 Yuyuan Huang1
Yuyuan Huang1 Suping Zhang1,2
Suping Zhang1,2 Yuanmeihui Tao1
Yuanmeihui Tao1 Jing Yang1,3
Jing Yang1,3 Shan Lu1,3
Shan Lu1,3 Dong Jin1,3
Dong Jin1,3 Ji Pu1
Ji Pu1 Wenbo Luo1
Wenbo Luo1 Han Zheng1
Han Zheng1 Liyun Liu1Jia-fu Jiang4*
Liyun Liu1Jia-fu Jiang4* Jianguo Xu1,2,3,5*
Jianguo Xu1,2,3,5*- 1State Key Laboratory of Infectious Disease Prevention and Control, National Institute for Communicable Disease Control and Prevention, Chinese Center for Disease Control and Prevention, Beijing, China
- 2Department of Epidemiology, Center for Global Health, School of Public Health, Nanjing Medical University, Nanjing, Jiangsu, China
- 3Research Units of Discovery of Unknown Bacteria and Function, Chinese Academy of Medical Sciences, Beijing, China
- 4Beijing Institute of Microbiology and Epidemiology, State Key Laboratory of Pathogen and Biosecurity, Beijing, China
- 5Research Institute of Public Health, Nankai University, Tianjin, China
Four Gram-staining-positive, aerobic, non-motile, circle-shaped bacteria were isolated from the faeces of bats (Rousettus leschenaultia and Taphozous perforates) collected from Guangxi autonomous region (E106°49′20″, N22°20′54″) and Yunnan province (E102°04′39″, N25°09′10″) of South China. Strains HY006T and HY008 shared highly 16S rRNA gene sequence similarity to those of Ornithinimicrobium pratense W204T (99.3%) and O. flavum CPCC 203535T (97.3%), while the strains HY1745 and HY1793T were closest to the type strains O. ciconiae H23M54T (98.7%), O. cavernae CFH 30183T (98.3%), and O. murale 01-Gi-040T (98.1%). Furthermore, when compared to the other members of the genus Ornithinimicrobium, the digital DNA-DNA hybridization and average nucleotide identity values of the four novel strains were within the ranges of 19.6–33.7% and 70.6–87.4%, respectively, both of which were below the respective recommended cutoff values of 70.0% and 95–96%. Significantly, strain HY006T was resistant to chloramphenicol and linezolid whereas strain HY1793T was resistant to erythromycin, clindamycin (intermediately), and levofloxacin (intermediately). The main cellular fatty acids (>20.0%) of our isolates were iso-C15:0 and iso-C16:0. Strains HY006T and HY1793T contained ornithine as the diagnostic diamino acid, also along with the alanine, glycine and glutamic acid in their cell wall. Based on phylogenetic, chemotaxonomic and phenotypic analyses, these four strains could be classified as two novel species of the genus Ornithinimicrobium, for which the names Ornithinimicrobium sufpigmenti sp. nov. and Ornithinimicrobium faecis sp. nov. are proposed. The type strains are HY006T (=CGMCC 1.16565T =JCM 33397T) and HY1793T (=CGMCC 1.19143T =JCM 34881T), respectively.
Introduction
Bats (order Chiroptera), only a group of flight-capable mammals, have sparked global attention due to their possible zoonotic association with severe acute respiratory syndrome coronavirus (SARS-CoV) and severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) (Ge et al., 2013; Zhou et al., 2020; Hu et al., 2021). Furthermore, many new viruses, such as adenovirus, hantaviruses, and Shimoni bat virus, have been discovered in bats (Kuzmin et al., 2010; Witkowski et al., 2014; Iglesias-Caballero et al., 2018). However, previous studies have primarily focused on viruses borne by bats (such as Artibeus lituratus, Pteropus giganteus, P. vampyrus, and so on.), leaving the prevalence and abundance of potentially pathogenic bacteria in bats largely neglected (Tan et al., 2021). Significantly, diverse filoviruses were detected in the visceral organs of Rousettus sp. bats, which were collected in Yunnan province (P.R. China) in 2009 and 2015 (Yang et al., 2017). In addition, Toxoplasma gondii infection was easily found in the Taphozous melanopogon collected from Guangxi, China (Jiang et al., 2014). We are curious if bat fecal samples contain any novel bacteria alongside the viruses and parasites. Interestingly, some novel bacterial species previously isolated from feces of the Rousettus leschenaultia and/or Taphozous perforates in our laboratory belong to the phylum Actinobacteria, such as Brevibacterium zhoupengii (Huang et al., 2022a), Microbacterium fandaimingii (Zhou et al., 2021), Tomitella gaofuii and T. fengzijianii (Huang et al., 2022d). In the present study, we continue to explore bat fecal microbiota and describe the phenotypic and genomic characterization of a novel species in the genus Ornithinimicrobium belonging to phylum Actinobacteria.
The genus Ornithinimicrobium which currently accommodates 14 validly published species (https://lpsn.dsmz.de/search?word=Ornithinimicrobium) was described by Groth et al. (Groth et al., 2001) and belongs to the family Ornithinimicrobiaceae (Nouioui et al., 2018) within the order Micrococcales. Strains belonging to the genus Ornithinimicrobium have been isolated from various sources, including soil (Groth et al., 2001; Liu et al., 2008), plants (Ramaprasad et al., 2015; Fang et al., 2017), water (Liu et al., 2013), and faeces of birds (Lee et al., 2021). Ornithinimicrobium is the genus of aerobic or facultatively anaerobic, non-motile Gram-stain-positive, catalase-positive, and oxidase-negative, with 68.3–72.5 mol% DNA G+C contents. The cellular fatty acid profile is dominated by iso-branched-chain acids (Fang et al., 2017; Zhang et al., 2019) and MK-8 (H4) is the major menaquinone. It is worth noting that O. kibberense was first isolated from the Indian Himalayas by Mayilraj in 2006 (Mayilraj et al., 2006), which showed high levels of cytotoxic activity against the murine mammary cell carcinoma 4T1 and human mammary adenocarcinoma MCF-7 cell lines in 2017 (Diaz-Cardenas et al., 2017). Additionally, O. pekingense was first isolated from the activated sludge by Liu in 2008 (Liu et al., 2008), which may be regarded as a causative agent for causing ocular infection (Borsali et al., 2011). These studies were in line with our previously proposed reverse microbial etiology (Xu, 2019), which was to predict and prevent a potential emerging infectious disease caused by an unknown microorganism. Moreover, Ornithinimicrobium may be used as one of the potential markers to distinguish Graves’ disease and Hashimoto’s thyroiditis from the healthy population for participating in the occurrence and development of the disease by regulating purine metabolism and pyrimidine metabolism (Zhao et al., 2022).
Here we report the polyphasic characterization of two novel species (designed strains HY006T/HY008 and HY1745/HY1793T) were isolated from the faeces of bats (Rousettus leschenaultia and Taphozous perforates), for which the names Ornithinimicrobium sufpigmenti sp. nov. and Ornithinimicrobium faecis sp. nov. are proposed, respectively. Strains HY006T (=CGMCC 1.16565T =JCM 33397T) and HY1793T (=CGMCC1.19143T =JCM 34881T) were obtained the certificates of deposition and availability from the China General Microbiological Culture Collection Center (CGMCC) and the Japan Collection of Microorganisms (JCM), respectively. Furthermore, MK-8(H4) was detected as the predominant menaquinone in strains HY006T and HY1793T, and the whole-cell peptidoglycan of both strains contained ornithine, alanine, glycine, and glutamic acid. They also contained glucose in the whole cell sugar. Significantly, strain HY006T was resistant to chloramphenicol, linezolid, and intermediately resistant to erythromycin and tetracycline, while strain HY1793T was resistant to erythromycin, intermediately resistant to clindamycin and levofloxacin. In addition, O. sufpigmenti HY006T accumulated two different carotenoids at significant concentrations by extraction with a solvent system while O. faecis could not detect them.
Materials and methods
Sample collection and strain isolation
The fecal samples from two bat genera were obtained from the Wuhan Institute of Virology, Chinese Academy of Sciences, which were collected from different far-apart sites in Yunnan and Guangxi provinces in October 2013 and July 2011, respectively. The specific latitude and longitude are E102°04′39″, N25°09′10″ (Yunnan) and E106°49′20″, N22°20′54″ (Guangxi) (Table S1). In addition, the species of bats have been identified as Rousettus leschenaultia and Taphozous perforates by blasting the cytochrome b gene sequence (Huang et al., 2022c). Sample collection, transportation and storage were almost like what was delineate antecedently (Ge et al., 2012; Huang et al., 2022a). Briefly, the clean plastic sheets (around 4 square meters) were placed under known bat roosting sites in their natural habitat (usually caves) at approximately six o’clock in the afternoon, and the fecal samples were collected from the sheets in the next morning (approximately 6:00 am) and placed into sterile tubes.
The samples were transported to the laboratory and stored at –80°C until use. Fecal samples obtained from the same bat species were mixed in the laboratory to minimize variations within the same species. The isolation of bacteria from the animal feces was conducted as described previously (Bai et al., 2016; Wang et al., 2018). Briefly, approximately 1 gram of the fecal sample was added in 1 mL of 0.85% (w/v) NaCl solution by lightly whirlpool oscillation, and 150 µL of the suspension was spread on brain-heart infusion (BHI) agar. After incubation at 28°C for 7 days under aerobic, emerging colonies of different morphology were selected, purified, and preliminarily identified by PCR amplification and sequencing of 16S rRNA gene sequence analysis. As previously described (Wang et al., 2018), the nearly full-length 16S rRNA gene fragments (1510 bp) of the four strains were obtained from TA clones using the pEASY-T3 cloning kit (Transgenes) after amplification with two bacterial universal primers, 27F and 1492R (Jin et al., 2013). The 16S rRNA gene sequences of our isolates were deposited in GenBank and calculated 16S rRNA gene sequence similarities with public 16S rRNA gene sequences of type strains using EzBioCloud (https://www.ezbiocloud.net/) 16S based ID service (Yoon et al., 2017a). Among these isolates, four circular, convex, smooth colonies (designated HY006T, HY008, HY1745, and HY1793T), undergoing an identical procedure as other colonies, were identified as potential novel species of genus Ornithinimicrobium and preserved at –80°C in BHI broth supplemented with glycerol (20%, v/v) for further analysis. Strains HY006T and HY1793T were isolated from feces of Rousettus leschenaultia whereas HY008 and HY1745 were isolated from feces of Taphozous perforates. According to the Rules 15, 18a, and 18b from Chapter 3 of International Code of Nomenclature of Prokaryotes (Charles et al., 2019), strains HY006T and HY1793T were designated as the type strain, both of them were deposited in the China General Microbiological Culture Collection Center (CGMCC) and the Japan Collection of Microorganisms (JCM) from which they would be available.
Phylogenetic analyses based on the 16S rRNA gene sequences
The assembled sequences were aligned with representative 16S rRNA gene sequences of phylogenetically related species using ClustalW software (Thompson et al., 1994). Phylogenetic trees were reconstructed by the type strain of the family Ornithinimicrobiaceae with three algorithms, neighbor-joining (Saitou and Nei, 1987), maximum-likelihood (Guindon and Gascuel, 2003) and maximum-parsimony (Kolaczkowski and Thornton, 2004), using MEGA version X (Kumar et al., 2018) based on bootstrap analysis with 1000 replications, as previously described (Huang et al., 2019; Zhu et al., 2019). Arthrobacter globiformis JCM 078085T (=NBRC 12137T) was used as the outgroup.
Whole-genome sequencing analyses
The genomic DNA of four strains was extracted using the Wizard Genomic DNA Purification Kit (Promega) according to the manufacturer’s instruction. Strains HY006T and HY1793T were sequenced by single molecule real-time (SMRT) technology (McCarthy, 2010) on the Pacific Biosciences (PacBio) sequencing platform, which was assembled using de novo and analyzed using the Hierarchical Genome Assembly Process (HGAP4) application (Castanera et al., 2019) to obtain its whole genome sequence without gaps. Meanwhile, the draft genome sequences of strains HY008 and HY1745 were sequenced on the Illumina HiSeq TM2000 platform and assembled using VELVET (Zerbino and Birney, 2008). The number of tRNAs, rRNAs, and protein-coding DNA sequences (CDSs) were predicted using GeneMarkS+4.2 (Berlin et al., 2015). Gene calling and annotation were performed with the Rapid Annotation using Subsystem Technology (RAST) server (https://rast.nmpdr.org/) (Aziz et al., 2008) and the SEED viewer framework (Overbeek et al., 2014). In addition, the antibiotic resistance genes of strains are obtained by comparison in the comprehensive antibiotic resistance database (https://card.mcmaster.ca/analyze) (Alcock et al., 2022). Using the virulence factor database (VFDB, http://www.mgc.ac.cn/VFs/main.htm) (Liu et al., 2019), we identified genome encoded virulence factor (VF) genes. The database of Clusters of Orthologous Groups of proteins (COGs) is a phylogenetic classification of proteins encoded in completely sequenced genomes and the COGs comprise a framework for functional and evolutionary genome analysis (Tatusov et al., 1997). Furthermore, The average nucleotide identity (ANI Calculator | Ezbiocloud.net; with a threshold of 95–96%) and digital DNA-DNA hybridization (dDDH) (http://ggdc.dsmz.de/ggdc.php; with a threshold of 70%) values of the whole genomes of our four isolates with closely related type strains were calculated using the OrthoANIu algorithm (Yoon et al., 2017b) and Genome-to-Genome Distance Calculator 3.0 (GGDC 3.0) (Meier-Kolthoff et al., 2022), respectively.
To further validate the taxonomic status of the four strains in the genus Ornithinimicrobium, a phylogenomic tree based on core genes was constructed using the FastTree program (Price et al., 2009). The core genes were extracted from 22 whole genomes (Table S2) from the NCBI GenBank database (including genomes of strains HY006T, HY008, HY1745, HY1793T, Arthrobacter globiformis NBRC 12137T serving as outgroup and 17 available genomes within the family of Ornithinimicrobiaceae) as aligned by Mafft and determined by CD-HIT (Fu et al., 2012) software (protein sequence identity threshold, 0.4). The output result was visualized in Dendroscope 3 (version 3.5.10) (Huson and Scornavacca, 2012) and modified by Interactive Tree of Life (https://itol.embl.de/) (Letunic and Bork, 2021). Moreover, the whole-genome comparison and annotation of orthologous clusters across multiple species were conducted with OrthoVenn2 (Xu et al., 2019). The pairwise and multiple whole genome alignments were generated with the progressiveMauve algorithm (version 2015-02-26) (Darling et al., 2010).
Phenotypic and biochemical characterization
The phenotypic and physiological tests were similar to our previously described (Huang et al., 2022a). Bacterial growth test was determined at a range of temperatures (4, 10, 15, 20, 25, 28, 30, 35, 37, 40, and 45°C) in BHI broth for 7 days. The pH levels (4.0–11.0, at intervals of 0.5) and NaCl concentrations (0–12%, w/v, at intervals of 0.5%) for growth were also examined using BHI broth at 28°C. The cells have an incubation period of up to 7 days for observing growth under microaerophilic (5.0% CO2 incubator) or anaerobic conditions (80.0% N2, 10.0% CO2, and 10.0% H2) by incubating the strains on identical plates. Cell morphology was observed by microscope (Light microscope, Eclipse 80i; Transmission electron microscope, HT7700) after incubating on BHI-1.0% NaCl (w/v) plates at 28°C for 5 days. Oxidase activity was examined by oxidase reagent (bioMérieux) and the catalase activity of strains was tested with bubble production in 3% (v/v) hydrogen peroxide. Gram staining was performed using a Gram-staining kit (Baso) (Austrian, 1960), and motility was tested by observing the spreading growth of cells inoculated by piercing into BHI semisolid (0.3% agar, w/v) medium in test tubes (Xu et al., 2013). Acid production from sucrose, glucose, and other physiological and biochemical tests were determined using API 50CH strips combined with API 50CHB medium and API 20NE, and qualitative enzyme tests were determined with the API ZYM system (bioMérieux). Additionally, Biolog GEN III MicroPlate (catalog No. 1030) with inoculating fluid (catalog No. 72401-IF-A) was also used to obtain the biochemical data following the manufacturers’ instructions (Biolog). Antibiotic sensitivity of strains HY006T and HY1793T was tested using the E-test stripes (bioMérieux) as previously described (Brown and Brown, 1991), on BHI supplemented with 5.0% sheep blood agar at 28°C containing the following antibiotics: ceftriaxone, chloramphenicol, clindamycin, erythromycin, levofloxacin, linezolid, meropenem, tetracycline, and vancomycin.
Chemotaxonomic characterization
For the biomass of chemotaxonomic assays, all the tested strains were harvested from BHI-1% NaCl plates after 5 days of incubation at 28°C. Cellular fatty acids were extracted, methylated, and identified by using the Sherlock Microbial Identification System (MIDI) according to the manufacturer’s standard protocols (Sasser, 1990). The respiratory isoprenoid quinones were extracted, purified, and analyzed by high performance liquid chromatography (HPLC) (Komagata and Suzuki, 1988) with various menaquinones and ubiquinones (Hu et al., 2001) as references. Polar lipids were analyzed using two-dimensional thin-layer chromatography (2D TLC) after hydrolysis with 6 M HCl at 100°C for 18 hours (Staneck and Roberts, 1974; Harper and Davis, 1979). Peptidoglycan amino acid composition was measured with a Hitachi-8900 high speed amino acid analyzer after hydrolyzing the cell wall, and whole-cell sugars were examined according to the method of Hasegawa et al. (Hasegawa et al., 1983).
Bacterial carotenoid extracts were analyzed at 450 nm using an UPLC system with DAD detector (UPLC, U3000; Thermo Scientific). The analytical conditions were as follows, UPLC: column, YMC Carotenoid S-3 μm (150 × 4.6 mm); column temperature, 40°C; flow rate, 1.0 mL/min; injection volume, 2 μL; solvent system (MeOH: MTBE: H2O=20: 75: 5); gradient program, 100:0 v/v at 0 min, 39:61 v/v at 15 min, 0:100 V/V at 25 min, 100:0 v/v at 25.1min, 100:0 v/v at 30 min. Data were acquired on the U3000 UPLC (Thermo Scientific) and processed using chromeleon 7.2 CDS (Thermo Scientific) (Meléndez-Martínez et al., 2010; Irakli et al., 2011). The carotenoid content in samples was calculated as the formulas: Carotenoid content (µg/100 mL) = Read concentration (µg/mL) × Dilution factor × 100.
Reference strains information
Five reference strains were respectively obtained from the Korean Collection for Type Cultures (O. cavernae CFH 30183T= KCTC 49018T; O. ciconiae H23M54T= KCTC 49151T), Canadian Phytoplankton Culture Collection (O. flavum CPCC 203535T), the Deutsche Sammlung von Mikroorganismen und Zellkulturen (O. murale 01-Gi-040T= DSM 22056T) and the Guangdong Microbial Culture Collection Center (O. pratense W204T= GDMCC 1.1391T).
Results
Phylogenetic and phylogenomic analyses
The 16S rRNA gene sequences of strains HY006T/HY008 (1,488 bp) and HY1745/HY1793T (1,510 bp) were obtained. According to the comparison results in the EzBioCloud database, strains HY006T and HY008 were highly similar to those of Ornithinimicrobium pratense W204T (99.3%) and O. flavum CPCC 203535T (97.3%), whereas strains HY1745 and HY1793T were closest to the type strains O. ciconiae H23M54T (98.7%), O. cavernae CFH 30183T (98.3%), and O. murale 01-Gi-040T (98.1%).
The phylogenetic trees constructed using various algorithms (NJ, ML, and MP) (Figure 1A) showed that strains HY1745 and HY1793T formed a separate phylogenetic sub-branch within a sub-clade in the Ornithinimicrobium clade encompassed by three recognized species of the genus (most closely related neighbors mentioned above). In addition, the closest relationship of strains HY006T/HY008 to O. pratense W204T was clearly shown in Figure 1A. Furthermore, the phylogenomic tree (based on 536 core genes) indicated that our four isolates belonged to the genus of Ornithinimicrobium, and strains HY006T/HY008 formed a distinct cluster with species O. pratense W204T and O. flavum CPCC 203535T. The strains HY1745 and HY1793T also formed a clade with O. ciconiae H23M54T, O. cavernae CFH 30183T, and O. murale 01-Gi-040T supported by high bootstrap value (Figure 1B), almost identical to the results of 16S rRNA gene phylogenetic trees.
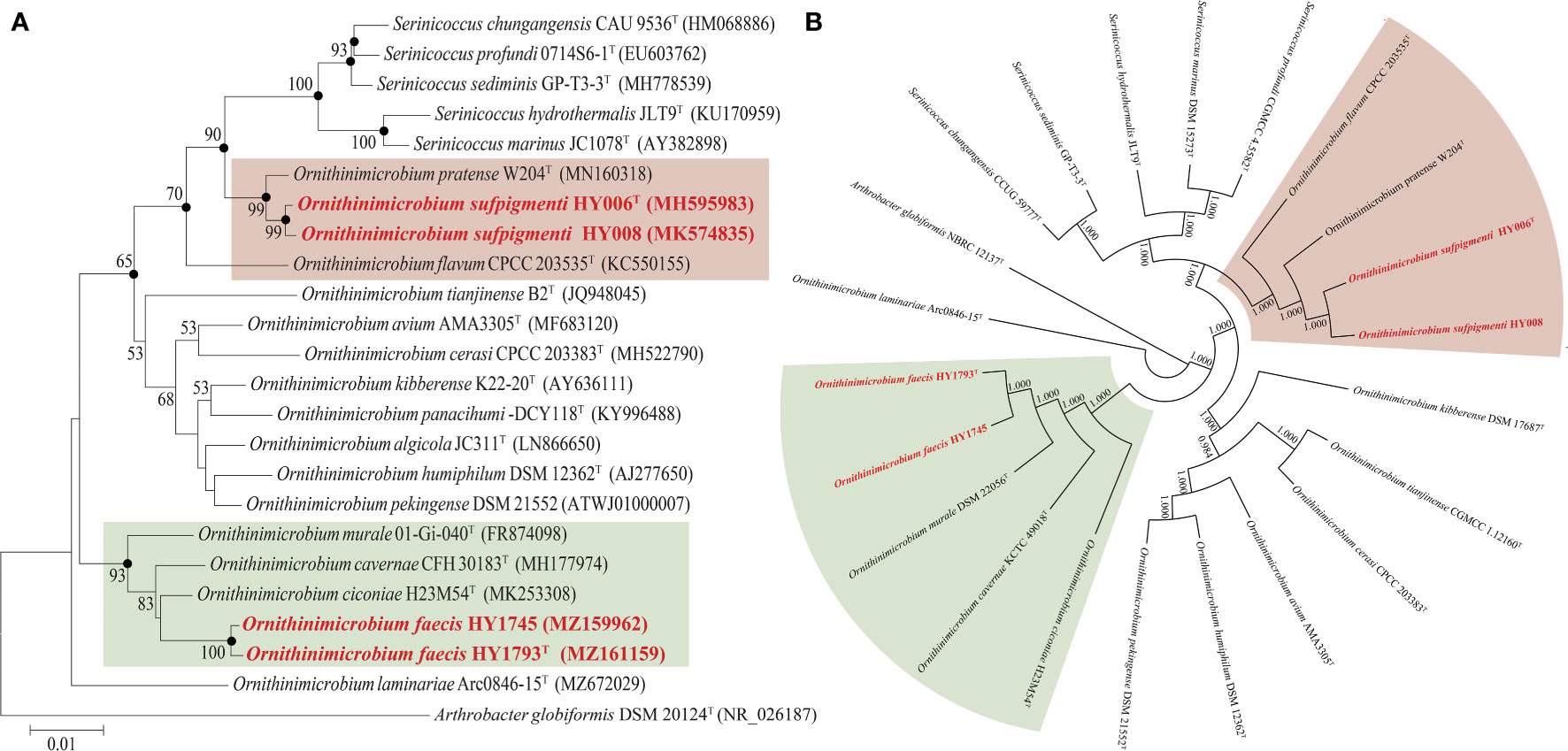
Figure 1 The phylogenetic (A) and phylogenomic tree (B) of O. sufpigmenti sp. nov., O. faecis sp. nov. and other species of family Ornithinimicrobiaceae. (A) Solid circles indicate the nodes supported by maximum-likelihood and maximum-parsimony analyses. Numbers on the tree indicate bootstrap values calculated for 1000 subsets for branch points >50%. Bar, 0.01 substitutions per nucleotide position. (B) Numbers on the tree indicate each split in the tree support values with the Shimodaira-Hasegawa test calculated for 1000 resamples.
Genome characteristics
The draft genome sequence of strain HY008 consisted of 35 contigs and yielded a genome of 4,147,990 bp (contained 3,817 genes) after assembly. The complete genome of type strain HY006T was 4,169,359 bp (N50: 4,169,359 bp; genome coverage: 240.0×) in length and contained 3,805 genes, with one plasmid (length: 46,843 bp). The plasmid contains 59 coding sequences (CDS) and the G + C content is 66.8 mol%. Most of the protein are hypothetical protein (88.1%), some are enzymes involved in DNA replication (6.8%), two uncharacterized protein (3.4%) and one structural protein (1.7%), no proteins with specific functions were found (Table S3). In addition, the DNA G+C content of strains HY006T and HY008 were 71.4 and 71.5 mol%, respectively. Strains HY1745 and HY1793T contained 4,421 and 4,262 genes, respectively. The number of tRNA genes in our four strains was 46, and the G + C content was between 68.7 and 71.5% (Table 1). Moreover, stains HY006T, HY008, HY1745 and HY1793T contained ten, nine, five, and ten predicted virulence factors, respectively (Table S4). Specifically, two virulence factor-like genes, namely sodA (superoxide dismutase, VFDB ID: VFG001421) and HSI-3 (type VI secretion system ATPase TssH, VFDB ID: VFG041043) were discovered in most our isolates except for strain HY1745. In addition, five virulence factor-like genes, including those encoding isocitrate lyase icl (VFG001381), nitrate reductase subunit alpha narG (VFG001391), nitrate reductase subunit beta narH (VFG001814), AlgW protein algW (VFG014984), and UDP-glucose 6-dehydrogenase ugd (VFG048797), were also dominantly found in the type strains HY006T and HY1793T, but were absent in strains HY008 and HY1745.
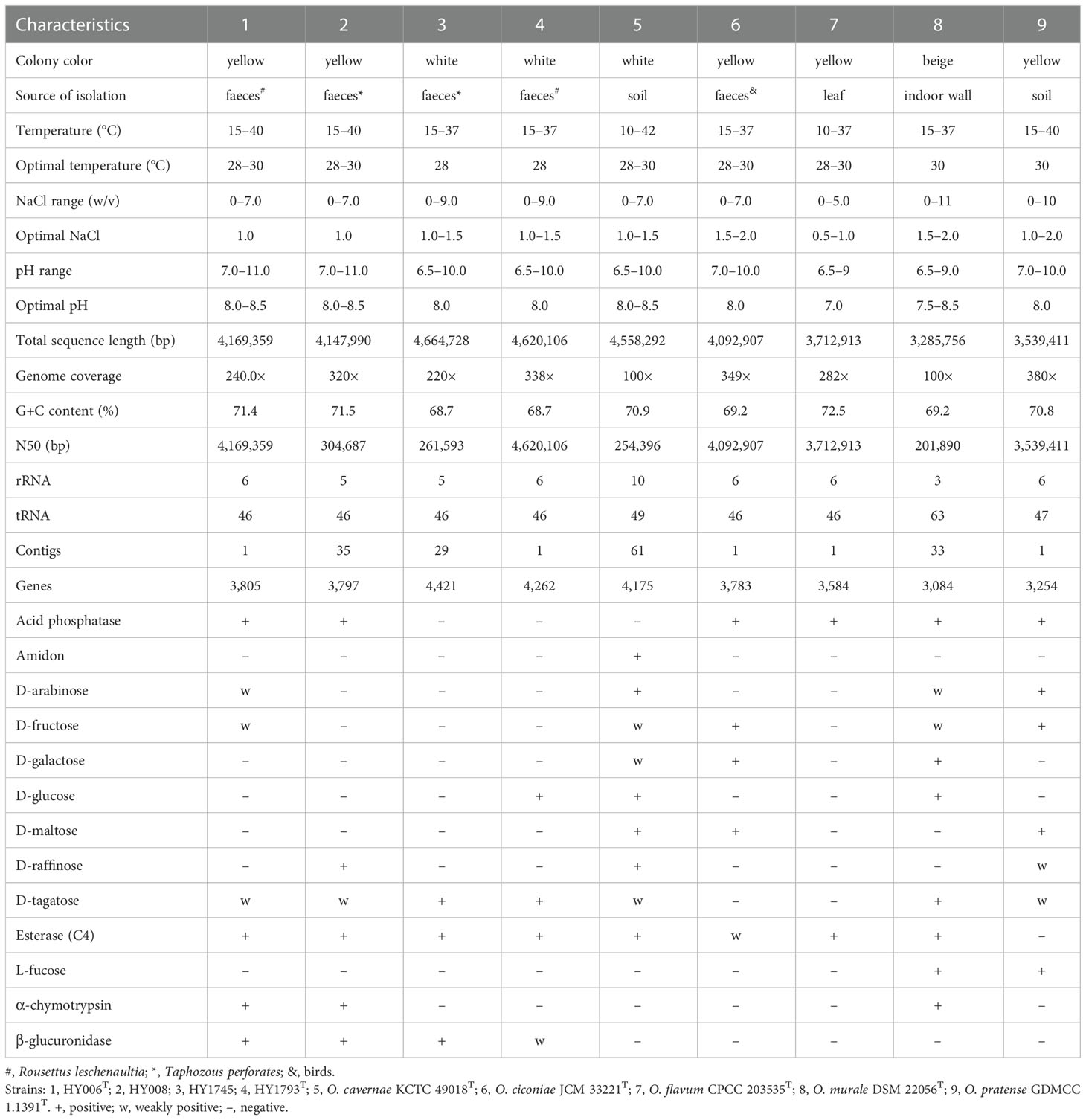
Table 1 Differential characteristics between our four isolates and the type strains of closely related species.
The values of dDDH between the four isolates and their closely related species or other available genome sequences in the family Ornithinimicrobiaceae were all below the 70% threshold (19.6–33.6%), by contrast to the finding that strains HY006T and HY008 had a dDDH value of 99.9% and strains HY1745 and HY1793T shared 78.9%, indicating that they belong to the same species (Table S2). Similarly, the ANI values of strain HY006T with strains O. flavum CPCC 203535T and O. pratense GDMCC 1.1391T were 79.3% and 87.4%, respectively, which were below the threshold value (95%) for species delineation; strain HY1793T with its three closely related species (O. ciconiae H23M54T, O. cavernae KCTC 49018T and O. murale DSM22056T) were between 80.5–82.9 and ranged from 71.2 to 75.0% with other species in the genus Ornithinimicrobium. However, the ANI relatedness within each strain pair is 99.9% (HY006T and HY008) and 97.5% (HY1745 and HY1793T). Moreover, the dDDH and ANI values between strains HY006T and HY1793T were 20.4% and 74.4% respectively, suggesting that they belong to different species. In short, these results support the notion that these four strains represent two different novel species in the genus Ornithinimicrobium.
Moreover, the Clusters of Orthologous Groups (COG) database was used for the classification of the genes in the sequenced genomes (Figure 2A). The results revealed that the highest numbers of genes contained in these genomes were associated with transcription (COG-K), translation (COG-J), and DNA replication and repair (COG-L) for information storage and processing. For cellular processes and signaling, the genes involved in cell wall/membrane/envelope biogenesis (COG-M) and signal transduction mechanisms (COG-T) were commonly abundant in sequenced genomes. Based on our analysis of the genes associated with metabolism, we found that the species of the genus Ornithinimicrobium focused on amino acids transport and metabolism (COG-E) and carbohydrate transport and metabolism (COG-G), additionally, the inorganic ion transport and metabolism (COG-P), energy production and conversion (COG-C) and coenzyme transport and metabolism (COG-H) were also abundant in the bacterial genomes. However, the proportion of secondary metabolites biosynthesis, transport and catabolism (COG-Q) and nucleotide transport and metabolism (COG-F) were relatively small. Notably, a large quantity of the genes in these bacterial genomes were poorly characterized, and their functions remain to be identified (COG-S) or general function prediction only (COG-R) (Figure 2A).
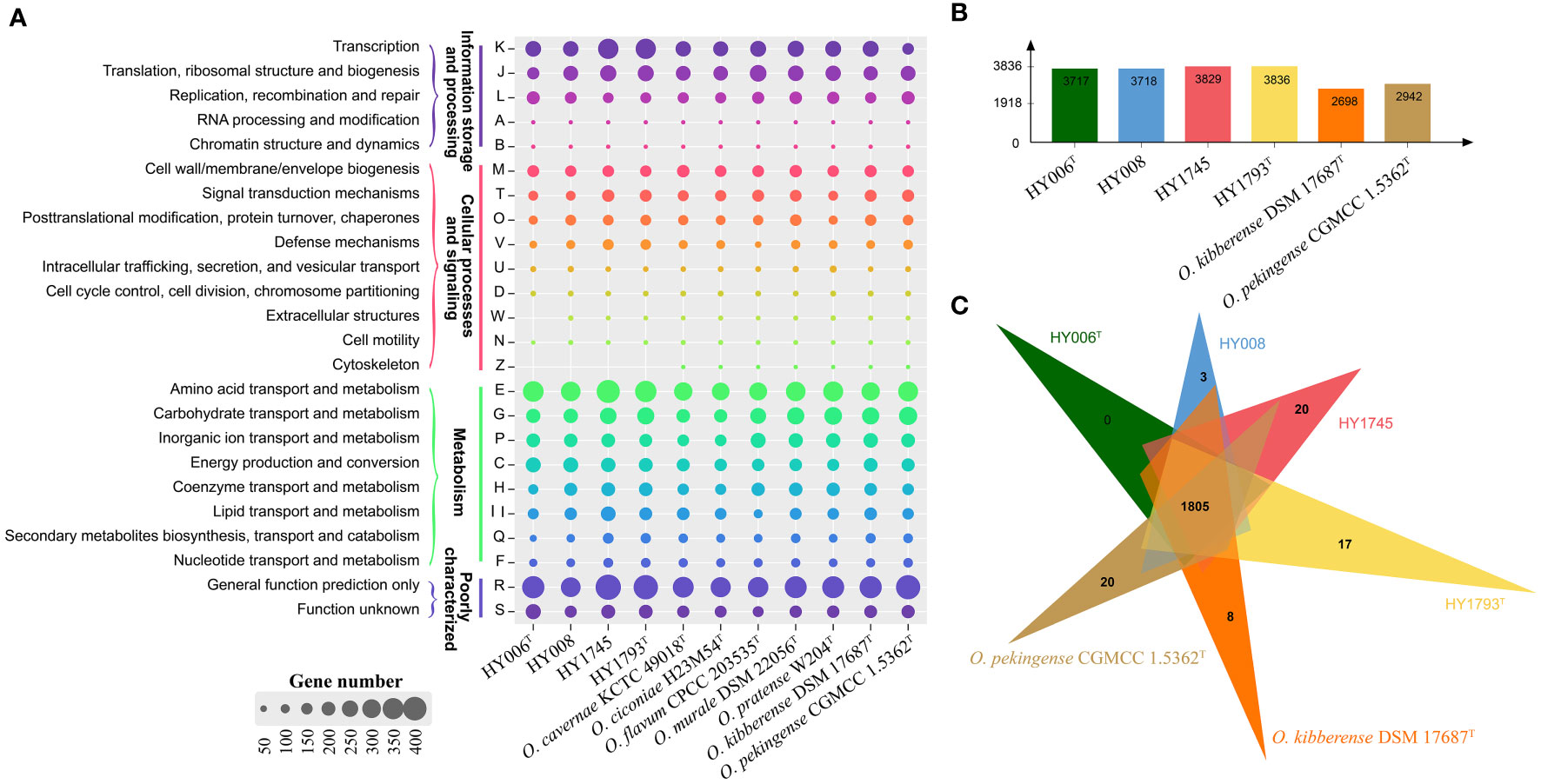
Figure 2 Whole-genome comparison and annotation of orthologous clusters across multiple strains. (A) Distributions of the COGs in the eleven sequenced genomes of genus Ornithinimicrobium; (B) The number of clusters in each strain; (C) Venn diagram displays the distribution of shared orthologous clusters among the six strains.
Furthermore, the Venn diagram displays the distribution of shared orthologous clusters among our four isolates and the other two strains (O. kibberense DSM 17687T and O. pekingense CGMCC 1.5362T, which should be deserved more attention because they were possessing cytotoxicity activity against to some mammary adenocarcinoma cell lines or causing ocular infection, respectively. In addition, MK-8(H4) was detected as the major predominant menaquinone, which was consistent with strains HY006T and HY1793T but different from the other strains isolated from Rousettus leschenaultia or Taphozous perforates. Furthermore, both of them were color in light yellow, which was also similar to species O. sufpigmenti). The six strains form clusters number between 2,698 to 3,836 (Figure 2B), and a total of 1805 clusters are shared with the above strains. In addition, there are 3, 20, 17, 8, and 20 single-copy gene clusters in strains HY008, HY1745, HY1793T, O. kibberense DSM 17687T, and O. pekingense CGMCC 1.5362T, respectively (Figure 2C). Additionally, our four strains and the above two strains have large segment sequence rearrangement. There are at least five locally collinear blocks, and the direction and arrange sequence are consistent, suggesting that genome evolution may experience a similar path (Figure S1).
Phenotypic, physiological and chemotaxonomic characteristics
Collectively, cells of strains HY006T, HY008, HY1745, and HY1793T were convex, opaque, and circular (0.5–1.0 mm in diameter) with smooth surfaces. Cells were Gram-stain-positive, catalase-positive, oxidase-negative, circle-shaped (HY006T 1.0 × 1.2 µm; HY1793T 1.0 × 1.2 µm) (Figure S2), non-motile. Interestingly, the color was totally different between the two type strains. The strain HY006T was yellow whereas strain HY1793T was cream, which may be associated to the production of carotenoids. Extraction of the HY006T cells with a solvent system showed that they accumulated two different carotenoids at significant concentrations (Figure S3). The contents of β-cryptoxanthin (C40H56O) and lycopene (C40H56) in strain HY006T were 26.0 µg/mL and 2.3 µg/mL, respectively. In the contrast, any carotenoid components were not found in the strain of HY1793T, which was consistent with its color (cream) but different from strain HY006T (yellow).
Optimal growth conditions for the strains HY006T and HY008 were 28–30°C (range, 15–40°C) for 5 days on BHI with 1.0% (w/v) NaCl [range, 0–7.0 (w/v)] at pH 8.0–8.5 (range, 7.0–11.0). Additionally, the optimal growth conditions of strains HY1745 and HY1793T were at 28°C (range, 15–37°C), pH 8.0 (range, pH 6.5–10.0), and with 1.0–1.5% (w/v) NaCl [up to 9.0% (w/v)] in aerobic. Notably, our strains were tolerant to 7.0% NaCl, while no growth of O. flavum CPCC 203535T was observed under this condition. However, O. cavernae KCTC 49018T, O. ciconiae JCM 33221T, O. murale DSM 22056T, and O. pratense GDMCC 1.1391T can grow well with 7.0% NaCl. The four isolates (HY006T, HY008, HY1745 and HY1793T) and their closely related strains (O. cavernae KCTC 49018T, O. ciconiae JCM 33221T, O. flavum CPCC 203535T, O. murale DSM 22056T and O. pratense GDMCC 1.1391T) were also not observed growth on pH value of lower than 6.0, which were different from strains O. pekingense DSM 21552T and O. kibberense DSM 17687T but consistent with most the species of Ornithinimicrobium (Huo et al., 2019; Fang et al., 2020; Cao et al., 2022). Also, strains HY006T and HY008 were positive for α-chymotrypsin, which were different from strains HY1745, HY1793T, O. cavernae KCTC 49018T, O. ciconiae JCM 33221T, O. flavum CPCC 203535T, O. pratense GDMCC 1.1391T but consistent with O. murale DSM 22056T. Conversely, alkaline phosphatase and cystine arylamidase were utilized by strains HY1745 and HY1793T whereas strains HY006T and HY008 were negative for them. The more differential biochemical characteristics of all the compared strains were shown in Table S5. As detailed in Figure S4, all strains positively assimilated aztreonam, d-serine and lithium chloride while tested with the Biolog GEN III MicroPlate. Of note is the strains HY006T and HY008 were positive for acetoacetic acid, d-arabitol, d-glucuronic acid, l-galactonic acid lactone, tetrazolium violet and α-hydroxy-butyric acid, distinctly different from the other two potential novel strains (HY1745 and HY1793T). Most notably, lincomycin was found to be utilized by strains HY1745 and HY1793T, which was consistent with strains O. cavernae CFH 30183T, O. murale DSM 22056T, O. kibberense K22-20T and O. panacihumi DCY118T (Mayilraj et al., 2006; Huo et al., 2019; Zhang et al., 2019). Conversely, strains HY006T, HY008 and the other four closely related strains (O. ciconiae JCM 33221T, O. flavum CPCC 203535T, and O. pratense GDMCC 1.1391T) were sensitive to lincomycin according to the results of Biolog GEN III MicroPlate (Figure S4). Furthermore, strain HY006T was resistant [minimum inhibitory concentration (MIC) ≥8.0 µg/mL] to chloramphenicol (MIC, >20.0 µg/mL), linezolid (MIC, 12 µg/mL), and intermediately resistant (MIC, 1.0–6.0 µg/mL) to erythromycin (MIC, 3.0 µg/mL) and tetracycline (MIC, 6.0 µg/mL), but susceptible to ceftriaxone, clindamycin levofloxacin, meropenem and vancomycin (MIC, ≤0.5 µg/mL). Additionally, strain HY1793T was resistant to erythromycin (MIC, 12.0 µg/mL), intermediately resistant to clindamycin (MIC, 2.0–3.0 µg/mL) and levofloxacin (MIC, 1.0 µg/mL) about the antibiotic sensitivity whereas susceptible to ceftriaxone, chloramphenicol, linezolid, meropenem, tetracycline and vancomycin (MIC, ≤0.5 µg/mL) (Figure S5). Of note, both strains contain the corresponding drug resistance genes in their genome but the value of identities is low (<60.0%), indicating that the resistant phenotype may be regulated by other different genes and will require more research to be identified.
The major cellular fatty acids (>20.0%) of our four isolates (HY006T, HY008, HY1745 and HY1793T) were iso-C15:0 and iso-C16:0, which were consistent with strain O. humiphilum HKI 0124T (Groth et al., 2001) and O. murale 01-Gi-040T (Kampfer et al., 2013). In addition, Summed Feature 9 (C16:010-methyl and/or iso-C17:1ω9C) was also accounted for a high proportion (>10.0%) in strains HY1745, HY1793T and other closely related strains (O. cavernae KCTC 49018T, O. ciconiae JCM 33221T, O. flavum CPCC 203535T, O. murale DSM 22056T, and O. pratense GDMCC 1.1391T) but lower in strains HY006T and HY008 (<6.5%). The detailed fatty acid profiles of the strains are presented in Table S6. MK-8(H4) was detected as the predominant menaquinone in strains HY006T and HY1793T, which was accounted for 59.0% and 93.3%, respectively. Furthermore, a significant amount of MK-8 (33.9%) was present in strain HY006T rather than MK-6 (6.7%) was shown in strain HY1793T, different from other species that had either simpler or more complicated compositions. For example, O. pekingense LW6T has been reported to contain partially saturated menaquinone MK-8(H2) (Liu et al., 2008); O. cerasi CPCC 203383T and O. flavum CPCC 203535T had MK-8(H4), MK-8(H2), and MK-8 as the predominant quinone (Fang et al., 2017; Fang et al., 2020). Both type strains (HY006T and HY1793T) had diphosphatidylglycerol (DPG), phosphatidylglycerol (PG), phosphatidyl inositol mannoside (PIM), two unidentified lipids (L1-2) and two unknown phosphoglycolipids (PGL1-2) in their polar lipid profiles (Figure S6), additionally, HY006T had two phospholipids (PL1-2) and two unidentified glycolipids (GL1-2) whereas HY1793T had five phospholipids (PL1-5) and five unidentified glycolipids (GL1-5). The whole-cell peptidoglycan of both type strains contained ornithine, alanine, glycine, and glutamic acid (Figure S7A), significantly, ornithine is the diagnostic diamino acid component within the cell wall of the genus Ornithinimicrobium. They also contained glucose in the whole cell sugar, however, ribose was only detected in strain HY1793T (Figure S7B).
Discussion
Compared with viruses, there are few studies on the microorganism carried by bats, particularly the reports of new bacterial species that are straightforward to be neglected (Muhldorfer, 2013). Our laboratory has investigated the intestinal and fecal flora of bats since 2018 to identify potentially pathogenic microbial species and dissect the process of disease transmission (Huang et al., 2022c). So far, we have published over ten new species of bacteria isolated from bat feces, among which Brevibacterium zhoupengii (Huang et al., 2022a), Microbacterium fandaimingii (Zhou et al., 2021), Gordonia zhenghanii, G. liuliyuniae (Huang et al., 2022b), Tomitella gaofuii and T. fengzijianii (Huang et al., 2022d) belong to the phylum Actinobacteria. As previously described, actinomycete bacteria are highly prevalent in Rousettus leschenaultia and Taphozous perforates through traditional isolation methods. Though O. sufpigmenti and O. faecis belongs to Actinobacteria, it differ from other actinobacterial strains isolated from R. leschenaultia and T. perforates by the presence of MK-8(H4) as major menaquinone. In addition, the optimal temperature of actinomycetes isolated from the faeces of Rousettus leschenaultia and/or Taphozous perforates is about 28°C, different from the strain Apibacter raozihei HY039 (belonged to Bacteroidota) best growth in 35°C. Furthermore, most of them are Gram-stain-positive, have high G+C content, and are creamy in color. However, Ornithinimicrobium sufpigmenti and O. faecis were observed to grow on the plates higher than 37°C, which was distinct from other actinomycetes but similarity to Apibacter raozihei (Table S7).
Moreover, the fecal microbiota in bats was found to be diverse with predominant bacterial phylum Proteobacteria and Firmicutes (Sun et al., 2020; Huang et al., 2022c). The presence of opportunistic pathogens Citrobacter freundii, Escherichia coli, Enterococcus faecalis, Serratia fonticola, Shigella flexneri, and Rahnella aquatilis were also recorded (Muhldorfer, 2013; Wolkers-Rooijackers et al., 2019). However, there is no proof that bats are a major vector for the transmission of bacterial zoonotic diseases (Wolkers-Rooijackers et al., 2019). Interestingly, Ornithinimicrobium pekingense has been reported to cause ocular infection by Borsali (Borsali et al., 2011). There are nine and ten predicted virulence factors in strains O. kibberense DSM 17687T and O. pekingense CGMCC 1.5362T, respectively (Table S4). Specifically, iron-dependent repressor and activator ideR (VFDB ID: VFG001406) was dominantly found in these two strains whereas absent in our four isolates. However, no specific virulence factor related genes or molecular mechanism can explain their potential pathogenicity according to searching against the virulence factor database (http://www.mgc.ac.cn/VFs/) and annotating by the RAST server. Two virulence factor-like genes, namely sodA (superoxide dismutase, VFDB ID: VFG001421) and HSI-3 (type VI secretion system ATPase TssH, VFDB ID: VFG041043) were discovered among all the compared strains except for strain HY1745. Interestingly, superoxide dismutase is important for Mycobacterium tuberculosis survival during infection (Edwards et al., 2001; Smith, 2003). The type VI secretion system (T6SS) participates in interbacterial competition as well as pathogenesis, it is a multiprotein complex widespread in Proteobacteria and dedicated to the delivery of toxins into both prokaryotic and eukaryotic cells, such as the type VI secretion system of Pseudomonas aeruginosa targets a toxin to bacteria (Hood et al., 2010; Journet and Cascales, 2016). Therefore, more research is needed to deepen the understanding of the genus Ornithinimicrobium. About the existence of Ornithinimicrobium sp. within these bats, we considered that these Ornithinimicrobium spp. might be a passing flora, probably not a long-term colonizer in the bat gut.
Conclusion
Based on evidence from the phylogenetic, genomic, physiological, biochemical, as well as chemotaxonomic findings suggest that our four strains represent two novel species of the genus Ornithinimicrobium, for which the names Ornithinimicrobium sufpigmenti (suf.pig.men’ti, N.L. neut. adj. sufpigmenti, referring to the production of pigment of β-cryptoxanthin and lycopene) sp. nov. (HY006T and HY008) and Ornithinimicrobiumfaecis (faeˊcis. L. gen. fem. n. faecis of faeces, as the organism was found in bat faeces) sp. nov. (HY1745 and HY1793T) are proposed, respectively. The type strains are HY006T (=CGMCC 1.16565T =JCM 33397T) and HY1793T (=CGMCC 1.19143T =JCM 34881T), respectively, were isolated from the feces of Rousettus leschenaultia in 2018, which were collected at Chuxiong Yi Autonomous Prefecture of Yunnan Province in 2013, China.
Data availability statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/Supplementary Material.
Ethics statement
The ethical practice was approved by the Ethical Committee of the National Institute for Communicable Disease Control and Prevention, Chinese Center for Disease Control and Prevention (NO: ICDC-2016004).
Author contributions
JX and JJ conceived the study. YH, SZ, YT, WL, SL and DJ performed the sampling, isolating and sequencing. YH, JY, JP, JJ and JX analyzed the data and drafted the manuscript. HZ and LL supervised the data analysis. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by grants from the National Key R&D Program of China (2019YFC1200501 and 2019YFC1200505) and Research Units of Discovery of Unknown Bacteria and Function (2018RU010).
Acknowledgments
We would like to express our appreciation to Professors Zhengli Shi and Peng Zhou (Wuhan Institute of Virology, Chinese Academy of Sciences) for sharing the bat fecal samples.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fcimb.2023.1093407/full#supplementary-material
Abbreviations
ANI, average nucleotide identity; BHI, brain-heart infusion; dDDH, digital DNA-DNA hybridization; ML, maximum likelihood; MP, maximum parsimony; NJ, neighbor-joining.
References
Alcock, B. P., Huynh, W., Chalil, R., Smith, K. W., Raphenya, A. R., Wlodarski, M. A., et al. (2022). CARD 2023: Expanded curation, support for machine learning, and resistome prediction at the comprehensive antibiotic resistance database. Nucleic Acids Res 51, D690–D699. doi: 10.1093/nar/gkac920
Austrian, R. (1960). The gram stain and the etiology of lobar pneumonia, an historical note. Bacteriol Rev. 24, 261–265. doi: 10.1128/br.24.3.261-265.1960
Aziz, R. K., Bartels, D., Best, A. A., DeJongh, M., Disz, T., Edwards, R. A., et al. (2008). The RAST server: rapid annotations using subsystems technology. BMC Genomics 9, 75. doi: 10.1186/1471-2164-9-75
Bai, X., Xiong, Y., Lu, S., Jin, D., Lai, X., Yang, J., et al. (2016). Streptococcus pantholopis sp. nov., isolated from faeces of the Tibetan antelope (Pantholops hodgsonii). Int. J. Syst. Evol. Microbiol. 66, 3281–3286. doi: 10.1099/ijsem.0.001189
Berlin, K., Koren, S., Chin, C. S., Drake, J. P., Landolin, J. M., Phillippy, A. M. (2015). Assembling large genomes with single-molecule sequencing and locality-sensitive hashing. Nat. Biotechnol. 33, 623–630. doi: 10.1038/nbt.3238
Borsali, E., Le Bouter, A., Abdiche, G., Goldschmdt, P., Legrand, P., Batellier, L., et al. (2011). Ornithinimicrobium pekingense ocular infection. Med. Mal Infect. 41, 345–346. doi: 10.1016/j.medmal.2010.12.015
Brown, D. F., Brown, L. (1991). Evaluation of the e test, a novel method of quantifying antimicrobial activity. J. Antimicrob. Chemother. 27, 185–190. doi: 10.1093/jac/27.2.185
Cao, W. R., Yu, X. R., Sun, Y. Y., Jiang, M. Y., Xu, X. D., Li, Y. J. (2022). Ornithinimicrobium laminariae sp. nov., isolated from the kelp Laminaria japonica. Int. J. Syst. Evol. Microbiol. 72. doi: 10.1099/ijsem.0.005187
Castanera, R., Ruggieri, V., Pujol, M., Garcia-Mas, J., Casacuberta, J. M. (2019). An improved melon reference genome with single-molecule sequencing uncovers a recent burst of transposable elements with potential impact on genes. Front. Plant Sci. 10. doi: 10.3389/fpls.2019.01815
Charles, T. P., Brian, J. T., George, M. G. (2019). International code of nomenclature of prokaryotes. Int. J. Syst. Evol. Microbiol. 69 (1A), S1–S111. doi: 10.1099/ijsem.0.000778
Darling, A. E., Mau, B., Perna, N. T. (2010). progressiveMauve: multiple genome alignment with gene gain, loss and rearrangement. PloS One 5, e11147. doi: 10.1371/journal.pone.0011147
Diaz-Cardenas, C., Cantillo, A., Rojas, L. Y., Sandoval, T., Fiorentino, S., Robles, J., et al. (2017). Microbial diversity of saline environments: searching for cytotoxic activities. AMB Express 7, 223. doi: 10.1186/s13568-017-0527-6
Edwards, K. M., Cynamon, M. H., Voladri, R. K., Hager, C. C., DeStefano, M. S., Tham, K. T., et al. (2001). Iron-cofactored superoxide dismutase inhibits host responses to mycobacterium tuberculosis. Am. J. Respir. Crit. Care Med. 164, 2213–2219. doi: 10.1164/ajrccm.164.12.2106093
Fang, X. M., Du, H. J., Bai, J. L., He, W. N., Li, J., Wang, H., et al. (2020). Ornithinimicrobium cerasi sp. nov., isolated from the fruit of Cerasus pseudocerasus and emended description of the genus Ornithinimicrobium. Int. J. Syst. Evol. Microbiol. 70, 1691–1697. doi: 10.1099/ijsem.0.003957
Fang, X. M., Yan, D., Bai, J. L., Su, J., Liu, H. Y., Ma, B. P., et al. (2017). Ornithinimicrobium flavum sp. nov., isolated from the leaf of Paris polyphylla. Int. J. Syst. Evol. Microbiol. 67, 4541–4545. doi: 10.1099/ijsem.0.002328
Fu, L., Niu, B., Zhu, Z., Wu, S., Li, W. (2012). CD-HIT: accelerated for clustering the next-generation sequencing data. Bioinformatics 28, 3150–3152. doi: 10.1093/bioinformatics/bts565
Ge, X. Y., Li, J. L., Yang, X. L., Chmura, A. A., Zhu, G., Epstein, J. H., et al. (2013). Isolation and characterization of a bat SARS-like coronavirus that uses the ACE2 receptor. Nature 503, 535–538. doi: 10.1038/nature12711
Ge, X., Li, Y., Yang, X., Zhang, H., Zhou, P., Zhang, Y. (2012). Metagenomic analysis of viruses from bat fecal samples reveals many novel viruses in insectivorous bats in China. J. Virol. 86, 4620–4630. doi: 10.1128/JVI.06671-11
Groth, I., Schumann, P., Weiss, N., Schuetze, B., Augsten, K., Stackebrandt, E. (2001). Ornithinimicrobium humiphilum gen. nov., sp. nov., a novel soil actinomycete with l-ornithine in the peptidoglycan. Int. J. Syst. Evol. Microbiol. 51, 81–87. doi: 10.1099/00207713-51-1-81
Guindon, S., Gascuel, O. (2003). A simple, fast, and accurate algorithm to estimate large phylogenies by maximum likelihood. Syst. Biol. 52, 696–704. doi: 10.1080/10635150390235520
Harper, J. J., Davis, G. H. G. (1979). Two-dimensional thin-layer chromatography for amino acid analysis of bacterial cell walls. Int. J. Syst. Evol. Microbiol. 29, 56–58. doi: 10.1099/00207713-29-1-56
Hasegawa, T., Takizawa, M., Tanida, S. (1983). A rapid analysis for chemical grouping of aerobic actinomycetes. J. Gen. Appl. Microbiol. 29, 319–322. doi: 10.2323/jgam.29.319
Hood, R. D., Singh, P., Hsu, F., Guvener, T., Carl, M. A., Trinidad, R. R., et al. (2010). A type VI secretion system of Pseudomonas aeruginosa targets a toxin to bacteria. Cell Host Microbe 7, 25–37. doi: 10.1016/j.chom.2009.12.007
Hu, B., Guo, H., Zhou, P., Shi, Z. L. (2021). Characteristics of SARS-CoV-2 and COVID-19. Nat. Rev. Microbiol. 19, 141–154. doi: 10.1038/s41579-020-00459-7
Hu, H. Y., Lim, B. R., Goto, N., Fujie, K. (2001). Analytical precision and repeatability of respiratory quinones for quantitative study of microbial community structure in environmental samples. J. Microbiol. Methods 47, 17–24. doi: 10.1016/s0167-7012(01)00286-x
Huang, Y., Dong, L., Gong, J., Yang, J., Lu, S., Lai, X. H., et al. (2022a). Phenotypic and genomic characteristics of Brevibacterium zhoupengii sp. nov., a novel halotolerant actinomycete isolated from bat feces. J. Microbiol 60, 977–985. doi: 10.1007/s12275-022-2134-8
Huang, Y., Gong, J., Dong, L., Yang, J., Lu, S., Lai, X. H., et al. (2022b). Gordonia zhenghanii sp. nov. and gordonia liuliyuniae sp. nov., isolated from bat faeces. Int. J. Syst. Evol. Microbiol. 72 (10). doi: 10.1099/ijsem.0.005579
Huang, Y., Sun, Y., Huang, Q., Lv, X., Pu, J., Zhu, W., et al. (2022c). The threat of potentially pathogenic bacteria in the feces of bats. Microbiol. Spectr. 10, e0180222. doi: 10.1128/spectrum.01802-22
Huang, Y., Wang, X., Yang, J., Lu, S., Lai, X.-H., Jin, D., et al. (2019). Nocardioides yefusunii sp. nov., isolated from Equus kiang (Tibetan wild ass) faeces. Int. J. Syst. Evol. Microbiol. 69, 3629–3635. doi: 10.1099/ijsem.0.003674
Huang, Y., Yang, J., Lu, S., Lai, X. H., Jin, D., Zhou, J., et al. (2022d). Morphological and genomic characteristics of two novel halotolerant actinomycetes, tomitella gaofuii sp. nov. and Tomitella fengzijianii sp. nov. isolated from bat faeces. Syst. Appl. Microbiol. 45 (2), 126294. doi: 10.1016/j.syapm.2022.126294
Huo, Y., Kang, J. P., Ahn, J. C., Yang, D. U., Yang, D. C. (2019). Ornithinimicrobium panacihumi sp. nov., antagonistic bacteria against root rot fungal pathogens, isolated from cultivated ginseng soil. Curr. Microbiol. 76, 22–28. doi: 10.1007/s00284-018-1579-9
Huson, D. H., Scornavacca, C. (2012). Dendroscope 3: An interactive tool for rooted phylogenetic trees and networks. Syst. Biol. 61, 1061–1067. doi: 10.1093/sysbio/sys062
Iglesias-Caballero, M., Juste, J., Vazquez-Moron, S., Falcon, A., Aznar-Lopez, C., Ibanez, C., et al. (2018). New adenovirus groups in Western palaearctic bats. Viruses 10, 443. doi: 10.3390/v10080443
Irakli, M. N., Samanidou, V. F., Papadoyannis, I. N. (2011). Development and validation of an HPLC method for the simultaneous determination of tocopherols, tocotrienols and carotenoids in cereals after solid-phase extraction. J. Sep Sci. 34, 1375–1382. doi: 10.1002/jssc.201100077
Jiang, H. H., Qin, S. Y., Wang, W., He, B., Hu, T. S., Wu, J. M., et al. (2014). Prevalence and genetic characterization of Toxoplasma gondii infection in bats in southern China. Vet. Parasitol. 203, 318–321. doi: 10.1016/j.vetpar.2014.04.016
Jin, D., Chen, C., Li, L., Lu, S., Li, Z., Zhou, Z., et al. (2013). Dynamics of fecal microbial communities in children with diarrhea of unknown etiology and genomic analysis of associated Streptococcus lutetiensis. BMC Microbiol. 13, 141. doi: 10.1186/1471-2180-13-141
Journet, L., Cascales, E. (2016). The type VI secretion system in Escherichia coli and related species. EcoSal Plus 7. doi: 10.1128/ecosalplus.ESP-0009-2015
Kampfer, P., Glaeser, S. P., Schafer, J., Lodders, N., Martin, K., Schumann, P. (2013). Ornithinimicrobium murale sp. nov., isolated from an indoor wall colonized by moulds. Int. J. Syst. Evol. Microbiol. 63, 119–123. doi: 10.1099/ijs.0.035873-0
Kolaczkowski, B., Thornton, J. W. (2004). Performance of maximum parsimony and likelihood phylogenetics when evolution is heterogeneous. Nature 431, 1980–1984. doi: 10.1038/nature02917
Komagata, K., Suzuki, K.-I. (1988). 4 lipid and cell-wall analysis in bacterial systematics. Methods Microbiol. 19, 161–207. doi: 10.1016/S0580-9517(08)70410-0
Kumar, S., Stecher, G., Li, M., Knyaz, C., Tamura, K. (2018). MEGA X: molecular evolutionary genetics analysis across computing platforms. Mol. Biol. Evol. 35, 1547–1549. doi: 10.1093/molbev/msy096
Kuzmin, I. V., Mayer, A. E., Niezgoda, M., Markotter, W., Agwanda, B., Breiman, R. F., et al. (2010). Shimoni bat virus, a new representative of the Lyssavirus genus. Virus Res. 149, 197–210. doi: 10.1016/j.virusres.2010.01.018
Lee, S. Y., Sung, H., Kim, P. S., Kim, H. S., Lee, J. Y., Lee, J. Y., et al. (2021). Description of Ornithinimicrobium ciconiae sp. nov., and Ornithinimicrobium avium sp. nov., isolated from the faeces of the endangered and near-threatened birds. J. Microbiol. 59, 978–987. doi: 10.1007/s12275-021-1323-1
Letunic, I., Bork, P. (2021). Interactive tree of life (iTOL) v5: an online tool for phylogenetic tree display and annotation. Nucleic Acids Res. 49 (W1), W293–W296. doi: 10.1093/nar/gkab301
Liu, L. Z., Liu, Y., Chen, Z., Liu, H. C., Zhou, Y. G., Liu, Z. P. (2013). Ornithinimicrobium tianjinense sp. nov., isolated from a recirculating aquaculture system. Int. J. Syst. Evol. Microbiol. 63, 4489–4494. doi: 10.1099/ijs.0.052514-0
Liu, X. Y., Wang, B. J., Jiang, C. Y., Liu, S. J. (2008). Ornithinimicrobium pekingense sp. nov., isolated from activated sludge. Int. J. Syst. Evol. Microbiol. 58, 116–119. doi: 10.1099/ijs.0.65229-0
Liu, B., Zheng, D., Jin, Q., Chen, L., Yang, J. (2019). VFDB 2019: a comparative pathogenomic platform with an interactive web interface. Nucleic Acids Res. 47, D687–D692. doi: 10.1093/nar/gky1080
Mayilraj, S., Saha, P., Suresh, K., Saini, H. S. (2006). Ornithinimicrobium kibberense sp. nov., isolated from the Indian Himalayas. Int. J. Syst. Evol. Microbiol. 56, 1657–1661. doi: 10.1099/ijs.0.64138-0
McCarthy, A. (2010). Third generation DNA sequencing: pacific biosciences’ single molecule real time technology. Chem. Biol. 17, 675–676. doi: 10.1016/j.chembiol.2010.07.004
Meier-Kolthoff, J. P., Carbasse, J. S., Peinado-Olarte, R. L., Goker, M. (2022). TYGS and LPSN: A database tandem for fast and reliable genome-based classification and nomenclature of prokaryotes. Nucleic Acids Res. 50, D801–D807. doi: 10.1093/nar/gkab902
Meléndez-Martínez, A. J., Escudero-Gilete, M. L., Vicario, I. M., Heredia, F. J. (2010). Study of the influence of carotenoid structure and individual carotenoids in the qualitative and quantitative attributes of orange juice color. Food Res. Int. 43, 1289–1296. doi: 10.1016/j.foodres.2010.03.012
Muhldorfer, K. (2013). Bats and bacterial pathogens: a review. Zoonoses Public Health 60, 93–103. doi: 10.1111/j.1863-2378.2012.01536.x
Nouioui, I., Carro, L., García-López, M., Meier-Kolthoff, J., Woyke, T., Kyrpides, N., et al. (2018). Genome-based taxonomic classification of the phylum actinobacteria. Front. Microbiol. 22. doi: 10.3389/fmicb.2018.02007
Overbeek, R., Olson, R., Pusch, G. D., Olsen, G. J., Davis, J. J., Disz, T., et al. (2014). The SEED and the rapid annotation of microbial genomes using subsystems technology (RAST). Nucleic Acids Res. 42, D206–D214. doi: 10.1093/nar/gkt1226
Price, M. N., Dehal, P. S., Arkin, A. P. (2009). FastTree: computing large minimum evolution trees with profiles instead of a distance matrix. Mol. Biol. Evol. 26, 1641–1650. doi: 10.1093/molbev/msp077
Ramaprasad, E. V., Sasikala, C., Ramana Ch, V. (2015). Ornithinimicrobium algicola sp. nov., a marine actinobacterium isolated from the green alga of the genus ulva. Int. J. Syst. Evol. Microbiol. 65, 4627–4631. doi: 10.1099/ijsem.0.000624
Saitou, N., Nei, M. (1987). The neighbor-joining method: a new method for reconstructing phylogenetic trees. Mol. Biol. Evol. 4, 406–425. doi: 10.1093/oxfordjournals.molbev.a040454
Sasser, M. (1990). Identification of bacteria by gas chromatography of cellular fatty acids. MIDI Technical Note 101. MIDI Inc., Newark, Delaware, USA. Available at: https://www.researchgate.net/publication/284025789.
Smith, I. (2003). Mycobacterium tuberculosis pathogenesis and molecular determinants of virulence. Clin. Microbiol. Rev. 16, 463–496. doi: 10.1128/CMR.16.3.463-496.2003
Staneck, J. L., Roberts, G. D. (1974). Simplified approach to identification of aerobic actinomycetes by thin-layer chromatography. Appl. Microbiol. 28, 226–231. doi: 10.1128/am.28.2.226-231.1974
Sun, D. L., Gao, Y. Z., Ge, X. Y., Shi, Z. L., Zhou, N. Y. (2020). Special features of bat microbiota differ from those of terrestrial mammals. Front. Microbiol. 11. doi: 10.3389/fmicb.2020.01040
Tan, C. W., Yang, X., Anderson, D. E., Wang, L. F. (2021). Bat virome research: the past, the present and the future. Curr. Opin. Virol. 49, 68–80. doi: 10.1016/j.coviro.2021.04.013
Tatusov, R. L., Koonin, E. V., Lipman, D. J. (1997). A genomic perspective on protein families. Science 278, 631–637. doi: 10.1126/science.278.5338.631
Thompson, J. D., Higgins, D. G., Gibson, T. J. (1994). CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 22, 4673–4680. doi: 10.1093/nar/22.22.4673
Wang, X., Yang, J., Lu, S., Lai, X. H., Jin, D., Pu, J., et al. (2018). Nocardioides houyundeii sp. nov., isolated from Tibetan antelope faeces. Int. J. Syst. Evol. Microbiol. 68, 3874–3880. doi: 10.1099/ijsem.0.003076
Witkowski, P. T., Klempa, B., Ithete, N. L., Auste, B., Mfune, J. K., Hoveka, J., et al. (2014). Hantaviruses in Africa. Virus Res. 187, 34–42. doi: 10.1016/j.virusres.2013.12.039
Wolkers-Rooijackers, J. C.M., Rebmann, K., Bosch, T., Hazeleger, W. C. (2019). Fecal bacterial communities in insectivorous bats from the Netherlands and their role as a possible vector for foodborne diseases. Acta Chiropterol. 20, 475–483. doi: 10.3161/15081109ACC2018.20.2.017
Xu, J. (2019). Reverse microbial etiology: A research field for predicting and preventing emerging infectious diseases caused by an unknown microorganism. J. Biosaf Biosecur 1, 19–21. doi: 10.1016/j.jobb.2018.12.005
Xu, L., Dong, Z., Fang, L., Luo, Y., Wei, Z., Guo, H., et al. (2019). OrthoVenn2: a web server for whole-genome comparison and annotation of orthologous clusters across multiple species. Nucleic Acids Res. 47, W52–W58. doi: 10.1093/nar/gkz333
Xu, Y., Xu, X., Lan, R., Xiong, Y., Ye, C., Ren, Z., et al. (2013). An O island 172 encoded RNA helicase regulates the motility of Escherichia coli O157:H7. PloS One 8, e64211. doi: 10.1371/journal.pone.0064211
Yang, X. L., Zhang, Y. Z., Jiang, R. D., Guo, H., Zhang, W., Li, B., et al. (2017). Genetically diverse filoviruses in Rousettus and eonycteris spp. bats, china 2009 and 2015. Emerg. Infect. Dis. 23, 482–486. doi: 10.3201/eid2303.161119
Yoon, S. H., Ha, S. M., Kwon, S., Lim, J., Kim, Y., Seo, H., et al. (2017a). Introducing EzBioCloud: A taxonomically united database of 16S rRNA gene sequences and whole-genome assemblies. Int. J. Syst. Evol. Microbiol. 67, 1613–1617. doi: 10.1099/ijsem.0.001755
Yoon, S. H., Ha, S. M., Lim, J., Kwon, S., Chun, J. (2017b). A large-scale evaluation of algorithms to calculate average nucleotide identity. Antonie Van Leeuwenhoek 110, 1281–1286. doi: 10.1007/s10482-017-0844-4
Zerbino, D. R., Birney, E. (2008). Velvet: algorithms for de novo short read assembly using de bruijn graphs. Genome Res. 18, 821–829. doi: 10.1101/gr.074492.107
Zhang, L. Y., Ming, H., Meng, X. L., Fang, B. Z., Jiao, J. Y., Salam, N., et al. (2019). Ornithinimicrobium cavernae sp. nov., an actinobacterium isolated from a karst cave. Antonie Van Leeuwenhoek 112, 179–186. doi: 10.1007/s10482-018-1141-6
Zhao, H., Yuan, L., Zhu, D., Sun, B., Du, J., Wang, J. (2022). Alterations and mechanism of gut microbiota in graves’ disease and hashimoto’s thyroiditis. Pol. J. Microbiol. 71, 173–189. doi: 10.33073/pjm-2022-016
Zhou, J., Huang, Y., Yang, J., Lai, X. H., Jin, D., Lu, S., et al. (2021). Microbacterium chengjingii sp. nov. and microbacterium fandaimingii sp. nov., isolated from bat faeces of Hipposideros and Rousettus species. Int. J. Syst. Evol. Microbiol., 71. doi: 10.1099/ijsem.0.004858
Zhou, P., Yang, X. L., Wang, X. G., Hu, B., Zhang, L., Zhang, W., et al. (2020). A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature 579, 270–273. doi: 10.1038/s41586-020-2012-7
Keywords: Ornithinimicrobium sufpimenti, Ornithinimicrobium faecis, bats, genome characteristics, carotenoids, antibiotic sensitivity
Citation: Huang Y, Zhang S, Tao Y, Yang J, Lu S, Jin D, Pu J, Luo W, Zheng H, Liu L, Jiang J-f and Xu J (2023) Morphological and genomic characteristics of two novel actinomycetes, Ornithinimicrobium sufpigmenti sp. nov. and Ornithinimicrobium faecis sp. nov. isolated from bat faeces (Rousettus leschenaultia and Taphozous perforates). Front. Cell. Infect. Microbiol. 13:1093407. doi: 10.3389/fcimb.2023.1093407
Received: 09 November 2022; Accepted: 31 January 2023;
Published: 14 February 2023.
Edited by:
Michael E. von Fricken, George Mason University, United StatesReviewed by:
A Alwin Prem Anand, University of Tübingen, GermanySung-min Ha, University of California, Los Angeles, United States
Copyright © 2023 Huang, Zhang, Tao, Yang, Lu, Jin, Pu, Luo, Zheng, Liu, Jiang and Xu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jianguo Xu, eHVqaWFuZ3VvQGljZGMuY24=; Jia-fu Jiang, amlhbmdqZjIwMDhAMTM5LmNvbQ==