 Jingyu Xu
Jingyu Xu Zihao Zhou
Zihao Zhou Yidan Zheng
Yidan Zheng Sai Yang2*
Sai Yang2* Kun Huang
Kun Huang Huili Li
Huili Li
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Cell. Infect. Microbiol. , 15 May 2023
Sec. Microbes and Innate Immunity
Volume 13 - 2023 | https://doi.org/10.3389/fcimb.2023.1149911
This article is part of the Research Topic The Role of Inflammasome in Viral Infection View all 7 articles
Viral myocarditis (VMC), characterized by viral infection-induced inflammation, is a life-threatening disease associated with dilated cardiomyopathy or heart failure. Innate immunity plays a crucial role in the progression of inflammation, in which inflammasomes provide a platform for the secretion of cytokines and mediate pyroptosis. Inflammasomes are rising stars gaining increasing attention. The nucleotide oligomerization domain-, leucine-rich repeat-, and pyrin domain-containing protein 3 (NLRP3) inflammasome, the caspase recruitment domain-containing protein 8 (CARD8) inflammasome, and the caspase-11 inflammasome are three inflammasomes that were reported to affect the process and prognosis of VMC. These inflammasomes can be activated by a wide range of cellular events. Accumulating evidence has suggested that inflammasomes are involved in different stages of VMC, including the trigger and progression of myocardial injury and remodeling after infection. In this review, we summarized the pathways involving inflammasomes in VMC and discussed the potential therapies targeting inflammasomes and related pathways.
Myocarditis is the inflammation and injury of the myocardium resulting from multiple infectious or non-infectious factors including viruses, immune system activation, or exposure to toxins/drugs. Viruses are the main infectious cause of myocarditis, and increasing evidence proves that excessive host immune responses probably play a more crucial part in the pathogenic process of VMC (Reyes and Lerner, 1985; Mason, 2003; Fairweather and Rose, 2007; Basso, 2022). In some patients with VMC, the developing or persistent myocardial injury can lead to dilated cardiomyopathy and even heart failure (Sagar et al., 2012). However, the pathogenic mechanisms of VMC have not been well demonstrated.
Inflammasomes are multimolecular protein complexes assembling in response to pathogen-associated molecular patterns (PAMPs) or damage/danger-associated molecular patterns (DAMPs), resulting in the maturation and release of interleukin-1β (IL-1β) and IL-18 and an inflammatory form of cell death named pyroptosis (Rathinam et al., 2012; Latz et al., 2013; Sharma and Kanneganti, 2021). The IL-1β and IL-18 are widely involved in inflammatory responses and myocardial injury.
In this review, we summarized relevant literature involving the roles of inflammasomes in different pathogenic stages of VMC. Besides, we discussed the possible roles of inflammasomes in COVID-19-related myocarditis. Furthermore, we proposed potential therapies targeting the NLRP3 inflammasome-IL-1β axis.
VMC is the main subtype of infectious myocarditis and a significant cause of dilated cardiomyopathy (DCM) (Cooper, 2009; Pollack et al., 2015; Basso, 2022). VMC can be induced by a variety of viruses, including coxsackievirus B3 (CVB3), encephalomyocarditis virus (EMCV), human immunodeficiency virus (HIV), human parvovirus B 19 (PVB-19) and angiotensin-converting enzyme 2-tropic cardiotoxic viruses such as severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) (Tschöpe et al., 2021). Laboratory VMC mouse models are commonly induced by CVB3 and EMCV. Moreover, COVID-19-related myocarditis has aroused increasing attention (Siripanthong et al., 2020; Chen et al., 2020; Inciardi et al., 2020; Long et al., 2020). Although the causative virus varies, the development of VMC is manifested as an infectious phase and a post-infectious phase.
The infectious phase is characterized by viral infection and subsequent direct damage to myocardium, including myocardial inflammation deriving from innate and adaptive immune reactions, necrosis, and apoptosis of cardiomyocytes.
The existing understanding of the pathogenic process of VMC is mainly derived from the CVB3-induced VMC mouse model. Coxsackieviruses enter cells through the combined effect of coxsackievirus-adenovirus receptor (CAR) and decay-accelerating factor (DAF) (Shenoy-Scaria et al., 1992; He et al., 2001), and subsequently affect cell function through a variety of viral proteases. Among the viral proteases, the roles of proteases 2A and 3C in cleaving cellular proteins stand out, which exacerbate the VMC (Liu and Mason, 2001; Xiong et al., 2007). The viral infection eventually leads to apoptosis and necrosis of the cardiomyocytes, increasing the release of viruses that infect the rest of the cardiomyocytes and causing damage to the myocardium.
Immune responses after viral infection play key roles in the development of VMC. On the one hand, the immune responses are prerequisites for the removal of pathogens; on the other hand, sustained and excessive immune responses may cause myocardial damage and even DCM (Sagar et al., 2012). In the context of VMC, viruses trigger innate immune responses by interacting with a variety of pathways involving melanoma differentiation-associated gene 5 (MDA5), toll-like receptors (TLRs), etc. MDA5 is essential for the production of maximal levels of interferon (IFN)-α in the early stage of infection. The absence of MDA5 inhibits the type I IFN production and exacerbates mortality in mice with CVB3-induced VMC (Wang et al., 2010; Hühn et al., 2010). Macrophages, neutrophils, dendritic cells, and other cells can recognize viruses through TLRs (Cooper, 2009; Rivadeneyra et al., 2018) and activate nuclear transcription factors such as NF-κB, promoting the production of proinflammatory cytokines including tumor necrosis factor (TNF), IL-1α, IL-1β, IL-2 and IFN-γ (Pollack et al., 2015; Lasrado and Reddy, 2020). This process involves the activation of inflammasomes such as the NLRP3 inflammasome. Furthermore, there is critical cooperation between the MDA5-mediated pathway and the TLR-mediated pathway on some occasions, such as the RIG-I/MDA5-type I IFN pathway and the TLR3-type II IFN pathway for efficient innate antiviral immune responses (Negishi et al., 2008). After the activation of innate immune responses, adaptive immune responses set off, initiating the activation and expansion of T cells and B cells and participating in the progress of VMC. The immune responses may promote cell necrosis, cardiac fibrosis, and remodeling, which can further result in severe arrhythmia, left ventricle dilation, and even heart failure (Cooper, 2009).
In EMCV-induced VMC, there is not much literature about viral internalization, but recently, a disintegrin and metalloproteinase 9 domain (ADAM9) has been identified as a major EMCV dependency factor (Bazzone et al., 2019). In the pathogenic process of EMCV-induced VMC, the viral protein 2B is crucial. The protein 2B activates the NLRP3 inflammasome by promoting calcium (Ca2+) flux from the Golgi apparatus and endoplasmic reticulum (ER) into the cytoplasm, along with K+ efflux out of the cytoplasm (De Jong et al., 2008). Inflammatory responses, including high levels of proinflammatory cytokines in the heart, can be detected in the early stage of EMCV-induced VMC and enhance the toxicity of EMCV, further impairing cardiac function (Shioi et al., 1996; Iwasaki et al., 1999; Matsumori et al., 2004). SARS-CoV-2 enters cardiomyocytes by binding its spike protein to angiotensin-converting enzyme 2 (ACE2) with the aid of transmembrane serine protease 2 (TMPRSS2) (Hoffmann et al., 2020), possibly providing the premise for the NLRP3 inflammasome activation and leading to subsequent inflammation and injury.
The main features of the post-infectious phase, taking CVB3 as an example, include cardiac remodeling, cardiac fibrosis, and cardiac dysfunctions. After the infectious phase, some patients will experience remission, including a reduction of viral titers, amelioration of inflammation, and complete resolution of myocardial damage. But some other patients may experience chronic inflammation and dilated cardiomyopathy due to the persistence of the viral genome or cross-reactive antibodies (such as antibodies targeting cardiac myosin) (Fairweather et al., 1998; Calabrese and Thiene, 2003). If inflammation persists, the release of proinflammatory cytokines can lead to the activation of matrix metalloproteinases (MMPs) and the production of pro-fibrotic cytokines, both of which can cause cardiac remodeling and fibrosis. Meanwhile, antigenic cross-reactions can also aggravate cell damage (Sagar et al., 2012).
Innate immunity is regarded as the first line of defense in the human immune system, responding to PAMPs and DAMPs with the assistance of pattern recognition receptors (PRRs) (Karasawa and Takahashi, 2017). A great number of PRRs are involved in the proinflammatory process via directly inducing the formation of the corresponding inflammasome (Figure 1).
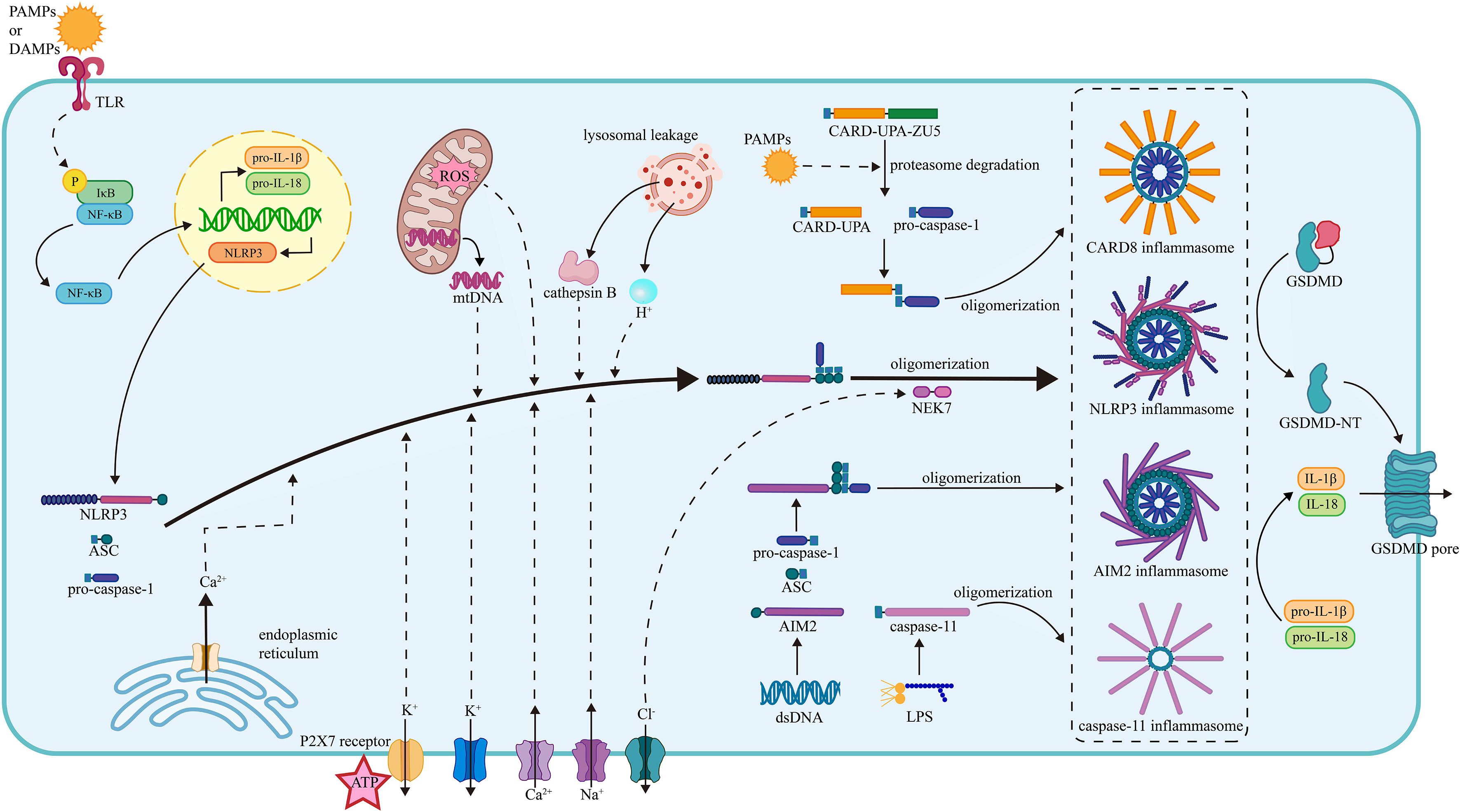
Figure 1 The mechanism of inflammasome activation. (1) The NLRP3 inflammasome activation. The activation of the NLRP3 inflammasome requires two steps of stimuli. The first signals are mainly PAMPs or DAMPs, interacting with the PRRs and activating the NF-κB pathway, thus promoting the transcription of NLRP3, pro-IL-1β and pro-IL-18. The second signals promote the assembly and oligomerization of the NLRP3 inflammasome. The efflux of K+, influx of Na+, mobilization of Ca2+ (from ER and extracellular), mtROS, mtDNA, lysosomal leakage of H+ and cathepsin B facilitate the assembly of NLRP3, ASC, and pro-caspase-1. The efflux of Cl- facilitates the NEK7 attachment and thus the oligomerization. (2) The CARD8 inflammasome activation. The PAMPs induce the degradation of CARD8 sensor in proteasome and facilitate the release of CARD-UPA domain, which recruits pro-caspase-1 and results in the formation of CARD8 inflammasome. (3) The caspase-11 inflammasome activation. Under the stimulation of LPS, the caspase-11 inflammasome is formed, gains proteolytic activity, and cleave. (4) The AIM2 inflammasome. The AIM2 senses the dsDNA and then recruits the ASC and pro-caspase-1, facilitating the assembly and oligomerization of the AIM2 inflammasome. The oligomerization of pro-caspase-1 facilitates the maturation of caspase-1. Caspase-1 cleaves pro-IL-1β and pro-IL-18 into mature IL-1β and IL-18. Besides, the caspase-1 splits GSDMD into two fragments and the N-terminal fragment form pores on plasma membrane promoting the release of cytokines. NLRP3, nucleotide oligomerization domain (NOD)-, leucine-rich repeat (LRR)- and pyrin domain (PYD)-containing protein3; PAMP, pathogen- associated molecular patterns; DAMP, damage-associated molecular patterns; TLR, toll-like receptor; NF-κB, nuclear factor κ-light-chain enhancer of activated B cells; ASC, apoptosis-associated speck-like protein containing a CARD; NEK7, NIMA-related kinase 7; NIMA, never in mitosis gene A; ROS, reactive oxygen species; CARD8, caspase recruitment domain-containing protein 8; LPS, lipopolysaccharide; AIM2, absent in melanoma 2; GSDMD, gasdermin D.
The NLRP3 inflammasome is an oligomeric intracellular multiprotein complex that contains three parts: NLRP3, apoptosis-associated speck-like protein containing a CARD (ASC), and pro-caspase-1 (Karasawa and Takahashi, 2017). NLRP3 has a general tripartite construction of NOD-like receptors (NLRs) proteins (Kim et al., 2016), comprising the central NOD domain (also known as NAIP, CIITA, HET-E, and TP-2 [NACHT]), the C-terminal leucine-rich repeat (LRR) domain, and the N-terminal pyrin as the effector domain (Ren et al., 2017). The LRR domain usually serves as an adaptor to the NACHT domain and detector of PAMPs and DAMPs in the NLR family (Kim et al., 2016). ASC is a speck-like protein consisting of a C-terminal CARD and an N-terminal PYD (Srinivasula et al., 2002) that performs as the connection between NLRP3 and pro-caspase-1. Pro-caspase-1 can perform auto-cleavage and generate cleaved caspase-1, which is a vital protease in human homeostasis and inflammatory reactions. Caspase-1 forms an active oligomer, cleaving pro-IL-1β and pro-IL-18 into mature IL-1β and IL-18 and promoting their release (Guo et al., 2015).
After TLRs detect PAMPs and DAMPs, the IκB kinase (IKK) complex in the cytoplasm is activated, leading to the phosphorylation, ubiquitination, and degradation of the inhibitor of NF-κB (IκB) and releasing the active NF-κB molecule (Akira et al., 2006; Solt and May, 2008). Then NF-κB enters the nucleus and initiates the transcription of the NLRP3, pro-IL-1β, and pro-IL-18 (Bauernfeind et al., 2009).
After the first stimuli of PAMPs and DAMPs, the NLRP3 just released into the cytoplasm is auto-suppressed and inactive. To generate the NLRP3 inflammasome with full proteolytic activity, the second stimuli are needed (Bauernfeind et al., 2009), mainly consisting of ionic flux, reactive oxygen species (ROS) and mitochondrial dysfunction, and lysosomal damage (Zhou et al., 2011). Under the second stimuli, NLRP3 tends to undergo the conformational change of self-oligomerization. The pyrin domain of NLRP3 binds to ASC, initiating the recruitment of ASC. CARD on ASC connects with pro-caspase-1 via the homotypic CARD–CARD interaction, and PYD on ASC also tightly connects with the PYD on the NLRP3 through the similar PYD-PYD interaction (Sborgi et al., 2015; Schmidt et al., 2016; Fernandes-Alnemri et al., 2007). The oligomerization of the pro-caspase-1 on ASC maturates the caspase-1, inducing subsequent inflammatory processes.
As mentioned above, the NLRP3 inflammasome activation requires two steps of stimulation. The first signals are mainly PAMPs or DAMPs, activating the PRRs and inducing the transcription of the NLRP3, pro-IL-1β, and pro-IL-18 via the NF-κB pathway. The second signals activate NLRP3 and promote the assembly of the inflammasome (Bauernfeind et al., 2009), mainly consisting of ionic flux, reactive oxygen species (ROS) and mitochondrial dysfunction, and lysosomal damage (Zhou et al., 2011).
Ionic flux is the main trigger of the NLRP3 inflammasome activation. The efflux of potassium (K+) has been considered a prevalent ion event triggering the activation of the NLRP3 inflammasome. It has been revealed that a low intercellular K+ concentration was capable of activating the NLRP3 inflammasome alone, while an increased extracellular K+ concentration held up the activation (Mohamed, 2015; Kwak et al., 2018). In addition, ATP is recognized as a vital activator of the NLRP3 inflammasome. However, previous research found that mice with a genetic lack of P2X7 receptor (P2X7R) did not release IL-1β in response to ATP (Solle et al., 2001; Mariathasan et al., 2004). This implies that ATP alone cannot activate the NLRP3 inflammasome. Instead, it relies on P2X7R to mediate the activation. P2X7R is a bi-functional ATP-gated plasma membrane ion channel that allows the efflux of K+ and the mobilization of calcium (Ca2+) while receiving the stimulus of ATP (Martínez-Cuesta et al., 2020). The mobilization of Ca2+ is also a common ionic event in the cell membrane system, participating in multiple intracellular signaling pathways (Clapham, 2007). Inositol 1,4,5-trisphosphate receptor (IP3R) is a Ca2+-release channel on the ER. It can be triggered by IP3, which is the downstream product of phospholipase C (PLC)-mediated phosphatidylinositol 4,5-bisphosphate (PIP2) cleavage. Previous evidence showed that inhibiting either IP3R or PLC could lead to the blockade of the NLRP3 inflammasome activation without external stimuli (Lee et al., 2012). Other than K+ and Ca2+, the ionic flux of sodium (Na+) and chloride (Cl-) plays a similar function in the NLRP3 inflammasome activation. Na+ influx usually leads to water influx and cellular swelling, lowering the intracellular K+ concerntration and promoting K+ efflux which activates the NLRP3 inflammasome (Schorn et al., 2011). Cl- efflux promotes NLRP3-NEK7 contact and the subsequent NLRP3-ASC complex formation and ASC oligomerization, thus facilitating the NLRP3 inflammasome assembly and activation (Tang et al., 2017).
Mitochondrial dysfunction results in the breakdown of the oxidation respiratory chain reaction, causing oxidative stress to accumulate in terms of mitochondrial ROS (mtROS). ROS is a broad group of substances decomposed by specialized cellular enzymes, including peroxidases and superoxide dismutases (Fidanboylu et al., 2011). It has been proved to be a trigger of the NLRP3 inflammasome activation through experiments with the application of chemical inhibitors (Dostert et al., 2008). Besides, another production of mitochondrial dysfunction is mitochondrial DNA (mtDNA). Multiple screens found that released mtDNA tended to interact with PRRs such as NLRP3 and AIM2. An in vitro study with synthesized mtDNA supported the view that released mtDNA in the cytoplasm would be oxidized into oxidized mtDNA, which promoted the NLRP3 inflammasome activation (Zhong et al., 2018).
Lysosomal damage causes two significant consequences: lysosomal acidification and lysosomal content leakage, both of which contribute to the NLRP3 inflammasome activation. Suppressing lysosomal H+ flux with a corresponding inhibitor resulted in the inhibition of particulate matter-induced NLRP3 inflammasome activation (Hornung et al., 2008), which indicated that lysosomal acidification was critical to the NLRP3 inflammasome activation. Lysosomal leakage matters are mainly lysosomal enzymes. Cathepsin B is an important lysosomal enzyme that is proven to be essential in the secretion of IL-1β, but not the transcription and maturation. Related findings were obtained from further studies on cathepsins such as cathepsin L, C, S, and X (Orlowski et al., 2015).
The caspase recruitment domain-containing protein 8 (CARD8) inflammasome consists of the sensor CARD8 and the effector pro-caspase-1 (Jin et al., 2022). The sensor CARD8 has two domains: the C-terminal CARD and the N-terminal function-to-find domain (FIIND), which comprises two subdomains named ZU5 and UPA (Taabazuing et al., 2020). Under the inducement of specific PAMPs, the FIIND of CARD8 undergoes degradation in proteasomes and leads to the release of the C-terminal bioactive subunit UPA-CARD, in which the CARD domain recruits pro-caspase-1 through CARD-CARD interaction and then results in the CARD8 inflammasome activation (Taabazuing et al., 2020; Wang and Shan, 2022). Specifically, it has been observed that when CVB3 infected aortic endothelial cells or cardiomyocytes, the CARD8 inflammasome would be activated and accelerate the viral release to neighboring target cells (Wang and Shan, 2022).
The non-canonical caspase-11 inflammasome robustly mediates anti-bacterial innate immune responses of murine by responding to the bacterial lipopolysaccharide (LPS) (Hagar et al., 2013; Kayagaki et al., 2013). Caspase-11 comprises two domains connected by a CARD domain linker (CDL): an N-terminal CARD and a protease domain, and the protease domain consists of a large subunit and a small subunit separated by an interdomain linker (IDL) (Ross et al., 2022). It has been reported that the CARD of caspase-11 could directly bind to LPS, leading to the oligomerization and activation of the caspase-11 inflammasome (Shi et al., 2014). In another research, caspase-11 gained its basal protease activity by LPS-induced dimerization, which was inadequate for the cleavage of GSDMD (Ross et al., 2018). Following the dimerization, the self-cleavage at site D285 on the IDL of caspase-11 generated the fully active protease species, which could cleave GSDMD and promote pyroptosis (Ross et al., 2018). Compared to canonical inflammasomes such as the NLRP3 inflammasome, the activation process of the caspase-11 inflammasome shows distinct features: the caspase-11 directly binds to the LPS and assembles without the aid of signaling sensors or adaptors (Shi et al., 2014; Ross et al., 2018). Furthermore, caspase-11 exists in murine, while caspase-4 and caspase-5 are the human orthologs of murine caspase-11 (Lamkanfi et al., 2002). It has been observed that caspase-4 and caspase-5 could also bind to LPS and be activated by the LPS inducement (Shi et al., 2014), which indicated that caspase-4 and caspase-5 might undergo their activation in similar ways as caspase-11.
Absent in melanoma 2 (AIM2), a double-stranded DNA sensor, comprises a PYD and a C-terminal HIN domain, which can recognize and combine with autologous or foreign DNA (Hornung et al., 2009). When the HIN domain combines with DNA, the PYD interacts with the PYD of the adaptor ASC, and the CARD of ASC links to the CARD of pro-caspase-1, promoting the assembly and activation of the AIM2 inflammasome (Bürckstümmer et al., 2009). The oligomerization of caspase-1 in the AIM2 inflammasome improves its ability to process pro-IL-1β into bioactive IL-1β. Besides, caspase-1 can also split gasdermin D (GSDMD) into two fragments, and the N-terminal fragment forms large pores on the plasma membrane, leading to IL-1β release and cell pyroptosis (Lugrin and Martinon, 2018).
During the infectious phase of VMC, multiple categories of molecules of either host cells or viruses participate in the regulation of the inflammasome activation and inhibition, thus influencing the development of VMC.
Extensive research has demonstrated that the NLRP3 inflammasome is the main inflammasome involved in CVB3-induced VMC (Wang et al., 2018; Wang et al., 2014; Tschöpe et al., 2017; Wang et al., 2019; Tong et al., 2020; Liu et al., 2022). The NLRP3 inflammasome is activated through a variety of pathways during CVB3 infection, promoting the production and secretion of IL-1β and IL-18 and exacerbating pyroptosis. Several mechanisms of the regulation of the NLRP3 inflammasome activation in CVB3-induced VMC are discussed below.
PRRs including nucleotide-binding oligomerization domain 2 (NOD2) and TLR4 may play significant roles in the NLRP3 inflammasome activation in CVB3-induced VMC. It has been reported that in the context of CVB3-induced VMC, NOD2 knockout mice manifested lower NLRP3 and ASC levels in the left ventricular and serum IL-1β levels in comparison with wild-type mice. Furthermore, the NOD2-mediated NLRP3 inflammasome activation has been demonstrated to be CVB3-dependent (Tschöpe et al., 2017). Besides, in mice with CVB3-induced VMC, TLR4 deficiency has been demonstrated to alleviate the production of two typical inflammasome related cytokines, IL-1β and IL-18 (Fairweather et al., 2003). Extensive colocalization between TLR4 and enterovirus capsid protein VP1 has been detected in the cytoplasm of cardiomyocytes obtained from DCM patients (Satoh et al., 2004), which indicated the possible interaction between the virus and TLR4 in cardiomyocytes. However, the detailed mechanisms of NOD2/TLR4-related NLRP3 inflammasome activation need further elucidation.
K+ efflux robustly promotes the NLRP3 inflammasome activation in CVB3-induced VMC. It has been demonstrated that in CVB3-infected cardiomyocytes, the NLRP3 inflammasome activation accounted for CVB3-induced IL-1β secretion, and once K+ efflux was inhibited by culturing cardiomyocytes in K+-rich medium, the CVB3-induced caspase-1 activity and IL-1β secretion were significantly down-regulated (Wang et al., 2014). Furthermore, to avoid the possibility that the functions of cardiomyocytes might be influenced not only by the K+ channels but also some voltage-gated Na+ channels, glibenclamide, an ATP-sensitive K+-channel inhibitor, was used to block K+ efflux and, in turn, robustly suppressed IL-1β secretion of CVB3-infected cardiomyocytes (Wang et al., 2014). These data demonstrated the role of K+ efflux in the NLRP3 inflammasome activation in CVB3-induced VMC.
The calpains are a conserved family of Ca2+-dependent cysteine proteases expressed generally in all cells (Goll et al., 2003), and calpain-1 has been demonstrated to be activated in CVB3-induced VMC to activate the NLRP3 inflammasome through promoting mitochondrial dysfunction and mtROS production (Liu et al., 2022). Liu et al. have found that in the cardiomyocytes of mice with CVB3-induced VMC, calpain was detected to be activated. The NLRP3 inflammasome activation triggered by CVB3 infection could be inhibited by overexpression of calpastatin, a natural and specific endogenous inhibitor of calpain activity. Meanwhile, the NLRP3 inflammasome activation could also be inhibited by mito−TEMPO, a mitochondrial-targeted antioxidant in cardiovascular conditions that could reduce CVB3-induced mtROS level (Liu et al., 2022). The relationship between calpain-1 and mtROS in CVB3-induced VMC could be further elucidated by the fact that mitochondrial function was improved when calpain activity was inhibited by calpastatin in vivo, and that the GSDMD N-terminus and caspase-1 exacerbated by CVB3-induced calpain-1 overexpression could be suppressed by mito-TEMPO (Liu et al., 2022). As the results above showed, excessive calpain-1 activity could increase mtROS, which activated the NLRP3 inflammasome in mice with CVB3-induced VMC (Liu et al., 2022). Furthermore, under CVB3 stimulation, calpain-1 was observed accumulating in the mitochondria and reducing the expression of ATP synthase-α (ATP5A1) (Liu et al., 2022), which was significant for mitochondrial function (Ni et al., 2016), and the reduction of ATP5A1 led to the NLRP3 inflammasome activation in cardiomyocytes (Liu et al., 2022). In summary, in the context of CVB3-induced VMC, CVB3-induced calpain-1 translocation from the cytoplasm to mitochondria reduces the expression of ATP5A1, a subunit of mitochondrial ATP synthase 1. The decrease of ATP5A1 impairs mitochondrial function and increases mtROS production, which subsequently activates the NLRP3 inflammasome and induces the pyroptosis of cardiomyocytes.
Cathepsins are a family of lysosomal cysteine proteases participating in multiple cellular processes, among which cathepsin B may play an important role in CVB3-induced VMC. Wang et al. have observed that cathepsin B was activated in both acute and chronic stages of CVB3-induced VMC in mice and intensified cardiomyocyte damage (Wang et al., 2018). Furthermore, they found that the activated cathepsin B elevated protein levels of NLRP3, ASC, caspase-1 p20, and IL-1β. More importantly, the activated cathepsin B exacerbated caspase-1 activity and myocardial pyroptosis, which were both significant outcomes of the NLRP3 inflammasome activation (Wang et al., 2018). The results above demonstrated that activated cathepsin B promoted the NLRP3 inflammasome activation and exacerbated myocardial symptoms in CVB3-induced VMC.
In recent years, microRNAs (miRs) have been identified to regulate gene expression at the transcriptional and post-transcriptional levels (Li et al., 2019). MiRs induce mRNA degradation or terminate transcription by binding to the 3’-untranslated region (UTR) of mRNAs (Bartel, 2004). Multiple miRs have been associated with VMC, including miR-1, miR-15, miR-21, miR-146, miR-155, miR-221, miR-222, and miR-223 (Chau et al., 2007; Rose, 2009; Blauwet and Cooper, 2010; Smith et al., 2017; Tong et al., 2020; Xue et al., 2020). Among them, miR-15 and miR-223 play important roles in regulating the NLRP3 inflammasome activation in CVB3-induced VMC.
An experiment demonstrated that the miR-15-NLRX1 axis was involved in the regulation of the NLRP3 inflammasome activation in CVB3-induced VMC (Tong et al., 2020). Nucleotide-binding domain and leucine-rich-repeat-containing family member X1(NLRX1), a member of the NOD-like receptor family, has been proved closely associated with inflammatory diseases (Karin et al., 2006; Kang et al., 2015). The expression of miR-15 dramatically increased after CVB3 infection, which down-regulated the expression of NLRX1. Once the expression of the NLRX1 protein was restrained by increased miR-15, the production of IL-1β and IL-18 increased through the NLRP3 inflammasome activation pathway, followed by intensified inflammatory responses, reduced cell viability, and promoted cell apoptosis. As a result, myocardial injury was aggravated. In summary, during the CVB3-induced VMC, the increased miR-15 represses NLRX1, contributing to the NLRP3 inflammasome activation. This implies a novel pathway for the NLRP3 inflammasome activation.
In addition to miR-15, the role of miR-223 is also remarkable. In CVB3-induced VMC mice, the researchers found that the levels of long non-coding RNA (lncRNA) maternally expressed gene 3 (MEG3) in the myocardium increased, resulting in miR-223 down-regulation. MiR-223 targets the mRNA of the TNF receptor‐associated factor 6 (TRAF6), a ubiquitin E3 ligase essential for IKK activation in the IL-1 and TLR pathways (Lomaga et al., 1999; Naito et al., 1999; Xue et al., 2020). Furthermore, the researchers found that the inhibition of miR-223 up-regulated TRAF6 (Xue et al., 2020). The up-regulation of TRAF6 activated the NF‐κB pathway to promote the NLRP3 inflammasome activation, elevating the protein levels of related inflammatory cytokines (Xue et al., 2020). Besides, another research demonstrated that in CVB3-infected mice, the treatment of A20, a TRAF6 inhibitor also known as tumor necrosis factor alpha induced protein 3 (TNFAIP3), effectively alleviated CVB3-induced VMC by down-regulating TRAF6 (Gui et al., 2012). The role of ubiquitination in the pathogenic process of VMC is further established, which is consistent with previous findings that ubiquitination plays significant roles in regulating the inflammasome function (Xu and Núñez, 2022).
As discussed above, multiple molecules participate in the regulation of the NLRP3 inflammasome activation in the VMC, including PRRs, K+, calpain-1, cathepsin B and microRNA, all of which participate in the development of the VMC. Interestingly, it has been reported that CVB3 protease 3C could degrade NLRP3 (Wang et al., 2019). Meanwhile, CVB3 protease 3C could also degrade receptor-interacting protein (RIP)1/RIP3, two molecules contributing to the NLRP3 inflammasome activation through the RIP1-RIP3-dynamin-related protein 1(DRP1)-NLRP3 inflammasome pathway (Wang et al., 2014; Wang et al., 2019). CVB3 might adopt this cleavage method to ameliorate the NLRP3 inflammasome activity in order to escape the host immune response. However, it is more widely observed that, upon CVB3 infection, various kinds of molecular events promote rather than inhibit the activation of the NLRP3 inflammasome, thereby exacerbating the symptoms of VMC.
Apart from the NLRP3 inflammasome, the CARD8 inflammasome can be activated by CVB3 proteases and participate in the progress of CVB3-induced VMC. Nadkarni et al. have demonstrated that in CVB3-infected human aortic endothelial cells (HAECs), CVB3 2A protease and 3C protease could cleave the CARD8 N-terminus. The cleaved CARD8 neo-N-terminus underwent degradation by proteasomes, releasing the bioactive UPA-CARD domain, which could interact with pro-caspase-1 and promote the CARD8 inflammasome assembly. Then the CARD8 inflammasome facilitated GSDMD cleavage, cell pyroptosis, and viral release to adjacent target cells (Nadkarni et al., 2022). Importantly, the researchers also demonstrated that CARD8 knockout HAECs protected the underlying cardiomyocytes from CVB3 infection (Nadkarni et al., 2022), indicating that the CARD8 inflammasome activity in CVB3-infected endothelial cells might increase the risk of viral infection in myocardium and even cause VMC. Furthermore, in CVB3-infected cardiomyocytes, knockout of CARD8 led to a reduction of cleaved GSDMD and ameliorated cell death (Nadkarni et al., 2022), suggesting that the CARD8 inflammasome also directly participated in the pyroptotic progress of CVB3-infected cardiomyocytes. In summary, the CARD8 inflammasome may participate in pyroptosis in different cell types and exacerbate the VMC after the viral infection.
Besides the canonical inflammasomes mentioned above, the non-canonical caspase-11 inflammasome also participates in the VMC. Calpain has been frequently observed up-regulated in hearts of mice with CVB3-induced VMC (Yu et al., 2020; Liu et al., 2022). Yu et al. have found that calpain was strongly activated and exacerbated pyroptosis in hearts of CVB3-infected mice (Yu et al., 2020), while in the calpastatin transgenic mouse strain (Tg-CAST) that overexpressed calpastatin, a natural and specific endogenous inhibitor of calpain, the severity of VMC was significantly lower than in the wild-type mice (Yu et al., 2020). The researchers further found that in the hearts of CVB3-infected mice, the inhibition of calpain not only down-regulated the expression of canonical NLRP3 inflammasome components, including NLRP3, ASC, and caspase-1, but also significantly suppressed the expression of caspase-11 (Yu et al., 2020). The results suggested that the activation of the non-canonical caspase-11 inflammasome might participate in the VMC as a downstream molecular event of calpain activation. However, more evidence is needed to elucidate the detailed mechanisms of caspase-11 inflammasome activation in the context of VMC. Although the involvement of the caspase-4 inflammasome in myocardial diseases such as myocardial reperfusion-induced microvascular injury has been reported (Sun et al., 2021), the roles of the caspase-4 inflammasome and the caspase-5 inflammasome in VMC have not been established and need more exploration.
The AIM2 inflammasome activation has been reported to exacerbate symptoms in multiple cardiac conditions, such as heart failure and myocardial infarction (Onódi et al., 2021; Liao et al., 2022). Although there has been no evidence that the AIM2 inflammasome directly participated in the VMC, Furrer et al. reported that AIM2 could inhibit NF-κB p65 acetylation and phosphorylation in cardiomyocytes, thus directly suppressing proinflammatory cytokine transcription (Furrer et al., 2016). The researchers also found that AIM2 inhibited inflammatory cytokine (including IL-6, IP-10, and TNF-α) transcription in cardiomyocytes indirectly via limiting phosphorylation of signal transducer and activator of transcription 1 (STAT1), an important regulator in the NF-κB pathway (Furrer et al., 2016). Interestingly, in cardiomyocytes, knockout of caspase-1 and AIM2 manifested the same profile of proinflammatory cytokine transcription as the mere knockout of AIM2 (Furrer et al., 2016), indicating that the AIM2-mediated suppression on proinflammatory cytokine production in cardiomyocytes possibly followed a caspase-1/inflammasome-independent manner (Furrer et al., 2016). The research above suggested that, apart from acting as an inflammasome sensor, AIM2 might play other potential roles in VMC. Besides, AIM-2 co-immunization has been reported to ameliorate symptoms of CVB3-induced VMC via multiple mechanisms, such as promoting specific multifunctional CD8 T cell induction, facilitating protective secretory immunoglobulin A (SIgA) response and increasing prophylactic efficacy of chitosan-DNA vaccine (Chai et al., 2014; Chai et al., 2015), all of which seemingly showed the potential of AIM2 in preventing VMC. However, the mechanisms underlying the AIM2 activity in VMC are not fully established, and further investigations are needed.
Among the downstream molecules of inflammasomes, IL-1β participates in multiple cellular events in both infectious and post-infectious phases, indicating that inflammasomes can influence the development of VMC through the secretion of IL-1β. (Figure 2).
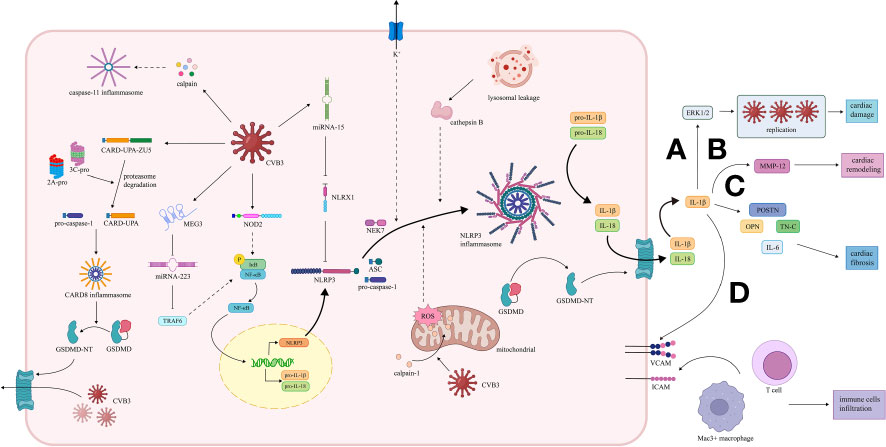
Figure 2 Activation and effect of inflammasome pathway in VMC induced by CVB3. (1) CARD8 inflammasome pathway. The 2A and 3C proteases of CVB3 promote the cleavage of CARD8 to form the CARD8 inflammasome, which provides a platform for the maturation of pro-caspase-1. Caspase-1 cleaves GSDMD, and the transfer of the GSDMD N-terminal fragment to the cell membrane increases the permeability of the cell membrane, thereby promoting the release of replication-generated CVB3. (2) NLRP3 inflammasome pathway. CVB3 is recognized by NOD2, activating the NF-κB pathway, leading to the transcription of NLRP3, pro-IL-1 and pro-IL-18. CVB3-induced NLRP3 inflammasome activation in VMC is mediated by potassium efflux, release of cathepsin B caused by lysosomal damage, and ROS and mitochondrial dysfunction due to the transfer of calpain-1 to mitochondria. The NLRP3 inflammasome promotes the maturation of pro-IL-1β and pro-IL-18 and the cleavage of GSDMD. IL-1β plays a significant role in the development of VMC. MicroRNAs, including miR-15/223, are also involved. MiR-15 is up-regulated to repress NLRX1, while miR-223 is down-regulated to elevate the TRAF6, both contributing to the NLRP3 inflammasome activation. (A) IL-1β induces high expression of ERK1/2, facilitating viral replication, leading to damage and inflammation of cardiac tissue. (B) IL-1β promotes transcription of MMP12 in myocytes, thus leading to cardiac remodeling. (C) IL-1β promotes the expression of OPN, TN-C, POSTN, and IL-6, contributing to cardiac fibrosis. (D) IL-1β enhances the expression of ICAM and VCAM, promoting the infiltration of immune cells, including Mac3+ macrophages and CD3+ T cells. (3) Caspase-11 inflammasome pathway. The calpain activation induced by CVB3 can also activates caspase-11 inflammasome, which may participate in the pathogenesis of VMC. VMC, viral myocarditis; CVB3, coxsackie virus B3; CARD8, caspase recruitment domain-containing protein 8; GSDMD, gasdermin D; NLRP3, nucleotide oligomerization domain-, leucine-rich repeat-, and pyrin domain-containing protein 3; NOD2, nucleotide-binding oligomerization domain 2; TLR, toll-like receptors; NF-κB, nuclear factor κ-light-chain enhancer of activated B cells; ROS, reactive oxygen species; miR, microRNA; NLRX1, Nucleotide-binding domain and leucine-rich-repeat-containing family member X1; TRAF6, TNF receptor‐associated factor 6; ERK 1/2, extracellular signal-regulated kinases 1 and 2; MMP12, matrix metalloproteinase 12; OPN, osteopontin; TN-C, tenascin C; POSTN, periostin; ICAM, intercellular cell adhesion molecule; VCAM, vascular cell adhesion molecule.
The activation of extracellular signal-regulated kinases 1 and 2 (ERK1/2), two significant molecules in IL-1β pathway, has been demonstrated to facilitate viral replication, exacerbate viral infectivity, and decrease cell survival in Hela cells (Luo et al., 2002; Weber et al., 2010). High levels of ERK1/2 were observed in the hearts of CVB3-infected mice, and ERK1/2 expression was detected spatially identical with myocardial damage and inflammation (Kraft et al., 2019), which were consistent with the results in Hela cells. However, in mice with CVB3-induced VMC, neutralization of IL-1β with IL-1β antibody led to the reduction of both viral replication and myocardial damage (Kraft et al., 2019). More importantly, IL-1β blockade robustly down-regulated the ERK1/2 expression (Kraft et al., 2019), which provided a reasonable explanation for the ameliorated viral replication. It can be deduced from the facts above that IL-1β matured by inflammasomes may exacerbate symptoms of CVB3-induced VMC by promoting viral replication.
The intercellular cell adhesion molecule (ICAM) and vascular cell adhesion molecule (VCAM) are vital for the adhesion and infiltration of leukocytes (Seko et al., 1996). It has been reported that both ICAM-1 and VCAM-1 were up-regulated in cardiomyocytes and participated in the development of myocarditis in mice with CVB3-induced VMC (Seko et al., 1993; Seko et al., 1996). Kraft et al. found that in the hearts of CVB3-infected mice, IL-1β antibody treatment in the acute stage of VMC down-regulated adhesion molecules, including ICAM-1 and VCAM-1, in the chronic stage of VMC, and inhibited infiltration of immune cells, including Mac3+ macrophages and CD3+ T cells, thus alleviating the CVB3-induced inflammation (Kraft et al., 2019). It is reasonable to deduce that in CVB3-induced VMC, IL-1β secretion via the inflammasome pathway can exacerbate the inflammatory responses by promoting the adhesion molecule expression and immune cell infiltration, which might promote enduring inflammation and cardiac symptoms.
In hearts, IL-6 is mainly produced by macrophages and fibroblasts, and it can be up-regulated under IL-1β stimulation (Ma et al., 2012; Althof et al., 2018; Kishimoto and Kang, 2022). The reduction of IL-6 has been reported to ameliorate cardiac fibrosis (Ma et al., 2012; Segers et al., 2018). Kraft et al. reported that in mice with CVB3-induced VMC, neutralization of IL-1β by IL-1β antibody in the acute stage of VMC robustly suppressed both the IL-6 expression and collagen type I deposition and inhibited the cardiac fibrosis process in the chronic stage (Kraft et al., 2019), which was consistent with previous publications. These studies indicate that the inflammasome-induced IL-1β secretion may promote IL-6 expression, thus exacerbating cardiac fibrosis during the chronic VMC.
Matricellular proteins are nonstructural proteins binding to the extracellular matrix and participate in the regulation of cell survival, differentiation, and mobilization (Zohar et al., 2004; Harris et al., 2011). Among them, osteopontin (OPN), tenascin C (TN-C), and periostin (POSTN) can accelerate cardiac fibrosis (López-Sánchez et al., 2017; Abdelaziz Mohamed et al., 2019; Mohammadzadeh et al., 2020). It has been demonstrated that the three matricellular proteins mentioned above were highly expressed in the hearts of CVB3-infected mice, while they were all down-regulated in the chronic stage by treating with IL-1β antibody during the acute stage of VMC (Kraft et al., 2019). Therefore, the inflammasome-induced IL-1β secretion may exacerbate cardiac fibrosis by up-regulating the matricellular proteins.
Matrix metalloproteinases (MMPs) are a group of proteolytic enzymes whose main function is the regulation of extracellular matrix reconstruction (Spinale, 2007). Previous studies showed vital involvement of MMPs in the cardiac remodeling process after the CVB3 infection (Westermann et al., 2010). Among multiple MMPs, MMP12 was observed remaining at a high transcription level while the other MMPs decreased after the acute stage of CVB3-induced VMC (Westermann et al., 2010), which indicated the possible involvement of MMP12 in the cardiac remodeling process. Kraft et al. found high expression levels of MMP12 in the hearts of mice during the chronic stage of CVB3-induced VMC, while MMP12 expression in the chronic stage was down-regulated significantly by IL-1β antibody in the acute stage (Kraft et al., 2019). The results above indicated that IL-1β secretion via the NLRP3 inflammasome pathway might contribute to the cardiac remodeling process by regulating the MMP12.
A wide variety of cardiovascular diseases after SARS-Cov-2 infection have been reported, including myocarditis (Boukhris et al., 2020; Irabien-Ortiz et al., 2020; Sawalha et al., 2021), and the NLRP3 inflammasome may be activated by SARS-Cov-2 infection and participate in the progression of Corona Virus Disease 2019 (COVID-19)-related myocarditis. Viral presence has been observed in the myocardium after SARS-Cov-2 infection by endomyocardial biopsy (EMB) and autopsy (Albert et al., 2020; Fox et al., 2020; Lindner et al., 2020), which indicated possible viral infection of cardiomyocytes in the COVID-19 context. Angiotensin-converting enzyme 2 (ACE2) is a host cellular receptor that can bind to the spike (S) protein of SARS-Cov-2 and facilitate viral entry into the host cell with the aid of TMPRSS2 (Fox et al., 2020; Quagliariello et al., 2020). Both ACE2 and TMPRSS2 have been demonstrated to be expressed in the heart (Feng et al., 2021; Chen et al., 2020; Clerkin et al., 2020; Nicin et al., 2020). Meanwhile, ACE2 can also cleave angiotensin (Ang) II to generate Ang- (1-7), thus reducing the level of Ang II (Santos et al., 2018). From the facts above, it is reasonable to deduce that with increasing viral infection and replication in myocardium, the S protein-ACE2 interaction and internalization of ACE2 after the interaction are enhanced, thus ameliorating the ACE2-Ang II interaction. The possible inhibition of the ACE2-Ang II interaction may suppress Ang II degradation and lead to Ang II accumulation. Importantly, it has been demonstrated that excessive Ang II could induce cardiomyocyte pyroptosis and cardiac fibrosis through activating the NLRP3 inflammasome (Pinar et al., 2020; Zhu et al., 2021), which manifested similar features of myocarditis. Thus, it is reasonable to suggest that SARS-Cov-2 infection may participate in COVID-19-related myocarditis by activating the NLRP3 inflammasome.
Cytokine release syndrome is a systemic inflammatory response triggered by adverse external factors such as viral infection (Shimabukuro-Vornhagen et al., 2018). The migration of activated CD8 T cells to the heart triggers cytokine release syndrome, leading to cardiac injury. This inflammatory cytokine storm is considered to be the main mechanism of acute COVID-19-related myocarditis (Chimenti et al., 2022). Furthermore, possible roles of the NLRP3 inflammasome in this process were also well-discussed. After SARS-CoV-2 infection, the NLRP3 inflammasome can be activated by multiple stimuli, including Ca2+ mobilization, K+ efflux, ROS production, Ang II accumulation induced by the S protein-ACE2 pathway, and the complement cascade induced by SARS-CoV-2 infection (Zhao et al., 2021). As the NLRP3 inflammasome-induced proinflammatory cascade progresses, IL-1β and IL-18 promote the release of other cytokines, including IL-6, which is hypothesized to be a central mediator of cytokine storm. IL-6 coordinates the proinflammatory responses of immune cells to further promote the release of inflammatory cytokines and achieve the positive feedback regulation that induces the cytokine storm, thus contributing to the cytokine release syndrome (Coomes and Haghbayan, 2020). Moreover, it has been suggested that suppressing the NLRP3 inflammasome with inhibitors such as MCC950 could effectively prevent the development of the cytokine storm. This study demonstrated the significant participation of the NLRP3 inflammasome in inducing the inflammatory cytokine storm, the main pathogenic mechanism of COVID-19-related myocarditis (Ratajczak et al., 2021), and provided ideas for the treatment of COVID-19-related myocarditis.
The well-established roles of inflammasomes, especially the NLRP3 inflammasome, in the VMC provide multiple ideas for possible therapies for VMC targeting the inflammasome pathway. Several drugs inhibiting the NLRP3 inflammasome or its downstream IL-1β are reported to be promising therapies for VMC in a variety of case reports, clinical trials, and animal experiments (Table 1).
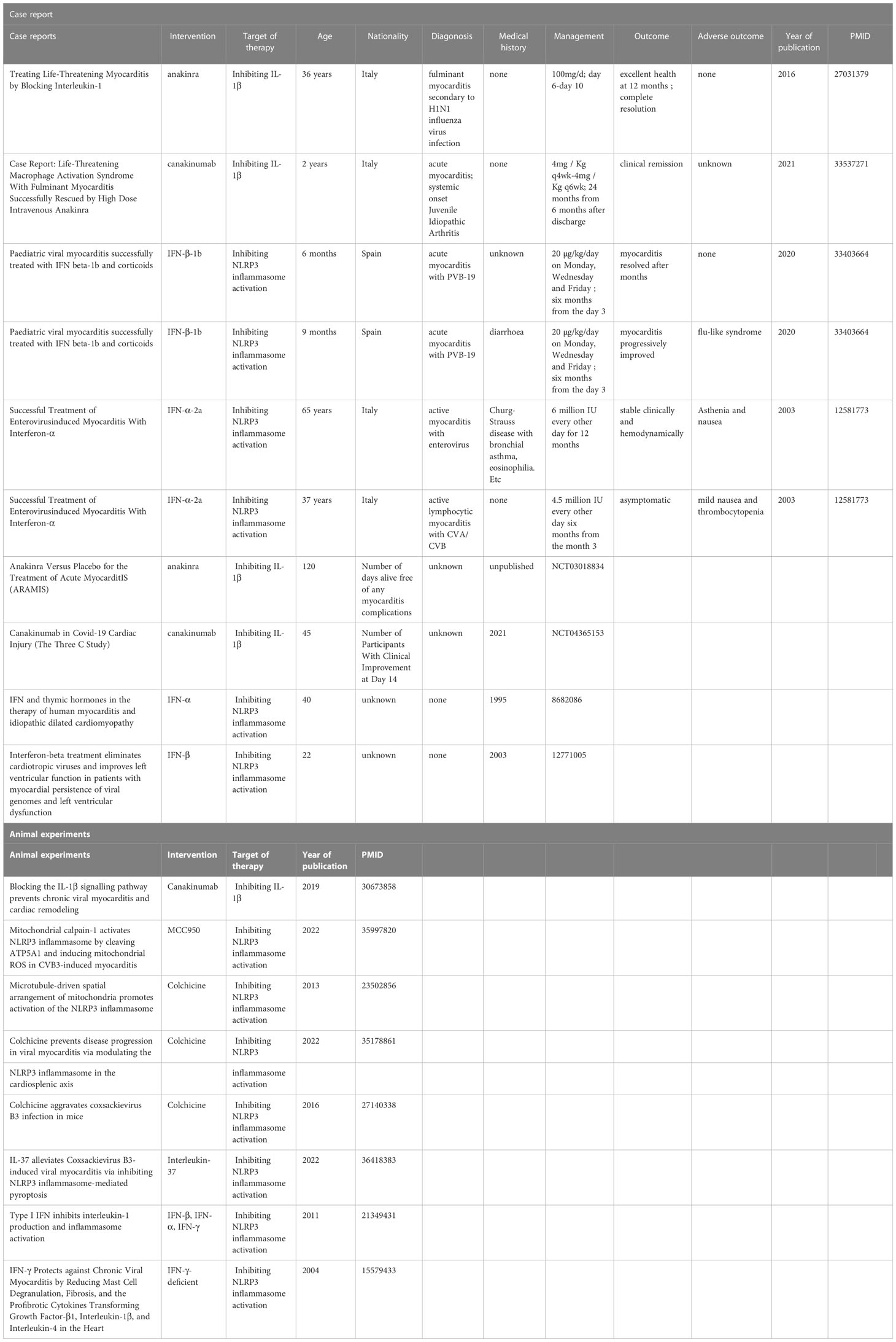
Table 1 Overview of case reports, clinical trials and animal experiments targeting the inflammasome pathway to treat VMC.
Anakinra is the recombinant form of the naturally occurring IL-1 receptor antagonist (IL-1Ra) and inhibits the activity of both IL-1α and IL-1β. The effectiveness of anakinra in the treatment of myocarditis has been revealed by many animal experiments and case reports, indicating the possible involvement of the NLRP3 inflammasome/IL-1β pathway. As far as we know, the earliest case report of anakinra applied to myocarditis dates back to 2016. A previously healthy 36-year-old Italian woman was admitted to the hospital due to fulminant myocarditis. Because previous treatment was ineffective, from the sixth day of admission, she was given anakinra 100 mg daily, and a dramatic remission was observed within 24 hours. The fever was alleviated and the neutrophil count reduced, along with the normalization of CRP, troponin T, ECG, and LVEF. With 4 days of anakinra treatment, sustained clinical improvement allowed weaning from ECMO and removal of the percutaneous LVAD (Cavalli et al., 2016). Yoshihiro Noji et al. accordingly suggested that anakinra targeted IL-1 to treat fulminant myocarditis (Noji, 2016).
The anakinra vs. Placebo for the Treatment of Acute Myocarditis (ARAMIS) trial (ClinicalTrials.gov identifier: NCT03018834) has been completed as of June 15, 2022. Although the results have not been posted yet, the clinical application of anakinra in acute myocarditis is promising.
In addition, the safety of anakinra has been demonstrated by extensive research. It is significant to know that anakinra is associated with an increased risk of infection (mainly upper respiratory infections), but not with increased infection-related mortality (Fleischmann et al., 2006). The evidence above indicates that anakinra may have great potential in the treatment of VMC.
Canakinumab is an IL-1β-neutralizing antibody that reduces inflammation in patients with autoimmune diseases. Compared with anakinra, it has a longer half-life and can be administered once a month, showing better clinical operability (Abbate et al., 2020). In the treatment of heart diseases, canakinumab is effective in reducing myocardial infarction in high-risk groups (Ridker et al., 2017).
In a CVB3-induced VMC mouse model, canakinumab’s counterpart was shown to prevent chronic VMC and cardiac remodeling. A total of 36 ABY/SnJ mice were divided into three control groups and three experimental groups. The control groups were infected with CVB3 alone, while the experimental groups were infected with CVB3 first, followed by neutralizing IL-1β with antibodies on days 1–14, 3–14, and 14–28, respectively. Significant reductions in myocardial injury and inflammation were observed in all three experimental groups. At 28 days post-infection, robust cardiac fibrosis and remodeling were observed in the three control groups, whereas the three corresponding experimental groups all showed a marked reduction in the degree of fibrosis during the same period (Kraft et al., 2019).
In a case report of fulminant myocarditis, significant remission was observed when anakinra was applied. After discharge, anakinra was switched to canakinumab because of the difficulty in maintaining daily injections of anakinra. Canakinumab was initiated six months after discharge at a dose of 4 mg/Kg q4wk and then gradually tapered down. Up to 24 months, the disease had been in clinical remission on medication (canakinumab 4 mg/Kg q6wk) (Meneghel et al., 2020). This case not only revealed canakinumab’s effectiveness, but also highlighted its advantage of less frequent injections than anakinra, which had a shorter half-life. In addition, a clinical trial, Canakinumab in COVID-19 Cardiac Injury (The Three C Study), was conducted in 2021 and revealed the role of canakinumab in the treatment of myocardial injury in the context of COVID-19 (ClinicalTrials.gov Identifier: NCT04365153). It indicated that canakinumab could ameliorate SARS-CoV-2-associated acute myocardial injury, but the doses remain to be explored and the side effects are less clear. In summary, it is suggested that canakinumab has great potential in the treatment of VMC. Nevertheless, more clinical studies are needed to explore the efficacy and safety of canakinumab in treating VMC, in order to better guide its application in the management of VMC patients.
MCC950, also known as CRID3 and CP-456,773, is a widely studied specific NLRP3 inflammasome inhibitor. Liu et al. proved that in mice with CVB3-induced VMC, intra-peritoneal injection of MCC950 could effectively inhibit CVB3-induced pyroptosis and inflammation by inhibiting the NLRP3 inflammasome activation pathway. Six 4 or 5-week-old male mice were equally divided into three groups: the sham group injected with PBS; the CVB3 group injected with CVB3; and the MCC950+CVB3 group injected with 10 mg/kg MCC950 daily extraperitoneal for six consecutive days and infected with CVB3 on the second day from MCC950 injection initiation. Western blot analysis indicated that the MCC950+CVB3 group showed a significant reduction in GSDMD N-terminus and cleaved caspase-1 compared with the CVB3 group. Moreover, cardiomyocyte viability was higher in the CVB3 + MCC950 group than in the CVB3 group (Liu et al., 2022). This revealed the promising prospect of MCC950 for the treatment of VMC as a specific inhibitor of the NLRP3 inflammasome activation.
Colchicine has been reported to inhibit the NLRP3 inflammasome activation via blocking microtubule assembly (Misawa et al., 2013). Pappritz et al. have reported that in C57BL6/j mice with CVB3-induced VMC, colchicine applied in the early stage of CVB3 infection could inhibit the splenic NLRP3 inflammasome activity, accompanied by alleviated immune cell infiltration, decreased cardiac troponin-1, and improved left ventricular function (Pappritz et al., 2022). These results could be explained by the fact that colchicine inhibited the infiltration of NLRP3-active inflammatory cells from the spleen to the heart (Pappritz et al., 2022). Furthermore, in vitro experiments demonstrated that colchicine suppressed the NLRP3 inflammasome activity in CVB3-infected HL-1 cardiomyocytes and fibroblasts, inhibiting their pro-inflammatory and pro-fibrotic capacities respectively (Pappritz et al., 2022). Importantly, colchicine applied to the CVB3-infected mice showed no exacerbation of CVB3 load, indicating that the colchicine might have little negative impact on anti-viral reactions of host cells and could be applied during viral persistence (Pappritz et al., 2022). However, previous research reported that colchicine injection could up-regulate the level of cardiac CVB3 mRNA and exacerbate CVB3-induced myocarditis, resulting in severe discomfort and higher mortality 3 days post-infection in CH3 mice (Smilde et al., 2016), which might partially be explained by the sensitivity difference between the two strains of mice, but indicated the potential cytotoxicity of colchicine. In summary, colchicine showed promising potential for balancing the modulation of excessive inflammation and the clearance of viruses. However, clinical studies, including follow-up biopsies to quantify myocardial inflammation and viral activity, are needed to evaluate the safety and efficacy of colchicine in human settings.
Interleukin-37 (IL-37), a member of the IL-1 cytokine family, has been demonstrated to alleviate CVB3-induced VMC in mice by suppressing the NF-κB pathway activation and inhibiting the NLRP3 inflammasome activity (Sun et al., 2022). In the well-constructed CVB3-induced VMC model of mice, IL-37 injection improved cardiac functions, manifesting as significantly higher LVEF, LVFS, IVSs and IVSd and significantly reduced the level of cardiac troponin I. Furthermore, the IL−37 treatment alleviated CVB3−induced inflammatory cell infiltration and fibrosis deposition in myocardium (Sun et al., 2022), which indicated the potential of IL-37 in ameliorating chronic inflammation and cardiac fibrosis. Though IL-37 has shown protective effects in CVB3-induced VMC, the mechanisms and possible side effects of IL-37 in treating VMC remain to be elucidated. More animal experiments are needed to confirm its safety and efficacy before admitting clinical application.
Interferons (IFNs) are a class of broad-spectrum antiviral agents that can limit the proliferation of many viruses in the human body and treat the diseases caused by virus infection. IFN-α and IFN-β exert antiviral effects mainly through IFN-stimulated genes (ISGs). ISGs respond to IFN-α and IFN-β produced upon infection, thus initiating the antiviral state in bystander cells. The main mechanisms involve myxovirus resistance 1 (MX1), IFN-inducible double-stranded RNA-dependent protein kinase (PKR; encoded by EIF2AK2), 2′-5′-oligoadenylate synthetase (OAS), etc (McNab et al., 2015).
IFNs have long been used in the treatment of VMC, and their effectiveness has been demonstrated in several clinical trials (Mirić et al., 1995; Kühl et al., 2003) and case reports (Daliento et al., 2003; Canales Siguero et al., 2021). In the past, people often attributed the effect of IFN treatment on VMC to its broad-spectrum antiviral activity. However, recent studies have shown complex interactions between IFNs and the NLRP3 inflammasome. IFNs inhibit the NLRP3 inflammasome activation by several mechanisms involving the phosphorylation of STAT1 transcription factor and the promotion of IL-10 transcription (Guarda et al., 2011), thus alleviating the inflammatory responses caused by the virus and its subsequent adverse symptoms. In addition, some experiments have shown that IFN-γ could rescue mice with chronic VMC by reducing IL-1β levels. In another experiment, IFN-γ-deficient and wild-type BALB/c mice were inoculated with CVB3 on day 0. Hearts were collected for analysis on day 35, and a dramatic increase in IL-1β level in IFN-γ-deficient mice was observed (Fairweather et al., 2004). This suggests that IFN-γ may achieve the purpose of alleviating VMC by inhibiting the NLRP3 inflammasome activity. The results above indicate that part of the demonstrated effectiveness of IFNs in treating VMC is through the NLRP3 inflammasome pathway.
From what we have discussed above, a variety of drugs suppressing the NLRP3 inflammasome activation or inhibiting the IL-1β activity may be potential therapies for VMC, while the safety and efficacy of some drugs need further elucidation by more comprehensive animal experiments and clinical trials. Apart from chemical drugs targeting the NLRP3 inflammasome-IL-1β axis, NLRP3 knockout might also be a potential therapy for VMC. However, Wang et al. demonstrated that NLRP3 knockout in CVB3-infected mice exacerbated viral infection, cardiac injury, and cardiac dysfunction (Wang et al., 2019), which indicated the potential risks of NLRP3 knockout in treating VMC. More research is needed to assess the safety and efficacy of NLRP3 knockout as a therapy for VMC.
At present, the roles of inflammasomes, mainly the NLRP3 inflammasome, in VMC are well established. During the infectious phase, the NLRP3 inflammasome is activated, induces the release of IL-1β and IL-18 and promotes pyroptosis, participating significantly in the development of VMC. Based on the inflammasome activation pathways, we proposed possible therapies. Although the efficacy and safety of some therapies remain to be further tested, we believe that therapies targeting the inflammasome pathways are promising in treating VMC.
Nowadays, the interaction between SARS-Cov-2 and myocarditis has received a lot of attention. Myocarditis is regarded as a rare complication of COVID-19, and the pathogenic process of COVID-19-related myocarditis also indicates the involvement of the NLRP3 inflammasome. However, there is a lack of direct evidence on the role of the NLRP3 inflammasome in COVID-19-related myocarditis, and further research is needed to better understand COVID-19-related myocarditis.
JX, ZZ and YZ were in charge of searching all the relative papers and writing this manuscript. HL, KH and SY gave their valuable and professional suggestions and guide in organizing and drafting this manuscript. All authors contributed to the article and approved the submitted version.
This research was funded by the National Natural Science Foundation of China, grant number 82170239 to KH; by the Natural Science Foundation of Hubei Province, grant number 2022CFB138 to HL; by the Scientific Research Foundation of Union Hospital to SY. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
The authors thank Dr. Shiyan Hu, School of Chemistry and Chemical Engineering, Huazhong University of Science and Technology, Wuhan, China. for providing professional and efficient assistance in creating high-quality figures.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
ACE2, angiotensin-converting enzyme 2; Ang II, angiotensin II; ASC, apoptosis-associated speck-like protein containing a CARD; ATP, adenosine triphosphate; ATP5A1, ATP synthase-α; CARD8, caspase recruitment domain-containing protein 8; CDL, CARD domain linker; COVID-19, Corona Virus Disease 2019; CVB3, Coxsackievirus B3; DAMP, damage/danger-associated molecular patterns; DCM, dilated cardiomyopathy; EMCV, encephalomyocarditis virus; ER, endoplasmic reticulum; ERK, extracellular signal-regulated kinases; GSDMD, gasdermin D; HAEC, human aortic endothelial cell; ICAM, intercellular cell adhesion molecule; IDL, interdomain linker; IFN, interferon; IL-18, interleukin-18; IL-1β, interleukin-1β; LPS, lipopolysaccharide; MEG3, maternally expressed gene 3; miR, microRNA; MMP, matrix metalloproteinase; mtDNA, mitochondrial DNA; mtROS, mitochondrial reactive oxygen species; NF-κB, nuclear factor κ-light-chain enhancer of activated B cells; NLRP3, nucleotide oligomerization domain (NOD)-, leucine-rich repeat (LRR)- and pyrin domain (PYD)-containing protein3; NLRX1, nucleotide-binding domain and leucine-rich-repeat-containing family member X1; NOD2, nucleotide-binding oligomerization domain 2; P2X7R, P2X7 receptor; PAMP, pathogen-associated molecular patterns; PRR, pattern recognition receptors; RIP, receptor-interacting protein; SARS-Cov-2, Severe Acute Respiratory Syndrome Coronavirus 2; STAT1, signal transducer and activator of transcription 1; TLR, toll-like receptor; TMPRSS2, transmembrane serine protease 2; TNF, tumor necrosis factor; TRAF6, TNF receptor‐associated factor 6; VCAM, vascular cell adhesion molecule; VMC, viral myocarditis.
Abbate, A., Toldo, S., Marchetti, C., Kron, J., Van Tassell, B. W., Dinarello, C. A. (2020). Interleukin-1 and the inflammasome as therapeutic targets in cardiovascular disease. Circ. Res. 126 (9), 1260–1280. doi: 10.1161/CIRCRESAHA.120.315937
Abdelaziz Mohamed, I., Gadeau, A. P., Hasan, A., Abdulrahman, N., Mraiche, F. (2019). Osteopontin: A promising therapeutic target in cardiac fibrosis. Cells 8 (12), 1558. doi: 10.3390/cells8121558
Akira, S., Uematsu, S., Takeuchi, O. (2006). Pathogen recognition and innate immunity. Cell 124 (4), 783–801. doi: 10.1016/j.cell.2006.02.015
Albert, C. L., Carmona-Rubio, A. E., Weiss, A. J., Procop, G. G., Starling, R. C., Rodriguez, E. R. (2020). The enemy within: Sudden-onset reversible cardiogenic shock with biopsy-proven cardiac myocyte infection by severe acute respiratory syndrome coronavirus 2. Circulation 142 (19), 1865–1870. doi: 10.1161/CIRCULATIONAHA.120.050097
Althof, N., Goetzke, C. C., Kespohl, M., Voss, K., Heuser, A., Pinkert, S., et al. (2018). The immunoproteasome-specific inhibitor ONX 0914 reverses susceptibility to acute viral myocarditis. EMBO Mol. Med. 10 (2), 200–218. doi: 10.15252/emmm.201708089
Bartel, D. P. (2004). MicroRNAs: genomics, biogenesis, mechanism, and function. Cell 116 (2), 281–297. doi: 10.1016/s0092-8674(04)00045-5
Bauernfeind, F. G., Horvath, G., Stutz, A., Alnemri, E. S., Macdonald, K., Speert, D., et al. (2009). Cutting edge: NF-kappaB activating pattern recognition and cytokine receptors license NLRP3 inflammasome activation by regulating NLRP3 expression. J. Immunol. 183 (2), 787–791. doi: 10.4049/jimmunol.0901363
Bazzone, L. E., King, M., MacKay, C. R., Kyawe, P. P., Meraner, P., Lindstrom, D., et al. (2019). A disintegrin and metalloproteinase 9 domain (ADAM9) is a major susceptibility factor in the early stages of encephalomyocarditis virus infection. mBio 10 (1), e02734–e02718. doi: 10.1128/mBio.02734-18
Blauwet, L. A., Cooper, L. T. (2010). Myocarditis. Prog. Cardiovasc. Dis. 52 (4), 274–288. doi: 10.1016/j.pcad.2009.11.006
Boukhris, M., Hillani, A., Moroni, F., Annabi, M. S., Addad, F., Ribeiro, M. H., et al. (2020). Cardiovascular implications of the COVID-19 pandemic: A global perspective. Can. J. Cardiol. 36 (7), 1068–1080. doi: 10.1016/j.cjca.2020.05.018
Bürckstümmer, T., Baumann, C., Blüml, S., Dixit, E., Dürnberger, G., Jahn, H., et al. (2009). An orthogonal proteomic-genomic screen identifies AIM2 as a cytoplasmic DNA sensor for the inflammasome. Nat. Immunol. 10 (3), 266–272. doi: 10.1038/ni.1702
Canales Siguero, D., García-Muñoz, C., Martínez de la Torre, F., Ferrari Piquero, J. M., Granados Ruíz, M. A. (2021). Paediatric viral myocarditis successfully treated with interferon beta-1b and corticoids. J. Clin. Pharm. Ther. 46 (3), 862–864. doi: 10.1111/jcpt.13357
Calabrese, F., Thiene, G. (2003). Myocarditis and inflammatory cardiomyopathy: microbiological and molecular biological aspects. Cardiovasc Res 60(1), 11–25. doi: 10.1016/s0008-6363(03)00475-9
Cavalli, G., Pappalardo, F., Mangieri, A., Dinarello, C. A., Dagna, L., Tresoldi, M. (2016). Treating life-threatening myocarditis by blocking interleukin-1. Crit. Care Med. 44 (8), e751–e754. doi: 10.1097/CCM.0000000000001654
Chai, D., Yue, Y., Xu, W., Dong, C., Xiong, S. (2015). AIM2 co-immunization favors specific multifunctional CD8(+) T cell induction and ameliorates coxsackievirus B3-induced chronic myocarditis. Antiviral Res. 119, 68–77. doi: 10.1016/j.antiviral.2015.04.015
Chai, D., Yue, Y., Xu, W., Dong, C., Xiong, S. (2014). Mucosal co-immunization with AIM2 enhances protective SIgA response and increases prophylactic efficacy of chitosan-DNA vaccine against coxsackievirus B3-induced myocarditis. Hum Vaccin Immunother 10 (5), 1284–1294. doi: 10.4161/hv.28333
Chau, D. H. W., Yuan, J., Zhang, H., Cheung, P., Lim, T., Liu, Z., et al. (2007). Coxsackievirus B3 proteases 2A and 3C induce apoptotic cell death through mitochondrial injury and cleavage of eIF4GI but not DAP5/p97/NAT1. Apoptosis 12 (3), 513–524. doi: 10.1007/s10495-006-0013-0
Chen, L., Li, X., Chen, M., Feng, Y., Xiong, C. (2020). The ACE2 expression in human heart indicates new potential mechanism of heart injury among patients infected with SARS-CoV-2. Cardiovasc. Res. 116 (6), 1097–1100. doi: 10.1093/cvr/cvaa078
Chen, C., Zhou, Y., Wang, D. W. (2020). SARS-CoV-2: a potential novel etiology of fulminant myocarditis. Herz 45 (3), 230–232. doi: 10.1007/s00059-020-04909-z
Chimenti, C., Magnocavallo, M., Ballatore, F., Bernardini, F., Alfarano, M., Della Rocca, D. G., et al. (2022). Prevalence and clinical implications of COVID-19 myocarditis. Card Electrophysiol Clin. 14 (1), 53–62. doi: 10.1016/j.ccep.2021.11.001
Clerkin, K. J., Fried, J. A., Raikhelkar, J., Sayer, G., Griffin, J.M., Masoumi, A., et al. (2020). COVID-19 and cardiovascular disease. Circulation 141 (20), 1648–1655. doi: 10.1161/circulationaha.120.046941
Coomes, E. A., Haghbayan, H. (2020). Interleukin-6 in covid-19: A systematic review and meta-analysis. Rev. Med. Virol. 30 (6), 1–9. doi: 10.1002/rmv.2141
Cooper, L. T., Jr. (2009). Myocarditis. N Engl. J. Med. 360 (15), 1526–1538. doi: 10.1056/NEJMra0800028
Daliento, L., Calabrese, F., Tona, F., Caforio, A. L., Tarsia, G., Angelini, A., et al. (2003). Successful treatment of enterovirus-induced myocarditis with interferon-alpha. J. Heart Lung Transplant 22 (2), 214–217. doi: 10.1016/s1053-2498(02)00565-x
De Jong, A. S., De Mattia, F., Van Dommelen, M. M., Lanke, K., Melchers, W. J., Willems, P. H., et al. (2008). Functional analysis of picornavirus 2B proteins: effects on calcium homeostasis and intracellular protein trafficking. J. Virol. 82 (7), 3782–3790. doi: 10.1128/JVI.02076-07
Dostert, C., Pétrilli, V., Van Bruggen, R., Steele, C., Mossman, B. T., Tschopp, J. (2008). Innate immune activation through Nalp3 inflammasome sensing of asbestos and silica. Science 320 (5876), 674–677. doi: 10.1126/science.1156995
Fairweather, D., Frisancho-Kiss, S., Yusung, S. A., Barrett, M. A., Davis, S. E., Gatewood, S. J., et al. (2004). Interferon-gamma protects against chronic viral myocarditis by reducing mast cell degranulation, fibrosis, and the profibrotic cytokines transforming growth factor-beta 1, interleukin-1 beta, and interleukin-4 in the heart. Am. J. Pathol. 165 (6), 1883–1894. doi: 10.1016/s0002-9440(10)63241-5
Fairweather, D., Lawson, C. M., Chapman, A. J., Brown, C. M., Booth, T. W., Papadimitriou, J. M., et al. (1998). Wild isolates of murine cytomegalovirus induce myocarditis and antibodies that cross-react with virus and cardiac myosin. Immunology 94 (2), 263–270. doi: 10.1046/j.1365-2567.1998.00500.x
Fairweather, D., Yusung, S., Frisancho, S., Barrett, M., Gatewood, S., Steele, R., et al. (2003). IL-12 receptor beta 1 and toll-like receptor 4 increase IL-1 beta- and IL-18-associated myocarditis and coxsackievirus replication. J. Immunol. 170 (9), 4731–4737. doi: 10.4049/jimmunol.170.9.4731
Fairweather, D., Rose, N. R. (2007). Coxsackievirus-induced myocarditis in mice: a model of autoimmune disease for studying immunotoxicity. Methods 41 (1), 118–122. doi: 10.1016/j.ymeth.2006.07.009
Feng, Y., Song, X., Huang, Y., Deng, W., Li, M., Guo, X., et al. (2021). SARS-CoV-2 leads to myocardial injury in rhesus macaque. Signal Transduct Target Ther 6 (1), 338. doi: 10.1038/s41392-021-00747-5
Fernandes-Alnemri, T., Wu, J., Yu, J. W., Datta, P., Miller, B., Jankowski, W., et al. (2007). The pyroptosome: a supramolecular assembly of ASC dimers mediating inflammatory cell death via caspase-1 activation. Cell Death Differ. 14 (9), 1590–1604. doi: 10.1038/sj.cdd.4402194
Fidanboylu, M., Griffiths, L. A., Flatters, S. J. L. (2011). Global inhibition of reactive oxygen species (ROS) inhibits paclitaxel-induced painful peripheral neuropathy. PloS One 6 (9), e25212. doi: 10.1371/journal.pone.0025212
Fleischmann, R. M., Tesser, J., Schiff, M. H., Schechtman, J., Burmester, G. R., Bennett, R., et al. (2006). Safety of extended treatment with anakinra in patients with rheumatoid arthritis. Ann. Rheum Dis. 65 (8), 1006–1012. doi: 10.1136/ard.2005.048371
Fox, S. E., Akmatbekov, A., Harbert, J. L., Li, G., Quincy Brown, J., Vander Heide, R. S. (2020). Pulmonary and cardiac pathology in African American patients with COVID-19: an autopsy series from new Orleans. Lancet Respir Med 8 (7), 681–686. doi: 10.1016/s2213-2600(20)30243-5
Furrer, A., Hottiger, M. O., Valaperti, A. (2016). Absent in melanoma 2 (AIM2) limits pro-inflammatory cytokine transcription in cardiomyocytes by inhibiting STAT1 phosphorylation. Mol. Immunol. 74, 47–58. doi: 10.1016/j.molimm.2016.04.009
Goll, D. E., Thompson, V. F., Li, H., Wei, W., Cong, J. (2003). The calpain system. Physiol. Rev. 83 (3), 731–801. doi: 10.1152/physrev.00029.2002
Guarda, G., Braun, M., Staehli, F., Tardivel, A., Mattmann, C., Förster, I., et al. (2011). Type I interferon inhibits interleukin-1 production and inflammasome activation. Immunity 34 (2), 213–223. doi: 10.1016/j.immuni.2011.02.006
Gui, J., Yue, Y., Chen, R., Xu, W., Xiong, S. (2012). A20 (TNFAIP3) alleviates CVB3-induced myocarditis via inhibiting NF-κB signaling. PloS One 7 (9), e46515. doi: 10.1371/journal.pone.0046515
Guo, H., Callaway, J. B., Ting, J. P. Y. (2015). Inflammasomes: mechanism of action, role in disease, and therapeutics. Nat. Med. 21 (7), 677–687. doi: 10.1038/nm.3893
Hagar, J. A., Powell, D. A., Aachoui, Y., Ernst, R. K., Miao, E. A. (2013). Cytoplasmic LPS activates caspase-11: implications in TLR4-independent endotoxic shock. Science 341 (6165), 1250–1253. doi: 10.1126/science.1240988
Harris, B. S., Zhang, Y., Card, L., Rivera, L. B., Brekken, R. A., Bradshaw, A. D. (2011). SPARC Regulates collagen interaction with cardiac fibroblast cell surfaces. Am. J. Physiol. Heart Circ. Physiol. 301 (3), H841–H847. doi: 10.1152/ajpheart.01247.2010
He, Y., Chipman, P. R., Howitt, J., Bator, C. M., Whitt, M. A., Baker, T. S., et al. (2001). Interaction of coxsackievirus B3 with the full length coxsackievirus-adenovirus receptor. Nat. Struct. Biol. 8 (10), 874–878. doi: 10.1038/nsb1001-874
Hoffmann, M., Kleine-Weber, H., Schroeder, S., Krüger, N., Herrler, T., Erichsen, S., et al. (2020). SARS-CoV-2 cell entry depends on ACE2 and TMPRSS2 and is blocked by a clinically proven protease inhibitor. Cell 181 (2), 271–280.e8. doi: 10.1016/j.cell.2020.02.052
Hornung, V., Ablasser, A., Charrel-Dennis, M., Bauernfeind, F., Horvath, G., Caffrey, D. R., et al. (2009). AIM2 recognizes cytosolic dsDNA and forms a caspase-1-activating inflammasome with ASC. Nature 458 (7237), 514–518. doi: 10.1038/nature07725
Hornung, V., Bauernfeind, F., Halle, A., Samstad, E. O., Kono, H., Rock, K. L., et al. (2008). Silica crystals and aluminum salts activate the NALP3 inflammasome through phagosomal destabilization. Nat. Immunol. 9 (8), 847–856. doi: 10.1038/ni.1631
Hühn, M. H., Mccartney, S. A., Lind, K., Svedin, E., Colonna, M., Flodström-Tullberg, M. (2010). Melanoma differentiation-associated protein-5 (MDA-5) limits early viral replication but is not essential for the induction of type 1 interferons after coxsackievirus infection. Virology 401 (1), 42–48. doi: 10.1016/j.virol.2010.02.010
Inciardi, R. M., Lupi, L., Zaccone, G., Italia, L., Raffo, M., Tomasoni, D., et al. (2020). Cardiac involvement in a patient with coronavirus disease 2019 (COVID-19). JAMA Cardiol. 5 (7), 819–824. doi: 10.1001/jamacardio.2020.1096
Irabien-Ortiz, Á, Carreras-Mora, J., Sionis, A., Pàmies, J., Montiel, J., Tauron, M. (2020). Fulminant myocarditis due to COVID-19. Rev. Esp Cardiol. (Engl Ed). 73 (6), 503–504. doi: 10.1016/j.rec.2020.04.005
Iwasaki, A., Matsumori, A., Yamada, T., Shioi, T., Wang, W., Ono, K., et al. (1999). Pimobendan inhibits the production of proinflammatory cytokines and gene expression of inducible nitric oxide synthase in a murine model of viral myocarditis. J. Am. Coll. Cardiol. 33 (5), 1400–1407. doi: 10.1016/s0735-1097(98)00692-5
Jin, X., Zhou, R., Huang, Y. (2022). Role of inflammasomes in HIV-1 infection and treatment. Trends Mol. Med. 28 (5), 421–434. doi: 10.1016/j.molmed.2022.02.010
Kang, M. J., Yoon, C. M., Kim, B. H., Lee, C. M., Zhou, Y., Sauler, M., et al. (2015). Suppression of NLRX1 in chronic obstructive pulmonary disease. J. Clin. Invest. 125 (6), 2458–2462. doi: 10.1172/JCI71747
Karasawa, T., Takahashi, M. (2017). Role of NLRP3 inflammasomes in atherosclerosis. J. Atheroscler Thromb. 24 (5), 443–451. doi: 10.5551/jat.RV17001
Karin, M., Lawrence, T., Nizet, V. (2006). Innate immunity gone awry: linking microbial infections to chronic inflammation and cancer. Cell 124 (4), 823–835. doi: 10.1016/j.cell.2006.02.016
Kayagaki, N., Wong, M. T., Stowe, I. B., Ramani, S. R., Gonzalez, L. C., Akashi-Takamura, S., et al. (2013). Noncanonical inflammasome activation by intracellular LPS independent of TLR4. Science 341 (6165), 1246–1249. doi: 10.1126/science.1240248
Kim, Y. K., Shin, J. S., Nahm, M. H. (2016). NOD-like receptors in infection, immunity, and diseases. Yonsei Med. J. 57 (1), 5–14. doi: 10.3349/ymj.2016.57.1.5
Kishimoto, T., Kang, S. (2022). IL-6 revisited: From rheumatoid arthritis to CAR T cell therapy and COVID-19. Annu. Rev. Immunol. 40, 323–348. doi: 10.1146/annurev-immunol-101220-023458
Kraft, L., Erdenesukh, T., Sauter, M., Tschöpe, C., Klingel, K. (2019). Blocking the IL-1β signalling pathway prevents chronic viral myocarditis and cardiac remodeling. Basic Res. Cardiol. 114 (2), 11. doi: 10.1007/s00395-019-0719-0
Kühl, U., Pauschinger, M., Schwimmbeck, P. L., Seeberg, B., Lober, C., Noutsias, M., et al. (2003). Interferon-beta treatment eliminates cardiotropic viruses and improves left ventricular function in patients with myocardial persistence of viral genomes and left ventricular dysfunction. Circulation 107 (22), 2793–2798. doi: 10.1161/01.CIR.0000072766.67150.51
Kwak, S. B., Koppula, S., In, E. J., Sun, X., Kim, Y. K., Kim, M. K., et al. (2018). Artemisia extract suppresses NLRP3 and AIM2 inflammasome activation by inhibition of ASC phosphorylation. Mediators Inflamm. 2018, 6054069. doi: 10.1155/2018/6054069
Lamkanfi, M., Declercq, W., Kalai, M., Saelens, X., Vandenabeele, P. (2002). Alice In caspase land. a phylogenetic analysis of caspases from worm to man. Cell Death Differ. 9 (4), 358–361. doi: 10.1038/sj.cdd.4400989
Lasrado, N., Reddy, J. (2020). An overview of the immune mechanisms of viral myocarditis. Rev. Med. Virol. 30 (6), 1–14. doi: 10.1002/rmv.2131
Latz, E., Xiao, T. S., Stutz, A. (2013). Activation and regulation of the inflammasomes. Nat. Rev. Immunol. 13 (6), 397–411. doi: 10.1038/nri3452
Lee, G. S., Subramanian, N., Kim, A. I., Aksentijevich, I., Goldbach-Mansky, R., Sacks, D. B., et al. (2012). The calcium-sensing receptor regulates the NLRP3 inflammasome through Ca2+ and cAMP. Nature 492 (7427), 123–127. doi: 10.1038/nature11588
Li, H., Tian, X., Wang, P., Huang, M., Xu, R., Nie, T. (2019). MicroRNA-582-3p negatively regulates cell proliferation and cell cycle progression in acute myeloid leukemia by targeting cyclin B2. Cell Mol. Biol. Lett. 24, 66. doi: 10.1186/s11658-019-0184-7
Liao, Y., Liu, K., Zhu, L. (2022). Emerging roles of inflammasomes in cardiovascular diseases. Front. Immunol. 13, 834289. doi: 10.3389/fimmu.2022.834289
Lindner, D., Fitzek, A., Bräuninger, H., Aleshcheva, G., Edler, C., Meissner, K., et al. (2020). Association of cardiac infection with SARS-CoV-2 in confirmed COVID-19 autopsy cases. JAMA Cardiol 5(11), 1281–1285. doi: 10.1001/jamacardio.2020.3551
Liu, X., Li, M., Chen, Z., Yu, Y., Shi, H., Yu, Y., et al. (2022). Mitochondrial calpain-1 activates NLRP3 inflammasome by cleaving ATP5A1 and inducing mitochondrial ROS in CVB3-induced myocarditis. Basic Res. Cardiol. 117 (1), 40. doi: 10.1007/s00395-022-00948-1
Liu, P. P., Mason, J. W. (2001). Advances in the understanding of myocarditis. Circulation 104 (9), 1076–1082. doi: 10.1161/hc3401.095198
Lomaga, M. A., Yeh, W. C., Sarosi, I., Duncan, G. S., Furlonger, C., Ho, A., et al. (1999). TRAF6 deficiency results in osteopetrosis and defective interleukin-1, CD40, and LPS signaling. Genes Dev. 13 (8), 1015–1024. doi: 10.1101/gad.13.8.1015
Long, B., Brady, W. J., Koyfman, A., Gottlieb, M. (2020). Cardiovascular complications in COVID-19. Am. J. Emerg. Med. 38 (7), 1504–1507. doi: 10.1016/j.ajem.2020.04.048
López-Sánchez, M., Muñoz-Esquerre, M., Huertas, D., Montes, A., Molina-Molina, M., Manresa, F., et al. (2017). Inflammatory markers and circulating extracellular matrix proteins in patients with chronic obstructive pulmonary disease and left ventricular diastolic dysfunction. Clin. Respir. J. 11 (6), 859–866. doi: 10.1111/crj.12428
Lugrin, J., Martinon, F. (2018). The AIM2 inflammasome: Sensor of pathogens and cellular perturbations. Immunol. Rev. 281 (1), 99–114. doi: 10.1111/imr.12618
Luo, H., Yanagawa, B., Zhang, J., Luo, Z., Zhang, M., Esfandiarei, M., et al. (2002). Coxsackievirus B3 replication is reduced by inhibition of the extracellular signal-regulated kinase (ERK) signaling pathway. J. Virol. 76 (7), 3365–3373. doi: 10.1128/jvi.76.7.3365-3373.2002
Ma, F., Li, Y., Jia, L., Han, Y., Cheng, J., Li, H., et al. (2012). Macrophage-stimulated cardiac fibroblast production of IL-6 is essential for TGF β/Smad activation and cardiac fibrosis induced by angiotensin II. PloS One 7 (5), e35144. doi: 10.1371/journal.pone.0035144
Mariathasan, S., Newton, K., Monack, D. M., Vucic, D., French, D. M., Lee, W. P., et al. (2004). Differential activation of the inflammasome by caspase-1 adaptors ASC and ipaf. Nature 430 (6996), 213–218. doi: 10.1038/nature02664
Martínez-Cuesta, MÁ, Blanch-Ruiz, M. A., Ortega-Luna, R., Sánchez-López, A., Álvarez, Á. (2020). Structural and functional basis for understanding the biological significance of P2X7 receptor. Int. J. Mol. Sci. 21 (22), 8454. doi: 10.3390/ijms21228454
Mason, J. W. (2003). Myocarditis and dilated cardiomyopathy: an inflammatory link. Cardiovasc. Res. 60 (1), 5–10. doi: 10.1016/s0008-6363(03)00437-1
Matsumori, A., Nunokawa, Y., Yamaki, A., Yamamoto, K., Hwang, M. W., Miyamoto, T., et al. (2004). Suppression of cytokines and nitric oxide production, and protection against lethal endotoxemia and viral myocarditis by a new NF-kappaB inhibitor. Eur. J. Heart Fail. 6 (2), 137–144. doi: 10.1016/j.ejheart.2003.10.007
McNab, F., Mayer-Barber, K., Sher, A., Wack, A., O’Garra, A. (2015). Type I interferons in infectious disease. Nat. Rev. Immunol. 15 (2), 87–103. doi: 10.1038/nri3787
Meneghel, A., Martini, G., Amigoni, A., Pettenazzo, A., Padalino, M., Zulian, F. (2020). Case report: Life-threatening macrophage activation syndrome with fulminant myocarditis successfully rescued by high dose intravenous anakinra. Front. Pediatr. 8. doi: 10.3389/fped.2020.635080
Mirić, M., Misković, A., Vasiljević, J. D., Keserović, N., Pesić, M. (1995). Interferon and thymic hormones in the therapy of human myocarditis and idiopathic dilated cardiomyopathy. Eur. Heart J. 16 (Suppl O), 150–152. doi: 10.1093/eurheartj/16.suppl_o.150
Misawa, T., Takahama, M., Kozaki, T., Lee, H., Zou, J., Saitoh, T., et al. (2013). Microtubule-driven spatial arrangement of mitochondria promotes activation of the NLRP3 inflammasome. Nat. Immunol. 14 (5), 454–460. doi: 10.1038/ni.2550
Mohamed, M. S. (2015). Calcium-activated potassium channels in ischemia-reperfusion: learning for the clinical application. Front. Med. (Lausanne). 2. doi: 10.3389/fmed.2015.00021
Mohammadzadeh, N., Melleby, A. O., Palmero, S., Sjaastad, I., Chakravarti, S., Engebretsen, K. V. T., et al. (2020). Moderate loss of the extracellular matrix proteoglycan lumican attenuates cardiac fibrosis in mice subjected to pressure overload. Cardiology 145 (3), 187–198. doi: 10.1159/000505318
Nadkarni, R., Chu, W. C., Lee, C. Q. E., Mohamud, Y., Yap, L., Toh, G. A., et al. (2022). Viral proteases activate the CARD8 inflammasome in the human cardiovascular system. J. Exp. Med. 219 (10), e20212117. doi: 10.1084/jem.20212117
Naito, A., Azuma, S., Tanaka, S., Miyazaki, T., Takaki, S., Takatsu, K., et al. (1999). Severe osteopetrosis, defective interleukin-1 signalling and lymph node organogenesis in TRAF6-deficient mice. Genes Cells 4 (6), 353–362. doi: 10.1046/j.1365-2443.1999.00265.x
Negishi, H., Osawa, T., Ogami, K., Ouyang, X., Sakaguchi, S., Koshiba, R., et al. (2008). A critical link between toll-like receptor 3 and type II interferon signaling pathways in antiviral innate immunity. Proc. Natl. Acad. Sci. U. S. A. 105 (51), 20446–20451. doi: 10.1073/pnas.0810372105
Ni, R., Zheng, D., Xiong, S., Hill, D. J., Sun, T., Gardiner, R. B., et al. (2016). Mitochondrial calpain-1 disrupts ATP synthase and induces superoxide generation in type 1 diabetic hearts: A novel mechanism contributing to diabetic cardiomyopathy. Diabetes 65 (1), 255–268. doi: 10.2337/db15-0963
Nicin, L., Abplanalp, W. T., Mellentin, H., Kattih, B., Tombor, L., John, D., et al. (2020). Cell type-specific expression of the putative SARS-CoV-2 receptor ACE2 in human hearts. Eur. Heart J. 41 (19), 1804–1806. doi: 10.1093/eurheartj/ehaa311
Noji, Y. (2016). Anakinra in fulminant myocarditis: Targeting interleukin-1 and the inflammasome formation. Crit. Care Med. 44 (8), 1630–1631. doi: 10.1097/CCM.0000000000001769
Onódi, Z., Ruppert, M., Kucsera, D., Sayour, A. A., Tóth, V. E., Koncsos, G., et al. (2021). AIM2-driven inflammasome activation in heart failure. Cardiovasc. Res. 117 (13), 2639–2651. doi: 10.1093/cvr/cvab202
Orlowski, G. M., Colbert, J. D., Sharma, S., Bogyo, M., Robertson, S. A., Rock, K. L. (2015). Multiple cathepsins promote pro-IL-1β synthesis and NLRP3-mediated IL-1β activation. J. Immunol. 195 (4), 1685–1697. doi: 10.4049/jimmunol.1500509
Pappritz, K., Lin, J., El-Shafeey, M., Fechner, H., Kühl, U., Alogna, A., et al. (2022). Colchicine prevents disease progression in viral myocarditis via modulating the NLRP3 inflammasome in the cardiosplenic axis. ESC Heart Fail. 9 (2), 925–941. doi: 10.1002/ehf2.13845
Pinar, A. A., Scott, T. E., Huuskes, B. M., Tapia Cáceres, F. E., Kemp-Harper, B. K., Samuel, C. S. (2020). Targeting the NLRP3 inflammasome to treat cardiovascular fibrosis. Pharmacol. Ther. 209, 107511. doi: 10.1016/j.pharmthera.2020.107511
Pollack, A., Kontorovich, A. R., Fuster, V., Dec, G. W. (2015). Viral myocarditis–diagnosis, treatment options, and current controversies. Nat. Rev. Cardiol. 12 (11), 670–680. doi: 10.1038/nrcardio.2015.108
Quagliariello, V., Bonelli, A., Caronna, A., Conforti, G., Iovine, M., Carbone, A., et al. (2020). SARS-CoV-2 infection and cardioncology: From cardiometabolic risk factors to outcomes in cancer patients. Cancers (Basel). 12 (11), 3316. doi: 10.3390/cancers12113316
Ratajczak, M. Z., Bujko, K., Ciechanowicz, A., Sielatycka, K., Cymer, M., Marlicz, W., et al. (2021). SARS-CoV-2 entry receptor ACE2 is expressed on very small CD45(-) precursors of hematopoietic and endothelial cells and in response to virus spike protein activates the Nlrp3 inflammasome. Stem Cell Rev Rep 17 (1), 266–277. doi: 10.1007/s12015-020-10010-z
Rathinam, V. A. K., Vanaja, S. K., Fitzgerald, K. A. (2012). Regulation of inflammasome signaling. Nat. Immunol. 13 (4), 333–342. doi: 10.1038/ni.2237
Ren, X. S., Tong, Y., Ling, L., Chen, D., Sun, H. J., Zhou, H., et al. (2017). NLRP3 gene deletion attenuates angiotensin II-induced phenotypic transformation of vascular smooth muscle cells and vascular remodeling. Cell Physiol. Biochem. 44 (6), 2269–2280. doi: 10.1159/000486061
Reyes, M. P., Lerner, A. M. (1985). Coxsackievirus myocarditis–with special reference to acute and chronic effects. Prog. Cardiovasc. Dis. 27 (6), 373–394. doi: 10.1016/0033-0620(85)90001-5
Ridker, P. M., Everett, B. M., Thuren, T., Macfadyen, J. G., Chang, W. H., Ballantyne, C., et al. (2017). Antiinflammatory therapy with canakinumab for atherosclerotic disease. N Engl. J. Med. 377 (12), 1119–1131. doi: 10.1056/NEJMoa1707914
Rivadeneyra, L., Charó, N., Kviatcovsky, D., de la Barrera, S., Gómez, R. M., Schattner, M. (2018). Role of neutrophils in CVB3 infection and viral myocarditis. J. Mol. Cell Cardiol. 125, 149–161. doi: 10.1016/j.yjmcc.2018.08.029
Rose, N. R. (2009). Myocarditis: infection versus autoimmunity. J. Clin. Immunol. 29 (6), 730–737. doi: 10.1007/s10875-009-9339-z
Ross, C., Chan, A. H., Von Pein, J., Boucher, D., Schroder, K. (2018). Dimerization and auto-processing induce caspase-11 protease activation within the non-canonical inflammasome. Life Sci. Alliance. 1 (6), e201800237. doi: 10.26508/lsa.201800237
Ross, C., Chan, A. H., von Pein, J. B., Maddugoda, M. P., Boucher, D., Schroder, K. (2022). Inflammatory caspases: Toward a unified model for caspase activation by inflammasomes. Annu. Rev. Immunol. 40, 249–269. doi: 10.1146/annurev-immunol-101220-030653
Sagar, S., Liu, P. P., Cooper, L. T., Jr. (2012). Myocarditis. Lancet 379 (9817), 738–747. doi: 10.1016/S0140-6736(11)60648-X
Santos, R. A. S., Sampaio, W. O., Alzamora, A. C., Motta-Santos, D., Alenina, N., Bader, M., et al. (2018). The ACE2/Angiotensin-(1-7)/MAS axis of the renin-angiotensin system: Focus on angiotensin-(1-7). Physiol. Rev. 98 (1), 505–553. doi: 10.1152/physrev.00023.2016
Satoh, M., Nakamura, M., Akatsu, T., Shimoda, Y., Segawa, I., Hiramori, K. (2004). Toll-like receptor 4 is expressed with enteroviral replication in myocardium from patients with dilated cardiomyopathy. Lab. Invest. 84 (2), 173–181. doi: 10.1038/labinvest.3700031
Sawalha, K., Abozenah, M., Kadado, A. J., Battisha, A., Al-Akchar, M., Salerno, C., et al. (2021). Systematic review of COVID-19 related myocarditis: Insights on management and outcome. Cardiovasc. Revasc Med. 23, 107–113. doi: 10.1016/j.carrev.2020.08.028
Sborgi, L., Ravotti, F., Dandey, V. P., Dick, M. S., Mazur, A., Reckel, S., et al. (2015). Structure and assembly of the mouse ASC inflammasome by combined NMR spectroscopy and cryo-electron microscopy. Proc Natl Acad Sci U. S. A. 112 (43), 13237–13242. doi: 10.1073/pnas.1507579112
Schmidt, F. I., Lu, A., Chen, J. W., Ruan, J., Tang, C., Wu, H., et al. (2016). A single domain antibody fragment that recognizes the adaptor ASC defines the role of ASC domains in inflammasome assembly. J Exp Med 213 (5), 771–790. doi: 10.1084/jem.20151790
Schorn, C., Frey, B., Lauber, K., Janko, C., Strysio, M., Keppeler, H., et al. (2011). Sodium overload and water influx activate the NALP3 inflammasome. J. Biol. Chem. 286 (1), 35–41. doi: 10.1074/jbc.M110.139048
Segers, V. F. M., Brutsaert, D. L., De Keulenaer, G. W. (2018). Cardiac remodeling: Endothelial cells have more to say than just NO. Front. Physiol. 9, 382. doi: 10.3389/fphys.2018.00382
Seko, Y., Matsuda, H., Kato, K., Hashimoto, Y., Yagita, H., Okumura, K., et al. (1993). Expression of intercellular adhesion molecule-1 in murine hearts with acute myocarditis caused by coxsackievirus B3. J. Clin. Invest. 91 (4), 1327–1336. doi: 10.1172/JCI116333
Seko, Y., Yagita, H., Okumura, K., Yazaki, Y. (1996). Expression of vascular cell adhesion molecule-1 in murine hearts with acute myocarditis caused by coxsackievirus B3. J. Pathol. 180 (4), 450–454. doi: 10.1002/(SICI)1096-9896(199612)180:4<450::AID-PATH693>3.0.CO;2-5
Sharma, B. R., Kanneganti, T. D. (2021). NLRP3 inflammasome in cancer and metabolic diseases. Nat. Immunol. 22 (5), 550–559. doi: 10.1038/s41590-021-00886-5
Shenoy-Scaria, A. M., Kwong, J., Fujita, T., Olszowy, M. W., Shaw, A. S., Lublin, D. M. (1992). Signal transduction through decay-accelerating factor. Interaction of glycosyl-phosphatidylinositol anchor and protein tyrosine kinases p56lck and p59fyn 1. J. Immunol. 149 (11), 3535–3541. doi: 10.4049/jimmunol.149.11.3535
Shi, J., Zhao, Y., Wang, Y., Gao, W., Ding, J., Li, P., et al. (2014). Inflammatory caspases are innate immune receptors for intracellular LPS. Nature 514 (7521), 187–192. doi: 10.1038/nature13683
Shimabukuro-Vornhagen, A., Gödel, P., Subklewe, M., Stemmler, H. J., Schlößer, H. A., Schlaak, M., et al. (2018). Cytokine release syndrome. J. Immunother. Cancer. 6 (1), 56. doi: 10.1186/s40425-018-0343-9
Shioi, T., Matsumori, A., Sasayama, S. (1996). Persistent expression of cytokine in the chronic stage of viral myocarditis in mice. Circulation 94 (11), 2930–2937. doi: 10.1161/01.cir.94.11.2930
Siripanthong, B., Nazarian, S., Muser, D., Deo, R., Santangeli, P., Khanji, M. Y., et al. (2020). Recognizing COVID-19-related myocarditis: The possible pathophysiology and proposed guideline for diagnosis and management. Heart Rhythm 17 (9), 1463–1471. doi: 10.1016/j.hrthm.2020.05.001
Smilde, B. J., Woudstra, L., Fong Hing, G., Wouters, D., Zeerleder, S., Murk, J. L., et al. (2016). Colchicine aggravates coxsackievirus B3 infection in mice. Int. J. Cardiol. 216, 58–65. doi: 10.1016/j.ijcard.2016.04.144
Smith, J. L., Jeng, S., McWeeney, S. K., Hirsch, A. J. (2017). A MicroRNA screen identifies the wnt signaling pathway as a regulator of the interferon response during flavivirus infection. J. Virol. 91 (8), e02388–e02316. doi: 10.1128/JVI.02388-16
Solle, M., Labasi, J., Perregaux, D. G., Stam, E., Petrushova, N., Koller, B. H., et al. (2001). Altered cytokine production in mice lacking P2X(7) receptors. J. Biol. Chem. 276 (1), 125–132. doi: 10.1074/jbc.M006781200
Solt, L. A., May, M. J. (2008). The IkappaB kinase complex: master regulator of NF-kappaB signaling. Immunol. Res. 42 (1-3), 3–18. doi: 10.1007/s12026-008-8025-1
Spinale, F. G. (2007). Myocardial matrix remodeling and the matrix metalloproteinases: influence on cardiac form and function. Physiol. Rev. 87 (4), 1285–1342. doi: 10.1152/physrev.00012.2007
Srinivasula, S. M., Poyet, J. L., Razmara, M., Datta, P., Zhang, Z., Alnemri, E. S. (2002). The PYRIN-CARD protein ASC is an activating adaptor for caspase-1. J Biol Chem 277 (24), 21119–21122. doi: 10.1074/jbc.C200179200
Sun, W., Lu, H., Dong, S., Li, R., Chu, Y., Wang, N., et al. (2021). Beclin1 controls caspase-4 inflammsome activation and pyroptosis in mouse myocardial reperfusion-induced microvascular injury. Cell Commun. Signal. 19 (1), 107. doi: 10.1186/s12964-021-00786-z
Sun, L., Yuan, H., Zhao, G. (2022). IL-37 alleviates coxsackievirus B3-induced viral myocarditis via inhibiting NLRP3 inflammasome-mediated pyroptosis. Sci Rep 12(1), 20077. doi: 10.1038/s41598-022-22617-y
Taabazuing, C. Y., Griswold, A. R., Bachovchin, D. A. (2020). The NLRP1 and CARD8 inflammasomes. Immunol. Rev. 297 (1), 13–25. doi: 10.1111/imr.12884
Tang, T., Lang, X., Xu, C., Wang, X., Gong, T., Yang, Y., et al. (2017). CLICs-dependent chloride efflux is an essential and proximal upstream event for NLRP3 inflammasome activation. Nat. Commun. 8 (1), 202. doi: 10.1038/s41467-017-00227-x
Tong, R., Jia, T., Shi, R., Yan, F. (2020). Inhibition of microRNA-15 protects H9c2 cells against CVB3-induced myocardial injury by targeting NLRX1 to regulate the NLRP3 inflammasome. Cell Mol. Biol. Lett. 25, 6. doi: 10.1186/s11658-020-00203-2
Tschöpe, C., Ammirati, E., Bozkurt, B., Caforio, A. L.P., Cooper, L. T., Felix, S. B., et al. (2021). Myocarditis and inflammatory cardiomyopathy: current evidence and future directions. Nat. Rev. Cardiol. 18 (3), 169–193. doi: 10.1038/s41569-020-00435-x
Tschöpe, C., Müller, I., Xia, Y., Savvatis, K., Pappritz, K., Pinkert, S., et al. (2017). NOD2 (Nucleotide-binding oligomerization domain 2) is a major pathogenic mediator of coxsackievirus B3-induced myocarditis. Circ. Heart Fail. 10 (9), e003870. doi: 10.1161/CIRCHEARTFAILURE.117.003870
Wang, C., Fung, G., Deng, H., Jagdeo, J., Mohamud, Y., Xue, Y. C., et al. (2019). NLRP3 deficiency exacerbates enterovirus infection in mice. FASEB J. 33 (1), 942–952. doi: 10.1096/fj.201800301RRR
Wang, Y., Gao, B., Xiong, S. (2014). Involvement of NLRP3 inflammasome in CVB3-induced viral myocarditis. Am. J. Physiol. Heart Circ. Physiol. 307 (10), H1438–H1447. doi: 10.1152/ajpheart.00441.2014
Wang, X., Jiang, W., Yan, Y., Gong, T., Han, J., Tian, Z., et al. (2014). RNA viruses promote activation of the NLRP3 inflammasome through a RIP1-RIP3-DRP1 signaling pathway. Nat. Immunol. 15 (12), 1126–1133. doi: 10.1038/ni.3015
Wang, Q., Shan, L. (2022). CARD8 makes coxsackievirus more “heartbreaking.” J. Exp. Med. 219 (10), e20221240. doi: 10.1084/jem.20221240
Wang, J. P., Cerny, A., Asher, D. R., Kurt-Jones, E. A., Bronson, R. T., Finberg, R. W. (2010). MDA5 and MAVS mediate type I interferon responses to coxsackie B virus. J Virol 84 (1), 254–260. doi: 10.1128/jvi.00631-09
Wang, Y., Jia, L., Shen, J., Wang, Y., Fu, Z., Su, S. A., et al. (2018). Cathepsin B aggravates coxsackievirus B3-induced myocarditis through activating the inflammasome and promoting pyroptosis. PLoS Pathog 14 (1), e1006872. doi: 10.1371/journal.ppat.1006872
Weber, A., Wasiliew, P., Kracht, M. (2010). Interleukin-1 (IL-1) pathway. Sci. Signal 3 (105), cm1. doi: 10.1126/scisignal.3105cm1
Westermann, D., Savvatis, K., Schultheiss, H. P., Tschöpe, C. (2010). Immunomodulation and matrix metalloproteinases in viral myocarditis. J. Mol. Cell Cardiol. 48 (3), 468–473. doi: 10.1016/j.yjmcc.2009.08.019
Xiong, D., Yajima, T., Lim, B. K., Stenbit, A., Dublin, A., Dalton, N. D., et al. (2007). Inducible cardiac-restricted expression of enteroviral protease 2A is sufficient to induce dilated cardiomyopathy. Circulation 115 (1), 94–102. doi: 10.1161/CIRCULATIONAHA.106.631093
Xu, J., Núñez, G. (2022). The NLRP3 inflammasome: activation and regulation. Trends Biochem. Sci. 48 (4), 331–344. doi: 10.1016/j.tibs.2022.10.002
Xue, Y. L., Zhang, S. X., Zheng, C. F., Li, Y. F., Zhang, L. H., Su, Q. Y., et al. (2020). Long non-coding RNA MEG3 inhibits M2 macrophage polarization by activating TRAF6 via microRNA-223 down-regulation in viral myocarditis. J. Cell Mol. Med. 24 (21), 12341–12354. doi: 10.1111/jcmm.15720
Yu, Y., Shi, H., Yu, Y., Liu, M., Li, M., Liu, X., et al. (2020). Inhibition of calpain alleviates coxsackievirus B3-induced myocarditis through suppressing the canonical NLRP3 inflammasome/caspase-1-mediated and noncanonical caspase-11-mediated pyroptosis pathways. Am. J. Transl. Res. 12 (5), 1954–1964.
Zhao, N., Di, B., Xu, L. L. (2021). The NLRP3 inflammasome and COVID-19: Activation, pathogenesis and therapeutic strategies. Cytokine Growth Factor Rev. 61, 2–15. doi: 10.1016/j.cytogfr.2021.06.002
Zhong, Z., Liang, S., Sanchez-Lopez, E., He, F., Shalapour, S., Lin, X. J., et al. (2018). New mitochondrial DNA synthesis enables NLRP3 inflammasome activation. Nature 560 (7717), 198–203. doi: 10.1038/s41586-018-0372-z
Zhou, R., Yazdi, A. S., Menu, P., Tschopp, J. (2011). A role for mitochondria in NLRP3 inflammasome activation. Nature 469 (7329), 221–225. doi: 10.1038/nature09663
Zhu, Y. F., Wang, R., Chen, W., Cao, Y. D., Li, L. P., Chen, X. (2021). miR-133a-3p attenuates cardiomyocyte hypertrophy through inhibiting pyroptosis activation by targeting IKKϵ. Acta Histochem. 123 (1), 151653. doi: 10.1016/j.acthis.2020.151653
Keywords: viral myocarditis, inflammasome, infection, cytokines, interleukin-1β
Citation: Xu J, Zhou Z, Zheng Y, Yang S, Huang K and Li H (2023) Roles of inflammasomes in viral myocarditis. Front. Cell. Infect. Microbiol. 13:1149911. doi: 10.3389/fcimb.2023.1149911
Received: 23 January 2023; Accepted: 28 March 2023;
Published: 15 May 2023.
Edited by:
Yongkui Li, Jinan University, ChinaCopyright © 2023 Xu, Zhou, Zheng, Yang, Huang and Li. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Huili Li, aHVpbGlfbGlAaHVzdC5lZHUuY24=; Kun Huang, a3VuX2h1YW5nQGh1c3QuZWR1LmNu; Sai Yang, eWFuZ3NhaUBtYWlsLmh1c3QuZWR1LmNu
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.