 Amanda Domingues de Araújo
Amanda Domingues de Araújo Omar dos Santos Carvalho
Omar dos Santos Carvalho Sandra Grossi Gava
Sandra Grossi Gava Roberta Lima Caldeira
Roberta Lima Caldeira- Helminthology and Medical Malacology Research Group, René Rachou Institute, Fiocruz Minas, Belo Horizonte, Minas Gerais, Brazil
Introduction: The genus Biomphalaria in Brazil includes 11 species and one subspecies, three of which are intermediate hosts of Schistosoma mansoni. Due to the recent evolution of this group, some species are difficult to identify based on morphological characters, making the use of genetic markers necessary for species identification. This study aimed to evaluate the use of partial sequences of the cytochrome c oxidase I (coi) gene for the identification of Biomphalaria species using phylogenetic reconstruction and species delimitation algorithms. The study tested the use of DNA barcoding technique for species delimitation within the genus.
Methods: DNA barcoding was performed by sequencing a partial region of the coi gene from specimens, and the sequences were analyzed using phylogenetic reconstruction and algorithms to delimit Operational Taxonomic Units (OTUs).
Results: The study found that the use of the coi gene in the reconstruction of the phylogeny of the genus might be an alternative for understanding the evolution and dispersion of species. However, this marker alone is not enough to solve complex taxonomic problems within the genus. A total of 223 sequences were analyzed, 102 of which could be separated using the barcode gap, enabling the correct identification of seven taxa.
Discussion: The study demonstrated that accurate mollusk identification is necessary for effective schistosomiasis control. The DNA barcoding methodology was found to be promising for accurate mollusk identification, which is crucial for concentrating schistosomiasis control efforts in places where it is needed.
1 Introduction
Schistosoma mansoni Sambon, 1907 is the etiologic agent of schistosomiasis mansoni, a disease that affects about 130 million people in South America, the Caribbean, Africa, Madagascar, and the Middle East (James et al., 2018; McManus et al., 2018).
Mollusks of the genus Biomphalaria (Preston, 1910) belong to the family Planorbidae, which has approximately 300 species, of which about 10% are within the Biomphalaria genus (Jarne et al., 2010). There are 26 morphospecies and one subspecies of the latter genus described in Latin America. In Brazil, in particular, there are 11 species and one subspecies, of which three are intermediate hosts of S. mansoni under natural conditions – Biomphalaria glabrata (Say, 1818), Biomphalaria tenagophila (d’Orbigny, 1835) and Biomphalaria straminea (Dunker, 1835). 1848) – while another three are considered potential hosts, as they can be infected under laboratory conditions – Biomphalaria peregrina (d’Orbigny, 1835), Biomphalaria amazonica Paraense, 1966 and Biomphalaria cousini Paraense, 1966 (Corrêa, 1971; Paraense and Corrêa, 1973; Teodoro et al., 2010).
In the genus Biomphalaria, there are two different species complexes, whose members are practically indistinguishable by the morphology of their shells and by most characters of the genital system: (i) the Biomphalaria straminea complex, containing the species B. straminea, Biomphalaria kuhniana (Clessin, 1883) and Biomphalaria intermedia Paraense & Deslandes, 1962; and (ii) the Biomphalaria tenagophila complex, containing the species/subspecies B. tenagophila tenagophila, Biomphalaria tenagophila guaibensis Paraense, 1984 and Biomphalaria occidentalis Paraense, 1981 (Paraense, 1988; Caldeira et al., 1998; Spatz et al., 1999). Distinguishing between these taxa is one of the main challenges of research that aims to elucidate the phylogeny of the genus Biomphalaria using molecular techniques, such as the present work (Caldeira et al., 2000; Vidigal et al., 2004).
The study of the Biomphalaria genus has always been motivated by the epidemiological and ecological aspects of the relationship between these planorbids and S. mansoni. Despite this, the amount of research effort devoted to studies of the phylogeny and systematics of these groups has always been relatively small, even though elucidating the phylogenetic relationships of this group could change our understanding of its host-parasite relationships, which is fundamental for the development and planning of schistosomiasis control programs (Jarne et al., 2011).
Little is known about the evolution of the genus Biomphalaria due to the difficulty of placing fossils in the history of this species group, mainly because the morphometry of the shell is not used as an essential taxonomic characteristic, due to its simplicity and lack of relevant attributes regarding phylogeny (Cabrera et al., 2016). What can be said is that the evolution of mollusks of this genus is very recent, with the ancestral taxa within this genus beginning to split between the Pliocene and Pleistocene, around 1.8 million years ago, a fact that is consistent with the fossil record (Kaesler and Parodiz, 1970; Wanninger and Wollesen, 2019). Precisely for this reason, the characteristics used to distinguish between the species is quite limited; especially among closely-related species such as those of the B. straminea complex (Jarne et al., 2011). The morphological differences between B. straminea and B. kuhniana are very subtle and practically indistinguishable, with differences being limited to the number of muscular layers in the penis wall (three in B. straminea and two in B. kuhniana) (Paraense, 1988) and distal segment of the spermiduct very winding in B. straminea and straight or slighetly wavy in B. kuhniana (Paraense, 1988). Additionally, the molecular technique used for species differentiation within the genus (PCR/RFLP) generates a similar banding pattern between the two species (Caldeira et al., 1998), which creates several controversies in their differentiation.
The similarities between B. tenagophila and B. occidentalis are also numerous, again confusing human differentiation between, and identification of, the two species. For a long time, it was believed that the rates of infection by S. mansoni in the state of São Paulo were very low since infected mollusks were not found in the field. However, in 1981, the researcher Lobato Paraense described a new species, B. occidentalis, refractory to infection by the parasite, and that was previously confused with B. tenagophila, as they were almost indistinguishable by the shell morphology (Paraense, 1981). As a result of this description, in a large area in western Brazil, the distribution map of this last species has been undergoing constant change and readjustment (Scholte et al., 2012; Fernandez et al., 2014; Carvalho et al., 2020).
Traditionally, the DNA barcode technique consists of the amplification and sequencing of a fragment of the cytochrome c oxidase I (coi) gene, using pairs of universal primers – most often those proposed by Folmer et al. in 1994 – and subsequent comparison for specific identification with a universal database (BOLD) (Ratnasingham and Hebert, 2007). Therefore, the main objective of this work was to generate new DNA barcodes for the species of Biomphalaria found in Brazil, and to evaluate the use of the coi gene as a potential molecular marker for the identification of species of the genus Biomphalaria.
One of the main advantages of DNA barcoding is its ability to rapidly and accurately identify species, in the most variable life stages and genders, as well as identifying organisms by parts or pieces and discriminating individuals in a matrix with various mixed species (Casiraghi et al., 2010; Leray and Knowlton, 2015). It is also possible to identify species that are difficult to distinguish based on traditional morphological characteristics (Hebert et al., 2003; Casiraghi et al., 2010).
DNA barcoding is a useful tool for taxonomists, with good cost-effectiveness and the ability to allow non-experts to have access to quick and accurate identifications. This tool can be used in four ways (McManus et al., 2018): screening in collections, based on existing sequences in databases (James et al., 2018); identification of specimens where morphological identification is not possible (immature, partial, damaged or very small individuals), or even to resolve inconsistencies (in dimorphic species where identification is only possible in one sex) (Paéibo, 1990; Hebert et al., 2003; Casiraghi et al., 2010) (Jarne et al., 2010); as a powerful supplementary tool to other forms of identification and thus help in delimiting closely related species phylogenetically (Schindel and Miller, 2005; Sun et al., 2016); and (Corrêa, 1971) the possibility of discovering previously undescribed species (Hebert et al., 2004). In addition, DNA barcoding is a relatively low-cost and scalable technique, with the potential for high-throughput analysis of large numbers of samples. This makes it particularly useful for large-scale biodiversity surveys and monitoring programs (Hebert et al., 2003; Leray and Knowlton, 2015).
2 Methods
2.1 Construction of the database and illustrative maps
Nucleotide sequences of the partial region of the coi gene were obtained from a search of the public GenBank database (https://www.ncbi.nlm.nih.gov/genbank). Additional new sequences were obtained by us through DNA Sanger sequencing of mollusks deposited in the Medical Malacology Collection (Fiocruz-CMM) located at the Instituto René Rachou (IRR – Fiocruz/Minas). These specimens were selected according to the known geographic distribution of each species that occurs within Brazilian territory, prioritizing the type localities.
Data referring to the exact geographic coordinates of each collection point were added to each specimen used from Fiocruz-CMM and for sequences obtained from GenBank for which this information was available. For sequences in the database without any indication of the exact location of collection, the coordinates of the city in which they were collected were assigned.
From the coordinates and cartographic data of the IBGE (2021), a thematic map was constructed to illustrate the geographic distribution of the specimens in this study using the free software QGIS version 3.20 (QGIS, 2021).
2.2 Molecular studies
2.2.1 - DNA extraction, amplification, and purification of the coi gene fragment
For specimens obtained from the Fiocruz-CMM collection, a cryopreserved fragment of the cephalopodal region was treated with proteinase K (0,1µg/µL) for at least 12 hours, precisely to degrade the proteins present in the mucus of the mollusk. Next, extraction is carried out with the commercial kit Wizard Genomic DNA Purification according to the manufacturer’s instructions
For the amplification of an ~600 bp region of the coi gene, the primers LCO1490 (5’-GGTCAACAAATCATAAAGATATTGG-3’) and HCO2198 (5’-TAAACTTCAGGGTGACCAAAAAATCA-3’) were used (Folmer et al., 1994). All PCR reactions were performed using the Platinum® Taq DNA Polymerase kit (Invitrogen) following the manufacturer’s instructions and according to amplification conditions described by Folmer (Folmer et al., 1994).
DNA purification was performed using the Wizard® SV Gel and PCR Clean-Up System Kit (Promega), according to the manufacturer’s instructions, using the PCR product directly in the purification columns. The purity and concentration of the purified PCR products were verified on a NanoDrop™ 2000 spectrophotometer (ThermoFisher Scientific).
2.2.2 - Sequencing
The samples were sequenced by the Sanger method on an ABI 3730xL automatic sequencer (Applied Biosystems), using the same primers described above for amplification, at the Capillary Electrophoresis DNA Sequencing Platform of the IRR.
2.3 Data analysis
2.3.1 Alignments
The reliability of the nucleotides obtained from sequencing was visually checked and verified using MEGA X software (Kumar et al., 2018) to obtain consensus sequences applying the MUSCLE algorithm (Edgar, 2004). Additionally, CodonCode Aligner© v. 9.0.2 (CodonCode Corporation, 2019) was used to assign Phred quality values to each base. The Phrap program was then used to construct consensus sequences using the assigned quality values, with a minimum quality value of 20. The resulting consensus sequences were compared with those obtained by visual inspection to eliminate ambiguous bases. The consensus sequences were submitted to Nucleotide BLAST (BLASTn) (Altschul et al., 1990), and the BOLD species identification system (Ratnasingham and Hebert, 2007) (https://www.boldsystems.org/index.php/IDS_OpenIdEngine) to confirm the species identification and detect contamination with other organisms.
Sequences generated for Biomphalaria species in this study were combined with previously published sequences obtained from the GenBank database. MAFFT online version 7 (https://mafft.cbrc.jp/alignment/server/ (Katoh et al., 2019),) was employed to align all sequences. The resulting alignment was subsequently exported in FASTA format and visually inspected and edited in MEGA X. After trimming, the resulting sequences had sizes ranging from 517 to 655 bp.
2.3.2 Phylogenetic reconstruction of the partial coi gene sequences
For the phylogenetic reconstruction, ModelFinder (Kalyaanamoorthy et al., 2017) implemented in PhyloSuite (Zhang et al., 2020) was used to find the evolutionary model that best fit the sequence alignment, according to the Corrected Akaike Information criterion (AICc).
Phylogenetic reconstruction was conducted using either Maximum Likelihood (ML) estimation (Felsenstein, 1981) or Bayesian Inference (BI) (Drummond et al., 2002). A coi sequence of the genus Helisoma sp. (Swainson, 1840), which also belongs to the Planorbidae family, was selected as an outgroup for tree rooting (GenBank accession number: KM612179.1).
ML trees were inferred using the IQ-TREE software v. 1.6.8 (Nguyen et al., 2015), with the evolutionary model K81u+R3+F for 5000 ultrafast bootstraps (Minh et al., 2013) and the Shimodaira-Hasegawa approximate likelihood ratio test (SH-aLRT) (Guindon et al., 2010) to determine the support values of the branches.
For BI the MrBayes software v. 3.2.6 (Ronquist et al., 2012) was used, with the evolutionary model HKY+I+G+F previously calculated by ModelFinder (Kalyaanamoorthy et al., 2017). Two independent parallel searches (Markov chains) were performed with 107 generations (sampled every 1000 generations) and 25% of the initial data were discarded as burn-in.
The ML and BI trees were visualized and edited using the software FigTree v. 1.4.4 (Rambaut, 2018), with final editing performed using Inkscape v.1.1 (Inkscape Project, 2021).
2.3.3 Verification of polymorphic sites and genetic divergence between populations
Genetic polymorphism indices were calculated for the partial region of the coi gene using the software DnaSP v. 6 (Rozas et al., 2017). The following indices of genetic diversity and their standard deviation were calculated: the number and diversity of mitochondrial haplotypes (h, Hd), nucleotide diversity (π) (Nei, 1987), both with and without the Jukes-Cantor correction (Jukes and Cantor, 1969; Nei, 1987; Lynch and Crease, 1990), the average number of differences between nucleotides between pairs of sequences (k) (Tajima, 1983; Nei, 1987). The number of singleton-type and parsimonious informative polymorphic sites were also estimated, in addition to the fixed divergence between populations (Hey, 1991) and nucleotide divergence between populations.
2.3.4 Specimen identification and species delimitation
In order to verify the accuracy of the partial coi sequences for the identification of Biomphalaria specimens using DNA barcoding methodology, BOLD Identification Criteria (thresh-ID) and Best Close Match (BCM) analyses were carried out (Meier et al., 2006) using the ape v. 5.5 (Paradis and Schliep, 2019) and spider v. 1.5.0 (Brown et al., 2012) packages of the R software v. 4.0.5 (R Foundation for Statistical Computing, 2021).
In order to define the barcode gap, the Kimura two-parameter (K2P) model (Tamura et al., 2004) was used to calculate the intraspecific and interspecific distances (Meyer and Paulay, 2005) between the species using the spider v. 1.5.0 (Brown et al., 2012) package in the R software v. 4.0.5 (R Foundation for Statistical Computing, 2021).
Additionally, the sequences were clustered/grouped into Operational Taxonomic Units (OTUs), grouping organisms by their similarity, in this case in relation to a genetic marker (Blaxter et al., 2005), using the criteria of four algorithms; Generalized Mixed Yule-Coalescent (GMYC) (Reid and Carstens, 2012; Fujisawa and Barraclough, 2013), Poisson Tree Processes (bPTP) (Zhang et al., 2013), Automatic Barcode Gap Discovery (ABGD) (Puillandre et al., 2012) and Assemble Species by Automatic Partitioning (ASAP) (Puillandre et al., 2021).
3 Results
3.1 coi sequence database and specimen distribution map
Overall, the molecular data set analyzed consisted of 223 Biomphalaria nucleotide sequences, comprised of 83 previously published sequences obtained from GenBank (Supplementary Table 1) and 140 sequences newly-generated in this study (Supplementary Table 2). In addition, a single sequence was selected as an outgroup for phylogenetic analysis (Helisoma sp. - KM612179.1). The 11 species and one subspecies from Brazil are represented among the samples obtained, and all type localities are represented among the samples (except for the species B. kuhniana). The new sequences obtained in this study were deposited in GenBank (under Accession Numbers MZ778865 - MZ778963) and their IDs are listed in Supplementary Table 2.
Based on the geographic location associated with each sequence, a map was constructed to illustrate the distribution in Central and South America of the specimens used in this study, not including those retrieved from GenBank databases located in Egypt and China (Figure 1).
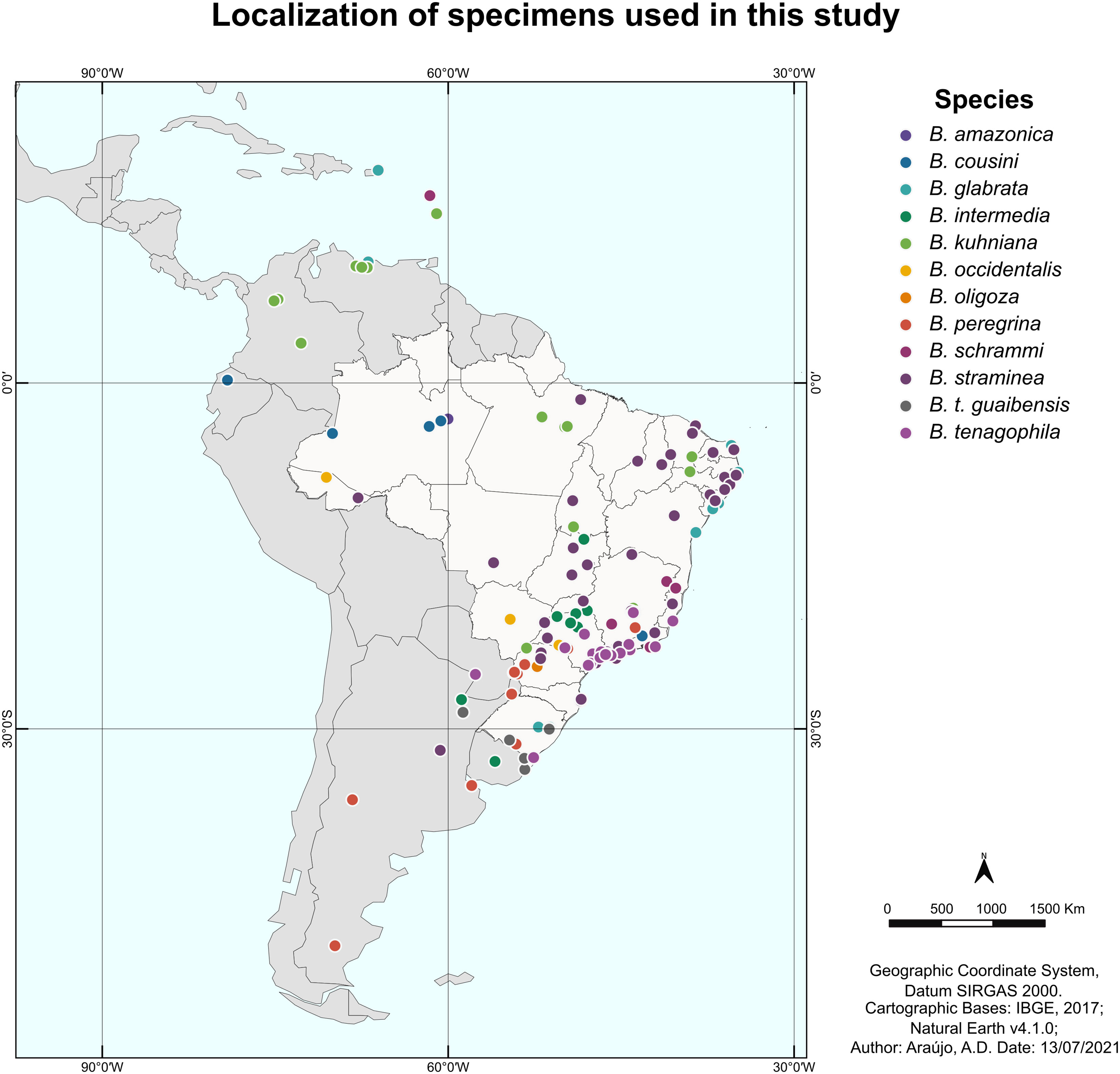
Figure 1 Map of Latin America showing the location of the specimens of the genus Biomphalaria used in this study. Each point represents the geographic location of a specimen, and the colors represent the different species used in the current study.
3.2 Phylogenetic reconstruction of mollusks of the genus Biomphalaria using the partial region of the coi gene
Phylogenetic inference produced well-supported clades that corresponded to the species studied, except for: (i) B. cousini, which formed two distantly-related lineages, one of which clustered with B. amazonica; (ii) B. kuhniana and B. straminea, whose sequences were polyphyletic, but together formed a single well-delimited monophyletic clade, within which the sequences of each of these two taxa were interspersed; (iii) The clades produced with sequences from B. oligoza and B. peregrina and B. tenagophila guaibensis were paraphyletic (Figure 2; Supplementary Figure 1). Since the topology of the trees are very similar, as well as the support values of the branches, we have provided only the representation of the tree from Bayesian Inference (Drummond et al., 2002) (Supplementary Figure 1).
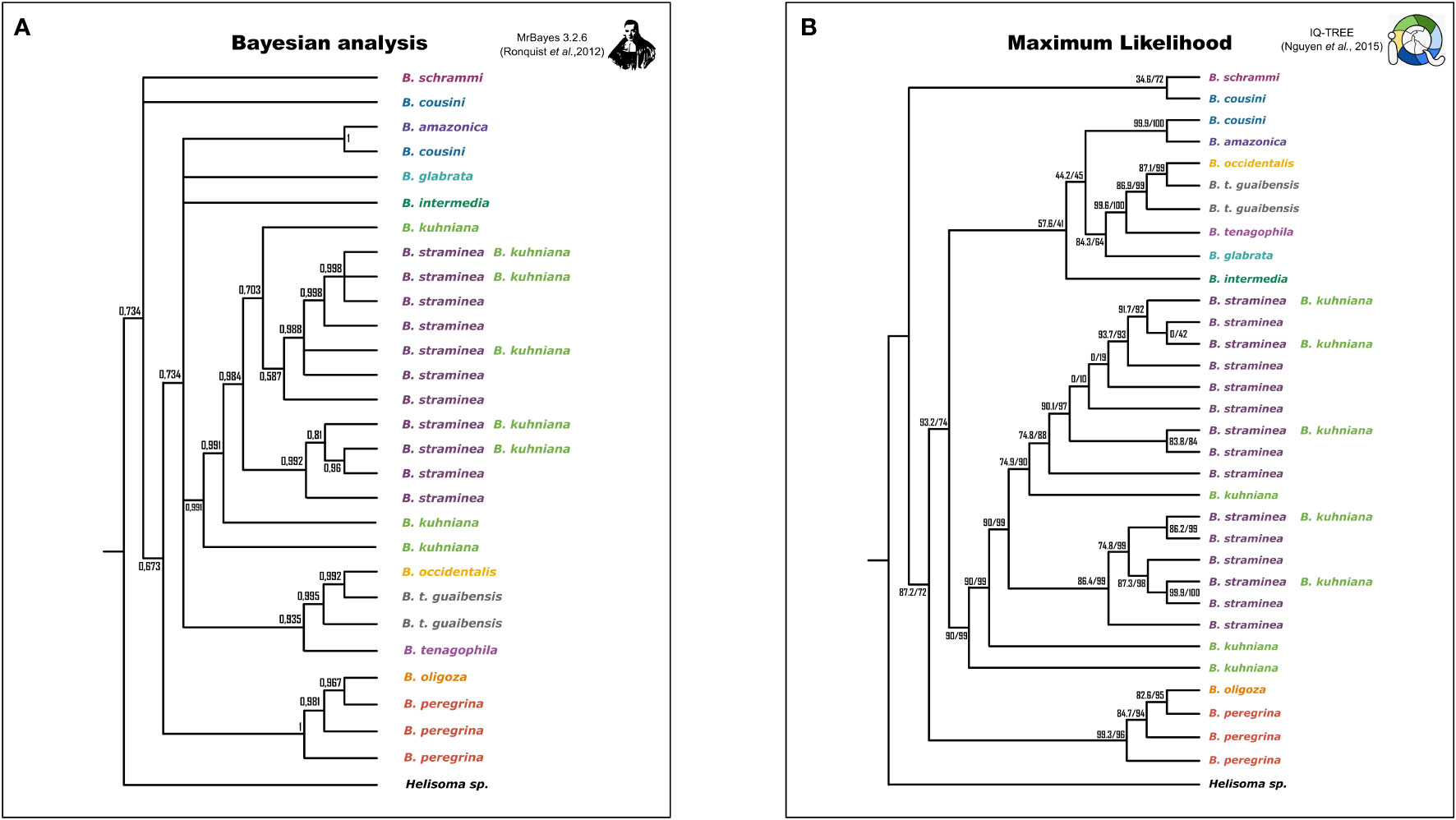
Figure 2 Simplified representation of the phylogenetic trees generated by Bayesian Inference (BI) and Maximum Likelihood (ML) criteria. Species are represented by different colors. Trees generated by MrBayes have posterior probability values represented on the branches (A). Trees generated by IQ-TREE have two support values for comparison, ultrafast bootstraps (MINH et al., 2013) and the Shimodaira-Hasegawa test (SH-aLRT) (Guindon et al., 2010) (B).
3.3 Polymorphic sites and genetic divergence between Biomphalaria populations
Analysis of invariant and variable sites showed that most of the sites are invariant and most of the variable sites are parsimony-informative sites for most species (Table 1). The genetic polymorphism indices were also obtained for partial sequences of the coi gene from populations of Biomphalaria (Table 2).
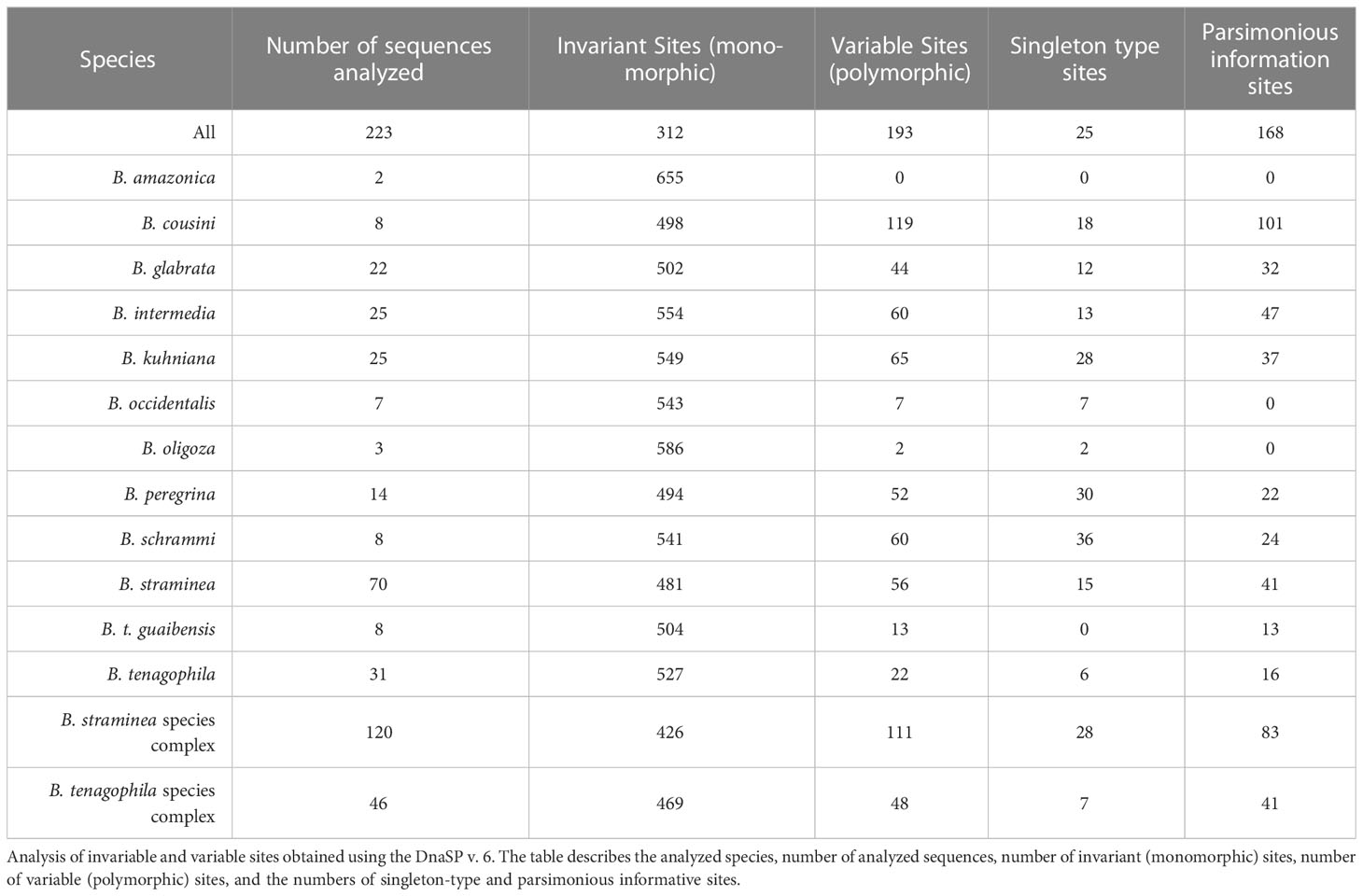
Table 1 Polymorphic sites in the partial sequences of the coi gene from populations of Biomphalaria.
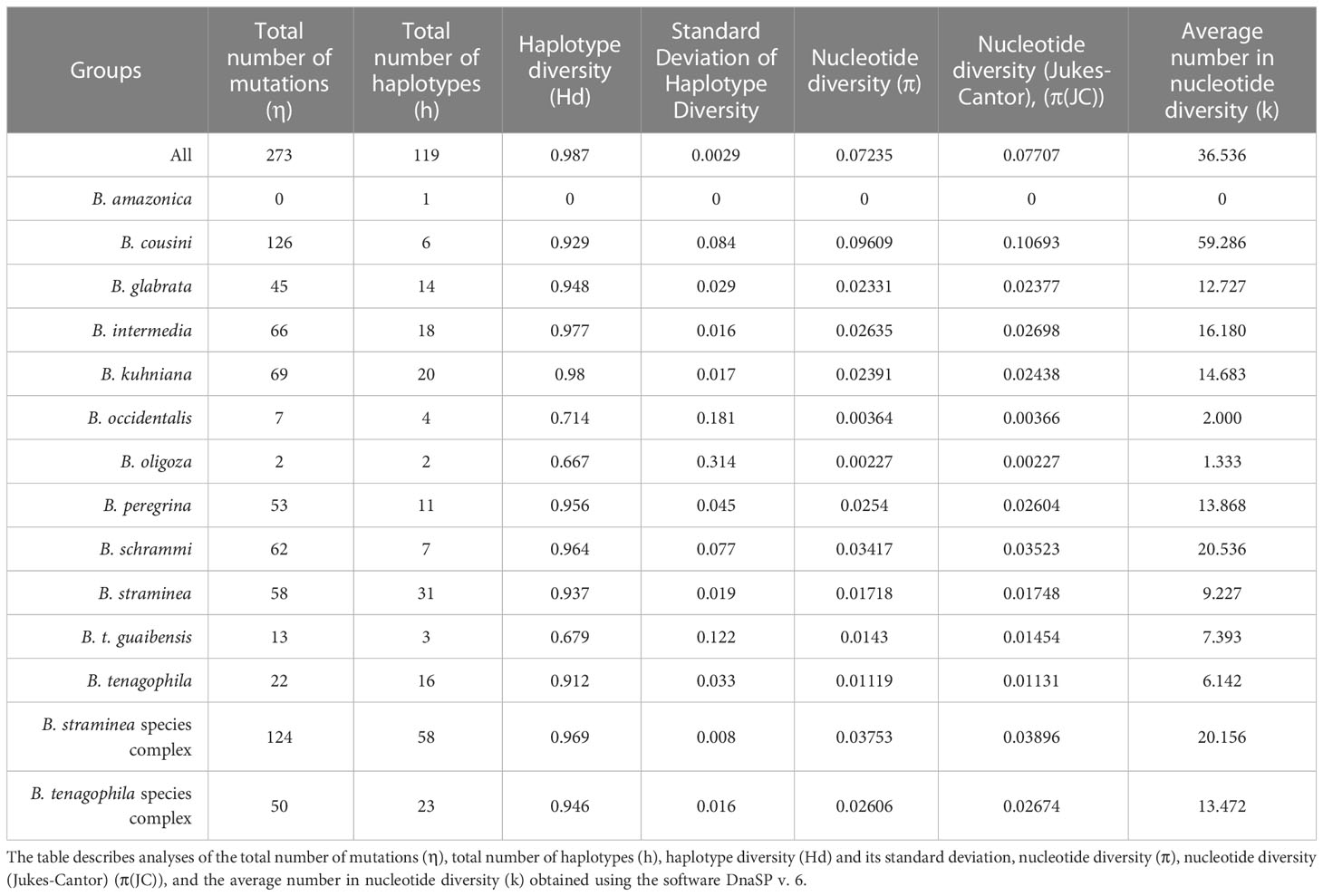
Table 2 Genetic polymorphism indices in partial sequences of the COI gene from populations of Biomphalaria.
The calculated divergence of DNA sequences between populations showed that: (i) there are no fixed differences between the species B. amazonica and B. cousini, (ii) fixed differences between the species within each of the different species complexes are smaller than those between these species and the other species which are not members of the species complex; and (iii) B. schrammi has the greatest number of differences when compared to most of the other species of the genus (Supplementary Figure 2). Also, in order to measure the distance between species, the average number of nucleotide divergences between populations (k) was calculated (Supplementary Figure 3). The greatest nucleotide divergences were observed for B. cousini and B. schrammi, with the divergence between the two species being the largest observed of the pairwise comparisons. The smallest nucleotide divergences from the pairwise comparisons were between the closest species already reported in the literature; that is, B. kuhniana and B. straminea, B. t. guaibensis and B. occidentalis, and B. peregrina and B. oligoza.
3.4 Biomphalaria species delimitation
BOLD Identification Criteria (thresh-ID) and Best Close Match (BCM) analyses showed similar results, with B. tenagophila, B. glabrata, B. t. guaibensis, B. occidentalis, and B. oligoza all showing 100% of the sequences correctly identified. In both analyses, B. intermedia had 24 (96%) correctly identified sequences, and one sequence without identification. Biomphalaria peregrina, had ten sequences correctly identified (71.4%), and four without identification. For B. schrammi, the two analyzes were able to correctly identify only two sequences (25%), and the other six sequences could not be identified. The biggest differences found between the two analyses were in the sequences of the two species B. straminea and B. kuhniana (Figures 3A, B). For B. straminea, using BCM analysis, 46 sequences were considered correct (65.7%), four were considered incorrect (5.7%), 19 were ambiguous (27.1%) and one sequence could not be identified. Using the BOLD analysis, 16 sequences were considered correct (22.9%), 53 were ambiguous (75.7%) and one could not be identified. For B. kuhniana, using the BCM analysis, 15 sequences were considered correct (60%), five incorrect (20%), two ambiguous (8%) and three without identification, while using the BOLD analysis, 15 sequences were considered correct (60%), three incorrect (12%), four ambiguous (16%) and three without identification.We calculated intra- and interspecific distances in order to define the barcode gap using the K2P model (Figure 4). The intraspecific distances calculated for all sequences presented a bimodal distribution, while the interspecific distances showed a trimodal distribution (Figure 4). The histograms of pairwise distances of each species (Figure 5) indicated the absence of a barcode gap for specific differentiation of B. straminea B. kuhniana B. intermedia B. t. guaibensis B. occidentalis B. peregrina and B. oligoza. Of the 223 analyzed sequences, it was possible to calculate the barcode gap for only 102, which corresponds to 45.7% of the analyzed sequences. Thus, 58.3% of the analyzed species have a barcode gap, which can be used to delimit them from one another using this methodology.
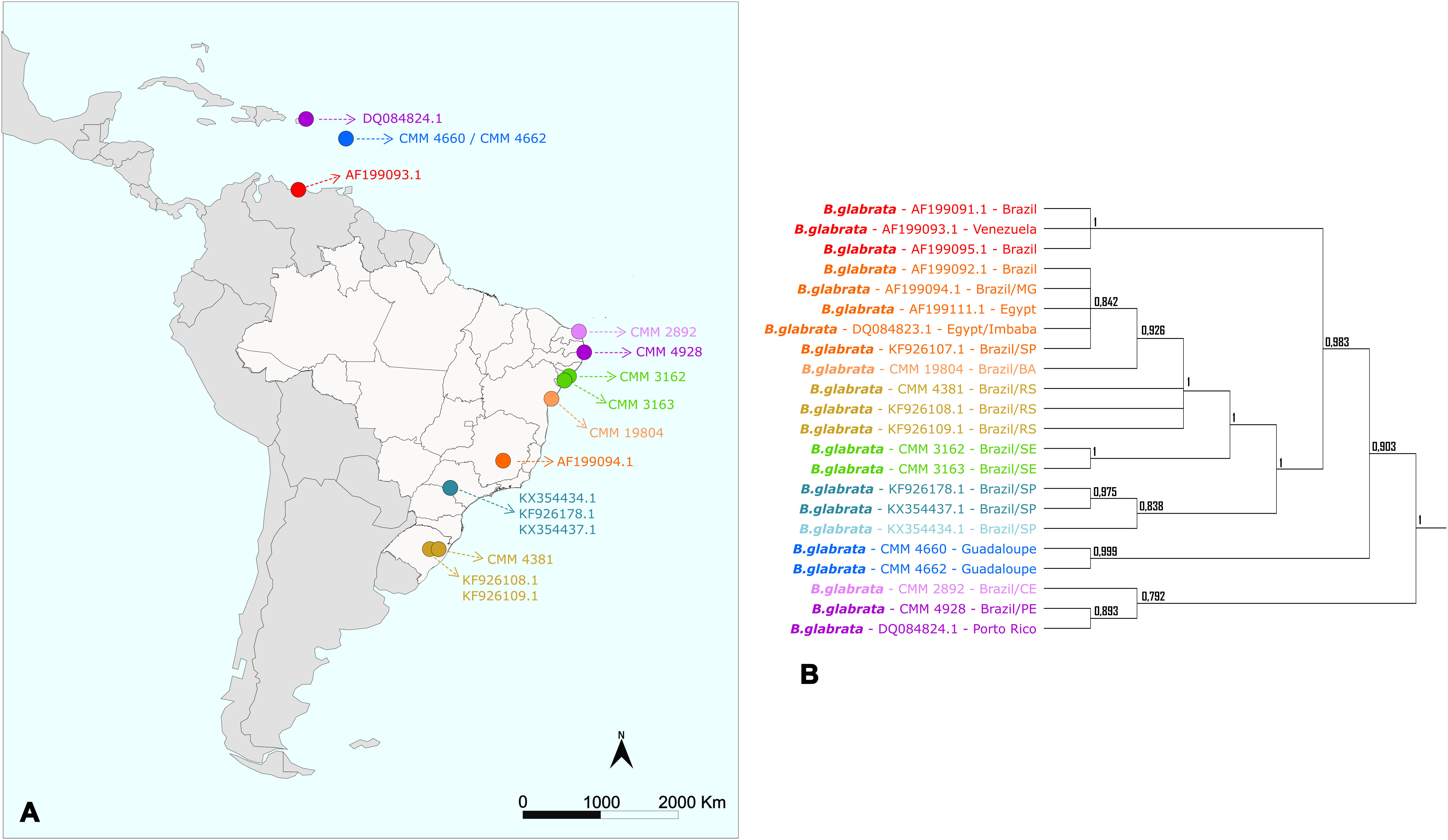
Figure 3 Number of sequences classified by the Best Close Match (BCM) and BOLD Identification Criteria. The bar graphs illustrate the absolute number of sequences identified using two different criteria: (A) Best Close Match (Meier et al., 2006) and (B) BOLD Identification Criteria, using the R software (version 4.0.5) and the package spider (version 1.5.0). The colors represent the four possible outcomes: correct (green), incorrect (red), ambiguous (yellow), and no identification (blue).
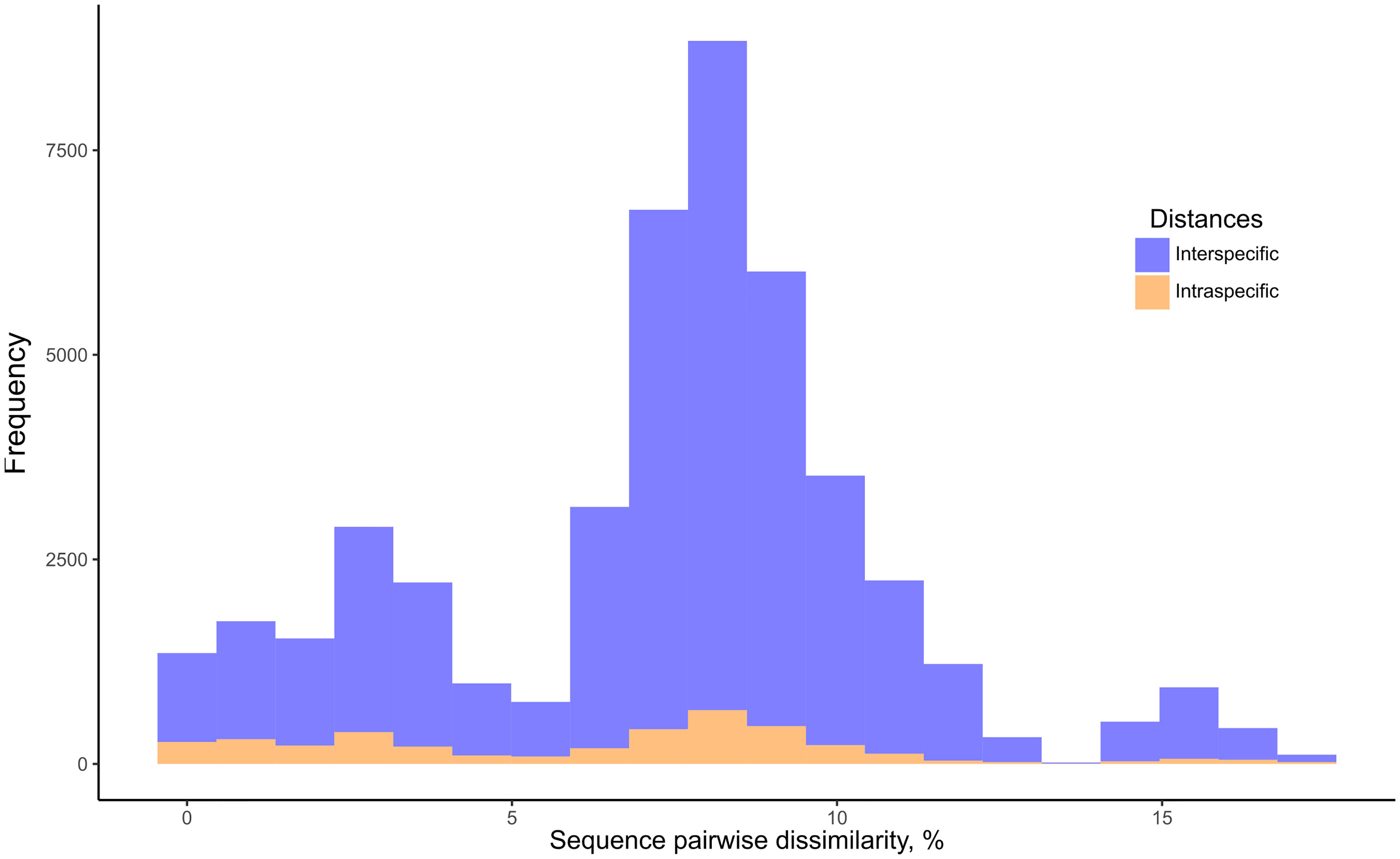
Figure 4 Frequency distribution of intraspecific and interspecific distances of Biomphalaria partial coi sequences. Histogram of intraspecific (orange) and interspecific (blue) distances based on the Biomphalaria partial coi sequences. Pairwise K2P distances were expressed as percentage dissimilarity (K2P * 100).
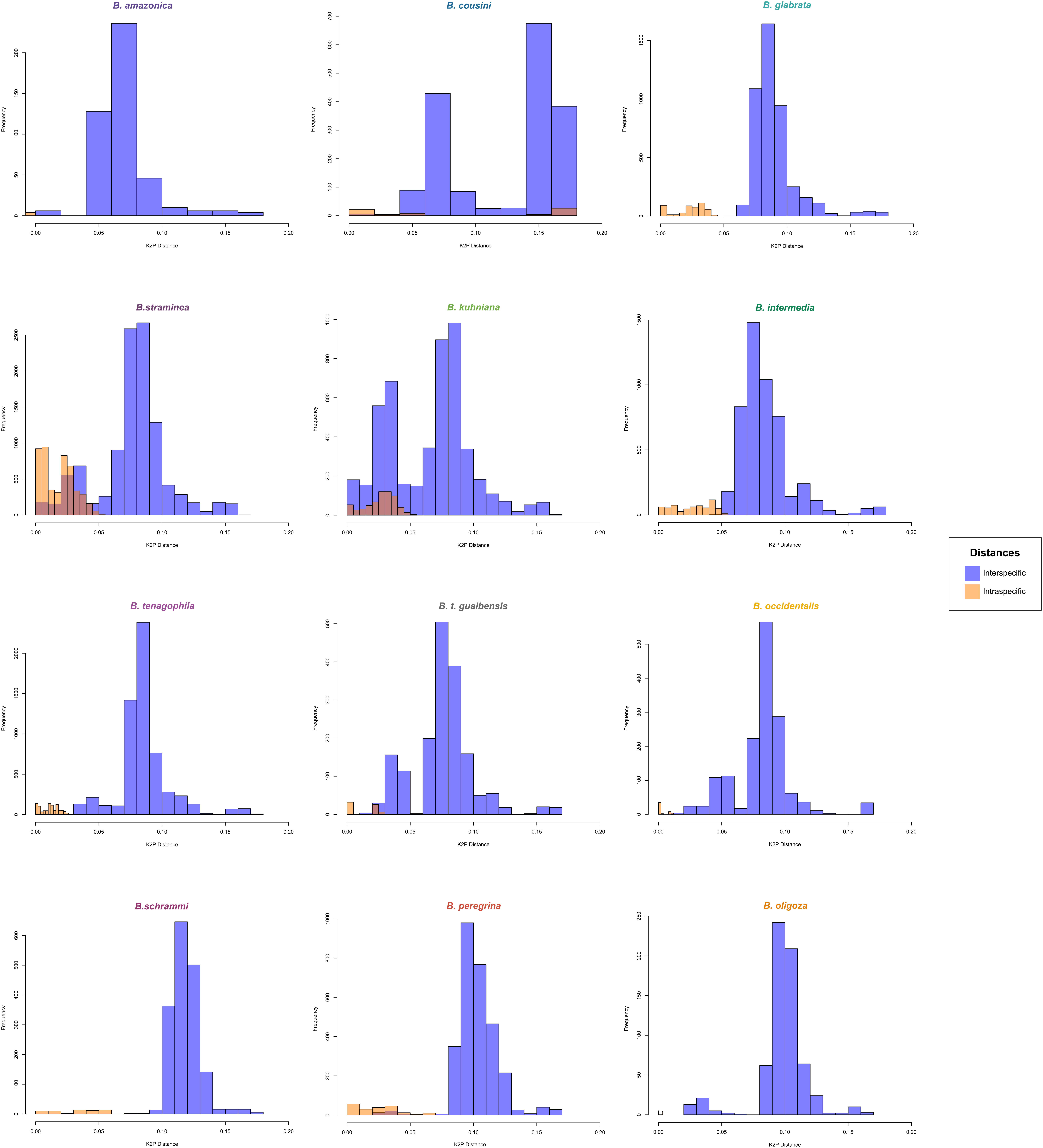
Figure 5 Frequency distribution of intraspecific and interspecific distances of Biomphalaria partial coi sequences according to species. Histogram of intraspecific (orange) and interspecific (blue) distances based on the Biomphalaria coi partial sequences. Pairwise distances were expressed as percentage dissimilarity for each species: B. amazonica, B. cousini, B. glabrata, B. straminea, B. kuhniana, B. intermedia, B. tenagophila, B. t. guaibensis, B. occidentalis, B. schrammi, B. peregrina, and B. oligoza.
In the formation of Operational Taxonomic Units (OTU), different clusters/clades were generated for each algorithm used (Table 3). The clusters/clades generated by ABGD, ASAP, and sGMYC were the closest to the current delineation of the species of the Biomphalaria genus. Despite the large number of groups created by bPTP, and mGMYC, they were congruent with the geographic locality of the specimens. The ABGD algorithm divided B. cousini into three different groups, with one of them being shared with the B. amazonica sequences, as inferred by phylogenetic reconstruction. Biomphalaria schrammi and B. peregrina were also divided into two groups, the latter sharing a cluster with the B. oligoza sequences. The sequences of B. kuhniana and B. straminea were pooled, as well as B. occidentalis, B. tenagophila, and B. t. guaibensis. ASAP was the algorithm that generated the least number of clusters. However, B. cousini was partitioned into three different clusters by the ASAP method, with the same division observed for the ABGD algorithm. These latter two methods, therefore, generated groups, each comprised of multiple species, as follows. ABGD: (i) B. amazonica and B. cousini, (ii) B. kuhniana and B. straminea, (iii) B. oligoza and B. peregrina, and (iv) all three species of the Biomphalaria tenagophila complex (B. occidentalis, B. tenagophila, and B. t. guaibensis). ASAP: (i) B. amazonica, B. cousini, B. intermedia, B. kuhniana, and B. straminea, (ii) B. oligoza and B. peregrina, and (iii) all three species of the Biomphalaria tenagophila complex. Using the sGMYC algorithm, 14 groups were formed, as follows. Biomphalaria cousini was divided into two groups, with one of them shared with B. schrammi sequences, while the other group was shared with a single large group comprised of B. amazonica, B. glabrata, B. intermedia, B. occidentalis, B. tenagophila and B. t. guaibensis. Biomphalaria kuhniana was divided into six different groups, sharing four of them with B. straminea, which was itself divided into eight groups (Supplementary Figure 1).
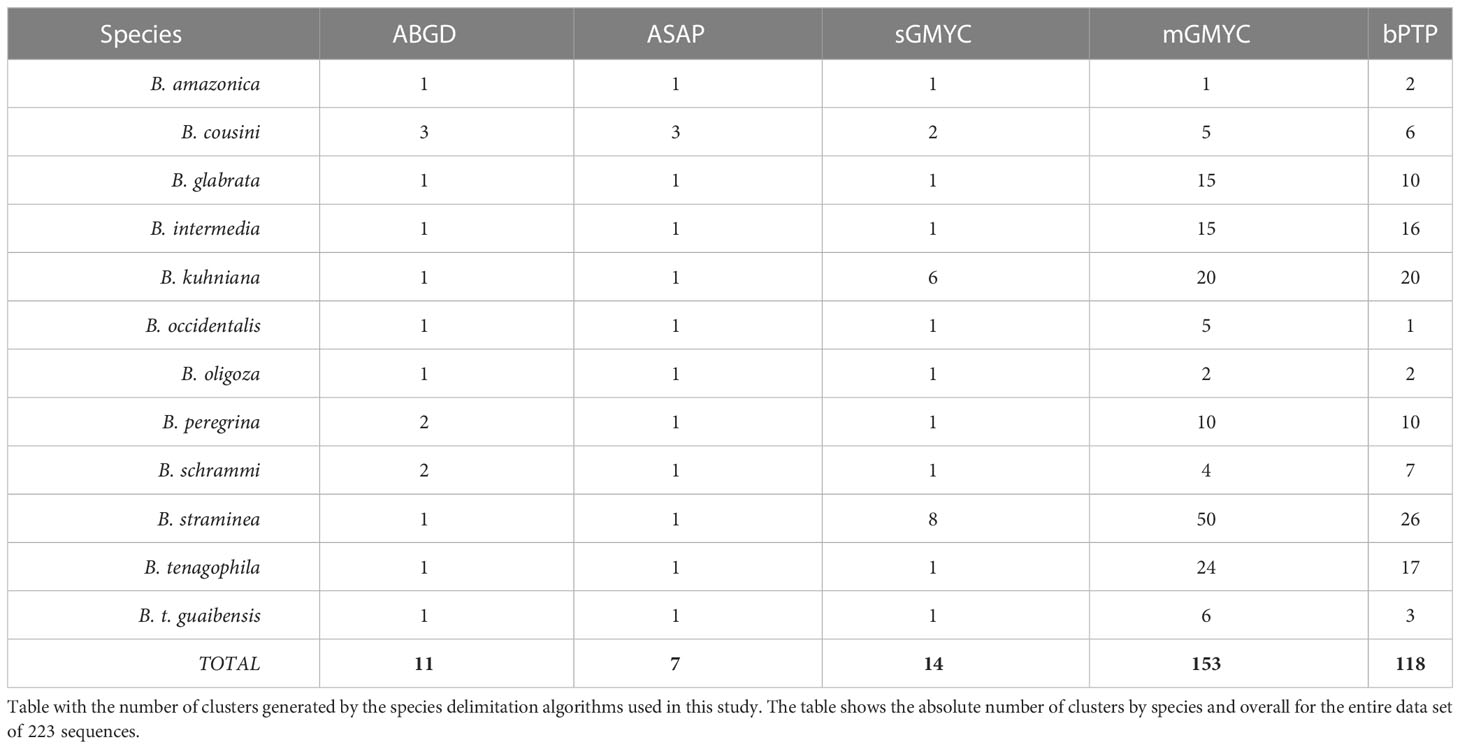
Table 3 Number of Operational Taxonomic Units (OTUs) according to Biomphalaria species.
4 Discussion
The correct identification of mollusks that are the intermediate hosts of Schistosoma is a fundamental prerequisite for understanding the epidemiology of schistosomiasis and mobilizing efforts for disease surveillance and control (Rollinson et al., 2009). The Fiocruz-CMM combines both classical taxonomy through (i) comparison of morphological characters of the shell, as well as male and female reproductive organs (Lobato Paraense and Deslandes, 1958a; Lobato Paraense and Deslandes, 1958b; Paraense, 1975; Paraense, 1981; Paraense, 1984; Paraense, 1988; Paraense, 1990; Paraense et al., 1992; Estrada et al., 2006), and (ii) use of molecular tools, such as PCR-RFLP of the internal transcribed spacer (ITS) of the ribosomal RNA gene, to distinguish Biomphalaria species (Vidigal et al., 2000; Teodoro et al., 2010). Despite this, some inconsistencies between the results of the identifications have already been reported (Aguiar-Silva et al., 2014). This highlights the importance of combining methodologies in the taxonomy of mollusks of the genus Biomphalaria for species identification following the Iterative taxonomy process, in order to refine and define the boundaries between species using multiple different lines of evidence (Yeates et al., 2011).
This study confirmed that the species identification within the genus Biomphalaria is not a simple task, a fact that had already been mentioned by Doctor Lobato Paraense, in the 1960s (Paraense and Deslandes, 1959). The anatomy of the reproductive tract has been an effective tool in mollusk identification within the genus (Paraense and Deslandes, 1959). However, a resolution limit is reached for groups of closely related species (Paraense, 1981; Paraense, 1988), such as the B. straminea complex, B. peregrina/B. oligoza, and the B. tenagophila complex, as a consequence of intraspecific variation or incomplete variation, incomplete speciation, or the “gray zone of speciation” that exists between species due to recent evolution (De Queiroz, 2007; Jarne et al., 2011).
Biomphalaria straminea is a species that originated from the northern region of South America and has been expanding its habitat throughout Brazil and surrounding countries, mainly due to human activity and its high capacity to survive long periods of drought combined with its high fertility (Wang et al., 2013; Attwood et al., 2015). It is known that this species was recently introduced into both China (Meier Brook, 1975; Dudgeon and Yipp, 1983; Tang, 1983; Yipp, 1990; Yang et al., 2018), and the Caribbean (Paraense et al., 1981; Pointier et al., 1993) from strains originating from South America. The reconstructed phylogenies in this work reflect this introduction, with northeastern Brazilian specimens grouped with specimens from China, and specimens from the Caribbean grouped with specimens from northern Brazil, Venezuela, and Colombia.
In the reconstructed phylogeny, the B. straminea complex indicated a low resolution between B. kuhniana and B. straminea, which formed a polyphyletic clade. The same was observed between B. cousini and B. amazonica. This polyphyly may be the result of clades with recent evolution or imperfect taxonomy, in which not all the genus species diversity has been documented (Meyer and Paulay, 2005). Furthermore, due to the proximity between the species, there is a possibility that there is some level of hybridization between them (DeJong et al., 2001; Teodoro et al., 2011). Mitochondrial gene flow via hybridization has already been reported for several groups of animals, including planorbid mollusks (Mello-Silva et al., 1998), although it is not well understood. Hybridization is one of the sources of non-monophyly between clades of different groups of living beings, causing introgression (Anderson and Hubricht, 1938).
The clade representing the B. glabrata species is monophyletic and well supported (100%) in both the BI and ML analyses, with subdivision into at least five different groups. This fact can be explained by the Refuge Theory (Haffer, 1969), which suggests that climatic oscillations in the Pleistocene period may have been responsible for the habitat fragmentation and consequent geographic separation of B. glabrata populations, forming genetically-distinct clades (Dejong et al., 2003).
The phylogenetic trees inferred in this study corroborate what was indicated by previous studies that: (i) the Greater Antilles – Cuba, Hispaniola (Haiti and the Dominican Republic), Jamaica, and Puerto Rico – were colonized very early in B. glabrata evolution; (ii) specimens from southeastern Brazil were probably introduced into northeastern Brazil; and (iii) the Lesser Antilles and Venezuela appear to have been colonized by B. glabrata more recently, probably due to deforestation and human occupation (Mavárez et al., 2002; Dejong et al., 2003). Furthermore, as reported in 1998, our work also presents evidence of colonization of the South Region of Brazil with lineages from the North Region, probably related to recent human dispersal (Figure 6) (Carvalho O dos et al., 1998).
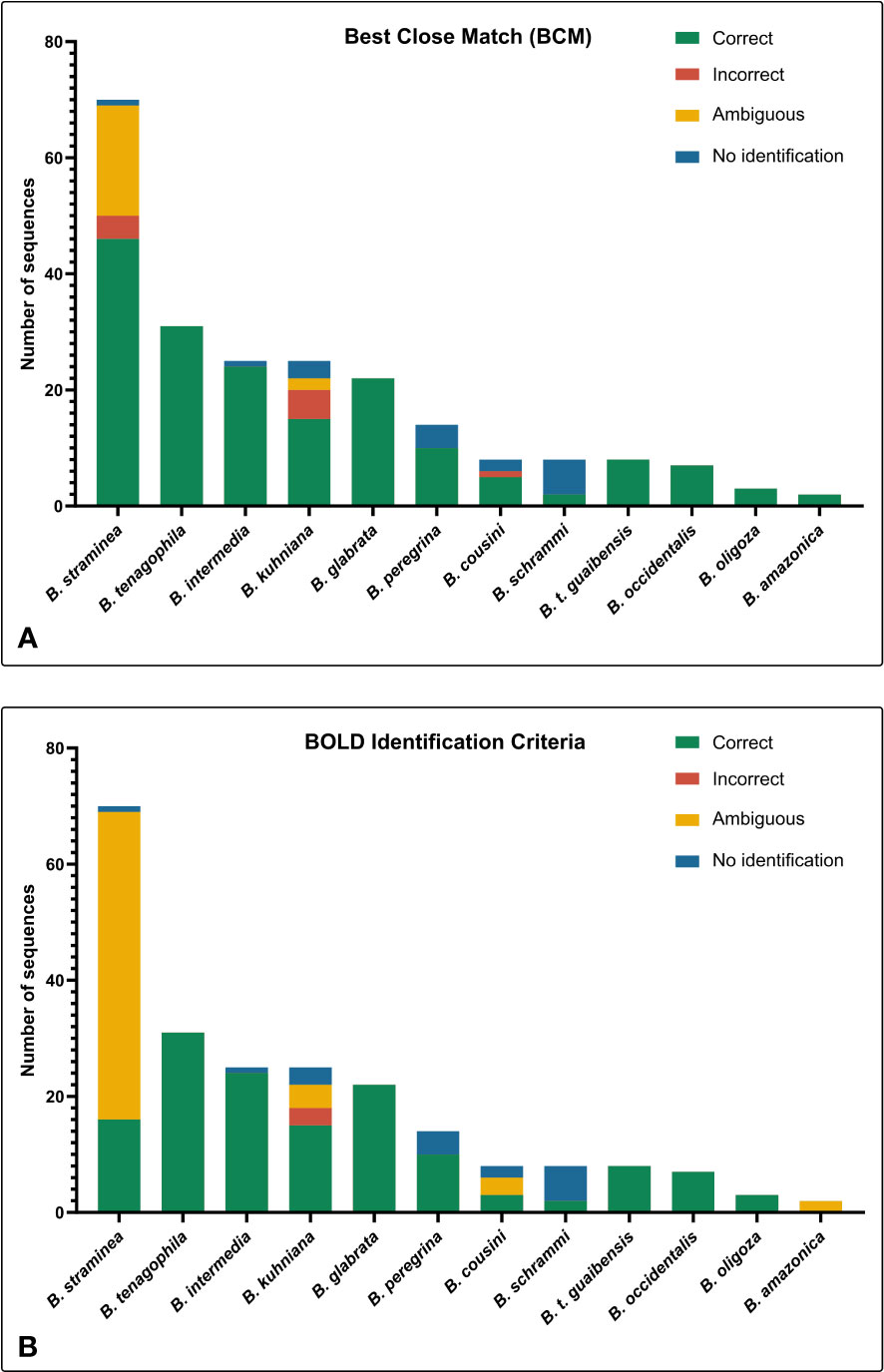
Figure 6 Distribution map and tree generated by Bayesian Inference of the Biomphalaria glabrata specimens used in this study. (A) Map with the geographical distribution of Biomphalaria specimens in Latin America. (B) Simplified phylogenetic tree obtained by Bayesian Inference (BI). The colors represent the large groups separated by the phylogeny of the species.
Of the 223 sequences used in this study, the barcode gap was successfully calculated for 102 (45.7%). However, of the 12 taxa studied, only seven (58.3%) can be differentiated from each other using DNA barcoding methodology. Among the invertebrate host species of S. mansoni in Brazil, only B. straminea could not be differentiated using this methodology, due to its genetic proximity to B. kuhniana. This result is promising regarding the use of DNA barcoding for taxonomy by the schistosomiasis epidemiology service in Brazil. In addition, due to the resolution observed using the Best Close Match (BCM) and BOLD Identification Criteria analyses, DNA barcoding methodology has a great potential to provide greater objectivity for mollusk identification, and can be used for the verification of identification performed by other methods (Ohlweiler et al., 2020). For species of the B. tenagophila complex, despite the proximity between the taxa, the DNA sequences were correctly identified by DNA barcoding methodology, with the analyses performed by BOLD Identification Criteria and BCM. Other authors also successfully separated the species of this complex, using DNA barcoding and phylogenetics, with high support values associated with each of the clades, and concluded that, for this species complex, molecular data may be more informative for species discrimination than morphology (Ohlweiler et al., 2020).
For some taxa, including B. kuhniana and B. straminea, the intraspecific divergence of the coi gene often surpassed the interspecific divergence, making coalescence methods for the formation of Operational Taxonomic Units (OTUs) inadequate for species delimitation. We observed the formation of many clusters within each species, some of them with only one sequence, others with sequences from different species within the same cluster, an artifact of the high variability observed for the coi gene. The use of nuclear markers with lower rates of evolution can be an alternative for the delimitation of species using these approaches.
The cluster analyses using species delimitation algorithms showed that the genetic divergence within populations was significant enough to generate substructuring within clades and differentiated clusters, which was also observed by other authors (Palasio et al., 2017). However, the genetic diversity observed among the species was not reflected in the morphological characters used in the classical taxonomy for distinguishing between species. Thus, it is evident that these morphological traits do not present enough characteristics to reflect the biodiversity of Biomphalaria in nature. The origin of such biodiversity inside the genus in South America may be a reflection of tropical forest fragmentation, which can cause speciation by allopatry result of the formation of distinct and isolated environments in recurrent episodes of vicariance (Colinvaux, 1987). This fragmentation of habitats has been substantially exacerbated by human intervention, particularly through the destruction of natural habitats through deforestation. (Barnosky et al., 2011).
The analysis of the variable sites revealed that B. cousini is the taxon with the highest number of polymorphic sites among the evaluated sequences. The number of polymorphic sites within the population indicates the intrapopulation genetic variability (Caldeira et al., 2001), showing that among all the analyzed species, B. cousini is the one that exhibits the greatest genetic diversity. Comparison of K2P genetic distances (Tamura et al., 2004) between the B. cousini sequences also showed values larger than 18%, values greater than the distance between this group and any other taxon in this study. One of the possible reasons for the diversity within this species may be the isolation of populations caused by the geographic barrier imposed by the Amazon rainforest, which also ends up preventing the dissemination of species from Venezuela, for example, Biomphalaria prona (Dejong et al., 2003; Jarne et al., 2011). Another relevant point for this discussion is that hybridization between B. cousini and B. amazonica has already been observed (Teodoro et al., 2011), which may have caused this high diversity among specimens of B. cousini and the proximity to B. amazonica, a fact already reported by Dr. Paraense in the description of the species, in 1966 (Paraense, 1966). These data may encourage discussion about the possibility of the existence of cryptic species within the currently recognized taxon B. cousini.
The systematics of mollusks is very nebulous and undergoes recurrent modifications (Wanninger and Wollesen, 2019). As observed for mussels of the genus Pecten (Canapa et al., 2000), this study showed that B. straminea and B. kuhniana are very similar, with small genetic distances, a fact that may raise the question of whether both may belong to the same species or are undergoing a recent speciation event.
5 Conclusions
Although the use of molecular data does not guarantee correct phylogenetic trees, and the study involving only one mitochondrial molecular marker is far from ideal and may not reliably reflect the evolution of the genus, this study demonstrated that the use of a region of the coi gene in the reconstruction of the phylogeny of the Biomphalaria genus can be an alternative to understanding the evolution and dispersion of the species. Despite the relationships established here, it must be considered that associating a single mitochondrial gene to the taxonomic history of a species is impossible, highlighting the need to expand studies with other molecular markers. The usefulness of the DNA barcoding methodology for mollusks of the genus Biomphalaria is well-suited to providing a better representation of this genus in public databases and the use of integrative methodologies for the correct identification of mollusks. This work contributed significantly to the number of partial sequences of the coi gene deposited in public databases, thus allowing a greater number of possible comparisons in future studies. Additionally, it was possible to obtain the correct delimitation of most of the Biomphalaria species whose occurrence is reported in Brazil using DNA barcoding and clustering/phylogenetic algorithms. Despite the high intraspecific diversity observed, two of the three species that act as intermediate hosts for S. mansoni in Brazil were successfully distinguished using the DNA barcoding technique based on the coi gene fragment. Further studies using both mitochondrial and nuclear markers are essential to elucidate the relationships between two of the most closely-related groups: B. kuhniana and B. straminea, and B. cousini and B. amazonica.
Data availability statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/Supplementary Material.
Author contributions
Conceptualization: RC, OC. Methodology & Investigation: AA, SG, RC. Formal analysis: AA, SG, RC. Resources: RC. Writing - original draft: AA. Writing – editing: AA, RC, SG, OC Writing – review: AA, RC, SG, OC. Visualisation: AA. Supervision: RC, SG. Project Administration: RC, SG. Funding acquisition: RC. All authors contributed to the article and approved the submitted version.
Funding
This work was funded by Fapemig (APQ-01533-22), Fiocruz (Oswaldo Cruz Foundation) and by the Coordenação de Aperfeiçoamento de Pessoal de Nível Superior–Brasil (CAPES) – Finance Code 001.
Acknowledgments
The authors would like to thank: the Medical Malacology Collection (Fiocruz-CMM, Instituto René Rachou, Oswaldo Cruz Foundation, Brazil) for providing samples; the Program for Technological Development in Tools for Health-PDTIS/FIOCRUZ for the use of its facilities; the Programa de Pós-graduação em Ciências da Saúde of the René Rachou Institute-Fiocruz; and Dr. Luke Baton for revising and commenting upon the manuscript.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fcimb.2023.1167787/full#supplementary-material
References
Aguiar-Silva, C., Mendonça, C. L., da Cunha Kellis Pinheiro, P., Mesquita, S., dos Carvalho, O., Caldeira, R. (2014). Evaluation and updating of the Medical Malacology Collection (Fiocruz-CMM) using molecular taxonomy. Springerplus 3, 446. doi: 10.1186/2193-1801-3-446
Altschul, S. F., Gish, W., Miller, W., Myers, E. W., Lipman, D. J. (1990). Basic Local Alignment Search Tool. J. Mol. Biol. 215, 403–410. doi: 10.1016/S0022-2836(05)80360-2
Anderson, E., Hubricht, L. (1938). Hybridization in tradescantia. III. the evidence for introgressive hybridization. Am. J. Bot. 25, 396–402. doi: 10.1002/j.1537-2197.1938.tb09237.x
Attwood, S. W., Huo, G.-N., Qiu, J.-W. (2015). Update on the distribution and phylogenetics of Biomphalaria (Gastropoda: Planorbidae) populations in Guangdong province, China. Acta Trop. 141, 258–270. doi: 10.1016/j.actatropica.2014.04.032
Inkscape Project (2021) Inkscape project. Available at: https://inkscape.org.
Barnosky, A. D., Matzke, N., Tomiya, S., Wogan, G., Swartz, B., Quental, T. B., et al. (2011). Has the earth’s sixth mass extinction already arrived? Nature 471, 51–57. doi: 10.1038/nature09678
Blaxter, M., Mann, J., Chapman, T., Thomas, F., Whitton, C., Floyd, R., et al. (2005). Defining operational taxonomic units using DNA barcode data. Philos. Trans. R Soc. B Biol. Sci. 360, 1935–1943. doi: 10.1098/rstb.2005.1725
Brown, S. D. J., Collins, R. A., Boyer, S., Lefort, M., Malumbres-Olarte, J., Vink, C. J., et al. (2012). Spider: An R package for the analysis of species identity and evolution, with particular reference to DNA barcoding. Mol. Ecol. Resour 12, 562–565. doi: 10.1111/j.1755-0998.2011.03108.x
Cabrera, F., Martinez, S., Norbis, W. (2016). Recognition of Biomphalaria species (Gastropoda: Planorbidae) using the spiral growth variability, with description of a new fossil species. Nautilus (Philadelphia) 130, 5–12.
Caldeira, R. L., Vidigal, T. H., Matinella, L., Simpson, A. J., Carvalho, O. S. (2000). Identification of planorbids from Venezuela by polymerase chain reaction amplification and restriction fragment length polymorphism of internal transcriber spacer of the RNA ribosomal gene. Mem Inst Oswaldo Cruz 95, 171–177. doi: 10.1590/S0074-02762000000200007
Caldeira, R. L., Vidigal, T. H. D. A., Paulinelli, S. T., Simpson, A. J. G., Carvalho, O. S. (1998). Molecular identification of similar species of the genus Biomphalaria (Mollusca: Planorbidae) determined by a polymerase chain reaction-restriction fragment length polymorphism. Mem Inst Oswaldo Cruz 93 (SUPPL), 219–225. doi: 10.1590/S0074-02761998000700039
Caldeira, R. L., Vidigal, T. H. D. A., Simpson, A. J. G., Carvalho, O. S. (2001). Genetic variability in Brazilian populations of Biomphalaria straminea complex detected by simple sequence repeat anchored polymerase chain reaction amplification. Mem Inst Oswaldo Cruz 96, 535–544. doi: 10.1590/S0074-02762001000400016
Canapa, A., Barucca, M., Marinelli, A., Olmo, E. (2000). Molecular data from the 16S rRNA gene for the phylogeny of Pectinidae (Mollusca: Bivalvia). J. Mol. Evol. 50, 93–97. doi: 10.1007/s002399910010
Carvalho, O. S., Mendonça, C. L. F., Teles, H. M. S., Finau, J., Caldeira, R. L., Scholte, R. G. C., et al. (2020). Moluscos hospedeiros intermediários de Schistosoma mansoni do Brasil. Belo Horizonte/Instituto René Rachou/Fiocruz.
Carvalho O dos, S., Nunes, I. M., Caldeira, R. L. (1998). First report of Biomphalaria glabrata in the state of Rio Grande do Sul, Brazil. Mem Inst Oswaldo Cruz 93, 39–40. doi: 10.1590/S0074-02761998000100008
Casiraghi, M., Labra, M., Ferri, E., Galimberti, A., Mattia, F. (2010). DNA Barcoding: theoretical aspects and practical applications. Tools Identifying Biodivers Prog. Probl, 269–273.
CodonCode Corporation (2019) Sequence assembly and alignment software - CodonCode. Available at: https://www.codoncode.com/index.htm (Accessed July 17, 2021).
Colinvaux, P. (1987). Amazon Diversity in light of the paleoecological record. Quat Sci. Rev. 6, 93–114. doi: 10.1016/0277-3791(87)90028-X
Corrêa, L. R. (1971). Susceptibility of Biomphalaria amazonica to infection with two strains of Schistosoma mansoni. Rev. Inst Med. Trop. Sao Paulo 13, 387–390.
DeJong, R. J., Morgan, J. A. T., Paraense, W. L., Pointier, J.-P., Amarista, M., Ayeh-Kumi, P. F. K., et al. (2001). Evolutionary relationships and biogeography of Biomphalaria (Gastropoda: Planorbidae) with implications regarding its role as host of the human bloodfluke, Schistosoma mansoni. Mol. Biol. Evol. 18, 2225–2239. doi: 10.1093/oxfordjournals.molbev.a003769
Dejong, R. J., Morgan, J. A. T., Wilson, W. D., Al-Jaser, M. H., Appleton, C. C., Coulibaly, G., et al. (2003). Phylogeography of Biomphalaria glabrata and B. pfeifferi, important intermediate hosts of Schistosoma mansoni in the new and old world tropics. Mol. Ecol. 12, 3041–3056. doi: 10.1046/j.1365-294X.2003.01977.x
De Queiroz, K. (2007). Species concepts and species delimitation. Syst. Biol. 56, 879–886. doi: 10.1080/10635150701701083
Drummond, A. J., Nicholls, G. K., Rodrigo, A. G., Solomon, W. (2002). Estimating mutation parameters, population history and genealogy simultaneously from temporally spaced sequence data. Genetics 161 (1307 LP), 1320. doi: 10.1093/genetics/161.3.1307
Dudgeon, D., Yipp, M. W. (1983). A report on the gastropod fauna of aquarium fish farms in Hong Kong, with special reference to an introduced human schistosome host species, Biomphalaria straminea (Pulmonata: Planorbidae). Malacol Rev. 16, 93–94.
Edgar, R. C. (2004). MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 32, 1792–1797. doi: 10.1093/nar/gkh340
Estrada, V. E., Velásquez, L. E., Caldeira, R. L., Bejarano, E. E., Rojas, W., Carvalho, O. S. (2006). Phylogenetics of south American Biomphalaria and description of a new species (Gastropoda: Planorbidae). J. Molluscan Stud. 72, 221–228. doi: 10.1093/mollus/eyl003
Felsenstein, J. (1981). Evolutionary trees from DNA sequences: A maximum likelihood approach. J. Mol. Evol. 17, 368–376. doi: 10.1007/BF01734359
Fernandez, M. A., de Mattos, A. C., da Silva, E. F., dos Santos, S. B., Thiengo, S. C. (2014). A malacological survey in the Manso Power Plant, state of Mato Grosso, Brazil: new records of freshwater snails, including transmitters of schistosomiasis and exotic species. Rev. Soc. Bras. Med. Trop. 47, 498–506. doi: 10.1590/0037-8682-0138-2014
Folmer, O., Black, M., Hoeh, W., Lutz, R., Vrijenhoek, R. (1994). DNA Primers for amplification of mitochondrial cytochrome c oxidase subunit I from diverse metazoan invertebrates. Mol. Mar. Biol. Biotechnol. 3, 294–299. doi: 10.1071/ZO9660275
Fujisawa, T., Barraclough, T. G. (2013). Delimiting species using single-locus data and the generalized mixed Yule coalescent approach: A revised method and evaluation on simulated data sets. Syst. Biol. 62, 707–724. doi: 10.1093/sysbio/syt033
Guindon, S., Dufayard, J.-F., Lefort, V., Anisimova, M., Hordijk, W., Gascuel, O. (2010). New algorithms and methods to estimate maximum-likelihood phylogenies: Assessing the performance of PhyML 3.0. Syst. Biol. 59, 307–321. doi: 10.1093/sysbio/syq010
Haffer, J. (1969). Speciation in Amazonian forest birds. Science 165, 131–137. doi: 10.1126/science.165.3889.131
Hebert, P. D. N., Cywinska, A., Ball, S. L., DeWaard, J. R. (2003). Biological identifications through DNA barcodes. Proc. R Soc. B Biol. Sci. 270, 313–321. doi: 10.1098/rspb.2002.2218
Hebert, P. D. N., Penton, E. H., Burns, J. M., Janzen, D. H., Hallwachs, W. (2004). Ten species in one: DNA barcoding reveals cryptic species in the neotropical skipper butterfly astraptes fulgerator. Proc. Natl. Acad. Sci. 101, 14812–14817. doi: 10.1073/pnas.0406166101
Hey, J. (1991). The structure of genealogies and the distribution of fixed differences between DNA sequence samples from natural populations. Genetics 128, 831–840. doi: 10.1093/genetics/128.4.831
IBGE (2021). Available at: https://www.ibge.gov.br/geociencias/downloads-geociencias.html (Accessed July 16, 2021).
James, S. L., Abate, D., Abate, K. H., Abay, S. M., Abbafati, C., Abbasi, N., et al. (2018). Global, regional, and national incidence, prevalence, and years lived with disability for 354 diseases and injuries for 195 countries and territories, 1990-2017: A systematic analysis for the global burden of disease study 2017. Lancet 392, 1789–1858. doi: 10.1016/S0140-6736(18)32279-7
Jarne, P., David, P., Pointier, J.-P., Koene, J. M. (2010). “Basommatophoran gastropods,” in The evolution of primary sexual characters in animals. Ed. Leonard, AC-AJL (Oxforf: Oxford University press), 173–196.
Jarne, P., Pointier, J.-P., David, P. (2011). “Biosystematics of Biomphalaria spp. with an emphasis on Biomphalaria glabrata,” in Biomphalaria snails and larval trematodes (New York, NY: Springer New York), 1–32. doi: 10.1007/978-1-4419-7028-2_1
Jukes, T. H., Cantor, C. R. (1969). Evolution of protein molecules. Mamm. Protein Metab., 21–132. doi: 10.1016/B978-1-4832-3211-9.50009-7
Kaesler, R. L., Parodiz, J. J. (1970). The tertiary non-marine Mollusca of South America. Syst. Zool 19, 306. doi: 10.2307/2412214
Kalyaanamoorthy, S., Minh, B. Q., Wong, T. K. F., von Haeseler, A., Jermiin, L. S. (2017). ModelFinder: fast model selection for accurate phylogenetic estimates. Nat. Methods 14, 587–589. doi: 10.1038/nmeth.4285
Katoh, K., Rozewicki, J., Yamada, K. D. (2019). MAFFT online service: multiple sequence alignment, interactive sequence choice and visualization. Brief Bioinform. 20, 1160–1166. doi: 10.1093/bib/bbx108
Kumar, S., Stecher, G., Li, M., Knyaz, C., Tamura, K. (2018). MEGA X: Molecular evolutionary genetics analysis across computing platforms. Mol. Biol. Evol. 35, 1547–1549. doi: 10.1093/molbev/msy096
Leray, M., Knowlton, N. (2015). DNA Barcoding and metabarcoding of standardized samples reveal patterns of marine benthic diversity. Proc. Natl. Acad. Sci. 112, 2076–2081. doi: 10.1073/pnas.1424997112
Lobato Paraense, W., Deslandes, N. (1958a). Observations on Taphius havanensis (Pulmonata, planorbidae). Rev. Bras. Biol. 18, 87–91.
Lobato Paraense, W., Deslandes, N. (1958b). Taphius pronus (Martens, 1873)(Pulmonata, planorbidae). Rev. Bras. Biol. 18, 367–373.
Lynch, M., Crease, T. J. (1990). The analysis of population survey data on DNA sequence variation. Mol. Biol. Evol. 7, 377–394.
Mavárez, J., Amarista, M., Pointier, J.-P., Jarne, P. (2002). Fine-scale population structure and dispersal in Biomphalaria glabrata, the intermediate snail host of Schistosoma mansoni, in Venezuela. Mol. Ecol. 11, 879–889. doi: 10.1046/j.1365-294X.2002.01486.x
McManus, D. P., Dunne, D. W., Sacko, M., Utzinger, J., Vennervald, B. J., Zhou, X.-N. (2018). Schistosomiasis. Nat. Rev. Dis. Prim 4, 13. doi: 10.1038/s41572-018-0013-8
Meier, R., Shiyang, K., Vaidya, G., Ng, P. K. L. (2006). DNA Barcoding and taxonomy in diptera: A tale of high intraspecific variability and low identification success. Syst. Biol. 55, 715–728. doi: 10.1080/10635150600969864
Meier Brook, C. (1975). A snail intermediate host of Schistosoma mansoni introduced into Hong Kong. Bull. World Health Organ 51, 661.
Mello-Silva, C. C., Grault, C. E., da, C. V. A., Barbosa, F. S. (1998). Possible hybridization of Brazilian planorbid snails and its importance in population dynamics. Mem Inst Oswaldo Cruz 93, 227–232. doi: 10.1590/S0074-02761998000700040
Meyer, C. P., Paulay, G. (2005). DNA Barcoding: Error rates based on comprehensive sampling. PloS Biol. 3, e422. doi: 10.1371/journal.pbio.0030422
Minh, B. Q., Nguyen, M. A. T., von Haeseler, A. (2013). Ultrafast approximation for phylogenetic bootstrap. Mol. Biol. Evol. 30, 1188–1195. doi: 10.1093/molbev/mst024
Nei, M. (1987). Molecular evolutionary genetics. (New York: Columbia University Press). doi: 10.7312/nei-92038
Nguyen, L.-T., Schmidt, H. A., von Haeseler, A., Minh, B. Q. (2015). IQ-TREE: A fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol. Biol. Evol. 32, 268–274. doi: 10.1093/molbev/msu300
Ohlweiler, F. P., Rossignoli T de, J., Palasio, R. G. S., Tuan, R. (2020). Taxonomic diversity of Biomphalaria (Planorbidae) in São Paulo state, Brazil. Biota Neotrop 20(2): e20200975. doi: 10.1590/1676-0611-bn-2020-0975
Pääbo, S. (1990). Amplifying ancient DNA. In MA Innis, DH Gelfand, JJ Sninsky, TJ White (eds). PCR Protocols: a Guide to Methods and Applications (San Diego: Academic Press), 159–166.
Palasio, R. G. S., Guimarães MC de, A., Ohlweiler, F. P., Tuan, R. (2017). Molecular and morphological identification of Biomphalaria species from the state of São Paulo, Brazil. Zookeys 2017, 11–32. doi: 10.3897/zookeys.668.10562
Paradis, E., Schliep, K. (2019). Ape 5.0: an environment for modern phylogenetics and evolutionary analyses in R. Bioinformatics 35, 526–528. doi: 10.1093/bioinformatics/bty633
Paraense, W. L. (1966). Biomphalaria amazonica and B. cousini, two new species of neotropical planorbid molluscs. Rev. Bras. Biol. 26, 115–126.
Paraense, W. L. (1975). Estado atual da sistemática dos planorbídeos brasileiros.(Mollusca, Gastropoda). Arq do Mus Nac Do Rio Janeiro 55, 105–128.
Paraense, W. L. (1981). Biomphalaria occidentalis sp. n. from South America (Mollusca basommatophora pulmonata). Mem Inst Oswaldo Cruz 76, 199–211. doi: 10.1590/S0074-02761981000200011
Paraense, W. L. (1984). Biomphalaria tenagophila guaibensis ssp. n. from southern Brazil and Uruguay (Pulmonata: Planorbidae). I. Morphology. Mem Inst Oswaldo Cruz 79, 465–469. doi: 10.1590/S0074-02761984000400012
Paraense, W. L. (1988). Biomphalaria kuhniana (Clessin, 1883), planorbid mollusc from South America. Mem Inst Oswaldo Cruz 83, 1–12. doi: 10.1590/s0074-02761988000100001
Paraense, W. L. (1990). Biomphalaria obstructa (Morelet, 1849): a study of topotypic specimens (Mollusca: Pulmonata: Planorbidae). Mem Inst Oswaldo Cruz 85, 391–399. doi: 10.1590/S0074-02761990000400002
Paraense, W. L., Corrêa, L. R. (1973). Susceptibility of Biomphalaria peregrina from Brazil and Ecuador to two strains of Schisotosoma mansoni. Rev. Inst Med. Trop. Sao Paulo 15, 127–130.
Paraense, W. L., Deslandes, N. (1959). The renal ridge as a reliable character for separating Taphius glabratus from Taphius tenagophilus. Am. J. Trop. Med. Hyg 8, 456–472. doi: 10.4269/ajtmh.1959.8.456
Paraense, W. L., Pointier, J. P., Delay, B., Pernot, A. F., Incani, R. N., Balzan, C., et al. (1992). Biomphalaria prona (Gastropoda: Planorbidae): a morphological and biochemical study. Mem Inst Oswaldo Cruz 87, 171–179. doi: 10.1590/S0074-02761992000200001
Paraense, W. L., Zeledón, R., Rojas, G., Zeledon, R. (1981). Biomphalaria straminea and other planorbid molluscs in Costa Rica. J. Parasitol. 67, 282. doi: 10.2307/3280654
Pointier, J. P., Paraense, W. L., Mazille, V. (1993). Introduction and spreading of Biomphalaria straminea (Dunker, 1848) (Mollusca: Pulmonata: Planorbidae) in Guadeloupe, French West Indies. Mem Inst Oswaldo Cruz 88, 449–455. doi: 10.1590/S0074-02761993000300016
Puillandre, N., Brouillet, S., Achaz, G. (2021). ASAP: assemble species by automatic partitioning. Mol. Ecol. Resour 21, 609–620. doi: 10.1111/1755-0998.13281
Puillandre, N., Lambert, A., Brouillet, S., Achaz, G. (2012). ABGD, automatic barcode gap discovery for primary species delimitation. Mol. Ecol. 21, 1864–1877. doi: 10.1111/j.1365-294X.2011.05239.x
QGIS (2021). Available at: https://www.qgis.org/en/site/ (Accessed July 16, 2021).
Rambaut, A. (2018) FigTree. Available at: http://tree.bio.ed.ac.uk/software/figtree/ (Accessed julho 15, 2021).
Ratnasingham, S., Hebert, P. D. N. (2007). BOLD: The barcode of life data system (http://www.barcodinglife.org). Mol. Ecol. Notes 7, 355–364. doi: 10.1111/j.1471-8286.2007.01678.x
Reid, N. M., Carstens, B. C. (2012). Phylogenetic estimation error can decrease the accuracy of species delimitation: a Bayesian implementation of the general mixed Yule-coalescent model. BMC Evol. Biol. 12, 196. doi: 10.1186/1471-2148-12-196
R Foundation for Statistical Computing (2021) R: A language and environment for statistical computing. Available at: https://www.r-project.org/.
Rollinson, D., Webster, J. P., Webster, B., Nyakaana, S., Jørgensen, A., Stothard, J. R. (2009). Genetic diversity of schistosomes and snails: implications for control. Parasitology 136, 1801–1811. doi: 10.1017/S0031182009990412
Ronquist, F., Teslenko, M., van der Mark, P., Ayres, D. L., Darling, A., Höhna, S., et al. (2012). MrBayes 3.2: Efficient Bayesian phylogenetic inference and model choice across a Large model space. Syst. Biol. 61, 539–542. doi: 10.1093/sysbio/sys029
Rozas, J., Ferrer-Mata, A., Sánchez-DelBarrio, J. C., Guirao-Rico, S., Librado, P., Ramos-Onsins, S. E., et al. (2017). DnaSP 6: DNA sequence polymorphism analysis of Large data sets. Mol. Biol. Evol. 34, 3299–3302. doi: 10.1093/molbev/msx248
Schindel, D. E., Miller, S. E. (2005). DNA Barcoding a useful tool for taxonomists. Nature 435, 17–17. doi: 10.1038/435017b
Scholte, R. G. C., Carvalho, O. S., Malone, J. B., Utzinger, J., Vounatsou, P. (2012). Spatial distribution of Biomphalaria spp., the intermediate host snails of Schistosoma mansoni, in Brazil. Geospat Health 6, 95. doi: 10.4081/gh.2012.127
Spatz, L., Vidigal, T. H. D. A., Caldeira, R. L., Neto, E. D., Cappa, S. M. G., Carvalho, O. S. (1999). Study of Biomphalaria tenagophila tenagophila, B. t. guaibensis and B. occidentalis by polymerase chain reaction amplification and restriction enzyme digestion of the ribosomal RNA intergenic spacer regions. J. Molluscan Stud. 65, 143–149. doi: 10.1093/mollus/65.2.143
Sun, S., Li, Q., Kong, L., Yu, H., Zheng, X., Yu, R., et al. (2016). DNA Barcoding reveal patterns of species diversity among northwestern pacific molluscs. Sci. Rep. 6, 1–17. doi: 10.1038/srep33367
Tajima, F. (1983). Evolutionary relationship of DNA sequences in finite populations. Genetics 105, 437–460. doi: 10.1093/genetics/105.2.437
Tamura, K., Nei, M., Kumar, S. (2004). Prospects for inferring very large phylogenies by using the neighbor-joining method. Proc. Natl. Acad. Sci. 101, 11030–11035. doi: 10.1073/pnas.0404206101
Tang, C. T. (1983). “A survey of Biomphalaria straminea (Dunker, 1848)(Planorbidae) for trematode infection, with a report on larval flukes from other Gastropoda in Hong Kong,” in Proceedings of the Second International Workshop in Malacology Hong Kong. 393–408.
Teodoro, T. M., Jannotti-Passos, L. K., Carvalho O dos, S., Grijalva, M. J., Baús, E. G., Caldeira, R. L. (2011). Hybridism between Biomphalaria cousini and Biomphalaria amazonica and its susceptibility to Schistosoma mansoni. Mem Inst Oswaldo Cruz 106, 851–855. doi: 10.1590/S0074-02762011000700011
Teodoro, T. M., Janotti-Passos, L. K., Carvalho O dos, S., Caldeira, R. L. (2010). Occurrence of Biomphalaria cousini (Mollusca: Gastropoda) in Brazil and its susceptibility to Schistosoma mansoni (Platyhelminths: Trematoda). Mol. Phylogenet Evol. 57, 144–151. doi: 10.1016/j.ympev.2010.05.019
Vidigal, T. H. D. A., Caldeira, R. L., Simpson, A. J. G., Carvalho, O. S. (2000). Further studies on the molecular systematics of Biomphalaria snails from Brazil. Mem Inst Oswaldo Cruz 95, 57–66. doi: 10.1590/S0074-02762000000100009
Vidigal, T. H. D. A., Spatz, L., Kissinger, J. C., Redondo, R. A. F., Pires, E. C. R., Simpson, A. J. G., et al. (2004). Analysis of the first and second internal transcribed spacer sequences of the ribosomal DNA in Biomphalaria tenagophila complex (Mollusca: Planorbidae). Mem Inst Oswaldo Cruz 99, 153–158. doi: 10.1590/S0074-02762004000200007
Wang, W., Liang, Y.-S., Hong, Q.-B., Dai, J.-R. (2013). African Schistosomiasis in mainland China: risk of transmission and countermeasures to tackle the risk. Parasit Vectors 6, 249. doi: 10.1186/1756-3305-6-249
Wanninger, A., Wollesen, T. (2019). The evolution of molluscs. Biol. Rev. 94, 102–115. doi: 10.1111/brv.12439
Yang, Y., Huang, S.-Y., Pei, F.-Q., Chen, Y., Jiang, Q.-W., Deng, Z.-H., et al. (2018). Spatial distribution and habitat suitability of Biomphalaria straminea, intermediate host of Schistosoma mansoni, in Guangdong, China. Infect. Dis. Poverty 7, 109. doi: 10.1186/s40249-018-0492-6
Yeates, D. K., Seago, A., Nelson, L., Cameron, S. L., Joseph, L., Trueman, J. W. H. (2011). Integrative taxonomy, or iterative taxonomy? Syst. Entomol 36, 209–217. doi: 10.1111/j.1365-3113.2010.00558.x
Yipp, M. W. (1990). Distribution of the schistosome vector snail, Biomphalaria straminea (Pulmonata: Planorbidae) in Hong Kong. J. Molluscan Stud. 56, 47–55. doi: 10.1093/mollus/56.1.47
Zhang, D., Gao, F., Jakovlić, I., Zou, H., Zhang, J., Li, W. X., et al. (2020). PhyloSuite: An integrated and scalable desktop platform for streamlined molecular sequence data management and evolutionary phylogenetics studies. Mol. Ecol. Resour 20, 348–355. doi: 10.1111/1755-0998.13096
Keywords: DNA barcode, mollusks, Biomphalaria, medical malacology collection, molecular idenlification, cytochrome c oxidase coi
Citation: de Araújo AD, Carvalho OS, Gava SG and Caldeira RL (2023) DNA barcoding as a valuable tool for delimiting mollusk species of the genus Biomphalaria Preston, 1910 (Gastropoda: Planorbidae). Front. Cell. Infect. Microbiol. 13:1167787. doi: 10.3389/fcimb.2023.1167787
Received: 17 February 2023; Accepted: 22 March 2023;
Published: 24 April 2023.
Edited by:
Omar Hamarsheh, Al-Quds University, PalestineCopyright © 2023 de Araújo, Carvalho, Gava and Caldeira. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Sandra Grossi Gava, c2FuZHJhLmdyb3NzaUBmaW9jcnV6LmJy; Roberta Lima Caldeira, cm9iZXJ0YS5jYWxkZWlyYUBmaW9jcnV6LmJy