 Viktoria Rex
Viktoria Rex Razieh Zargari
Razieh Zargari Markus Stempel1,3
Markus Stempel1,3 Stephan Halle
Stephan Halle- 1Institute of Genetics, Technische Universität Braunschweig, Braunschweig, Germany
- 2Institute of Immunology, Hannover Medical School, Hannover, Germany
- 3Virology and Innate Immunity Research Group, Helmholtz Centre for Infection Research, Braunschweig, Germany
- 4Institute of Clinical Chemistry, Hannover Medical School, Hannover, Germany
Immediately after entry into host cells, viruses are sensed by the innate immune system, leading to the activation of innate antiviral effector mechanisms including the type I interferon (IFN) response and natural killer (NK) cells. This innate immune response helps to shape an effective adaptive T cell immune response mediated by cytotoxic T cells and CD4+ T helper cells and is also critical for the maintenance of protective T cells during chronic infection. The human gammaherpesvirus Epstein-Barr virus (EBV) is a highly prevalent lymphotropic oncovirus that establishes chronic lifelong infections in the vast majority of the adult population. Although acute EBV infection is controlled in an immunocompetent host, chronic EBV infection can lead to severe complications in immunosuppressed patients. Given that EBV is strictly host-specific, its murine homolog murid herpesvirus 4 or MHV68 is a widely used model to obtain in vivo insights into the interaction between gammaherpesviruses and their host. Despite the fact that EBV and MHV68 have developed strategies to evade the innate and adaptive immune response, innate antiviral effector mechanisms still play a vital role in not only controlling the acute infection but also shaping an efficient long-lasting adaptive immune response. Here, we summarize the current knowledge about the innate immune response mediated by the type I IFN system and NK cells, and the adaptive T cell-mediated response during EBV and MHV68 infection. Investigating the fine-tuned interplay between the innate immune and T cell response will provide valuable insights which may be exploited to design better therapeutic strategies to vanquish chronic herpesviral infection.
Introduction
The oncogenic human gammaherpesviruses EBV and KSHV
The Herpesviridae are a large family of DNA viruses that infect a wide range of host species. The family comprises three subfamilies, the Alpha-, Beta-, and Gammaherpesvirinae, and they all share the ability to establish chronic, lifelong infections in their hosts. Mostly, herpesviruses cause severe disease only in naïve or immunosuppressed individuals. After primary infection, they establish a state called latency, with the hallmark of minimal viral gene expression and absence of de novo synthesis of viral particles (Cohen, 2020). Disruptions can induce reactivation and transition from latency to the lytic replication cycle, which results in viral gene expression and production of new virus progeny and virus dissemination (Davison et al., 2009). Unlike the Alpha- and Betaherpesviruses, the Gammaherpesviruses are oncogenic and are associated with the development of lymphoproliferative diseases and lymphomas as well as multiple other cancers (Wen et al., 2021). The two Gammaherpesviruses known to infect humans are Epstein-Barr virus (EBV or Human Herpesvirus 4) and Kaposi’s sarcoma-associated herpesvirus (KSHV or Human Herpesvirus 8).
KSHV was initially discovered through its tight association with Kaposi’s sarcoma (KS) (Chang et al., 1994; Cesarman et al., 2019) and since then has been associated with a wide spectrum malignancies (Mariggiò et al., 2017). These include B cell lymphoproliferative disorders like primary effusion lymphoma (Nador et al., 1996), multicentric Castleman disease (Bélec et al., 1999; Carbone et al., 2021), diffuse large B cell lymphoma (Dupin et al., 2000), and germinotropic lymphoproliferative disorder (Du et al., 2002). In recent years, further KSHV-associated diseases were added to this list, including KSHV-positive reactive lymphoid hyperplasia and plasmablastic proliferation of the splenic red pulp (Gonzalez-Farre et al., 2017), bone marrow failure in immunosuppressed patients after transplantation (Luppi et al., 2000), and KSHV inflammatory cytokine syndrome (Uldrick et al., 2010). KSHV infection is not ubiquitous, with seroprevalence varying among different populations from high-level endemic areas (mainly occurring in Sub-Saharan Africa with seropositivity rates >50%), intermediate-level endemic areas (Mediterranean countries with seroprevalence rates between 10-30%), and non-endemic areas (most parts of Europe, Asia, and the US with <10%) (Yan et al., 2019). Conversely, EBV infection is ubiquitous, with about 95% of older adults worldwide being infected (Andrei et al., 2019). While EBV is mostly unnoticeably acquired in childhood, it can cause a diverse range of diseases (Damania et al., 2022). For example, EBV is associated with the endemic form of Burkitt’s lymphoma and is involved in the genesis of another geographically restricted cancer, nasopharyngeal carcinoma, as well as a subset of Hodgkin’s lymphoma and gastric carcinoma (Crawford et al., 2014). Chronic EBV infection or reactivation in patients who have been immunocompromised due to organ transplantation can lead to the development of various B cell malignancies known as post-transplant lymphoproliferative disease (PTLD) (Nijland et al., 2016). In some rare cases, individuals are not able to resolve and control EBV infection, which leads to the development of chronic active EBV (CAEBV) disease (Kimura and Cohen, 2017). During CAEBV disease, EBV-positive lymphocytes infiltrate different organs and the viral load in blood is elevated which is often accompanied by fever and enlargement of the spleen (splenomegaly) (Kimura and Cohen, 2017). Very recently, EBV infection has been suggested as a possible cause of multiple sclerosis (MS), a neurodegenerative autoimmune disease, a finding that may open up new directions for clinical trials of MS treatment (Bjornevik et al., 2022).
In the immunocompetent host, EBV and KSHV persist for many years without causing noticeable pathology. However, when the host becomes immunocompromised, long-term persistence via latency is postulated to contribute to cancer, and a subset of proteins expressed during the lytic viral life cycle have also been suggested to enhance transformation, possibly via auto- and paracrine effects (Manners et al., 2018; Wen et al., 2021).
The restricted host range of EBV and KSHV requires animal models
The very narrow host range of EBV and KSHV is challenging for studying their pathogenesis, but over the past decades animal models were established that provided important insights into the interaction with the host’s immune system of these oncogenic viruses. Different animal models are required to study EBV and KSHV infection as these viruses are the cause of a diverse set of diseases. To analyze the formation of KS-like tumors, a New-World non-human primate model was established, while humanized mice can be used to simulate PEL-like lymphomas (Fujiwara and Nakamura, 2020), as well as KS (Dubich et al., 2019).
Very few species of laboratory animals can be infected with EBV, including cotton-top tamarins (Saguinus Oedipus) and common marmosets (Callithrix jacchus). Rabbits inoculated with EBV also exhibit infection, which results in viral DNA load in peripheral blood lymphocytes and serum antibodies specific to EBV. Humanized mice harboring reconstituted human B, T and natural killer (NK) cells, macrophages, and dendritic cells (DCs) after transplantation of human hematopoietic stem cells exhibit infectious mononucleosis-like symptoms, B cell lymphoproliferative disease and latency (Fujiwara, 2018).
As infection of the animals often results in some, but not all, specific disease conditions resembling human EBV- or KSHV-associated disorders, the use of homologous viruses as surrogate models for EBV and KSHV has been explored. Most prominently, the rhesus lymphocryptovirus (rhLCV, Macacine gammaherpesvirus 4), rhesus macaque rhadinovirus (RRV), and murid herpesvirus 4, also known as murine gammaherpesvirus 68 (MHV68) (Fujiwara, 2018; Fujiwara and Nakamura, 2020) have been studied. While rhLCV and RRV infect non-human primates, MHV68 has been widely used as a model virus to study aspects of gammaherpesvirus infection in mice and lies in the focus of this review (Barton et al., 2011; Wang et al., 2021).
Similar to EBV and KSHV, MHV68 infects and exploits B cells to establish latency (Barton et al., 2011; Wang et al., 2021). Mice infected with MHV68 show symptoms similar to infectious mononucleosis including CD8+ T cell lymphocytosis and splenomegaly (Tripp et al., 1997; Flaño et al., 2002). Additionally, MHV68 infection can drive tumor development in immunocompromised mice (Barton et al., 2011). Besides, MHV68 is a valuable model to study coinfections with other pathogens such as Plasmodium falciparum, helminths or bacteria (Reese, 2016).
The innate and adaptive immune responses are tightly interlinked
The innate antiviral immune response initiates with the recognition of viruses by specific sensors called pattern recognition receptors (PRRs). Upon activation, induced mostly by virus-derived or aberrantly localized nucleic acids, PRR signaling leads to the initiation of an antiviral inflammatory response (de Oliveira Mann and Hornung, 2021; Guy and Bowie, 2022). This includes secretion of proinflammatory cytokines such as interleukins, tumor necrosis factor (TNF), as well as type I interferons (IFN), resulting in the recruitment of innate immune cells including monocytes, DCs, and NK cells (McNab et al., 2015; Carty et al., 2021). The antiviral innate immune response also initiates and shapes the adaptive immune response with its central players, the cytotoxic CD8+ T cells (CTL), CD4+ T helper cells, and B cells (Tomalka et al., 2022). CTL can directly attack and kill virus-infected cells, T helper cells secrete soluble factors with antiviral functions, and the antibodies produced by B cells contribute to neutralization of free virus particles.
An inefficient innate immune response to viral infection results in the development of a non-protective adaptive immune response, higher virus replication and antigen burden, which in turn can lead to T cell exhaustion, a mechanism that also might protect the host from severe inflammation (Panetti et al., 2022). Exhaustion of T cells is identified by loss of effector functions including decreased production of IL-2, TNF, and IFNγ cytokines, as well as elevated and persistent expression of inhibitory receptors such as Programmed cell death protein 1 (PD-1). Different factors can lead to T cell exhaustion, with the presence of high antigen burden in addition to long and persistent antigen stimulation being the main contributors during chronic viral infections (McLane et al., 2019).
In this review, we will highlight both the innate immune response, with focus on the type I IFN and NK cell response, and the adaptive immune response mediated by T cells to EBV and its murine homologue MHV68.
The type I interferon response is crucial to control herpesvirus infections
Host cells are equipped with PRRs that are expressed at the cell surface, the cytoplasm, the endolysosomal compartment and the nucleus, where they recognize molecular structures of invading pathogens or sense cellular alterations induced by pathogens (Figure 1). Several classes of PRRs are involved in innate immune sensing of gammaherpesviruses, including plasma membrane or endosomal Toll-like receptors (TLRs), cytosolic retinoic acid inducible gene I (RIG-I)-like receptors (RLRs), as well as nuclear and cytosolic DNA sensors, which will be discussed in this review. Upon sensing viral infection, PRRs activate downstream signaling cascades leading to the secretion of type I IFN and proinflammatory cytokines (de Oliveira Mann and Hornung, 2021; Guy and Bowie, 2022). Upon secretion, type I IFN exert their activity in an autocrine and paracrine manner by activation of the type I IFN α/β receptor (IFNAR). Binding of type I IFN to the IFNAR leads to the phosphorylation and activation of the transcription factors signal transducers and activators of transcription 1 and 2 (STAT1 and STAT2), carried out by the IFNAR-associated kinases tyrosine kinase 2 (TYK2) (Yan et al., 1996) and Janus kinase 1 (JAK1) (Domanski et al., 1997). Activated STAT1 and STAT2 form a trimeric complex with interferon regulatory factor 9 (IRF9) and translocate into the nucleus, resulting in the induction of interferon-stimulated gene (ISG) expression, whose products mediate broad antiviral activities (Stark and Darnell, 2012).
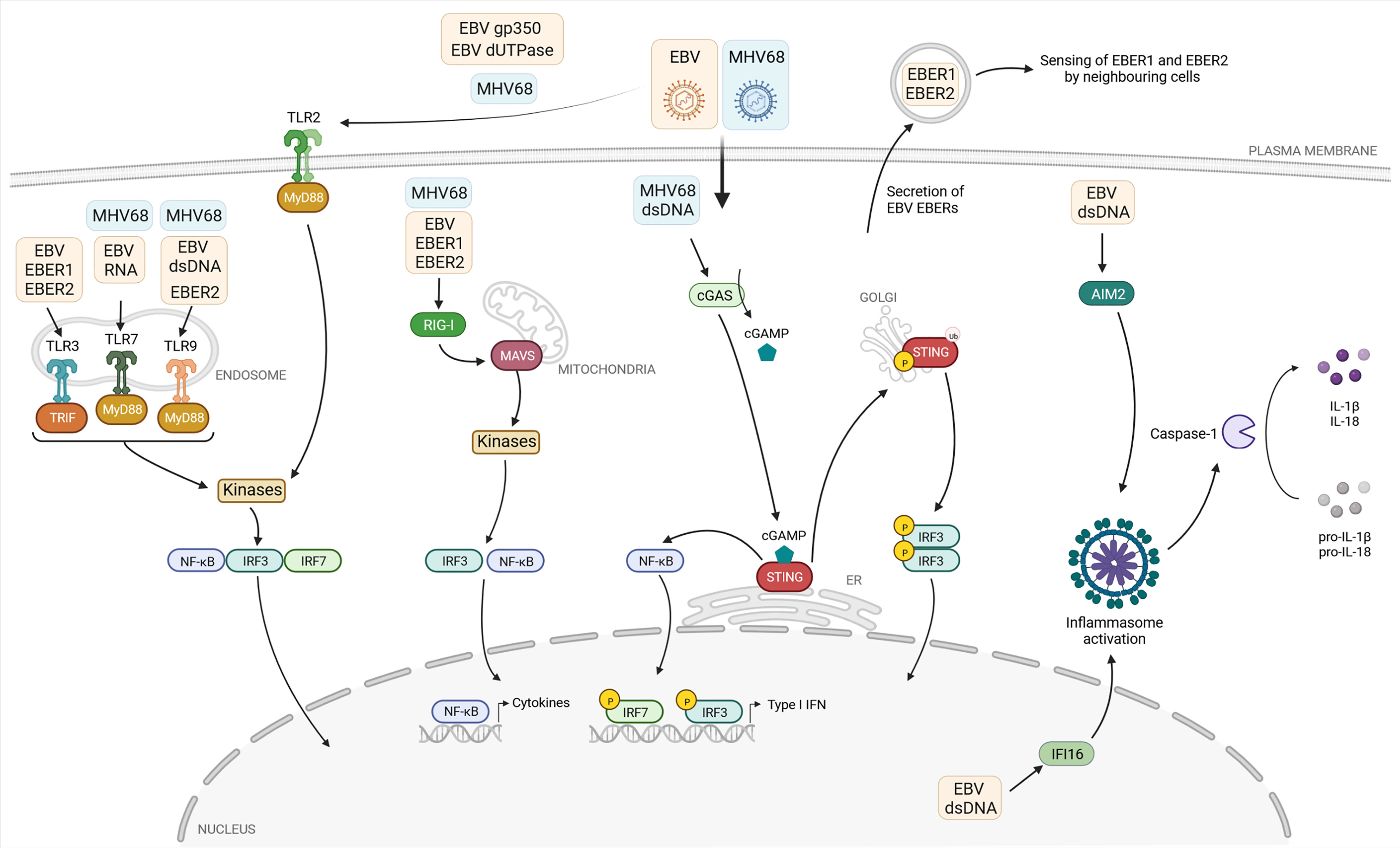
Figure 1 Current knowledge about the detection of the gammaherpesviruses EBV and MHV68 by pattern recognition receptors. The pattern recognition receptors (PRR) Toll-like receptors (TLR) 2, 3, 7, and 9, the cytoplasmic RNA sensor retinoic-acid-inducible protein 1 (RIG-I), and the DNA sensor cyclic GMP-AMP (cGAMP) synthase (cGAS) sense viral proteins or viral nucleic acids associated with EBV (yellow box) or MHV68 (blue box). If known, the viral ligands are depicted in the respective boxes. After binding their ligands, these PRR initiate a signaling cascade, leading to recruitment and/or activation of adaptor molecules: TIR domain-containing adaptor-inducing interferon-β (TRIF) and myeloid differentiation primary-response protein 88 (MyD88) in the case of TLRs, mitochondrial antiviral signaling protein (MAVS) for RIG-I, and stimulator of interferon genes (STING) in the case of cGAS. Subsequently, the transcription factors interferon regulatory factor 3 and 7 (IRF3, IRF7) and nuclear factor-κB (NF-κB) are activated, translocate to the nucleus and induce type I IFN and proinflammatory cytokine expression. The two DNA sensors absent in melanoma 2 (AIM2) and gamma-interferon-inducible protein 16 (IFI16) sense EBV-derived DNA and activate the formation of the inflammasome, leading to caspase-1-dependent formation of the proinflammatory cytokines IL-1β and IL-18. EBER: EBV-encoded small RNA. Created with BioRender.com.
The type I IFN response plays a vital role not only for protecting single cells from viral infection, but also for initiating inflammation and shaping an effective innate and adaptive immune response (Le Bon and Tough, 2002; Ivashkiv and Donlin, 2014). Therefore, type I IFN deficiencies, including non-functional PRR or IFNAR signaling, are usually detrimental and lead to fatalities due to massive viral spread and the inability of the host immune system to control the acute infection as revealed by in vivo studies in mice (Meyts and Casanova, 2021). In humans, such deficiencies or inborn errors in the type I IFN response or its induction lead to complications with different severities, following vaccination with live-attenuated viruses, or infection with herpesviruses and respiratory viruses such as SARS-Coronavirus 2 and Influenza Virus (Casrouge et al., 2006; Hernandez et al., 2019; Bastard et al., 2020; Carter-Timofte et al., 2020; Zhang et al., 2020; Bastard et al., 2021; Andreakos et al., 2022; Gothe et al., 2022; Manry et al., 2022; Mogensen, 2022; Zhang et al., 2022b; Zhang et al., 2022a). To date, the knowledge about type I IFN deficiencies and the outcome of infections with human gammaherpesvirus is limited, probably due to their rare appearance in human populations or due to the absence of clinical complications following most infections (Meyts and Casanova, 2021).
While in vivo evidence for EBV and KSHV control by the type I IFN system is sparse, the MHV68 mouse model provides important insights into its crucial role. Although MHV68 antagonizes the type I IFN response in various ways, the type I IFN response still remains crucial for controlling acute MHV68 infection, latency and reactivation in mice (Wang et al., 2021; Schwerk et al., 2022). The transcription factor interferon regulatory factor 3 (IRF3), which is activated by PRRs, has been shown to be responsible for the induction of type I IFN following MHV68 lytic infection in primary macrophages in an IFNAR-dependent manner (Wood et al., 2013). While WT mice efficiently control acute infection, Ifnar1-/- mice are highly susceptible depending on the viral dose. While 80-90% of Ifnar1-/- mice succumb to MHV68 infection following a high dose intranasal infection (4x106 PFU), ~50% survive low dose infection (4x103 PFU), with viral titers in the lungs being 100-1000 fold higher in KO mice at both high and low doses. In addition, MHV68 disseminates systemically faster in Ifnar1-/- mice (Dutia et al., 1999). Interestingly, genetically modified MHV68 recombinant viruses producing mIFNα1 are attenuated in vivo while they can still establish latency in the spleen (Lenschow et al., 2007; Aricò et al., 2011).
Toll-like receptors: Sensing viral infection at the plasma membrane and in endolysosomes
To date, 10 human (TLR1-TLR10) and 12 murine members (TLR1-TLR9 and TLR11-TLR13) of the TLR family have been identified (El-Zayat et al., 2019; Duan et al., 2022). TLRs are composed of an extracellular domain in charge of sensing pathogen-associated molecular patterns (PAMPs), one transmembrane domain and a cytoplasmic C-terminal domain which mediates the signaling activity via cytoplasmic adapter proteins. TLRs localize to the cell surface or within the intracellular endolysosomal compartment. Cell-surface TLRs include TLR1, TLR2, TLR4, TLR5, TLR6, and TLR10, while intracellular TLRs comprise TLR3, TLR7, TLR8, TLR9, TLR11, TLR12, and TLR13 (Kawasaki and Kawai, 2014). Upon binding their specific ligand, conformational changes of the transmembrane receptors lead to the recruitment and binding of the adaptor molecules myeloid differentiation primary-response protein 88 (MyD88), MyD88-adaptor-like protein (MAL) and TIR domain-containing adaptor-inducing interferon-β (TRIF), with MyD88 being the key adaptor for most TLRs. Upon activation, MyD88 recruits kinases to induce a complex signaling response, eventually leading to the phosphorylation and activation of transcription factors including nuclear factor-κB (NF-κB), IRF3, and IRF7. TLR3 does not recruit MyD88, but instead recruits TRIF to drive an individual signaling pathway, resulting in the similar activation of NF-κB signaling (Figure 1) (Sartorius et al., 2021; Duan et al., 2022).
TLR2, TLR3, TLR7, and TLR9 have been described to contribute to the detection of gammaherpesviruses (Table 1). Upon infection of TLR2-transfected HEK293 cells, UV-inactivated EBV particles strongly induce NF-κB activation and secretion of the chemokine monocyte chemoattractant protein-1 (Gaudreault et al., 2007). Furthermore, by pre-treating TLR2-expressing cells with the viral DNA polymerase inhibitor phosphonoacetic acid and infection with UV-inactivated EBV, NF-κB levels were also increased, indicating that recognition by TLR2 is not dependent on viral replication, but likely induced by binding of EBV particles on the cell surface. Further experiments suggested that TLR2 may recognize the viral surface glycoprotein gp350 (Figure 1), which mediates viral entry into the host cell. Another study based on HEK293 cells identified the EBV-encoded deoxyuridine triphosphate nucleotidohydrolase (dUTPase) as a PAMP, also thought to be recognized by TLR2 (Figure 1) (Ariza et al., 2009). However, further investigations are needed to verify the reported connection between TLR2 and EBV-encoded dUTPase, starting with the examination of possible dUTPase release to the extracellular milieu from EBV infected cells. For MHV68, NF-κB activation in TLR2-transfected HEK293 cells was shown and experiments in primary mouse embryonic fibroblasts (MEF) from WT and TLR2 KO mice showed a TLR2-dependent IL-6 and IFNα response (Figure 1) (Michaud et al., 2010a). Further, MHV68-infected TLR2-deficient mice show decreased IFNα level in their lungs after intranasal infection and increased viral titers. However, upon intravenous infection we could not find evidence for a role of TLR2 during acute MHV68 infection (Bussey et al., 2019). To date, TLR2 is the only cell surface TLR proposed to recognize EBV and MHV68, but the precise nature of the viral ligands was not shown convincingly yet, and its role during EBV infection of its human host is not known.
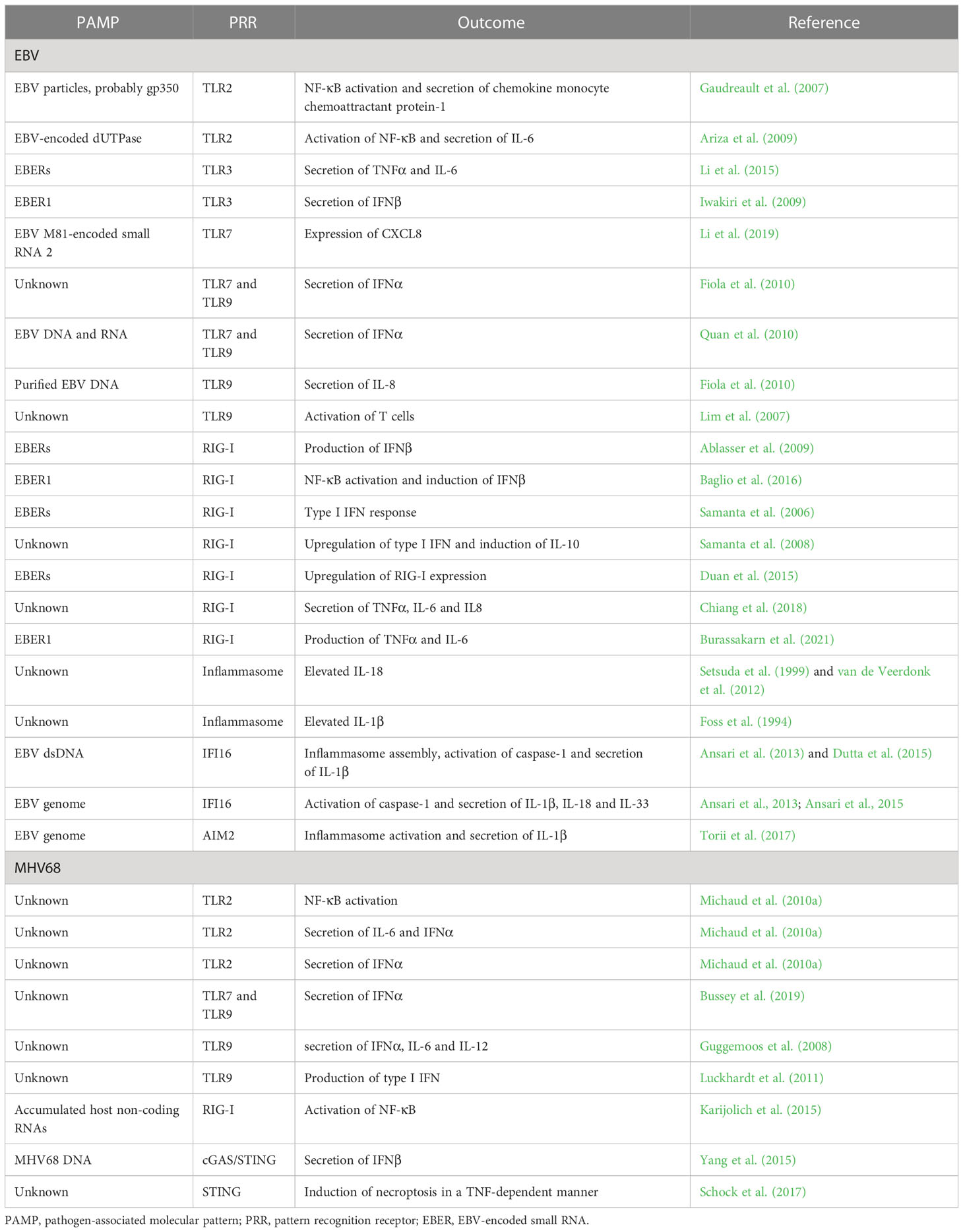
Table 1 Activated innate immunity pathways after EBV and MHV68 infection.
The endosomal TLRs TLR3, TLR7, and TLR9 are responsible for detection of viral nucleic acids. While double-stranded RNA (dsRNA) is recognized by TLR3, TLR7 senses fragments of single-stranded RNA (ssRNA) and TLR9 preferentially recognizes ssDNA containing unmethylated CpG motifs (Alexopoulou et al., 2001; Bauer et al., 2001; Heil et al., 2004). The recognition of gammaherpesviral dsRNA by TLR3 will be illuminated in the RNA-sensing section.
During EBV infection, TLR7 and TLR9 were shown to sense EBV nucleic acids and induce an antiviral response in primary monocytes, plasmacytoid dendritic cells (pDC), and B cells (Figure 1) (Lim et al., 2007; Fiola et al., 2010; Quan et al., 2010; Li et al., 2019). By applying an inhibitor of endosomal TLR activation, TLR9 is shown to recognize purified EBV DNA in primary monocytes, resulting in IL-8 secretion. pDC stimulated with EBV secrete IFNα, which can be reduced by adding specific inhibitors of TLR9 or, to a lesser extent, TLR7 (Fiola et al., 2010). Besides, TLR9- and TLR7-dependent IFNα production induced by EBV DNA and RNA, respectively, was demonstrated in pDC (Quan et al., 2010). In humanized mice, it was shown that EBV-stimulated pDC contribute to the activation of T cells in a TLR9-dependent manner (Lim et al., 2007). A recent study showed a strain-specific effect of EBV strain M81, which was originally isolated from a nasopharyngeal carcinoma. EBV M81-encoded small RNA 2 (EBER2) increases TLR7-dependent expression of the chemokine CXCL8, leading to spontaneous lytic replication in infected B cells, which is not observed with EBERs transcribed from the EBV B95-8 or Akata strains (Li et al., 2019). This suggests that strain-specific polymorphisms may results in different outcomes of the antiviral immune response, enhancing the complexity of studying virus-host interactions.
Consistent with the findings in human cells, TLR9 is also involved in the detection of MHV68 in murine DCs, being responsible for IFNα, IL-6, and IL-12 secretion (Figure 1) (Guggemoos et al., 2008). Moreover, the authors observed an increased viral load in the spleen of TLR9-depleted mice after intraperitoneal infection. In another model, TLR9 expression is shown to be involved in the protection from MHV68-induced lung fibrosis, and required for type I IFN production in the lungs of intranasally infected animals (Luckhardt et al., 2011). Our own work confirmed the important role of TLR9, but also highlighted that TLR7 contributes to the IFNα response of pDC to MHV68 infection (Figure 1). While IFNα secretion is reduced in Tlr9-/- cells compared to WT cells, it is only completey abolished in Tlr7-/-Tlr9-/- double-knockout pDC (Bussey et al., 2019). Thus, the only PRRs contributing to the IFN-α response to MHV68 in pDC are TLR7 and TLR9, but the contribution of TLR7 is masked by the presence of TLR9. Congruently, lytic replication of MHV68 after intravenous infection is enhanced in the liver and spleen of Tlr7-/-Tlr9-/- mice. In addition, latent viral loads and reactivation of MHV68 are enhanced in latently infected Tlr7-/-Tlr9-/- splenocytes (Bussey et al., 2019).
In summary, TLR7 and TLR9 both contribute to detection and control of MHV68 infection in vivo. The exact nature of the TLR7 and TLR9 ligands during MHV68 infection has not been shown yet. For EBV, the data on TLR2, TLR7 and TLR9 are scarce but can now be generated with the tools of the Cas9 and genomics era, at least in vitro.
How RNA sensors detect DNA viruses: TLR3 and the cytoplasmic sensor RIG-I
At first sight it seems counterintuitive that DNA viruses, which replicate their DNA genome in the nucleus and transcribe their genes with the cellular nuclear transcription machinery, are sensed by cytosolic or endosomal RNA sensors. However, several studies suggest that cellular sensors of dsRNA, namely TLR3 and RIG-I, indeed play a role for detection of gammaherpesviruses (Table 1).
EBV encodes non-coding and non-polyadenylated RNAs called EBERs 1 and 2. These small RNAs can adopt secondary structures containing multiple intramolecular stem-loops which resemble dsRNA structures (Rosa et al., 1981; Glickman et al., 1988) and are transcribed by the host DNA-dependent RNA polymerase III from the EBV genome (Rosa et al., 1981; Arrand and Rymo, 1982). TLR3 senses dsRNA in endosomes, and two studies have shown that it seems to detect EBERs (Figure 1) (Iwakiri et al., 2009; Li et al., 2015). One of them shows that exogenously expressed EBERs induce inflammatory responses through TLR3 in nasopharyngeal carcinoma cells (Li et al., 2015). The other study addresses the possible underlying mechanism of TLR3-sensing of EBERs: EBER1 binds the cellular lupus erythematosus-associated antigen (La) to evade degradation, and this interaction induces the active secretion of EBER1-La complexes with the possibility of being secreted as an exosome during EBV infection (Figure 1). Upon endocytosis of EBER1-La-containing exosomes, TLR3 can recognize EBER1 and induce downstream signaling (Iwakiri et al., 2009). However, further studies are needed to clarify how the EBER1-La complex is recognized by TLR3 and the exact mechanism of EBER1 release.
The cytoplasmic RNA helicases retinoic-acid-inducible protein 1 (RIG-I, also known as Ddx58) and melanoma differentiation-associated protein 5 (MDA5) belong to the RIG-I-like receptor (RLR) family. While RIG-I preferentially binds short, 5’ di- and triphosphorylated ssRNAs, as well as dsRNA, MDA5 preferentially recognizes long dsRNA in the cytoplasm (Hornung et al., 2006; Wang et al., 2010; Peisley et al., 2011). Binding of RIG-I or MDA5 to their respective ligands results in the activation of the mitochondrial antiviral signaling protein (MAVS), which is associated with mitochondria. Activated MAVS induces downstream signaling of TBK1/IRF3 or IKK/NF-κB leading to the production of type I IFN and proinflammatory cytokines, respectively. Several studies highlight the role of EBV-encoded EBER transcripts, which are sensed by RIG-I (Figure 1).
RNA polymerase III, responsible for producing the EBER transcripts as well as other cellular small RNAs such as rRNAs and tRNAs, was shown to induce RIG-I-dependent IFNβ production in EBV-infected cells (Ablasser et al., 2009). It does so by converting cytosolic poly(dA-dT) DNA into the RIG-I ligand 5’-phosphorylated dsRNA (Ablasser et al., 2009; Chiu et al., 2009). Since inhibition of RNA polymerase III leads to suppressed EBER1/2 RNA transcription, resulting in a lower RIG-I-dependent activation of IFNβ production (Ablasser et al., 2009). RNA polymerase III seems to contribute to the detection of EBV infection by transcribing EBV-derived small RNAs. This is also supported by the finding that EBER1 can be transferred via exosomes to uninfected DCs and trigger antiviral immunity in a RIG-I-dependent manner (Baglio et al., 2016).
Additional studies highlight RIG-I as a critical sensor for EBER1 and EBER2. The direct interaction of RIG-I and both EBERs was shown after transfection of RIG-I-expressing plasmids into EBER-positive EBV-infected cells followed by RIG-I immunoprecipitation and reverse-transcription PCR (RT-PCR) for EBERs (Samanta et al., 2006). Furthermore, a RIG-I-dependent type I IFN response can be detected after reintroducing EBERs in EBER-knockout EBV. Besides the RIG-I-dependent upregulation of type I IFN upon EBV infection in EBV-infected cancer cells, it is demonstrated that IL-10 is induced by RIG-I. Knockdown of RIG-I downregulated IL-10 secretion in EBER-positive EBV-infected cells, which is dependent on the transcription factor IRF3 (Samanta et al., 2008). Furthermore, it is shown that EBV activates RIG-I by disrupting binding of nuclear 5S rRNA pseudogene transcripts with binding partners leading to their unshielding, and thereby recognition by RIG-I, but not MDA5 (Figure 1) (Chiang et al., 2018). While the exact mechanism driving the re-localization of host nuclear 5S rRNA is unknown, it was hypothesized that the EBV-mediated host shut-off of the cellular translation machinery plays an important role in inhibiting expression of host proteins which regulate the localization of the nuclear RNA pseudogene transcripts. The link between transcription of EBERs and RIG-I is furthermore shown in the nasopharyngeal carcinoma-derived HNE2 cell line transfected with EBERs which resulted in upregulation of RIG-I expression in a dose-dependent manner (Duan et al., 2015). Aside from IL-10 and type I IFN, TNFα, IL-6, and IL-8 transcripts are also upregulated in EBV-reactivated gastric adenocarcinoma cells in a RIG-I-dependent manner (Chiang et al., 2018).
While human B lymphocytes and epithelial cells are the major targets of lytic and latent EBV infection, several studies have demonstrated the involvement of monocytes in EBV infection (Savard et al., 2000; Masy et al., 2002; Michaud et al., 2010b). One mechanism showing the interplay between EBV and the innate immune response of human monocyte-derived macrophages (MDM) includes incoming exosomes containing EBER1 transcripts, which induce TNFα and IL-6 production in a RIG-I-dependent manner, promoting indoleamine 2, 3-diocygenase (IDO) expression in the cells. Activation of IDO creates an immunosuppressive microenvironment, which negatively affects T cell responses by suppressing the proliferation and cytolytic activity of CD4+ and CD8+ T cells (Burassakarn et al., 2021), suggesting a possible role of EBERs for inhibition of the adaptive immune response.
In summary, it is not clear how exactly EBERs are secreted into the supernatant of infected cells, either by active secretion in complex with a host protein or in form of extracellular vesicles such as exosomes or microvesicles (Zhao et al., 2019). To date, the properties of these EBER-containing vesicles and their specific roles in the viral life cycle remain largely unclear and the exact mechanism of innate immune detection needs further validation.
Similar to EBV, the involvement of RNA polymerase III was reported in the context of MHV68 infection. RNA polymerase III-dependent transcription of host non-coding RNAs, such as nucleolar protein 14 and Go-Ichi-Ni-San complex subunit 1, can be sensed by RIG-I and activate NF-κB. Infection with MHV68 results in an accumulation of these stimulatory host RNAs, showing an indirect mechanism for RIG-I-dependent sensing of gammaherpesvirus infection (Figure 1) (Karijolich et al., 2015). In line with this, MEF lacking functional RIG-I are found to be more permissive to MHV68 infection compared to cells expressing functional RIG-I (Inn et al., 2011).
Taken together, these data point to a critical role of the RNA sensors TLR3 and RIG-I to detect human and murine gammaherpesvirus infection through the recognition of stimulatory virus- as well as host-derived non-coding RNAs.
DNA sensors: Potent activators of the antiviral response
Intracellular DNA is recognized by different sensors, including the proteins type I IFN inducible protein absent in melanoma 2 (AIM2) (Hornung et al., 2009), gamma-interferon-inducible protein 16 (IFI16) (Unterholzner et al., 2010), and cyclic GMP-AMP synthase (cGAS) (Wu et al., 2013) (Table 1). Upon binding to dsDNA in a length-dependent manner, cGAS catalyzes the formation of the second messenger 2’3’-cGAMP which binds to the endoplasmic reticulum (ER)-resident adaptor protein stimulator of IFN genes (STING) leading to its activation (Liu et al., 2015). Originally, cGAS was identified as a cytosolic sensor, but recent studies indicate that cGAS also resides in the nucleus (Volkman et al., 2019; Pathare et al., 2020; Sun et al., 2021). Upon activation, STING translocates from the ER to the Golgi apparatus (Ishikawa and Barber, 2011). Located at the Golgi, STING undergoes several poly-ubiquitinations (Tsuchida et al., 2010; Zhang et al., 2012; Wang et al., 2014), which lead to the phosphorylation of STING followed by the phosphorylation and activation of IRF3 and subsequent type I IFN expression (Liu et al., 2015). Moreover, upon activation, ER-resident STING activates NF-κB prior to its translocation to the Golgi apparatus, mediating the expression of proinflammatory cytokines (Stempel et al., 2019b).
While many studies highlight cGAS/STING signaling during herpesvirus infection and herpesviral evasion of this pathway (Stempel et al., 2019a), research into the role of DNA sensors in the context of EBV infection is sparse. For EBV, most studies are restricted to B cells, as the virus exploits B cells as a reservoir for persistent infection. Interestingly, uninfected B cells were found to lack detectable STING expression, while EBV-infected cells did express cGAS and STING, yet were not able to produce type I IFN upon dsDNA stimulation (Gram et al., 2017). Correlating with this observation, EBV has been shown to induce the E3 ligase TRIM29 in epithelial cells, which regulates K48-linked ubiquitination and degradation of STING, preventing the activation of cGAS-STING signaling (Xing et al., 2017). However, if this scenario contributes to the dysfunction of cGAS-STING signaling in EBV-infected B cells or if EBV inhibits cGAS/STING activation by a hitherto unrecognized mechanism warrants further investigation.
In contrast to EBV, the cGAS/STING pathway was found to be relevant during MHV68 infection (Figure 1). Upon intraperitoneal infection, viral titers are increased in the spleens and lungs of cGAS-deficient mice, confirming the importance of this signaling pathway for the antiviral immune response (Schoggins et al., 2014). Following stimulation with MHV68 DNA, cGAS/STING-dependent signaling is activated in mesenchymal stem cells resulting in IFNβ secretion (Yang et al., 2015). Furthermore, MHV68 infection is capable of inducing necroptosis in a murine fibrosarcoma cell line through STING in a TNF-dependent manner (Schock et al., 2017), proposing distinct roles of STING in the response to gammaherpesvirus infection in mice.
Inflammasomes: Multiprotein complexes engaging in cytokine secretion
Inflammasomes are intracellular multiprotein complexes that are assembled upon pathogen recognition or danger signals. Members of the NOD-like-receptor (NLR) family, i.e. NLRP1, NLPR3, or NLRC4, are associated with inflammasome formation. AIM2 is another cytoplasmic sensor for inflammasome activation that recognizes dsDNA (Hornung et al., 2009; Hu et al., 2016). Different herpesviruses activate distinct inflammasome-activating sensors, e.g. HSV-1 was demonstrated to activate the NLRP3 inflammasome (Karaba et al., 2020), HCMV activates the AIM2 inflammasome (Botto et al., 2019), while KSHV is shown to induce IFI16-dependent inflammasome activation (Kerur et al., 2011). For EBV and MHV68, multiple inflammasome-activating pathways were identified, which will be discussed in this section (Table 1).
Canonical inflammasomes are composed of three major components: a sensor protein, a complex called adaptor-apoptosis-associated-speck-like-protein-containing-a-caspase-recruitment domain (ASC) and caspase-1 (Figure 1) (Evavold and Kagan, 2019). Upon activation, the sensor protein recruits ASC molecules which undergo oligomerization, followed by the recruitment of pro-caspase-1. Pro-caspase-1 catalyzes autolysis to produce the active caspase-1, followed by caspase-dependent cleavage of immature forms of the proinflammatory cytokines IL-1β and IL-18. Mainly myeloid cells such as macrophages produce IL-1β to mediate immune responses against pathogens and tissue damage (Hornung et al., 2009). While IL-1β is shown to be a potent proinflammatory cytokine that is crucial for host-defense to infection and injury, but also for the polarization of CD4+ T cells and the activation and differentiation of antigen-specific CTL (Dinarello, 1996; Nambu et al., 2006; Ben-Sasson et al., 2013; Garlanda et al., 2013), IL-18 is demonstrated to induce the differentiation of CD4+ to T helper cells 1 (Th1) and Th2, regulating their immune responses, and to drive NK cell and CTL activity through the promotion of IFNγ production (Xu et al., 2000; Nakanishi et al., 2001; Dupaul-Chicoine et al., 2015).
Previous studies observed elevated IL-18 levels in EBV-induced infectious mononucleosis, indicating the activation of the inflammasome (Setsuda et al., 1999; van de Veerdonk et al., 2012). Similarly, IL-1β is found to be elevated in the tonsils of children infected with EBV (Foss et al., 1994). In infected B cells, IFI16-dependent and AIM2-independent inflammasome assembly, production of active caspase-1, and IL-1β secretion is shown, which relies on episomal dsDNA binding of IFI16 in the nucleus (Figure 1) (Ansari et al., 2013; Dutta et al., 2015). The colocalization of IFI16 and the EBV genome is demonstrated by immunofluorescence of latently infected B cells and this colocalization results in the acetylation of IFI16 and association with ASC in de novo infected primary B cells, followed by cleavage of pro-caspase-1 and secretion of IL-1β, IL-18, and IL-33 (Ansari et al., 2013; Ansari et al., 2015). Moreover, Epstein-Barr nuclear antigen 1 (EBNA1) and EBERs could be excluded from being responsible for inflammasome activation. Interestingly, the knockdown of endogenous IFI16 results in enhanced levels of EBV lytic gene expression as well as an increase in EBV genome abundance, while overexpression of IFI16 reverses these effects (Pisano et al., 2017; Torii et al., 2017), suggesting that IFI16 is critical for controlling EBV replication and gene expression.
Apart from nuclear IFI16, the cytoplasmic dsDNA sensor AIM2 is involved in EBV genome sensing and inflammasome activation (Figure 1). In the human monocytic cell line THP-1 and primary human monocytes, EBV infection leads to the release of IL-1β. AIM2 expression is upregulated in infected cells, and knockdown of AIM2 attenuates IL-1β release (Torii et al., 2017), indicating that AIM2-dependent activation of the inflammasome is triggered by EBV infection.
In summary, multiple inflammasome pathways play a role during EBV infection, but the cell type-specificity of inflammasome activation and its effects on the adaptive immune response during the different stages of EBV infection have yet to be analyzed.
Too strong to be tolerated: EBV and MHV68 evade the PRR-mediated type I IFN response
The fact that herpesviruses evolved multiple strategies to modulate their host’s immune response clearly highlights the power of the antiviral response to control infection. Studies analyzing the role and mechanism of virally encoded immune evasions clearly substantiated our knowledge about the essential role of the type I IFN response to control viral infection. MHV68 evolved different strategies to evade the type I IFN response to promote the primary lytic infection, obvious by hardly detectable type I IFN secretion in in vitro infected cells (Bussey et al., 2018). Herpesviral immune evasion appears at multiple levels of the innate immune response. To avoid the DNA-sensing pathway after entry into the host cell, MHV68-encoded ORF64 efficiently delivers viral DNA to the nucleus, while loss of ORF64 results in accumulated localization of viral DNA in the cytoplasm, leading to the activation of STING and AIM2 (Sun et al., 2015). Furthermore, MHV68-encoded ORF11, a virion-associated tegument protein, binds to the kinase TBK1 and disrupts its interaction with IRF3, thereby inhibiting IRF3-mediated induction of ifnb1 transcription (Kang et al., 2014). MHV68 ORF36, a conserved herpesviral kinase, inhibits IFNβ production by interacting with the activated form of IRF3 inside the nucleus, thereby suppressing the recruitment of RNA polymerase II to the IFNβ gene promoter. The lack of ORF36 leads to the attenuation of the virus in vitro and in vivo, causing delayed but not completely impaired establishment of latency (Hwang et al., 2009). Moreover, the MHV68 M2 latency protein contributes to inhibition of the type I IFN response by downregulating STAT1 and STAT2 expression in fibroblasts and B lymphocytes (Liang et al., 2004). The EBV-encoded microRNA BamHI fragment A rightward transcript 16 (BART16) suppresses type I IFN signaling via directly targeting Cyclic adenosine monophosphate Response Element Binding protein (CREB), which is a key transcriptional activator of the type I IFN signaling pathway (Hooykaas et al., 2017). Additionally, the EBV early protein nuclear egress protein 2 suppresses IFNβ transcription by inhibiting IRF3 activation (Wang et al., 2020). The EBV tegument protein BGLF2 suppresses type I IFN signaling by binding to Tyk2 and suppressing JAK-STAT signaling through recruitment of Src homology region 2 domain-containing phosphatase-1 (SHP1) phosphatase, promoting STAT2 degradation, which leads to decreased expression of ISGs including IRF1, IRF7, and MxA (Liu et al., 2020; Jangra et al., 2021). This large portfolio of herpesviral immune modulators highlights the necessity to dampen the innate immune response at multiple levels and allow the virus to gain a foothold in its host.
The role of the type I IFN response in shaping the immune response to gammaherpesvirus infection
Type I IFN and proinflammatory cytokines are critical for the maturation of other innate and adaptive immune cells, such as NK and cytotoxic T cells, and guide their recruitment to the site of infection, where they contribute to recognition of virus infection, cytokine production, and killing of infected cells (Wedekind et al., 2020). In addition to directly impeding viral replication, type I IFN also helps to shape an effective adaptive immune response (Figure 2), however, this appears to be context-dependent and complex.
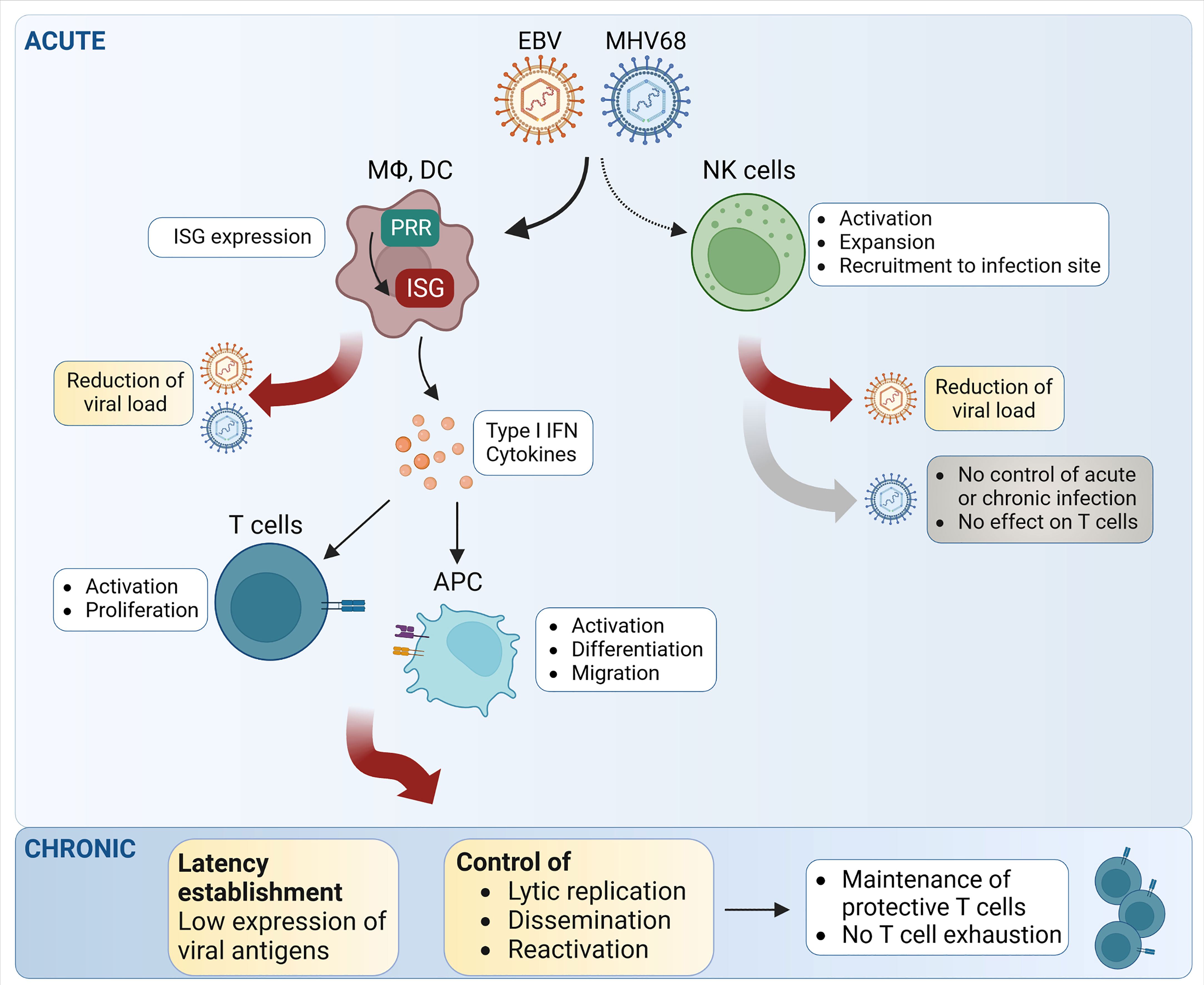
Figure 2 Schematic representation of the main steps of NK- and T cell-mediated control of EBV and MHV68 infection. During acute infection, both EBV and MHV68 trigger natural killer (NK) cell activation, recruitment and expansion. However, the relative impact on the control of MHV68 infection and MHV68-triggered T cell responses appears limited, while NK cells seem to have an effect on EBV replication. Upon detection of EBV and MHV68, pattern recognition receptors (PRR) induce the expression of interferon-stimulated genes (ISG), which exert direct effects on critical steps of the viral life cycle. By inducing type I IFN and proinflammatory cytokine secretion, EBV and MHV68 can trigger important downstream effects leading to T cell activation and T cell proliferation. During chronic infection, the type I IFN response and T cells both contribute to the control of latency and reactivation. APC, antigen-presenting cells; DC, dendritic cells; MΦ, macrophages. Created with BioRender.com.
Type I IFN can impact the priming of adaptive cell-mediated immune responses in many ways such as by promoting differentiation of myeloid precursors into DCs (Santini et al., 2000; Dauer et al., 2003), upregulating the expression of major histocompatibility complex (MHC) and co-stimulatory molecules such as CD80 and CD86 (Montoya et al., 2002), and promoting the migration of DCs from tissues to lymph nodes by upregulation of chemokine receptor 7 expression, which ultimately promotes antigen presentation and priming of T cells in the lymph nodes (Parlato et al., 2001; Rouzaut et al., 2010). In addition to indirectly affecting T cell priming and activation, the type I IFN response can directly promote or inhibit T cell proliferation and cytokine production depending on the activation status of T cells and their antigen specificity (Marshall et al., 2011; Keppler et al., 2012; Welsh et al., 2012).
The impact of the type I IFN response on gammaherpesvirus infection and the developing adaptive immune response has been more intensively studied for MHV68 than for EBV. The type I IFN response is essential for the control of acute and latent MHV68 infection (Mboko et al., 2017; Schwerk et al., 2022). Following MHV68 infection of Ifnar1-/- mice, the expression of TNF-α, IFNγ, and IL-2 is decreased in antigen-specific CD8+ T cells, showing clear hallmarks of T cell exhaustion in the absence of type I IFN signaling (Jennings et al., 2014). Thus, intact type I IFN signaling is important for the CD8+ T cell response following MHV68 infection (Figure 2). However, the effects of the type I IFN response on the CD8+ T cell response are probably mediated by non-T cells, because T cell-specific abrogation of type I IFN signaling shows no effect (Jennings et al., 2014). Hence, the type I IFN response may control the MHV68-specific T cell response in an indirect manner by regulating the extent of MHV68 replication.
Similarly, it has recently been shown that the type I IFN response contributes significantly to the control of MHV68 latency (Figure 2). Adoptive transfer of MHV68 latently- splenocytes into Ifnar1-/- recipient mice led to higher virus propagation and dissemination as well as higher risk of virus reactivation in Ifnar1-/- mice (Schwerk et al., 2022). Accordingly, type I IFN-deficient mice can only survive low dose MHV68 infection (Barton et al., 2005). During EBV infection, type I IFN responses by pDCs can transiently dampen virus replication and thereby suppress CD8+ T cell proliferation (Gujer et al., 2019). But even without type I IFN signaling, T cells can infiltrate the brain and spinal cord of MHV68-infected Ifnar1-/- mice, revealing the impact of IFN-independent inflammatory pathways for T cell migration (Márquez et al., 2022).
The contribution of central downstream signaling factors of the type I IFN response has also been described. For example, interferon regulatory factor 1 (IRF1) is an antiviral transcription factor and tumor suppressor (Tamura et al., 2008; Panda et al., 2019). IRF1 expression is robustly induced in epithelial cells in response to IFNβ, and IRF1-deficient mice are more susceptible to viral infections (Novatt et al., 2016). Expression of IRF1 leads to selective attenuation of the MHV68-driven germinal center reaction in a global and T cell intrinsic manner, hence restricting the expansion of the latent MHV68 reservoir (Jondle et al., 2021). IRF1 deficiency leads to an increase and expansion of IL17A expressing CD4+ T cells as well as follicular T helper cells which are critical for the initiation of the germinal center reaction, indicating a role for IRF1 in suppressing subpopulations of CD4+ T cells that support chronic MHV68 infection. Accordingly, IRF1 deficiency resulted in an increase in the frequency of germinal center B cells, Tfh cells as well as the latent MHV68 reservoir in the spleen and peritoneal cavity of mice following MHV68 infection (Jondle et al., 2021).
Together, these recent studies highlight that the gammaherpesvirus-triggered type I IFN response influences the T cell response on different levels. Firstly, type I IFN acts directly on antigen-presenting cells and on responding T cells themselves. Secondly, type I IFN activity helps to minimize gammaherpesvirus spread and thereby keeps the viral load under control (Figure 2). In this way, a reduced gammaherpesvirus load in the host leads to reduced overall lytic gene expression, thereby limiting the available amount of antigen for T cell stimulation. This tuning of viral antigen availability by type I IFN is therefore a major factor for T cell proliferation and activation.
The NK cell-mediated response during EBV and MHV68 infection
Natural killer cells are major players in the host response to viral infection. Known as a member of innate lymphoid cells (ILC) with cytotoxic properties, they were first identified by their ability to lyse tumor cells in vitro (Kiessling et al., 1975; Oldham, 1983). NK cells derive from common lymphocyte progenitors and reside in peripheral blood, lymphoid organs, and various other tissues (Carrega and Ferlazzo, 2012). Although they have similar functions as cytotoxic CD8+ T cells, NK cells do not need prior antigen exposure and priming. They express germline-encoded receptors to sense their environment, including inhibitory receptors, e.g., Killer Ig-Like Receptors (KIR) in humans and members of the Ly49 family in mice, and activating receptors, e.g., NKG2D, DNAX accessory molecule-1 (DNAM-1) in mice and humans and Natural cytotoxicity receptors (NCRs) (e.g. NKp46) in mice (Nabekura et al., 2014; Abel et al., 2018). NK cells sense inflammatory signals and become activated and expanded through various pathways: via their cytokine receptors (IFNAR1, IL-12R, IL-15R, and IL-18R) (Vivier et al., 2008), via their Fc gamma RIII (CD16) receptor (Lee et al., 2015), and by activating receptors expressed by the majority of NK cells (Bottino et al., 2005).
During viral infection, NK cells are alerted by cytokines, such as type I IFN, IL-12, or IL-18 (Hammer et al., 2018). After activation, NK cells exert their role via two effector functions, first by contact-dependent cytotoxicity (Krzewski and Strominger, 2008). This function includes recognizing, contacting, and establishing an immunological synapse with the target cell and inducing apoptosis by ligation of FasL and TRAIL ligands (Khosravi-Far and Esposti, 2004), or by degranulation of Granzyme B and Perforins (Bhat and Watzl, 2007). As a second effector function, NK cells also secrete a range of proinflammatory cytokines and chemokines, including IFNγ, TNF, and Granulocyte-macrophage colony-stimulating factor, which further activate other immune cells (Fauriat et al., 2010; Vivier et al., 2011).
NK cells probably play a role during gammaherpesvirus infection (Münz, 2021). Accordingly, to counteract this arm of the immune response, human gammaherpesviruses have developed various strategies to evade NK cell-mediated immune responses, mostly by suppressing the signaling of activating receptors and triggering the signaling of inhibitory receptors (Münz, 2021). Multiple observations support the theory that NK cells contribute to the cellular immune response following human gammaherpesvirus infections. For example, EBV-specific CD8+ T cells and human NK cells expand during infectious mononucleosis (IM) (Chijioke et al., 2016). Also, genetic polymorphisms of the NKG2D receptor gene axis have been associated with susceptibility to develop EBV-induced nasopharyngeal carcinoma (Viet et al., 2021). Additionally, decreased frequency of cytotoxic NK cells (CD56dim CD16+) causes impairment in antibody-dependent NK cell cytotoxicity in patients with EBV+ classical Hodgkin lymphoma (Pánisová et al., 2022).
Interestingly, increased activation of the PI3K/Akt pathway and increased levels of STAT1 were observed in NK cells from patients with chronically active EBV infection (Howe et al., 2020). This indicates that NK cells are continuously activated by lytic EBV infection and contribute to the innate immune response against EBV (Figure 2). In addition, IFNβ treatment increases the cytotoxicity of NK cells against nasopharyngeal carcinoma cells in vitro in a TNF-related apoptosis-inducing ligand (TRAIL)-dependent manner as TRAIL expression levels on the cell surface of NK cells increased following IFNβ treatment in patients (Makowska et al., 2019).
Only few studies on the role of NK cells in the context of MHV68 infection are published yet, and the mechanisms of MHV68-mediated evasion of the NK cell response are not well understood. Although NK cells are activated, expand, and get recruited to the site of infection following MHV68 infection in C57BL/6 mice, they do not significantly contribute to the control of MHV68 acute or latent infection (Figure 2) (Usherwood et al., 2005; Thomson et al., 2008). Depletion of NK cells does not lead to significantly higher viral loads in the lungs compared to control mice following intranasal infection. Additionally, NK cells do not seem to play an important role for the development of the adaptive immune response during MHV68 infection, in particular for the expansion of virus-specific CD8+ T cells (Usherwood et al., 2005; Thomson et al., 2008). However, another study showed that after subcutaneous footpad MHV68 infection, NK cells restrict the lytic infection of sub-capsular (SCS) macrophages in the infected lymph nodes, suggesting that NK cells may contribute to the immune response in a tissue-specific manner (Lawler et al., 2016). In another study, the authors proposed a CD4+ T cell-NK cell axis that is contributing to the control of MHV68 infection in the lungs (Lawler and Stevenson, 2020). In this case, primed virus-specific CD4+ T cells migrate to the lungs and drive the activation of local antigen-presenting cells (APC) via IFN-γ secretion. The activated APC then recruit and activate NK cells, presumably by secreting IL-12 and IL-18. Subsequently, activated NK cells contribute to the killing of infected cells and suppress further viral replication via IFN-γ secretion (Lawler and Stevenson, 2020). Regarding NK cell-mediated contact-dependent cytotoxicity, it was suggested that MHV68-infected cells circumvent this by up-regulating the inhibitory receptor CEACAM1 on the surface of infected cells (Adler et al., 2009; Adler et al., 2014). However, this needs further investigation. Taken together, there is clear evidence in both humans and mice that NK cells are activated following gammaherpesvirus infection. Nonetheless, a dominant role of NK cells in the control of gammaherpesvirus infection has not been found, indicating that the NK cell immune response alone is not strong enough to control gammaherpesvirus infections.
Mechanisms of T cell-mediated control of EBV and MHV68 infection
The conventional thymus-derived T cells express alpha and beta T cell receptor chains that allow them to specifically bind to short peptides presented in the context of either MHC class I or II. Most mammalian cells express MHC class I to present peptides to CD8-expressing T cells, and CD4-expressing T cells can be activated by peptide recognition by MHC class II expressed on the surface of antigen-presenting cells (Chopp et al., 2023).
CD8+ T cells play a crucial role in controlling different phases of MHV68 infection including acute and latent infection as well as reactivation via secretion of perforin, granzymes and IFNγ (Topham et al., 2001; Tibbetts et al., 2002; Loh et al., 2004). Likewise, CD8+ T cells play a central role in controlling EBV primary infection (Taylor et al., 2015). However, both MHV68 and EBV can establish latency in the host, irrespective of the very strong cellular immune response (Torti and Oxenius, 2012).
Gammaherpesvirus immune evasion: The role of CD8+ T cells and surface MHC class I expression
How MHV68 and EBV evade the MHC class I-restricted CD8+ T cell response remains an important question for both immunological and virological studies. Gammaherpesvirus proteins like the K3 protein mediate MHC class I downregulation in KSHV (Brulois et al., 2014) and MHV68 (Stevenson et al., 2002). Furthermore, gammaherpesvirus genome maintenance proteins (GMP) have recently been established to play a role in both latency maintenance and evasion of CD8+ T cell immunity (Sorel and Dewals, 2019), following infection with both MHV68 and EBV (Bennett et al., 2005; Ressing et al., 2008). Additionally, the presence of gammaherpesvirus-infected B cells in the thymus could alter T cell development in this primary lymphoid organ, and thereby cause evasion of viral epitopes by depletion of virus-specific CD8+ T cells even before such T cells could be activated in lymph nodes that drain gammaherpesvirus-infected tissues (Yamano et al., 2019). To treat latently infected patients, a specific reduction of gammaherpesvirus-mediated MHC class I immune evasion may allow more efficient CD8+ T cell immunity. This concept was recently tested in KSHV-infected human umbilical vein endothelial cells in vitro, where treatment with CDK4/6 inhibitors was shown to counteract KSHV-triggered MHC class I down-modulation (Wu et al., 2022). Such direct inhibition of viral immune evasion could be beneficial in the context of adoptive CD8+ T cell therapy of chronic gammaherpesvirus-infected patients to allow more complete recognition and killing of infected target cells.
The contribution of CD4+ helper vs. CD8+ T cells during gammaherpesvirus infection
CD4+ T cells also play an important role in immune-cell mediated control of gammaherpesvirus infection. Heterogeneous clones of CD4-expressing T helper cells are generated during MHV68 infection (Hu et al., 2015). It is proposed that CD4+ T cell activation during MHV68 infection is mediated by uninfected myeloid cells that present MHV68-derived peptides loaded on MHC class II (Lawler and Stevenson, 2020). This indirect cellular control of herpesvirus infection has been previously shown for MCMV infection models (Lueder et al., 2018) and following different routes of MCMV infection (Xie et al., 2022). By recognition of different gammaherpesvirus-infected cell types, CD4+ and CD8+ T cells can cooperate to control the infection (Tan et al., 2017). However, further insights into the regulation of CD4+ and CD8+ T cell responses are needed to allow a better understanding of infection control and host damage limitation during persistent infection.
Direct contact-dependent killing of MHV68- or EBV-infected cells is an important mechanism how the adaptive immune system controls the viral spread in the host (Ressing et al., 2015). In light of this mechanism, adoptive T cell therapy in EBV-infected patients aims to selectively kill EBV-infected cells (Lammoglia Cobo et al., 2022). In ongoing trials, EBV-specific T cells are injected with the purpose to reduce numbers of EBV-infected B cells and thus lower overall disease severity. It is well established that T cells recognize many different gammaherpesvirus epitopes during primary infection and latency (Gredmark-Russ et al., 2008; Freeman et al., 2010). However, it remains unclear (1) which of these epitopes are presented in vivo on the different types of MHV68- and EBV-infected cells, and (2) when they are presented during infection. Strong T cell-mediated responses might be triggered against cross-presented epitopes that may be not present at high levels on the actual target cells. i.e. latently infected B cells. Adding additional complexity, the cell type- and tissue-specific innate sensing and resistance mechanisms can determine which cells are productively infected by MHV68 and EBV (Fujiwara and Nakamura, 2020). In this context, it is important to keep in mind that T cell-mediated responses likely differ between control of acute virus infection versus chronic viral infection (Sarawar et al., 2020). This distinction may depend on the different inflammatory milieu during acute and chronic phases of gammaherpesvirus infection, and further studies are necessary to gain knowledge about the infected cell types, triggered innate immune mechanisms, and different MHC class I and class II T cell epitopes presented in vivo.
Gammaherpesvirus control by T cells: Role of T cell activation and costimulation
Costimulatory signals are important in shaping the magnitude and phenotype of virus-specific T cells. In the case of EBV, innate immunity and downstream inflammation can affect T cell responses by shaping signaling in antigen-presenting cells (Figure 2). For example, CD27 costimulatory signals from antigen-presenting B cells are needed for effective EBV-specific CD8+ T cell immune responses (Deng et al., 2021). Antigen-presenting cells can either stimulate T cell responses (costimulatory pathways) or inhibit T cell responses (coinhibitory pathways), a phenomenon with important roles in e.g. autoimmunity or transplantation (Kean et al., 2017). In the context of gammaherpesvirus-mediated diseases, therapeutic interventions that target co-inhibitory PD-1 signaling might have severe consequences for the patients, because PD-1 sends important inhibitory signals to prevent T cells from causing immunopathology (Volk et al., 2021). Taken together, future studies should determine which stimulatory and inhibitory signals are sent out by gammaherpesvirus-infected antigen-presenting cells. At the current stage, how these mechanisms can be optimized to enhance infection control without risking overt immunopathology is unknown.
In chronic viral infection, constant expression of viral antigens often leads to the loss of T cell functions, a phenomenon well known as T cell exhaustion (Kanev and Zehn, 2021). How T cells interact with persistent pathogens like gammaherpesviruses is determined by the amount of T cell exhaustion as a hallmark of chronic infections (Saeidi et al., 2018). Interestingly, in latently gammaherpesvirus-infected patients, the virus-specific T cells remain largely functional (Figure 2) (Cannons et al., 2018). In contrast, in patients with EBV-associated lymphoproliferative disorders, EBV-specific T cells show signs of T cell exhaustion (Nakid-Cordero et al., 2021). Also, in immunodeficient transplant patients, high EBV viral load was associated with T cell exhaustion (Macedo et al., 2011). Future studies are needed to better understand how chronic antigen stimulation can lead to T cell exhaustion in the context of latent or chronically active EBV and MHV68 infections.
Taken together, the antiviral effects mediated by gammaherpesvirus-specific T cells are important resistance factors for the host, but even a fully activated T cells response cannot eradicate chronic gammaherpesvirus infection, even in fully immunocompetent patients. Therefore, a better understanding of how gammaherpesviruses evade the antiviral T cell immune response is needed, especially since gammaherpesviruses have evolved multiple ways how to evade the underlying innate immune responses.
Final remarks
Clear evidence has accumulated over the past decades that gammaherpesviruses are sensed by the immune system, evoke an immune response, and modulate it to avoid their elimination and achieve the establishment of a lifelong chronic infection. Since the innate immune response plays a crucial role in orchestrating and maintaining a functional and efficient adaptive immune response, herpesviruses shape the adaptive response not only directly, but also by their inhibition of the type I IFN and NK cell response. Clearly, more in vivo studies are needed to assess the consequences of viral modulation of the type I IFN and NK cell response on the quality of the adaptive immune response, during acute as well as chronic infection. However, this endeavor is not straightforward considering that viral proteins are often multifunctional, with some of them targeting multiple arms of the immune system or regulating viral replication or gene expression. In addition, although an animal model for EBV exists with the murine MHV68 model, not all findings with MHV68 can be transferred to the human gammaherpesviruses and their human host, raising the importance of developing novel model systems. So far, probably only very few of the major players that determine the magnitude of the T cell response against gammaherpesviral infection have been described in vivo. Further, future studies will have to disentangle the role of the immune system for antiviral defense as opposed to its role for efficient viral spread and latency establishment. In addition, the protective efficiency of CD4+ and CD8+ T cells during gammaherpesvirus infection probably differs greatly with the type of virus-infected target cell, as gammaherpesviruses are known to infect both macrophages, epithelial cells and B cells. Thus, systematic studies are needed in the future to show in a quantitative manner which pathways of the innate immune system are most relevant for controlling gammaherpesvirus infection and how an optimal T cell response can be generated.
Author contributions
All authors contributed to the article and approved the submitted version.
Funding
This work was funded by the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) – Projektnummer 158989968 - SFB 900 (MS, RZ, SH, MMB), the SMART BIOTECS alliance between the Technische Universität Braunschweig and the Leibniz Universität Hannover, an initiative supported by the Ministry of Science and Culture of Lower Saxony, Germany (VR, MMB), and the Helmholtz Association (W2/W3-090) (MMB).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Abel, A. M., Yang, C., Thakar, M. S., Malarkannan, S. (2018). Natural killer cells: Development, maturation, and clinical utilization. Front. Immunol. 9. doi: 10.3389/fimmu.2018.01869
Ablasser, A., Bauernfeind, F., Hartmann, G., Latz, E., Fitzgerald, K. A., Hornung, V. (2009). RIG-i-dependent sensing of poly(dA:dT) through the induction of an RNA polymerase III–transcribed RNA intermediate. Nat. Immunol. 10, 1065–1072. doi: 10.1038/ni.1779
Adler, H., El-Gogo, S., Guggemoos, S., Zimmermann, W., Beauchemin, N., Kammerer, R. (2009). Perturbation of lytic and latent gammaherpesvirus infection in the absence of the inhibitory receptor CEACAM1. PloS One. 4, e6317. doi: 10.1371/journal.pone.0006317
Adler, H., Steer, B., Juskewitz, E., Kammerer, R. (2014). To the Editor murine gammaherpesvirus 68 (MHV-68) escapes from NK-cell-mediated immune surveillance by a CEACAM1-mediated immune evasion mechanism. Eur. J. Immunol. 44, 2521–2522. doi: 10.1002/eji.201444593
Alexopoulou, L., Holt, A. C., Medzhitov, R., Flavell, R. A. (2001). Recognition of double-stranded RNA and activation of NF-κB by toll-like receptor 3. Nature 413, 732–738. doi: 10.1038/35099560
Andreakos, E., Abel, L., Vinh, D. C., Kaja, E., Drolet, B. A., Zhang, Q., et al. (2022). A global effort to dissect the human genetic basis of resistance to SARS-CoV-2 infection. Nat. Immunol. 23, 159–164. doi: 10.1038/s41590-021-01030-z
Andrei, G., Trompet, E., Snoeck, R. (2019). Novel therapeutics for Epstein–Barr virus. Molecules 24, 997. doi: 10.3390/molecules24050997
Ansari, M. A., Dutta, S., Veettil, M. V., Dutta, D., Iqbal, J., Kumar, B., et al. (2015). Herpesvirus genome recognition induced acetylation of nuclear IFI16 is essential for its cytoplasmic translocation, inflammasome and IFN-β responses. PloS Pathog. 11, e1005019. doi: 10.1371/journal.ppat.1005019
Ansari, M. A., Singh, V. V., Dutta, S., Veettil, M. V., Dutta, D., Chikoti, L., et al. (2013). Constitutive interferon-inducible protein 16-inflammasome activation during Epstein-Barr virus latency I, II, and III in b and epithelial cells. J. Virol. 87, 8606–8623. doi: 10.1128/JVI.00805-13
Aricò, E., Monque, D. M., D’Agostino, G., Moschella, F., Venditti, M., Kalinke, U., et al. (2011). MHV-68 producing mIFNα1 is severely attenuated in vivo and effectively protects mice against challenge with wt MHV-68. Vaccine. 29, 3935–3944. doi: 10.1016/j.vaccine.2011.03.092
Ariza, M.-E., Glaser, R., Kaumaya, P. T. P., Jones, C., Williams, M. V. (2009). The EBV-encoded dUTPase activates NF-κB through the TLR2 and MyD88-dependent signaling pathway. J. Immunol. 182, 851–859. doi: 10.4049/jimmunol.182.2.851
Arrand, J. R., Rymo, L. (1982). Characterization of the major Epstein-Barr virus-specific RNA in burkitt lymphoma-derived cells. J. Virol. 41, 376–389. doi: 10.1128/jvi.41.2.376-389.1982
Baglio, S. R., van Eijndhoven, M. A. J., Koppers-Lalic, D., Berenguer, J., Lougheed, S. M., Gibbs, S., et al. (2016). Sensing of latent EBV infection through exosomal transfer of 5′pppRNA. Proc. Natl. Acad. Sci. 113, E587–E596. doi: 10.1073/pnas.1518130113
Barton, E. S., Lutzke, M. L., Rochford, R., Virgin, H. W. (2005). Alpha/Beta interferons regulate murine gammaherpesvirus latent gene expression and reactivation from latency. J. Virol. 79, 14149–14160. doi: 10.1128/JVI.79.22.14149-14160.2005
Barton, E., Mandal, P., Speck, S. H. (2011). Pathogenesis and host control of gammaherpesviruses: Lessons from the mouse. Annu. Rev. Immunol. 29, 351–397. doi: 10.1146/annurev-immunol-072710-081639
Bastard, P., Manry, J., Chen, J., Rosain, J., Seeleuthner, Y., AbuZaitun, O., et al. (2021). Herpes simplex encephalitis in a patient with a distinctive form of inherited IFNAR1 deficiency. J. Clin. Invest. 131, e139980. doi: 10.1172/JCI139980
Bastard, P., Rosen, L. B., Zhang, Q., Michailidis, E., Hoffmann, H.-H., Zhang, Y., et al. (2020). Autoantibodies against type I IFNs in patients with life-threatening COVID-19. Sci. (1979) 370. doi: 10.1126/science.abd4585
Bauer, S., Kirschning, C. J., Häcker, H., Redecke, V., Hausmann, S., Akira, S., et al. (2001). Human TLR9 confers responsiveness to bacterial DNA via species-specific CpG motif recognition. Proc. Natl. Acad. Sci. 98, 9237–9242. doi: 10.1073/pnas.161293498
Bélec, L., Mohamed, A. S., Authier, F.-J., Hallouin, M.-C., Soe, A. M., Cotigny, S., et al. (1999). Human herpesvirus 8 infection in patients with POEMS syndrome–associated multicentric castleman’s disease. Blood 93, 3643–3653. doi: 10.1182/blood.V93.11.3643
Bennett, N. J., May, J. S., Stevenson, P. G. (2005). Gamma-herpesvirus latency requires T cell evasion during episome maintenance. PloS Biol. 3, e120. doi: 10.1371/journal.pbio.0030120
Ben-Sasson, S. Z., Wang, K., Cohen, J., Paul, W. E. (2013). IL-1b strikingly enhances antigen-driven CD4 and CD8 T-cell responses. Cold Spring Harb. Symp Quant Biol. 78, 117–124. doi: 10.1101/sqb.2013.78.021246
Bhat, R., Watzl, C. (2007). Serial killing of tumor cells by human natural killer cells–enhancement by therapeutic antibodies. PloS One 2, e326. doi: 10.1371/journal.pone.0000326
Bjornevik, K., Cortese, M., Healy, B. C., Kuhle, J., Mina, M. J., Leng, Y., et al. (2022). Longitudinal analysis reveals high prevalence of Epstein-Barr virus associated with multiple sclerosis. Sci. (1979) 375, 296–301. doi: 10.1126/science.abj8222
Bottino, C., Castriconi, R., Moretta, L., Moretta, A. (2005). Cellular ligands of activating NK receptors. Trends Immunol. 26, 221–226. doi: 10.1016/j.it.2005.02.007
Botto, S., Abraham, J., Mizuno, N., Pryke, K., Gall, B., Landais, I., et al. (2019). Human cytomegalovirus immediate early 86-kDa protein blocks transcription and induces degradation of the immature interleukin-1β protein during virion-mediated activation of the AIM2 inflammasome. mBio 10, e02510-18. doi: 10.1128/mBio.02510-18
Brulois, K., Toth, Z., Wong, L.-Y., Feng, P., Gao, S.-J., Ensser, A., et al. (2014). Kaposi’s sarcoma-associated herpesvirus K3 and K5 ubiquitin E3 ligases have stage-specific immune evasion roles during lytic replication. J. Virol. 88, 9335–9349. doi: 10.1128/JVI.00873-14
Burassakarn, A., Srisathaporn, S., Pientong, C., Wongjampa, W., Vatanasapt, P., Patarapadungkit, N., et al. (2021). Exosomes-carrying Epstein-Barr virus-encoded small RNA-1 induces indoleamine 2, 3-dioxygenase expression in tumor-infiltrating macrophages of oral squamous-cell carcinomas and suppresses T-cell activity by activating RIG-I/IL-6/TNF-α pathway. Oral. Oncol. 117, 105279. doi: 10.1016/j.oraloncology.2021.105279
Bussey, K. A., Lau, U., Schumann, S., Gallo, A., Osbelt, L., Stempel, M., et al. (2018). The interferon-stimulated gene product oligoadenylate synthetase-like protein enhances replication of kaposi’s sarcoma-associated herpesvirus (KSHV) and interacts with the KSHV ORF20 protein. PloS Pathog. 14, e1006937. doi: 10.1371/journal.ppat.1006937
Bussey, K. A., Murthy, S., Reimer, E., Chan, B., Hatesuer, B., Schughart, K., et al. (2019). Endosomal toll-like receptors 7 and 9 cooperate in detection of murine gammaherpesvirus 68 infection. J. Virol. 93, e01173-18. doi: 10.1128/JVI.01173-18
Cannons, J. L., Preite, S., Kapnick, S. M., Uzel, G., Schwartzberg, P. L. (2018). Genetic defects in phosphoinositide 3-kinase δ influence CD8+ T cell survival, differentiation, and function. Front. Immunol. 9. doi: 10.3389/fimmu.2018.01758
Carbone, A., Borok, M., Damania, B., Gloghini, A., Polizzotto, M. N., Jayanthan, R. K., et al. (2021). Castleman disease. Nat. Rev. Dis. Primers 7, 84. doi: 10.1038/s41572-021-00317-7
Carrega, P., Ferlazzo, G. (2012). Natural killer cell distribution and trafficking in human tissues. Front. Immunol. 3. doi: 10.3389/fimmu.2012.00347
Carter-Timofte, M. E., Jørgensen, S. E., Freytag, M. R., Thomsen, M. M., Brinck Andersen, N.-S., Al-Mousawi, A., et al. (2020). Deciphering the role of host genetics in susceptibility to severe COVID-19. Front. Immunol. 11. doi: 10.3389/fimmu.2020.01606
Carty, M., Guy, C., Bowie, A. G. (2021). Detection of viral infections by innate immunity. Biochem. Pharmacol. 183, 114316. doi: 10.1016/j.bcp.2020.114316
Casrouge, A., Zhang, S.-Y., Eidenschenk, C., Jouanguy, E., Puel, A., Yang, K., et al. (2006). Herpes simplex virus encephalitis in human UNC-93B deficiency. Sci. (1979) 314, 308–312. doi: 10.1126/science.1128346
Cesarman, E., Damania, B., Krown, S. E., Martin, J., Bower, M., Whitby, D. (2019). Kaposi sarcoma. Nat. Rev. Dis. Primers 5, 9. doi: 10.1038/s41572-019-0060-9
Chang, Y., Cesarman, E., Pessin, M. S., Lee, F., Culpepper, J., Knowles, D. M., et al. (1994). Identification of herpesvirus-like DNA sequences in AIDS-sssociated kaposi’s sarcoma. Sci. (1979) 266, 1865–1869. doi: 10.1126/science.7997879
Chiang, J. J., Sparrer, K. M. J., van Gent, M., Lässig, C., Huang, T., Osterrieder, N., et al. (2018). Viral unmasking of cellular 5S rRNA pseudogene transcripts induces RIG-i-mediated immunity. Nat. Immunol. 19, 53–62. doi: 10.1038/s41590-017-0005-y
Chijioke, O., Landtwing, V., Münz, C. (2016). NK cell influence on the outcome of primary Epstein–Barr virus infection. Front. Immunol. 7. doi: 10.3389/fimmu.2016.00323
Chiu, Y.-H., MacMillan, J. B., Chen, Z. J. (2009). RNA Polymerase III detects cytosolic DNA and induces type I interferons through the RIG-I pathway. Cell 138, 576–591. doi: 10.1016/j.cell.2009.06.015
Chopp, L., Redmond, C., O’Shea, J. J., Schwartz, D. M. (2023). From thymus to tissues and tumors: A review of T-cell biology. J. Allergy Clin. Immunol. 151, 81–97. doi: 10.1016/j.jaci.2022.10.011
Crawford, D. H., Rickinson, A., Johannessen, I. (2014). Cancer virus: the story of Epstein-Barr virus (Oxford, United Kingdom: Oxford University Press).
Damania, B., Kenney, S. C., Raab-Traub, N. (2022). Epstein-Barr Virus: Biology and clinical disease. Cell 185, 3652–3670. doi: 10.1016/j.cell.2022.08.026
Dauer, M., Pohl, K., Obermaier, B., Meskendahl, T., Robe, J., Schnurr, M., et al. (2003). Interferon-alpha disables dendritic cell precursors: Dendritic cells derived from interferon-alpha-treated monocytes are defective in maturation and T-cell stimulation. Immunology 110, 38–47. doi: 10.1046/j.1365-2567.2003.01702.x
Davison, A. J., Eberle, R., Ehlers, B., Hayward, G. S., McGeoch, D. J., Minson, A. C., et al. (2009). The order herpesvirales. Arch. Virol. 154, 171–177. doi: 10.1007/s00705-008-0278-4
Deng, Y., Chatterjee, B., Zens, K., Zdimerova, H., Müller, A., Schuhmachers, P., et al. (2021). CD27 is required for protective lytic EBV antigen–specific CD8+ T-cell expansion. Blood 137, 3225–3236. doi: 10.1182/blood.2020009482
de Oliveira Mann, C. C., Hornung, V. (2021). Molecular mechanisms of nonself nucleic acid recognition by the innate immune system. Eur. J. Immunol. 51, 1897–1910. doi: 10.1002/eji.202049116
Dinarello, C. A. (1996) Biologic basis for interleukin-l in disease. Available at: http://ashpublications.org/blood/article-pdf/87/6/2095/620593/2095.pdf.
Domanski, P., Fish, E., Nadeau, O. W., Witte, M., Platanias, L. C., Yan, H., et al. (1997). A region of the β subunit of the interferon α receptor different from box 1 interacts with Jak1 and is sufficient to activate the jak-stat pathway and induce an antiviral state. J. Biol. Chem. 272, 26388–26393. doi: 10.1074/jbc.272.42.26388
Du, M.-Q., Diss, T. C., Liu, H., Ye, H., Hamoudi, R. A., Cabeçadas, J., et al. (2002). KSHV- and EBV-associated germinotropic lymphoproliferative disorder. Blood 100, 3415–3418. doi: 10.1182/blood-2002-02-0487
Duan, T., Du, Y., Xing, C., Wang, H. Y., Wang, R.-F. (2022). Toll-like receptor signaling and its role in cell-mediated immunity. Front. Immunol. 13. doi: 10.3389/fimmu.2022.812774
Duan, Y., Li, Z., Cheng, S., Chen, Y., Zhang, L., He, J., et al. (2015). Nasopharyngeal carcinoma progression is mediated by EBER-triggered inflammation via the RIG-I pathway. Cancer Lett. 361, 67–74. doi: 10.1016/j.canlet.2015.02.037
Dubich, T., Lieske, A., Santag, S., Beauclair, G., Rückert, J., Herrmann, J., et al. (2019). An endothelial cell line infected by kaposi’s sarcoma–associated herpes virus (KSHV) allows the investigation of kaposi’s sarcoma and the validation of novel viral inhibitors in vitro and in vivo. J. Mol. Med. 97, 311–324. doi: 10.1007/s00109-018-01733-1
Dupaul-Chicoine, J., Arabzadeh, A., Dagenais, M., Douglas, T., Champagne, C., Morizot, A., et al. (2015). The Nlrp3 inflammasome suppresses colorectal cancer metastatic growth in the liver by promoting natural killer cell tumoricidal activity. Immunity 43, 751–763. doi: 10.1016/j.immuni.2015.08.013
Dupin, N., Diss, T. L., Kellam, P., Tulliez, M., Du, M.-Q., Sicard, D., et al. (2000). HHV-8 is associated with a plasmablastic variant of castleman disease that is linked to HHV-8–positive plasmablastic lymphoma. Blood 95, 1406–1412. doi: 10.1182/blood.V95.4.1406.004k26_1406_1412
Dutia, B. M., Allen, D. J., Dyson, H., Nash, A. A. (1999). Type I interferons and IRF-1 play a critical role in the control of a gammaherpesvirus infection. Virology 261, 173–179. doi: 10.1006/viro.1999.9834
Dutta, D., Dutta, S., Veettil, M. V., Roy, A., Ansari, M. A., Iqbal, J., et al. (2015). BRCA1 regulates IFI16 mediated nuclear innate sensing of herpes viral DNA and subsequent induction of the innate inflammasome and interferon-β responses. PloS Pathog. 11, e1005030. doi: 10.1371/journal.ppat.1005030
El-Zayat, S. R., Sibaii, H., Mannaa, F. A. (2019). Toll-like receptors activation, signaling, and targeting: an overview. Bull. Natl. Res. Cent 43, 187. doi: 10.1186/s42269-019-0227-2
Evavold, C. L., Kagan, J. C. (2019). Inflammasomes: Threat-assessment organelles of the innate immune system. Immunity 51, 609–624. doi: 10.1016/j.immuni.2019.08.005
Fauriat, C., Long, E. O., Ljunggren, H.-G., Bryceson, Y. T. (2010). Regulation of human NK-cell cytokine and chemokine production by target cell recognition. Blood 115, 2167–2176. doi: 10.1182/blood-2009-08-238469
Fiola, S., Gosselin, D., Takada, K., Gosselin, J. (2010). TLR9 contributes to the recognition of EBV by primary monocytes and plasmacytoid dendritic cells. J. Immunol. 185, 3620–3631. doi: 10.4049/jimmunol.0903736
Flaño, E., Woodland, D. L., Blackman, M. A. (2002). A mouse model for infectious mononucleosis. Immunol. Res. 25, 201–218. doi: 10.1385/IR:25:3:201
Foss, H., Herbst, H., Hummel, M., Araujo, I., Latza, U., Rancso, C., et al. (1994). Patterns of cytokine gene expression in infectious mononucleosis. Blood 83, 707–712. doi: 10.1182/blood.V83.3.707.707
Freeman, M. L., Lanzer, K. G., Cookenham, T., Peters, B., Sidney, J., Wu, T.-T., et al. (2010). Two kinetic patterns of epitope-specific CD8 T-cell responses following murine gammaherpesvirus 68 infection. J. Virol. 84, 2881–2892. doi: 10.1128/JVI.02229-09
Fujiwara, S. (2018). Animal models of human gammaherpesvirus infections. Adv. Exp. Med. Biol. 1045, 413–436. doi: 10.1007/978-981-10-7230-7_19
Fujiwara, S., Nakamura, H. (2020). Animal models for gammaherpesvirus infections: Recent development in the analysis of virus-induced pathogenesis. Pathogens 9, 116. doi: 10.3390/pathogens9020116
Garlanda, C., Dinarello, C. A., Mantovani, A. (2013). The interleukin-1 family: Back to the future. Immunity 39, 1003–1018. doi: 10.1016/j.immuni.2013.11.010
Gaudreault, E., Fiola, S., Olivier, M., Gosselin, J. (2007). Epstein-Barr Virus induces MCP-1 secretion by human monocytes via TLR2. J. Virol. 81, 8016–8024. doi: 10.1128/JVI.00403-07
Glickman, J. N., Howe, J. G., Steitz, J. A. (1988). Structural analyses of EBER1 and EBER2 ribonucleoprotein particles present in Epstein-Barr virus-infected cells. J. Virol. 62, 902–911. doi: 10.1128/jvi.62.3.902-911.1988
Gonzalez-Farre, B., Martinez, D., Lopez-Guerra, M., Xipell, M., Monclus, E., Rovira, J., et al. (2017). HHV8-related lymphoid proliferations: A broad spectrum of lesions from reactive lymphoid hyperplasia to overt lymphoma. Modern Pathol. 30, 745–760. doi: 10.1038/modpathol.2016.233
Gothe, F., Hatton, C. F., Truong, L., Klimova, Z., Kanderova, V., Fejtkova, M., et al. (2022). A novel case of homozygous interferon Alpha/Beta receptor alpha chain (IFNAR1) deficiency with hemophagocytic lymphohistiocytosis. Clin. Infect. Dis. 74, 136–139. doi: 10.1093/cid/ciaa1790
Gram, A. M., Sun, C., Landman, S. L., Oosenbrug, T., Koppejan, H. J., Kwakkenbos, M. J., et al. (2017). Human b cells fail to secrete type I interferons upon cytoplasmic DNA exposure. Mol. Immunol. 91, 225–237. doi: 10.1016/j.molimm.2017.08.025
Gredmark-Russ, S., Cheung, E. J., Isaacson, M. K., Ploegh, H. L., Grotenbreg, G. M. (2008). The CD8 T-cell response against murine gammaherpesvirus 68 is directed toward a broad repertoire of epitopes from both early and late antigens. J. Virol. 82, 12205–12212. doi: 10.1128/JVI.01463-08
Guggemoos, S., Hangel, D., Hamm, S., Heit, A., Bauer, S., Adler, H. (2008). TLR9 contributes to antiviral immunity during gammaherpesvirus infection. J. Immunol. 180, 438–443. doi: 10.4049/jimmunol.180.1.438
Gujer, C., Murer, A., Müller, A., Vanoaica, D., Sutter, K., Jacque, E., et al. (2019). Plasmacytoid dendritic cells respond to Epstein-Barr virus infection with a distinct type I interferon subtype profile. Blood Adv. 3, 1129–1144. doi: 10.1182/bloodadvances.2018025536
Guy, C., Bowie, A. G. (2022). Recent insights into innate immune nucleic acid sensing during viral infection. Curr. Opin. Immunol. 78, 102250. doi: 10.1016/j.coi.2022.102250
Hammer, Q., Rückert, T., Romagnani, C. (2018). Natural killer cell specificity for viral infections. Nat. Immunol. 19, 800–808. doi: 10.1038/s41590-018-0163-6
Heil, F., Hemmi, H., Hochrein, H., Ampenberger, F., Kirschning, C., Akira, S., et al. (2004). Species-specific recognition of single-stranded RNA via toll-like receptor 7 and 8. Sci. (1979) 303, 1526–1529. doi: 10.1126/science.1093620
Hernandez, N., Bucciol, G., Moens, L., Le Pen, J., Shahrooei, M., Goudouris, E., et al. (2019). Inherited IFNAR1 deficiency in otherwise healthy patients with adverse reaction to measles and yellow fever live vaccines. J. Exp. Med. 216, 2057–2070. doi: 10.1084/jem.20182295
Hooykaas, M. J. G., van Gent, M., Soppe, J. A., Kruse, E., Boer, I. G. J., van Leenen, D., et al. (2017). EBV MicroRNA BART16 suppresses type I IFN signaling. J. Immunol. 198, 4062–4073. doi: 10.4049/jimmunol.1501605
Hornung, V., Ablasser, A., Charrel-Dennis, M., Bauernfeind, F., Horvath, G., Caffrey, D., et al. (2009). AIM2 recognizes cytosolic dsDNA and forms a caspase-1-activating inflammasome with ASC. Nature 458, 514–518. doi: 10.1038/nature07725
Hornung, V., Ellegast, J., Kim, S., Brzózka, K., Jung, A., Kato, H., et al. (2006). 5’-triphosphate RNA is the ligand for RIG-I. Sci. (1979) 314, 994–997. doi: 10.1126/science.1132505
Howe, M. K., Dowdell, K., Kuehn, H. S., Li, Q., Hart, G. T., Garabedian, D., et al. (2020). Patients with natural killer (NK) cell chronic active Epstein-Barr virus have immature NK cells and hyperactivation of PI3K/Akt/mTOR and STAT1 pathways. J. Infect. Dis. 222, 1170–1179. doi: 10.1093/infdis/jiaa232
Hu, Z., Blackman, M. A., Kaye, K. M., Usherwood, E. J. (2015). Functional heterogeneity in the CD4+ T cell response to murine γ-herpesvirus 68. J. Immunol. 194, 2746–2756. doi: 10.4049/jimmunol.1401928
Hu, B., Jin, C., Li, H.-B., Tong, J., Ouyang, X., Cetinbas, N. M., et al. (2016). The DNA-sensing AIM2 inflammasome controls radiation-induced cell death and tissue injury. Sci. (1979) 354, 765–768. doi: 10.1126/science.aaf7532
Hwang, S., Kim, K. S., Flano, E., Wu, T.-T., Tong, L. M., Park, A. N., et al. (2009). Conserved herpesviral kinase promotes viral persistence by inhibiting the IRF-3-Mediated type I interferon response. Cell Host Microbe 5, 166–178. doi: 10.1016/j.chom.2008.12.013
Inn, K.-S., Lee, S.-H., Rathbun, J. Y., Wong, L.-Y., Toth, Z., Machida, K., et al. (2011). Inhibition of RIG-I-Mediated signaling by kaposi’s sarcoma-associated herpesvirus-encoded deubiquitinase ORF64. J. Virol. 85, 10899–10904. doi: 10.1128/JVI.00690-11
Ishikawa, H., Barber, G. N. (2011). The STING pathway and regulation of innate immune signaling in response to DNA pathogens. Cell. Mol. Life Sci. 68, 1157–1165. doi: 10.1007/s00018-010-0605-2
Ivashkiv, L. B., Donlin, L. T. (2014). Regulation of type I interferon responses. Nat. Rev. Immunol. 14, 36–49. doi: 10.1038/nri3581
Iwakiri, D., Zhou, L., Samanta, M., Matsumoto, M., Ebihara, T., Seya, T., et al. (2009). Epstein-Barr Virus (EBV)–encoded small RNA is released from EBV-infected cells and activates signaling from toll-like receptor 3. J. Exp. Med. 206, 2091–2099. doi: 10.1084/jem.20081761
Jangra, S., Bharti, A., Lui, W.-Y., Chaudhary, V., Botelho, M. G., Yuen, K.-S., et al. (2021). Suppression of JAK-STAT signaling by Epstein-Barr virus tegument protein BGLF2 through recruitment of SHP1 phosphatase and promotion of STAT2 degradation. J. Virol. 95, e0102721. doi: 10.1128/JVI.01027-21
Jennings, R. N., Grayson, J. M., Barton, E. S. (2014). Type I interferon signaling enhances CD8+ T cell effector function and differentiation during murine gammaherpesvirus 68 infection. J. Virol. 88, 14040–14049. doi: 10.1128/JVI.02360-14
Jondle, C. N., Johnson, K. E., Mboko, W. P., Tarakanova, V. L. (2021). T Cell-intrinsic interferon regulatory factor 1 expression suppresses differentiation of CD4 + T cell populations that support chronic gammaherpesvirus infection. J. Virol. 95, e0072621. doi: 10.1128/JVI.00726-21
Kanev, K., Zehn, D. (2021). Origin and fine-tuning of effector CD8 T cell subpopulations in chronic infection. Curr. Opin. Virol. 46, 27–35. doi: 10.1016/j.coviro.2020.10.003
Kang, H.-R., Cheong, W.-C., Park, J.-E., Ryu, S., Cho, H.-J., Youn, H., et al. (2014). Murine gammaherpesvirus 68 encoding open reading frame 11 targets TANK binding kinase 1 to negatively regulate the host type I interferon response. J. Virol. 88, 6832–6846. doi: 10.1128/JVI.03460-13
Karaba, A. H., Figueroa, A., Massaccesi, G., Botto, S., DeFilippis, V. R., Cox, A. L. (2020). Herpes simplex virus type 1 inflammasome activation in proinflammatory human macrophages is dependent on NLRP3, ASC, and caspase-1. PloS One 15, e0229570. doi: 10.1371/journal.pone.0229570
Karijolich, J., Abernathy, E., Glaunsinger, B. A. (2015). Infection-induced retrotransposon-derived noncoding RNAs enhance herpesviral gene expression via the NF-κB pathway. PloS Pathog. 11, e1005260. doi: 10.1371/journal.ppat.1005260
Kawasaki, T., Kawai, T. (2014). Toll-like receptor signaling pathways. Front. Immunol. 5. doi: 10.3389/fimmu.2014.00461
Kean, L. S., Turka, L. A., Blazar, B. R. (2017). Advances in targeting co-inhibitory and co-stimulatory pathways in transplantation settings: the yin to the yang of cancer immunotherapy. Immunol. Rev. 276, 192–212. doi: 10.1111/imr.12523
Keppler, S. J., Rosenits, K., Koegl, T., Vucikuja, S., Aichele, P. (2012). Signal 3 cytokines as modulators of primary immune responses during infections: The interplay of type I IFN and IL-12 in CD8 T cell responses. PloS One 7, e40865. doi: 10.1371/journal.pone.0040865
Kerur, N., Veettil, M. V., Sharma-Walia, N., Bottero, V., Sadagopan, S., Otageri, P., et al. (2011). IFI16 acts as a nuclear pathogen sensor to induce the inflammasome in response to kaposi sarcoma-associated herpesvirus infection. Cell Host Microbe 9, 363–375. doi: 10.1016/j.chom.2011.04.008
Khosravi-Far, R., Esposti, M. D. (2004). Death receptor signals to mitochondria. Cancer Biol. Ther. 3, 1051–1057. doi: 10.4161/cbt.3.11.1173
Kiessling, R., Klein, E., Pross, H., Wigzell, H. (1975). “Natural” killer cells in the mouse. II. cytotoxic cells with specificity for mouse moloney leukemia cells. characteristics of the killer cell. Eur. J. Immunol. 5, 117–121. doi: 10.1002/eji.1830050209
Kimura, H., Cohen, J. I. (2017). Chronic active Epstein–Barr virus disease. Front. Immunol. 8. doi: 10.3389/fimmu.2017.01867
Krzewski, K., Strominger, J. L. (2008). The killer’s kiss: the many functions of NK cell immunological synapses. Curr. Opin. Cell Biol. 20, 597–605. doi: 10.1016/j.ceb.2008.05.006
Lammoglia Cobo, M. F., Ritter, J., Gary, R., Seitz, V., Mautner, J., Aigner, M., et al. (2022). Reconstitution of EBV-directed T cell immunity by adoptive transfer of peptide-stimulated T cells in a patient after allogeneic stem cell transplantation for AITL. PloS Pathog. 18, e1010206. doi: 10.1371/journal.ppat.1010206
Lawler, C., Stevenson, P. G. (2020). A CD4 + T cell-NK cell axis of gammaherpesvirus control. J. Virol. 94, e01545-19. doi: 10.1128/jvi.01545-19
Lawler, C., Tan, C. S. E., Simas, J. P., Stevenson, P. G. (2016). Type I interferons and NK cells restrict gammaherpesvirus lymph node infection. J. Virol. 90, 9046–9057. doi: 10.1128/jvi.01108-16
Le Bon, A., Tough, D. F. (2002). Links between innate and adaptive immunity via type I interferon. Curr. Opin. Immunol. 14, 432–436. doi: 10.1016/S0952-7915(02)00354-0
Lee, J., Zhang, T., Hwang, I., Kim, A., Nitschke, L., Kim, M., et al. (2015). Epigenetic modification and antibody-dependent expansion of memory-like NK cells in human cytomegalovirus-infected individuals. Immunity 42, 431–442. doi: 10.1016/j.immuni.2015.02.013
Lenschow, D. J., Lai, C., Frias-Staheli, N., Giannakopoulos, N. V., Lutz, A., Wolff, T., et al. (2007). IFN-stimulated gene 15 functions as a critical antiviral molecule against influenza, herpes, and sindbis viruses. Proc. Natl. Acad. Sci. 104, 1371 LP–1376. doi: 10.1073/pnas.0607038104
Li, Z., Duan, Y., Cheng, S., Chen, Y., Hu, Y., Zhang, L., et al. (2015). EBV-encoded RNA via TLR3 induces inflammation in nasopharyngeal carcinoma. Oncotarget 6, 24291–24303. doi: 10.18632/oncotarget.4552
Li, Z., Tsai, M.-H., Shumilov, A., Baccianti, F., Tsao, S. W., Poirey, R., et al. (2019). Epstein–Barr Virus ncRNA from a nasopharyngeal carcinoma induces an inflammatory response that promotes virus production. Nat. Microbiol. 4, 2475–2486. doi: 10.1038/s41564-019-0546-y
Liang, X., Shin, Y. C., Means, R. E., Jung, J. U. (2004). Inhibition of interferon-mediated antiviral activity by murine gammaherpesvirus 68 latency-associated M2 protein. J. Virol. 78, 12416–12427. doi: 10.1128/JVI.78.22.12416-12427.2004
Lim, W. H., Kireta, S., Russ, G. R., Coates, P. T. H. (2007). Human plasmacytoid dendritic cells regulate immune responses to Epstein-Barr virus (EBV) infection and delay EBV-related mortality in humanized NOD-SCID mice. Blood 109, 1043–1050. doi: 10.1182/blood-2005-12-024802
Liu, S., Cai, X., Wu, J., Cong, Q., Chen, X., Li, T., et al. (2015). Phosphorylation of innate immune adaptor proteins MAVS, STING, and TRIF induces IRF3 activation. Sci. (1979) 347, aaa2630. doi: 10.1126/science.aaa2630
Liu, X., Sadaoka, T., Krogmann, T., Cohen, J. I. (2020). Epstein-Barr Virus (EBV) tegument protein BGLF2 suppresses type I interferon signaling to promote EBV reactivation. J. Virol. 94, e00258-20. doi: 10.1128/JVI.00258-20
Loh, J., Thomas, D. A., Revell, P. A., Ley, T. J., Virgin, H. W. (2004). Granzymes and caspase 3 play important roles in control of gammaherpesvirus latency. J. Virol. 78, 12519–12528. doi: 10.1128/JVI.78.22.12519-12528.2004
Luckhardt, T. R., Coomes, S. M., Trujillo, G., Stoolman, J. S., Vannella, K. M., Bhan, U., et al. (2011). TLR9-induced interferon β is associated with protection from gammaherpesvirus-induced exacerbation of lung fibrosis. Fibrogenesis Tissue Repair. 4, 18. doi: 10.1186/1755-1536-4-18
Lueder, Y., Heller, K., Ritter, C., Keyser, K. A., Wagner, K., Liu, X., et al. (2018). Control of primary mouse cytomegalovirus infection in lung nodular inflammatory foci by cooperation of interferon-gamma expressing CD4 and CD8 T cells. PloS Pathog. 14, e1007252. doi: 10.1371/journal.ppat.1007252
Luppi, M., Barozzi, P., Schulz, T. F., Setti, G., Staskus, K., Trovato, R., et al. (2000). Bone marrow failure associated with human herpesvirus 8 infection after transplantation. New Engl. J. Med. 343, 1378–1385. doi: 10.1056/NEJM200011093431905
Macedo, C., Webber, S. A., Donnenberg, A. D., Popescu, I., Hua, Y., Green, M., et al. (2011). EBV-specific CD8+ T cells from asymptomatic pediatric thoracic transplant patients carrying chronic high EBV loads display contrasting features: Activated phenotype and exhausted function. J. Immunol. 186, 5854–5862. doi: 10.4049/jimmunol.1001024
Makowska, A., Franzen, S., Braunschweig, T., Denecke, B., Shen, L., Baloche, V., et al. (2019). Interferon beta increases NK cell cytotoxicity against tumor cells in patients with nasopharyngeal carcinoma via tumor necrosis factor apoptosis-inducing ligand. Cancer Immunol. Immunother. 68, 1317–1329. doi: 10.1007/s00262-019-02368-y
Manners, O., Murphy, J. C., Coleman, A., Hughes, D. J., Whitehouse, A. (2018). Contribution of the KSHV and EBV lytic cycles to tumourigenesis. Curr. Opin. Virol. 32, 60–70. doi: 10.1016/j.coviro.2018.08.014
Manry, J., Bastard, P., Gervais, A., Le Voyer, T., Rosain, J., Philippot, Q., et al. (2022). The risk of COVID-19 death is much greater and age dependent with type I IFN autoantibodies. Proc. Natl. Acad. Sci. 119, e2200413119. doi: 10.1073/pnas.2200413119
Mariggiò, G., Koch, S., Schulz, T. F. (2017). Kaposi sarcoma herpesvirus pathogenesis. Philos. Trans. R. Soc. B: Biol. Sci. 372, 20160275. doi: 10.1098/rstb.2016.0275
Márquez, A. C., Croft, C., Shanina, I., Horwitz, M. S. (2022). Influence of type I interferons in gammaherpesvirus-68 and its influence on EAE enhancement. Front. Immunol. 13. doi: 10.3389/fimmu.2022.858583
Marshall, H. D., Urban, S. L., Welsh, R. M. (2011). Virus-induced transient immune suppression and the inhibition of T cell proliferation by type I interferon. J. Virol. 85, 5929–5939. doi: 10.1128/JVI.02516-10
Masy, E., Adriaenssens, E., Montpellier, C., Crépieux, P., Mougel, A., Quatannens, B., et al. (2002). Human monocytic cell lines transformed In vitro by Epstein-Barr virus display a type II latency and LMP-1-Dependent proliferation. J. Virol. 76, 6460–6472. doi: 10.1128/JVI.76.13.6460-6472.2002
Mboko, W. P., Rekow, M. M., Ledwith, M. P., Lange, P. T., Schmitz, K. E., Anderson, S., et al. (2017). Interferon regulatory factor 1 and type I interferon cooperate to control acute gammaherpesvirus infection. J. Virol. 91, e01444-16. doi: 10.1128/JVI.01444-16
McLane, L. M., Abdel-Hakeem, M. S., Wherry, E. J. (2019). CD8 T cell exhaustion during chronic viral infection and cancer. Annu. Rev. Immunol. 37, 457–495. doi: 10.1146/annurev-immunol-041015-055318
McNab, F., Mayer-Barber, K., Sher, A., Wack, A., O’Garra, A. (2015). Type I interferons in infectious disease. Nat. Rev. Immunol. 15, 87–103. doi: 10.1038/nri3787
Meyts, I., Casanova, J. (2021). Viral infections in humans and mice with genetic deficiencies of the type I IFN response pathway. Eur. J. Immunol. 51, 1039–1061. doi: 10.1002/eji.202048793
Michaud, F., Coulombe, F., Gaudreault, É, Kriz, J., Gosselin, J. (2010a). Involvement of TLR2 in recognition of acute gammaherpesvirus-68 infection. PloS One 5, e13742. doi: 10.1371/journal.pone.0013742
Michaud, F., Coulombe, F., Gaudreault, E., Paquet-Bouchard, C., Rola-Pleszczynski, M., Gosselin, J. (2010b). Epstein-Barr Virus interferes with the amplification of IFNα secretion by activating suppressor of cytokine signaling 3 in primary human monocytes. PloS One 5, e11908. doi: 10.1371/journal.pone.0011908
Mogensen, T. H. (2022). Genetic susceptibility to viral disease in humans. Clin. Microbiol. Infection 28, 1411–1416. doi: 10.1016/j.cmi.2022.02.023
Montoya, M., Schiavoni, G., Mattei, F., Gresser, I., Belardelli, F., Borrow, P., et al. (2002). Type I interferons produced by dendritic cells promote their phenotypic and functional activation. Blood 99, 3263–3271. doi: 10.1182/blood.V99.9.3263
Münz, C. (2021). Natural killer cell responses during human γ-herpesvirus infections. Vaccines (Basel) 9, 655. doi: 10.3390/vaccines9060655
Nabekura, T., Kanaya, M., Shibuya, A., Fu, G., Gascoigne, N. R. J., Lanier, L. L. (2014). Costimulatory molecule DNAM-1 is essential for optimal differentiation of memory natural killer cells during mouse cytomegalovirus infection. Immunity 40, 225–234. doi: 10.1016/j.immuni.2013.12.011
Nador, R., Cesarman, E., Chadburn, A., Dawson, D., Ansari, M., Sald, J., et al. (1996). Primary effusion lymphoma: a distinct clinicopathologic entity associated with the kaposi’s sarcoma-associated herpes virus. Blood 88, 645–656. doi: 10.1182/blood.V88.2.645.bloodjournal882645
Nakanishi, K., Yoshimoto, T., Tsutsui, H., Okamura, H. (2001). Interleukin-18 regulates both Th1 and Th2 responses. Annu. Rev. Immunol. 19, 423–474. doi: 10.1146/annurev.immunol.19.1.423
Nakid-Cordero, C., Choquet, S., Gauthier, N., Balegroune, N., Tarantino, N., Morel, V., et al. (2021). Distinct immunopathological mechanisms of EBV-positive and EBV-negative posttransplant lymphoproliferative disorders. Am. J. Transplant. 21, 2846–2863. doi: 10.1111/ajt.16547
Nambu, A., Nakae, S., Iwakura, Y. (2006). IL-1β, but not IL-1α, is required for antigen-specific T cell activation and the induction of local inflammation in the delayed-type hypersensitivity responses. Int. Immunol. 18, 701–712. doi: 10.1093/intimm/dxl007
Nijland, M. L., Kersten, M. J., Pals, S. T., Bemelman, F. J., ten Berge, I. J. M. (2016). Epstein-Barr Virus–positive posttransplant lymphoproliferative disease after solid organ transplantation. Transplant. Direct 2, e48. doi: 10.1097/TXD.0000000000000557
Novatt, H., Theisen, T. C., Massie, T., Massie, T., Simonyan, V., Voskanian-Kordi, A., et al. (2016). Distinct patterns of expression of transcription factors in response to interferonβ and Interferonλ1. J. Interferon Cytokine Res. 36, 589–598. doi: 10.1089/jir.2016.0031
Oldham, R. K. (1983). Natural killer cells: Artifact to reality. Cancer Metastasis Rev. 2, 323–336. doi: 10.1007/BF00048565
Panda, D., Gjinaj, E., Bachu, M., Squire, E., Novatt, H., Ozato, K., et al. (2019). IRF1 maintains optimal constitutive expression of antiviral genes and regulates the early antiviral response. Front. Immunol. 10. doi: 10.3389/fimmu.2019.01019
Panetti, C., Kao, K., Joller, N. (2022). Dampening antiviral immunity can protect the host. FEBS J. 289, 634–646. doi: 10.1111/febs.15756
Pánisová, E., Lünemann, A., Bürgler, S., Kotur, M., Lazarovici, J., Danu, A., et al. (2022). Reduced frequency of cytotoxic CD56(dim) CD16(+) NK cells leads to impaired antibody-dependent degranulation in EBV-positive classical Hodgkin lymphoma. Cancer Immunol. Immunother. 71, 13–24. doi: 10.1007/s00262-021-02956-x
Parlato, S., Santini, S. M., Lapenta, C., Di Pucchio, T., Logozzi, M., Spada, M., et al. (2001). Expression of CCR-7, MIP-3β, and Th-1 chemokines in type I IFN-induced monocyte-derived dendritic cells: importance for the rapid acquisition of potent migratory and functional activities. Blood 98, 3022–3029. doi: 10.1182/blood.V98.10.3022
Pathare, G. R., Decout, A., Glück, S., Cavadini, S., Makasheva, K., Hovius, R., et al. (2020). Structural mechanism of cGAS inhibition by the nucleosome. Nature 587, 668–672. doi: 10.1038/s41586-020-2750-6
Peisley, A., Lin, C., Wu, B., Orme-Johnson, M., Liu, M., Walz, T., et al. (2011). Cooperative assembly and dynamic disassembly of MDA5 filaments for viral dsRNA recognition. Proc. Natl. Acad. Sci. 108, 21010–21015. doi: 10.1073/pnas.1113651108
Pisano, G., Roy, A., Ahmed Ansari, M., Kumar, B., Chikoti, L., Chandran, B. (2017). Interferon-γ-inducible protein 16 (IFI16) is required for the maintenance of Epstein-Barr virus latency. Virol. J. 14, 221. doi: 10.1186/s12985-017-0891-5
Quan, T. E., Roman, R. M., Rudenga, B. J., Holers, V. M., Craft, J. E. (2010). Epstein-Barr Virus promotes interferon-α production by plasmacytoid dendritic cells. Arthritis Rheum 62, 1693–1701. doi: 10.1002/art.27408
Reese, T. A. (2016). Coinfections: Another variable in the herpesvirus latency-reactivation dynamic. J. Virol. 90, 5534–5537. doi: 10.1128/JVI.01865-15
Ressing, M. E., Horst, D., Griffin, B. D., Tellam, J., Zuo, J., Khanna, R., et al. (2008). Epstein-Barr Virus evasion of CD8+ and CD4+ T cell immunity via concerted actions of multiple gene products. Semin. Cancer Biol. 18, 397–408. doi: 10.1016/j.semcancer.2008.10.008
Ressing, M. E., van Gent, M., Gram, A. M., Hooykaas, M. J. G., Piersma, S. J., Wiertz, E. J. H. J. (2015). Immune evasion by Epstein-Barr virus. Curr Top Microbiol Immunol. 391, 355–381. doi: 10.1007/978-3-319-22834-1_12
Rosa, M. D., Gottlieb, E., Lerner, M. R., Steitz, J. A. (1981). Striking similarities are exhibited by two small Epstein-Barr virus-encoded ribonucleic acids and the adenovirus-associated ribonucleic acids VAI and VAII. Mol. Cell Biol. 1, 785–796. doi: 10.1128/mcb.1.9.785-796.1981
Rouzaut, A., Garasa, S., Teijeira, Á, González, I., Martinez-Forero, I., Suarez, N., et al. (2010). Dendritic cells adhere to and transmigrate across lymphatic endothelium in response to IFN-α. Eur. J. Immunol. 40, 3054–3063. doi: 10.1002/eji.201040523
Saeidi, A., Zandi, K., Cheok, Y. Y., Saeidi, H., Wong, W. F., Lee, C. Y. Q., et al. (2018). T-Cell exhaustion in chronic infections: Reversing the state of exhaustion and reinvigorating optimal protective immune responses. Front. Immunol. 9. doi: 10.3389/fimmu.2018.02569
Samanta, M., Iwakiri, D., Kanda, T., Imaizumi, T., Takada, K. (2006). EB Virus-encoded RNAs are recognized by RIG-I and activate signaling to induce type I IFN. EMBO J. 25, 4207–4214. doi: 10.1038/sj.emboj.7601314
Samanta, M., Iwakiri, D., Takada, K. (2008). Epstein–Barr Virus-encoded small RNA induces IL-10 through RIG-i-mediated IRF-3 signaling. Oncogene 27, 4150–4160. doi: 10.1038/onc.2008.75
Santini, S. M., Lapenta, C., Logozzi, M., Parlato, S., Spada, M., Di Pucchio, T., et al. (2000). Type I interferon as a powerful adjuvant for monocyte-derived dendritic cell development and activity in vitro and in hu-Pbl-Scid mice. J. Exp. Med. 191, 1777–1788. doi: 10.1084/jem.191.10.1777
Sarawar, S. R., Shen, J., Dias, P. (2020). Insights into CD8 T cell activation and exhaustion from a mouse gammaherpesvirus model. Viral Immunol. 33, 215–224. doi: 10.1089/vim.2019.0183
Sartorius, R., Trovato, M., Manco, R., D’Apice, L., De Berardinis, P. (2021). Exploiting viral sensing mediated by toll-like receptors to design innovative vaccines. NPJ Vaccines 6, 127. doi: 10.1038/s41541-021-00391-8
Savard, M., Bélanger, C., Tardif, M., Gourde, P., Flamand, L., Gosselin, J. (2000). Infection of primary human monocytes by Epstein-Barr virus. J. Virol. 74, 2612–2619. doi: 10.1128/JVI.74.6.2612-2619.2000
Schock, S. N., Chandra, N. V., Sun, Y., Irie, T., Kitagawa, Y., Gotoh, B., et al. (2017). Induction of necroptotic cell death by viral activation of the RIG-I or STING pathway. Cell Death Differ 24, 615–625. doi: 10.1038/cdd.2016.153
Schoggins, J. W., MacDuff, D. A., Imanaka, N., Gainey, M. D., Shrestha, B., Eitson, J. L., et al. (2014). Pan-viral specificity of IFN-induced genes reveals new roles for cGAS in innate immunity. Nature 505, 691–695. doi: 10.1038/nature12862
Schwerk, J., Kemper, L., Bussey, K. A., Lienenklaus, S., Weiss, S., Čičin-Šain, L., et al. (2022). Type I interferon signaling controls gammaherpesvirus latency In vivo. Pathogens 11, 1554. doi: 10.3390/pathogens11121554
Setsuda, J., Teruya-Feldstein, J., Harris, N. L., Ferry, J. A., Sorbara, L., Gupta, G., et al. (1999). Interleukin-18, interferon-γ, IP-10, and mig expression in Epstein-Barr virus-induced infectious mononucleosis and posttransplant lymphoproliferative disease. Am. J. Pathol. 155, 257–265. doi: 10.1016/S0002-9440(10)65119-X
Sorel, O., Dewals, B. G. (2019). The critical role of genome maintenance proteins in immune evasion during gammaherpesvirus latency. Front. Microbiol. 9. doi: 10.3389/fmicb.2018.03315
Stark, G. R., Darnell, J. E. (2012). The JAK-STAT pathway at twenty. Immunity 36, 503–514. doi: 10.1016/j.immuni.2012.03.013
Stempel, M., Chan, B., Brinkmann, M. M. (2019a). Coevolution pays off: Herpesviruses have the license to escape the DNA sensing pathway. Med. Microbiol. Immunol. 208, 495–512. doi: 10.1007/s00430-019-00582-0
Stempel, M., Chan, B., Juranić Lisnić, V., Krmpotić, A., Hartung, J., Paludan, S. R., et al. (2019b). The herpesviral antagonist m152 reveals differential activation of STING -dependent IRF and NF-κB signaling and STING’s dual role during infection. EMBO J. 38, e100983. doi: 10.15252/embj.2018100983
Stevenson, P. G., May, J. S., Smith, X. G., Marques, S., Adler, H., Koszinowski, U. H., et al. (2002). K3-mediated evasion of CD8+ T cells aids amplification of a latent γ-herpesvirus. Nat. Immunol. 3, 733–740. doi: 10.1038/ni818
Sun, H., Huang, Y., Mei, S., Xu, F., Liu, X., Zhao, F., et al. (2021). A nuclear export signal is required for cGAS to sense cytosolic DNA. Cell Rep. 34, 108586. doi: 10.1016/j.celrep.2020.108586
Sun, C., Schattgen, S. A., Pisitkun, P., Jorgensen, J. P., Hilterbrand, A. T., Wang, L. J., et al. (2015). Evasion of innate cytosolic DNA sensing by a gammaherpesvirus facilitates establishment of latent infection. J. Immunol. 194, 1819–1831. doi: 10.4049/jimmunol.1402495
Tamura, T., Yanai, H., Savitsky, D., Taniguchi, T. (2008). The IRF family transcription factors in immunity and oncogenesis. Annu. Rev. Immunol. 26, 535–584. doi: 10.1146/annurev.immunol.26.021607.090400
Tan, C. S. E., Lawler, C., Stevenson, P. G. (2017). CD8+ T cell evasion mandates CD4+ T cell control of chronic gamma-herpesvirus infection. PloS Pathog. 13, e1006311. doi: 10.1371/journal.ppat.1006311
Taylor, G. S., Long, H. M., Brooks, J. M., Rickinson, A. B., Hislop, A. D. (2015). The immunology of Epstein-Barr virus–induced disease. Annu. Rev. Immunol. 33, 787–821. doi: 10.1146/annurev-immunol-032414-112326
Thomson, R. C., Petrik, J., Nash, A. A., Dutia, B. M. (2008). Expansion and activation of NK cell populations in a gammaherpesvirus infection. Scand. J. Immunol. 67, 489–495. doi: 10.1111/j.1365-3083.2008.02100.x
Tibbetts, S. A., van Dyk, L. F., Speck, S. H., Virgin, H. W. (2002). Immune control of the number and reactivation phenotype of cells latently infected with a gammaherpesvirus. J. Virol. 76, 7125–7132. doi: 10.1128/JVI.76.14.7125-7132.2002
Tomalka, J. A., Suthar, M. S., Diamond, M. S., Sekaly, R. P. (2022). Innate antiviral immunity: how prior exposures can guide future responses. Trends Immunol. 43, 696–705. doi: 10.1016/j.it.2022.07.001
Topham, D. J., Cardin, R. C., Christensen, J. P., Brooks, J. W., Belz, G. T., Doherty, P. C. (2001). Perforin and fas in murine gammaherpesvirus-specific CD8+ T cell control and morbidity. J. Gen. Virol. 82, 1971–1981. doi: 10.1099/0022-1317-82-8-1971
Torii, Y., Kawada, J., Murata, T., Yoshiyama, H., Kimura, H., Ito, Y. (2017). Epstein-Barr Virus infection-induced inflammasome activation in human monocytes. PloS One 12, e0175053. doi: 10.1371/journal.pone.0175053
Torti, N., Oxenius, A. (2012). T Cell memory in the context of persistent herpes viral infections. Viruses 4, 1116–1143. doi: 10.3390/v4071116
Tripp, R. A., Hamilton-Easton, A. M., Cardin, R. D., Nguyen, P., Behm, F. G., Woodland, D. L., et al. (1997). Pathogenesis of an infectious mononucleosis-like disease induced by a murine γ-herpesvirus: Role for a viral superantigen? J. Exp. Med. 185, 1641–1650. doi: 10.1084/jem.185.9.1641
Tsuchida, T., Zou, J., Saitoh, T., Kumar, H., Abe, T., Matsuura, Y., et al. (2010). The ubiquitin ligase TRIM56 regulates innate immune responses to intracellular double-stranded DNA. Immunity 33, 765–776. doi: 10.1016/j.immuni.2010.10.013
Uldrick, T. S., Wang, V., O’Mahony, D., Aleman, K., Wyvill, K. M., Marshall, V., et al. (2010). An Interleukin-6–related systemic inflammatory syndrome in patients Co-infected with kaposi sarcoma–associated herpesvirus and HIV but without multicentric castleman disease. Clin. Infect. Dis. 51, 350–358. doi: 10.1086/654798
Unterholzner, L., Keating, S. E., Baran, M., Horan, K. A., Jensen, S. B., Sharma, S., et al. (2010). IFI16 is an innate immune sensor for intracellular DNA. Nat. Immunol. 11, 997–1004. doi: 10.1038/ni.1932
Usherwood, E. J., Meadows, S. K., Crist, S. G., Bellfy, S. C., Sentman, C. L. (2005). Control of murine gammaherpesvirus infection is independent of NK cells. Eur. J. Immunol. 35, 2956–2961. doi: 10.1002/eji.200526245
van de Veerdonk, F. L., Wever, P. C., Hermans, M. H. A., Fijnheer, R., Joosten, L. A. B., van der Meer, J. W. M., et al. (2012). IL-18 serum concentration is markedly elevated in acute EBV infection and can serve as a marker for disease severity. J. Infect. Dis. 206, 197–201. doi: 10.1093/infdis/jis335
Viet, N. H., Trung, N. Q., Dong, L. T., Trung, L. Q., Espinoza, J. L. (2021). Genetic variants in NKG2D axis and susceptibility to Epstein-Barr virus-induced nasopharyngeal carcinoma. J. Cancer Res. Clin. Oncol. 147, 713–723. doi: 10.1007/s00432-020-03475-5
Vivier, E., Raulet, D. H., Moretta, A., Caligiuri, M. A., Zitvogel, L., Lanier, L. L., et al. (2011). Innate or adaptive immunity? the example of natural killer cells. Science 331, 44–49. doi: 10.1126/science.1198687
Vivier, E., Tomasello, E., Baratin, M., Walzer, T., Ugolini, S. (2008). Functions of natural killer cells. Nat. Immunol. 9, 503–510. doi: 10.1038/ni1582
Volk, V., Theobald, S. J., Danisch, S., Khailaie, S., Kalbarczyk, M., Schneider, A., et al. (2021). PD-1 blockade aggravates Epstein–Barr virus+ post-transplant lymphoproliferative disorder in humanized mice resulting in central nervous system involvement and CD4+ T cell dysregulations. Front. Oncol. 10, e47491. doi: 10.3389/fonc.2020.614876
Volkman, H. E., Cambier, S., Gray, E. E., Stetson, D. B. (2019). Tight nuclear tethering of cGAS is essential for preventing autoreactivity. Elife 8, e47491. doi: 10.7554/eLife.47491
Wang, P., Deng, Y., Guo, Y., Xu, Z., Li, Y., Ou, X., et al. (2020). Epstein-Barr Virus early protein BFRF1 suppresses IFN-β activity by inhibiting the activation of IRF3. Front. Immunol. 11. doi: 10.3389/fimmu.2020.513383
Wang, Q., Liu, X., Cui, Y., Tang, Y., Chen, W., Li, S., et al. (2014). The E3 ubiquitin ligase AMFR and INSIG1 bridge the activation of TBK1 kinase by modifying the adaptor STING. Immunity 41, 919–933. doi: 10.1016/j.immuni.2014.11.011
Wang, Y., Ludwig, J., Schuberth, C., Goldeck, M., Schlee, M., Li, H., et al. (2010). Structural and functional insights into 5′-ppp RNA pattern recognition by the innate immune receptor RIG-I. Nat. Struct. Mol. Biol. 17, 781–787. doi: 10.1038/nsmb.1863
Wang, Y., Tibbetts, S. A., Krug, L. T. (2021). Conquering the host: Determinants of pathogenesis learned from murine gammaherpesvirus 68. Annu. Rev. Virol. 8, 349–371. doi: 10.1146/annurev-virology-011921-082615
Wedekind, A., Fritzsch, D., Kröger, A. (2020). Add on the next level–the time point of the type I IFN response orchestrates the immune response. Cell Mol. Immunol. 17, 791–793. doi: 10.1038/s41423-020-0442-7
Welsh, R. M., Bahl, K., Marshall, H. D., Urban, S. L. (2012). Type 1 interferons and antiviral CD8 T-cell responses. PloS Pathog. 8, e1002352. doi: 10.1371/journal.ppat.1002352
Wen, K. W., Wang, L., Menke, J. R., Damania, B. (2021). Cancers associated with human gammaherpesviruses. FEBS J. 289, 7631–7669. doi: 10.1111/febs.16206
Wood, B. M., Mboko, W. P., Mounce, B. C., Tarakanova, V. L. (2013). Mouse gammaherpesvirus-68 infection acts as a rheostat to set the level of type I interferon signaling in primary macrophages. Virology. 443, 123–133. doi: 10.1016/j.virol.2013.04.036
Wu, Y., Shrestha, P., Heape, N. M., Yarchoan, R. (2022). CDK4/6 inhibitors sensitize gammaherpesvirus-infected tumor cells to T-cell killing by enhancing expression of immune surface molecules. J. Transl. Med. 20, 217. doi: 10.1186/s12967-022-03400-z
Wu, J., Sun, L., Chen, X., Du, F., Shi, H., Chen, C., et al. (2013). Cyclic GMP-AMP is an endogenous second messenger in innate immune signaling by cytosolic DNA. Sci. (1979) 339, 826–830. doi: 10.1126/science.1229963
Xie, W., Lee, B., Bruce, K., Lawler, C., Farrell, H. E., Stevenson, P. G. (2022). CD4+ T cells control murine cytomegalovirus infection indirectly. J. Virol. 96, e0007722. doi: 10.1128/jvi.00077-22
Xing, J., Zhang, A., Zhang, H., Wang, J., Li, X. C., Zeng, M.-S., et al. (2017). TRIM29 promotes DNA virus infections by inhibiting innate immune response. Nat. Commun. 8, 945. doi: 10.1038/s41467-017-00101-w
Xu, D., Trajkovic, V., Hunter, D., Leung, B. P., Schulz, K., Gracie, J. A., et al. (2000). IL-18 induces the differentiation of Th1 or Th2 cells depending upon cytokine milieu and genetic background. Eur. J. Immunol. 30, 3147–3156. doi: 10.1002/1521-4141(200011)30:11<3147::AID-IMMU3147>3.0.CO;2-J
Yamano, T., Steinert, M., Steer, B., Klein, L., Hammerschmidt, W., Adler, H. (2019). B cells latently infected with murine gammaherpesvirus 68 (MHV-68) are present in the mouse thymus-a step toward immune evasion? Eur. J. Immunol. 49, 351–352. doi: 10.1002/eji.201847886
Yan, H., Krishnan, K., Lim, J. T., Contillo, L. G., Krolewski, J. J. (1996). Molecular characterization of an alpha interferon receptor 1 subunit (IFNaR1) domain required for TYK2 binding and signal transduction. Mol. Cell Biol. 16, 2074–2082. doi: 10.1128/MCB.16.5.2074
Yan, L., Majerciak, V., Zheng, Z.-M., Lan, K. (2019). Towards better understanding of KSHV life cycle: From transcription and posttranscriptional regulations to pathogenesis. Virol. Sin. 34, 135–161. doi: 10.1007/s12250-019-00114-3
Yang, K., Wang, J., Wu, M., Li, M., Wang, Y., Huang, X. (2015). Mesenchymal stem cells detect and defend against gammaherpesvirus infection via the cGAS-STING pathway. Sci. Rep. 5, 7820. doi: 10.1038/srep07820
Zhang, Q., Bastard, P., Karbuz, A., Gervais, A., Tayoun, A. A., Aiuti, A., et al. (2022a). Human genetic and immunological determinants of critical COVID-19 pneumonia. Nature 603, 587–598. doi: 10.1038/s41586-022-04447-0
Zhang, Q., Bastard, P., Liu, Z., Le Pen, J., Moncada-Velez, M., Chen, J., et al. (2020). Inborn errors of type I IFN immunity in patients with life-threatening COVID-19. Sci. (1979) 370, eabd4570. doi: 10.1126/science.abd4570
Zhang, J., Hu, M.-M., Wang, Y.-Y., Shu, H.-B. (2012). TRIM32 protein modulates type I interferon induction and cellular antiviral response by targeting MITA/STING protein for K63-linked ubiquitination. J. Biol. Chem. 287, 28646–28655. doi: 10.1074/jbc.M112.362608
Zhang, Q., Pizzorno, A., Miorin, L., Bastard, P., Gervais, A., Le Voyer, T., et al. (2022b). Autoantibodies against type I IFNs in patients with critical influenza pneumonia. J. Exp. Med. 219, e20220514. doi: 10.1084/jem.20220514
Keywords: EBV, MHV68, gammaherpesvirus, innate immunity, interferon, NK cells, T cells, CTL
Citation: Rex V, Zargari R, Stempel M, Halle S and Brinkmann MM (2023) The innate and T-cell mediated immune response during acute and chronic gammaherpesvirus infection. Front. Cell. Infect. Microbiol. 13:1146381. doi: 10.3389/fcimb.2023.1146381
Received: 17 January 2023; Accepted: 20 March 2023;
Published: 31 March 2023.
Edited by:
M. Zeeshan Chaudhry, Diamantina Institute, The University of Queensland, AustraliaReviewed by:
Heather Koehler, Washington State University, United StatesPratyusha Mandal, City College of New York (CUNY), United States
Copyright © 2023 Rex, Zargari, Stempel, Halle and Brinkmann. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Stephan Halle, aGFsbGUuc3RlcGhhbkBNSC1IYW5ub3Zlci5kZQ==; Melanie M. Brinkmann, bS5icmlua21hbm5AdHUtYnJhdW5zY2h3ZWlnLmRl
†These authors have contributed equally to this work and share first authorship