
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Cell. Infect. Microbiol., 14 April 2023
Sec. Parasite and Host
Volume 13 - 2023 | https://doi.org/10.3389/fcimb.2023.1159814
 Miriam Rodi1†
Miriam Rodi1† Caspar Gross2†
Caspar Gross2† Thaisa Lucas Sandri1,3
Thaisa Lucas Sandri1,3 Lilith Berner1Marina Marcet-Houben4,5
Lilith Berner1Marina Marcet-Houben4,5 Ersoy Kocak6
Ersoy Kocak6 Michaela Pogoda2,6
Michaela Pogoda2,6 Nicolas Casadei2,6Carsten Köhler1,7
Nicolas Casadei2,6Carsten Köhler1,7 Andrea Kreidenweiss1,7,8Selidji Todagbe Agnandji1,8
Andrea Kreidenweiss1,7,8Selidji Todagbe Agnandji1,8 Toni Gabaldón4,5,9,10
Toni Gabaldón4,5,9,10 Stephan Ossowski2‡
Stephan Ossowski2‡ Jana Held1,7,8*‡
Jana Held1,7,8*‡Introduction: Mansonella species are filarial parasites that infect humans worldwide. Although these infections are common, knowledge of the pathology and diversity of the causative species is limited. Furthermore, the lack of sequencing data for Mansonella species, shows that their research is neglected. Apart from Mansonella perstans, a potential new species called Mansonella sp “DEUX” has been identified in Gabon, which is prevalent at high frequencies. We aimed to further determine if Mansonella sp “DEUX” is a genotype of M. perstans, or if these are two sympatric species.
Methods: We screened individuals in the area of Fougamou, Gabon for Mansonella mono-infections and generated de novo assemblies from the respective samples. For evolutionary analysis, a phylogenetic tree was reconstructed, and the differences and divergence times are presented. In addition, mitogenomes were generated and phylogenies based on 12S rDNA and cox1 were created.
Results: We successfully generated whole genomes for M. perstans and Mansonella sp “DEUX”. Phylogenetic analysis based on annotated protein sequences, support the hypothesis of two distinct species. The inferred evolutionary analysis suggested, that M. perstans and Mansonella sp “DEUX” separated around 778,000 years ago. Analysis based on mitochondrial marker genes support our hypothesis of two sympatric human Mansonella species.
Discussion: The results presented indicate that Mansonella sp “DEUX” is a new Mansonella species. These findings reflect the neglect of this research topic. And the availability of whole genome data will allow further investigations of these species
Infections with nematodes of the genus Mansonella are among the most neglected diseases, even though these parasites belong to the most widespread human filaria species. It has been estimated that more than 100 million people are infected with Mansonella species in Africa only and around 600 million people are at risk of infection (Simonsen et al., 2011). Three species are known to be prevalent in humans, Mansonella perstans, M. ozzardi, and M. streptocerca with varying geographic distribution. While M. perstans is prevalent in most parts of Sub-Saharan Africa and some regions in South and Central America, M. streptocerca is restricted to the African continent and M. ozzardi to Latin America. In 2015 a potential new species provisionally called Mansonella sp “DEUX” was described in febrile children in a hospital-based study in Gabon (Mourembou et al., 2015). Further investigations showed that this was the most frequent filarial species, as during a cross-sectional study in rural Fougamou, Gabon, 35% of individuals carried this parasite (Sandri et al., 2021).
In addition to the nuclear and the mitochondrial genomes, most filarial species comprise an additional genome - the genome of their bacterial endosymbiont Wolbachia. These intracellular bacteria can be found in most filarial parasites (Ferri et al., 2011). For M. perstans the obligate presence of Wolbachia was under debate for a long time (Grobusch et al., 2003; Gehringer et al., 2014; Batsa Debrah et al., 2019). However, improved molecular diagnostics, as well as results from treatment studies demonstrating the efficacy of antibiotics, indicate that M. perstans contains these bacteria (Keiser et al., 2008; Coulibaly et al., 2009; Gehringer et al., 2014; Batsa Debrah et al., 2019). Wolbachia were also detected in individuals mono infected with Mansonella sp “DEUX”, confirming carriage of endosymbionts also by this new species/genotype (Sandri et al., 2021). As Wolbachia are usually transferred maternally and because of the known genetic variability of this endosymbiont among nematodes, Wolbachia genomes could give a further hint on the genetic relatedness of their host species (Bordenstein et al., 2009).
Until recently, no whole genome for any Mansonella species infecting humans was available, reflecting the neglect of these infections. Fortunately, this has been recognized and sequences of M. perstans and M. ozzardi have been generated in the last years (Crainey et al., 2018; Chung et al., 2020; Sinha et al., 2023). A recently published study from Tamarozzi et al. has confirmed the lack of consensus in the management of Mansonella infections (Tamarozzi et al., 2022). There are no clear treatment guidelines and also information on specific symptoms and burden of disease is not well described. To contribute to a better evolutionary understanding of Mansonella species, we generated and compared the whole genomes as well as the mitogenomes of M. perstans and Mansonella sp “DEUX” isolated from circulating microfilariae from infected individuals. We aimed to assess the differences between the two sympatric variants to identify if they are two separate species. In addition, we also obtained and analyzed the genomes of the Wolbachia symbionts.
The study protocol was approved by the Institutional Ethics Committee at the Centre de Recherches Médicales de Lambaréné, Gabon (CEI-CERMEL 002/2018). Adolescents were asked to give written assent, written informed consent was obtained from their legal guardian and of all adult individuals taking part in this study.
Individuals ≥ 16 years living in the area of Fougamou, Gabon, with a confirmed Mansonella species microfilaria count ≥ 50 parasites/ml and no microscopical detectable Loa loa infection were invited to take part in the study. Diagnosis was based on the microscopic reading of two Giemsa-stained thick blood smears (2x 10 µl blood). 18 ml of venous blood was collected in EDTA tubes from participating individuals. The sample was diluted 1:2 with phosphate buffered saline (PBS; Merck) and filtered through a 5 µm filter paper (Whatman) with a syringe to enrich the microfilariae. Filtration was repeated with 30 ml of PBS to remove remaining blood cells. Subsequently the Whatman filter was removed, the sticking microfilariae were washed off with 10 ml PBS, and the mixture was centrifuged (10 minutes, 1800 rpm) to remove the supernatant. The pellet was transferred to a cryotube with RNAlater and frozen at -80°C.
DNA was extracted from a 25 µl pellet using NEB Monarch Genomic Purification Kit (NEB). Sample lysis was performed according to the animal tissue protocol provided by the manufacturer. Incubation was increased to 3 h at 56°C to improve the yield. DNA was eluted with 50 µl elution buffer. Loa loa infection as well as coinfection with either M. perstans or Mansonella sp “DEUX” based on the internal transcribed spacer region (ITS1 region) was excluded by qPCR according to previously published protocols (Sandri et al., 2021). These procedures resulted in three eligible samples, named mperst1, mdeux2 and mdeux3.
Genomic DNA was quantified using Qubit dsDNA HS assay kit with a Qubit fluorometer (ThermoFisher). DNA library preparation was performed using the Nextera DNA Flex kit (Illumina) following the manufacturers instruction. Briefly, 4 to 20 ng of genomic DNA was diluted in a total volume of 30 µl of nuclease-free water and fragmented at 55°C for 15 minutes using tagmentation. The enzymatic reaction was stopped, and the adapter-tagged DNA was purified using magnetic-bead clean-up. The resulting DNA was amplified using 8 to 12 cycles of PCR, allowing the required sequencing adapters to anneal to the DNA. The resulting libraries were purified using a double-sided beads purification. Library molarity was determined by measuring the library size (approximately 600 bp) using the Fragment Analyzer with the High NGS Fragment 1-6000bp assay (Agilent) and the library concentration (approximately 10 ng/µl) using the Infinite 200Pro (Tecan) and the Quant-iT HS Assay Kit (Thermo Fisher Scientific). The libraries were denaturated, diluted to 162 pM and sequenced as paired-end 250 bp reads on an Illumina NovaSeq6000 (Illumina, San Diego, CA, USA) with a sequencing depth of approximately 200 million clusters per sample.
The first step in the de novo assembly of the Mansonella species genomes was to remove the reads from the human host from all generated reads. Therefore, Kraken2 (Wood et al., 2019) was used to classify the reads taxonomically since unmapped reads may also indicate the presence of Wolbachia sequences. The reads reported as unclassified by Kraken were assumed to be Mansonella species reads and used for the de novo assembly. Information about the pre-built Kraken database used can be found in the Supplementary File 1. The assembly was performed using the SPAdes De Novo Assembler (Bankevich et al., 2012) without error correction, using the default parameters. The quality of the scaffolds assembled by SPAdes were assessed using the genome assembly evaluation tool QUAST (Gurevich et al., 2013) and the genome completeness was assessed using BUSCO (Simao et al., 2015). Also, the N50 values and coverage values of the scaffolds were calculated in this way. BLAST (Altschul et al., 1990) was used to investigate the similarity of the potential Mansonella species scaffolds to closely related species. If at least one of the 39 listed species (Supplementary Table 1) occurred in the top 10 BLAST hits of a scaffold, the scaffold was counted as a fragment of the Mansonella species genome. Estimated genome length was calculated by summing the lengths of these filtered scaffolds. Intron hints for the gene annotation were generated using the protein sequences of B. malayi with the freely available sequence alignment program, Exonerate (Slater and Birney, 2005). Subsequently, AUGUSTUS (Stanke and Morgenstern, 2005) gene prediction tool was used to predict the genes on the scaffolds using the hints and to annotate the predicted genes Supplementary File 1. The predicted genes were aligned to the NCBI protein sequence database using DIAMOND (Buchfink et al., 2021) to find similar genes.
The evolutionary analysis was performed using genome assemblies from 10 nematodes, that were downloaded from NCBI and Uniprot (Supplementary Table 2), in addition to mperst1 and mdeux3. Genome completeness was assessed using BUSCO v4.0.2 (Supplementary Table 2) (Simao et al., 2015). The phylome of Mansonella sp “DEUX” was reconstructed from mdeux3 using a set of 11 additional Nematode proteomes, including mperst1. The genomes of Brugia timori and Onchocerca flexuosa were not used in the phylome reconstruction due to their poor completeness, as well as mdeux2. An automated pipeline was used that applies the same process to reconstruct gene trees as one would do manually (Fuentes et al., 2022). First the proteome database was reconstructed using the 12 species and formatting the codes to phylomeDB format. Then a blastp search was performed between each gene in the genome of Mansonella sp “DEUX” and this proteome database. Blast results were filtered using an e-value threshold of 1e-05 and an overlap threshold of 50%. The number of hits was limited to the 150 best hits for each protein. Next, six different multiple sequence alignments were reconstructed using three programs [Muscle v3.8.1551 (Edgar, 2004), mafft v7.407 (Katoh et al., 2005) and kalign v2.04 (Lassmann and Sonnhammer, 2005)] and aligning the sequences in forward and in reverse direction. From this group of alignments, a consensus alignment was obtained using M-coffee from the T-coffee package v12.0 (Wallace et al., 2006). Alignments were then trimmed using trimAl v1.4.rev15 (consistency-score cut-off 0.1667, gap-score cut-off 0.9) (Capella-Gutierrez et al., 2009). IQTREE v1.6.9 (Nguyen et al., 2015) was used to reconstruct a maximum likelihood phylogenetic tree. Model selection was limited to 5 models (DCmut, JTTDCMut, LG, WAG, VT) with freerate categories set to vary between 4 and 10. The best model according to the BIC criterion was used. 1000 rapid bootstraps were calculated. All trees and alignments were stored in phylomedb (Fuentes et al., 2022) with phylomeID 97 (http://phylomedb.org).
A species tree was reconstructed using a supertree approach as implemented in duptree v1.48 (Wehe et al., 2008) using all the trees reconstructed in the phylome as input. A second species tree was reconstructed using a gene concatenation approach, adding the three genome assemblies from the species that were not initially included in the phylome reconstruction process (B. timori and O. flexuosa, as well as the second Mansonella sp “DEUX” sample mdeux2). In order to do that, genes in the phylome were selected that were found in single copy in all species. 1068 such genes were selected, and a blast search was performed between these genes and the proteomes of the three newly added assemblies. Only those genes were kept that had only one hit in each of the three new assemblies, reducing the set to 562 genes. The alignment was redone for each selected protein family, adding the three new sequences, and using the same pipeline applied during phylome reconstruction. These alignments were subsequently concatenated into a single multiple sequence alignment which was trimmed using trimAl v1.4.rev15 (Capella-Gutierrez et al., 2009), producing an alignment of 260.126 amino acid positions. IQTREE v1.6.9 (Nguyen et al., 2015) was used to reconstruct the species tree using the model selection option without restrictions. The best model according to BIC was JTT+F+R4.
Nucleotide divergence was calculated by performing a best bidirectional hit analysis between all pairs of Brugia, Onchocerca and Mansonella species. For pairs of orthologs, the percent of identity was calculated by aligning 100 random pairs of nucleotide sequences using Muscle (Edgar, 2004) and using trimAl (Capella-Gutierrez et al., 2009) to calculate the pairwise identity without considering gaps.
Fossil records for Nematodes were obtained from Paleobiology Database (https://paleobiodb.org/). Four fossils fell within the species tree used in this paper: Cascofilaria baltica (33.9 - 38.0 MyA) and Cascofilaria dominicana (13.82 - 20.44 MyA) which belong to the Filariidae family, as well as Ascarites gerus (129.4 - 122.46 MyA) and Ascarites rufferi (237 - 242 MyA), which belong to the Ascarididae family. SortaDate (Smith et al., 2018) was used to select the 20 best genes to use for dating based on their clock-likeness, tree length and topological similarity to the species tree. Trees were used from the phylome, despite having three genome assemblies not included. Alignments were then concatenated, a species tree was reconstructed as detailed above, and phylobayes was used to calculate divergence times using the two calibration points based on the fossil records. The rooting point was set to have happened at least 242 MyA without detailing an upper limit. The second calibration point was set at the base of Filarioidea and was set to have happened between 13 and 38 MyA. Phylobayes 4.1 (Lartillot et al., 2009) was run on the species tree and with those calibration points using two chains, the CAT model and the Birth and Death model. It was run until both chains converged as seen with tracecomp from the Phylobayes package. Readdiv was run to obtain the chronogram using a burn-in of 10.000 and a subsampling frequency of 10.
The mitogenomes were extracted from the whole genome assembly of the individual samples. Using the visual tool Bandage (Wick et al., 2015) the M. perstans mitogenome was blasted against the assembly graph to identify contigs with mitochondrial genomic sequences. In case of multiple matching contigs these were linked based on coverage and sequence similarity to extract one or more complete mitogenomes from the assembly.
The analysis follows the companion repository from Chung et al. (2020). First, a multiple sequence alignment including the mitogenomes of mperst1, mdeux2 and mdeux3 as well as Mansonella related species from NCBI (Supplementary Table 3) was created using CLUSTAL OMEGA v.1.2.3. Then the phylogeny was calculated using IQTREE v2.1.4 (Nguyen et al., 2015) and visualized using iTOL (Letunic and Bork, 2021). For the 12S rDNA phylogenies, rRNA sequences were extracted from the genome assemblies using BARRNAP v0.9 Seemann, T (2013) and then combined with published 12S rDNA sequences from other Mansonella species. 12S rDNA and cox1 phylogenies were created using the same methods described above for the metagenome data.
Genome assemblies were generated from Mansonella microfilaria isolated from three individuals living in the area of Fougamou, Gabon. They presented with a qPCR-confirmed mono-infections of either M. perstans (individual 1: mperst1) or Mansonella sp “DEUX” (individual 2: mdeux2 and individual 3: mdeux3) (Table 1). Reads for all three samples were obtained with a coverage of 7.4x (mperst1), 32.4x (mdeux2), and 65.5x (mdeux3), resulting in a total assembly size of 76.2 Mb, 80 Mb and 78.5 Mb, respectively (Table 2 and Supplementary Table 4). Although the generated whole genome sizes are comparable, the N50 from mdeux3 is twice and three times the size of the other two assemblies respectively. This makes mdeux3 the most contiguous assembly generated thus far. To further verify the quality of the assemblies, the completeness of the data was quantified in terms of the expected gene content, using BUSCO (Benchmarking Single-Copy Orthologs) tool. For all three samples, more than 90% of conserved eukaryotic gene orthologs were identified. GC content as well as the number of predicted genes are comparable among all three samples.
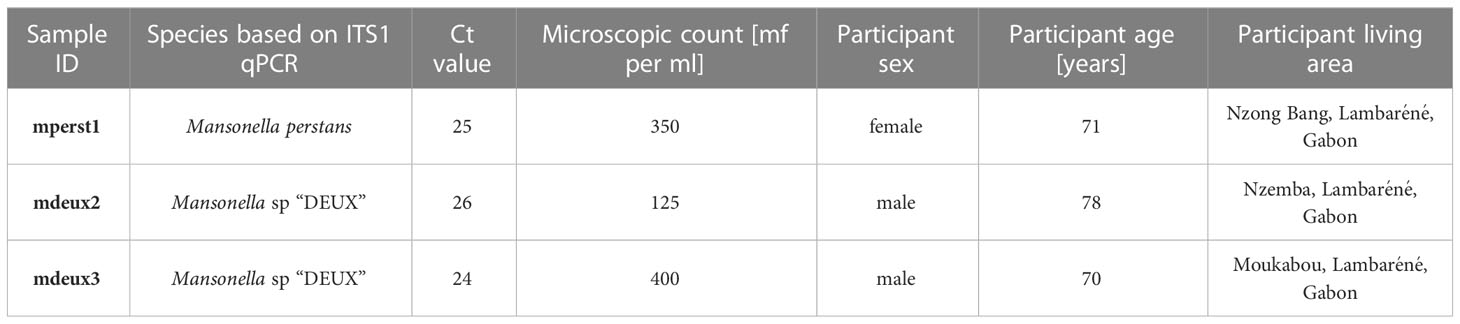
Table 1 Infection status, parasitemia and demographic data of individuals for mperst1, mdeux2 and mdeux3.
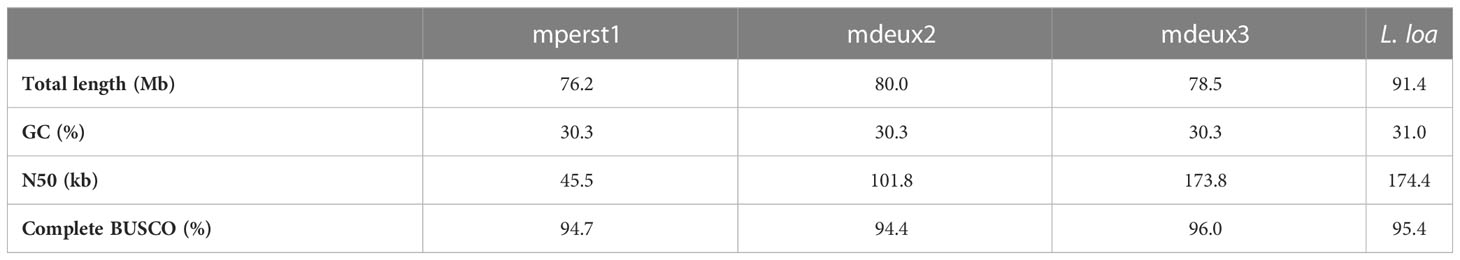
Table 2 Comparison of genome assemblies mperst1, mdeux2 and mdeux3 to L. loa. A more detailed comparison to other filarial species can be found in Supplementary Table 4.
The respective mitochondrial and Wolbachia genomes were assembled independently for all three samples. Wolbachia reads were sequenced and assembled from all three samples called wMpe1 for mperst1, wMde2 for mdeux2, and wMde3 for mdeux3. The Wolbachia assemblies are of comparable low quality (Supplementary Table 5). As a comparison, Wolbachia assemblies from other hosts were included. Among them are Wolbachia from supergroup F, as well as from supergroup D. Overall the here generated Wolbachia assemblies show a slightly lower GC content and a comparably high number of small contigs. In general, the assemblies are shorter than those used for comparison, especially wMpe1 is four times smaller than the reference wMpe (JACZHU01.1). Due to the poor quality, further analyses are not presented here.
To provide further information on the orthology and paralogy relationships across the genes of the nematode species, a phylome – i.e., a complete catalogue of gene evolutionary histories - was reconstructed for M. perstans from mperst1 and Mansonella sp “DEUX” from mdeux3, including 10 other sequenced nematode species. This allowed the selection of widespread genes that were present in single copy in the selected species. Subsequently, these genes were also identified in the second Mansonella sp “DEUX” sample mdeux2 as well as in two additional nematode species which were not included in the phylome analyses due to the fragmented nature of their assemblies (Brugia timori and Onchocerca flexuosa). This set of widespread genes was used to build a concatenated alignment and then a species tree was reconstructed (Figure 1). Using the inferred branch lengths from this tree, the phylogenetic distances within groups of species or strains from the same genus (Mansonella, Brugia and Onchocerca) were compared. As seen in Figure 1, branch lengths are very short for all three species groups, though the branches are shortest between the two Mansonella sp “DEUX” strains mdeux2 and mdeux3. The branch separating Mansonella sp “DEUX” and M. perstans is similar in length to the one separating Onchocerca ochengi and O. volvulus and the one separating B. timori and B. malayi, indicating that, at a genomic level, they have a similar genetic divergence. These findings are further confirmed by the calculations of nucleotide and amino acid diversity between the species in the three genera Mansonella, Brugia and Onchocerca (Supplementary Table 6). These analyses support the separation of Mansonella sp “DEUX” and M. perstans into two different species, as their genetic divergence is similar to that between established nematode species.
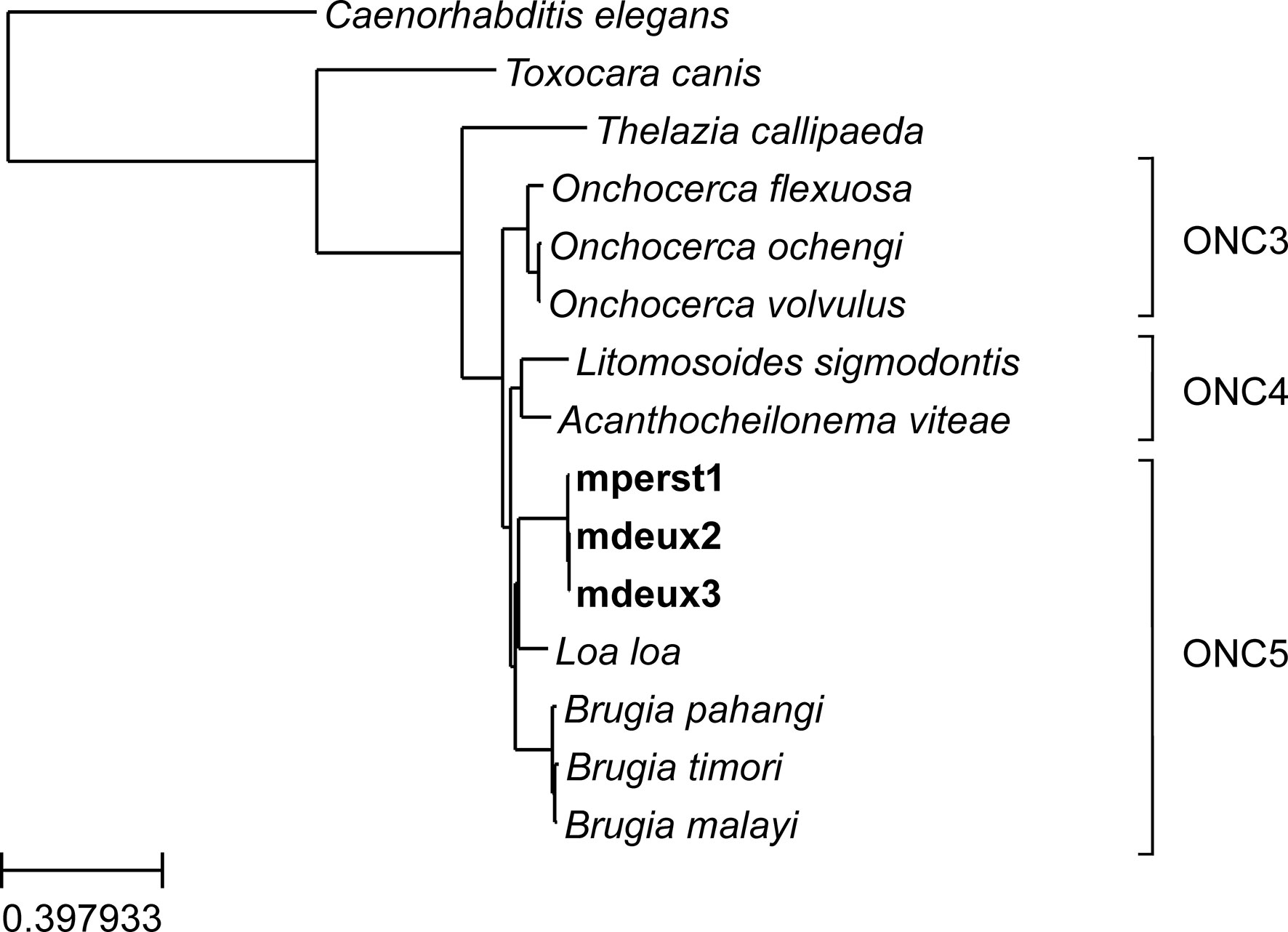
Figure 1 A phylogenetic tree reconstructed for M. perstans assembly mperst1 and the two Mansonella sp “DEUX” assemblies mdeux2 and mdeux3, including 12 other nematode species based on the concatenation of 562 protein alignments. Onchocercidae clades ONC3, ONC4 and ONC5 are shown next to the corresponding species.
Our phylogenetic analysis supports the results of the multi-locus sequence typing analysis by Lefoulon et al. (2015), which suggests a division of Onchocercidae into distinct phylogenetic clades (ONC1-ONC5). Figure 1 additionally shows the phylogenetic Onchocercidae clades ONC3, ONC4, and ONC5. Consistent with Lefoulon et al., all marked species are placed into the same clades. O. flexuosa, O. ochengi, and O. volvulus into clade ONC3, Litomosoides sigmodontis and Acanthocheilonema vitae into clade ONC4. Loa loa, B. pahangi, B. timori, B. malayi as well as the three Mansonella assemblies from mperst1, mdeux2 and mdeux3 are placed into clade ONC5.
To calculate the divergence time of mperst1 from mdeux2 and mdeux3, a chronogram was generated (Figure 2). Two calibration points were set based on fossil records, one placed at the root of the tree and the other at the base of the Filarioidea super family. Using this calibration, the divergence time between Mansonella sp “DEUX” and M. perstans is estimated at 778,000 years. To better classify these time spans, we give the divergence time of the closest members of Brugia (1.6 MyA) and Onchocerca (1.8 MyA) for comparison. The divergence time of the two strains of Mansonella sp “DEUX” is 33,000 years.
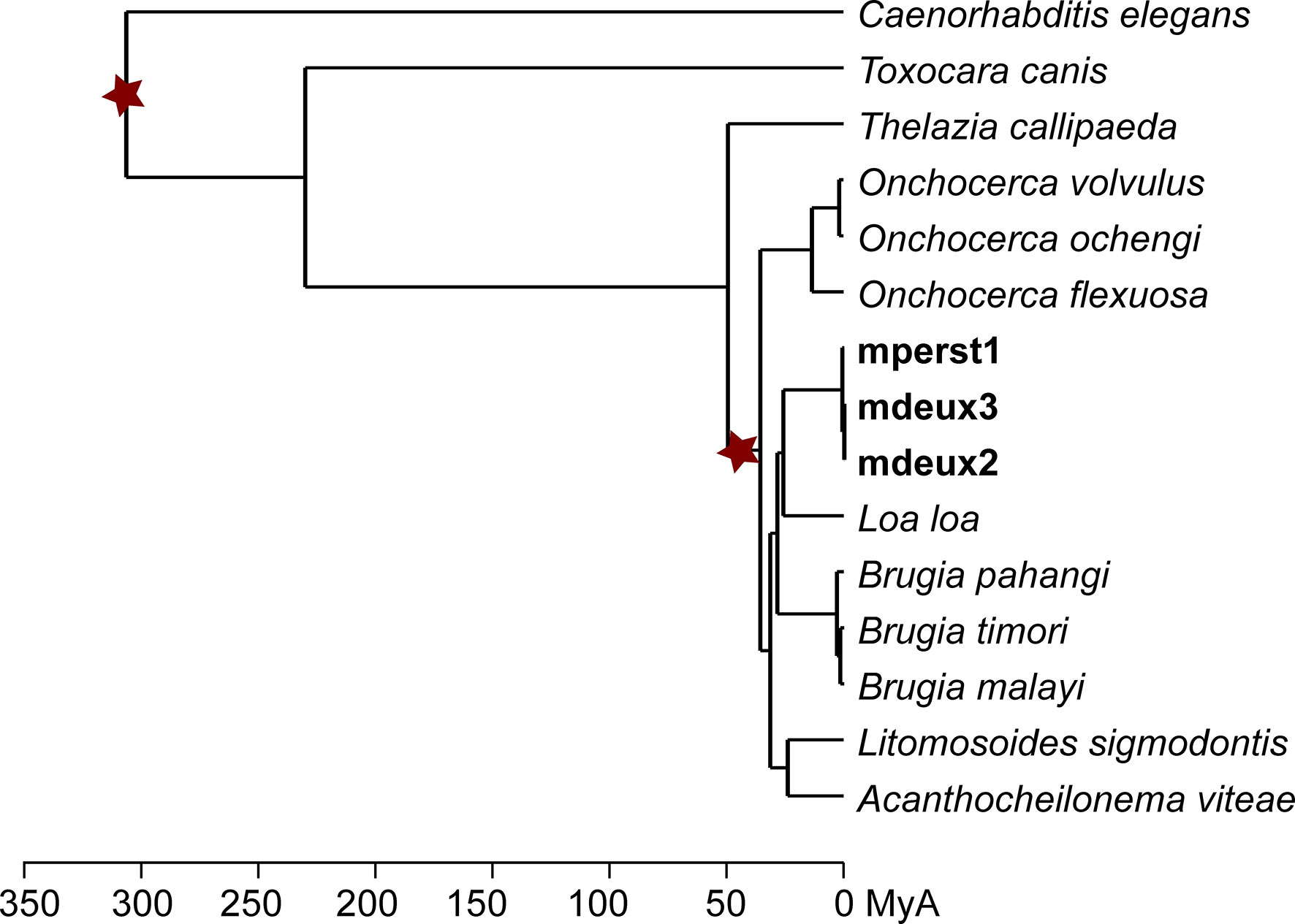
Figure 2 Evolutionary analysis of Mansonella sp “DEUX” and M. perstans. Chronogram detailing the divergence times of 15 nematode species. Stars indicate calibration points derived from recorded fossil evidence.
Sequence information for filaria species is rare, however a mitochondrial genome of M. ozzardi and M. perstans has been published recently (Crainey et al., 2018; Chung et al., 2020). Therefore, a phylogenetic analysis based on the mitogenomes was performed to further understand the relationship between M. perstans and Mansonella sp “DEUX”.
Figure 3A shows the generated phylogeny based on the mitogenomes of 17 nematode species, including the newly generated mitogenomes from the three assemblies. In total, three distinct mitogenome clusters from different species were identified from the assemblies. The first mitogenome cluster, designated as mMpe (mitogenome M. perstans), can be assigned to M. perstans, as it clusters with the published M. perstans mitogenome (GenBank: MT361687). It is found in mperst1 with a coverage of 40x. Unexpectedly, although identified as mono-infected with Mansonella sp “DEUX” via qPCR, assemblies of the individuals mdeux2 and mdeux3 were found to be mixed infected at low coverage with M. perstans. Both have in addition one mMpe mitogenome each, with lower coverage depths of 7x and 12x compared to the published M. perstans mitogenome, respectively. However, there were also two additional mitogenomes identified in each of these two assemblies that cluster together, both with a high coverage of 140x and 290x respectively. They show no similarity to any published mitogenomes and were therefore assigned to Mansonella sp “DEUX” and designated as mMde (mitogenome Mansonella sp “DEUX”). Surprisingly, a third mitogenome not clustering to the other mitogenomes or any other published sequence was identified in assembly mdeux2. Though it was sequenced at low coverage of 8x, it was assembled into a single contig, therefore confidence in the sequence is high. As the phylogeny suggests that this mitogenome originates from a separate species we provisionally call it “Mansonella cermeli” and the respective mitogenome mMce.
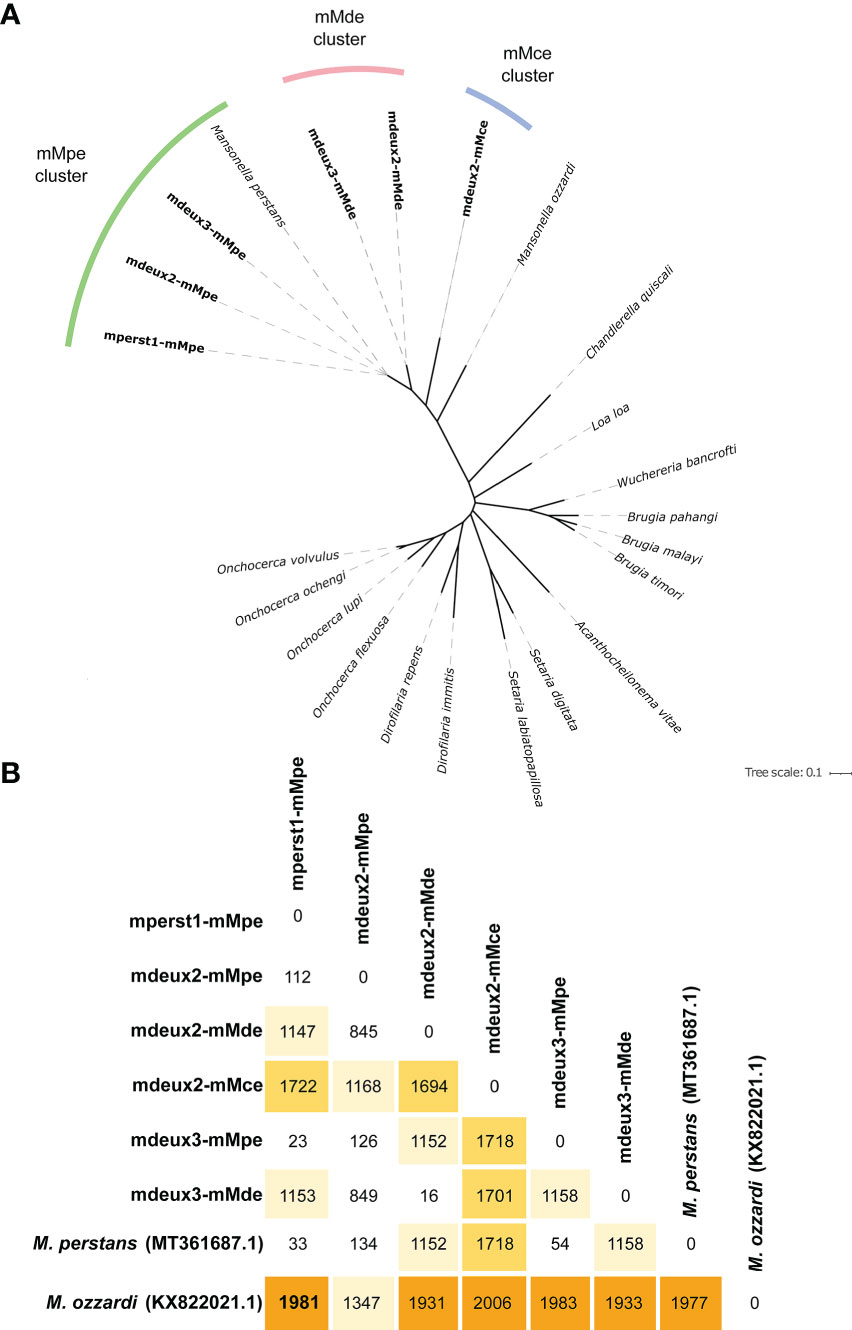
Figure 3 Mitogenome analysis. (A) Mitogenome phylogeny of different nematode species, including the newly generated mitogenomes from the three assemblies forming three Mansonella mitogenome clusters: Mitogenome of M. perstans mMpe marked in green, comprising of the mitogenomes from mperst1, and additionally two mitogenomes at low coverage of the individuals mdeux2 and mdeux3. Mansonella sp “DEUX” mMde marked in orange, with two mitogenomes of high coverage of mdeux2 and mdeux3. And the surprising third mitogenome of “M. cermeli” mMce with a single mitogenome of individual 2 (mdeux2). (B) SNP variant call between the three mitogenomes of each individual generated by us and the published mitogenomes of M. perstans (GenBank: MT361687) and M. ozzardi (GenBank: KX822021).
To compare the similarity of the generated mitogenomes, a SNP variant call was performed, including the reference mitogenomes of M. perstans (MT361687) and M. ozzardi (KX822021) (Figure 3B). The highest number of SNPs is found between each mitogenome and M. ozzardi. Whereas the lowest number of SNPs is found between the mitogenomes within one cluster as well as the mitogenomes from the cluster mMpe and M. perstans. Both samples from the mMde cluster (mdeux2-mMde and mdeux3-mMde) as well as the mMce cluster have a high number of SNPs compared to the mMpe cluster and the two references from M. perstans and M. ozzardi. This further supports the findings from the phylogenetic tree. Overall, genome size and GC content from all generated clusters are comparable to the published mitogenomes from M. perstans and M. ozzardi for all mitogenomes that were generated (Table 3). Both Mansonella sp “DEUX” mitogenomes mdeux2-mMde and mdeux3-mMde have a high coverage and are very similar to each other, showing a 99.8% pairwise identity. The same applies for the three M. perstans mitogenomes mperst1-mMpe, mdeux2-mMpe and mdeux3-mMpe. However, mdeux2_mMpe has a lower coverage and could not be completely assembled. The other two M. perstans mitogenomes though show a high similarity to the published M. perstans mitogenome. These data further support our hypothesis of two sympatric human Mansonella species.
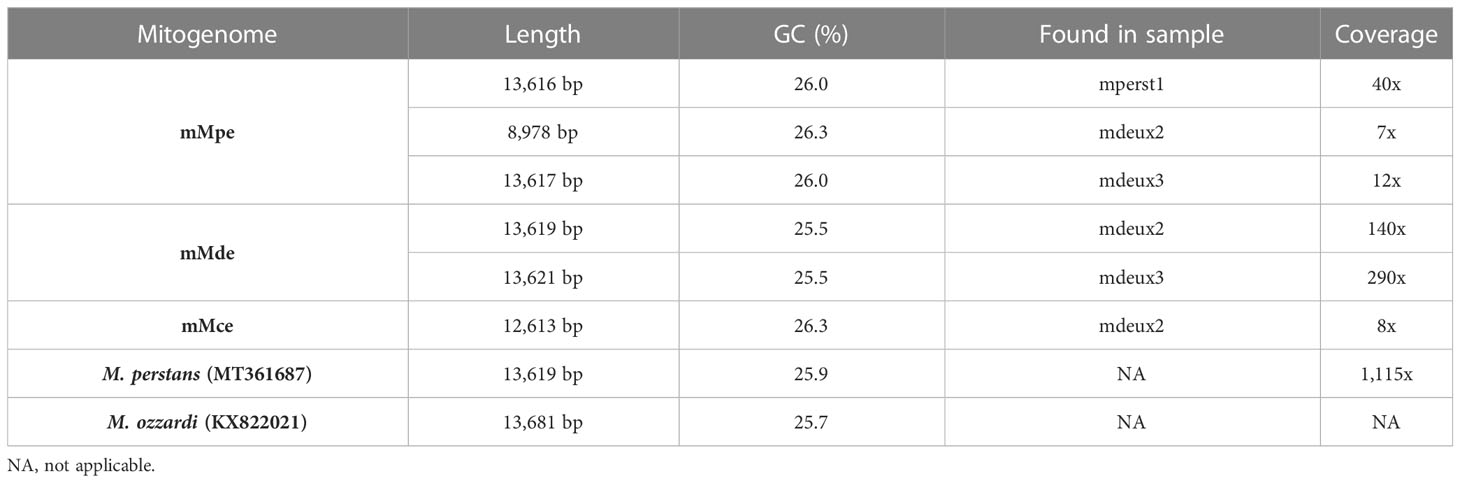
Table 3 Comparison of different characteristics of the generated mitogenome clusters, including the published mitogenomes from M. perstans (MT361687) and M. ozzardi (KX822021).
Parasites from the genus Mansonella are also known to infect non-human primates (NHPs). Only recently, filarial DNA extracted from NHPs faecal samples from Gabon and the neighbouring country Cameroon was used to investigate the Onchocercidae phylogeny based on two mitochondrial genes, cytochrome c oxidase subunit 1 (cox1) and 12S rDNA (Gaillard et al., 2020). These phylogenetic analyses showed that sequences obtained from chimpanzee cluster with samples from humans infected with M. perstans, concluding that chimpanzees can also be infected with M. perstans. In addition, they found NHPs to be infected with sequences of filaria that fall within the Mansonella genus, but cluster separately and are different from all published sequences.
We extended the published phylogenetic trees from Gaillard et al. with the respective cox1 (Figure 4) and 12S rDNA (Figure 5) sequences derived from the newly generated mitogenomes. As expected, the cox1 sequences of three mitogenomes of the mMpe cluster, map to the published sequences by Gaillard et al., that cluster to M. perstans. The cox1 sequences of the Mansonella sp “DEUX” mitogenomes (mMde) map to the unclassified cox1 sequences of NHPs that could not be assigned to a Mansonella species by Gaillard et al. Therefore, it can be assumed that Mansonella sp “DEUX” infects humans and great apes. Cox1 sequences of the mMce mitogenome cluster separately. Overall, we see the same five monophyletic groups, as depicted in Gaillard et al. The phylogenetic tree (Figure 5) based on 12S rDNA clusters corresponds very well to the clusters seen for cox1. We see separate clusters for the potential different species, as sequences from the M. perstans assemblies (mMpe assemblies: mperst1-mMpe, mdeux2-mMpe and mdeux3-mMpe) cluster to the published M. perstans sequences. This cluster can therefore be assigned to M. perstans. The 12S rDNA sequence from the mMce mitogenome forms a separate cluster. A third cluster is generated from two 12S rDNA sequences potentially originating from the Mansonella sp “DEUX” mitogenome assemblies mMde and additional 12S rDNA sequences generated from Gaillard et al., that were formerly grouped together with M. perstans sequences.
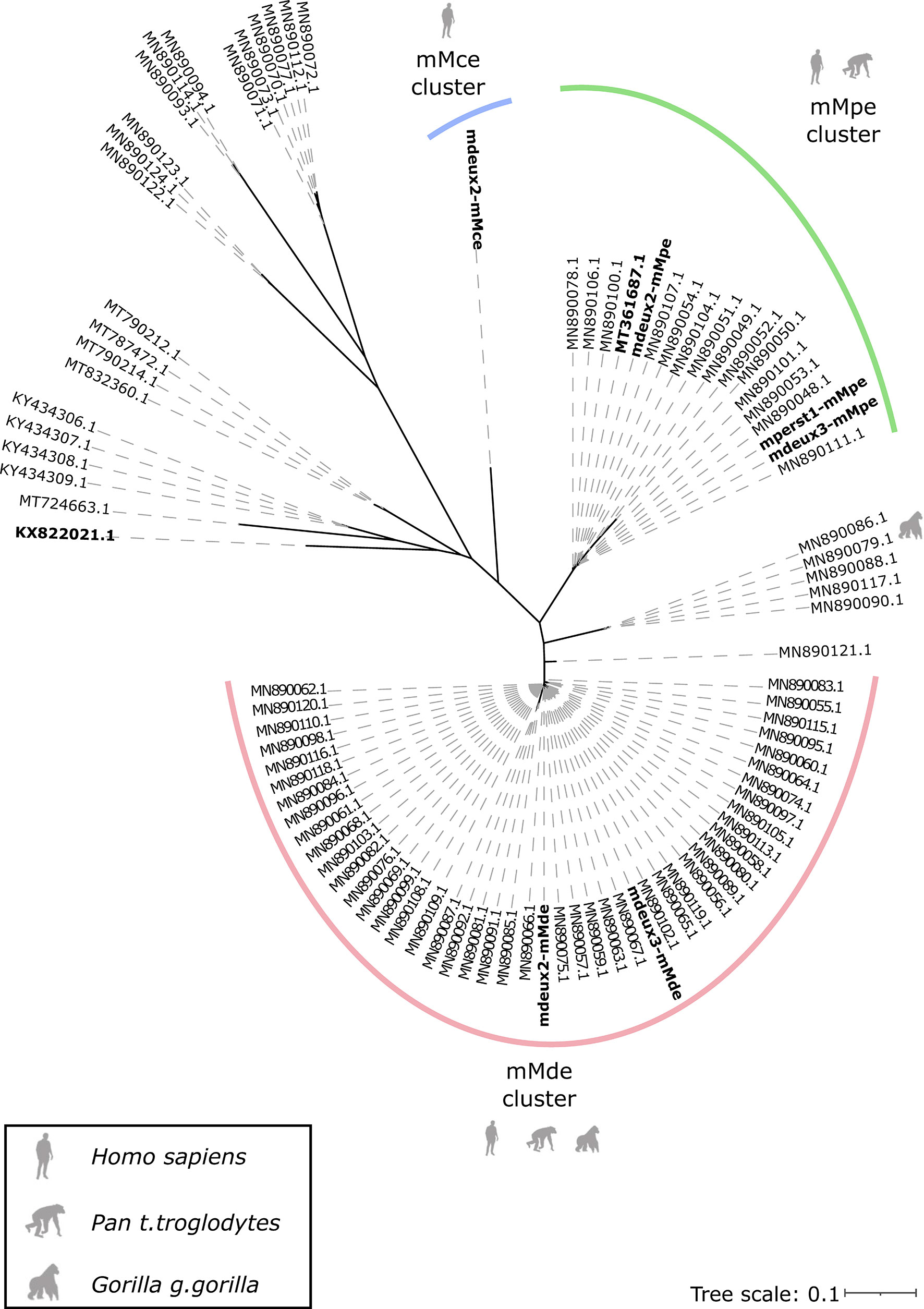
Figure 4 Phylogeny based on cox1 gene sequences. The three mitogenome clusters mMpe, mMde and mMce are highlighted in green, red and blue respectively. The generated mitogenomes, as well as the published mitogenomes from M. perstans (GenBank: MT361687) and M. ozzardi (GenBank: KX822021) are highlighted in bold.
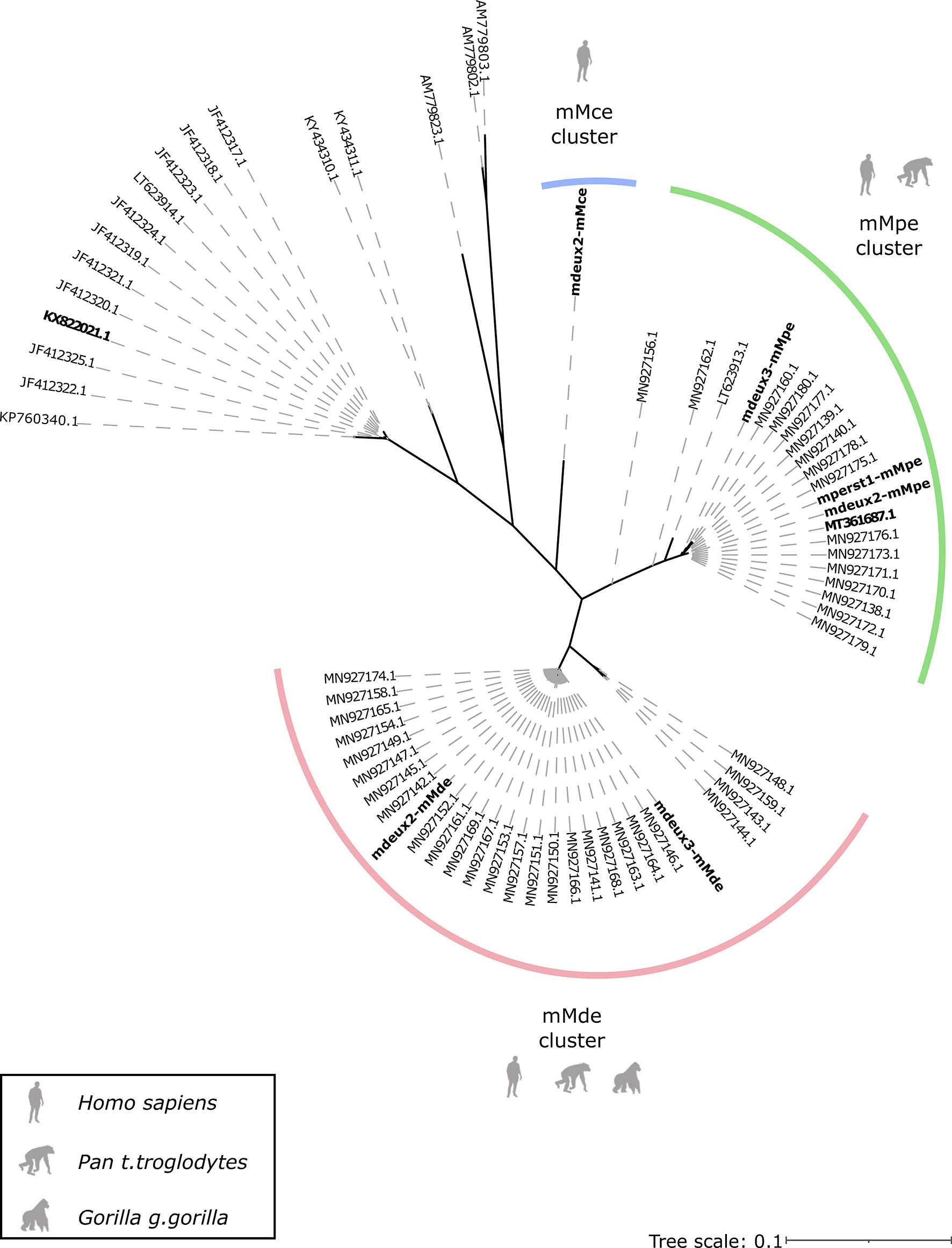
Figure 5 Phylogeny based on 12S rDNA gene sequences. The three mitogenome clusters mMpe, mMde and mMce are highlighted in green, red and blue respectively. The generated mitogenomes, as well as the published mitogenomes from M. perstans (GenBank: MT361687) and M. ozzardi (GenBank: KX822021) are highlighted in bold.
Infections with Mansonella species have been neglected in all areas, this is also reflected by the fact, that until recently no whole genome of any of the Mansonella species was available. We generated whole genomes of Mansonella perstans and the related Mansonella sp “DEUX” from microfilaria of infected individuals from Gabon. All assemblies show a high level of quality, as is indicated by high coverage, N50 values and BUSCO scores. The whole genomes are comparable in size, number of predicted genes and GC content to other filarial species. Phylogenetic analysis of the whole genomes as well as the mitogenomes of these two Mansonella suggest that these are two distinct species. Evolutionary analysis of the whole genome comparisons supported these findings as they give estimates that these sympatric species have diverged 778,000 years ago.
However, we found sites of recombination between the whole genomes of M. perstans and Mansonella sp “DEUX”. This can be a true finding, indicating that these species are still able to cross in rare occasions. On the other hand, this can also be due to our whole genome assemblies. Based on mitogenomic data, we discovered that the Mansonella sp “DEUX” samples, which we originally assumed to be mono-infected, are in fact co-infected with M. perstans and/or an unknown potential third species “M. cermeli” at a low coverage. These co-infections have influenced our analysis, and it might be that they are responsible for the observed recombination regions that could lead to less accurate estimates. The relatively high divergence time of 33,000 years between the two Mansonella sp “DEUX” assemblies could also be explained by the influence of the co-infections in those samples. However, the overall results provide a good starting point for further research. With the collection of more Mansonella samples from different regions in the future, the genetic differences between samples of the same species can be estimated and can be included in the equations, and the divergence times can be calculated more accurately.
The first previously sequenced and published mitogenome of M. perstans from a sample from Cameroon (Chung et al., 2020) clusters closely to our M. perstans mitogenome and its inclusion in the phylogenetic analysis additionally supports the finding of two distinct species. Since sequence data on Mansonella species from different regions are limited, we additionally investigated the genes for which the most sequence data are available: cox1 and 12S rDNA. Based on both genes, we have identified two Mansonella species clusters that correspond to either M. perstans or Mansonella sp “DEUX”; the same clustering was also seen by Gaillard et al. for cox1 sequences (Gaillard et al., 2020). We can confirm these two separate clusters also based on the 12S rDNA sequences. Only one cluster for 12S rDNA is shown by Gaillard et al., however, when we reanalyzed Gaillard’s data, we identified two clusters.
Molecular markers based on mitochondrial sequences are a commonly used tool in phylogenetic studies due to the conserved nature among species, and several markers have assisted in recent years to differentiate filarial species and determine the phylogeny of the Onchocercidae family (Xie et al., 1994; Lefoulon et al., 2015; Yilmaz et al., 2016; Crainey et al., 2018). The investigated markers 12S rDNA and cox1 overall support the evolutionary relationships and classification into different clades, as suggested by Lefoulon et al. (2015). Based on multi locus sequence typing they had identified five strongly supported clades, whereby all Mansonella species fall within clade ONC5. The affiliation to the different ONC clades is also reflected in our phylogeny based on the complete mitogenome and the phylogeny based on the concatenation of the 562 protein alignments as seen in Figure 1.
Our analysis of the cox1 and 12S rDNA genes additionally showed that our Mansonella sp “DEUX” sequences align to Mansonella sequences obtained from great apes from the same country (Gabon) and the neighboring country Cameroon (Gaillard et al., 2020), suggesting that Mansonella sp “DEUX” is infecting humans as well as NHPs. There are a few Mansonella species that have been described to occur in great apes based on morphology, as M. vanhoofi (Peel, 1946), M. gorillae (van den Berghe and Chardome, 1949), M. leopoldi (van den Berghe et al., 1957) and M. loopensis (Bain et al., 1995). Most of them have been described in the form of a case report. One ape related species has been described to occasionally infect humans in Gabon – M. rodhaini (Richard-Lenoble et al., 1988). There is no sequence information for any of these species available to compare them on a genetic level to our assemblies. However, M. rodhaini is described to be a skin dwelling species like M. streptocerca and not a blood dwelling species like M. perstans or Mansonella sp “DEUX”. We also compared our obtained sequences to the few available sequences of the skin dwelling M. streptocerca, but they did not map. Therefore, there is no indication that these skin dwelling species are the same as Mansonella sp “DEUX”. To determine if one of the morphologically described great ape infecting Mansonella species could be Mansonella sp “DEUX”, microscopic investigations of mono-infected samples could be done.
We additionally identified Wolbachia reads in all three sequenced samples. Wolbachia are intracellular bacteria (order Rickettsiales) found in a range of arthropod and nematode species (Taylor et al., 2018). In filarial nematodes Wolbachia are usually symbionts, beneficial for the host survival and reproduction (Bandi et al., 1999; Hoerauf et al., 1999; Volkmann et al., 2003). Antibiotic treatment of filarial infections targets the Wolbachia and are promising alternative treatment options, especially for M. perstans infections, where standard filarial treatments are not efficacious (Coulibaly et al., 2009; Raoult, 2010; Batsa Debrah et al., 2019). Currently, Wolbachia are taxonomically subdivided into 16 different so-called supergroups. Wolbachia of filaria fall in supergroups C, D, and J with the exception of Wolbachia of Mansonella species that fall in supergroup F. Interestingly, all other Wolbachia of supergroup F have arthropod hosts (Covacin and Barker, 2007; Ferri et al., 2011). We could confirm that Wolbachia reads from Mansonella sp “DEUX” also belong to supergroup F, similar to Wolbachia of M. perstans and M. ozzardi (Casiraghi et al., 2001; Keiser et al., 2008). Whole genomes as well as sequence comparisons of the Wolbachia of these two Mansonella species (M. perstans and M. ozzardi) are described in a preprint (Sinha et al., 2021). Wolbachia are usually inherited maternally and differences between Wolbachia of different species can give further evidence on their relatedness. The read coverage of our assembled Wolbachia genomes was low (<10x) leading to a high fragmentation with many small contigs. However, the finding of Wolbachia reads in all three samples, further supports the carriage of Wolbachia in M. perstans and Mansonella sp “DEUX”.
Based on mitogenomic analysis, we found a third Mansonella mitogenome that did not cluster with any of the available sequences and might therefore even present another new Mansonella species, that we named “Mansonella cermeli”. As read coverage was low, we could not construct a separate whole genome, but because of the multicopy nature of mitogenomic DNA in general, we could construct a separate mitogenome. Evidence of this separate genome is high, as the whole mitogenomes could be assembled into a single circular contig. This is the first time that sequences of “M. cermeli” are detected, and we do not have a morphologic proof of its existence. Also, we do not know if this is a common species/infection in that region. Further studies must be conducted to analyze if this is another common Mansonella species infecting humans. The mitogenomes reported here show that the species diversity within Mansonella might even be higher than previously thought.
Overall, the results of our study suggest that Mansonella sp “DEUX” represents a new Mansonella species. It is most probably not restricted to the human host, but also infects great apes. However, given the high prevalence of this mansonellosis in the population in this area, one can assume that it is also a true human infection. The detection of the additional “M. cermeli” mitogenome further illustrates the lack of knowledge about this genus. Fortunately, awareness is increasing and more data are being published, such as a preprint this year describing the entire genomes of M. perstans and M. ozzardi (Sinha et al., 2023). The authors discuss clinical differences in response to antifungal drugs in the context of molecular differences in selected genes of the different species. These publications together with the sequence data are of great value for further research on Mansonella. There are many open questions on Mansonella infections not only related to phylogeny and number of infecting species, but also on the burden of disease, adequate treatment and impact on the immune system. However, to properly address these questions, one prerequisite is to be able to identify, name, and differentiate the infecting species.
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found below: https://www.ebi.ac.uk/ena, PRJEB57801 and PRJNA942613.
JH, TS, SO, SA, NC designed the study. LB collected samples. MR, LB, MP performed lab experiments. MR, CG, TS, MM-H, EK, TG, SO, JH analyzed data. TS, NC, AK, SA interpreted results. TG, NC, CK, AK, JH provided resources. MR, CG, JH wrote the first draft. All authors contributed to the article and approved the submitted version.
We acknowledge funding by DZIF (DZIF sequencing project number: TTU 03.820_00) and BMBF (ELIMONCHO-Project: 01DG20021). TG group acknowledges support from the Spanish Ministry of Science and Innovation for grant PID2021-126067NB-I00, cofounded by European Regional Development Fund (ERDF), from the Catalan Research Agency (AGAUR) SGR423; from the European Union’s Horizon 2020 research and innovation program (ERC-2016-724173); from the Gordon and Betty Moore Foundation (Grant GBMF9742); from the “La Caixa” foundation (Grant LCF/PR/HR21/00737), and from the Instituto de Salud Carlos III (IMPACT Grant IMP/0019 and CIBERINFEC CB21/13/00061-ISCIII-SGEFI/ERDF). NGS sequencing methods were performed with the support of the DFG-funded NGS Competence Center Tübingen (INST 37/1049-1). We acknowledge support by Open Access Publishing Fund of University of Tübingen.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fcimb.2023.1159814/full#supplementary-material
Altschul, S. F., Gish, W., Miller, W., Myers, E. W., Lipman, D. J. (1990). Basic local alignment search tool. J. Mol. Biol. 215 (3), 403–410. doi: 10.1016/S0022-2836(05)80360-2
Bain, O., Moisson, P., Huerre, M., Landsoud-Soukate, J., Tutin, C. (1995). Filariae from a wild gorilla in Gabon with description of a new species of mansonella. Parasite. 2 (3), 315–322. doi: 10.1051/parasite/1995023315
Bandi, C., McCall, J. W., Genchi, C., Corona, S., Venco, L., Sacchi, L. (1999). Effects of tetracycline on the filarial worms brugia pahangi and dirofilaria immitis and their bacterial endosymbionts wolbachia. Int. J. Parasitol. 29 (2), 357–364. doi: 10.1016/S0020-7519(98)00200-8
Bankevich, A., Nurk, S., Antipov, D., Gurevich, A. A., Dvorkin, M., Kulikov, A. S., et al. (2012). SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 19 (5), 455–477. doi: 10.1089/cmb.2012.0021
Batsa Debrah, L., Phillips, R. O., Pfarr, K., Klarmann-Schulz, U., Opoku, V. S., Nausch, N., et al. (2019). The efficacy of doxycycline treatment on. Am. J. Trop. Med. Hyg. 101 (1), 84–92. doi: 10.4269/ajtmh.18-0491
Bordenstein, S. R., Paraskevopoulos, C., Dunning Hotopp, J. C., Sapountzis, P., Lo, N., Bandi, C., et al. (2009). Parasitism and mutualism in wolbachia: what the phylogenomic trees can and cannot say. Mol. Biol. Evol. 26 (1), 231–241. doi: 10.1093/molbev/msn243
Buchfink, B., Reuter, K., Drost, H. G. (2021). Sensitive protein alignments at tree-of-life scale using DIAMOND. Nat. Methods 18 (4), 366–368. doi: 10.1038/s41592-021-01101-x
Capella-Gutierrez, S., Silla-Martinez, J. M., Gabaldon, T. (2009). trimAl: a tool for automated alignment trimming in large-scale phylogenetic analyses. Bioinformatics. 25 (15), 1972–1973. doi: 10.1093/bioinformatics/btp348
Casiraghi, M., Favia, G., Cancrini, G., Bartoloni, A., Bandi, C. (2001). Molecular identification of wolbachia from the filarial nematode mansonella ozzardi. Parasitol. Res. 87 (5), 417–420. doi: 10.1007/s004360000368
Chung, M., Aluvathingal, J., Bromley, R. E., Nadendla, S., Fombad, F. F., Kien, C. A., et al. (2020). Complete mitochondrial genome sequence of mansonella perstans. Microbiol. Resour Announc 9 (30). doi: 10.1128/MRA.00490-20
Coulibaly, Y. I., Dembele, B., Diallo, A. A., Lipner, E. M., Doumbia, S. S., Coulibaly, S. Y., et al. (2009). A randomized trial of doxycycline for mansonella perstans infection. N Engl. J. Med. 361 (15), 1448–1458. doi: 10.1056/NEJMoa0900863
Covacin, C., Barker, S. C. (2007). Supergroup f wolbachia bacteria parasitise lice (Insecta: Phthiraptera). Parasitol. Res. 100 (3), 479–485. doi: 10.1007/s00436-006-0452-0
Crainey, J. L., Marín, M. A., Silva, T. R. R. D., de Medeiros, J. F., Pessoa, F. A. C., Santos, Y. V., et al. (2018). Mansonella ozzardi mitogenome and pseudogene characterisation provides new perspectives on filarial parasite systematics and CO-1 barcoding. Sci. Rep. 8 (1), 6158. doi: 10.1038/s41598-018-24382-3
Edgar, R. C. (2004). MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 32 (5), 1792–1797. doi: 10.1093/nar/gkh340
Ferri, E., Bain, O., Barbuto, M., Martin, C., Lo, N., Uni, S., et al. (2011). New insights into the evolution of wolbachia infections in filarial nematodes inferred from a large range of screened species. PloS One 6 (6), e20843. doi: 10.1371/journal.pone.0020843
Fuentes, D., Molina, M., Chorostecki, U., Capella-Gutiérrez, S., Marcet-Houben, M., Gabaldón, T. (2022). PhylomeDB V5: an expanding repository for genome-wide catalogues of annotated gene phylogenies. Nucleic Acids Res. 50 (D1), D1062–D10D8. doi: 10.1093/nar/gkab966
Gaillard, C. M., Pion, S. D., Hamou, H., Sirima, C., Bizet, C., Lemarcis, T., et al. (2020). Detection of DNA of filariae closely related to mansonella perstans in faecal samples from wild non-human primates from Cameroon and Gabon. Parasit Vectors. 13 (1), 313. doi: 10.1186/s13071-020-04184-1
Gehringer, C., Kreidenweiss, A., Flamen, A., Antony, J. S., Grobusch, M. P., Bélard, S. (2014). Molecular evidence of wolbachia endosymbiosis in mansonella perstans in Gabon, central Africa. J. Infect. Dis. 210 (10), 1633–1638. doi: 10.1093/infdis/jiu320
Grobusch, M. P., Kombila, M., Autenrieth, I., Mehlhorn, H., Kremsner, P. G. (2003). No evidence of wolbachia endosymbiosis with loa loa and mansonella perstans. Parasitol. Res. 90 (5), 405–408. doi: 10.1007/s00436-003-0872-z
Gurevich, A., Saveliev, V., Vyahhi, N., Tesler, G. (2013). QUAST: quality assessment tool for genome assemblies. Bioinformatics. 29 (8), 1072–1075. doi: 10.1093/bioinformatics/btt086
Hoerauf, A., Nissen-Pähle, K., Schmetz, C., Henkle-Dührsen, K., Blaxter, M. L., Büttner, D. W., et al. (1999). Tetracycline therapy targets intracellular bacteria in the filarial nematode litomosoides sigmodontis and results in filarial infertility. J. Clin. Invest. 103 (1), 11–18. doi: 10.1172/JCI4768
Katoh, K., Kuma, K., Toh, H., Miyata, T. (2005). MAFFT version 5: improvement in accuracy of multiple sequence alignment. Nucleic Acids Res. 33 (2), 511–518. doi: 10.1093/nar/gki198
Keiser, P. B., Coulibaly, Y., Kubofcik, J., Diallo, A. A., Klion, A. D., Traoré, S. F., et al. (2008). Molecular identification of wolbachia from the filarial nematode mansonella perstans. Mol. Biochem. Parasitol. 160 (2), 123–128. doi: 10.1016/j.molbiopara.2008.04.012
Lartillot, N., Lepage, T., Blanquart, S. (2009). PhyloBayes 3: a Bayesian software package for phylogenetic reconstruction and molecular dating. Bioinformatics. 25 (17), 2286–2288. doi: 10.1093/bioinformatics/btp368
Lassmann, T., Sonnhammer, E. L. (2005). Kalign–an accurate and fast multiple sequence alignment algorithm. BMC Bioinf. 6, 298. doi: 10.1186/1471-2105-6-298
Lefoulon, E., Bain, O., Bourret, J., Junker, K., Guerrero, R., Cañizales, I., et al. (2015). Shaking the tree: Multi-locus sequence typing usurps current onchocercid (Filarial nematode) phylogeny. PloS Negl. Trop. Dis. 9 (11), e0004233. doi: 10.1371/journal.pntd.0004233
Letunic, I., Bork, P. (2021). Interactive tree of life (iTOL) v5: an online tool for phylogenetic tree display and annotation. Nucleic Acids Res. 49 (W1), W293–W2W6. doi: 10.1093/nar/gkab301
Mourembou, G., Fenollar, F., Lekana-Douki, J. B., Ndjoyi Mbiguino, A., Maghendji Nzondo, S., Matsiegui, P. B., et al. (2015). Mansonella, including a potential new species, as common parasites in children in Gabon. PloS Negl. Trop. Dis. 9 (10), e0004155. doi: 10.1371/journal.pntd.0004155
Nguyen, L. T., Schmidt, H. A., von Haeseler, A., Minh, B. Q. (2015). IQ-TREE: a fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol. Biol. Evol. 32 (1), 268–274. doi: 10.1093/molbev/msu300
Peel, E. C. Sur des Filarides de Chimpanzés Pan piscus et Pan satyrus au Congo Belge. Annales de la société belge de médecine tropicale (1946), 26, 117–150.
Raoult, D. (2010). Doxycycline for mansonella perstans infection. N Engl. J. Med. 362 (3), 272; author reply–3. doi: 10.1056/NEJMc0910675
Richard-Lenoble, D., Kombila, M., Bain, O., Chandenier, J., Mariotte, O. (1988). Filariasis in Gabon: human infections with microfilaria rodhaini. Am. J. Trop. Med. Hyg. 39 (1), 91–92. doi: 10.4269/ajtmh.1988.39.91
Sandri, T. L., Kreidenweiss, A., Cavallo, S., Weber, D., Juhas, S., Rodi, M., et al. (2021). Molecular epidemiology of mansonella species in Gabon. J. Infect. Dis. 223 (2), 287–296. doi: 10.1093/infdis/jiaa670
Seemann, T. (2013). Available at: https://github.com/tseemann/barrnap.
Simao, F. A., Waterhouse, R. M., Ioannidis, P., Kriventseva, E. V., Zdobnov, E. M. (2015). BUSCO: assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics. 31 (19), 3210–3212. doi: 10.1093/bioinformatics/btv351
Simonsen, P. E., Onapa, A. W., Asio, S. M. (2011). Mansonella perstans filariasis in Africa. Acta Trop. 120 Suppl 1), S109–S120. doi: 10.1016/j.actatropica.2010.01.014
Sinha, A., Li, Z., Poole, C. B., Ettwiller, L., Lima, N. F., Ferreira, M. U., et al. (2021). Multiple origins of nematode-wolbachia symbiosis in supergroup f and convergent loss of bacterioferritin in filarial wolbachia. doi: 10.1101/2021.03.23.436630
Sinha, A., Li, Z., Poole, C. B., Morgan, R. D., Ettwiller, L., Lima, N. F., et al. (2023). Genomes of the human filarial parasites mansonella perstans and mansonella ozzardi. doi: 10.1101/2023.01.06.523030
Slater, GS, Birney, E. (2018). Automated generation of heuristics for biological sequence comparison. BMC Bioinformatics (2005) 6, 31. doi: 10.1186/1471-2105-6-31
Smith, S. A., Brown, J. W., Walker, J. F. (2018). So many genes, so little time: A practical approach to divergence-time estimation in the genomic era. PloS One 13 (5), e0197433. doi: 10.1371/journal.pone.0197433
Stanke, M., Morgenstern, B. (2005). AUGUSTUS: a web server for gene prediction in eukaryotes that allows user-defined constraints. Nucleic Acids Res. 33 (Web Server issue), W465–W467. doi: 10.1093/nar/gki458
Tamarozzi, F., Rodari, P., Salas-Coronas, J., Bottieau, E., Salvador, F., Soriano-Pérez, M. J., et al. (2022). A large case series of travel-related mansonella perstans (vector-borne filarial nematode): a TropNet study in Europe. J. Travel Med. 29 (7). doi: 10.1093/jtm/taac048
Taylor, M. J., Bordenstein, S. R., Slatko, B. (2018). Microbe profile: Wolbachia: a sex selector, a viral protector and a target to treat filarial nematodes. Microbiol. (Reading). 164 (11), 1345–1347. doi: 10.1099/mic.0.000724
van den Berghe, L., Chardome, M. (1949). Une microfilaire du gorille. Microfilaria gorillae. Annales la Société Belge Médecine tropicale. Folia Scientif. Afr. Centr. 29, 495–499.
van den Berghe, L., Chardome, M., Peel, E. (1957). Microfilaria leopoldi n. sp. chez gorilla gorilla au Congo belge. 87.
Volkmann, L., Fischer, K., Taylor, M., Hoerauf, A. (2003). Antibiotic therapy in murine filariasis (Litomosoides sigmodontis): comparative effects of doxycycline and rifampicin on wolbachia and filarial viability. Trop. Med. Int. Health 8 (5), 392–401. doi: 10.1046/j.1365-3156.2003.01040.x
Wallace, I. M., O’Sullivan, O., Higgins, D. G., Notredame, C. (2006). M-coffee: combining multiple sequence alignment methods with T-coffee. Nucleic Acids Res. 34 (6), 1692–1699. doi: 10.1093/nar/gkl091
Wehe, A., Bansal, M. S., Burleigh, J. G., Eulenstein, O. (2008). DupTree: a program for large-scale phylogenetic analyses using gene tree parsimony. Bioinformatics. 24 (13), 1540–1541. doi: 10.1093/bioinformatics/btn230
Wick, R. R., Schultz, M. B., Zobel, J., Holt, K. E. (2015). Bandage: interactive visualization of de novo genome assemblies. Bioinformatics. 31 (20), 3350–3352. doi: 10.1093/bioinformatics/btv383
Wood, D. E., Lu, J., Langmead, B. (2019). Improved metagenomic analysis with kraken 2. Genome Biol. 20 (1), 257. doi: 10.1186/s13059-019-1891-0
Xie, H., Bain, O., Williams, S. A. (1994). Molecular phylogenetic studies on filarial parasites based on 5S ribosomal spacer sequences. Parasite. 1 (2), 141–151. doi: 10.1051/parasite/1994012141
Yilmaz, E., Fritzenwanker, M., Pantchev, N., Lendner, M., Wongkamchai, S., Otranto, D., et al. (2016). The mitochondrial genomes of the zoonotic canine filarial parasites dirofilaria (Nochtiella) repens and candidatus dirofilaria (Nochtiella) hongkongensis provide evidence for presence of cryptic species. PloS Negl. Trop. Dis. 10 (10), e0005028. doi: 10.1371/journal.pntd.0005028
Keywords: Mansonella sp “DEUX”, Mansonella perstans, whole genome sequencing, phylogeny, Wolbachia
Citation: Rodi M, Gross C, Sandri TL, Berner L, Marcet-Houben M, Kocak E, Pogoda M, Casadei N, Köhler C, Kreidenweiss A, Agnandji ST, Gabaldón T, Ossowski S and Held J (2023) Whole genome analysis of two sympatric human Mansonella: Mansonella perstans and Mansonella sp “DEUX”. Front. Cell. Infect. Microbiol. 13:1159814. doi: 10.3389/fcimb.2023.1159814
Received: 06 February 2023; Accepted: 15 March 2023;
Published: 14 April 2023.
Edited by:
Omar Hamarsheh, Al-Quds University, PalestineReviewed by:
Kenneth Pfarr, University Hospital Bonn, GermanyCopyright © 2023 Rodi, Gross, Sandri, Berner, Marcet-Houben, Kocak, Pogoda, Casadei, Köhler, Kreidenweiss, Agnandji, Gabaldón, Ossowski and Held. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jana Held, amFuYS5oZWxkQHVuaS10dWViaW5nZW4uZGU=
†These authors share first authorship
‡These authors share senior authorship
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.