
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Cell. Infect. Microbiol. , 27 February 2023
Sec. Molecular Bacterial Pathogenesis
Volume 13 - 2023 | https://doi.org/10.3389/fcimb.2023.1113992
This article is part of the Research Topic New Insights on the Transmission and Pathogenicity of rickettsiae View all 8 articles
 Jie Zhang1
Jie Zhang1 Yuan-Chun Zheng2Yan-Li Chu2Xiao-Ming Cui1Ran Wei3Cai Bian2Hong-Bo Liu4Nan-Nan Yao2
Yuan-Chun Zheng2Yan-Li Chu2Xiao-Ming Cui1Ran Wei3Cai Bian2Hong-Bo Liu4Nan-Nan Yao2 Rui-Ruo Jiang5Qiu-Bo Huo2Ting-Ting Yuan6Jie Li1Lin Zhao7Lian-Feng Li7Qian Wang1Wei Wei1Jin-Guo Zhu8Mei-Chao Chen8Yan Gao8Fei Wang8Jin-Ling Ye2Ju-Liang Song2Jia-Fu Jiang1
Rui-Ruo Jiang5Qiu-Bo Huo2Ting-Ting Yuan6Jie Li1Lin Zhao7Lian-Feng Li7Qian Wang1Wei Wei1Jin-Guo Zhu8Mei-Chao Chen8Yan Gao8Fei Wang8Jin-Ling Ye2Ju-Liang Song2Jia-Fu Jiang1 Tommy Tsan-Yuk Lam9*Xue-Bing Ni9*
Tommy Tsan-Yuk Lam9*Xue-Bing Ni9* Na Jia1*
Na Jia1*Introduction: Ticks are the most important obligate blood-feeding vectors of human pathogens. With the advance of high-throughput sequencing, more and more bacterial community and virome in tick has been reported, which seems to pose a great threat to people.
Methods: A total of 14 skin specimens collected from tick-bite patients with mild to severe symptoms were analyzed through meta-transcriptomic sequencings.
Results: Four bacteria genera were both detected in the skins and ticks, including Pseudomonas, Acinetobacter, Corynebacterium and Propionibacterium, and three tick-associated viruses, Jingmen tick virus (JMTV), Bole tick virus 4 (BLTV4) and Deer tick mononegavirales-like virus (DTMV) were identified in the skin samples. Except of known pathogens such as pathogenic rickettsia, Coxiella burnetii and JMTV, we suggest Roseomonas cervicalis and BLTV4 as potential new agents amplified in the skins and then disseminated into the blood. As early as 1 day after a tick-bite, these pathogens can transmit to skins and at most four ones can co-infect in skins.
Discussion: Advances in sequencing technologies have revealed that the diversity of tick microbiome and virome goes far beyond our previous understanding. This report not only identifies three new potential pathogens in humans but also shows that the skin barrier is vital in preventing horizontal transmissions of tick-associated bacteria or virus communities to the host. It is the first research on patients’ skin infectome after a tick bite and demonstrates that more attention should be paid to the cutaneous response to prevent tick-borne illness.
Ticks are the most important vectors of human pathogens, causing increasing public health burdens worldwide. Ticks can cause a variety of diseases, such as tick-borne viral illness, Lyme disease, spotted fever group rickettsiosis, human anaplasmosis, and human babesiosis (Jia et al., 2020b). Ticks transmit infectious agents through continuous attachment to the host’s skin and blood feeding for days. The skin plays a key role in vector-borne diseases because it is the site where the arthropod co-inoculates pathogens and its saliva. It is becoming increasingly evident that the skin is not simply a physical barrier, but is also an organ important in innate immunity, as the first barrier met by the tick and the pathogen (Wikel, 2013; Boulanger and Wikel, 2021). The cutaneous response contributes to the tick-borne pathogen multiplying and sometimes persisting before disseminating, as in Lyme borreliosis (Bernard et al., 2020), spotted fever rickettsiosis (Heinze et al., 2014; Jia et al., 2020a), and Jingmen tick virus infection (Jia et al., 2019). Although various pathogens have been detected in skin lesions, the skin infectome after a tick bite remains largely unknown. This knowledge will undoubtedly provide a deep understanding of the risks presented by tick bites in humans.
In recent years, next-generation sequencing has been successfully used to characterize the complete tick microbiome, identify novel and unexpected pathogens, and elucidate tick microbiome–host interactions, with the goal of developing antivectorial control measures (Carpi et al., 2011; Hawlena et al., 2013; Nakao et al., 2013; Williams-Newkirk et al., 2014). We collected 14 skin lesions from patients in a sentinel hospital who had a history of a tick bite. The microbiome and virome of skin biopsy specimens were identified utilizing a meta-transcriptomics approach, and pathogens that could potentially be transmitted from skin to blood were analyzed, providing valuable data illustrating the horizontal transmission of pathogens from ticks to humans.
Patients who had been bitten by ticks and sought medical treatment at Mudanjiang Forestry Central Hospital in the Heilongjiang Province of China were included in this study. Ticks were aseptically removed from the patient’s body, and an area of skin tissue measuring 1 cm × 1 cm was excised from the site of the tick bite in a sterile operation conducted by hospital surgeons, and then stored in RNase-free microfuge tubes at –80°C. Venous blood samples of volume 2 mL were collected into vacuum blood collection tubes. The medical information of each patient was recorded with their consent.
All participants provided written informed consent for this study, which was approved by the Mudanjiang Forestry Central Hospital Review Board and the Academy of Military Medical Sciences Review Board, China.
Total RNA extraction was performed using an AllPrep DNA/RNA Mini Kit (lot no. 80204; Qiagen, USA), with modifications. Briefly, tick and skin samples were quickly thawed and homogenized in an RLT solution in liquid nitrogen. The tick homogenate, skin homogenate, and blood samples were incubated at 55°C for 10 min with proteinase K (Qiagen) and centrifuged for 30 s at 12,000 × g. The homogenized lysate was transferred to an AllPrep DNA spin column and centrifuged for 30s at 8,000 × g. The flowthrough and the AllPrep RNA spin column were used for further RNA purification, in accordance with the manufacturer’s instructions.
The extracted RNA was used for transcriptome sequencing (RNA-seq) after RNA quantification and qualification (Thermo Scientific NanoDrop2000). The rRNA was removed using RiBo-Zero Gold rRNA Removal Reagents (human/mouse/rat) (Illumina), then the sequencing library was prepared following Illumina’s standard protocol (Modi et al., 2021). Paired-end (2 × 150-bp) sequencing of the RNA library was performed on an Illumina HiSeq 4000 platform at Novogene Tech (Beijing, China).
RNA sequencing reads were assembled de novo using the Trinity program (Grabherr et al., 2011). Assembled contigs were subject to Basic Local Alignment Search Tool—Nucleotide (BLASTn) analysis and compared with a non-redundant nucleotide (nt) database and to BLASTx analysis and compared with all non-redundant protein (nr) databases downloaded from GenBank, and the threshold E-value was set to 1e–5. The contigs that best matched the virus were kept for downstream analysis. Putative viral contigs were further merged by high-identity overlaps using the SeqMan program of Lasergene package v.7.1 (DNASTAR, Madison, WI, USA). Original reads were aligned to the contigs again using Bowtie2 (Langmead and Salzberg, 2012) to close the remaining gaps and verify the assembly in the Integrated Genomics Viewer (Thorvaldsdóttir et al., 2013).
All host reads were first removed by mapping against Homo sapiens (NW_012132914.1) using Bowtie2 (Langmead and Salzberg, 2012). The remaining reads were analyzed against the SILVA 16s rRNA database (https://www.arb-silva.de/) using BLASTn with a threshold E-value of < 1e–5. After annotation, the Ribosome Database Project (RDP) taxonomic database was selected for taxonomic assignment. The bacterial read counts were summarized at genus level, and histograms were plotted to visualize the results. The contigs of pathogenic bacteria were identified through BLASTn analysis based on the criterion that more than 80% of base pairs aligned to the reference sequence with similarity higher than 85% (nucleotides) or 90% (amino acids). Read counts were estimated by mapping reads to the reference genome of the same species as classified above using Bowtie2 (Langmead and Salzberg, 2012).
Nucleotide alignments were prepared with the reference sequences at the species level for pathogenic bacteria based on the 16s rRNA gene and for the virus based on the RNA-dependent RNA polymerase (RdRp) gene using the E-INS-i algorithm in MAFFT (multiple alignment using fast Fourier transformation), version 7 (Katoh and Standley, 2013). Ambiguously aligned regions were removed using trimAl (Capella-Gutiérrez et al., 2009). The GTR+I+G model was identified to be the best-fit nucleotide substitution model using ProtTest 3.4 (Darriba et al., 2011), which was used for maximum likelihood (ML) phylogeny reconstruction with bootstrap tests (1,000 replicates) in PhyML v.3.0 (Guindon and Gascuel, 2003). The ML trees were visualized with FigTree v.1.4.2.
The above filtered non-host reads were subject to GhostKOALA (https://www.kegg.jp/ghostkoala/) against both the prokaryotic Kyoto Encyclopedia of Genes and Genomes (KEGG) genes database at the genus level and the eukaryotic KEGG genes database at the family level using CD-HIT (Cluster Database at High Identity with Tolerance) clusters with 50% identity cut-off value.
A total of 14 patients with a tick bite were enrolled in this study. Nine patients saw a doctor while the tick was still on their bodies (four Ixodes persulcatus, four Dermacentor silvarum, and one Haemaphysalis concinna). The ticks were most likely to be found on the scalp (4/9) or the back (2/9). The interval between the tick biting and sample collection ranged from 1 to 30 days, with a median of 8 days. The necrotic cells were distributed in the epidermis of eschars, while inflammatory infiltration by lymphocytes was identified in some skin lesions by hematoxylin and eosin (HE) staining (Figure S1). The clinical symptoms varied from mild to severe and included local itching, headache, rash, lymphadenectasis, and fever, with laboratory tests showing that some patients had an abnormal liver function and neutropenia (Table 1).
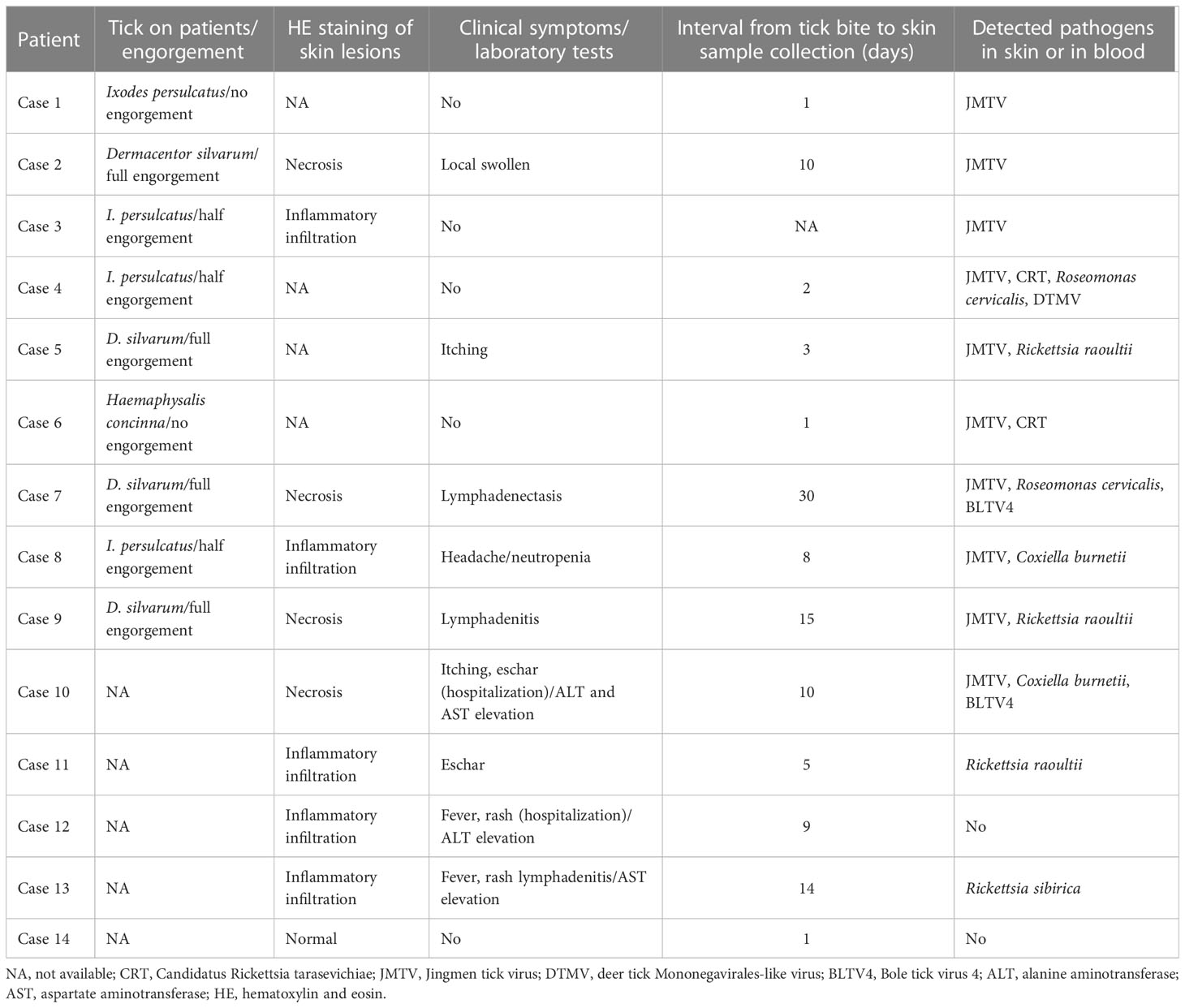
Table 1 The tick bite history and clinical manifestation of patients enrolled in the study.
The predominant bacterial genus isolated from the skin around a tick bite was in most cases Staphylococcus, but in four cases (i.e., cases 1, 4, 6, and 12) it was Lactobacillus, Sphigobacterium, or Mycoplasma (Figure 1A). Examining the microbiome in ticks detached from the patients (Figure S2) revealed that four bacterial genera were common to the skin and tick: Pseudomonas, Acinetobacter, Corynebacterium, and Propionibacterium.
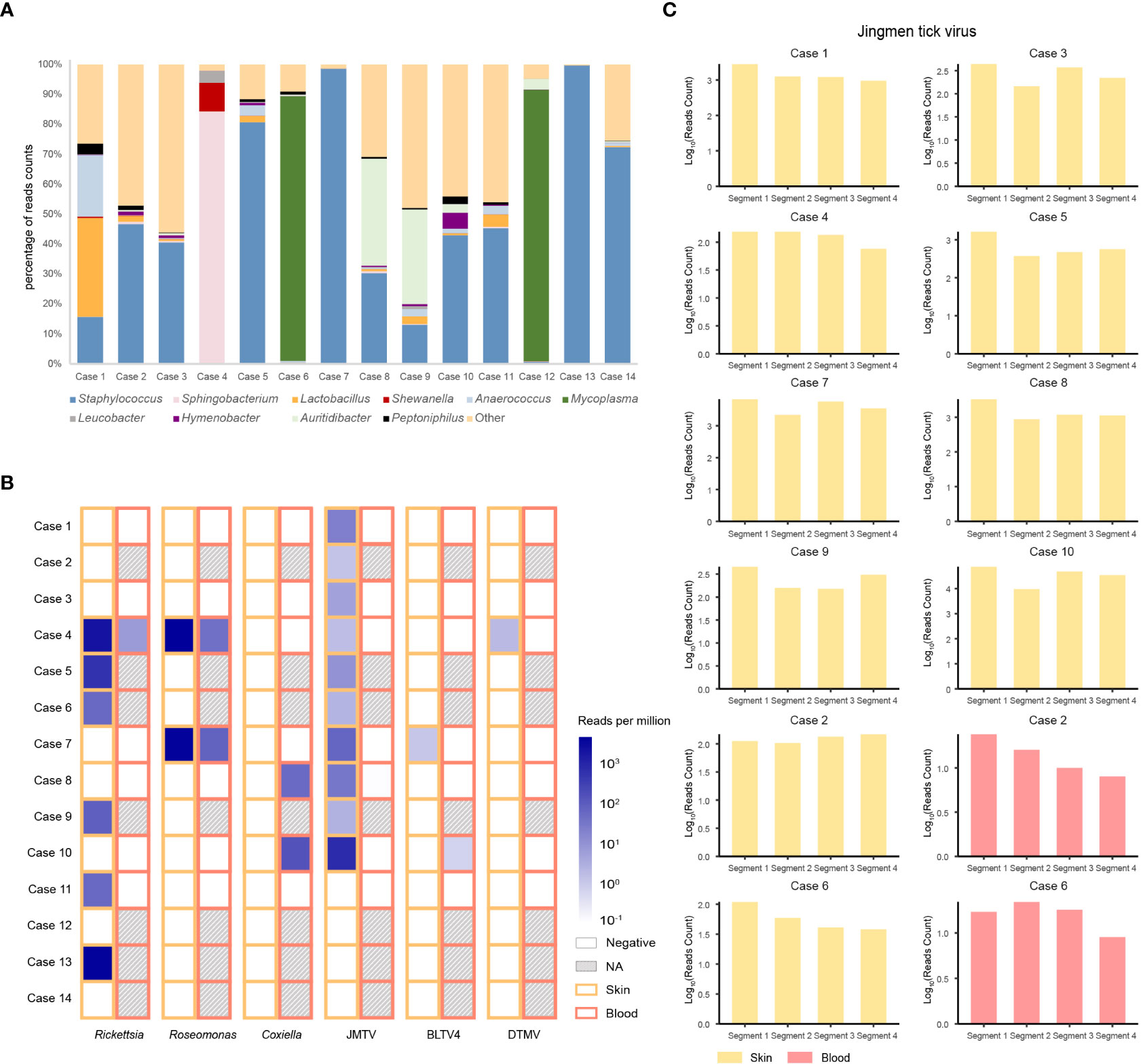
Figure 1 Bacteria and viruses identified in skin samples from 14 patients with a tick bite as determined by metatranscriptomic sequencing. (A) Percentage of bacterial read counts in the skin samples after a tick bite. (B) Comparisons of the bacterial and viral read counts of six tick-borne pathogens in skin and blood samples. (C) Viral abundance of the Jingmen tick virus by viral segments in the positive skin and blood samples.
We then focused on pathogenic bacteria and found that Coxiella burnetii, Candidatus Rickettsia tarasevichiae (CRT), Rickettsia raoultii, Rickettsia sibirica, and Roseomonas cervicalis could be detected among tick bite patients’ skin and blood samples by RNA-seq (Table 1, Figure 1B). The 16s rRNA gene sequences of the above pathogens showed, respectively, 87.3%–87.5%, 93.8%–99.9%, 92.5%–98.2%, 99.7%–100%, and 88.5%–90.0% Nucleotide (nt) similarity with the reference strains (Table S1). Interestingly, Roseomonas could be identified in both skin and blood, whereas Coxiella was detected only in blood samples (cases 8 and 10; Figure 1B). Bacterial contigs were successfully assembled from case 4, who was co-infected with CRT and R. cervicalis, and were used for constructing phylogenic trees. The sequences fell into the clade with R. cervicalis, or with CRT, respectively (Figure 2). The rickettsial pathogens could be horizontally transmitted from ticks to skin as early as 1 day after a tick bite and could be amplified for at least 15 days. The interval between tick bite and skin lesion collection was 2–30 days for R. cervicalis infections (Table 1).
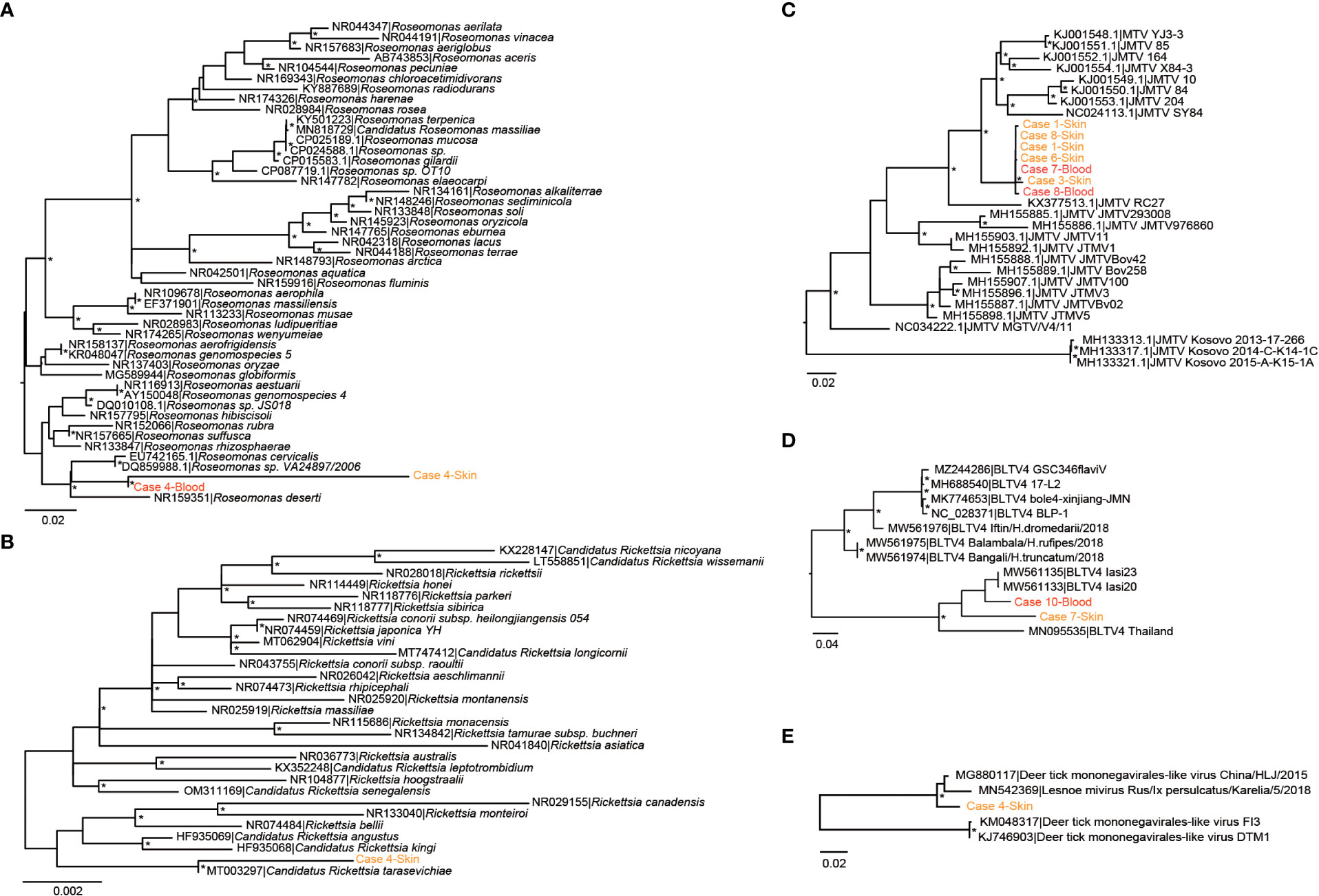
Figure 2 Phylogenetic analyses of (potential) pathogens detected in patients in this study. (A) Roseomonas, (B) Rickettsia, (C) Jingmen tick virus, (D) Bole tick virus 4, and (E) deer tick Mononegavirales-like virus. The symbol * mean the value of bootstrap for this branch >70.
The KEGG pathways of the microbiomes on patients’ skin and blood were analysed to understand the microbiome response. In general, microbial communities in the skin showed higher activity than those in the blood, owing to the fact most patients did not have bacteremia. The metabolic pathways of the microbiome were mostly activated, leading to the biosynthesis of secondary metabolites and microbial metabolism in diverse environments. However, the cofactor and vitamin metabolic pathways, as well as the immune system of the microbiome, were activated to a lesser extent.
Three viruses, Jingmen tick virus (JMTV), Bole tick virus 4 (BLTV4), and deer tick Mononegavirales-like virus (DTMV), could be detected in the skin, whereas JMTV and BLTV4 could also be detected in blood (Figure 1B).
Segment 1 of JMTV, encoding the NS5-like protein with the conserved motifs for methyltransferase and RNA-dependent RNA polymerase, was found in nine skin samples and was the most abundant segment, followed by segment 3 of NS2b–NS3-like protein, which relates to the serine protease domain and helicase domain, and was found in seven patients (Figure 1C). Segments 2 and 4, which encode structural proteins, were less abundant (Figure 1C). The viral abundance of segments of JMTV varied between two blood samples (Figure 1C). JMTV isolated from patients showed 94.9%–100% nt similarity in the RdRp gene, with ticks collected in China, and formed a separate clade with other strains detected from ticks (Table S1, Figure 2). BLTV4 belongs to the unclassified Riboviria, and has not previously been reported in skin or blood from patients with a tick bite history. BLTV4 in the skin (case 7) and in the blood (case 10) showed 92.4% and 90.9% nt similarity, respectively, to strains previously identified from Dermacentor reticulatus in Romania (Table S1, Figure 2). DTMV is an unclassified virus and has not yet been reported in samples from patients. DTMV was detected from case 4’s skin sample, with an abundance of 257 reads, showing 99.3% nt similarity to DTMV isolate China/HLJ/2015 (Table S1, Figure 2). This reference strain in the phylogenic tree was identified in I. persulcatus from Heilongjiang province in China, where our patients were living.
We noticed that JMTV and DTMV could be transmitted to patients’ skin as soon as 1 or 2 days after a tick bite, and BLTV4 could be detected in the skin until 30 days after a tick bite (Table 1). Co-infections with tick-borne viruses and bacteria in the skin were found in half of the patients, and up to four pathogens could be detected in one skin lesion specimen.
Advances in sequencing technologies have revealed that the diversity of tick microbiome and virome goes far beyond our previous understanding (Li et al., 2021; Narasimhan et al., 2021); it is, therefore, interesting to characterize the host skin infectome after a tick feeds. Transcriptome analysis of skin lesions in patients with a clear tick bite history revealed that four bacterial genera, Pseudomonas, Acinetobacter, Corynebacterium, and Propionibacterium, were common in ticks and patients’ skin. However, we cannot determine whether the detected bacteria were in the tick or on the tick, or whether pathogens or bacteria were transmitted from the tick’s surface. In addition, excluding known bacterial pathogens, such as the spotted fever group rickettsia C. burnetii (Huang et al., 2021; Fu et al., 2022), a potentially new pathogen, R. cervicalis, was detected in both the blood and skin samples of patients. Although more than 10 viral families were found in ticks by RNA-seq in our previous study (Li et al., 2021), only three viruses (i.e., JMTV, BLTV4, and DTMV) could be detected in skin biopsy specimens. This report not only identifies three new potential pathogens in humans but also shows that the skin barrier is vital in preventing horizontal transmissions of tick-associated bacteria or virus communities to the host.
The co-transmission of vector microbiomes and vector-borne pathogens has been previously suspected in insects (Finney et al., 2015). However, regarding the transmission of tick-borne pathogens, very few studies have been performed to investigate the potential transmission of microbiota during the process of a tick bite. In addition, very few studies have investigated the role of skin microbiota in pathogen transmission at the skin interface. At the skin interface, the microbiome also contributes to the regulation of inflammation and is likely to affect the host’s responses to the tick bite. The skin might play a key role in the process of tolerance as the first interface met by the tick and the pathogen (Bernard et al., 2020). Our study also implies that the duration of tick blood feeding is important for the successful passage of an infectious agent into the bite site and the subsequent establishment of infection, although a larger sample size is required to confirm our results. Furthermore, this observation has practical implications for disease prevention.
Rosemonas is thought to be opportunistically pathogenic to humans, particularly immunocompromised patients with underlying diseases such as acute leukemia, cancer, or rheumatoid arthritis (Dé et al., 2004). A previous study found that Rosemonas sp. (R. cervicalis) could be isolated from adult Dermacentor nuttalli and its larval progeny, and obtained evidence of transovarial transmission (Liu et al., 2010). This study further demonstrates the role of the tick in transmitting R. cervicalis to humans.
Tick-associated DTMV was also detected in the skin of case 4, but not from the same patient’s blood sample. It showed high similarity with DTMV strain China/HLJ/2015, which was identified in I. persulcatus in the same province as our patients, and with Lesnoe mivirus, which is from the same tick species but is found in Russia. Another horizontally transmitted tick virus is BLTV4. It is particularly noteworthy that we detected BLTV4 in both skin and blood samples from two patients. BLTV4 was first identified in a metaviromic study carried out in China in 2012 and was subsequently reported in Hyalomma asiaticum, Hyalomma truncatum, Dermacentor reticulatus, and Rhipicephalus microplus collected in Romania and Kenya (Shi et al., 2016; Zhang et al., 2021). We cannot provide evidence of the pathogenicity of these viruses; however, considering their wide distribution in terms of both tick species and geographic location, we stress the importance of continuous active surveillance of patients with a tick bite history.
JMTV was first identified in R. microplus ticks sampled from Jingmen in Hubei Province and Wenzhou in Zhejiang Province, China, in 2010 (Qin et al., 2014). The genome of JMTV comprises four segments of single-stranded positive-sense RNA. Two segments exhibit homology with the non-structural protein NS3 and NS5 sequences of flaviviruses, whereas the other two segments are unique and have no known homologs (Qin et al., 2014). Since its initial identification, JMTV has been identified in several regions of China as well as in Brazil, Kosovo, Uganda, Turkey, the French Antilles, and Trinidad and Tobago (Ladner et al., 2016; Emmerich et al., 2018; Souza et al., 2018; Dinçer et al., 2019; Pascoal et al., 2019; Sameroff et al., 2019; Temmam et al., 2019; Guo et al., 2020). JMTV can infect a broad range of animal hosts, including cattle, rodents, bats, and primates (Souza et al., 2018; Sameroff et al., 2019; Guo et al., 2020). It has been reported as the causative agent of febrile disease in China and Kosovo (Emmerich et al., 2018; Jia et al., 2019). In our study, among 10 patients with JMTV infection, seven had been co-infected with either bacteria or other viruses in the skin. More in-depth laboratory studies and long-term epidemiological studies are needed to clarify the pathogenicity of JMTV in humans.
We found that some pathogenic bacteria could infest patients very quickly after a tick bite, as exemplified by CRT and R. cervicalis (e.g., cases 4 and 6). However, it appears that viruses took several days (i.e., 8–30 days) to successfully cause viremia (e.g., JMTV and BLTV4 in cases 7, 8, and 10). This may be related to the difference in the abundance of bacteria and viruses in ticks, as well as to differences in humans’ immune responses to bacteria and viruses. These are possible directions for future study.
This work is limited by the small sample size. In addition, the pathogenicity of new agents deserves further verification. The detection of seroconversion or a fourfold increase in antibodies in reaction to these bacteria and viruses would be useful to clarify their pathogenicity. Nevertheless, this work is the first research on patients’ skin infectome after a tick bite and demonstrates that more attention should be paid to the cutaneous response to prevent tick-borne illness.
The data presented in the study are deposited in the Gene Expression Omnibus data repository, accession number GSE141235.
The studies involving human participants were reviewed and approved by Mudanjiang Forestry Central Hospital. The patients/participants provided their written informed consent to participate in this study.
NJ contributed to the conception and design of the study. XBN performed the statistical analysis. NJ, X-BN, and T-YL reviewed the draft. JZ wrote the first draft of the manuscript. X-MC, R-RJ, and RW investigated the hospital. X-MC, J-FJ, J-LS, J-LY, FW, YG, M-CC, J-GZ, WW, JL, T-TY, Q-BH, N-NY, CB, Y-LC, and Y-CZ collected the samples. H-BL and LZ contributed to the methodology. L-FL organized the database. QW contributed to the data curation. All authors contributed to the article and approved the submitted version.
This study was supported by the Natural Science Foundation of China (81773492).
We thank all staff members at Mudanjiang Forestry Central Hospital, Mudanjiang, China, who helped with sample and medical records collection.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fcimb.2023.1113992/full#supplementary-material
Bernard, Q., Grillon, A., Lenormand, C., Ehret-Sabatier, L., Boulanger, N. (2020). Skin interface, a key player for borrelia multiplication and persistence in Lyme borreliosis. Trends Parasitol. 36, 304–314. doi: 10.1016/j.pt.2019.12.017
Boulanger, N., Wikel, S. (2021). Induced transient immune tolerance in ticks and vertebrate host: A keystone of tick-borne diseases? Front. Immunol. 12. doi: 10.3389/fimmu.2021.625993
Capella-Gutiérrez, S., Silla-Martínez, J. M., Gabaldón, T. (2009). trimAl: a tool for automated alignment trimming in large-scale phylogenetic analyses. Bioinformatics 25, 1972–1973. doi: 10.1093/bioinformatics/btp348
Carpi, G., Cagnacci, F., Wittekindt, N. E., Zhao, F., Qi, J., Tomsho, L. P., et al. (2011). Metagenomic profile of the bacterial communities associated with ixodes ricinus ticks. PloS One 6, e25604. doi: 10.1371/journal.pone.0025604
Darriba, D., Taboada, G. L., Doallo, R., Posada, D. (2011). ProtTest 3: fast selection of best-fit models of protein evolution. Bioinformatics 27, 1164–1165. doi: 10.1093/bioinformatics/btr088
Dé, I., Rolston, K. V., Han, X. Y. (2004). Clinical significance of roseomonas species isolated from catheter and blood samples: analysis of 36 cases in patients with cancer. Clin. Infect. Dis. 38, 1579–1584. doi: 10.1086/420824
Dinçer, E., Hacıoğlu, S., Kar, S., Emanet, N., Brinkmann, A., Nitsche, A., et al. (2019). Survey and characterization of jingmen tick virus variants. Viruses 11, 1071. doi: 10.3390/v11111071
Emmerich, P., Jakupi, X., Von Possel, R., Berisha, L., Halili, B., Günther, S., et al. (2018). Viral metagenomics, genetic and evolutionary characteristics of Crimean-Congo hemorrhagic fever orthonairovirus in humans, Kosovo. Infect. Genet. Evol. 65, 6–11. doi: 10.1016/j.meegid.2018.07.010
Finney, C. A., Kamhawi, S., Wasmuth, J. D. (2015). Does the arthropod microbiota impact the establishment of vector-borne diseases in mammalian hosts? PloS Pathog. 11, e1004646. doi: 10.1371/journal.ppat.1004646
Fu, M., Liu, Y., Wang, G., Wang, P., Zhang, J., Chen, C., et al. (2022). A protein-protein interaction map reveals that the coxiella burnetii effector CirB inhibits host proteasome activity. PloS Pathog. 18, e1010660. doi: 10.1371/journal.ppat.1010660
Grabherr, M. G., Haas, B. J., Yassour, M., Levin, J. Z., Thompson, D. A., Amit, I., et al. (2011). Full-length transcriptome assembly from RNA-seq data without a reference genome. Nat. Biotechnol. 29, 644–652. doi: 10.1038/nbt.1883
Guindon, S., Gascuel, O. (2003). A simple, fast, and accurate algorithm to estimate large phylogenies by maximum likelihood. Syst. Biol. 52, 696–704. doi: 10.1080/10635150390235520
Guo, J. J., Lin, X. D., Chen, Y. M., Hao, Z. Y., Wang, Z. X., Yu, Z. M., et al. (2020). Diversity and circulation of jingmen tick virus in ticks and mammals. Virus Evol. 6, veaa051. doi: 10.1093/ve/veaa051
Hawlena, H., Rynkiewicz, E., Toh, E., Alfred, A., Durden, L. A., Hastriter, M. W., et al. (2013). The arthropod, but not the vertebrate host or its environment, dictates bacterial community composition of fleas and ticks. Isme J. 7, 221–223. doi: 10.1038/ismej.2012.71
Heinze, D. M., Carmical, J. R., Aronson, J. F., Alarcon-Chaidez, F., Wikel, S., Thangamani, S. (2014). Murine cutaneous responses to the rocky mountain spotted fever vector, dermacentor andersoni, feeding. Front. Microbiol. 5. doi: 10.3389/fmicb.2014.00198
Huang, M., Ma, J., Jiao, J., Li, C., Chen, L., Zhu, Z., et al. (2021). The epidemic of q fever in 2018 to 2019 in zhuhai city of China determined by metagenomic next-generation sequencing. PloS Negl. Trop. Dis. 15, e0009520. doi: 10.1371/journal.pntd.0009520
Jia, N., Liu, H. B., Ni, X. B., Bell-Sakyi, L., Zheng, Y. C., Song, J. L., et al. (2019). Emergence of human infection with jingmen tick virus in China: A retrospective study. EBioMedicine 43, 317–324. doi: 10.1016/j.ebiom.2019.04.004
Jia, N., Liu, H. B., Zheng, Y. C., Shi, W. Q., Wei, R., Chu, Y. L., et al. (2020a). Cutaneous immunoprofiles of three spotted fever group rickettsia cases. Infect. Immun. 88, e00686-19. doi: 10.1128/iai.00686-19
Jia, N., Wang, J., Shi, W., Du, L., Sun, Y., Zhan, W., et al. (2020b). Large-Scale comparative analyses of tick genomes elucidate their genetic diversity and vector capacities. Cell 182, 1328–1340.e1313. doi: 10.1016/j.cell.2020.07.023
Katoh, K., Standley, D. M. (2013). MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol. Biol. Evol. 30, 772–780. doi: 10.1093/molbev/mst010
Ladner, J. T., Wiley, M. R., Beitzel, B., Auguste, A. J., Dupuis, A. P., 2nd, Lindquist, M. E., et al. (2016). A multicomponent animal virus isolated from mosquitoes. Cell Host Microbe 20, 357–367. doi: 10.1016/j.chom.2016.07.011
Langmead, B., Salzberg, S. L. (2012). Fast gapped-read alignment with bowtie 2. Nat. Methods 9, 357–359. doi: 10.1038/nmeth.1923
Li, L. J., Ning, N. Z., Zheng, Y. C., Chu, Y. L., Cui, X. M., Zhang, M. Z., et al. (2021). Virome and blood meal-associated host responses in ixodes persulcatus naturally fed on patients. Front. Microbiol. 12. doi: 10.3389/fmicb.2021.728996
Liu, W., Zhang, F., Qiu, E. C., Yang, J., Xin, Z. T., Wu, X. M., et al. (2010). Roseomonas sp. isolated from ticks, China. Emerg. Infect. Dis. 16, 1177–1178. doi: 10.3201/eid1607.090166
Modi, A., Vai, S., Caramelli, D., Lari, M. (2021). The illumina sequencing protocol and the NovaSeq 6000 system. Methods Mol. Biol. 2242, 15–42. doi: 10.1007/978-1-0716-1099-2_2
Nakao, R., Abe, T., Nijhof, A. M., Yamamoto, S., Jongejan, F., Ikemura, T., et al. (2013). A novel approach, based on BLSOMs (Batch learning self-organizing maps), to the microbiome analysis of ticks. Isme J. 7, 1003–1015. doi: 10.1038/ismej.2012.171
Narasimhan, S., Swei, A., Abouneameh, S., Pal, U., Pedra, J. H. F., Fikrig, E. (2021). Grappling with the tick microbiome. Trends Parasitol. 37, 722–733. doi: 10.1016/j.pt.2021.04.004
Pascoal, J. O., Siqueira, S. M., Maia, R. D. C., Juan Szabó, M. P., Yokosawa, J. (2019). Detection and molecular characterization of mogiana tick virus (MGTV) in rhipicephalus microplus collected from cattle in a savannah area, uberlândia, Brazil. Ticks Tick Borne Dis. 10, 162–165. doi: 10.1016/j.ttbdis.2018.10.002
Qin, X. C., Shi, M., Tian, J. H., Lin, X. D., Gao, D. Y., He, J. R., et al. (2014). A tick-borne segmented RNA virus contains genome segments derived from unsegmented viral ancestors. Proc. Natl. Acad. Sci. U S A. 111, 6744–6749. doi: 10.1073/pnas.1324194111
Sameroff, S., Tokarz, R., Charles, R. A., Jain, K., Oleynik, A., Che, X., et al. (2019). Viral diversity of tick species parasitizing cattle and dogs in Trinidad and Tobago. Sci. Rep. 9, 10421. doi: 10.1038/s41598-019-46914-1
Shi, M., Lin, X. D., Vasilakis, N., Tian, J. H., Li, C. X., Chen, L. J., et al. (2016). Divergent viruses discovered in arthropods and vertebrates revise the evolutionary history of the flaviviridae and related viruses. J. Virol. 90, 659–669. doi: 10.1128/jvi.02036-15
Souza, W. M., Fumagalli, M. J., Torres Carrasco, A. O., Romeiro, M. F., Modha, S., Seki, M. C., et al. (2018). Viral diversity of rhipicephalus microplus parasitizing cattle in southern Brazil. Sci. Rep. 8, 16315. doi: 10.1038/s41598-018-34630-1
Temmam, S., Bigot, T., Chrétien, D., Gondard, M., Pérot, P., Pommelet, V., et al. (2019). Insights into the host range, genetic diversity, and geographical distribution of jingmenviruses. mSphere 4, e00645-19. doi: 10.1128/mSphere.00645-19
Thorvaldsdóttir, H., Robinson, J. T., Mesirov, J. P. (2013). Integrative genomics viewer (IGV): high-performance genomics data visualization and exploration. Brief Bioinform. 14, 178–192. doi: 10.1093/bib/bbs017
Wikel, S. (2013). Ticks and tick-borne pathogens at the cutaneous interface: host defenses, tick countermeasures, and a suitable environment for pathogen establishment. Front. Microbiol. 4. doi: 10.3389/fmicb.2013.00337
Williams-Newkirk, A. J., Rowe, L. A., Mixson-Hayden, T. R., Dasch, G. A. (2014). Characterization of the bacterial communities of life stages of free living lone star ticks (Amblyomma americanum). PloS One 9, e102130. doi: 10.1371/journal.pone.0102130
Keywords: ticks, skin, patients, infectome, metatranscriptomic sequencing
Citation: Zhang J, Zheng Y-C, Chu Y-L, Cui X-M, Wei R, Bian C, Liu H-B, Yao N-N, Jiang R-R, Huo Q-B, Yuan T-T, Li J, Zhao L, Li L-F, Wang Q, Wei W, Zhu J-G, Chen M-C, Gao Y, Wang F, Ye J-L, Song J-L, Jiang J-F, Lam TT-Y, Ni X-B and Jia N (2023) Skin infectome of patients with a tick bite history. Front. Cell. Infect. Microbiol. 13:1113992. doi: 10.3389/fcimb.2023.1113992
Received: 02 December 2022; Accepted: 09 February 2023;
Published: 27 February 2023.
Edited by:
Hua Niu, Affiliated Hospital of Guilin Medical University, ChinaReviewed by:
Shu Shen, Wuhan Institute of Virology (CAS), ChinaCopyright © 2023 Zhang, Zheng, Chu, Cui, Wei, Bian, Liu, Yao, Jiang, Huo, Yuan, Li, Zhao, Li, Wang, Wei, Zhu, Chen, Gao, Wang, Ye, Song, Jiang, Lam, Ni and Jia. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Na Jia, amlhbmE3OV80MUBob3RtYWlsLmNvbQ==; Xue-Bing Ni, bml4dWViaW5nNzJAMTYzLmNvbQ==; Tommy Tsan-Yuk Lam, dHR5bGFtQGhrdS5oaw==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.