 Diana Salomi Ponraj1Michael Lund2Jeppe Lange1,3
Diana Salomi Ponraj1Michael Lund2Jeppe Lange1,3 Anja Poehlein4
Anja Poehlein4 Axel Himmelbach5Thomas Falstie-Jensen6Nis Pedersen Jørgensen7
Axel Himmelbach5Thomas Falstie-Jensen6Nis Pedersen Jørgensen7 Christen Ravn6
Christen Ravn6 Holger Brüggemann2*
Holger Brüggemann2*- 1Department of Clinical Medicine, Aarhus University, Aarhus, Denmark
- 2Department of Biomedicine, Aarhus University, Aarhus, Denmark
- 3Department of Orthopaedic Surgery, Regional Hospital, Horsens, Denmark
- 4Department of Genomic and Applied Microbiology, Institute of Microbiology and Genetics, University of Göttingen, Göttingen, Germany
- 5Leibniz Institute of Plant Genetics and Crop Plant Research (IPK), Gatersleben, Germany
- 6Department of Orthopaedic Surgery, Aarhus University Hospital, Aarhus, Denmark
- 7Department of Infectious Diseases, Aarhus University Hospital, Aarhus, Denmark
Orthopaedic implant-associated infections (OIAIs) due to Cutibacterium acnes can be difficult to diagnose. The aim of this pilot study was to determine if metagenomic next-generation sequencing (mNGS) can provide additional information to improve the diagnosis of C. acnes OIAIs. mNGS was performed on sonication fluid (SF) specimens derived from 24 implants. These were divided into three groups, based on culture results: group I, culture-negative (n = 4); group II, culture-positive for C. acnes (n = 10); and group III, culture-positive for other bacteria (n = 10). In group I, sequence reads from C. acnes were detected in only one SF sample, originating from a suspected case of OIAIs, which was SF and tissue culture-negative. In group II, C. acnes sequences were detected in 7/10 samples. In group III, C. acnes sequence reads were found in 5/10 samples, in addition to sequence reads that matched the bacterial species identified by culture. These samples could represent polymicrobial infections that were missed by culture. Taken together, mNGS was able to detect C. acnes DNA in more samples compared to culture and could be used to identify cases of suspected C. acnes OIAIs, in particular regarding possible polymicrobial infections, where the growth of C. acnes might be compromised due to a fast-growing bacterial species. However, since SF specimens are usually low-biomass samples, mNGS is prone to DNA contamination, possibly introduced during DNA extraction or sequencing procedures. Thus, it is advisable to set a sequence read count threshold, taking into account project- and NGS-specific criteria.
1 Introduction
Cutibacterium acnes (formerly known as Propionibacterium acnes) is a Gram-positive slow-growing anaerobic bacterium (SGAB). It is a skin commensal that has also been implicated in various implant-associated infections, including orthopaedic implant-associated infections (OIAIs) (Zeller et al., 2007; McDowell et al., 2013; Achermann et al., 2014; Aubin et al., 2014; Ponraj et al., 2021). However, the diagnosis of OIAIs caused by SGAB such as C. acnes is complicated because of non-specific or even absent clinical, radiological, histopathological, and laboratory diagnostic features (Achermann et al., 2014; Aubin et al., 2014; Ponraj et al., 2021). Microbiological diagnosis based on culture can also be constrained by the need for prolonged incubation, due to the bacterium’s slow-growing nature, along with the attendant increased risk of sample contamination (Butler-Wu et al., 2011).
Culture-independent methods can potentially help to offset this problem (Gu et al., 2019). Culture-independent microbiological methods contain polymerase chain reaction (PCR)-based methods and sequencing-based methods; the latter comprise amplicon-based next-generation sequencing (aNGS) and metagenomic NGS (mNGS) (Tande and Patel, 2014; Ponraj et al., 2021). The utility of mNGS in the diagnosis of OIAIs from various specimens including synovial fluid aspirates and tissue samples has been reported previously (Ivy et al., 2018; Weaver et al., 2019; Cai et al., 2020; Fang et al., 2020; Huang et al., 2020; Kildow et al., 2021; Tan et al., 2022). However, only a few mNGS studies have so far been reported regarding the use of sonication fluid (SF) to support the diagnosis of OIAIs (Street et al., 2017; He et al., 2021) and, to the best of our knowledge, none with a specific focus on C. acnes.
In our previous study, we reported on the sonication of 100 orthopaedic implants that were removed for both presumed aseptic reasons and suspected infection (Ponraj et al., 2022). SF samples were further subjected to culture-dependent and culture-independent analyses. Thereby, an aNGS approach was employed to detect and phylotype C. acnes, and this along with the patients’ clinical information as well as whole-genome sequencing of the C. acnes isolates was used to differentiate C. acnes contamination from true infection.
The aim of this pilot study was to determine if mNGS of SF provides an additional value in the diagnosis of OIAIs due to C. acnes. Therefore, mNGS of SF from 24 implants was carried out, and the mNGS data were compared to other data available regarding these implants, including aNGS and tissue culture results. The results show that C. acnes was more often detected by mNGS than by culture and could potentially help to identify cases of C. acnes OIAIs that might be missed by culture alone.
2 Methods
2.1 Sample selection
In a previous study, 100 implants removed during revision surgery were collected between August 2019 and September 2020 and processed by sonication (Ponraj et al., 2022). SF specimens from 24 of the 100 implants were included in this pilot study. The selection was based on the SF culture results, and the included specimens were divided into three groups: group I, culture-negative (n = 4); group II, culture-positive for C. acnes (n = 10); and group III, culture-positive for other bacteria, i.e., staphylococci and Finegoldia magna (n = 10). In addition, normal saline that was sonicated and processed like SF from implants was included as the negative control.
2.2 DNA extraction from sonication fluid
Implants removed during revision surgeries were processed by the previously described vortex-sonication method (Borens et al., 2013). The resultant SF was split into two samples, one for culture-dependent analyses and another for culture-independent analyses. The samples for culture-independent analyses were stored at −20°C until processing. DNA extraction of the SF and the negative control was described previously (Ponraj et al., 2022). In brief, SF (40 ml) stored at −20°C was thawed overnight at 4°C, concentrated by centrifugation (15,000 × g for 1 h at 16°C using rotor F 13–14 × 50 Cy, Thermo Scientific™ Sorvall™ RC 6 Plus Centrifuge), and DNA extraction was performed using the DNeasy PowerSoil Kit (QIAGEN, Hilden, Germany) as per manufacturer’s instructions. DNA concentrations were measured using the Qubit dsDNA HS Assay (Thermo Fisher Scientific, Waltham, MA, USA) with a Qubit fluorometer. The same DNA extraction kit was used for the 24 samples to exclude any possible batch effect.
2.3 Library preparation and Illumina HiSeq sequencing
Illumina shotgun libraries were prepared using the Illumina DNA Prep Tagmentation Kit and Nextera DNA CD Indexes for multiplexing as recommended by the manufacturer (Illumina, San Diego, CA, USA). To assess the quality and size of the libraries, samples were run on an Agilent Bioanalyzer 2100 using an Agilent High Sensitivity DNA Kit as recommended by the manufacturer (Agilent Technologies, Waldbronn, Germany). The concentration of the libraries was determined using the Qubit dsDNA HS Assay Kit as recommended by the manufacturer (Life Technologies GmbH, Darmstadt, Germany). Sequencing was performed by using the NovaSeq6000 instrument (Illumina Inc., San Diego, CA, USA) using the NovaSeq6000 SP Reagent Kit (v. 1.5) and the NovaSeq XP 2-Lane Kit (v. 1.5) for sequencing in the paired-end mode 2 × 250 cycles.
2.4 Bioinformatics
The raw sequencing data were paired, and the paired-end reads along with metadata were uploaded and analysed using the publicly available web-based MG-RAST pipeline (https://www.mg-rast.org/) (Meyer et al., 2019). The workflow involves the following steps: 1) normalization or quality control where artificial duplicate reads are removed along with quality- and length-based trimming; 2) screening of sequences for potential protein-coding genes using a BLASTX search against multiple databases; 3) functional assignments and taxonomic distributions using the matches to external databases. To further analyse sequence reads that were assigned to Cutibacterium sp. or Finegoldia sp., the reads were extracted in MG-RAST and mapped to their corresponding genomes. Complete genomes of C. acnes KPA171202, Cutibacterium avidum 44067, F. magna ATCC29328, and “Finegoldia nericia” 09T494 were used as reference genomes and subsequently indexed using Bowtie2 (v. 2.5.0) (Langmead and Salzberg, 2012). The extracted query reads were mapped to the reference genomes using Bowtie2, and the resulting SAM-files were converted into BAM-files and indexed using Samtools (v. 1.6) (Li et al., 2009). Resulting BAM-files and reference genomes were imported into Geneious Prime (v. 2023.0.1) to visualize mapping results.
In an additional analysis, the mNGS data were analysed using Kraken2/Bracken. First, human DNA reads were removed as follows: paired-end reads were mapped against the Genome Reference Consortium human genome build 38 (GRCh38) using Bowtie2 (v. 2.5.1) (Langmead and Salzberg, 2012). Any reads concordantly or discordantly mapped to the human reference genome were removed from the samples using Samtools (v. 1.6) and Bedtools (v. 2.30.0). The resulting filtered FASTQ-files were classified using Kraken2 (v. 2.1.2) (Wood et al., 2019) using a database consisting of bacteria, archaea, viruses, and plasmids with a minimum hit group of 3. The classification was subsequently re-estimated using Bracken (v. 2.5.0) (Lu et al., 2017). Since the Kraken2/Bracken analysis is more sensitive, resulting in more taxonomically assigned reads than MG-RAST, we introduced a minimum read count threshold of 150 (corresponding to approx. 0.1% of the total amount of bacterial reads per sample). Four samples had C. acnes reads <150, i.e., samples 2 and 3 in group II, and samples 9 and 10 in group III.
2.5 Amplicon NGS control PCR
To test the sensitivity of the aNGS approach (in detail described previously in Ponraj et al., 2022), a PCR was performed with the single-locus sequence typing (SLST) primers (5′-TTGCTCGCAACTGCAAGCA-3′ and 5′-CCGGCTGGCAAATGAGGCAT-3′) and different amounts (1,000 to 0.001 pg) of C. acnes genomic DNA (strain 266). The PCR contained 5 μl of genomic DNA template, 2.5 μl of AccuPrime PCR Buffer II (Invitrogen, Waltham, MA, USA), 1.5 μl of each primer (10 μM; DNA Technology, Risskov, Denmark), 0.15 μl of AccuPrime Taq DNA Polymerase High Fidelity (Invitrogen, Waltham, MA, USA), and 14.35 μl of PCR-grade water. The PCR was performed using the following cycle conditions: initial denaturation at 94°C for 2 min, 35 cycles of denaturation at 94°C for 20 s, annealing at 55°C for 30 s, elongation at 68°C for 1 min, and final elongation step at 72°C for 5 min.
2.6 Ethical approval
The study was registered with Region Midtjylland with reference number 661624. The Central Denmark Region ethical committee waived the need for ethical approval.
2.7 Statistical analyses
Data were entered in Excel, and graphs and charts were prepared using Prism GraphPad.
3 Results
Sequencing (mNGS) of SF specimens from 24 implants, divided into three groups (group I, culture-negative (n = 4); group II, culture-positive for C. acnes (n = 10); group III, culture-positive for other bacteria (n = 10)) was performed. The anatomical locations of the selected 24 implants from the three groups are shown in Figure 1. The majority of the implants were from the shoulder, followed by the hip.
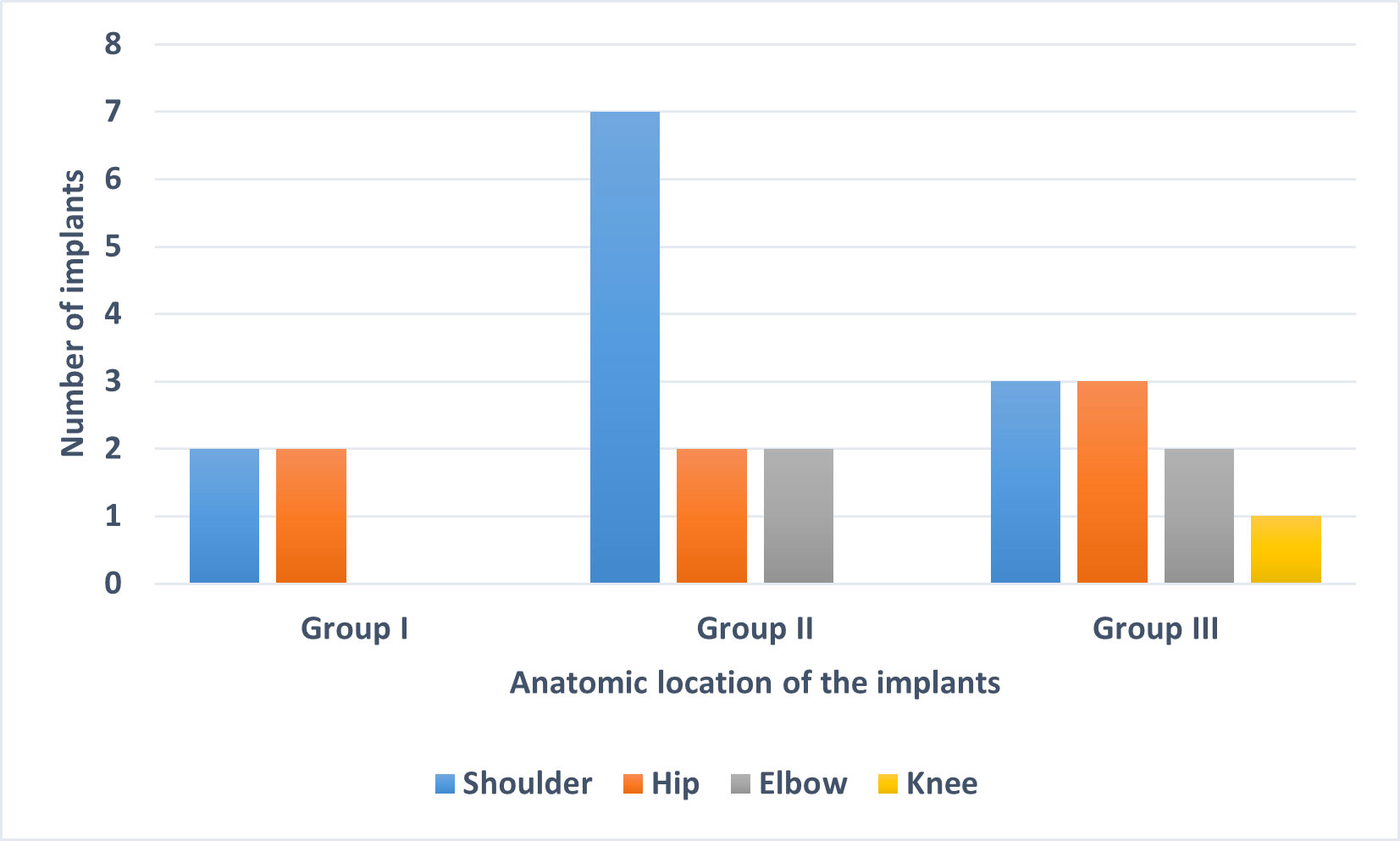
Figure 1 Anatomic location of the 24 implants used in this study. The implants were divided into three groups based on sonication fluid culture results: Group I: culture-negative, Group II: culture-positive for C. acnes and Group III: culture-positive for organisms other than C. acnes.
Shotgun sequencing of DNA extracted from the 24 SF samples resulted in an average of 8,437,406 sequence reads per sample (range, 5,898,825–12,907,838). The data were first analysed with MG-RAST for taxonomic assignment of sequence reads. The total numbers of sequence reads, and sequence reads that passed quality control are given in Table S1. The average number of reads that passed quality control was 778,496 (range, 443,276–1,424,198) (9.2%). The top 10 genera assigned by MG-RAST for the 24 SF samples as well as the number of reads and percentage of total reads for each genus are given in Table S2. No sequence reads were obtained from the saline control sample.
Overall, the percentage of human DNA across the 24 SF samples in the three groups was higher than the percentage of bacterial DNA (Figures 2A, B). Reads that matched non-bacterial genera like Canis, Macaca, Drosophila, and Danio were also detected in all SF samples (Table S2). In addition, several SF samples showed the apparent presence of DNA from gut bacteria like the genera Coprobacillus (n = 23), Bacteroides (n = 13), Prevotella (n = 10), and Clostridium (n = 7). We also used an alternative approach to analyse the data, by first filtering and discarding reads that matched the human genome and subsequently analysing the remaining reads with Kraken2/Bracken for taxonomic assignment (Table 1).
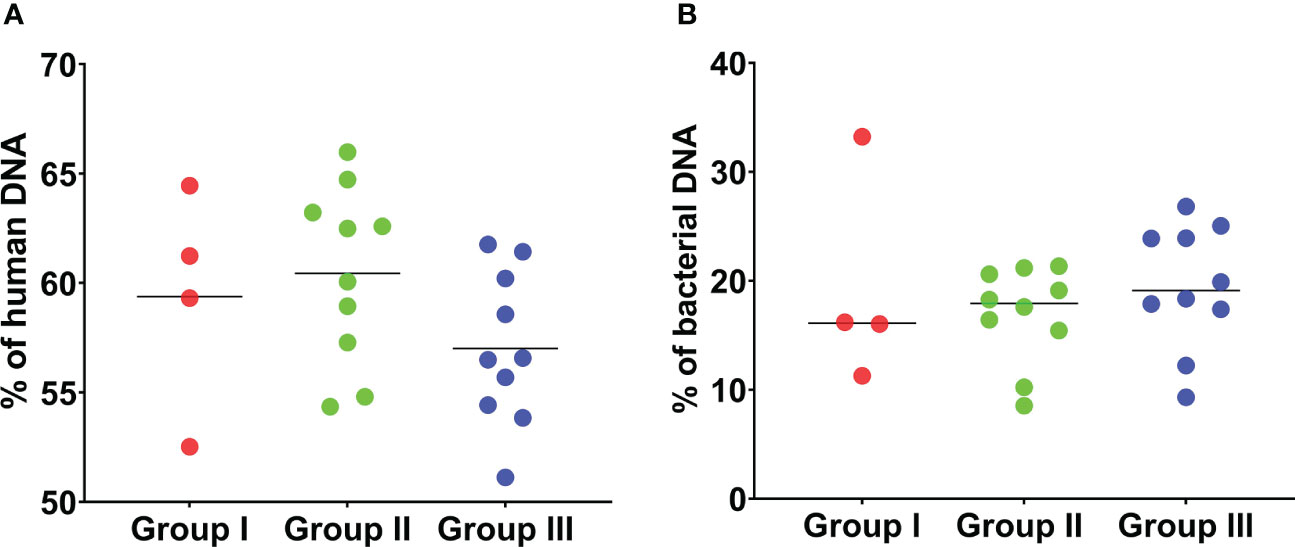
Figure 2 Percentage of human and bacterial DNA detected by mNGS of 24 sonication fluid samples. (A) The percentage of human DNA detected by mNGS in the 24 SF samples in the three groups is shown. The percentage of human DNA is similar across the three groups. (B) The percentage of bacterial DNA detected by mNGS is shown. The percentage of bacterial DNA across the three groups is much lower than the percentage of human DNA in the three groups. This analysis is based on MG-RAST assignment results.
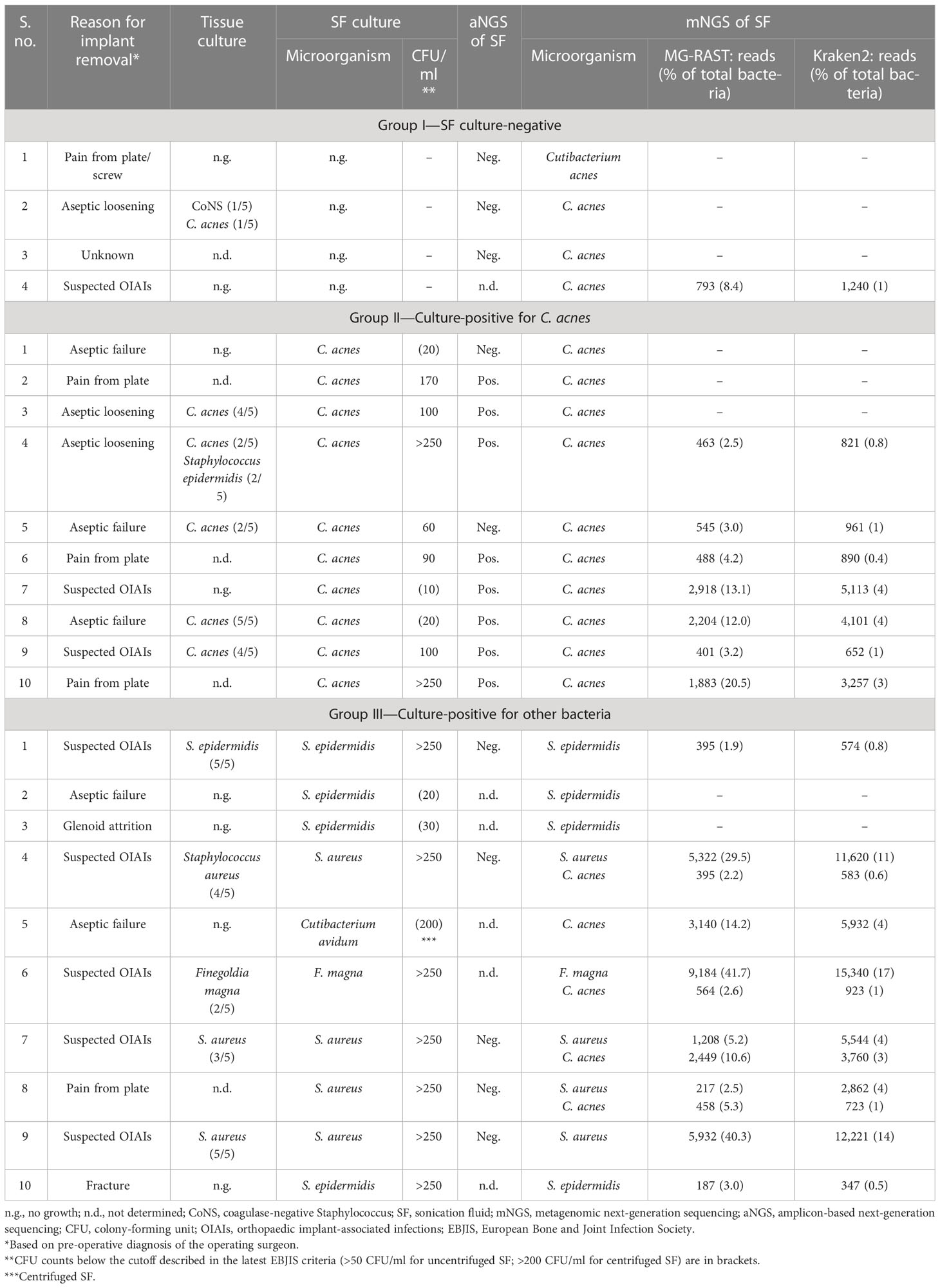
Table 1 Comparison of SF culture results with results of mNGS and aNGS of SF of the 24 implants included in this pilot study.
In the following, mNGS results are outlined for the three groups and presented in the context of other results, i.e., SF culture and aNGS data regarding the detection of C. acnes.
● Group I—culture negative: in three out of four SF specimens in group I, no sequence reads matching C. acnes were detected by mNGS. This corresponded with aNGS results, as all three samples were negative for C. acnes by SLST PCR (Ponraj et al., 2022). In one SF sample (group I #4), C. acnes DNA was detected (793 reads (MG-RAST); 1,240 reads (Kraken2/Bracken)) (Table 1). The corresponding implant was both SF and tissue culture-negative; aNGS was not performed for this SF specimen. The implant was removed due to suspected infection diagnosed by the presence of a sinus tract, so C. acnes could potentially represent the causative agent that was missed by culture.
● Group II—culture-positive for C. acnes: group II included 10 samples that were SF culture-positive for C. acnes (Table 1). mNGS detected sequence reads matching C. acnes in seven of the 10 samples in this group. In these seven samples, 401 to 2,918 C. acnes sequence reads (average 1,271 reads) were identified with MG-RAST, corresponding to 2.5% to 20.5% of all detected bacterial reads. A slightly higher number of C. acnes reads were found with the Kraken2/Bracken approach (average 2,256 reads). Regarding the three samples, for which no C. acnes reads were obtained, one of them (group II #1) was also aNGS-negative (Table 1). In addition, the SF culture showed C. acnes growth with a low colony-forming unit (CFU) count of only 20 CFU/ml, and the five tissue cultures from that implant had no bacterial growth. Thus, C. acnes in this case could likely be a contaminant, possibly obtained at the cultivation step. The second sample (group II #2) with no Cutibacterium sp. reads was aNGS-positive. However, tissue culture results were not available for this sample (Table 1). The third sample (group II #3) was tissue culture-positive for C. acnes. This sample had a low C. acnes read count (49 and 81 reads assigned by MG-RAST and Kraken2/Bracken, respectively), which was under the applied read count threshold.
● Group III—culture-positive for other bacteria: 10 SF samples that were SF culture-positive for bacteria other than C. acnes were included in group III. The bacterial species identified from SF culture (Staphylococcus epidermidis, Staphylococcus aureus, F. magna, and C. avidum) and their corresponding tissue culture results are listed in Table 1. In eight out of 10 samples in this group, the bacterial genus determined by mNGS matched the bacterial genus detected by SF culture (Table 1). The two samples (group III #2 and #3) with discordant results had both growth of S. epidermidis on SF culture, albeit with a low CFU count of 20–30 CFU/ml. Interestingly, in four samples (group III #4, #6, #7, and #8), in addition to the bacteria detected by SF culture (3 × S. aureus; 1 × F. magna), sequence reads from C. acnes (395 to 2,449 reads (MG-RAST); 2.2% to 10.6% of bacterial reads) were detected. In two of these samples (group III #7 and #8), there were twice as many C. acnes reads detected than staphylococcal reads, according to the MG-RAST analysis (Table 1). Three of the samples (group III #4, #7, and #8) were negative by aNGS, while aNGS was not performed in the remaining sample (group III #6). This raised the question of how sensitive the aNGS approach is. We therefore tested the aNGS PCR with different amounts of genomic DNA of C. acnes. The PCR detection limit was determined to be a minimum amount of 0.1 pg C. acnes template DNA, which corresponded to ca. 37 genome copies (Supplementary Figure 1).
To test whether the obtained sequence reads by mNGS were originating from the entire bacterial genome or were potential artefacts, we mapped extracted sequence reads to reference genomes. Four samples (group I #4 and group II #5, #7, and #8) with C. acnes reads were selected and mapped against the C. acnes reference genome of strain KPA171202 (Figure 3A). The results showed that the sequence reads obtained by mNGS originated from the entire C. acnes genome and not only from certain conserved parts, such as rRNA genes. In one sample (group III #5), whose SF was culture-positive for C. avidum, mNGS detected 3,140 and 5,932 sequence reads assigned to C. acnes based on MG-RAST and Kraken2/Bracken analysis, respectively. To test if these reads originated from C. acnes or C. avidum, as culture results suggested, sequence reads were mapped against the genomes of C. avidum 44067 and C. acnes KPA171202 (Figure 3B). Results indicated that most reads originated from C. acnes and not from C. avidum. In another sample (group III #6), F. magna was identified by culture. F. magna is closely related to the tentative species “F. nericia” (Brüggemann et al., 2018). The 9,184 sequence reads assigned to Finegoldia sp. by MG-RAST were mapped against the genomes of F. magna ATCC29328 and “F. nericia” 09T494 (Figure 3C). Results showed that the detected DNA originated mainly, if not entirely, from “F. nericia”.
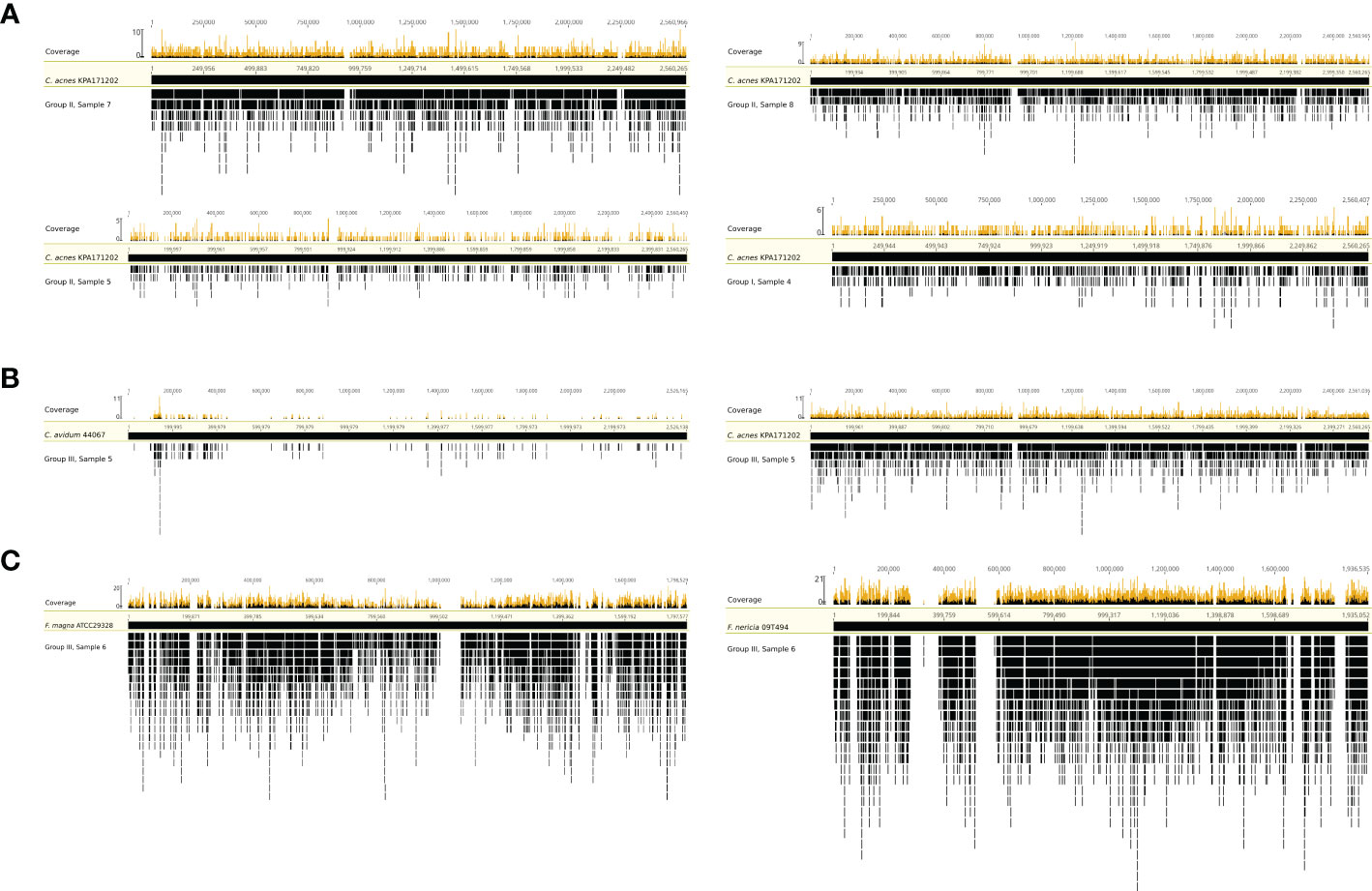
Figure 3 Mapping of sequence reads assigned to Cutibacterium sp. and/or Finegoldia sp. to their corresponding reference genomes. (A) Mapping of sequence reads assigned to Cutibacterium sp. from four samples (sample 4 from group I, samples 5,7 and 8 from group II) to the reference genome of C. acnes KPA171202. (B) Mapping of sequence reads assigned to Cutibacterium sp. from sample 5 in group III to the genome of C. avidum 44067 and to the genome of C. acnes KPA171202. (C) Mapping of sequence reads corresponding to Finegoldia sp. from sample 6 in group III to the genome of Finegoldia magna ATCC29328 and “Finegoldia nericia” 09T494. This analysis is based on MG-RAST assignment results.
4 Discussion
The significance of C. acnes isolated from removed orthopaedic implants and their surrounding tissues, including the possibility of overdiagnosis due to the interpretation of contamination or commensal colonization as infection, has often been debated (Hudek et al., 2014; Patel et al., 2020; Falstie-Jensen et al., 2021; Hudek et al., 2021). At the same time, the risk of underdiagnosing C. acnes OIAIs because of the slow-growing nature of the bacterium has also been discussed (Butler-Wu et al., 2011; Aubin et al., 2014; Bossard et al., 2016).
In this pilot study, a shotgun sequencing approach of SF was used that could potentially help to identify cases of C. acnes OIAIs that were not detected by culture. A total of 24 SF samples, based on their SF culture results, were included and divided into three groups. A large number of microbial sequence reads were obtained in all SF samples including the SF culture-negative samples. As pointed out in several recent studies, particular care needs to be taken when applying mNGS to low-biomass samples, as low-biomass samples are prone to contamination derived from DNA that could be present in liquids or reagents used, such as DNA extraction kits or reagents used for NGS library preparations (Weiss et al., 2014; Street et al., 2017; Thoendel et al., 2017; Thoendel et al., 2018; Eisenhofer et al., 2019). In our study, no DNA was detected by mNGS when a mock sample was sequenced, i.e., the saline used for sonication.
In all SF specimens, sequence reads that originated from gut bacteria were detected by mNGS (Table S2). The origin of these sequence reads in SF samples remains to be investigated. DNA of the gut microbiome in SF could potentially be due to gut permeability, which has been linked to OIAIs via the “gut–immune–joint axis” (Chisari et al., 2022), but this does not explain the presence of gut microbial DNA in culture-negative samples with no clinical indication of infection. Another possibility is that DNA of gut bacteria could be present in the normal joint microenvironment, as some studies suggested the presence of bacteria or DNA thereof in native joints (Torchia et al., 2020; Clarkson et al., 2022).
Regarding C. acnes in particular, several previous studies suggested that C. acnes or rather its DNA is a common contaminant in mNGS data, where it is assumed that C. acnes DNA is present in reagents needed for DNA isolation (e.g., DNA extraction kits) and sequence library preparation (Mollerup et al., 2016; Street et al., 2017). However, we did not detect C. acnes DNA in the control sample (saline). Moreover, C. acnes DNA was only detected in 14/24 samples and not in all samples, as would be expected if C. acnes DNA is a common contaminant. This speaks against a general contamination issue with C. acnes DNA in the used saline solution or DNA extraction kit or sequencing reagents. However, it cannot be excluded that C. acnes could have contaminated the implant in the process of surgery/implant removal in some but not all cases. Such surgery-related contamination is very difficult to prove or disprove. It has previously been reported that carriage of C. acnes on human skin can vary substantially from patient to patient and from body site to body site, which can lead to C. acnes wound contamination in some but not all patients (McLorinan et al., 2005; Patrick et al., 2017; Patel et al., 2020; Shroff et al., 2023). For example, the shoulder site is usually more heavily colonized with C. acnes, especially in male patients (Patel et al., 2020; Hudek et al., 2021; Shroff et al., 2023). A potential possibility to reduce wound contamination could be to wash the removed implants before sonication; this was not performed in this study due to logistic reasons. Here, the implants were placed in sterile, airtight, single-use plastic containers in the operating room and covered with sterile saline before being transported to the lab (Ponraj et al., 2022).
In the culture-negative group, sequence reads from C. acnes were obtained in only one sample that was both SF and tissue culture-negative. Clinically, the respective implant was defined as infected due to the presence of a sinus tract. Thus, it could potentially represent a C. acnes infection that was missed by culture. Alternatively, C. acnes or its DNA contaminated the sample at the DNA extraction step or later. Both explanations are possible, as the risk of contamination in low-biomass mNGS studies (as outlined above) as well as the ability of mNGS to identify organisms not detected by culture has been described previously in multiple studies (Street et al., 2017; Ivy et al., 2018; Sanderson et al., 2018; Weaver et al., 2019; Cai et al., 2020; Wang et al., 2020; He et al., 2021; Noone et al., 2021; Street et al., 2022; Tan et al., 2022).
Of the 10 samples in group II that had growth of C. acnes in SF culture, reads from C. acnes were obtained in seven samples. C. acnes isolates from two of the remaining three samples (#1 and #2) had been classified as likely contamination in our previous study (Ponraj et al., 2022), in which a combined analysis of genome sequencing and SLST typing of C. acnes isolates, aNGS of SF, tissue culture results, and pre-operative clinical diagnosis was used to determine the significance of C. acnes detection.
In four samples from group III that included SF culture-positive samples for bacteria other than C. acnes, mNGS detected C. acnes reads in addition to the reads from the bacteria also detected by SF culture, i.e., S. aureus in three cases and F. magna in one case. This could either represent C. acnes DNA contamination during mNGS or be a polymicrobial infection that was not detected in culture because the slow-growing C. acnes was overgrown or inhibited by the other faster-growing organism, i.e., S. aureus and F. magna. Interestingly, in two samples, there were twice as many Cutibacterium sp. reads detected than S. aureus reads (using the MG-RAST pipeline), albeit only S. aureus was identified by SF culture. This suggests that a polymicrobial infection could be present, which was missed by culture since S. aureus grows rapidly and possibly inhibits the growth of C. acnes.
It is challenging to determine and compare the detection limit of the two methods, mNGS and aNGS. Here, we determined in a mock setting that the aNGS method has a detection limit of ca. 37 C. acnes genome copies. However, in real samples, such as SF specimens, which potentially contain much DNA of human origin that could interfere with the C. acnes SLST PCR, the number of C. acnes genome copies needed to obtain a positive PCR result will likely be higher. Regarding the mNGS method, the detection limit has not been experimentally determined so far; it will depend on many internal and external factors, such as the sequencing specificities (platform, materials, sequencing depths, and bioinformatics pipeline) as well as the relative abundance of C. acnes DNA in a given sample. Our data here suggested that mNGS is more sensitive than aNGS since four out of 10 aNGS-negative samples had actually C. acnes reads above the read count threshold. Like in other mNGS projects, a read count threshold was set (150 reads, corresponding to approx. 0.1% of the total amount of bacterial reads per sample) to eliminate potential false positives (Street et al., 2017). Some samples had read counts below the threshold, e.g., MG-RAST and Kraken2/Bracken detected one and four samples with low C. acnes read count (on average 51 reads) and four and 13 samples with low S. epidermidis read count (on average 37 reads). This might represent NGS-derived contamination, as low counts of S. epidermidis and C. acnes reads were previously reported in negative controls used in shotgun sequencing projects (Sanabria et al., 2020).
This pilot study has several limitations. The number of samples is limited due to the exploratory nature of the study. Optimal read count thresholds to filter out potential contaminant reads were not established in the study due to the lack of mock community mNGS data (with defined C. acnes genome copy numbers) and also hampered by the lack of detecting bacterial reads in negative control samples. Tissue sample culture and aNGS were not performed for all samples included in the study, and finally, due to lack of clinical follow-up, the significance of C. acnes reads detected by mNGS, especially in groups I and III, could not be determined. Further studies with the inclusion of clinical follow-up are needed before mNGS can be recommended for the diagnosis of OIAIs due to C. acnes.
5 Conclusions
The current study shows that C. acnes DNA was detected in more samples by mNGS compared to the culture of SF. Additional identification of C. acnes was seen in one culture-negative SF sample and in four SF samples that were culture-positive for other bacteria. Thus, mNGS could possibly be used to identify cases of potential C. acnes OIAIs, especially in polymicrobial infections that might be overlooked or misinterpreted by culture-based detection alone.
Data availability statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found below: SRA with the accession number PRJNA940664.
Ethics statement
The studies involving human participants were reviewed and approved by Ethical Committee Region Midtjylland, Denmark. The patients/participants provided their written informed consent to participate in this study.
Author contributions
DP, JL, TF-J, NJ, CR, and HB contributed to the conception and design of the study. DP performed wet lab benchwork. DP analysed the data. ML, AP, AH, and HB contributed to sequence data generation and data analyses. DP and HB wrote the manuscript. All authors contributed to the article and approved the submitted version.
Funding
This research was supported with funds from the A. P. Møller Foundation (no. 30903) for running costs and the “Fabrikant Vilhelm Pedersen og Hustrus Legat” (by the recommendation from the Novo Nordisk Foundation) for running costs and personnel (no. 30658).
Acknowledgments
The authors would like to thank Kristine Schweinshaut and Lise Hald Schultz for their excellent technical assistance.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fcimb.2023.1165017/full#supplementary-material
Supplementary Figure 1 | SLST-PCR with different concentrations of C. acnes genomic DNA as template. C. acnes strain 266 was used. The lanes: 1, DNA ladder (EasyLadder I); 2, 1000 pg; 3, 100 pg; 4, 10 pg; 5, 1 pg; 6, 0.1 pg; 7, 0.01 pg; 8, 0.001 pg; 9, negative control; 10, positive control (1 ng C. acnes DNA).
References
Achermann, Y., Goldstein, E. J., Coenye, T., Shirtliff, M. E. (2014). Propionibacterium acnes: from commensal to opportunistic biofilm-associated implant pathogen. Clin. Microbiol. Rev. 27, 419–440. doi: 10.1128/CMR.00092-13
Aubin, G. G., Portillo, M. E., Trampuz, A., Corvec, S. (2014). Propionibacterium acnes, an emerging pathogen: from acne to implant-infections, from phylotype to resistance. Med. Mal. Infect. 44, 241–250. doi: 10.1016/j.medmal.2014.02.004
Borens, O., Yusuf, E., Steinrücken, J., Trampuz, A. (2013). Accurate and early diagnosis of orthopedic device-related infection by microbial heat production and sonication. J. Orthop. Res. 31, 1700–1703. doi: 10.1002/jor.22419
Bossard, D. A., Ledergerber, B., Zingg, P. O., Gerber, C., Zinkernagel, A. S., Zbinden, R., et al. (2016). Optimal length of cultivation time for isolation of Propionibacterium acnes in suspected bone and joint infections is more than 7 days. J. Clin. Microbiol. 54, 3043–3049. doi: 10.1128/JCM.01435-16
Brüggemann, H., Jensen, A., Nazipi, S., Aslan, H., Meyer, R. L., Poehlein, A., et al. (2018). Pan-genome analysis of the genus Finegoldia identifies two distinct clades, strain-specific heterogeneity, and putative virulence factors. Sci. Rep. 8, 266. doi: 10.1038/s41598-017-18661-8
Butler-Wu, S. M., Burns, E. M., Pottinger, P. S., Magaret, A. S., Rakeman, J. L., Matsen, F. A., et al. (2011). Optimization of periprosthetic culture for diagnosis of Propionibacterium acnes prosthetic joint infection. J. Clin. Microbiol. 49, 2490–2495. doi: 10.1128/JCM.00450-11
Cai, Y., Fang, X., Chen, Y., Huang, Z., Zhang, C., Li, W., et al. (2020). Metagenomic next generation sequencing improves diagnosis of prosthetic joint infection by detecting the presence of bacteria in periprosthetic tissues. Int. J. Infect. Dis. 96, 573–578. doi: 10.1016/j.ijid.2020.05.125
Chisari, E., Cho, J., Wouthuyzen-Bakker, M., Parvizi, J. (2022). Gut permeability may be associated with periprosthetic joint infection after total hip and knee arthroplasty. Sci. Rep. 12, 15094. doi: 10.1038/s41598-022-19034-6
Clarkson, S. J., Goswami, K., Parvizi, J. (2022). “The microbiome of the joint,” in Essentials of cemented knee arthroplasty. Eds. Hansen, E., Kühn, K.-D. (Berlin, Heidelberg: Springer Berlin Heidelberg), 101–107.
Eisenhofer, R., Minich, J. J., Marotz, C., Cooper, A., Knight, R., Weyrich, L. S. (2019). Contamination in low microbial biomass microbiome studies: issues and recommendations. Trends Microbiol. 27, 105–117. doi: 10.1016/j.tim.2018.11.003
Falstie-Jensen, T., Lange, J., Daugaard, H., Sørensen, A. K. B., Ovesen, J., Søballe, K. (2021). Unexpected positive cultures after revision shoulder arthroplasty: does it affect outcome? J. Shoulder Elbow Surg. 30, 1299–1308. doi: 10.1016/j.jse.2020.12.014
Fang, X., Cai, Y., Shi, T., Huang, Z., Zhang, C., Li, W., et al. (2020). Detecting the presence of bacteria in low-volume preoperative aspirated synovial fluid by metagenomic next-generation sequencing. Int. J. Infect. Dis. 99, 108–116. doi: 10.1016/j.ijid.2020.07.039
Gu, W., Miller, S., Chiu, C. Y. (2019). Clinical metagenomic next-generation sequencing for pathogen detection. Annu. Rev. Pathol. 14, 319–338. doi: 10.1146/annurev-pathmechdis-012418-012751
He, R., Wang, Q., Wang, J., Tang, J., Shen, H., Zhang, X. (2021). Better choice of the type of specimen used for untargeted metagenomic sequencing in the diagnosis of periprosthetic joint infections. Bone Joint J. 103-B, 923–930. doi: 10.1302/0301-620X.103B5.BJJ-2020-0745.R1
Huang, Z., Li, W., Lee, G. C., Fang, X., Xing, L., Yang, B., et al. (2020). Metagenomic next-generation sequencing of synovial fluid demonstrates high accuracy in prosthetic joint infection diagnostics: mNGS for diagnosing PJI. Bone Joint Res. 9, 440–449. doi: 10.1302/2046-3758.97.BJR-2019-0325.R2
Hudek, R., Brobeil, A., Brüggemann, H., Sommer, F., Gattenlöhner, S., Gohlke, F. (2021). Cutibacterium acnes is an intracellular and intra-articular commensal of the human shoulder joint. J. Shoulder Elbow Surg. 30, 16–26. doi: 10.1016/j.jse.2020.04.020
Hudek, R., Sommer, F., Kerwat, M., Abdelkawi, A. F., Loos, F., Gohlke, F. (2014). Propionibacterium acnes in shoulder surgery: true infection, contamination, or commensal of the deep tissue? J. Shoulder Elbow Surg. 23, 1763–1771. doi: 10.1016/j.jse.2014.05.024
Ivy, M. I., Thoendel, M. J., Jeraldo, P. R., Greenwood-Quaintance, K. E., Hanssen, A. D., Abdel, M. P., et al. (2018). Direct detection and identification of prosthetic joint infection pathogens in synovial fluid by metagenomic shotgun sequencing. J. Clin. Microbiol. 56, e00402–e00418. doi: 10.1128/JCM.00402-18
Kildow, B. J., Ryan, S. P., Danilkowicz, R., Lazarides, A. L., Penrose, C., Bolognesi, M. P., et al. (2021). Next-generation sequencing not superior to culture in periprosthetic joint infection diagnosis. Bone Joint J. 103-B, 26–31. doi: 10.1302/0301-620X.103B1.BJJ-2020-0017.R3
Langmead, B., Salzberg, S. L. (2012). Fast gapped-read alignment with bowtie 2. Nat. Methods 9, 357–359. doi: 10.1038/nmeth.1923
Li, H., Handsaker, B., Wysoker, A., Fennell, T., Ruan, J., Homer, N., et al. (2009). The sequence Alignment/Map format and SAMtools. Bioinformatics 25, 2078–2079. doi: 10.1093/bioinformatics/btp352
Lu, J., Breitwieser, F. P., Thielen, P., Salzberg, S. L. (2017). Bracken: estimating species abundance in metagenomics data. PeerJ Comput. Sci. 3, e104. doi: 10.7717/peerj-cs.104
McDowell, A., Nagy, I., Magyari, M., Barnard, E., Patrick, S. (2013). The opportunistic pathogen Propionibacterium acnes: insights into typing, human disease, clonal diversification and CAMP factor evolution. PloS One 8, e70897. doi: 10.1371/journal.pone.0070897
McLorinan, G. C., Glenn, J. V., McMullan, M. G., Patrick, S. (2005). Propionibacterium acnes wound contamination at the time of spinal surgery. Clin. Orthop. Relat. Res. 437, 67–73. doi: 10.1097/00003086-200508000-00012
Meyer, F., Bagchi, S., Chaterji, S., Gerlach, W., Grama, A., Harrison, T., et al. (2019). MG-RAST version 4-lessons learned from a decade of low-budget ultra-high-throughput metagenome analysis. Brief. Bioinform. 20, 1151–1159. doi: 10.1093/bib/bbx105
Mollerup, S., Friis-Nielsen, J., Vinner, L., Hansen, T. A., Richter, S. R., Fridholm, H., et al. (2016). Propionibacterium acnes: disease-causing agent or common contaminant? detection in diverse patient samples by next-generation sequencing. J. Clin. Microbiol. 54, 980–987. doi: 10.1128/JCM.02723-15
Noone, J. C., Helmersen, K., Leegaard, T. M., Skråmm, I., Aamot, H. V. (2021). Rapid diagnostics of orthopaedic-Implant-Associated infections using nanopore shotgun metagenomic sequencing on tissue biopsies. Microorganisms 9, 97. doi: 10.3390/microorganisms9010097
Patel, M. S., Singh, A. M., Gregori, P., Horneff, J. G., Namdari, S., Lazarus, M. D. (2020). Cutibacterium acnes: a threat to shoulder surgery or an orthopedic red herring? J. Shoulder Elbow Surg. 29, 1920–1927. doi: 10.1016/j.jse.2020.02.020
Patrick, S., McDowell, A., Lee, A., Frau, A., Martin, U., Gardner, E., et al. (2017). Antisepsis of the skin before spinal surgery with povidone iodine-alcohol followed by chlorhexidine gluconate-alcohol versus povidone iodine-alcohol applied twice for the prevention of contamination of the wound by bacteria: a randomised controlled trial. Bone Joint J. 99-B, 1354–1365. doi: 10.1302/0301-620X.99B10.BJJ-2017-0291.R1
Ponraj, D. S., Falstie-Jensen, T., Jørgensen, N. P., Ravn, C., Brüggemann, H., Lange, J. (2021). Diagnosis of orthopaedic-implant-associated infections caused by slow-growing gram-positive anaerobic bacteria – a clinical perspective. J. Bone Joint Infect. 6, 367–378. doi: 10.5194/jbji-6-367-2021
Ponraj, D. S., Lange, J., Falstie-Jensen, T., Jørgensen, N. P., Ravn, C., Poehlein, A., et al. (2022). Amplicon-based next-generation sequencing as a diagnostic tool for the detection of phylotypes of Cutibacterium acnes in orthopedic implant-associated infections. Front. Microbiol. 13, 866893. doi: 10.3389/fmicb.2022.866893
Sanabria, A., Hjerde, E., Johannessen, M., Sollid, J. E., Simonsen, G. S., Hanssen, A. M. (2020). Shotgun-metagenomics on positive blood culture bottles inoculated with prosthetic joint tissue: a proof of concept study. Front. Microbiol. 11, 1687. doi: 10.3389/fmicb.2020.01687
Sanderson, N. D., Street, T. L., Foster, D., Swann, J., Atkins, B. L., Brent, A. J., et al. (2018). Real-time analysis of nanopore-based metagenomic sequencing from infected orthopaedic devices. BMC Genomics 19, 714. doi: 10.1186/s12864-018-5094-y
Shroff, J. B., Hanna, P., Levy, B. J., Jimenez, A. E., Grimm, N. L., Cote, M. P., et al. (2023). Is there value in the routine practice of discarding the incision scalpel from the surgical field to prevent deep wound contamination with cutibacterium acnes? an update. J. Shoulder Elbow Surg., S1058–S2746. doi: 10.1016/j.jse.2023.02.130
Street, T. L., Sanderson, N. D., Atkins, B. L., Brent, A. J., Cole, K., Foster, D., et al. (2017). Molecular diagnosis of orthopedic-Device-Related infection directly from sonication fluid by metagenomic sequencing. J. Clin. Microbiol. 55, 2334–2347. doi: 10.1128/JCM.00462-17
Street, T. L., Sanderson, N. D., Kolenda, C., Kavanagh, J., Pickford, H., Hoosdally, S., et al. (2022). Clinical metagenomic sequencing for species identification and antimicrobial resistance prediction in orthopedic device infection. J. Clin. Microbiol. 60, e0215621. doi: 10.1128/jcm.02156-21
Tan, J., Liu, Y., Ehnert, S., Nüssler, A. K., Yu, Y., Xu, J., et al. (2022). The effectiveness of metagenomic next-generation sequencing in the diagnosis of prosthetic joint infection: a systematic review and meta-analysis. Front. Cell. Infect. Microbiol. 12, 875822. doi: 10.3389/fcimb.2022.875822
Tande, A. J., Patel, R. (2014). Prosthetic joint infection. Clin. Microbiol. Rev. 27, 302–345. doi: 10.1128/CMR.00111-13
Thoendel, M., Jeraldo, P., Greenwood-Quaintance, K. E., Yao, J., Chia, N., Hanssen, A. D., et al. (2017). Impact of contaminating DNA in whole-genome amplification kits used for metagenomic shotgun sequencing for infection diagnosis. J. Clin. Microbiol. 55, 1789–1801. doi: 10.1128/JCM.02402-16
Thoendel, M. J., Jeraldo, P. R., Greenwood-Quaintance, K. E., Yao, J. Z., Chia, N., Hanssen, A. D., et al. (2018). Identification of prosthetic joint infection pathogens using a shotgun metagenomics approach. Clin. Infect. Dis. 67, 1333–1338. doi: 10.1093/cid/ciy303
Torchia, M. T., Amakiri, I., Werth, P., Moschetti, W. (2020). Characterization of native knee microorganisms using next-generation sequencing in patients undergoing primary total knee arthroplasty. Knee 27, 1113–1119. doi: 10.1016/j.knee.2019.12.013
Wang, C., Huang, Z., Li, W., Fang, X., Zhang, W. (2020). Can metagenomic next-generation sequencing identify the pathogens responsible for culture-negative prosthetic joint infection? BMC Infect. Dis. 20, 253. doi: 10.1186/s12879-020-04955-2
Weaver, A. A., Hasan, N. A., Klaassen, M., Karathia, H., Colwell, R. R., Shrout, J. D. (2019). Prosthetic joint infections present diverse and unique microbial communities using combined whole-genome shotgun sequencing and culturing methods. J. Med. Microbiol. 68, 1507–1516. doi: 10.1099/jmm.0.001068
Weiss, S., Amir, A., Hyde, E. R., Metcalf, J. L., Song, S. J., Knight, R. (2014). Tracking down the sources of experimental contamination in microbiome studies. Genome Biol. 15, 564. doi: 10.1186/s13059-014-0564-2
Wood, D. E., Lu, J., Langmead, B. (2019). Improved metagenomic analysis with kraken 2. Genome Biol. 20, 257. doi: 10.1186/s13059-019-1891-0
Keywords: Cutibacterium acnes, shotgun sequencing, metagenomics, orthopaedic implant-associated infections, prosthetic infections, sonication fluid
Citation: Ponraj DS, Lund M, Lange J, Poehlein A, Himmelbach A, Falstie-Jensen T, Jørgensen NP, Ravn C and Brüggemann H (2023) Shotgun sequencing of sonication fluid for the diagnosis of orthopaedic implant-associated infections with Cutibacterium acnes as suspected causative agent. Front. Cell. Infect. Microbiol. 13:1165017. doi: 10.3389/fcimb.2023.1165017
Received: 13 February 2023; Accepted: 02 May 2023;
Published: 17 May 2023.
Edited by:
Michael Marceau, Université Lille Nord de France, FranceReviewed by:
Andrew McDowell, Ulster University, United KingdomLlanos Salar Vidal, University Hospital Fundación Jiménez Díaz, Spain
Copyright © 2023 Ponraj, Lund, Lange, Poehlein, Himmelbach, Falstie-Jensen, Jørgensen, Ravn and Brüggemann. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Holger Brüggemann, YnJ1ZWdnZW1hbm5AYmlvbWVkLmF1LmRr