the challenge with typing due to non-identical mompS copies remained unsolved (Moran-Gilad et al., 2015). In 2017, the mompS tool (https://github.com/bioinfo-core-BGU/mompS) was developed to solve the challenge by accepting only reads that also flanked the mompS2 gene and thus ensuring correct allele calls, and to ensure backwards compatibility with the Sanger SBT scheme (Gordon et al., 2017). However, in our experience the tool often failed, due to too low read coverage of the 352 bp mompS2 sequence once the reads that do not overlap the gene and a flanking region have been filtered out. It is therefore not included as part of our standard bioinformatic analyses for L. pneumophila typing. In 2018, the first release of legsta was published (https://github.com/tseemann/legsta). This in silico approach for L. pneumophila SBT takes assemblies as input and uses the L. pneumophila SBT database together with primer sequences to identify the correct alleles for the SBT scheme. For assemblies from short-reads, long-reads, and hybrid assemblies, we unfortunately experienced that this tool was sometimes unable to call alleles or called the incorrect mompS allele. Neither the mompS tool nor legsta have been maintained or updated for several years.
Our analyses of short-read L. pneumophila genomes also showed that different assemblers or even different versions of the same assembler affected the typeability of the mompS2 allele, adding yet another layer of complexity to the challenge of identifying STs from short-reads. When taking together the challenges of resolving the mompS-region from short-reads and the issues with identifying the mompS2 allele with existing tools, it was clear that an alternative method was needed, which is why we explored using ONT long-reads and developed ONTmompS to perform in silico L. pneumophila SBT on ONT-only or hybrid assembled genomes.
With ONT-only reads and the ONTmompS tool, we were able to resolve the mompS-region and to perform in silico L. pneumophila SBT on all 81 genomes that were tested (the initial 35 genomes + the 46 that were used for validation). Both the Kit9/R9.4.1 and Kit12/R10.4 chemistry/flow cells with Guppy’s super accurate basecalling model were used. The assemblies that were created with Trycycler followed by long-read polishing with Medaka (Trycycler+Medaka) were assigned the same ST as with Sanger SBT, EQA or hybrid assemblies for all isolates where these results were available, and an ST was assigned when using ONTmompS in all of the tested genomes. With Trycycler-only, Flye+Medaka and Unicycler+Medaka, the STs of a few of the 35 genomes did not match with the Sanger/EQA or hybrid assemblies. There were no apparent quality issues with these genomes. We therefore recommend using Trycycler with Medaka long-read polishing for ONT-only assemblies for in silico SBT of L. pneumophila.
There was no difference in the typeability of the genomes that were ONT sequenced with Kit9/R9.4.1 and Kit12/R10.4, indicating that both chemistries/kits are appropriate for determining STs of L. pneumophila. This is consistent with two recent studies of ONT sequencing: Wagner et al. (2023) showed that genomic analyses of Bordetella pertussis from ONT-only assemblies from Kit12/R10.4 yielded results with comparable accuracy as from short-reads. Similarly, Foster-Nyarko et al. (2023) found that ONT-only reads from Kit10/R9.4, basecalled with the super accuracy model, with or without Medaka long-read polishing, were sufficient for calling ST, capsule type and AMR determinants for Klebsiella pneumoniae, but not sufficient for defining outbreak clusters. It was outside the scope of our study to investigate if the R10.4 ONT-only reads could be used for defining outbreak clusters of L. pneumophila. However, Sanderson et al. (2023) recently compared the raw read accuracy and assembly accuracy of ONT Kit10/R9.4, Kit12/R10.3, Kit12/R10.4, Kit12/R10.4 with duplex reads, and Illumina sequencing of four bacterial pathogens (not L. pneumophila). Sereika et al. (2022) did a similar comparison. Both studies found that ONT Kit12/R10.4 duplex reads that were basecalled with the super accuracy model could be used for complete reconstruction of bacterial genomes without the use of Illumina reads. However, Sanderson et al. (2023) noted that recovery of small plasmids was inconsistent and that hybrid assemblies still remain the most cost-effective and robust approach for bacterial whole-genome reconstruction.
By combining real-time basecalling and the ONT Kit12/R10.4, Wagner et al. (2023) recently showed that it is possible to perform highly accurate and fast high-resolution typing of bacterial pathogens while sequencing is ongoing, highlighting the time-saving potential of the ONT-technology in outbreak situations. Furthermore, the flexibility in the number of samples and/or flow cells that can be run on for instance ONT’s GridIon device, makes ONT well-suited and efficient for analyzing both a single isolate and larger collections, e.g. in suspected outbreaks. In our laboratory, the cost of generating ONT-only assemblies is overall lower than for Illumina-only assemblies, with the difference being more profound for lower sample numbers. However, the cost per isolate for both technologies are dependent on the kit used, number of samples analyzed and how well the capacity of the sequencing kit/flow cell is utilized. ONT has recently launched Kit14/R10.4.1 flow cell promising an even more improved accuracy. Given our results with Kit9 and Kit12 we expect to obtain similar results with Trycycler+Medaka and hopefully even better results with the other assembly methods with the improved ONT technology.
L. pneumophila SBT is important for the characterization of L. pneumophila isolates and for standardized comparison of results over time and between different laboratories (Raphael et al., 2016). In source investigations, the SBT serves well to identify potential sources in the initial phase of the investigation. However, due to the lower discriminatory power of this method compared to whole genome genetic relatedness analyses, further analyses are usually needed to confirm a potential source (Raphael et al., 2016; Ricci et al., 2022). By utilizing long-read WGS, SBT can easily be assessed in an initial analysis, and in-depth whole-genome analyses may also be performed, either together with complementary short-reads to correct ONT read errors, or ONT reads on their own if and when these have been proven sufficient to use in whole genome analyses, for which evidence is starting to emerge (Sereika et al., 2022; Sanderson et al., 2023; Wagner et al., 2023).
To conclude, ONT-only sequencing is sufficient for identifying mompS2 and L. pneumophila ST. Our analyses show that ONT-only assemblies provide a cost- and time-efficient solution for determining L. pneumophila ST from WGS, where Illumina short-reads often fail to identify mompS2. For the best results, we recommend basecalling with Guppy using the super accurate basecalling model, assembly with Trycycler and polishing with Medaka, before identifying the ST with ONTmompS.
Data availability statement
The datasets presented in this study are available online in the European Nucleotide Archive under accession numbers PRJEB58776 and PRJEB50383. Please see Tables S1 and S2 for individual accession numbers.
Author contributions
AK, MH, EB, and IL conceptualized the study. AK and EB performed genome sequencing. MH and MS developed ONTmompS. AK and AB analyzed Sanger and WGS results. IL provided resources and supervision. AK, MH and EB wrote the first draft of the manuscript. All authors contributed to data interpretation, reviewed and edited the manuscript, and have read and agreed to the published version of the manuscript.
Funding
This study has been funded by the Department of Medical Microbiology at Stavanger University Hospital, Norway.
Acknowledgments
We would like to thank Celine Bergum Nilsen, Maria Helle Spence, Ragna-Johanne Bakksjø, Mona Øye Lütcherath for excellent laboratory assistance, and Monica Regine Romstad and Benjamin Silvester for valuable discussions and proofreading of the manuscript.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fcimb.2023.1176182/full#supplementary-material
References
Bankevich, A., Nurk, S., Antipov, D., Gurevich, A. A., Dvorkin, M., Kulikov, A. S., et al. (2012). SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 19 (5), 455–477. doi: 10.1089/cmb.2012.0021
Bartram, J., Chartier, Y., Lee, J. V., Pond, K., Surmann-Lee, S. (2007) WHO. legionella and the prevention of prevention of legionellosis. Available at: https://www.who.int/water_sanitation_health/publications/legionella/en/.
Ben Khedher, M., Ghedira, K., Rolain, J. M., Ruimy, R., Croce, O. (2022). Application and challenge of 3rd generation sequencing for clinical bacterial studies. Int. J. Mol. Sci. 23 (3), 1395. doi: 10.3390/ijms23031395
Camacho, C., Coulouris, G., Avagyan, V., Ma, N., Papadopoulos, J., Bealer, K., et al. (2009). BLAST+: architecture and applications. BMC Bioinf. 15 (10), 421. doi: 10.1186/1471-2105-10-421
Chauhan, D., Shames, S. R. (2021). Pathogenicity and virulence of legionella: intracellular replication and host response. Virulence 12 (1), 1122–1144. doi: 10.1080/21505594.2021.1903199
Edgar, R. C. (2004). MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 32 (5), 1792–1797. doi: 10.1093/nar/gkh340
Foster-Nyarko, E., Cottingham, H., Wick, R. R., Judd, L. M., Lam, M. M. C., Wyres, K. L., et al. (2023). Nanopore-only assemblies for genomic surveillance of the global priority drug-resistant pathogen, klebsiella pneumoniae. Microb. Genom 9 (2), mgen000936. doi: 10.1099/mgen.0.000936
Gaia, V., Fry, N. K., Afshar, B., Lück, P. C., Meugnier, H., Etienne, J., et al. (2005). Consensus sequence-based scheme for epidemiological typing of clinical and environmental isolates of legionella pneumophila. J. Clin. Microbiol. 43 (5), 2047–2052. doi: 10.1128/JCM.43.5.2047-2052.2005
Gordon, M., Yakunin, E., Valinsky, L., Chalifa-Caspi, V., Moran-Gilad, J. (2017). A bioinformatics tool for ensuring the backwards compatibility of legionella pneumophila typing in the genomic era. Clin. Microbiol. Infect. 23 (5), 306–310. doi: 10.1016/j.cmi.2017.01.002
Herwaldt, L. A., Marra, A. R. (2018). Legionella: a reemerging pathogen. Curr. Opin. Infect. Dis. 31 (4), 325–333. doi: 10.1097/QCO.0000000000000468
Khodr, A., Kay, E., Gomez-Valero, L., Ginevra, C., Doublet, P., Buchrieser, C., et al. (2016). Molecular epidemiology, phylogeny and evolution of legionella. Infect. Genet. Evol. 43, 108–122. doi: 10.1016/j.meegid.2016.04.033
Kolmogorov, M., Yuan, J., Lin, Y., Pevzner, P. A. (2019). Assembly of long, error-prone reads using repeat graphs. Nat. Biotechnol. 37 (5), 540–546. doi: 10.1038/s41587-019-0072-8
Krøvel, A. V., Bernhoff, E., Austerheim, E., Soma, M. A., Romstad, M. R., Löhr, I. H. (2022). Legionella pneumophila in municipal shower systems in stavanger, norway; a longitudinal surveillance study using whole genome sequencing in risk management. Microorganisms 10 (3), 536. doi: 10.3390/microorganisms10030536
Lam, M. M. C., Wick, R. R., Watts, S. C., Cerdeira, L. T., Wyres, K. L., Holt, K. E. (2021). A genomic surveillance framework and genotyping tool for klebsiella pneumoniae and its related species complex. Nat. Commun. 12 (1), 4188. doi: 10.1038/s41467-021-24448-3
Legionella-SBT (2023). Database ESCMID study group for legionella infections (ESGLI) (United Kingdom: United Kingdom Health and Safety Agency (UKHSA)). Available at: https://legionella-sbt[at]ukhsa.gov.uk
Li, H. (2016). Minimap and miniasm: fast mapping and de novo assembly for noisy long sequences. Bioinf. (Oxford England) 32 (4), 2103–2110. doi: 10.1093/bioinformatics/btw152
Moran-Gilad, J., Prior, K., Yakunin, E., Harrison, T. G., Underwood, A., Lazarovitch, T., et al. (2015). Design and application of a core genome multilocus sequence typing scheme for investigation of legionnaires' disease incidents. Euro Surveill 20 (28), 21186. doi: 10.2807/1560-7917.ES2015.20.28.21186
Parte, A. C., Sardà Carbasse, J., Meier-Kolthoff, J. P., Reimer, L. C., Göker, M. (2020). List of prokaryotic names with standing in nomenclature (LPSN) moves to the DSMZ. Int. J. Syst. Evolutionary Microbiol. 70, 5607–5612. doi: 10.1099/ijsem.0.004332
Raphael, B. H., Baker, D. J., Nazarian, E., Lapierre, P., Bopp, D., Kozak-Muiznieks, N. A., et al. (2016). Genomic resolution of outbreak-associated legionella pneumophila serogroup 1 isolates from new York state. Appl. Environ. Microbiol. 82 (12), 3582–3590. doi: 10.1128/AEM.00362-16
Ratzow, S., Gaia, V., Helbig, J. H., Fry, N. K., Lück, P. C. (2007). Addition of neuA, the gene encoding n-acylneuraminate cytidylyl transferase, increases the discriminatory ability of the consensus sequence-based scheme for typing legionella pneumophila serogroup 1 strains. J. Clin. Microbiol. 45 (6), 1965–1968. doi: 10.1128/JCM.00261-07
Ricci, M. L., Fillo, S., Ciammaruconi, A., Lista, F., Ginevra, C., Jarraud, S., et al. (2022). Genome analysis of Legionella pneumophila ST23 from various countries reveals highly similar strains. Life Sci. Alliance 5 (6), e202101117. doi: 10.26508/lsa.202101117
Robinson, J. T., Mesirov, J. P., Thorvaldsdóttir, H., Winckler, W., Guttman, M., Lander, E. S., et al. (2011). Integrative genomics viewer. Nat. Biotechnol. 29 (1), 24–26. doi: 10.1038/nbt.1754
Sanderson, N. D., Kapel, N., Rodger, G., Webster, H., Lipworth, S., Street, T. L., et al. (2023). Comparison of R9.4.1/Kit10 and R10/Kit12 Oxford nanopore flowcells and chemistries in bacterial genome reconstruction. Microb. Genom 9 (1), mgen000910. doi: 10.1099/mgen.0.000910
Sereika, M., Kirkegaard, R. H., Karst, S. M., Michaelsen, T. Y., Sørensen, E. A., Wollenberg, R. D., et al. (2022). Oxford Nanopore R10.4 long-read sequencing enables the generation of near-finished bacterial genomes from pure cultures and metagenomes without short-read or reference polishing. Nat. Methods 19, 823–826. doi: 10.1016/0022-2836(81)90087-5
Smith, T. F., Waterman, M. S. (1981). Identification of common molecular subsequences. J Mol Biol. 147 (1), 195–197. doi: 10.1016/0022-2836(81)90087-5
Souvorov, A., Agarwala, R., Lipman, D. J. (2018). SKESA: strategic k-mer extension for scrupulous assemblies. Genome Biol. 19 (1), 153. doi: 10.1186/s13059-018-1540-z
Vaser, R., Šikić, M. (2021). Time- and memory-efficient genome assembly with raven. Nat. Comput. Sci. 1 (5), 332–336. doi: 10.1038/s43588-021-00073-4
Wagner, G. E., Dabernig-Heinz, J., Lipp, M., Cabal, A., Simantzik, J., Kohl, M., et al. (2023). Real-time nanopore Q20+ sequencing enables extremely fast and accurate core genome MLST typing and democratizes access to high-resolution bacterial pathogen surveillance. J. Clin. Microbiol. 29, e0163122. doi: 10.1128/jcm.01631-22
Whiley, H., Keegan, A., Fallowfield, H., Ross, K. (2014). Uncertainties associated with assessing the public health risk from legionella. Front. Microbiol. 5. doi: 10.3389/fmicb.2014.00501
Wick, R. R., Holt, K. E. (2019). Benchmarking of long-read assemblers for prokaryote whole genome sequencing. F1000Res 8, 2138. doi: 10.12688/f1000research.21782.4
Wick, R. R., Judd, L. M., Cerdeira, L. T., Hawkey, J., Méric, G., Vezina, B., et al. (2021). Trycycler: consensus long-read assemblies for bacterial genomes. Genome Biol. 22 (1), 266. doi: 10.1186/s13059-021-02483-z
Wick, R. R., Judd, L. M., Gorrie, C. L., Holt, K. E. (2017). Unicycler: resolving bacterial genome assemblies from short and long sequencing reads. PloS Comput. Biol. 13 (6), e1005595. doi: 10.1371/journal.pcbi.1005595
Wick, R. R., Judd, L. M., Holt, K. E. (2023). Assembling the perfect bacterial genome using Oxford nanopore and illumina sequencing. PLoS Comput. Biol. 19 (3), e1010905. doi: 10.1371/journal.pcbi.1010905
Keywords: Legionella pneumophila SBT, mompS, WGS, Illumina, short-reads, ONT, long-reads
Citation: Krøvel AV, Hetland MAK, Bernhoff E, Bjørheim AS, Soma MA and Löhr IH (2023) Long-read sequencing for reliably calling the mompS allele in Legionella pneumophila sequence-based typing. Front. Cell. Infect. Microbiol. 13:1176182. doi: 10.3389/fcimb.2023.1176182
Received: 28 February 2023; Accepted: 21 April 2023;
Published: 15 May 2023.
Edited by:
Paul Edelstein, University of Pennsylvania, United StatesReviewed by:
Maria Scaturro, National Institute of Health (ISS), ItalyKimberlee A. Musser, Wadsworth Center, United States
Copyright © 2023 Krøvel, Hetland, Bernhoff, Bjørheim, Soma and Löhr. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Anne Vatland Krøvel, YW5uZS52YXRsYW5kLmtyb3ZlbEBzdXMubm8=; Marit A. K. Hetland, bWFyaXQuaGV0bGFuZEB1aWIubm8=
†These authors have contributed equally to this work and share first authorship