
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Cell. Infect. Microbiol. , 01 March 2023
Sec. Parasite and Host
Volume 13 - 2023 | https://doi.org/10.3389/fcimb.2023.1105126
This article is part of the Research Topic Advances in Molecular Biology, Pathogenesis, Diagnosis, Vaccines, and Treatment of Diseases Caused by Apicomplexan Parasites View all 14 articles
 Hailong Dong1†
Hailong Dong1† Xiushuang Chen2,3†
Xiushuang Chen2,3† Xiaoxiao Zhao2,3Chenxi Zhao2,3
Xiaoxiao Zhao2,3Chenxi Zhao2,3 Khalid Mehmood4Muhammad Fakhar-e-Alam Kulyar5
Khalid Mehmood4Muhammad Fakhar-e-Alam Kulyar5 Zeeshan Ahmad Bhutta6
Zeeshan Ahmad Bhutta6 Jiangyong Zeng7
Jiangyong Zeng7 Shah Nawaz8Qingxia Wu1*
Shah Nawaz8Qingxia Wu1* Kun Li2,3*
Kun Li2,3*Diarrhea is a severe bovine disease, globally prevalent in farm animals with a decrease in milk production and a low fertility rate. Cryptosporidium spp. are important zoonotic agents of bovine diarrhea. However, little is known about microbiota and short-chain fatty acids (SCFAs) changes in yaks infected with Cryptosporidium spp. Therefore, we performed 16S rRNA sequencing and detected the concentrations of SCFAs in Cryptosporidium-infected yaks. Results showed that over 80,000 raw and 70,000 filtered sequences were prevalent in yak samples. Shannon (p<0.01) and Simpson (p<0.01) were both significantly higher in Cryptosporidium-infected yaks. A total of 1072 amplicon sequence variants were shared in healthy and infected yaks. There were 11 phyla and 58 genera that differ significantly between the two yak groups. A total of 235 enzymes with a significant difference in abundance (p<0.001) were found between healthy and infected yaks. KEGG L3 analysis discovered that the abundance of 43 pathways was significantly higher, while 49 pathways were significantly lower in Cryptosporidium-infected yaks. The concentration of acetic acid (p<0.05), propionic acid (p<0.05), isobutyric acid (p<0.05), butyric acid (p<0.05), and isovaleric acid was noticeably lower in infected yaks, respectively. The findings of the study revealed that Cryptosporidium infection causes gut dysbiosis and results in a significant drop in the SCFAs concentrations in yaks with severe diarrhea, which may give new insights regarding the prevention and treatment of diarrhea in livestock.
The long-haired ruminant yak is a plateau bovine species living in the 3000-5000 m high-altitude regions and is mostly found on the Qinghai Tibet plateau (Li et al., 2022a). Diarrhea is a serious bovine problem detected globally in livestock farms associated with a decrease in fertility rate and milk production, especially neonatal diarrhea is usually found with high morbidity and mortality (Han et al., 2017; Li et al., 2019a; Lan et al., 2021)
Previously, studies revealed that diarrhea contributed to more than 50% of calf mortality in Canada (Smith et al., 2014), and affected 19% of the cattle population in the USA (Smulski et al., 2020), which indeed was the cause of huge economic detriment. Like other bovine animals, diarrhea has been commonly reported in yaks (Diao et al., 2020; Cui et al., 2022; Li et al., 2022a). There have been many biological factors which are associated for diarrhea and leading cause of death in calves (Kim et al., 2021). Many pathogens like bovine viral diarrhea virus, Noroviruses, Escherichia coli, Salmonella spp., and Cryptosporidium spp. have been commonly observed in infected cattle (Meganck et al., 2014; Cui et al., 2022). Among others, Cryptosporidium spp. are important zoonotic protozoa infecting various animal species (Li et al., 2019b; Kandeel et al., 2022), and are also generally recognized as the primary agent of cattle diarrhea (Li et al., 2019a; Li et al., 2019b). A previous study reported that the infection of Cryptosporidium spp. was an important issue in UK and Scotland (Smith et al., 2014). As yaks and cattle species are economically important for native herdsmen in China (Cheng et al., 2022), infectious diseases like those caused by Cryptosporidium spp. may not only affect animal health but are also potential threats leading to public health concerns.
Intestine microbiota is composed of millions of complex and diverse microorganisms, which contribute greatly to host health, nutrition absorption, host metabolism, and immunological development (Zeineldin et al., 2018). Previous studies demonstrated that this bacteria was related to various diseases like Type 2 diabetes (Martinez-Lopez et al., 2022), acute pancreatitis (Mei et al., 2022), obesity (Salazar et al., 2022), and diarrhea (Han et al., 2017; Zeineldin et al., 2018; Li et al., 2022b). Short-chain fatty acids are metabolic products of microbiota, which contribute to the cellular metabolism of the host (Bachem et al., 2019), regulating immune function and suppressing inflammatory reactions (Abdalkareem Jasim et al., 2022). In our previous study, we observed prominent changes in intestinal microbiota in a horse infected with Cryptosporidium spp. (Wang et al., 2022). However, scarce information is available about microbiota and SCFAs changes in plateau yaks infected with Cryptosporidium spp. Therefore, this study was conducted to explore intestinal microbiota and SCFAs response to natural Cryptosporidium infection in plateau yaks.
Fecal samples (n=40) were collected from free-ranged yaks in Xining, Qinghai (North latitude 31˚36´-39˚19´, east longitude 89˚35´-103˚04´) and examined for Cryptosporidium spp. by employing nested PCR (Chen et al., 2022) and positive samples were saved for further analysis. In this study, all the Cryptosporidium spp. positive samples (n=4) with equal number of negative samples (n=4) were sequenced and divided into infected (INF) and healthy (H) groups, respectively.
The extraction of total genomic DNA was performed by utilizing a commercial TIANamp Stool DNA Kit (Tiangen Biotech (Beijing) Co., Ltd, China) according to the product’s specifications. Fecal DNA concentration, purification, and quality examination were performed through NanoDrop 2000 UV-Vis spectrophotometer (Thermo Scientific, USA) and 1.2% agarose gel electrophoresis, respectively. Then the hypervariable regions of bacterial 16S rRNA gene (V3-V4) were amplified using primers 338F and 806R as described in a previous study (Wang et al., 2019). All PCR products were individually subjected to agarose gel electrophoresis, gel extraction, and purification using the PureLink™ PCR Purification kit (Invitrogen™, USA). Finally, the purified DNA products were quantified by piloting QuantiFluor™-ST as guided by the instruction manual (Promega, USA).
Library construction was carried out by employing commercial Hieff NGS® OnePot II DNA Library Prep Kit for Illumina® (Yeasen, China) according to the product’s instructions, and sequenced through the Illumina NovaSeq platform (Illumina, San Diego, USA). Quality control of sequencing data was performed by employing QIIME2 (https://docs.qiime2.org/2019.1/) to generate amplicon sequence variant (ASV) (Callahan et al., 2016) and taxonomy table (Bokulich et al., 2018). Analysis of variance was performed using ANCOM (Analysis of Composition of Microbiomes), One-way ANOVA, Kruskal Wallis, LEfSe (LDA (Linear Discriminant Analysis) score >2), DEseq2 (p<0.05 and log2 (FoldChange) > 2), clustering heatmap (with Z-score > 0.5 or < -0.5) and evolutionary tree (p<0.05) methods to reveal differences in bacterial abundance among yak samples (Segata et al., 2011; Love et al., 2014; Mandal et al., 2015). Microbial alpha diversities analyses were performed through QIIME2 by calculating indices including observed OTUs, Chao1, Shannon, and Faith’s. Microbial beta diversities of principal coordinate analysis (PCoA), nonmetric multidimensional scaling (NMDS) (Vazquez-Baeza et al., 2013), and partial least squares discriminant analysis (PLS-DA) were carried out to explore the structural variation of microbial communities across yak samples. The evolutionary relation tree was constructed by using ggtree in R package.
The potential KEGG Ortholog (KO) functional profiles of yak microbiota was predicted with PICRUSt (Langille et al., 2013) by annotating with MetaCyc and ENZYME database. One-way ANOVA was used to analyze the data, while Duncan test was used as post-hoc test to measure the individual differences in microbial function between the yak groups with a p<0.05 as statistically significant.
The concentrations of SCFAs in fecal samples were detected by employing GC-MS (Hsu et al., 2019; Zhang et al., 2019), and the differences between yak groups were explored via t-test.
The differences between different yak groups were calculated by the chi-square test piloting IBM SPSS Statistics (SPSS 22.0). P values < 0.05 were considered as statistically significant.
In the current study, over 80,000 raw and 70,000 filtered sequences were obtained in yak samples. The non-chimeric sequences ranged from 62,133 to 73,453 in healthy yaks, and 68,173 to 74,350 in infected yaks (Table 1). There were a total of 1072 shared ASVs between the healthy (group H) and infected (group INF) groups. (Figure 1A). Alpha diversity index analysis showed that there was no significant difference in chao1, faith, and observed features between group H and INF, respectively. Shannon (p<0.01) and Simpson (p<0.01) were both significantly higher in group INF than in group H (Figure 1B).
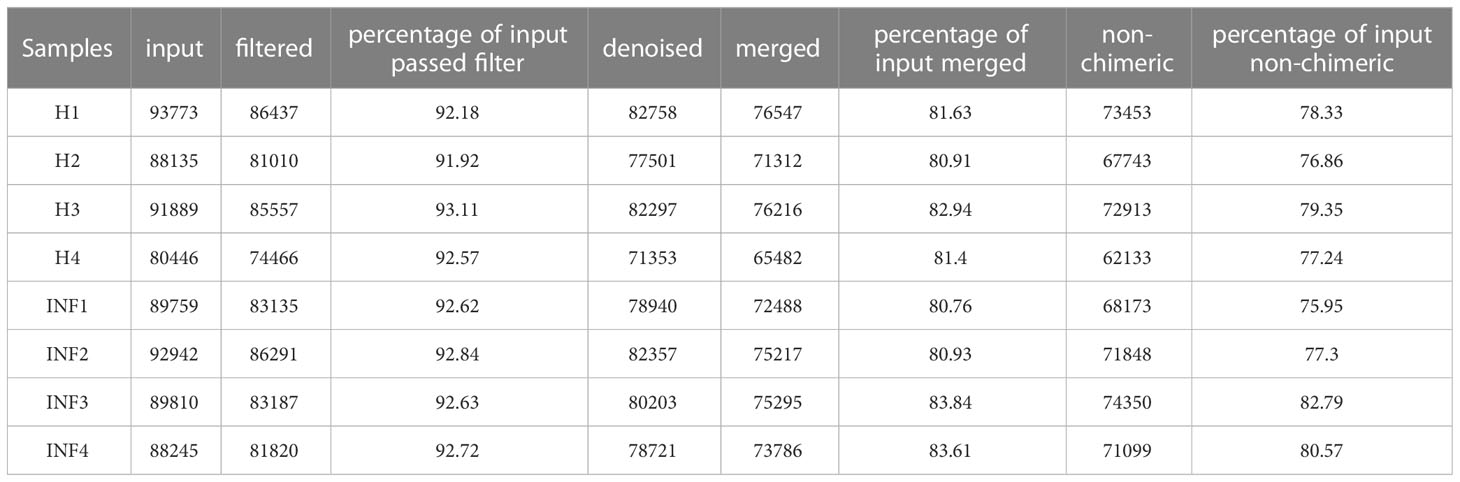
Table 1 The sequence data statistic analysis.
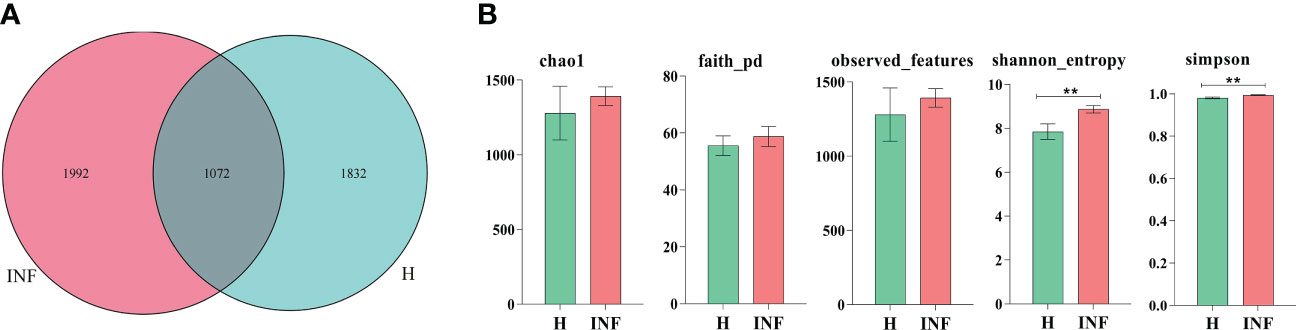
Figure 1 ASV venn map and Alpha diversity index analysis. (A) Venn map, (B) Alpha diversity index. ** refers to significance level, p<0.05.
The sequence percentage in different taxa of group H and INF is shown in Figure 2A. At the phylum level, the dominant phyla were Firmicutes (69.61%), Proteobacteria (8.97%), and Actinobacteria (8.72%) in group H, while Firmicutes (56.38%) and Bacteroidetes (29.83%) were the main phyla in group INF (Figure 2B). At the class level, Clostridia (51.13%) and Bacilli (17.28%) were the primary classes in healthy yaks, while Clostridia (51.13%) and Bacteroidia (29.83%) were the major classes in infected yaks (Figure 2C). At the order level, Clostridiales (51.13%), Lactobacillales (8.20%), and Bacillales (8.10%) were the primary orders in healthy yaks, while Clostridiales (51.04%) and Bacteroides (29.83%) were the main orders in infected yaks (Figure 2D). At the family level, the main families were unclassified, Ruminococcaceae and Lachnospiraceae in groups H and INF (Figure 2E). At the genus level, unclassified (52.06%), Pseudomonadaceae Pseudomonas (6.13%), and Lactobacillus (6.00%) were the dominating genera in healthy yaks, while unclassified (69.25%), Prevotellaceae Prevotella (5.13%) and Arthrobacter (2.45%) were the main genera in infected yaks (Figure 2F). At the species level, the main bacteria in group H were unclassified (87.85%), Veronii (6.11%), and Alactolyticus (1.66%), while unclassified (95.15%), Flavefaciens (1.50%) and Veronii (1.12%) were the main bacteria in group INF (Figure 2G).
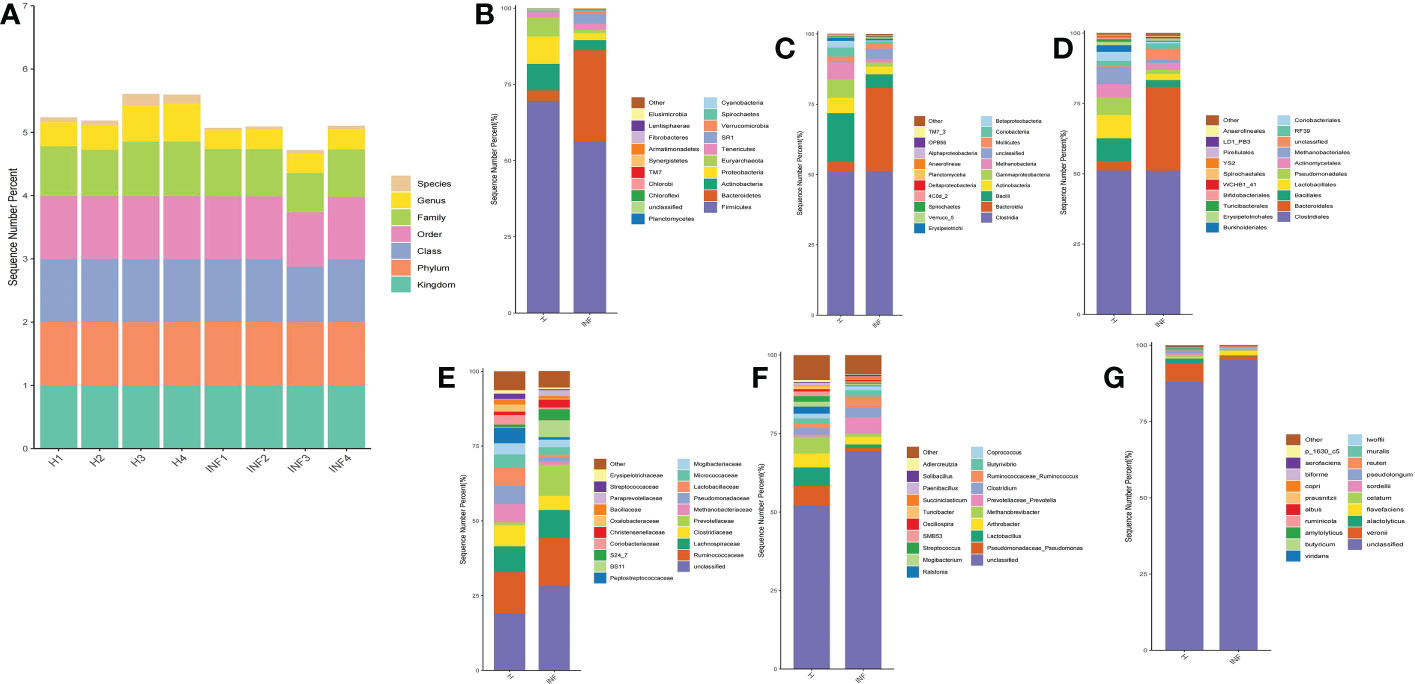
Figure 2 Statistical analysis of yak microbiota in different taxa. (A) Sequence percentages in different taxa, (B) Phylum, (C) Class, (D) Order, (E) Family, (F) Genus, (G) Species.
To reveal the microbiota difference between healthy and infected yaks, beta diversity analysis was carried out through NMDS, PCoA, Qiime 2β, and PCA analysis. The results showed a huge difference in composition and structure between samples from group H and group INF animals (Figure 3). To explore the microbiota changes caused by Cryptosporidium in different taxa, a clustering heatmap (top 20 abundance) and evolutionary tree (top 50 abundance) with heat map analysis were plotted. The results revealed that at the order level, infected yaks showed an abundance of Bacteroidia and Deltaproteobacteria, while healthy animals showed abundance of Bacilli, Erysipelotrichi, Betaproteobacteria, Alphaproteobacteria, and Nitriliruptoria as expressed in the clustering heatmap. The evolutionary tree also showed an obvious abundance difference in Betaproteobacteria, Fibrobacteria, SJA_176, 4C0d_2, Nitriliruptoria, Clostridia, and Bacilli between groups H and INF (Figure 4A). At the order level, the clustering heatmap revealed significant differences in the abundance of Bacteroidales, Lactobacillales, Burkholderiales, Erysipelotrichales, YS2, Turicibacterales and Enterobacteriales between healthy and infected animals. Evolutionary tree detected remarkable differences in the abundance of Oceanospirillales, Burkholderiales, Enterobacteriales, Fibrobacterales, Turicibacterales, RB046, YS2, Nitriliruptorales, Clostridiales and Lactobacillales between healthy and infected animals (Figure 4B). At the family level, there was a noteworthy difference of Clostridiaceae, Prevotellaceae, Lactobacillaceae, Peptostreptococcaceae, BS11, Christensenellaceae, Oxalobacteraceae, Paraprevotellaceae, Streptococcaceae and Erysipelotrichaceae between groups H and INF as revealed by the clustering heatmap. Evolutionary tree analysis showed a clear difference of Halomonadaceae, Oxalobacteraceae, Enterobacteriaceae, Streptococcaceae, Peptostreptococcaceae, Turicibacteraceae, Dietziaceae, Sanguibacteraceae, Nitriliruptoraceae, Christensenellaceae, Clostridiaceae and Lactobacillaceae between healthy and infected yaks (Figure 4C). At the genus level, interesting difference of Lactobacillus, Prevotellaceae_Prevotella, Ralstonia, Streptococcus, SMB53, Turicibacter, and Adlercreutzia was found between the two yak groups. Evolutionary tree analysis demonstrated that the abundance of Halomonadaceae, Oxalobacteraceae, Streptococcaceae, Clostridiaceae, Turicibacteraceae, Planococcaceae, Erysipelotrichaceae, Sanguibacteraceae, Coriobacteriaceae, Paraprevotellaceae, Ruminococcaceae, Lachnospiraceae, Clostridiaceae, Lactobacillaceae and Lachnospiraceae were significantly different between the two yak groups (Figure 4D). At the species level, the abundance of alactolyticus, celatum, reuteri, butyricum, ruminicola, prausnitzii, biforme, p_1630_c5, and aerofaciens were noticeably different in groups H and INF. Evolutionary tree analysis uncovered that the abundance of alactolyticus, ruminis, p_1630_c5, biforme, umbonata, aerofaciens, prausnitzii, butyricum, celatum and reuteri were significantly different between healthy and infected animals (Figure 4E).

Figure 3 Beta diversity analysis between yak groups. (A) NMDS, (B) PCoA, (C) Qiime 2β, (D) PCA.
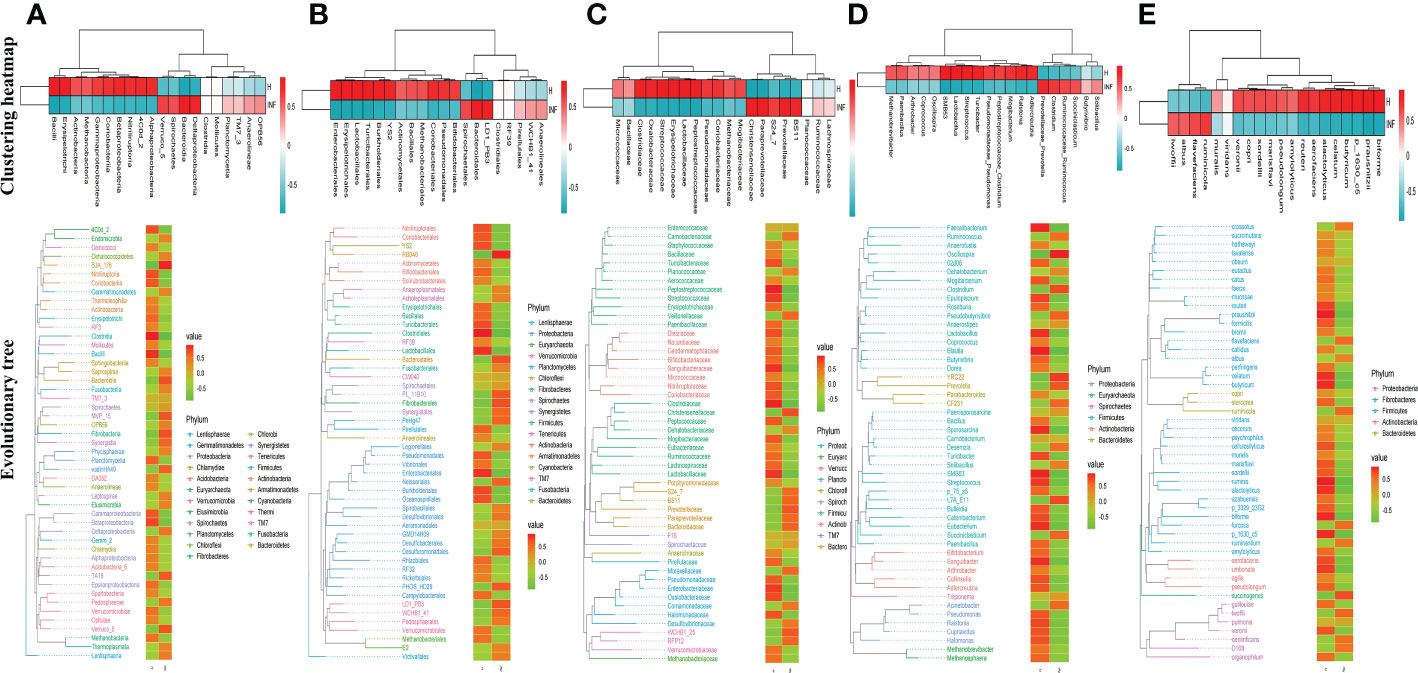
Figure 4 Clustering heatmap and evolutionary tree with heat map analysis of yak microbiota in different taxa. (A) Class, (B) Order, (C) Family, (D) Genus, (E) Species.
To further uncover the marker bacteria between healthy and Cryptosporidium-infected yaks, we performed one-way ANOVA and Kruskal Wallis tests to determine the significance of the difference and depicted results by DESeq 2 volcano diagram and LEfSe chart, respectively. Results showed that at the phylum level, the abundance of SR1 (p<0.0001), Bacteroidetes (p<0.0001), Armatimonadetes (p<0.0001), Fibrobacteres (p<0.01), and Synergistetes (p<0.01) were visibly higher in infected yaks, while Cyanobacteria (p<0.0001), Proteobacteria (p<0.0001), Armatimonadetes (p<0.0001), Euryarchaeota (p<0.0001), Actinobacteria (p<0.01), Firmicutes (p<0.01), and Elusimicrobia (p<0.05) were significantly lower (Figure 5A). At the genus level, the abundance of YRC22 (p<0.0001), Prevotellaceae_Prevotella (p<0.0001), CF231 (p<0.0001), L7A_E11 (p<0.0001), BF311 (p<0.0001), Desulfovibrio (p<0.0001), Succiniclasticum (p<0.0001), Desemzia (p<0.0001), Anaerovorax (p<0.0001), Pseudobutyrivibrio (p<0.0001), Acinetobacter (p<0.0001), Fibrobacter (p<0.0001), Ruminococcaceae_Ruminococcus (p<0.0001), Anaerorhabdus (p<0.0001), Treponema (p<0.0001), Selenomonas (p<0.001), Clostridium (p<0.001), Shuttleworthia (p<0.001), Dehalobacterium (p<0.001), TG5 (p<0.01), unclassified (p<0.01), Anaerostipes (p<0.01), Syntrophomonas (p<0.01), Brachymonas (p<0.01), Pyramidobacter (p<0.01), SHD_231 (p<0.05), Butyrivibrio (p<0.05), Desulfobulbus (p<0.05), RFN20 (p<0.05), and Anaerofustis (p<0.05) were significantly higher in infected yaks, while Turicibacter (p<0.0001), Lactobacillus (p<0.0001), Sporosarcina (p<0.0001), Ralstonia (p<0.0001), Akkermansia (p<0.001), Streptococcus (p<0.001), Methylobacterium (p<0.01), Adlercreutzia (p<0.01), Faecalibacterium (p<0.01), Roseburia (p<0.01), Paenibacillus (p<0.01), Methanosphaera (p<0.01), Pseudomonadaceae_Pseudomonas (p<0.01), Peptostreptococcaceae_Clostridium (p<0.01), Slackia (p<0.01), Cupriavidus (p<0.01), Halomonas (p<0.01), Gemmiger (p<0.01), Dietzia (p<0.01), Blautia (p<0.05), Agrobacterium (p<0.05), Nesterenkonia (p<0.05), Sanguibacter (p<0.05), Phascolarctobacterium (p<0.05), Actinomycetospora (p<0.05), Bifidobacterium (p<0.05), SMB53 (p<0.05), and Dorea (p<0.05) were significantly lower in infected animals (Figure 5B).
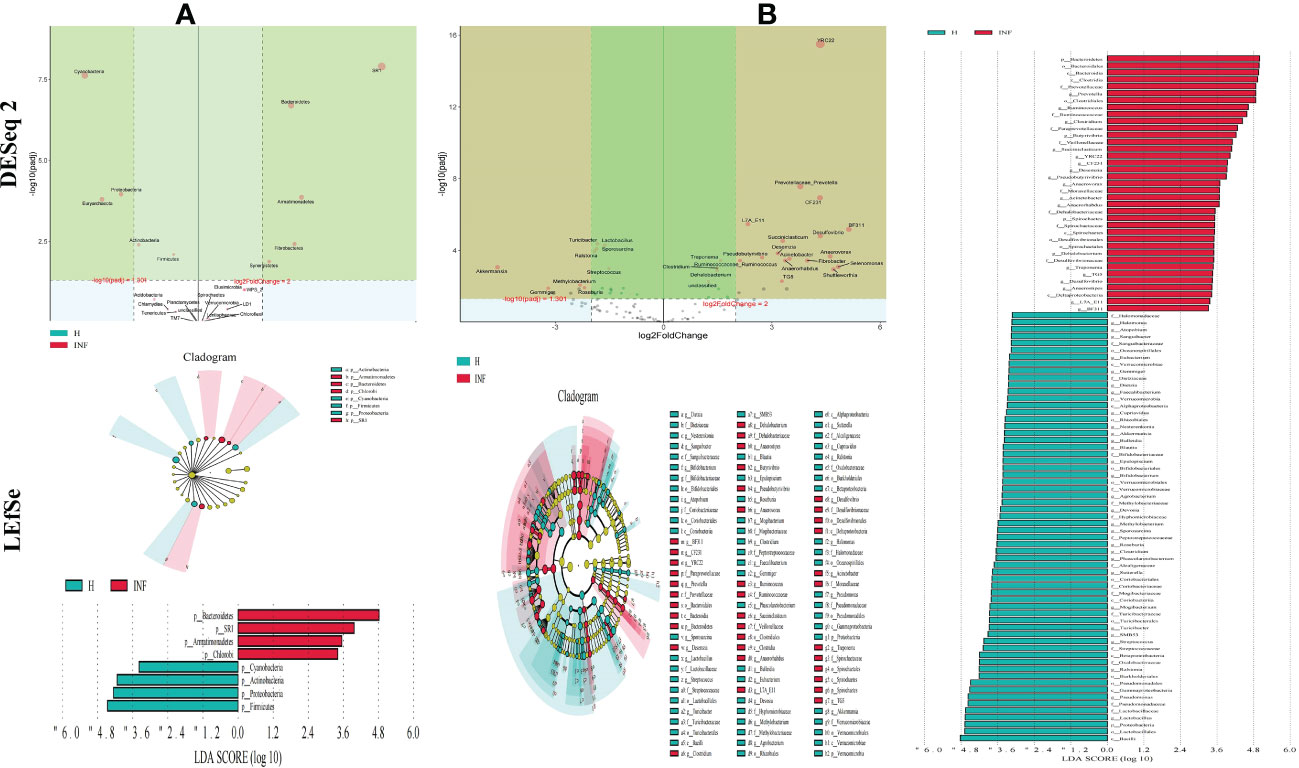
Figure 5 Cryptosporidium infection changes microbiota in different taxa through DESeq 2 volcano plot and LEFSe analysis. (A) Phylum, (B) Genus.
The prediction of yaks’ microbiota function was carried out by PICRUSt2, and the functional difference between yaks was explored by using one-way ANOVA and Duncan test through R language as previously reported (Zhai et al., 2020). A total of 235 enzymes with a significant difference in abundance (p<0.001) were found between healthy and infected yaks, with 119 higher and 116 lower abundance enzymes in INF yaks (Figure 6A). Only one different MetaCys pathway of pentose phosphate pathway (non-oxidative branch) was found between the two yak groups (Figure 6B). KEGG L1 analysis found that the abundance of genetic information processing was prominently higher in infected yaks, while cellular processes and environmental information processing were significantly lower (Figure 7A). KEGG L2 analysis revealed that the abundance of biosynthesis of other secondary metabolites, glycan biosynthesis, and metabolism, metabolism of cofactors and vitamins, and nucleotide metabolism were remarkably higher in INF yaks, while amino acid metabolism, chemical structure transformation maps, lipid metabolism, metabolism of other amino acids, xenobiotics biodegradation, and metabolism were conspicuously lower (Figure 7B). KEGG L3 analysis discovered that the abundance of 43 pathways was significantly higher in INF yaks, while 49 pathways were significantly lower (Figure 7C).
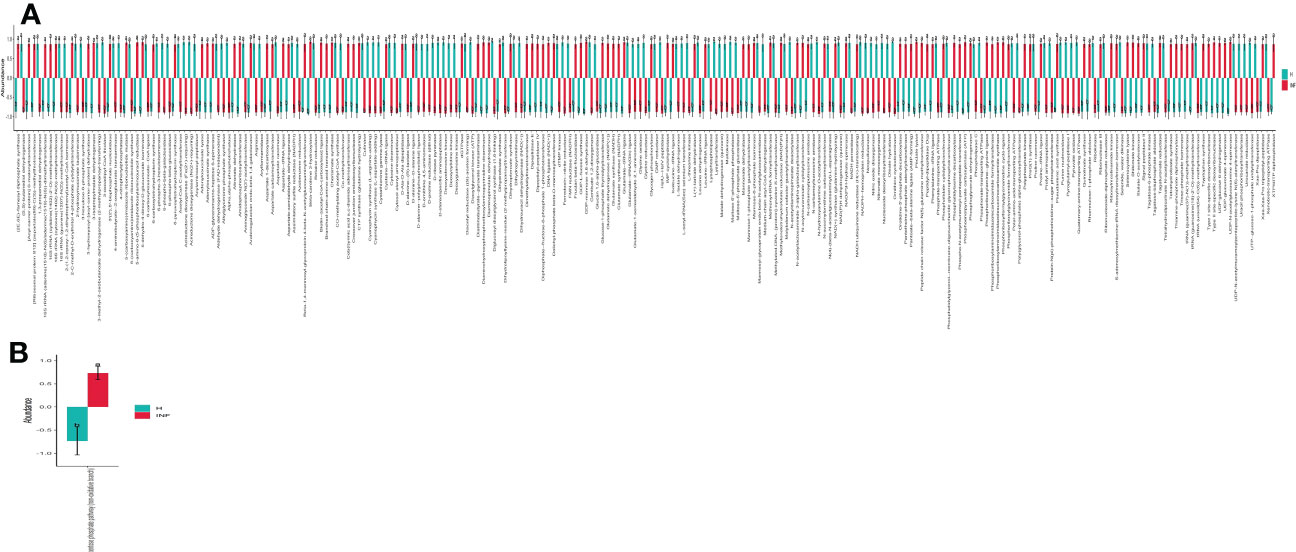
Figure 6 Cryptosporidium infection affected enzyme and MetaCys pathway abundance of yaks. (A) Enzyme (p<0.001), (B) MetaCys (p<0.05). "a, b" are showing significance relation.
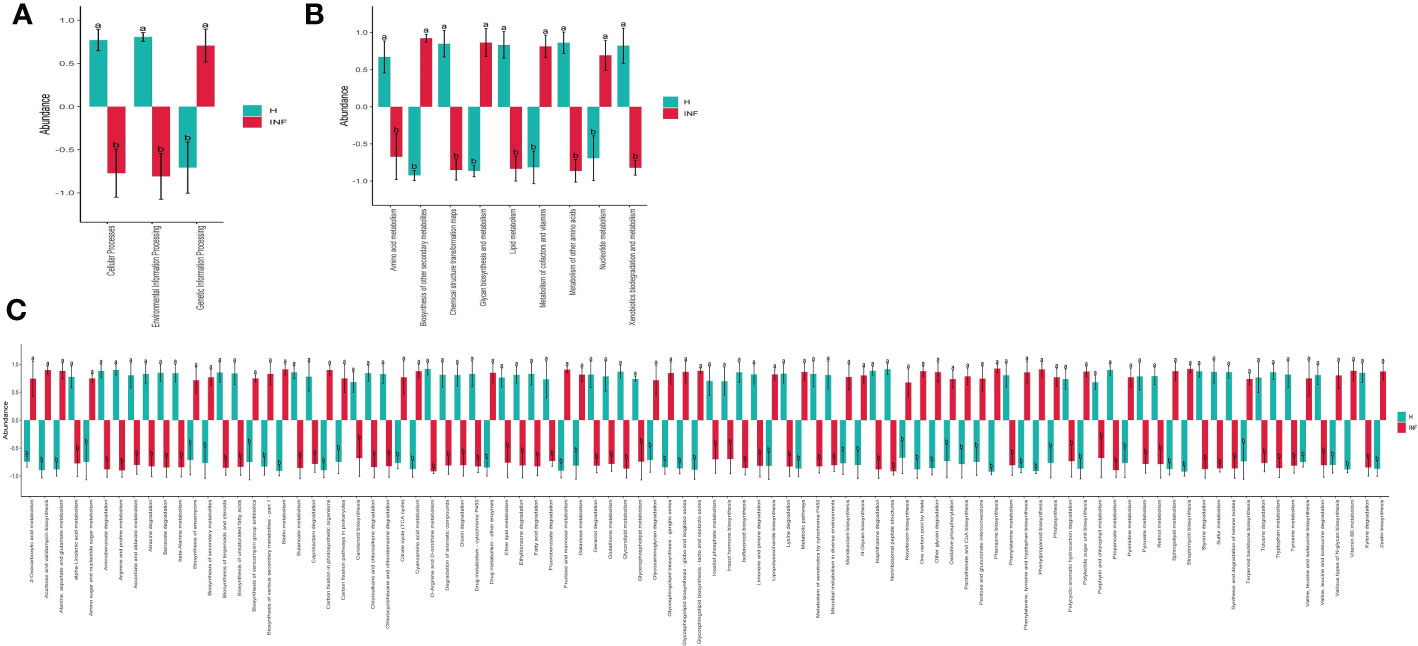
Figure 7 Cryptosporidium infection potentially affected the microbiota function of yaks. (A) KEGG L1 (p<0.05), (B) KEGG L2 (p<0.05), (C) KEGG L3 (p<0.05). "a, b" are showing significance relation.
The concentration of acetic acid (p<0.05), propionic acid (p<0.05), isobutyric acid (p<0.05), butyric acid (p<0.05) and isovaleric acid was significantly lower in infected yaks, respectively, while there was no significant difference of valeric acid and caproic acid between H and INF groups (Figure 8).
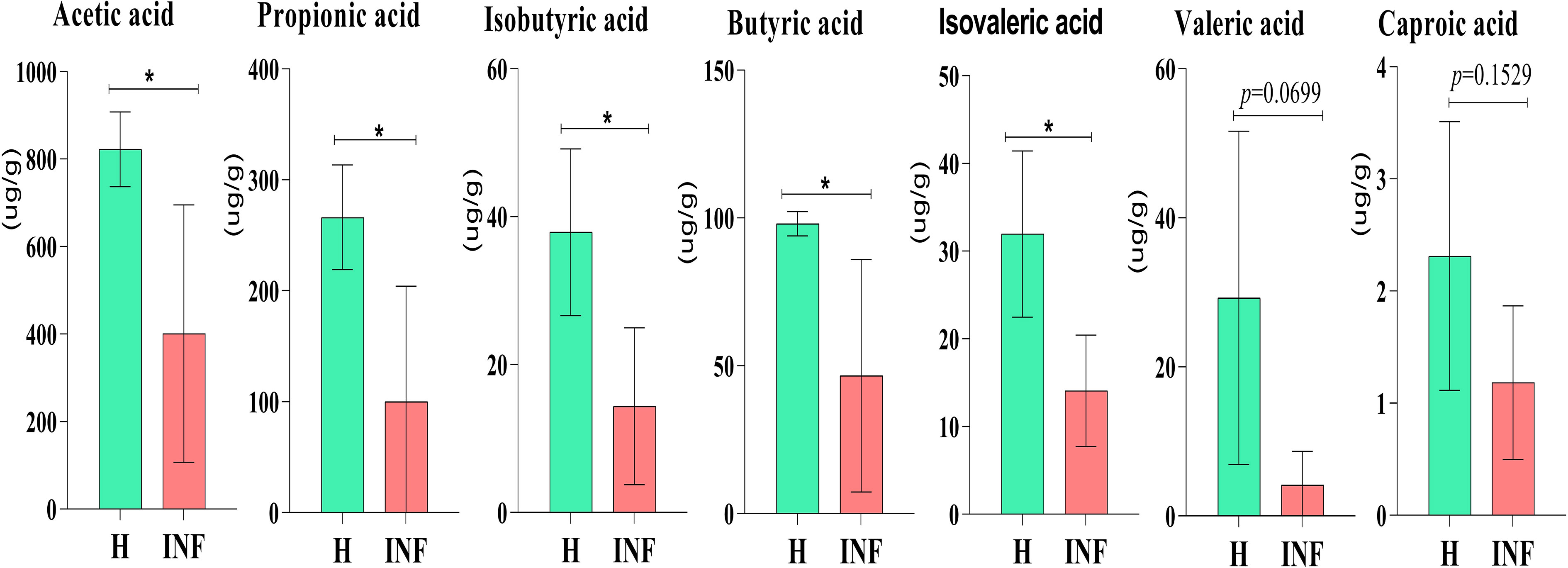
Figure 8 Concentration of SCFAs in yaks. Significance is presented as *p < 0.05; data are presented as the mean ± SEM (n = 4).
Cattle diarrhea is still an important worldwide issue on farms, despite observing advanced preventive measures such as herd management, animal facilities and care, feeding and nutrition, and timely medication (Wei et al., 2021). The infectious Cryptosporidium was one of the main causative agents of diarrhea with limited available effective treatments (Li et al., 2019a). The harsh climatic conditions with heavy snowfall in the long frigid season (from October to May, with average temperature −15 to −5°C) didn’t permit collection of many samples in the Plateau region. Also, very few positive samples (n=4) were observed out of total collected samples (n=40) in the present study. However, a prevalence as low as 1.3% of Cryptosporidium spp. positive samples has been reported in yaks in China region (Li et al., 2020). Moreover, despite the harsh climatic conditions and the low number of positive samples available for analysis, this number was above the minimum required for high throughput sequencing, and validation of changes of the microbiota (Ray et al., 2019). In the current study, we performed 16S rDNA sequencing of fecal samples collected from healthy and Cryptosporidium-infected yaks. Results showed that Cryptosporidium infection increased the alpha diversity index of Shannon (p<0.01) and Simpson (p<0.01) (Figure 1B), which demonstrated the increased microbiota complexity of infected animals. The current results are in line with our previous results found in Cryptosporidium-infected horses (Wang et al., 2022). Beta diversity analysis through NMDS, PCoA, Qiime 2β, and PCA analysis revealed microbiota differences between healthy and infected yaks (Figure 3), which were confirmed by comparing the dominating gut microbiota in different taxa (Figures 2, 4). Then we explored the significantly different bacteria between the H and INF groups through DESeq 2 volcano diagram and LEfSe chart analysis. The results showed that a total of 11 phyla and 58 genera were significantly different (Figure 5), which is in accordance with the previously reported results in a study conducted on infected people and horses (Chappell et al., 2016; Wang et al., 2022). The increased genera in yaks were in line with previous studies that found a higher abundance of Desulfovibrio and Butyrivibrio in colitis patients (Berry and Reinisch, 2013; Gryaznova et al., 2021), Prevotellaceae_Prevotella in diarrheic pigs (Yang et al., 2017), Anaerovorax in slow growth performers in nursery pigs (Zhai et al., 2020), Succiniclasticum in LPS induced dual-flow continuous culture system (Dai et al., 2019), Pseudobutyrivibrio in chronic kidney people (Wu et al., 2020), Anaerorhabdus in pulmonary fibrosis persons (Tong et al., 2019), Selenomonas in gastric cancer patients (Zhang et al., 2021), Anaerostipes in diabetic nephropathy patients (Du et al., 2021), Pyramidobacter in endoscopic sphincterotomy surgery gallstone patients (Shen et al., 2021), and Anaerofustis in Alzheimer people (Hou et al., 2021). The genus of Acinetobacter is an underrated food-borne pathogen (Amorim and Nascimento, 2017). A previous study found Acinetobacter in acute diarrhea of children (Polanco and Manzi, 2008). The genus of Treponema is the main pathogen in bovine dermatitis (Mamuad et al., 2020), Clostridia are clinical species and some of them may cause severe infections like colitis (Sanchez Ramos and Rodloff, 2018). Those increased genera may have contributed greatly to diarrhea caused by Cryptosporidium. The lower abundance of genera in yaks was in accordance with the results revealing Turicibacter and Lactobacillus in Salmonella-infected pigs (Garrido et al., 2021), Akkermansia and Roseburiain in colitis in mouse (Bu et al., 2021; Li et al., 2021), Adlercreutzia in influenza virus-infected mouse (Lu et al., 2021), Faecalibacterium in pre-eclampsia people (Chen et al., 2020), Methanosphaera in sheep without treatment of anthelmintic (Moon et al., 2021), Slackia in Vogt-Koyanagi-Harada patients (Li et al., 2022a; Li et al., 2022b), Gemmiger in immune-mediated inflammatory people (Forbes et al., 2018), and Dorea in HIV patients (Xu et al., 2021). Those deficient genera in Cryptosporidium-infected animals may be the reason for diarrhea in yaks. The genus of Cupriavidus was related to mycotoxin biodegradation (AL-Nussairawi et al., 2020), and the dropped Cupriavidus in yaks may affect mycotoxin metabolism in yaks. The previous study uncovered probiotics of Dietzia as a new therapy for Crohn’s disease (Click, 2015), and Blautia, Phascolarctobacterium, and Bifidobacterium are probiotic genera (Papizadeh et al., 2017; Chen et al., 2021; Liu et al., 2021), which demonstrated that Cryptosporidium led diarrhea may be due to the decrease of probiotics in the microbiota.
The shifted intestine microflora also changed their functions, as 235 significantly different enzymes were found between healthy and infected yaks (p<0.001) (Figure 6A). Only one obvious different MetaCys pathway of pentose phosphate pathway (non-oxidative branch) was found between the two yak groups (Figure 6B). Also, KEGG L3 analysis discovered that the abundance of 92 pathways was significantly different between healthy and infected animals (Figure 7C). Those results may infer that Cryptosporidium broke the balance of gut microbiota, which affected the microbiota function and caused diarrhea in yaks.
In the present study, significantly lower concentrations of SCFAs were found in Cryptosporidium-infected animals (Figure 8), consistent with yak diarrhea (Li et al., 2022a), LPS-induced piglets (Yang et al., 2021), and dextran sulfate sodium-induced colitis in mouse (Xu et al., 2020). SCFAs play very important roles in host physiology and energy homeostasis (Chambers et al., 2018). Among them, acetate and propionate can provide energy to peripheral tissues (den Besten et al., 2013). A previous study reported that acetate was responsible for maintaining intestine barrier integrity by inhibiting pathogens infection (Skonieczna-Żydecka et al., 2018). In a recent study, it was found that acetate could regulate IgA reactivity (Takeuchi et al., 2021), and propionate contributed to intestinal epithelial turnover and repair (Bilotta et al., 2021). Butyrate is highly related to intestine structure, energy providing to epithelial cells, and regulates immune function (Abdalkareem Jasim et al., 2022). Isobutyric acid and isovaleric acid may be related to mucosal and inflammation responses (Li et al., 2022a). Therefore, the decreased SCFAs in Cryptosporidium-infected yaks might have affected the intestinal barrier and immunity of the host (Aho et al., 2021), which potentially caused diarrhea in plateau yaks.
In conclusion, Cryptosporidium is an important zoonotic protozoon causing severe diarrhea in young animals; however, limited treatment measures are available. Here we reveal that Cryptosporidium infection causes dysbiosis and results in reduced SCFAs in yaks with severe diarrhea, which may give new insights regarding the prevention and treatment of diarrhea in livestock. The low sample size remains the limitation of our study.
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found below: https://www.ncbi.nlm.nih.gov/, PRJNA880359.
The animal study was reviewed and approved by ethics committee of Nanjing Agricultural University.
KL and QW, research idea and methodology. HD, XC, XZ, and CZ, reagents, materials, and analysis tools. KL, writing-original draft and preparation. KM, MF-E-A, ZB, QW, JZ, SN, and KL, writing-review and editing. KL, JZ, and QW, visualization and supervision. All authors contributed to the article and approved the submitted version.
The study was partially supported by the National Natural Science Foundation of China (32102692), the Start-up fund of Nanjing Agricultural University (804131), and the Yak Germplasm innovation and healthy breeding project: Research on the prevention and control of yak infectious diseases for establishing rapid detection methods, prevention and control techniques (XZ202101ZD0002N-05).
We thank Bioyi Biotechnology Co., Ltd. (Wuhan, China) for providing sequencing help in our research.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Abdalkareem Jasim, S., Jade Catalan Opulencia, M., Alexis Ramírez-Coronel, A., Kamal Abdelbasset, W., Hasan Abed, M., Markov, A., et al. (2022). The emerging role of microbiota-derived short-chain fatty acids in immunometabolism. Int. IMMUNOPHARMACOL 110, 108983. doi: 10.1016/j.intimp.2022.108983
Aho, V. T. E., Houser, M. C., Pereira, P. A. B., Chang, J., Rudi, K., Paulin, L., et al. (2021). Relationships of gut microbiota, short-chain fatty acids, inflammation, and the gut barrier in parkinson’s disease. Mol. Neurodegener. 16. doi: 10.1186/s13024-021-00427-6
AL-Nussairawi, M., Risa, A., Garai, E., Varga, E., Szabó, I., Csenki-Bakos, Z., et al. (2020). Mycotoxin biodegradation ability of the cupriavidus genus. Curr. Microbiol. 77, 2430–2440. doi: 10.1007/s00284-020-02063-7
Amorim, A. M., Nascimento, J. D. (2017). Acinetobacter: an underrated foodborne pathogen? J. Infect. Dev. Ctries 11, 111–114. doi: 10.3855/jidc.8418
Bachem, A., Makhlouf, C., Binger, K. J., de Souza, D. P., Tull, D., Hochheiser, K., et al. (2019). Microbiota-derived short-chain fatty acids promote the memory potential of antigen-activated CD8+ T cells. IMMUNITY 51, 285–297. doi: 10.1016/j.immuni.2019.06.002
Berry, D., Reinisch, W. (2013). Intestinal microbiota: A source of novel biomarkers in inflammatory bowel diseases? Best Pract. Res. Clin. Gastroenterol. 27, 47–58. doi: 10.1016/j.bpg.2013.03.005
Bilotta, A. J., Ma, C., Yang, W., Yu, Y., Yu, Y., Zhao, X., et al. (2021). Propionate enhances cell speed and persistence to promote intestinal epithelial turnover and repair. Cell Mol. Gastroenterol. Hepatol. 11, 1023–1044. doi: 10.1016/j.jcmgh.2020.11.011
Bokulich, N. A., Kaehler, B. D., Rideout, J. R., Dillon, M., Bolyen, E., Knight, R., et al. (2018). Optimizing taxonomic classification of marker-gene amplicon sequences with QIIME 2’s q2-feature-classifier plugin. MICROBIOME 6. doi: 10.1186/s40168-018-0470-z
Bu, F., Ding, Y., Chen, T., Wang, Q., Wang, R., Zhou, J., et al. (2021). Total flavone of abelmoschus manihot improves colitis by promoting the growth of akkermansia in mice. Sci. REP-UK 11. doi: 10.1038/s41598-021-00070-7
Callahan, B. J., McMurdie, P. J., Rosen, M. J., Han, A. W., Johnson, A. J. A., Holmes, S. P. (2016). DADA2: High-resolution sample inference from illumina amplicon data. Nat. Methods 13, 581–583. doi: 10.1038/nmeth.3869
Chambers, E. S., Preston, T., Frost, G., Morrison, D. J. (2018). Role of gut microbiota-generated short-chain fatty acids in metabolic and cardiovascular health. Curr. Nutr. Rep. 7, 198–206. doi: 10.1007/s13668-018-0248-8
Chappell, C. L., Darkoh, C., Shimmin, L., Farhana, N., Kim, D., Okhuysen, P. C., et al. (2016). Fecal indole as a biomarker of susceptibility to cryptosporidium infection. Infect. Immun. 84, 2299–2306. doi: 10.1128/IAI.00336-16
Chen, G., Hu, P., Xu, Z., Peng, C., Wang, Y., Wan, X., et al. (2021). The beneficial or detrimental fluoride to gut microbiota depends on its dosages. Ecotoxicol Environ. Saf. 209, 111732. doi: 10.1016/j.ecoenv.2020.111732
Chen, X., Li, P., Liu, M., Zheng, H., He, Y., Chen, M., et al. (2020). Gut dysbiosis induces the development of pre-eclampsia through bacterial translocation. GUT 69, 513–522. doi: 10.1136/gutjnl-2019-319101
Chen, X., Saeed, N. M., Ding, J., Dong, H., Kulyar, M. F., Bhutta, Z. A., et al. (2022). Molecular epidemiological investigation of cryptosporidium sp., giardia duodenalis, enterocytozoon bieneusi and blastocystis sp. Infection Free-ranged Yaks Tibetan Pigs Plateau. Pak Vet. J. 2022, 533–539. doi: 10.29261/pakvetj/2022.060
Cheng, H., Ao, S., Yun, L., Weihong, S., Hong, L., Jianbo, L., et al. (2022). RNA-Seq transcriptome analysis to unravel the gene expression profile of ovarian development in xiangxi cattle. Pak Vet. J. 42, 222–228. doi: 10.29261/pakvetj/2022.004
Click, R. E. (2015). Crohn's disease therapy with dietzia: the end of anti-inflammatory drugs. Future Microbiol. 10, 147–150. doi: 10.2217/fmb.14.133
Cui, Y., Chen, X., Yue, H., Tang, C. (2022). First detection and genomic characterization of bovine norovirus from yak. Pathogens 11, 192. doi: 10.3390/pathogens11020192
Dai, X., Paula, E. M., Lelis, A. L. J., Silva, L. G., Brandao, V. L. N., Monteiro, H. F., et al. (2019). Effects of lipopolysaccharide dosing on bacterial community composition and fermentation in a dual-flow continuous culture system. J. DAIRY Sci. 102, 334–350. doi: 10.3168/jds.2018-14807
den Besten, G., van Eunen, K., Groen, A. K., Venema, K., Reijngoud, D., Bakker, B. M. (2013). The role of short-chain fatty acids in the interplay between diet, gut microbiota, and host energy metabolism. J. Lipid Res. 54, 2325–2340. doi: 10.1194/jlr.R036012
Diao, N., Gong, Q., Li, J., Zhao, D., Li, D., Zhao, B., et al. (2020). Prevalence of bovine viral diarrhea virus (BVDV) in yaks between 1987 and 2019 in mainland China: A systematic review and meta-analysis. Microb. PATHOGENESIS 144, 104185. doi: 10.1016/j.micpath.2020.104185
Du, X., Liu, J., Xue, Y., Kong, X., Lv, C., Li, Z., et al. (2021). Alteration of gut microbial profile in patients with diabetic nephropathy. ENDOCRINE 73, 71–84. doi: 10.1007/s12020-021-02721-1
Forbes, J. D., Chen, C., Knox, N. C., Marrie, R., El-Gabalawy, H., de Kievit, T., et al. (2018). A comparative study of the gut microbiota in immune-mediated inflammatory diseases–does a common dysbiosis exist? MICROBIOME 6. doi: 10.1186/s40168-018-0603-4
Garrido, V., Migura-García, L., Gaitán, I., Arrieta-Gisasola, A., Martínez-Ballesteros, I., Fraile, L., et al. (2021). Prevalence of salmonella in free-range pigs: Risk factors and intestinal microbiota composition. Foods 10, 1410. doi: 10.3390/foods10061410
Gryaznova, M. V., Solodskikh, S. A., Panevina, A. V., Syromyatnikov, M. Y., Dvoretskaya, Y. D., Sviridova, T. N., et al. (2021). Study of microbiome changes in patients with ulcerative colitis in the central European part of Russia. Heliyon 7, e6432. doi: 10.1016/j.heliyon.2021.e06432
Han, Z., Li, K., Shahzad, M., Zhang, H., Luo, H., Qiu, G., et al. (2017). Analysis of the intestinal microbial community in healthy and diarrheal perinatal yaks by high-throughput sequencing. Microb. PATHOGENESIS 111, 60–70. doi: 10.1016/j.micpath.2017.08.025
Hou, M., Xu, G., Ran, M., Luo, W., Wang, H. (2021). APOE-ϵ4 carrier status and gut microbiota dysbiosis in patients with Alzheimer disease. Front. NEUROSCI-SWITZ 15. doi: 10.3389/fnins.2021.619051
Hsu, Y., Chen, C., Lin, Y., Wu, W., Chang, L., Lai, C., et al. (2019). Evaluation and optimization of sample handling methods for quantification of short-chain fatty acids in human fecal samples by GC–MS. J. Proteome Res. 5, 1948–1957. doi: 10.1021/acs.jproteome.8b00536
Kandeel, M., Akhtar, T., Zaheer, T., Ahmad, S., Ashraf, U., Omar, M. (2022). Anti-parasitic applications of nanoparticles: A review. PAK Vet. J. 42, 135–140. doi: 10.29261/pakvetj/2022.040
Kim, S., Yu, D. H., Jung, S., Kang, J., Park, K., Chae, J. B., et al. (2021). Biological factors associated with infectious diarrhea in calves. PAK Vet. J. 41, 531–537. doi: 10.29261/pakvetj/2021.078
Lan, Y., Li, K., Mehmood, K. (2021). Molecular investigation of important protozoal infections in yaks. PAK Vet. J. 41 (4), 557–561. doi: 10.29261/pakvetj/2020.048
Langille, M. G. I., Zaneveld, J., Caporaso, J. G., McDonald, D., Knights, D., Reyes, J. A., et al. (2013). Predictive functional profiling of microbial communities using 16S rRNA marker gene sequences. Nat. Biotechnol. 31, 814–821. doi: 10.1038/nbt.2676
Li, B., Du, P., Du, Y., Zhao, D., Cai, Y., Yang, Q., et al. (2021). Luteolin alleviates inflammation and modulates gut microbiota in ulcerative colitis rats. Life Sci. 269, 119008. doi: 10.1016/j.lfs.2020.119008
Li, K., Li, Z., Zeng, Z., Li, A., Mehmood, K., Shahzad, M., et al. (2020). Prevalence and molecular characterization of cryptosporidium spp. in yaks (Bos grunniens) in naqu, China. Microb. Pathog. 144, 104190. doi: 10.1016/j.micpath.2020.104190
Li, K., Nader, S. M., Zhang, X., Ray, B. C., Kim, C. Y., Das, A., et al. (2019a). Novel lactate dehydrogenase inhibitors with in vivo efficacy against cryptosporidium parvum. PloS Pathog. 15, e1007953. doi: 10.1371/journal.ppat.1007953
Li, N., Wang, R., Cai, M., Jiang, W., Feng, Y., Xiao, L. (2019b). Outbreak of cryptosporidiosis due to cryptosporidium parvum subtype IIdA19G1 in neonatal calves on a dairy farm in China. Int. J. Parasitol. 49, 569–577. doi: 10.1016/j.ijpara.2019.02.006
Li, M., Yang, L., Cao, J., Liu, T., Liu, X. (2022b). Enriched and decreased intestinal microbes in active VKH patients. Invest. Ophthalmol. Vis. Sci. 63, 21. doi: 10.1167/iovs.63.2.21
Li, K., Zeng, Z., Liu, J., Pei, L., Wang, Y., Li, A., et al. (2022a). Effects of short-chain fatty acid modulation on potentially diarrhea-causing pathogens in yaks through metagenomic sequencing. Front. Cell Infect. MI 12. doi: 10.3389/fcimb.2022.805481
Liu, X., Mao, B., Gu, J., Wu, J., Cui, S., Wang, G., et al. (2021). Blautia-a new functional genus with potential probiotic properties? Gut Microbes 13, 1–21. doi: 10.1080/19490976.2021.1875796
Love, M. I., Huber, W., Anders, S. (2014). Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15. doi: 10.1186/s13059-014-0550-8
Lu, W., Fang, Z., Liu, X., Li, L., Zhang, P., Zhao, J., et al. (2021). The potential role of probiotics in protection against influenza a virus infection in mice. Foods 10, 902. doi: 10.3390/foods10040902
Mamuad, L. L., Seo, B. J., Faruk, M. S. A., Espiritu, H. M., Jin, S. J., Kim, W., et al. (2020). Treponema spp., the dominant pathogen in the lesion of bovine digital dermatitis and its characterization in dairy cattle. Vet. Microbiol. 245, 108696. doi: 10.1016/j.vetmic.2020.108696
Mandal, S., Van Treuren, W., White, R. A., Eggesbø, M., Knight, R., Peddada, S. D. (2015). Analysis of composition of microbiomes: a novel method for studying microbial composition. Microbial Ecol. Health Dis. 26, 27663. doi: 10.3402/mehd.v26.27663
Martinez-Lopez, Y. E., Esquivel-Hernandez, D. A., Sanchez-Castaneda, J. P., Neri-Rosario, D., Guardado-Mendoza, R., Resendis-Antonio, O. (2022). Type 2 diabetes, gut microbiome, and systems biology: A novel perspective for a new era. Gut Microbes 14, 2111952. doi: 10.1080/19490976.2022.2111952
Meganck, V., Hoflack, G., Opsomer, G. (2014). Advances in prevention and therapy of neonatal dairy calf diarrhoea: a systematical review with emphasis on colostrum management and fluid therapy. Acta Vet. Scand. 56, 75. doi: 10.1186/s13028-014-0075-x
Mei, Q., Fu, Y., Huang, Z., Yin, N., Wang, R., Xu, B., et al. (2022). Intestinal TLR4 deletion exacerbates acute pancreatitis through gut microbiota dysbiosis and paneth cells deficiency. Gut Microbes 14. doi: 10.1080/19490976.2022.2112882
Moon, C. D., Carvalho, L., Kirk, M. R., McCulloch, A. F., Kittelmann, S., Young, W., et al. (2021). Effects of long-acting, broad spectra anthelmintic treatments on the rumen microbial community compositions of grazing sheep. Sci. REP-UK 11. doi: 10.1038/s41598-021-82815-y
Papizadeh, M., Rohani, M., Nahrevanian, H., Javadi, A., Pourshafie, M. R. (2017). Probiotic characters of bifidobacterium and lactobacillus are a result of the ongoing gene acquisition and genome minimization evolutionary trends. Microb. PATHOGENESIS 111, 118–131. doi: 10.1016/j.micpath.2017.08.021
Polanco, N., Manzi, L. (2008). Oxigenic effect of acinetobacter baumanniiisolated from children with acute diarrhea. Invest. Clin. 49, 59–67.
Ray, K. J., Cotter, S. Y., Arzika, A. M., Kim, J., Boubacar, N., Zhou, Z., et al. (2019). High-throughput sequencing of pooled samples to determine community-level microbiome diversity. Ann. Epidemiol. 39, 63–68. doi: 10.1016/j.annepidem.2019.09.002
Salazar, N., Ponce-Alonso, M., Garriga, M., Sanchez-Carrillo, S., Hernandez-Barranco, A. M., Redruello, B., et al. (2022). Fecal metabolome and bacterial composition in severe obesity: Impact of diet and bariatric surgery. Gut Microbes 14, 2106102. doi: 10.1080/19490976.2022.2106102
Sanchez Ramos, L., Rodloff, A. C. (2018). Identification of clostridium species using the VITEK® MS. ANAEROBE 54, 217–223. doi: 10.1016/j.anaerobe.2018.01.007
Segata, N., Izard, J., Waldron, L., Gevers, D., Miropolsky, L., Garrett, W. S., et al. (2011). Metagenomic biomarker discovery and explanation. Genome Biol. 12, R60. doi: 10.1186/gb-2011-12-6-r60
Shen, H., Zhu, J., Ye, F., Xu, D., Fang, L., Yang, J., et al. (2021). Biliary microbial structure of gallstone patients with a history of endoscopic sphincterotomy surgery. Front. Cell Infect. MI 10. doi: 10.3389/fcimb.2020.594778
Skonieczna-Żydecka, K., Grochans, E., Maciejewska, D., Szkup, M., Schneider-Matyka, D., Jurczak, A., et al. (2018). Faecal short chain fatty acids profile is changed in polish depressive women. NUTRIENTS 10, 1939. doi: 10.3390/nu10121939
Smith, R. P., Clifton-Hadley, F. A., Cheney, T., Giles, M. (2014). Prevalence and molecular typing of cryptosporidium in dairy cattle in England and Wales and examination of potential on-farm transmission routes. Vet. Parasitol. 204, 111–119. doi: 10.1016/j.vetpar.2014.05.022
Smulski, S., Turlewicz-Podbielska, H., Wylandowska, A., Włodarek, J. (2020). Non-antibiotic possibilities in prevention and treatment of calf diarrhoea. J. Vet. Res. 64, 119–126. doi: 10.2478/jvetres-2020-0002
Takeuchi, T., Miyauchi, E., Kanaya, T., Kato, T., Nakanishi, Y., Watanabe, T., et al. (2021). Acetate differentially regulates IgA reactivity to commensal bacteria. NATURE 595, 560–564. doi: 10.1038/s41586-021-03727-5
Tong, X., Su, F., Xu, X., Xu, H., Yang, T., Xu, Q., et al. (2019). Alterations to the lung microbiome in idiopathic pulmonary fibrosis patients. Front. Cell Infect. MI 9. doi: 10.3389/fcimb.2019.00149
Vazquez-Baeza, Y., Pirrung, M., Gonzalez, A., Knight, R. (2013). EMPeror: a tool for visualizing high-throughput microbial community data. GIGASCIENCE 2, 16. doi: 10.1186/2047-217X-2-16
Wang, Y., Li, X., Chen, X., Kulyar, M. F., Duan, K., Li, H., et al. (2022). Gut fungal microbiome responses to natural cryptosporidium infection in horses. Front. Microbiol. 13. doi: 10.3389/fmicb.2022.877280
Wang, W., Zhai, S., Xia, Y., Wang, H., Ruan, D., Zhou, T., et al. (2019). Ochratoxin a induces liver inflammation: involvement of intestinal microbiota. MICROBIOME 7. doi: 10.1186/s40168-019-0761-z
Wei, X., Wang, W., Dong, Z., Cheng, F., Zhou, X., Li, B., et al. (2021). Detection of infectious agents causing neonatal calf diarrhea on two Large dairy farms in yangxin county, Shandong province, China. Front. Veterinary Sci. 7. doi: 10.3389/fvets.2020.589126
Wu, I., Lin, C., Chang, L., Lee, C., Chiu, C., Hsu, H., et al. (2020). Gut microbiota as diagnostic tools for mirroring disease progression and circulating nephrotoxin levels in chronic kidney disease: Discovery and validation study. Int. J. Biol. Sci. 16, 420–434. doi: 10.7150/ijbs.37421
Xu, H., Ou, Z., Zhou, Y., Li, Y., Huang, H., Zhao, H., et al. (2021). Intestinal mucosal microbiota composition of patients with acquired immune deficiency syndrome in guangzhou, China. Exp. Ther. Med. 21, 391. doi: 10.3892/etm.2021.9822
Xu, Z., Tang, H., Huang, F., Qiao, Z., Wang, X., Yang, C., et al. (2020). Algal oil rich in n-3 PUFA alleviates DSS-induced colitis via regulation of gut microbiota and restoration of intestinal barrier. Front. Microbiol. 11, 615404. doi: 10.3389/fmicb.2020.615404
Yang, Q., Huang, X., Zhao, S., Sun, W., Yan, Z., Wang, P., et al. (2017). Structure and function of the fecal microbiota in diarrheic neonatal piglets. Front. Microbiol. 8. doi: 10.3389/fmicb.2017.00502
Yang, C., Wang, M., Tang, X., Yang, H., Li, F., Wang, Y., et al. (2021). Effect of dietary Amylose/Amylopectin ratio on intestinal health and cecal microbes’ profiles of weaned pigs undergoing feed transition or challenged with escherichia coli lipopolysaccharide. Front. Microbiol. 12. doi: 10.3389/fmicb.2021.693839
Zeineldin, M., Aldridge, B., Lowe, J. (2018). Dysbiosis of the fecal microbiota in feedlot cattle with hemorrhagic diarrhea. Microb. PATHOGENESIS 115, 123–130. doi: 10.1016/j.micpath.2017.12.059
Zhai, H., Luo, Y., Ren, W., Schyns, G., Guggenbuhl, P. (2020). The effects of benzoic acid and essential oils on growth performance, nutrient digestibility, and colonic microbiota in nursery pigs. Anim. FEED Sci. TECH 262, 114426. doi: 10.1016/j.anifeedsci.2020.114426
Zhang, X., Li, C., Cao, W., Zhang, Z. (2021). Alterations of gastric microbiota in gastric cancer and precancerous stages. Front. Cell Infect. MI 11. doi: 10.3389/fcimb.2021.559148
Keywords: Cryptosporidium, yaks, diarrhea, microbiota, SCFAs
Citation: Dong H, Chen X, Zhao X, Zhao C, Mehmood K, Kulyar MFA, Bhutta ZA, Zeng J, Nawaz S, Wu Q and Li K (2023) Intestine microbiota and SCFAs response in naturally Cryptosporidium-infected plateau yaks. Front. Cell. Infect. Microbiol. 13:1105126. doi: 10.3389/fcimb.2023.1105126
Received: 22 November 2022; Accepted: 16 February 2023;
Published: 01 March 2023.
Edited by:
Ivana Klun, Institute for Medical Research, University of Belgrade, SerbiaReviewed by:
Iti Saraav, Washington University in St. Louis, United StatesCopyright © 2023 Dong, Chen, Zhao, Zhao, Mehmood, Kulyar, Bhutta, Zeng, Nawaz, Wu and Li. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Qingxia Wu, Z29vZHdxeEAxNjMuY29t; Kun Li, bGszMDA1QG5qYXUuZWR1LmNu
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.