 Wellington Owusu1
Wellington Owusu1 Arnoud H. M. van Vliet2
Arnoud H. M. van Vliet2 Natalie E. Riddell3
Natalie E. Riddell3 Graham Stewart1
Graham Stewart1 Winifred C. Akwani1
Winifred C. Akwani1 Sherihane Aryeetey4
Sherihane Aryeetey4 Rejoice Agyeiwaa Arthur4
Rejoice Agyeiwaa Arthur4 Augustina Angelina Sylverken4,5*
Augustina Angelina Sylverken4,5* Suzanne M. Hingley-Wilson1*
Suzanne M. Hingley-Wilson1*- 1Department of Microbial Sciences, School of Biosciences, Faculty of Health and Medical Sciences, University of Surrey, Guildford, United Kingdom
- 2Department of Comparative Biomedical Sciences, School of Veterinary Medicine, Faculty of Health and Medical Sciences, University of Surrey, Guildford, United Kingdom
- 3Department of Biochemical Sciences, School of Biosciences and Medicine, University of Surrey, Guildford, United Kingdom
- 4Kumasi Centre for Collaborative Research in Tropical Medicine, Kwame Nkrumah University of Science and Technology, Kumasi, Ghana
- 5Department of Theoretical and Applied Biology, Kwame Nkrumah University of Science and Technology, Kumasi, Ghana
In low-resource settings with high tuberculosis (TB) burdens, lack of rapid diagnostic methods for detection and differentiation of Mycobacterium tuberculosis complex (MTBC) is a major challenge affecting TB management. This study utilized comparative genomic analyses of MTBC lineages; M. tuberculosis, M. africanum Lineages 5/6 and M. bovis to identify lineage-specific genes. Primers were designed for the development of a Multiplex PCR assay which was successful in differentiating the MTBC lineages. There was no cross-reaction with other respiratory pathogens tested. Validation of the assay using clinical samples was performed with sputum DNA extracts from 341 clinically confirmed active TB patients. It was observed that 24.9% of cases were caused by M. tuberculosis, while M. africanum L5 & L6 reported 9.0% and 14.4%, respectively. M. bovis infection was the least frequently detected lineage with 1.8%. Also, 27.0% and 17.0% of the cases were PCR negative and unspeciated, respectively. However, mixed-lineage TB infections were recorded at a surprising 5.9%. This multiplex PCR assay will allow speciation of MTBC lineages in low-resource regions, providing rapid differentiation of TB infections to select appropriate medication at the earliest possible time point. It will also be useful in epidemiological surveillance studies providing reliable information on the prevalence of TB lineages as well as identifying difficult to treat cases of mixed-lineage tuberculosis infections.
Introduction
Human tuberculosis (TB) is a communicable disease caused by some members of the Mycobacterium tuberculosis complex (MTBC), mainly; Mycobacterium tuberculosis (Mtb), Mycobacterium africanum (Maf) and Mycobacterium bovis (Mbo). It is one of the leading causes of death from a single infectious organism, infecting about a quarter of the world’s population (WHO, 2020). It remains a global pandemic, despite the availability of interventional control measures such as the use of a live attenuated vaccine (BCG) and multi-drug therapy. The situation has been further aggravated by the lack of rapid and reliable, point-of-care diagnostic methods for low-resource areas, and the use of various forms of insufficient treatment procedures among poor resource countries (WHO, 2020).
TB in an individual is often assumed to be caused by a single clonal MTBC lineage, although mixed infections have been previously noted (Hingley-Wilson et al., 2013). Advances in molecular-based approaches in TB studies also demonstrated multiple lineages causing TB in the same patient (Van Rie et al., 2005; Huyen et al., 2012; Zetola et al., 2014) and the occurrence of mixed-lineage TB infections in high TB endemic regions has been reported (Cohen et al., 2011). In TB management, mixed-lineage TB infections have been strongly associated with poor treatment outcome (Zetola et al., 2014).
West-Africa has one of the highest incidences of TB world-wide with a unique set of circulating MTBC species namely: M. tuberculosis, M. africanum and M. bovis. While M. tuberculosis is generally the predominant pathogen for human TB, unusually almost 50% of all TB cases in West Africa are caused by M. africanum (Mostowy et al., 2004). In The Gambia, 39% of TB cases are caused by M. africanum (de Jong et al., 2010a). In Ghana, M. africanum rates remain stable at around 20%, with one of the highest rates of infections in the Northern part of Ghana (De Jong et al., 2009). While the reservoir of infection for M. tuberculosis is the latently infected human population, a non-human reservoir of infection for M. africanum in Ghana has been postulated, likely to be more concentrated in Northern Ghana (Otchere et al., 2018).
The gold standard of TB diagnosis is the isolation of MTBC by culture and the use of biochemical tests (Gholoobi et al., 2014). However, these methods are very laborious and time-consuming which further risk aggravating the condition of patients due to delayed treatment. In addition, with culturing-based techniques in a mixed infection, the fastest growing is often noted as a single infection (Hingley-Wilson et al., 2013). Species differentiation is often challenged with misidentification. For instance, M. africanum Lineage 5 (MafL5) and Lineage 6 (MafL6) exhibit growth characteristics which are intermediates of both M. tuberculosis and M. bovis (de Jong et al., 2010b). Since 2010, WHO recommended the use of GeneXpert assay in diagnostic facilities as a first-line TB diagnostic tool (Goig et al., 2019). It detects MTBC through the identification of insertion sequence (IS6110) as well as identifying rifampicin resistant genes. Although an improved modified GeneXpert Ultra version has been produced with high sensitivity and specificity, it is unable to differentiate the individual MTBC lineages to inform selection of appropriate medication.
In low-resource regions, MTBC lineages are often not differentiated prior to treatment due to reasons such as unavailability of high cost, non-portable genome sequencing machines and length of time for culture results. This can lead to inappropriate treatment regimens, for example, M. bovis is intrinsically resistant to pyrazinamide, one of the frontline drugs used collectively for standard TB treatment (Oryan et al., 2022). Indeed, patient exposure to prolonged pyrazinamide treatment can result in hepatotoxicity and polyarthralgia (Papastavros et al., 2002) and should therefore be avoided if not required. Additionally, antibiotic treatment duration of M. bovis infections is recommended for 9 months (rather than the standard 6 months) because of the absence of pyrazinamide efficacy (Lan et al., 2016). In general, TB treatment durations shorter than recommendation may lead to incomplete sterilization of an infection and increase the risk of the development of antibiotic resistance (Khalif Ali et al., 2017; Ali et al., 2019). It is therefore important to investigate and identify lineage-specific TB molecular markers for designing diagnostic assays with high level of sensitivity and specificity to inform selection of appropriate medication to limit morbidity and drug resistance.
Using the comparative genomics workflow previously described by Akwani et al., 2022, MTBC lineage-specific genes identified were transferred into the development of multiplex PCR assay for TB lineage differentiation. This will enhance precise disease diagnosis, improve epidemiological surveillance studies and help inform selection of appropriate TB drug regimens at early time point especially in low resource settings with high TB incidence.
Materials and methods
Selection and processing of genome sequences
Genome sequences of M. tuberculosis, M. africanum and M. bovis in the form of sequence reads and assembled genomes were obtained from NCBI, Genbank and EMBL-EBI repositories using fastq-dump instructions (SRA-Tools-NCBI, 2021). In addition, reference sequences were also obtained. An overall total of 7,456 genome sequences comprising M. tuberculosis (6802), M. africanum (244), M. bovis (391) and other animal-adapted MTBCs (19) were assessed (Supplementary Table S1). Genome assembly was performed with Shovil Megahit toolkit version 1.2.9. To evaluate the consistency of the assembled genomes, quality assessment was performed with QUAST version. 4.6.3 (Gurevich et al., 2013). The inclusion criteria for checks included: largest contig must be greater than 100kb, N50 >25kb, L50 < 50 and the genomic size between 4.0 and 4.8 Mbp. A total of 120 genomes of M. tuberculosis, M. africanum and M. bovis were used for the pangenome analysis leading to the identification of lineage-specific genes as shown in Figure 1.
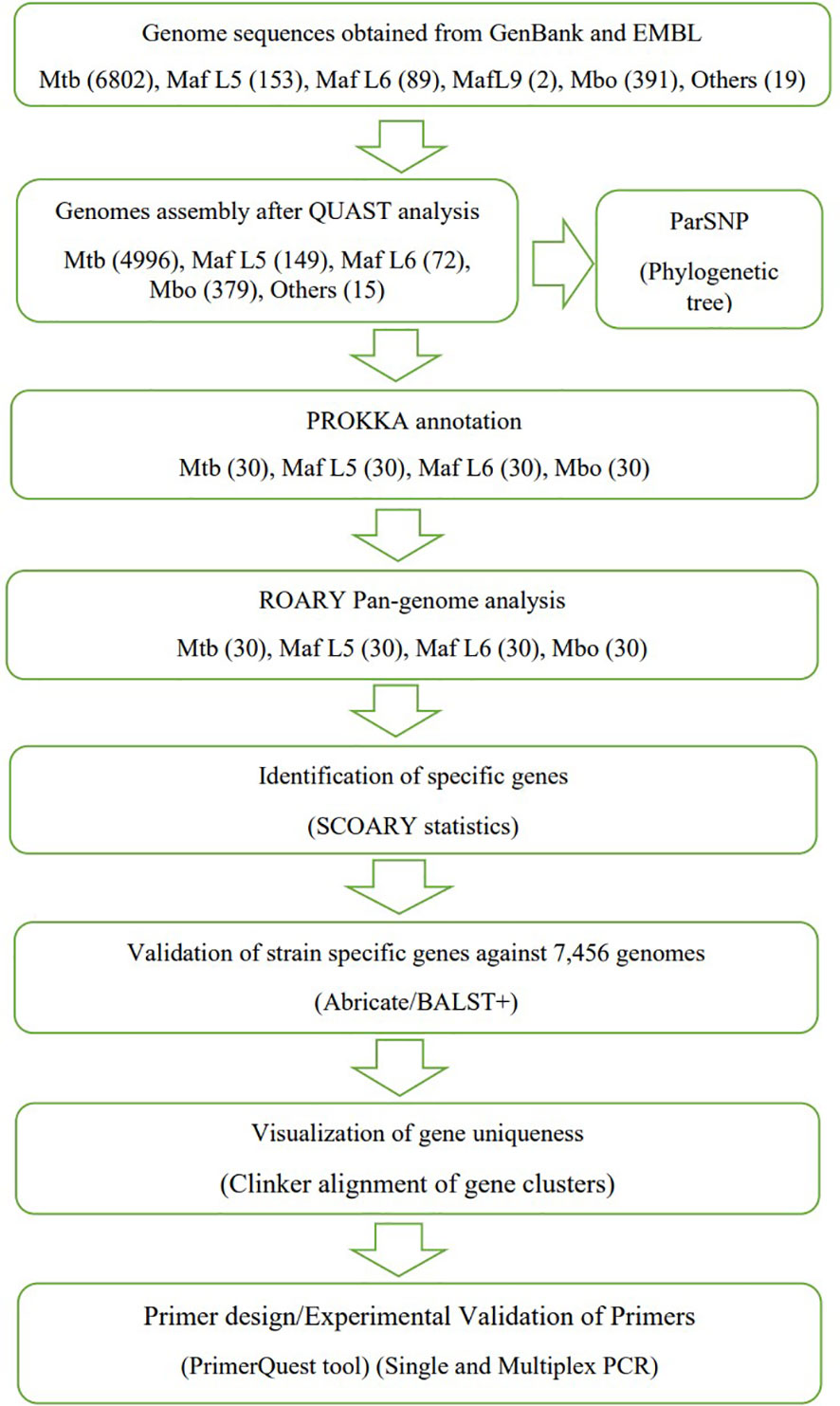
Figure 1 An overview of Scheme of work towards identification of lineage-specific genes for PCR primers development (similar to comparative genomic workflow previously described by Akwani et al., 2022).
Phylogenetic analysis of MTBC
ParSNP v. 1.2 was used as described previously (Pornsukarom et al., 2018) using the “-a 13 – x” settings to generate a phylogenetic tree of MTBC lineages. Classification of lineages was achieved based on the phylogenetic tree constructed. The output was visualised with FigTree version 1.4.3.
Comparative genomic analysis and identification of lineage-specific genes
Genomes were annotated using Prokka v1.14 (Seemann, 2014), while pangenomes were analyzed using Roary v3.12 (Page et al., 2015) at default settings and 90% BLAST cut-offusing randomly selected 120 genomes comprising M. tuberculosis (30), M. africanum L5 (30), M. africanum L6 (30) and M. bovis (30) (Supplementary Table S1). Scoary analysis (Brynildsrud et al., 2016) was used to examine the association between accessory (lineage-specific) genes and phenotypic traits. The number of lineage-specific genes was trimmed using statistical results from Bonferroni corrected p-value of ≤ 0.05. Also, lineage-specific genes were selected only if present in greater than 90% of the respective lineages and less than 10% in the other lineages. Further screening of the lineage-specific genes was performed by BLAST+ version 2.13.0 against all 7,456 MTBC genomes via Abricate v.1.0.9 (https://github.com/tseemann/abricate) with minimum coverage of 70% and minimum identity of 80% for a correct match. Genomic regions were compared to identify uniqueness using Clinker alignment of complete genomes (Gilchrist & Chooi, 2021).
Isolation of genomic DNA
The following reagents were obtained through BEI Resources, NIAID, NIH: genomic DNA from M. africanum strains NLA009502090, NR-49655 and M. africanum strain NLA000017316, NR-49652. Heat-killed M. tuberculosis (H37Rv) and M. bovis (AF2122/97) were obtained from liquid cultures prepared in the containment level 3 (CL3) lab before being transferred to the CL2 lab for DNA extraction. Genomic DNA of mycobacterial strains was extracted using the cetyltrimethylammonium bromide (CTAB)-chloroform method as described previously (Belisle et al., 2009). The concentration and purity of DNA was determined by the NanoDrop 2000 at absorbance of 260nm and purity A260/A280 ratio of 1.7 to 2.0.
Primer design
Candidate genes identified were selected for primer design, using the PrimerQuest Tool developed by Integrated DNA Technologies (https://eu.idtdna.com/Primerquest). The FASTA format of each nucleotide sequence was inputted with PCR 2 primer options. Each primer was assigned a specific product size ranging from 100 to 1000 bp. Details of primers have been shown in Table 1.
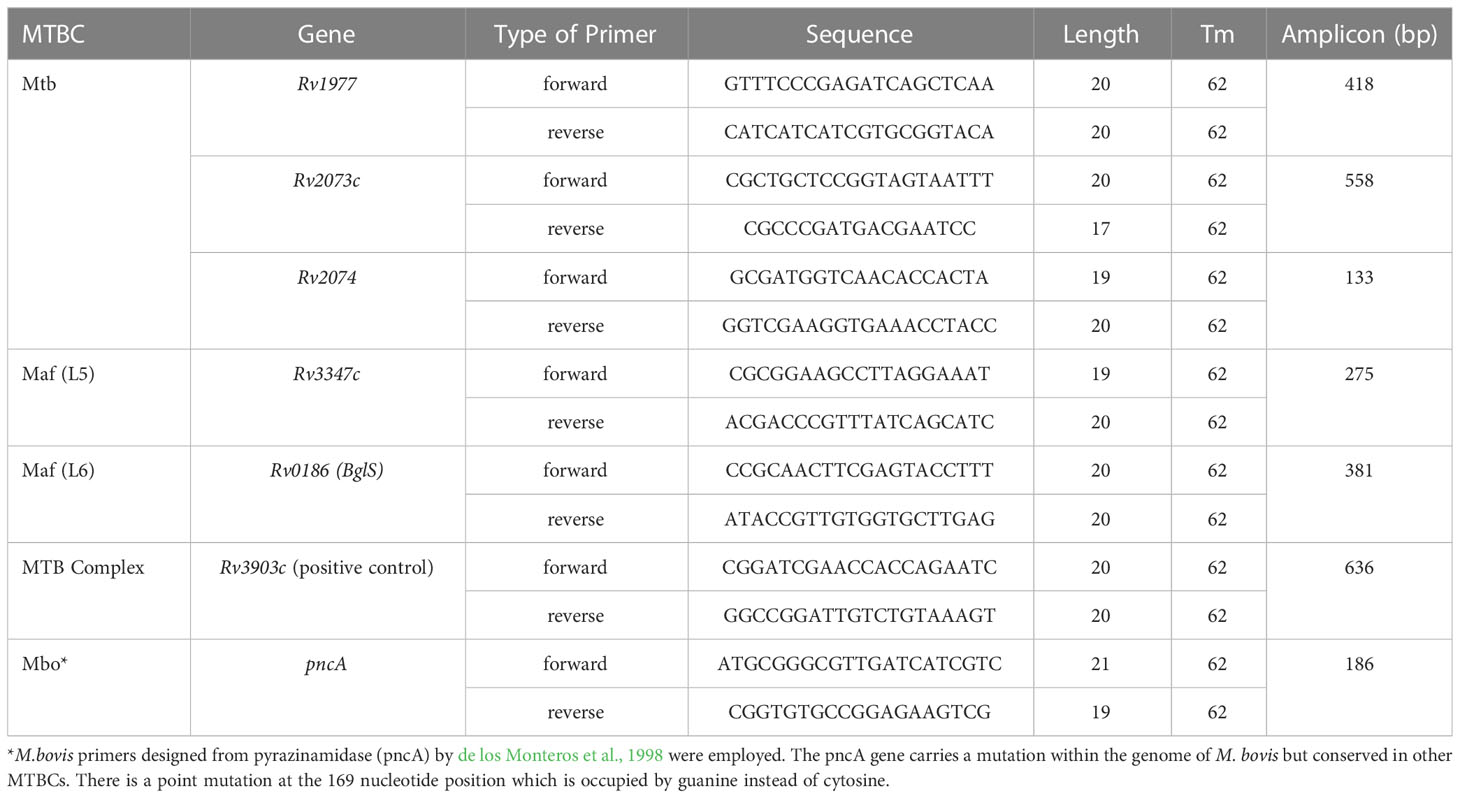
Table 1 MTBC lineage-specific genes and primer sequences.
Preparation of PCR assay
For the single PCR assays, a final volume of 25 µl was setup. Each setup contained 12.5 µl of 2x GoTaq® Hot Start Green Master Mix (400 µM polymerase, 400 µM of dNTPs, 4 mM MgCl2 and pH 8.5 buffer), produced by Promega, UK, 1µl each of 10µM forward and reverse primers, 1 µl DNA (< 250 ng), 1 µl DMSO and nuclease free water. The non-template control consisted of the master mix, specific primers and nuclease free water, while 1 µl of E.coli DNA was used as negative control. For the multiplex PCR assays, a 50µl reaction volume was achieved with the following constituents: 25 µl of 2x GoTaq® Hot Start Green Master Mix, 5 µl of 10 µM of forward/reverse primers (1 µl of each lineage-specific primer), 1µl DNA (<250 ng), 2 µl DMSO and nuclease free water. An all-in-one multiplex PCR had 4 µl of DNA (1µl from each lineage). The reaction mix contained an excess of primers and nucleotides to ensure reaction continuity without limitation. The amplification was carried out in the SimpliAmp Thermal cycler at an initial denaturation of 2 mins at 95°C; 30 cycles of 30 sec at 95°C; 1 min at 62°C; 1 min at 72°C and a final extension at 72°C for 5 min. The separation of PCR products was performed using 2% gel agarose electrophoresis at 80 V for 1.30 hrs. A 100 bp DNA ladder was used as indicator. Visualization of gel was performed under ultraviolet light of Microtek MiBio Fluo version1.04.
Ethical clearance
Ethical approval for the use of human sputum samples was granted by the Committee on Human Research and Publication Ethics (CHRPE) at the School of Medical Science of Kwame Nkrumah University of Science and Technology (KNUST), Ghana: (CHRPE/AP/396/22).
Results
Stratification and identification of lineage-specific genes of the MTBC
A selection of 120 MTBC genomes (30 M. tuberculosis, 30 M. africanum L5, 30 M. africanum L6 and 30 M. bovis) from GenBank and EMBL repositories, were subjected to comparative genomic analysis. The phylogenetic relationship between the MTBCs was established with ParSNP which constructs a phylogenetic tree using core genome SNPs. In Figure 2, divisions were observed in four large clusters representing M. tuberculosis, M. africanum L5, M. africanum L6 and M. bovis. Pangenome analysis was performed on the same set of genomes to obtain the distribution of gene families within the MTBCs. A Roary matrix shows the clustering of 7,610 genes into either core genes (commonly shared by all members) or accessory genes (found in only few members) (Figure 1). It could be seen that almost all the genes are skewed toward the core gene section while only a few were categorised as accessory genes. This type of gene distribution highlights the high level of clonality of the MTBC.
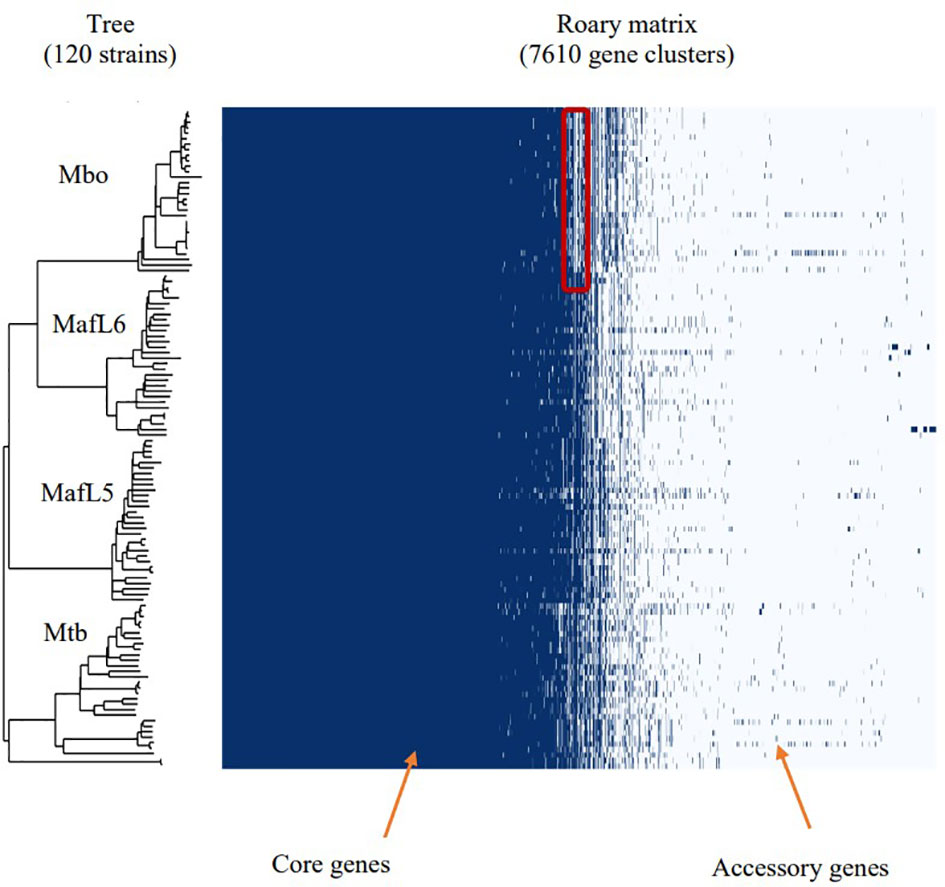
Figure 2 Phylogenetic tree together with pangenome distribution of genes within the MTBC – M. tuberculosis, M. africanum and M. bovis. The phylogenetic tree was constructed via identification of core genome SNPs using ParSNP tool. Roary matrix obtained from pangenome analysis shows the distribution of core and accessory genes within the MTBC. The highlighted section of the M. bovis genomes shows deletions which are characteristic of M. bovis as RD4, RD7, RD8, RD9 and RD12 deletions.
Further analysis was conducted on the pangenome outcome to ascertain the relationship between accessory genes and trait (lineages) using Scoary statistics. The definition of lineage-specific genes was set as being present in more than 90% of specific species and less than 10% in the other lineages. A total of 56 lineage-specific genes were obtained comprising 16 M. africanum L5, 10 M. africanum L6, 10 M. tuberculosis and 20 M. bovis specific genes (Supplementary Table S2). A final screening of these lineage-specific genes was performed by BLAST against 7,456 MTBC genomes via Abricate with a minimum coverage of 70% and minimum identity of 80% for a correct match as shown in Table 2. The candidate genes specific for M. tuberculosis were Rv1977, Rv2073c and Rv2074. The Rv0186-betaglucosidase was unique for M. africanum L6 while Rv3903c was conserved in all the MTBCs, thus serving as positive control marker. The Rv3347c was unique for M. africanum L5 via Clinker alignment of gene clusters shown in Figure 3. Although BLAST hits did not show any unique gene for M. bovis, the pncA gene highlighted to be distinctive in M. bovis by de los Monteros et al., 1998 was used.

Table 2 Summary of Abricate BLAST results showing MTBC lineage-specific genes.
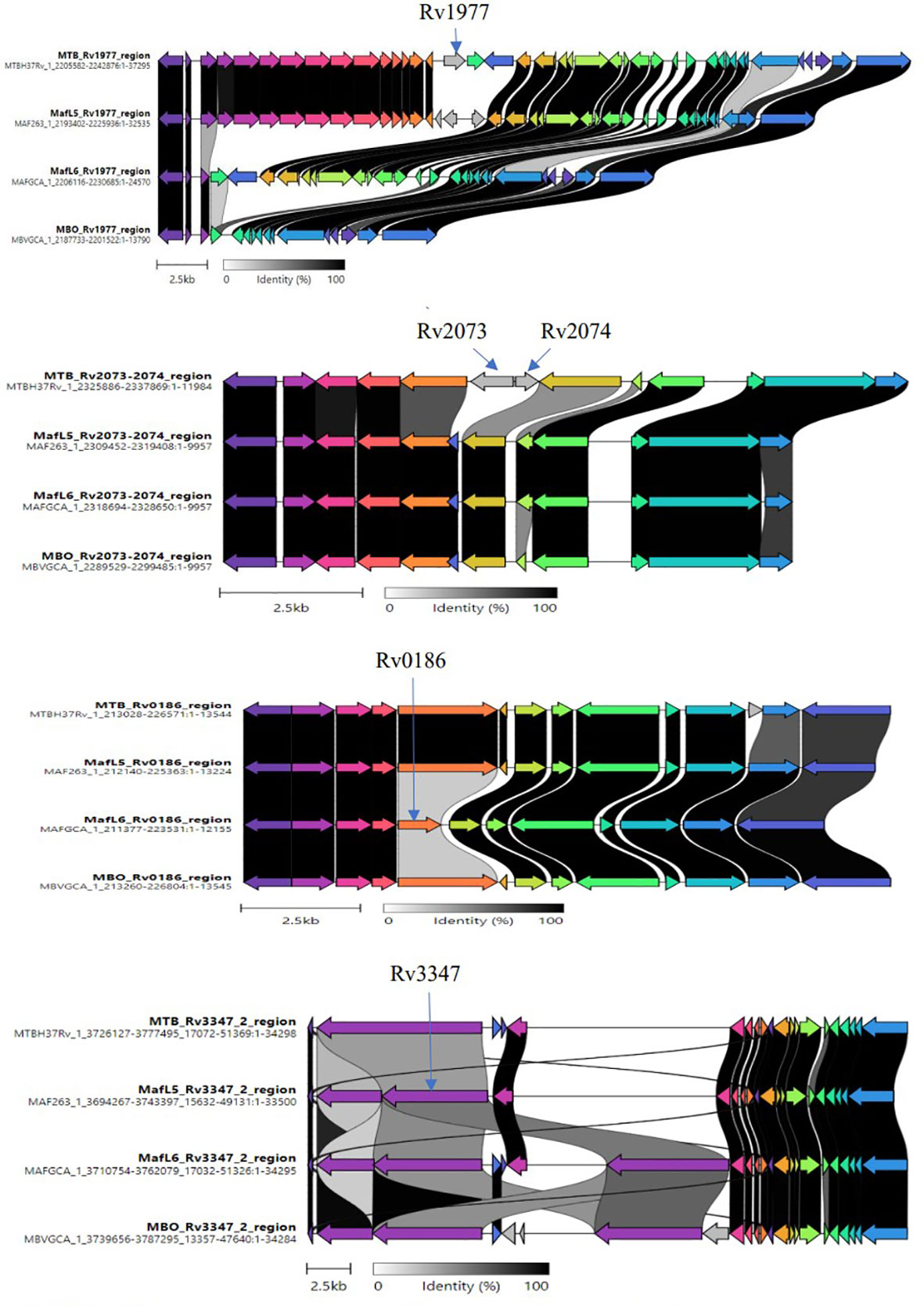
Figure 3 Clinker showing variations of lineage-specific genes via alignment of gene clusters. Variable regions of the identified unique genes were examined through the alignment of gene clusters from members of the MTBC (M. tuberculosis, M. africanum L5/L6, M. bovis).
Comparison of genomic regions by clinker
The uniqueness of lineage-specific genes was visualized by comparing gene clusters via Clinker software as shown in Figure 3. Variable regions of genes were observed to aid primers design.
Single PCR assays showing MTBC lineage-specificity
The primerQuest tool was used to design and assign all primers to different PCR product sizes for the purpose of differentiating the MTBCs in a multiplex PCR assay. Primers were screened and selected on the bases of sensitivity, specificity and compatibility. The M. tuberculosis specific primers designed from Rv1977, Rv2073c and Rv2074 produced single amplification products of 418 bp, 558 bp and 133 bp specifically in reactions with M. tuberculosis DNA and not with other members of the MTBC (Figures 4A–C). Primers to the Rv3347c gene unique to M. africanum L5 produced a product band size of 275 bp specifically in reaction with M. africanum L5 DNA (Figure 4D), while M. africanum L6- BgIS primers amplified a fragment of 381 bp specifically from M. africanum L6 DNA (Figure 4E). For M. bovis pncA primers designed by de los Monteros et al., 1998 were used and produced an M. bovis-specific amplicon of 186 bp (Figure 4F). The positive control primers (Rv3903c) were also assigned to 636 bp (Figure 4G).
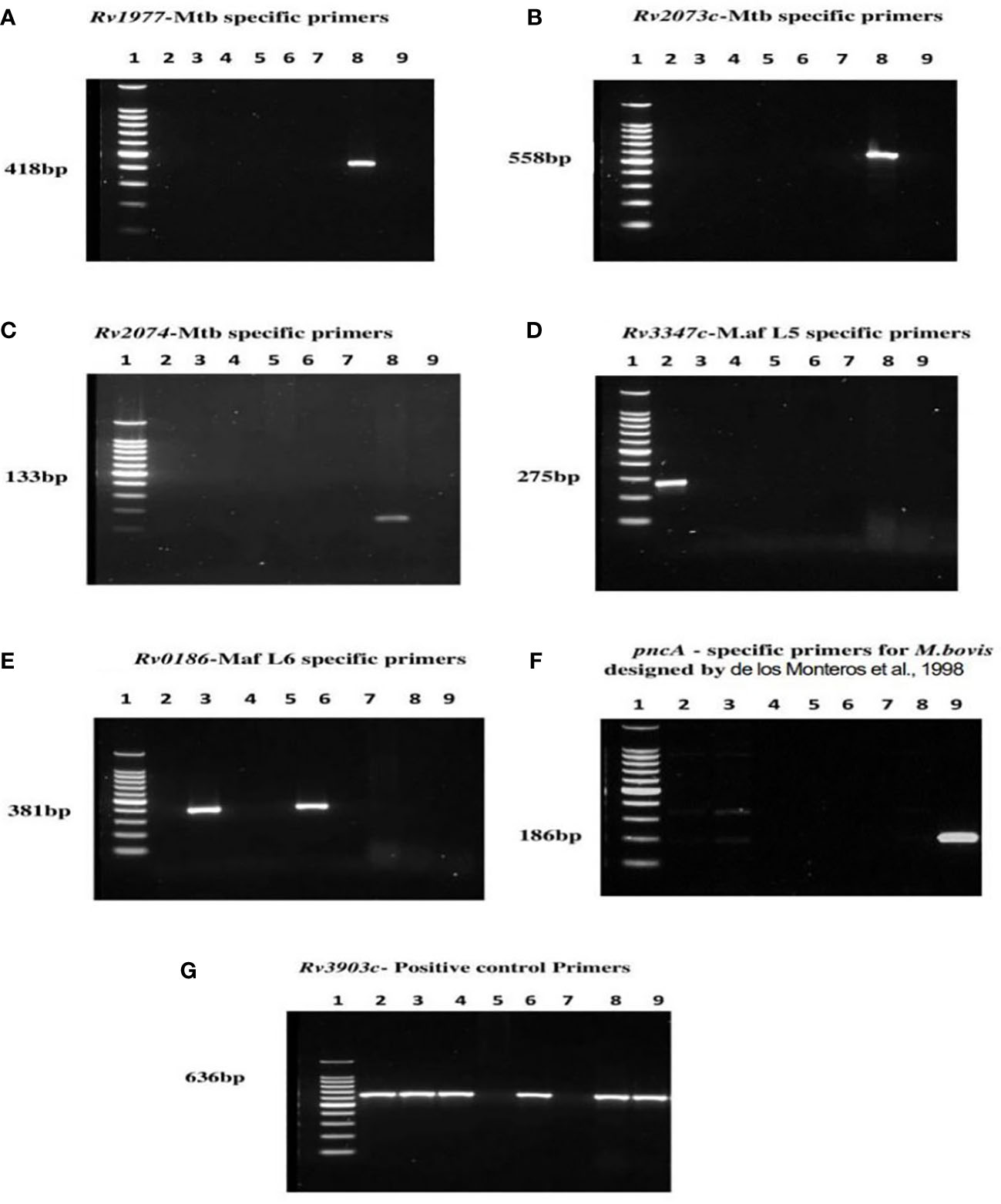
Figure 4 PCR results of MTBC specific lineages after optimization of various product sizes; Lane 1= DNA ladder, 2= M. africanum L5, 3= M. africanum L6 (isolate a), 4=BCG, 5= E. coli, 6= M. africanum L6 (isolate b), 7= water, 8= M. tuberculosis and 9= M. bovis. Each of the primers was tested against genomic DNA of all MTBCs for specificity. DNA of E. coli and nuclease-free water were used as negative and non-template controls respectively. Optimum separation of PCR products was achieved with 2% agarose gel at 80V, 1hr:30mins. Primers designed from genes; (A) Rv1977, (B) Rv2073c, and (C) Rv2074 amplifying at 418 bp, 558 bp and 133 bp respectively were specific for Mtb. The M. africanum L5 and M. africanum L6 primers were set at 275 bp and 381bp respectively as shown in (D, E). Primers developed from pncA gene by de los Monteros et al., 1998 were used for M. bovis identification (F) at 186 bp. The (G) Rv3903c primers at 636 bp served as positive control since it was conserved in all MTBCs.
Multiplex PCR assay differentiating MTBC
Two forms of multiplex PCR assays were performed in a 50 µl reaction for each: Multiplex primers tested on each DNA sample (Figure 5A) and an “All in one” reaction i.e., combination of all primers with mixture of all DNA samples (Figure 5B). All expected amplification products were observed without any extra products formations.
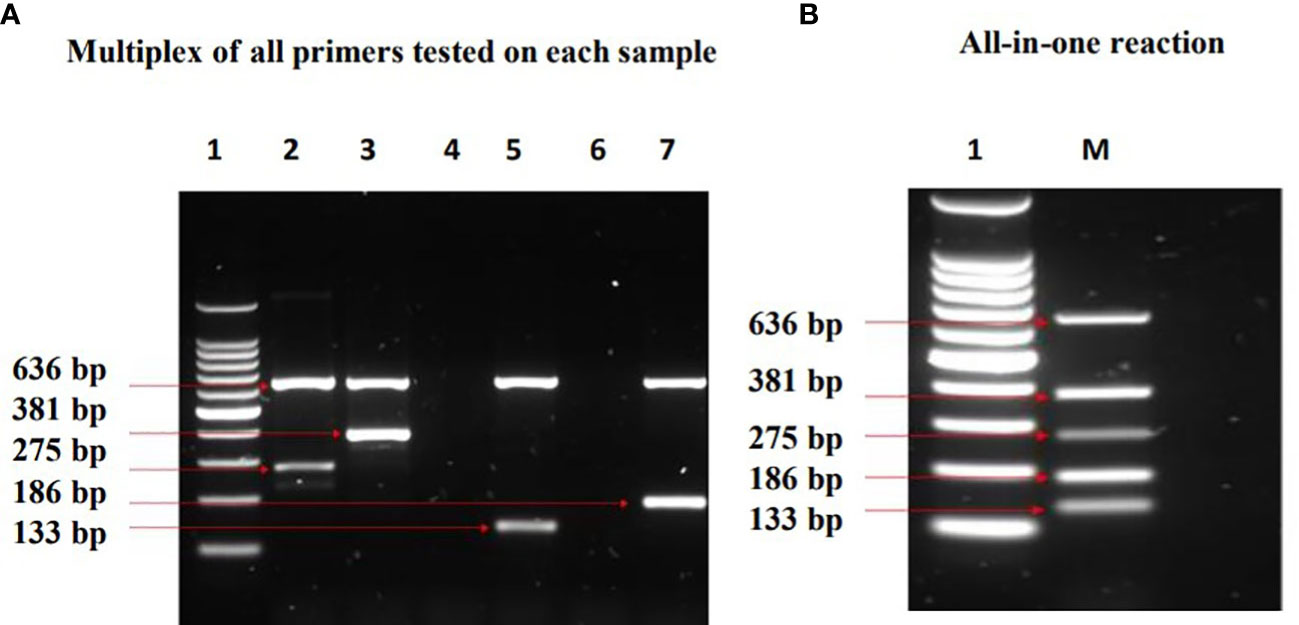
Figure 5 Results of Multiplex PCR assays which show identification of the MTBC lineages investigated. Lane 1= DNA ladder, 2= M. africanum L5, 3= M. africanum L6 (isolate a), 4= E. coli, 5= M. tuberculosis, 6= water, 7= M. bovis, M=mixture of all samples. In (A) multiplex of all primers tested on each DNA sample, two bands were observed in each of the MTBC as expected. The band at 636 bp (positive control) is conserved in all the MTBCs, whereas the other band depicts the respective lineage-specific band. Bands at 275 bp and 381 bp denote M. africanum L5 and L6 specificity respectively, while Mtb was represented at 133 bp. M. bovis was identified at 186 bp. In (B) All in one reaction; the compatibility and specificity of the assay was ascertained by combining all primers with mixtures of M. tuberculosis, M. africanum L5 & L6, and M. bovis. The outcome depicts a successful differentiation of the MTBCs without any inhibition.
Limits of detection (LOD) of PCR assays
PCR experiments were performed using the identified lineage-specific primers against their respective DNA samples to identify the least amount of DNA required for amplification. M. africanum L5, M. africanum L6 and M. bovis recorded LOD of 0.003 ng/µl which equates to 620 genome copy numbers, while M. tuberculosis was detected at 0.012 ng/µl or 2479 copy numbers as shown in Table 3.
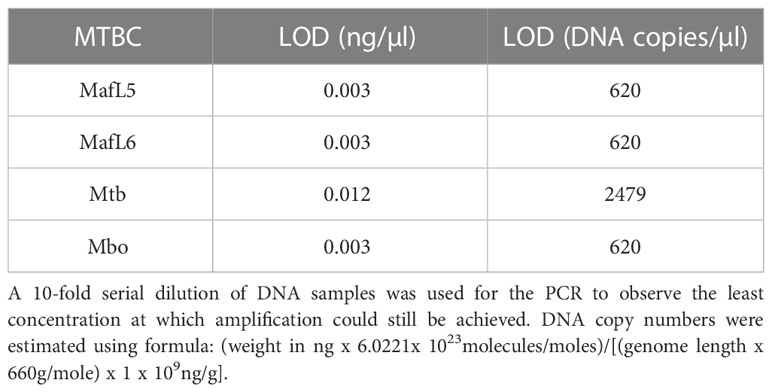
Table 3 Results showing LOD of PCR assays.
Specificity of MTBC primers against other pathogens
In view of misdiagnoses of tuberculosis with other respiratory pathogens such as a range of non-tuberculous mycobacteria (NTM) (Yılmaz et al., 2017), cross-reactivity experiments involving testing primers against other microorganisms was conducted. NTMs obtained from Reference Centre for Mycobacteria, Borstel-Germany were used for the cross-reactivity study. The MTBC primers did not show any cross reactivity since negative PCR test results were obtained against all non-MTBC DNAs (Supplementary Table S4). Furthermore, PCR test results of other respiratory pathogens comprising a cocktail of bacteria and viruses (22 targets) also recorded negative (Supplementary Table S5). Details of various bacterial and viral analytes are shown in Supplementary Table S3.
Validation of multiplex PCR assay using clinical samples
A total of 341 retrospective sputum samples from TB patients in Ghana were used for the validation of PCR assays. These samples have been confirmed TB positive using sputum smear microscopy, GeneXpert MTB/RIF assay and culture (BD BACTEC Mycobacterium Growth Indicator Tube- MGIT) methods based on previous studies conducted on TB drug resistance surveillance in Ghana (Sylverken et al., 2021). Sputum samples were decontaminated by treating with 4% N-Acetyl-L-Cysteine-Sodium-Hydroxide (NALC-NaOH) before neutralizing with 1X phosphate-buffered saline (PBS). DNA extraction was performed using the GenoLYSE extraction kit and followed by the multiplex PCR assay procedure described earlier. The results showed that M. tuberculosis contributes to a quarter (24.9%) of the cases, M. africanum L5 and M. africanum L6 were identified with 9.1% and 14.4% respectively, while M. bovis recorded only 1.8% of the cases. Interestingly, there was an observation of mixed-lineage TB infections at 5.9%. Also, 27.0% and 17.0% of the cases were PCR negative and unspeciated respectively, which may have been due to the extremely low concentration of DNA in some samples.
Discussion
The ability to differentiate between the lineages of the MBTC is very important in TB management because it provides reliable information for epidemiological surveillance and treatment choice. In this study, MTBC have been phylogenetically classified leading to the identification of lineage-specific genes. These lineage-specific genes have been explored for the development of a multiplex PCR assay which distinguishes between members of the MTBC.
In low resource regions, Ziehl-Neelsen acid-fast staining microscopy is the most common technique used to diagnose TB (Denkinger et al., 2013). It requires about 5,000 – 10,000 bacilli per ml of sputum for successful detection (Ausina Ruiz et al., 2013). Thus, its limitations are low sensitivity as well as the inability to differentiate between different mycobacterial species. Although culture, biochemical tests and sequencing are considered gold standard for identification and differentiation (Gholoobi et al., 2014), these are expensive, laborious and time-consuming. Additionally, obtaining results from these methods are sometimes unreliable due to difficulty in identification of some lineages (M. africanum lineages exhibit growth characteristics which are intermediates of M. tuberculosis and M. bovis) (de Jong et al., 2010a). The advent of genome sequencing techniques has provided relevant data for performing extensive genomic analyses. As a result, several molecular-based assays have been designed to detect MTBCs. These methods are highly sensitive and specific because unique gene sequences are targeted for amplification. Researchers have discovered gene markers such as IS6110, hsp65, dnaJ, psbA, lepA and MPT64 to detect MTBCs against other respiratory pathogens such NTMs (Chin et al., 2018). A recent multiplex PCR assay (Akwani et al., 2022) demonstrated successful separation of Mycobacterium abscessus complex subspecies from other NTMs as well as M. tuberculosis, although evaluation of assay performance in clinical samples needs to be carried out. Since 2010, WHO has recommended the use of GeneXpert MTB/RIF assay as the first-line diagnostic tool which detects MTBC together with rifampicin resistance (Goig et al., 2019). This is a molecular approach based on detection of the repetitive elements IS6110 and IS1081 and rifampicin resistance region. However, misdiagnoses of TB using the GeneXpert assay have been observed in NTM species at a high bacterial load (Pang et al., 2017). In TB endemic areas with infections caused by a diversity of MTBC species, a suitable differential diagnostic approach will be required since GeneXpert lacks the ability to distinguish between MTBC lineages.
In West Africa, MTBC classification has been achieved using spoligotyping technique which involves the amplification of direct repeat copies, followed by hybridization into intergenic spacers experiments (De Jong et al., 2009; de Jong et al., 2010b; Ofori-Anyinam et al., 2016; Otchere et al., 2018; Otchere et al., 2019). This is a two-step approach which is expensive, laborious and time-consuming. In Ghana, a single multiplex PCR experiment was conducted on the MTBC differentiation using primers from spacer regions 33 and 34 of the DR copies of MTBC, IS6110 and the hsp65 (Yeboah-Manu et al., 2001). Although this assay is not successful in separation of M. tuberculosis from M. africanum L6, the assay could still be used to complement biochemical testing.
However, the present study introduces a successful differentiation of MTBCs via a single multiplex PCR method which is rapid, cost effective and has a short turnaround time. The different PCR product sizes can be easily used to distinguish between lineages without the need for sequencing. One advantage of this PCR assay is the ease of adapting it to the available hardware as it will work on any PCR platform. This new assay provides a reliable solution to misdiagnoses with other NTM infections reported in some endemic regions (Brown-Elliott et al., 2012; Yılmaz et al., 2017; He et al., 2022). Indeed, our assay did not cross-react with a range of NTMs, respiratory bacterial and viral pathogens (Supplementary Tables S4, S5). We tested our assay using clinical samples (Table 4; Supplementary Figure S1) to demonstrate its utility at revealing the diversity of MTBC lineages in Ghana. The highest number of cases (24.9%) was caused by M. tuberculosis, followed by M. africanum L5 & L6 (23.5%). M. bovis recorded 1.8% which is comparable to 1.5% observed by Otchere et al., 2019. Negative PCR results (27.0%) and unspeciated lineages (17.0%) may require further confirmation via genome sequencing, although samples have been previously detected as MTBCs by liquid cultures, followed by confirmations using purity tests (on blood agar) and rapid test kit (TB cID) (Sylverken et al., 2021). However, since these are retrospective samples stored over time, sample integrity may have been compromised through repeated freeze/thaw cycles which were beyond our control. Following the reports of mixed MTBC infections among high TB burden settings (Van Rie et al., 2005; Huyen et al., 2012; Zetola et al., 2014), this study detected 20 (5.9%) cases of mixed-lineage TB infections. Poor treatment outcomes have been strongly associated with mixed-lineage TB infections (Zetola et al., 2014). Therefore, the effect of mixed-lineage TB infections in TB management cannot be overlooked as treatment failures are often observed in various regions of Ghana (Agyare et al., 2021).
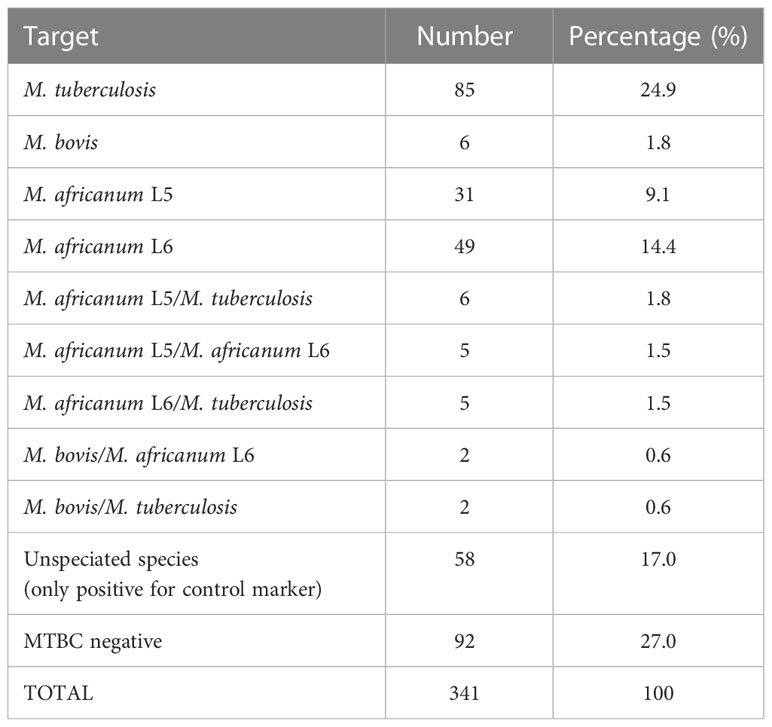
Table 4 Results of multiplex PCR validation using samples from 341 active TB patients in Ghana.
In summary, this assay is not an alternate replacement for GeneXpert which is currently the first-line TB diagnostic tool recommended by WHO. However, it will be beneficial to low-resource regions where TB is caused by diverse members of the MTBC providing rapid diagnosis to inform appropriate TB drug selection, reduce treatment relapse and the development of antimicrobial resistance. It will also be useful in epidemiological surveillance studies providing reliable information on TB lineage prevalence as well as identifying cases of mixed-lineage tuberculosis infections.
Data availability statement
The original contributions presented in the study are included in the article/Supplementary Material. Further inquiries can be directed to the corresponding authors.
Author contributions
The concept and study design were established by SH-W, AS, NR, and GS. Bioinformatic analyses was performed by AvV. Assay optimization by WO, WA, and SH-W. Assay validation using clinical samples was performed in Ghana by RA, SA, and AS. Manuscript writing and editing were done by WO, AS, AvV, GS, and SH-W. All authors contributed to the article and approved the submitted version.
Funding
This research was financially supported by the Vice Chancellor’s Studentship Award from the University of Surrey and the Global Challenges Research Fund.
Acknowledgments
We would like to say thank you to Reference Centre for Mycobacteria, Borstel-Germany for providing NTM strains used in the cross-reactivity experiment as well as the management of TB DRS Ghana project for supporting with clinical sputum samples. Thank you to BEI resources for providing the M. africanum DNA samples used in this study.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fcimb.2023.1125079/full#supplementary-material
References
Agyare, S. A., Osei, F. A., Odoom, S. F., Mensah, N. K., Amanor, E., Martyn-Dickens, C., et al. (2021). Treatment outcomes and associated factors in tuberculosis patients at atwima nwabiagya district, Ashanti region, Ghana: A ten-year retrospective study. Tuberc. Res. Treat. 2021, 1–9. doi: 10.1155/2021/9952806
Akwani, W. C., van Vliet, A. H. M., Joel, J. O., Andres, S., Diricks, M., Maurer, F. P., et al. (2022). The use of comparative genomic analysis for the development of subspecies-specific PCR assays for mycobacterium abscessus. Front. Cell. Infect. Microbiol. 12. doi: 10.3389/FCIMB.2022.816615/BIBTEX
Ali, M. H., Alrasheedy, A. A., Kibuule, D., Godman, B., Hassali, M. A. (2019). Assessment of multidrug-resistant tuberculosis (MDR-TB) treatment outcomes in sudan; findings and implications. Expert Rev. Anti Infect. Ther. 17, 927–937. doi: 10.1080/14787210.2019.1689818
Ausina Ruiz, V., Fernández-Rivas, G., Vilaplana Messeguer, C. (2013). Selected culture and drug-susceptibility testing methods for drug-resistant mycobacterium tuberculosis screening in resource-constrained settings. Expert Rev. Mol. Diagn. 13, 247–249. doi: 10.1586/erm.13.10
Belisle, J. T., Mahaffey, S. B., Hill, P. J. (2009). “Isolation of mycobacterium species genomic DNA,” in Mycobacteria protocols, 2nd ed. Eds. Parish, T., Brown, A. C. (Totowa, NJ: Humana Press), 1–12.
Brown-Elliott, B. A., Nash, K. A., Wallace, R. J. (2012). Antimicrobial susceptibility testing, drug resistance mechanisms, and therapy of infections with nontuberculous mycobacteria. Clin. Microbiol. Rev. 25, 545–582. doi: 10.1128/CMR.05030-11
Brynildsrud, O., Bohlin, J., Scheffer, L., Eldholm, V. (2016). Rapid scoring of genes in microbial pan-genome-wide association studies with scoary. Genome Biol. 17, 1–9. doi: 10.1186/S13059-016-1108-8/FIGURES/4
Chin, K. L., Sarmiento, M. E., Norazmi, M. N., Acosta, A. (2018). DNA Markers for tuberculosis diagnosis. Tuberculosis 113, 139–152. doi: 10.1016/j.tube.2018.09.008
Cohen, T., Wilson, D., Wallengren, K., Samuel, E. Y., Murray, M. (2011). Mixed-strain mycobacterium tuberculosis infections among patients dying in a hospital in KwaZulu-natal, south Africa. J. Clin. Microbiol. 49, 385. doi: 10.1128/JCM.01378-10
de Jong, B. C., Adetifa, I., Walther, B., Hill, P. C., Antonio, M., Ota, M., et al. (2010a). Differences between tuberculosis cases infected with Mycobacterium africanum , West African type 2, relative to Euro-American Mycobacterium tuberculosis: An update. FEMS Immunol. Med. Microbiol. 58, 102–105. doi: 10.1111/j.1574-695X.2009.00628.x
De Jong, B. C., Antonio, M., Awine, T., Ogungbemi, K., De Jong, Y. P., Gagneux, S., et al. (2009). Use of spoligotyping and large sequence polymorphisms to study the population structure of the mycobacterium tuberculosis complex in a cohort study of consecutive smear-positive tuberculosis cases in the Gambia. J. Clin. Microbiol. 47, 994–1001. doi: 10.1128/JCM.01216-08
de Jong, B. C., Antonio, M., Gagneux, S. (2010b). Mycobacterium africanum-review of an important cause of human tuberculosis in West Africa. PloS Negl. Trop. Dis. 4, e744. doi: 10.1371/journal.pntd.0000744
de los Monteros, L. E. E., Galán, J. C., Gutiérrez, M., Samper, S., García Marín, J. F., Martín, C, et al. (1998). Allele-specific PCR method based on pncA and oxyR sequences for distinguishing mycobacterium bovis from mycobacterium tuberculosis: Intraspecific m. bovis pncA sequence polymorphism. J. Clin. Microbiol. 36, 239–242. doi: 10.1128/JCM.36.1.239-242.1998
Denkinger, C. M., Kik, S. V., Pai, M. (2013). Robust, reliable and resilient: designing molecular tuberculosis tests for microscopy centers in developing countries. Expert Rev. Mol. Diagn. 13, 763–767. doi: 10.1586/14737159.2013.850034
Gholoobi, A., Masoudi-Kazemabad, A., Meshkat, Z. (2014). Comparison of culture and PCR methods for diagnosis of mycobacterium tuberculosis in different clinical specimens. Jundishapur. J. Microbiol. 7, 8939. doi: 10.5812/jjm.8939
Gilchrist, C. L. M., Chooi, Y. H. (2021). Clinker & clustermap.js: Automatic generation of gene cluster comparison figures. Bioinformatics 37, 2473–2475. doi: 10.1093/BIOINFORMATICS/BTAB007
Goig, G. A., Torres-Puente, M., Mariner-Llicer, C., Villamayor, L. M., Chiner-Oms, Á., Gil-Brusola, A., et al. (2019). Towards next-generation diagnostics for tuberculosis: identification of novel molecular targets by large-scale comparative genomics. Bioinformatics 36, 985–989. doi: 10.1093/bioinformatics/btz729
Gurevich, A., Saveliev, V., Vyahhi, N., Tesler, G. (2013). QUAST: quality assessment tool for genome assemblies. Bioinformatics 29, 1072–1075. doi: 10.1093/BIOINFORMATICS/BTT086
He, Y., Wang, J. L., Zhang, Y. A., Wang, M. S. (2022). Prevalence of culture-confirmed tuberculosis among patients with nontuberculous mycobacterial disease. Infect. Drug Resist. 15, 3097–3101. doi: 10.2147/IDR.S363765
Hingley-Wilson, S. M., Casey, R., Connell, D., Bremang, S., Evans, J. T., Hawkey, P. M., et al. (2013). Undetected multidrug-resistant tuberculosis amplified by first-line therapy in mixed infection. Emerg. Infect. Dis. 19, 1138–1141. doi: 10.3201/1907.130313
Huyen, M. N. T., Kremer, K., Lan, N. T. N., Cobelens, F. G. J., Buu, T. N., Dung, N. H., et al. (2012). Mixed tuberculosis infections in rural south Vietnam. J. Clin. Microbiol. 50, 1586–1592. doi: 10.1128/JCM.00434-12
Khalif Ali, M., Karanja, S., Karama, M., Kenyatta, J. (2017). Factors associated with tuberculosis treatment outcomes among tuberculosis patients attending tuberculosis treatment centres in 2016-2017 in Mogadishu, Somalia. Pan. Afr. Med. J. 28, 197. doi: 10.4314/pamj.v28i1
Lan, Z., Bastos, M., Menzies, D. (2016). Treatment of human disease due to mycobacterium bovis: A systematic review. Eur. Respir. J. 48, 1500–1503. doi: 10.1183/13993003.00629-2016
Mostowy, S., Onipede, A., Gagneux, S., Niemann, S., Kremer, K., Desmond, E. P., et al. (2004). Genomic analysis distinguishes mycobacterium africanum. J. Clin. Microbiol. 42, 3594–3599. doi: 10.1128/JCM.42.8.3594-3599.2004
Ofori-Anyinam, B., Kanuteh, F., Agbla, S. C., Adetifa, I., Okoi, C., Dolganov, G., et al. (2016). Impact of the mycobaterium africanum West Africa 2 lineage on TB diagnostics in West Africa: Decreased sensitivity of rapid identification tests in the Gambia. PloS Negl. Trop. Dis. 10, e0004801. doi: 10.1371/journal.pntd.0004801
Oryan, A., Yazdi, H. S., Alidadi, S., Doostmohammadi, S. (2022). Use of a gyrB PCR-RFLP method to diagnose tuberculosis and identify the causative mycobacterium sp. in cattle and humans. Comp. Immunol. Microbiol. Infect. Dis. 82, 101767. doi: 10.1016/J.CIMID.2022.101767
Otchere, I. D., Coscollá, M., Sánchez-Busó, L., Asante-Poku, A., Brites, D., Loiseau, C., et al. (2018). Comparative genomics of mycobacterium africanum lineage 5 and lineage 6 from Ghana suggests distinct ecological niches. Sci. Rep. 8, 1–11. doi: 10.1038/s41598-018-29620-2
Otchere, I. D., van Tonder, A. J., Asante-Poku, A., Sánchez-Busó, L., Coscollá, M., Osei-Wusu, S., et al. (2019). Molecular epidemiology and whole genome sequencing analysis of clinical mycobacterium bovis from Ghana. PloS One 14, e0209395. doi: 10.1371/journal.pone.0209395
Page, A. J., Cummins, C. A., Hunt, M., Wong, V. K., Reuter, S., Holden, M. T. G., et al. (2015). Roary: Rapid large-scale prokaryote pan genome analysis. Bioinformatics 31, 3691–3693. doi: 10.1093/BIOINFORMATICS/BTV421
Pang, Y., Lu, J., Su, B., Zheng, H., Zhao, Y. (2017). Misdiagnosis of tuberculosis associated with some species of nontuberculous mycobacteria by GeneXpert MTB/RIF assay. Infection 45, 677–681. doi: 10.1007/s15010-017-1044-x
Papastavros, T., Dolovich, L. R., Holbrook, A., Whitehead, L., Loeb, M. (2002). Adverse events associated with pyrazinamide and levofloxacin in the treatment of latent multidrug-resistant tuberculosis. Can. Med. Assoc. J. 167, 131.
Pornsukarom, S., van Vliet, A. H. M., Thakur, S. (2018). Whole genome sequencing analysis of multiple salmonella serovars provides insights into phylogenetic relatedness, antimicrobial resistance, and virulence markers across humans, food animals and agriculture environmental sources. BMC Genomics 19, 801. doi: 10.1186/s12864-018-5137-4
PrimerQuest Tool IDT. Available at: https://eu.idtdna.com/Primerquest/Home/Index (Accessed June 18, 2022).
Seemann, T. (2014). Prokka: Rapid prokaryotic genome annotation. Bioinformatics 30, 2068–2069. doi: 10.1093/BIOINFORMATICS/BTU153
SRA-Tools-NCBI (2021). Available at: https://ncbi.github.io/sra-tools/fastq-dump.html (Accessed June 22, 2021).
Sylverken, A. A., Kwarteng, A., Twumasi-Ankrah, S., Owusu, M., Arthur, R. A., Dumevi, R. M., et al. (2021). The burden of drug resistance tuberculosis in ghana; results of the first national survey. PloS One 16, e0252819. doi: 10.1371/JOURNAL.PONE.0252819
Van Rie, A., Victor, T. C., Richardson, M., Johnson, R., van der Spuy, G. D., Murray, E. J., et al. (2005). Reinfection and mixed infection cause changing mycobacterium tuberculosis drug-resistance patterns. Am. J. Respir. Crit. Care Med. 172, 636. doi: 10.1164/RCCM.200503-449OC
WHO (2020) Global tuberculosis report 2020: executive summary. Available at: http://apps.who.int/bookorders (Accessed January 5, 2021).
Yeboah-Manu, D., Yates, M. D., Wilson, S. M. (2001). Application of a simple multiplex PCR to aid in routine work of the mycobacterium reference laboratory. J. Clin. Microbiol. 39, 4166–4168. doi: 10.1128/JCM.39.11.4166-4168.2001
Yılmaz, N., Uçar, E. Y., Sağlam, L. (2017). Mycobacterium tuberculosis and nontuberculous mycobacteria coinfection of the lungs. Turkish Thorac. J. 18, 23. doi: 10.5152/TURKTHORACJ.2017.16034
Zetola, N. M., Shin, S. S., Tumedi, K. A., Moeti, K., Ncube, R., Nicol, M., et al. (2014). Mixed mycobacterium tuberculosis complex infections and false-negative results for rifampin resistance by GeneXpert MTB/RIF are associated with poor clinical outcomes. J. Clin. Microbiol. 52, 2422. doi: 10.1128/JCM.02489-13
Keywords: Mycobacterium tuberculosis complex, bioinformatic analyses, multiplex polymerase chain reaction, mixed-lineage tuberculosis infections, tuberculosis diagnosis, Ghana
Citation: Owusu W, van Vliet AHM, Riddell NE, Stewart G, Akwani WC, Aryeetey S, Arthur RA, Sylverken AA and Hingley-Wilson SM (2023) A multiplex PCR assay for the differentiation of Mycobacterium tuberculosis complex reveals high rates of mixed-lineage tuberculosis infections among patients in Ghana. Front. Cell. Infect. Microbiol. 13:1125079. doi: 10.3389/fcimb.2023.1125079
Received: 15 December 2022; Accepted: 17 March 2023;
Published: 03 April 2023.
Edited by:
Michael McClelland, University of California, Irvine, United StatesReviewed by:
Nadine Christine Lemaitre, Centre Hospitalier Universitaire (CHU) d’Amiens, FranceYolanda González Hernández, National Institute for Respiratory Diseases, Mexico
Copyright © 2023 Owusu, van Vliet, Riddell, Stewart, Akwani, Aryeetey, Arthur, Sylverken and Hingley-Wilson. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Suzanne M. Hingley-Wilson, cy5oaW5nbGV5LXdpbHNvbkBzdXJyZXkuYWMudWs=; Augustina Angelina Sylverken, YXN5bHZlcmtlbkBrbnVzdC5lZHUuZ2g=