 Rui Yang
Rui Yang Xiaojuan Zhang
Xiaojuan Zhang
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Cell. Infect. Microbiol., 15 September 2022
Sec. Bacteria and Host
Volume 12 - 2022 | https://doi.org/10.3389/fcimb.2022.984835
Sepsis is a significant cause of mortality in critically ill patients. Acute lung injury (ALI) is a leading cause of death in these patients. Endothelial cells exposed to the bacterial endotoxin lipopolysaccharide (LPS) can progress into pyroptosis, a programmed lysis of cell death triggered by inflammatory caspases. It is characterized by lytic cell death induced by the binding of intracellular LPS to caspases 4/5 in human cells and caspase-11 in mouse cells. In mice,caspase-11-dependent pyroptosis plays an important role in endotoxemia. HMGB1 released into the plasma binds to LPS and is internalized into lysosomes in endothelial cells via the advanced glycation end product receptor. In the acidic lysosomal environment, HMGB1 permeates the phospholipid bilayer, which is followed by the leakage of LPS into the cytoplasm and the activation of caspase-11. Heparin is an anticoagulant widely applied in the treatment of thrombotic disease. Previous studies have found that heparin could block caspase-11-dependent inflammatory reactions, decrease sepsis-related mortality, and reduce ALI, independent of its anticoagulant activity. Heparin or modified heparin with no anticoagulant property could inhibit the alarmin HMGB1-LPS interactions, minimize LPS entry into the cytoplasm, and thus blocking caspase-11 activation. Heparin has been studied in septic ALI, but the regulatory mechanism of pulmonary endothelial cell pyroptosis is still unclear. In this paper, we discuss the potential novel role of heparin in the treatment of septic ALI from the unique mechanism of pulmonary endothelial cell pyroptosis.
Sepsis is a significant cause of mortality. Acute lung injury (ALI) is a leading cause of death in critically ill patients with sepsis. Acute respiratory distress syndrome (ARDS) is the most serious form of ALI (Ashbaugh et al., 1967; Matthay et al., 2019). Its in-hospital mortality rate is approximately 35% to 46% (Bellani et al., 2016; Sauer et al., 2021; Peukert et al., 2021). During sepsis, infiltration of inflammatory cells in the lung can lead to irreversible damage, which finally develops into ALI or even ARDS (Kumar and Chhibber, 2011). Both direct lung epithelial injury and indirect endothelial cell injury can cause sepsis-related ALI/ARDS (Englert et al., 2019; Huppert et al., 2019). Pro-inflammatory cytokines can activate the pulmonary endothelial cells with subsequent increased expressions of e-selectin or endothelium-endothelial adhesion molecule-1 (ELam-1), or leucocyte adhesion molecule-2 (lecMA-2), which further induces neutrophil infiltration. This, together with the extensive endothelial cell death and disintegration of endothelial adhesion junctions, can contribute to pulmonary endothelial barrier disruption and the development of ALI (Mehta and Malik, 2006; Gong et al., 2015; Matthay et al., 2019).
Recent studies suggested that the caspase-11 signaling pathway participates in the pathogenesis of sepsis (Kayagaki et al., 2011; Kayagaki et al., 2015; Cheng et al., 2017; Deng et al., 2018). Caspase-11 can be found in various cell types, including pulmonary endothelial cells and macrophages (Cheng et al., 2017; Deng et al., 2018). Caspase-11 activated by the intracellular endotoxin lipopolysaccharide (LPS) cleaves Gasdermin D (GSDMD) into polypeptides to form a nanopore on the cytoplasmic membrane (Kayagaki et al., 2015; He et al., 2015; Shi et al., 2015; Ding et al., 2016). This process not only results in a lysis form of programmed cell death named pyroptosis but also activates living cells to secret interleukin (IL)-1 (He et al., 2015; Ding et al., 2016; Zanoni et al., 2016; Evavold et al., 2018). ECs demonstrated lytic cell death, activation of Gsdmd, and release of the proinflammatory cytokine IL-1β, processes that are dependent on caspase-4/5 in human ECs and caspase-11 in mouse Ecs (Cheng et al., 2017).
Activations of caspase-11 and GSDMD are facilitated by high mobility group box 1 (HMGB1). HMGB1 is a 25 kD nuclear protein found in all cell types and is highly conserved in mammals. The intracellular functions of HMGB1 include gene transcription and chromatin repair regulation (Müller et al., 2001; Lotze and Tracey, 2005; Davalos et al., 2013). Extracellular HMGB1 carries the characteristic alarm protein functions to activate innate immunity, and HMGB1 can be actively or passively released after cell death during endotoxemia or sepsis. During an infection, LPS triggers hepatocytes to release HMGB1 into the circulation (Deng et al., 2018). The plasma HMGB1 attaches to extracellular LPS to form advanced glycation end products (RAGE) in the cytoplasm, which induces lysosome rupture by receptor-mediated internalization of LPS. HMGB1 then causes lysosomal rupture and ultimately activates cleavage of caspase-11 and GSDMD (Deng et al., 2018). Animal studies have shown that either deficiency in caspase-11, loss of GSDMD, neutralizing extracellular HMGB1, or deletion of HMGB1 in hepatocytes can improve survival in experimental septic models (Cheng et al., 2017; Deng et al., 2018).
Heparin was discovered in 1916 and is traditionally used as an anticoagulant in the treatment of thrombotic diseases, such as various venous thromboembolism (Hirsh and Levine, 1992; Li and Ma, 2017). Heparin can bind to the lysine residues in antithrombin and induce irreversible conformational changes in arginine reaction sites (Rezaie et al., 2004; Yang et al., 2004), which leads to a more than 100-fold increase in antithrombin activity (Chuang et al., 2001; Rezaie et al., 2004). In addition to its anticoagulant function, heparin was found to have other properties, such as protease modulation and anti-complement and anti-inflammatory activities (Davidson et al., 2002; Hoppensteadt et al., 2008). Heparin therapy has been shown to improve the prognosis of sepsis and alleviate lung injury by inhibiting caspase-11 signaling (Cornet et al., 2007; Li et al., 2011; Liu et al., 2014; Wang et al., 2014; Zarychanski et al., 2015; Fan et al., 2016; Li and Ma, 2017; Liu et al., 2019; Tang et al., 2021). This article proposes a mechanism for how heparin alleviates sepsis-induced ALI from the perspective of pulmonary endothelial cell pyroptosis.
The pathophysiology of lung injury mainly includes inflammation disorder and increased permeability of the lung endothelium and epithelium. In ARDS, the endothelium permeability is increased allowing movement of fluid and protein through the pulmonary vascular endothelium, leading to interstitial edema. Increased permeability of neutrophils and red blood cells (causing them to accumulate in the alveolar space) is a hallmark of ARDS (Matthay et al., 2019; Lindsey et al., 2019; Gerber et al., 2020). Cardiac outflow goes directly into the lungs and the pulmonary tissues. As a result, pulmonary endothelial cells are constantly the subject of injury from circulating pathogens and bacterial endotoxins such as LPS. This is essential for the development of ALI (Maniatis and Orfanos, 2008). Pulmonary vascular permeability and leukocyte recruitment are maintained by an intact endothelial barrier (Mehta and Malik, 2006; Nourshargh and Alon, 2014). Thus, disruption of the endothelial barrier can lead to pre-clotting pathway activation, pro-inflammatory cytokine release (such as IL-1β), neutrophil influx, and tissue edema (Mehta and Malik, 2006). During inflammation, there is an adaptive increase in endothelial permeability increase to allow white blood cell chemotaxis into the lung to overcome the infection. Severe uncontrolled infection can have massive inflammatory reactions and endothelial barrier destruction, which eventually leads to death in patients with septic ALI. Recent evidence suggests that endothelial injury might be related to pyroptosis of endothelial cells (Cheng et al., 2017).
Pyroptosis, apoptosis, and necrosis are different types of programmed cell death. Each type of programmed cell death is regulated by a unique individual set of host proteins that mediate multiple biological outcomes (Vanden Berghe et al., 2014; Blander, 2014; Chan et al., 2015; Wallach et al., 2016; Man and Kanneganti, 2016; Vande Walle and Lamkanfi, 2016; Weinlich et al., 2017). Caspase activation participates in both apoptosis and pyroptosis. Apoptosis starts with initiator caspases-2, -8, -9, and -10 and ends with executioner caspases-3, -6, and -7 (Fink and Cookson, 2005). Different from apoptosis, pyroptosis is an inflammatory caspase-induced form of necrosis and inflammatory programmed cell death (Vande Walle and Lamkanfi, 2016). Pyroptosis is also distinct from another necrotic and inflammatory form of programmed cell death called necroptosis, because the execution of pyroptosis requires inflammatory caspases (Vercammen et al., 1998; Holler et al., 2000; Weinlich et al., 2017). The role of inflammatory caspases-4/5/11 in the cytoplasmic LPS signal transduction pathway and pyroptosis has been studied mostly in dendritic cells and macrophages (Miao et al., 2011; Kayagaki et al., 2013; Aachoui et al., 2013; Kayagaki et al., 2015; Shi et al., 2015; Zanoni et al., 2016). Furthermore,endothelial cells could have highly sensitive and complex intracellular LPS induction mechanisms that could lead to caspase-4/5/11-dependent endothelial lysis (Shi et al., 2014; Shi et al., 2015). Characteristic presentations include plasma membrane rupture and LDH release, maturation, release of Il-1β, and cleavage of perforin GSDMD by constriction effects (Kayagaki et al., 2015; Shi et al., 2015; Ding et al., 2016; Liu et al., 2016), which causes extensive pulmonary endothelial death and damages the endothelial barrier to induce ALI.
In Gram-negative bacterial infections, bacterial lysis leads to septic cascades that release large amounts of endotoxin (LPS) into the circulation (Baumgartner et al., 1985). The typical mechanism by which mammalian host cells detect LPS is achieved by Toll-like receptor 4 (TLR4) on the cell surface (Medzhitov et al., 1997; Poltorak et al., 1998). However, recent studies in macrophages suggested that another LPS-mediated intracellular pathway might play a role in the development of septic shock, namely triggering pyroptosis (Kayagaki et al., 2013; Hagar et al., 2013). The endotoxin LPS could damage the cell membrane and bind to caspase-4/5/11 to activate macrophage pyroptosis, leading to rapid cell lysis (Kayagaki et al., 2013; Hagar et al., 2013; Kayagaki et al., 2015; Jorgensen and Miao, 2015).
Caspase-11 is an intracellular LPS receptor that mediates pyroptosis. Sepsis-related mortality induced by endotoxemia is largely due to caspase-11 activation (Wang et al., 1998; Kayagaki et al., 2011; Kayagaki et al., 2013; Hagar et al., 2013; Kayagaki et al., 2015), which can cleave GSDMD to unleash active membrane pore-forming peptides. The latter can cause cell pyroptosis and release the leukotriene-like LTB4 via cyclooxygenase-1 and Alarmins, including IL-1α (Hagar et al., 2013). Studies have shown improved survival in endotoxemia mice with Gasdmd deletion or cyclooxyg (Kayagaki et al., 2013; Hagar et al., 2013; Kayagaki et al., 2015; Jorgensen and Miao, 2015)enase-1 (COX-1) inhibition (Hagar et al., 2013; Kayagaki et al., 2015). Caspase-11-mediated pyroptosis could trigger the local immune defense and disrupt intracellular niches by promoting vascular permeability and releasing chemokines (Kayagaki et al., 2011; Kayagaki et al., 2013; Aachoui et al., 2013; Hagar et al., 2013; Shi et al., 2014).
Caspase-11-dependent lysis of GSDMD and subsequent release of active membrane pore-forming peptides results in cell swelling, lysis, and death (Shi et al., 2015; Jorgensen and Miao, 2015; Broz, 2015; de Gassart and Martinon, 2015; Ding et al., 2016; Liu et al., 2016; Sborgi et al., 2016; Aglietti et al., 2016; Yuan et al., 2016). Microvesicles containing LPS could be shed from the gram-negative bacteria and fuse with the host cellular membrane to release LPS intracellularly and activate inflammatory cysteine signaling and cell pyroptosis (Vanaja et al., 2016; Man et al., 2016). In mice, endothelial cell pyroptosis is characterized by the caspase-11-dependent cell lysis, GSDMD activation, and release of pro-inflammatory cytokine IL-1β. LPS enters the endothelial cytoplasm through the bacterial microvesicles or destructed inner cortical membrane, subsequently activating caspase-4/5/11 to trigger cell pyroptosis through GSDMD lysis. Extensive endothelial cell lysis results in massive destruction of the pulmonary endothelial barrier (Cheng et al., 2017). Pro-inflammatory cytokine release, leukocyte influx, and pulmonary edema emerge as characteristic signs of ALI (Aird, 2003; Andonegui et al., 2003; Matthay et al., 2019) (Figure 1). Interestingly, although endothelial cells could also undergo pyroptosis without activated caspase-11 expression, this required long-term exposure to LPS (Cheng et al., 2017).
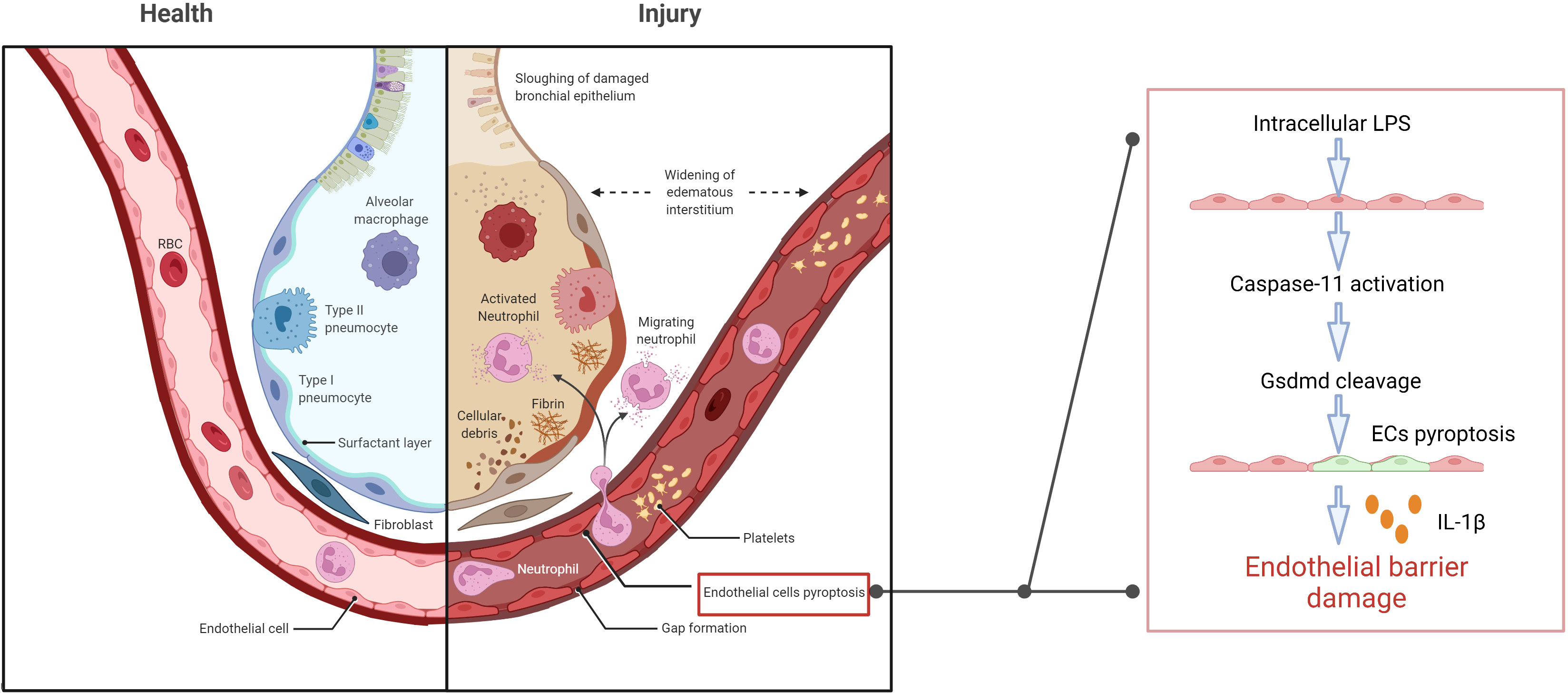
Figure 1 Extracellular LPS enters the endothelial cytoplasm through the cell membrane, and then triggers caspase-11-dependent endothelial cell pyroptosis and damages the pulmonary endothelial barrier, resulting in pulmonary edema, proinflammatory cytokine release, fluid and protein leakage, and massive influx of leukocytes.
Extracellular LPS could trigger pyroptosis of endothelial cells and immune cells only after LPS binds to intracellular caspase-11 (Kayagaki et al., 2013; Aachoui et al., 2013; Hagar et al., 2013; Cheng et al., 2017). Activation of extracellular LPS enhances the expression of caspase-11 in mouse endothelial cells and similarly enhances the expression of caspase-4 and -5 in human endothelial cells, which has been shown to increase the binding of intracellular LPS to these inflammatory caspases (Cheng et al., 2017).
HMGB1 is named due to its high electrophoretic mobility in an agarose and polyacrylamide gel (Klune et al., 2008). It is a highly conserved and unanimously expressed nuclear protein that participates in the nucleosome structure maintenance and the regulation process of DNA replication, recombination, transcription, and repair (Travers, 2003). HMGB1 is released extracellularly after cell activation, injury, stress, or death and functions as a transporter to promote inflammation (Andersson et al., 2018; Qu et al., 2019). In 1999, it was first reported that HMGB1 could act as a proinflammatory cytokine to stimulate macrophages, which were activated by LPS in sepsis (Wang et al., 1999). Subsequently, it has been reported that HMGB1 could be actively secreted by natural killer cells, monocytes, platelets, endothelial cells, and dendritic cells, and passively released from the nuclei of damaged/necrotic cells during infection. Studies have shown that, after LPS stimulation, HMGB1 was secreted into the extracellular space where it bound to its receptors, including TLR2, TLR4, and RAGE, to induce the expressions of cytokines, adhesion molecules, and chemokines, which further exacerbated the inflammation and injury (Wolfson et al., 2011; Qu et al., 2019). Deletion of the HMGB1 gene or neutralizing circulation of HMGB1 has been shown to have a protective effect against fatal endotoxemia and sepsis (Wang et al., 1999; Wang et al., 2004; Qin et al., 2006; Rittirsch et al., 2008; Lamkanfi et al., 2010; Andersson and Tracey, 2011).
During the signal cascade in sepsis, HMGB1 utilizes a variety of membrane receptors. Its bindings to RAGE and TLR4 occur at its residues 150-183 and 89-108, respectively (Huttunen et al., 2002; Yang et al., 2010). Bioactivity of HMGB1 depends on the redox state of its three cysteine residues (Kazama et al., 2008; Lu et al., 2012). The disulfide isoforms could activate cytokine production and TLR4, while the fully reduced isoforms could not (Lu et al., 2012).
Recent studies have shown that LPS relies on HMGB1 and RAGE to assist cell transportation. These molecules could facilitate LPS leakage into the cytoplasm with subsequent activation of the key receptor caspase-11 (Deng et al., 2018). In sepsis, the circulating pathogen-associated molecular patterns (PAMPs), including LPS, can trigger hepatocytes to release HMGB1 into the bloodstream. Extracellular LPS can physically bind to HMGB1, and the HMGB1-LPS complex can be internalized into lysosomes within endothelial cells and macrophages via RAGE, disrupting lysosome stability via HMGB1 (Deng et al., 2018). HMGB1 has a unique ability to act as a detergent in the lysosomal membranes owing to the acidic conditions of the lysosomal environment (Xu et al., 2014; Deng et al., 2018; Yuan et al., 2020). Thus, partner molecules transported by HMGB1 will avoid the expected degradation in the lysosome and instead leak into the cytoplasm, reaching homologous cytoplasmic receptors and potentiating the pro-inflammatory response. HMGB1 then penetrates directly into the phospholipid bilayer of the lysosome. Using the liposome leakage test and whole-cell patch clamp analysis, it was found that the phospholipid bilayer penetrating ability of HMGB1 was enhanced under acidic conditions, demonstrating the pH-dependent ability of HMGB1 to induce lysosomal rupture in organelles. In addition, it also explained the reason why HMGB1 does not cause cytoplasm or nuclear membrane damages under normal physiological conditions (Deng et al., 2018). These effects of HMGB1 in the lysosome result in the leakage of LPS into the cytoplasm and activation of caspase-11 and pulmonary endothelial cell pyroptosis (Figure 2).
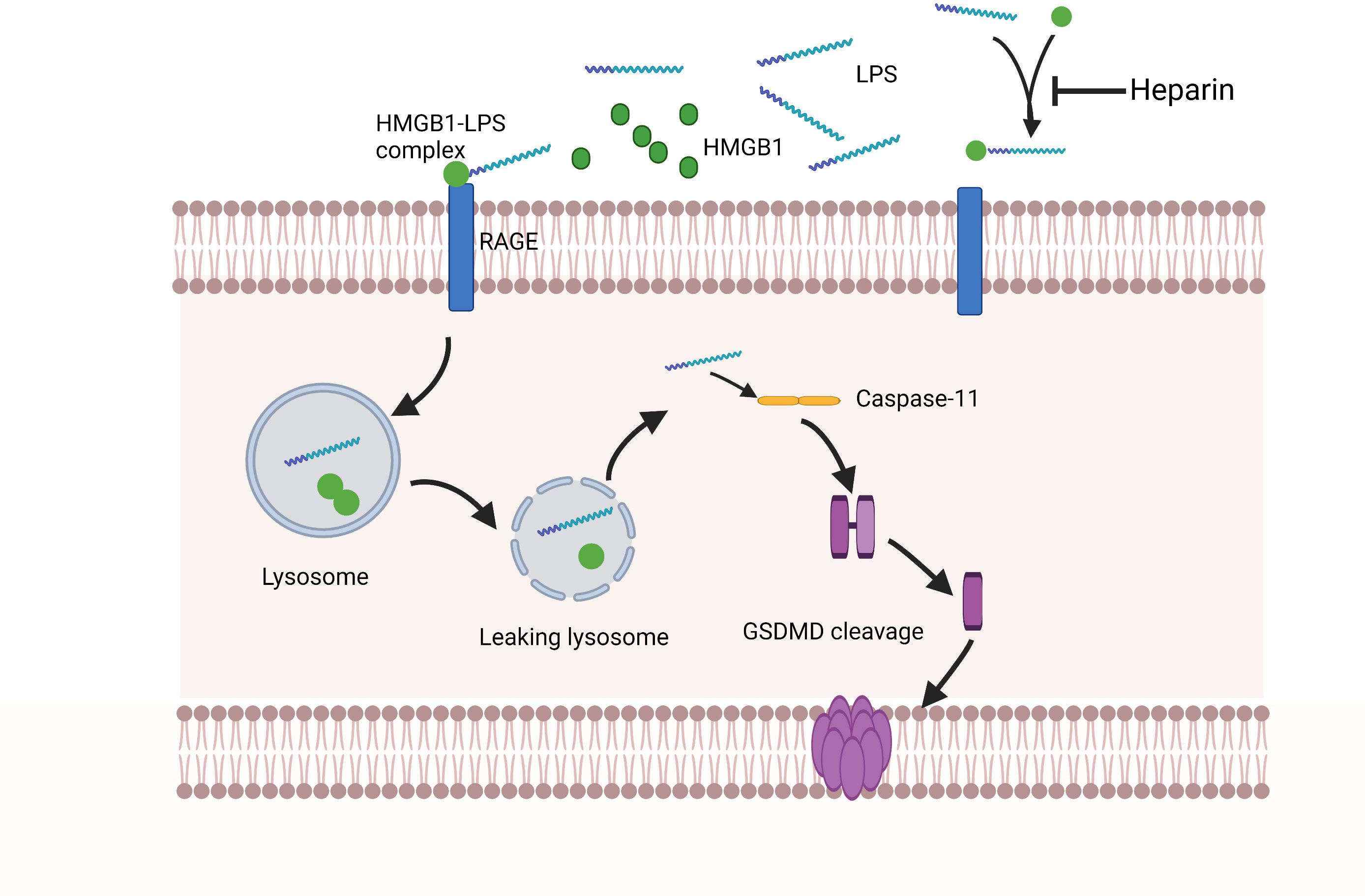
Figure 2 Extracellular HMGB1 and LPS form complexes. These complexes bind to the RAGE expressed by pulmonary endothelial cells, which is followed by the internalization of the HMGB1-LPS complex into lysosomes. Under acidic conditions, HMGB1 acts as a detergent to destroy the lysosome membrane, allowing LPS to enter the cytoplasm and activate caspase-11. Subsequently, activated caspase-11 mediates GSDMD lysis to form pores on the cell membrane, ultimately leading to pulmonary endothelial cell pyroptosis. Heparin has a high affinity for HMGB1, which can compete with the formation of HMGB1-LPS complexes and prevent pulmonary endothelial cell pyroptosis upstream of caspase-11 activation.
Both myeloid and endothelial cells can express RAGE and caspase-11 (Hofmann et al., 1999; Liliensiek et al., 2004). Endothelial caspase-11 participates in pyroptosis induced by endotoxemia (Cheng et al., 2017). Reduced HMGB1 release from hepatocytes, RAGE deficiency, and extracellular HMGB1 neutralization might prevent caspase-11-dependent pyroptosis during endotoxemia and bacterial sepsis (Deng et al., 2018). Interestingly, another study reported that HMGB1 alone could activate ASC-dependent pyroptosis, which was independent of the caspase-11 pathway (Yuan et al., 2020). HMGB1 could also damage the lysosomes and activate the NLRP3 inflammasome independently from the caspase-11 pathway in the macrophages with TLR agonist primers. This response could be the result of HMGB1 oxidation that is followed by the intramolecular bond formation between cysteine 23 and cysteine 45, which causes TLR4-MD2-mediated HMGB1 mobilization to function as a trigger for NLRP3 inflammasome activation (Hornung et al., 2008; Yang et al., 2015; Frank et al., 2016). However, in other studies, HMGB1 alone could not induce pyroptosis. The different redox states of the recombinant HMGB1 protein in these studies might explain the different experimental results (Deng et al., 2018).
Previous studies have found that heparin could attenuate the caspase-11-dependent inflammatory reactions and reduce mortality from sepsis in mice. Both heparin administration and caspase-11 deletion have been shown to reduce lung injury in mice (Tang et al., 2021). Most ligands bind to heparin/heparan sulfate through electrostatic interactions of the positively charged arginine and lysine residues in the ligand with the negatively charged uronic acids and sulfate groups in heparin (Xu et al., 2011). HMGB1 has many basic residues: eight arginine and 43 lysine residues, which altogether account for 24% of HMGB1 amino acids. Among them, six basic residues are important for heparan sulfate binding (Xu et al., 2011). Arg97, Lys87, Lys88, Lys90, and Lys96 residues are within the latter part of the long ring segment that connects A- and B-boxes. As a separate residue on the last spiral of the B-box, Lys150 also participates in heparin-ligand binding (Xu et al., 2011). Heparin can bind to HMGB1 with a high affinity and induce spatial conformational changes in the latter (Ling et al., 2011). These conformational changes in HMGB1 might lower its binding affinity to its receptors. In addition, some studies demonstrated that HMGB1 alone would not cause inflammatory reactions. Purified rHMGB1 could only induce limited cytokine secretions (Rouhiainen et al., 2007; Sha et al., 2008; Youn et al., 2008; Hreggvidsdottir et al., 2009). HMGB1 binds with other mediators, such as IL-1B, DNA, LPS, or nucleosomes, to form complexes associated with inflammation (Sha et al., 2008). HMGB1 also has the allosteric effects to change the conformation of cytokines or their interactions with cytokine receptors (Bianchi, 2009). Tang et al. found that heparin could bind to HMGB1 and block hMGB1-LPS interactions (Tang et al., 2021). In a mouse model of sepsis, it was found that heparin intervention could minimize IL-1α and IL-1β release, as well as GSDMD distributions in the lungs after LPS stimulation (Tang et al., 2021). Meanwhile, in a clinical study of 20 sepsis patients who received heparin and 21 sepsis patients who did not, activation of caspase-4 (the human homologue of caspase-11) was measured (Tang et al., 2021). Markers of caspase-4 activation, including serum levels of IL-1α and IL-1β, were significantly lower in patients who received the heparin treatment compared to those who did not (Tang et al., 2021). RAGE has been shown to be highly expressed in endothelial cells and binds to HMGB1 and other ligands, such as AGEs, S100 proteins, and amyloid proteins, via its two N-terminal IgG-like domains (Basta, 2008; Yan et al., 2010). RAGE has also been reported to bind to heparin (Xu et al., 2011), but this mechanism needs to be further investigated. In another study, heparin sulfate, a chemically modified heparin, did not demonstrate any anticoagulant properties, indicating that heparin sulfate could dose-dependently block caspase-11 as a non-anticoagulant heparin (NAH) (Tang et al., 2021). As such, heparin may block caspase-11 by a mechanism independent of its anticoagulant properties, by which it can inhibit pyroptosis of pulmonary endothelial cells and ameliorate ALI during the sepsis (Figure 2).
In pulmonary endothelial cells, there is a highly sensitive and complex intracellular LPS-sensing mechanism that leads to caspase-4/5/11 dependent pyroptosis (Cheng et al., 2017), which is characterized by plasma membrane rupture and LDH release, IL-1β maturation and secretion, and division of constriction-effector perforin GSDMD (Shi et al., 2014; Kayagaki et al., 2015; Shi et al., 2015; Ding et al., 2016). Pyroptosis is an effective method of eliminating the intracellular bacterial niche and releasing inflammatory mediators such as IL-1β, while retaining uninfected adjacent cells (Aachoui et al., 2013; Hagar et al., 2013; Maltez et al., 2015; Jorgensen et al., 2016). Therefore, the innate immune response induced by caspases-1/4/5/11 is different from the typical inflammasome activation pathway mediated through the cell surface TLR4 (Kayagaki et al., 2011; Miao et al., 2011; Kayagaki et al., 2013; Man and Kanneganti, 2016; Broz and Dixit, 2016). On the other hand, In the study of mouse sepsis model, caspase-11 may also be immunopathologic (Napier et al., 2016). Caspase-11 can be activated when LPS is delivered intracellularly into the target cells, such as endothelial cells and macrophages, by HMGB1 secreted by hepatocytes or microvesicles released by bacteria (Vanaja et al., 2016; Deng et al., 2018). This leads to immune cell death with subsequent endothelial barrier disruptions (Vanaja et al., 2016; Cheng et al., 2017; Deng et al., 2018). Endothelial cell-specific deletion of caspase-11 was shown to ameliorate LPS-dependent pulmonary vascular endothelial permeability and significantly improve survival in both endotoxemia models and cecal ligation and puncture-induced ALI models of multi-microbial sepsis (Cheng et al., 2017). Activation of extracellular LPS enhances the expression of caspase-11 in mouse endothelial cells and similarly enhances the expression of caspase-4 and -5 in human endothelial cells (Cheng et al., 2017).
Heparin treatment has been found to block caspase-11-dependent inflammatory reactions and is an effective inhibitor in the caspase-11 pathway during sepsis (Tang et al., 2021). In animal models of fatal endotoxemia or severe sepsis, heparin inhibited the LPS cytoplasmic transmission by blocking the hMGB1-LPS interactions and decreasing the heparinase-induced glycocalygeal degradation in macrophages, thereby attenuating the overactivation of this harmful cascade (Tang et al., 2021). In the mouse experiment,it was found that heparin could block caspase-11 by a mechanism unrelated to its anticoagulant activity, thereby inhibiting cell pyroptosis (Tang et al., 2021). Meanwhile, in the clinical study, markers of caspase-4 activation, including serum levels of IL-1α and IL-1β, were significantly lower in patients who received the heparin treatment compared to those who did not (Tang et al., 2021). A study has also showed that nonanticoagulant heparin, purified from clinical grade heparin, binds histones and prevents histone-mediated cytotoxicity in vitro and reduces mortality from sterile inflammation and sepsis in mouse models without increasing the risk of bleeding (Wildhagen et al., 2014). As such this evidence suggests that low dose heparin or modified heparin without anticoagulant properties could be used to achieve a therapeutic effect and reduce the risk of bleeding. This will mean that we find a solution to the conflict between heparin treatment of sepsis ALI and its side effects. This is the purpose of this review.However, other studies show that heparin treatment does not significantly reduce the 28-day mortality of septic patients (Jaimes et al., 2009; Li and Ma, 2017). The discrepancy between these studies might be due to the difference in infected pathogens.Gram-positive sepsis causes death through mechanisms distinct from that of Gram-negative sepsis (Popescu et al., 2018).Thus,further research is needed to better understand this potential role of heparin in treatment of sepsis.
RY: collected and analyzed the data; drafted the article. XZ: revised and submitted the manuscript. All authors have read and approved the final version of manuscript.
The present review was supported by the Liaoning Natural Science Foundation (grant No. 2021-MS-188).
Figures 1 and 2 were created using BioRender.com.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Aachoui, Y., Leaf, I. A., Hagar, J. A., Fontana, M. F., Campos, C. G., Zak, D. E., et al. (2013). Caspase-11 protects against bacteria that escape the vacuole. Science 339 (6122), 975–978. doi: 10.1126/science.1230751
Aglietti, R. A., Estevez, A., Gupta, A., Ramirez, M. G., Liu, P. S., Kayagaki, N., et al. (2016). GsdmD p30 elicited by caspase-11 during pyroptosis forms pores in membranes. Proc. Natl. Acad. Sci. U. S. A. 113 (28), 7858–7863. doi: 10.1073/pnas.1607769113
Aird, W. C. (2003). The role of the endothelium in severe sepsis and multiple organ dysfunction syndrome. Blood 101 (10), 3765–3777. doi: 10.1182/blood-2002-06-1887
Andersson, U., Tracey, K. J. (2011). HMGB1 is a therapeutic target for sterile inflammation and infection. Annu. Rev. Immunol. 29, 139–162. doi: 10.1146/annurev-immunol-030409-101323
Andersson, U., Yang, H., Harris, H. (2018). Extracellular HMGB1 as a therapeutic target in inflammatory diseases. Expert Opin. Ther. Targets. 22 (3), 263–277. doi: 10.1080/14728222.2018.1439924
Andonegui, G., Bonder, C. S., Green, F., Mullaly, S. C., Zbytnuik, L., Raharjo, E., et al. (2003). Endothelium-derived Toll-like receptor-4 is the key molecule in LPS-induced neutrophil sequestration into lungs [published correction appears in J Clin Invest. J. Clin. Invest. 111 (7), 1011–1020. doi: 10.1172/JCI16510
Ashbaugh, D. G., Bigelow, D. B., Petty, T. L., Levine, B. E. (1967). Acute respiratory distress in adults. Lancet 2 (7511), 319–323. doi: 10.1016/s0140-6736(67)90168-7
Basta, G. (2008). Receptor for advanced glycation endproducts and atherosclerosis: From basic mechanisms to clinical implications. Atherosclerosis 196 (1), 9–21. doi: 10.1016/j.atherosclerosis.2007.07.025
Baumgartner, J. D., Glauser, M. P., McCutchan, J. A., Ziegler, E. J., van Melle, G., Klauber, M. R., et al. (1985). Prevention of gram-negative shock and death in surgical patients by antibody to endotoxin core glycolipid. Lancet 2 (8446), 59–63. doi: 10.1016/s0140-6736(85)90176-x
Bellani, G., Laffey, J. G., Pham, T., Fan, E., LUNG SAFE Investigators and the ESICM Trials Group (2016). The LUNG SAFE study: a presentation of the prevalence of ARDS according to the Berlin Definition! Crit. Care 20 (1), 268. doi: 10.1186/s13054-016-1443-x
Bianchi, M. E. (2009). HMGB1 loves company. J. Leukoc. Biol. 86 (3), 573–576. doi: 10.1189/jlb.1008585
Blander, J. M. (2014). A long-awaited merger of the pathways mediating host defence and programmed cell death. Nat. Rev. Immunol. 14 (9), 601–618. doi: 10.1038/nri3720
Broz, P. (2015). Immunology: Caspase target drives pyroptosis. Nature 526 (7575), 642–643. doi: 10.1038/nature15632
Broz, P., Dixit, V. M. (2016). Inflammasomes: mechanism of assembly, regulation and signalling. Nat. Rev. Immunol. 16 (7), 407–420. doi: 10.1038/nri.2016.58
Chan, F. K., Luz, N. F., Moriwaki, K. (2015). Programmed necrosis in the cross talk of cell death and inflammation. Annu. Rev. Immunol. 33, 79–106. doi: 10.1146/annurev-immunol-032414-112248
Cheng, K. T., Xiong, S., Ye, Z., Hong, Z., Di, A., Tsang, K. M., et al. (2017). Caspase-11-mediated endothelial pyroptosis underlies endotoxemia-induced lung injury. J. Clin. Invest. 127 (11), 4124–4135. doi: 10.1172/JCI94495
Chuang, Y. J., Swanson, R., Raja, S. M., Olson, S. T. (2001). Heparin enhances the specificity of antithrombin for thrombin and factor Xa independent of the reactive center loop sequence. Evidence for an exosite determinant of factor Xa specificity in heparin-activated antithrombin. J. Biol. Chem. 276 (18), 14961–14971. doi: 10.1074/jbc.M011550200
Cornet, A. D., Smit, E. G., Beishuizen, A., Groeneveld, A. B. (2007). The role of heparin and allied compounds in the treatment of sepsis. Thromb. Haemost. 98 (3), 579–586. doi: 10.1160/TH07-01-0006
Davalos, A. R., Kawahara, M., Malhotra, G. K., Schaum, N., Huang, J., Ved, U., et al. (2013). p53-dependent release of Alarmin HMGB1 is a central mediator of senescent phenotypes. J. Cell Biol. 201 (4), 613–629. doi: 10.1083/jcb.201206006
Davidson, B. L., Geerts, W. H., Lensing, A. W. (2002). Low-dose heparin for severe sepsis. N Engl. J. Med. 347 (13), 1036–1037. doi: 10.1056/NEJM200209263471316
de Gassart, A., Martinon, F. (2015). Pyroptosis: Caspase-11 Unlocks the Gates of Death. Immunity 43 (5), 835–837. doi: 10.1016/j.immuni.2015.10.024
Deng, M., Tang, Y., Li, W., Wang, X., Zhang, R., Zhang, X., et al. (2018). The Endotoxin Delivery Protein HMGB1 Mediates Caspase-11-Dependent Lethality in Sepsis. Immunity 49 (4), 740–753.e7. doi: 10.1016/j.immuni.2018.08.016
Ding, J., Wang, K., Liu, W., She, Y., Sun, Q., Shi, J., et al. (2016). Pore-forming activity and structural autoinhibition of the gasdermin family [published correction appears in Nature. Nature 535 (7610), 111–116. doi: 10.1038/nature18590
Englert, J. A., Bobba, C., Baron, R. M. (2019). Integrating molecular pathogenesis and clinical translation in sepsis-induced acute respiratory distress syndrome [published online ahead of print, 2019 Jan 24]. JCI Insight 4 (2), e124061. doi: 10.1172/jci.insight.124061
Evavold, C. L., Ruan, J., Tan, Y., Xia, S., Wu, H., Kagan, J. C. (2018). The Pore-Forming Protein Gasdermin D Regulates Interleukin-1 Secretion from Living Macrophages. Immunity 48 (1), 35–44.e6. doi: 10.1016/j.immuni.2017.11.013
Fan, Y., Jiang, M., Gong, D., Zou, C. (2016). Efficacy and safety of low-molecular-weight heparin in patients with sepsis: a meta-analysis of randomized controlled trials. Sci. Rep. 6, 25984. doi: 10.1038/srep25984
Fink, S. L., Cookson, B. T. (2005). Apoptosis, pyroptosis, and necrosis: mechanistic description of dead and dying eukaryotic cells. Infect. Immun. 73 (4), 1907–1916. doi: 10.1128/IAI.73.4.1907-1916.2005
Frank, M. G., Weber, M. D., Fonken, L. K., Hershman, S. A., Watkins, L. R., Maier, S. F. (2016). The redox state of the alarmin HMGB1 is a pivotal factor in neuroinflammatory and microglial priming: A role for the NLRP3 inflammasome. Brain Behav. Immun. 55, 215–224. doi: 10.1016/j.bbi.2015.10.009
Gerber, A., Goldklang, M., Stearns, K., Ma, X., Xiao, R., Zelonina, T., et al. (2020). Attenuation of pulmonary injury by an inhaled MMP inhibitor in the endotoxin lung injury model. Am. J. Physiol. Lung Cell Mol. Physiol. 319 (6), L1036–L1047. doi: 10.1152/ajplung.00420.2019
Gong, H., Rehman, J., Tang, H., Wary, K., Mittal, M., Chaturvedi, P., et al. (2015). HIF2α signaling inhibits adherens junctional disruption in acute lung injury [published correction appears in. J. Clin. Invest. 125 (3), 1364. doi: 10.1172/JCI77701
Hagar, J. A., Powell, D. A., Aachoui, Y., Ernst, R. K., Miao, E. A. (2013). Cytoplasmic LPS activates caspase-11: implications in TLR4-independent endotoxic shock. Science 341 (6151), 1250–1253. doi: 10.1126/science.1240988
He, W. T., Wan, H., Hu, L., Chen, P., Wang, X., Huang, Z., et al. (2015). Gasdermin D is an executor of pyroptosis and required for interleukin-1β secretion. Cell Res. 25 (12), 1285–1298. doi: 10.1038/cr.2015.139
Hofmann, M. A., Drury, S., Fu, C., Qu, W., Taguchi, A., Lu, Y., et al. (1999). RAGE mediates a novel proinflammatory axis: a central cell surface receptor for S100/calgranulin polypeptides. Cell 97 (7), 889–901. doi: 10.1016/s0092-8674(00)80801-6
Holler, N., Zaru, R., Micheau, O., Thome, M., Attinger, A., Valitutti, S., et al. (2000). Fas triggers an alternative, caspase-8-independent cell death pathway using the kinase RIP as effector molecule. Nat. Immunol. 1 (6), 489–495. doi: 10.1038/82732
Hoppensteadt, D., Fareed, J., Klein, A. L., Jasper, S. E., Apperson-Hansen, C., Lieber, EA., et al. (2008). Comparison of anticoagulant and anti-inflammatory responses using enoxaparin versus unfractionated heparin for transesophageal echocardiography-guided cardioversion of atrial fibrillation. Am. J. Cardiol. 102 (7), 842–846. doi: 10.1016/j.amjcard.2008.05.025
Hornung, V., Bauernfeind, F., Halle, A., Samstad, E. O., Kono, H., Rock, K. L., et al. (2008). Silica crystals and aluminum salts activate the NALP3 inflammasome through phagosomal destabilization. Nat. Immunol. 9 (8), 847–856. doi: 10.1038/ni.1631
Hreggvidsdottir, H. S., Ostberg, T., Wähämaa, H., Schierbeck, H., Aveberger, A. C., Klevenvall, L., et al. (2009). The alarmin HMGB1 acts in synergy with endogenous and exogenous danger signals to promote inflammation. J. Leukoc. Biol. 86 (3), 655–662. doi: 10.1189/jlb.0908548
Huppert, L. A., Matthay, M. A., Ware, L. B. (2019). Pathogenesis of Acute Respiratory Distress Syndrome. Semin. Respir. Crit. Care Med. 40 (1), 31–39. doi: 10.1055/s-0039-1683996
Huttunen, H. J., Fages, C., Kuja-Panula, J., Ridley, A. J., Rauvala, H. (2002). Receptor for advanced glycation end products-binding COOH-terminal motif of amphoterin inhibits invasive migration and metastasis. Cancer Res. 62 (16), 4805–4811.
Jaimes, F., de la Rosa, G., Morales, C., Fortich, F., Arango, C., Aguirre, D., et al. (2009). Unfractioned heparin for treatment of sepsis: A randomized clinical trial (The HETRASE Study). Crit. Care Med. 37, 1185–1196. doi: 10.1097/CCM.0b013e31819c06bc
Jorgensen, I., Miao, E. A. (2015). Pyroptotic cell death defends against intracellular pathogens. Immunol. Rev. 265 (1), 130–142. doi: 10.1111/imr.12287
Jorgensen, I., Zhang, Y., Krantz, B. A., Miao, E. A. (2016). Pyroptosis triggers pore-induced intracellular traps (PITs) that capture bacteria and lead to their clearance by efferocytosis. J. Exp. Med. 213 (10), 2113–2128. doi: 10.1084/jem.20151613
Kayagaki, N., Stowe, I. B., Lee, B. L., O'Rourke, K., Anderson, K., Warming, S., et al. (2015). Caspase-11 cleaves gasdermin D for non-canonical inflammasome signalling. Nature 526 (7575), 666–671. doi: 10.1038/nature15541
Kayagaki, N., Warming, S., Lamkanfi, M., Vande Walle, L., Louie, S., Dong, J., et al. (2011). Non-canonical inflammasome activation targets caspase-11. Nature 479 (7371), 117–121. doi: 10.1038/nature10558
Kayagaki, N., Wong, M. T., Stowe, I. B., Ramani, S. R., Gonzalez, L. C., Akashi-Takamura, S., et al. (2013). Noncanonical inflammasome activation by intracellular LPS independent of TLR4. Science 341 (6151), 1246–1249. doi: 10.1126/science.1240248
Kazama, H., Ricci, J. E., Herndon, J. M., Hoppe, G., Green, D. R., Ferguson, T. A. (2008). Induction of immunological tolerance by apoptotic cells requires caspase-dependent oxidation of high-mobility group box-1 protein. Immunity 29 (1), 21–32. doi: 10.1016/j.immuni.2008.05.013
Klune, J. R., Dhupar, R., Cardinal, J., Billiar, T. R., Tsung, A. (2008). HMGB1: endogenous danger signaling. Mol. Med. 14 (7-8), 476–484. doi: 10.2119/2008-00034.Klune
Kumar, V., Chhibber, S. (2011). Acute lung inflammation in Klebsiella pneumoniae B5055-induced pneumonia and sepsis in BALB/c mice: a comparative study. Inflammation 34 (5), 452–462. doi: 10.1007/s10753-010-9253-9
Lamkanfi, M., Sarkar, A., Vande Walle, L., Vitari, A. C., Amer, A. O., Wewers, M. D., et al. (2010). Inflammasome-dependent release of the alarmin HMGB1 in endotoxemia. J. Immunol. 185 (7), 4385–4392. doi: 10.4049/jimmunol.1000803
Liliensiek, B., Weigand, M. A., Bierhaus, A., Nicklas, W., Kasper, M., Hofer, S., et al. (2004). Receptor for advanced glycation end products (RAGE) regulates sepsis but not the adaptive immune response. J. Clin. Invest. 113 (11), 1641–1650. doi: 10.1172/JCI18704
Li, X., Ma, X. (2017). The role of heparin in sepsis: much more than just an anticoagulant. Br. J. Haematol. 179 (3), 389–398. doi: 10.1111/bjh.14885
Lindsey, A. S., Sullivan, L. M., Housley, N. A., Koloteva, A., King, J. A., Audia, J. P., et al. (2019). Analysis of pulmonary vascular injury and repair during Pseudomonas aeruginosa infection-induced pneumonia and acute respiratory distress syndrome. Pulm Circ. 9 (1), 2045894019826941. doi: 10.1177/2045894019826941
Ling, Y., Yang, Z. Y., Yin, T., Li, L., Yuan, W. W., Wu, H. S., et al. (2011). Heparin changes the conformation of high-mobility group protein 1 and decreases its affinity toward receptor for advanced glycation endproducts. vitro. Int. Immunopharmacol. 11 (2), 187–193. doi: 10.1016/j.intimp.2010.11.014
Li, Y., Sun, J. F., Cui, X., Mani, H., Danner, R. L., Li, X., et al. (2011). The effect of heparin administration in animal models of sepsis: a prospective study in Escherichia coli-challenged mice and a systematic review and metaregression analysis of published studies. Crit. Care Med. 39 (5), 1104–1112. doi: 10.1097/CCM.0b013e31820eb718
Liu, Y., Mu, S., Li, X., Liang, Y., Wang, L., Ma, X. (2019). Unfractionated Heparin Alleviates Sepsis-Induced Acute Lung Injury by Protecting Tight Junctions. J. Surg. Res. 238, 175–185. doi: 10.1016/j.jss.2019.01.020
Liu, X., Zhang, Z., Ruan, J., Pan, Y., Magupalli, V. G., Wu, H., et al. (2016). Inflammasome-activated gasdermin D causes pyroptosis by forming membrane pores. Nature 535 (7610), 153–158. doi: 10.1038/nature18629
Liu, Z., Zhu, H., Ma, X. (2014). Heparin for treatment of sepsis: a systemic review. Zhonghua Wei Zhong Bing Ji Jiu Yi Xue. 26 (3), 135–141. doi: 10.3760/cma.j.issn.2095-4352.2014.03.003
Lotze, M. T., Tracey, K. J. (2005). High-mobility group box 1 protein (HMGB1): nuclear weapon in the immune arsenal. Nat. Rev. Immunol. 5 (4), 331–342. doi: 10.1038/nri1594
Lu, B., Nakamura, T., Inouye, K., Li, J., Tang, Y., Lundbäck, P., et al. (2012). Novel role of PKR in inflammasome activation and HMGB1 release. Nature 488 (7413), 670–674. doi: 10.1038/nature11290
Maltez, V. I., Tubbs, A. L., Cook, K. D., Aachoui, Y., Falcone, E. L., Holland, S. M., et al. (2015). Inflammasomes Coordinate Pyroptosis and Natural Killer Cell Cytotoxicity to Clear Infection by a Ubiquitous Environmental Bacterium. Immunity 43 (5), 987–997. doi: 10.1016/j.immuni.2015.10.010
Maniatis, N. A., Orfanos, S. E. (2008). The endothelium in acute lung injury/acute respiratory distress syndrome. Curr. Opin. Crit. Care 14 (1), 22–30. doi: 10.1097/MCC.0b013e3282f269b9
Man, S. M., Kanneganti, T. D. (2016). Converging roles of caspases in inflammasome activation, cell death and innate immunity. Nat. Rev. Immunol. 16 (1), 7–21. doi: 10.1038/nri.2015.7
Man, S. M., Karki, R., Sasai, M., Place, D. E., Kesavardhana, S., Temirov, J., et al. (2016). IRGB10 Liberates Bacterial Ligands for Sensing by the AIM2 and Caspase-11-NLRP3 Inflammasomes. Cell 167 (2), 382–396.e17. doi: 10.1016/j.cell.2016.09.012
Matthay, M. A., Zemans, R. L., Zimmerman, G. A., Arabi, Y. M., Beitler, J. R., Mercat, A., et al. (2019). Acute respiratory distress syndrome. Nat. Rev. Dis. Primers. 5 (1), 18. doi: 10.1038/s41572-019-0069-0
Medzhitov, R., Preston-Hurlburt, P., Janeway, C. A., Jr. (1997). A human homologue of the Drosophila Toll protein signals activation of adaptive immunity. Nature 388 (6640), 394–397. doi: 10.1038/41131
Mehta, D., Malik, A. B. (2006). Signaling mechanisms regulating endothelial permeability. Physiol. Rev. 86 (1), 279–367. doi: 10.1152/physrev.00012.2005
Miao, E. A., Rajan, J. V., Aderem, A. (2011). Caspase-1-induced pyroptotic cell death. Immunol. Rev. 243 (1), 206–214. doi: 10.1111/j.1600-065X.2011.01044.x
Müller, S., Scaffidi, P., Degryse, B., Bonaldi, T., Ronfani, L., Agresti, A., et al. (2001). New EMBO members’ review: the double life of HMGB1 chromatin protein: architectural factor and extracellular signal. EMBO J. 20 (16), 4337–4340. doi: 10.1093/emboj/20.16.4337
Napier, B. A., Brubaker, S. W., Sweeney, T. E., Monette, P., Rothmeier, G. H., Gertsvolf, N. A., et al. (2016). Complement pathway amplifies caspase-11-dependent cell death and endotoxin-induced sepsis severity. J. Exp. Med. 213 (11), 2365–2382. doi: 10.1084/jem.20160027
Nourshargh, S., Alon, R. (2014). Leukocyte migration into inflamed tissues. Immunity 41 (5), 694–707. doi: 10.1016/j.immuni.2014.10.008
Peukert, K., Fox, M., Schulz, S., Feuerborn, C., Frede, S., Putensen, C., et al. (2021). Inhibition of Caspase-1 with Tetracycline Ameliorates Acute Lung Injury. Am. J. Respir. Crit. Care Med. 204 (1), 53–63. doi: 10.1164/rccm.202005-1916OC
Poltorak, A., He, X., Smirnova, I., Liu, M. Y., Van Huffel, C., Du, X., et al. (1998). Defective LPS signaling in C3H/HeJ and C57BL/10ScCr mice: mutations in Tlr4 gene. Science 282 (5396), 2085–2088. doi: 10.1126/science.282.5396.2085
Popescu, N. I., Silasi, R., Keshari, R. S., Girton, A., Burgett, T., Zeerleder, S. S., et al. (2018). Peptidoglycan induces disseminated intravascular coagulation in baboons through activation of both coagulation pathways. Blood 132, 849–860. doi: 10.1182/blood-2017-10-813618
Qin, S., Wang, H., Yuan, R., Li, H., Ochani, M., Ochani, K., et al. (2006). Role of HMGB1 in apoptosis-mediated sepsis lethality. J. Exp. Med. 203 (7), 1637–1642. doi: 10.1084/jem.20052203
Qu, L., Chen, C., Chen, Y., Li, Y., Tang, F., Huang, H., et al. (2019). High-Mobility Group Box 1 (HMGB1) and Autophagy in Acute Lung Injury (ALI): A Review. Med. Sci. Monit. 25, 1828–1837. doi: 10.12659/MSM.912867
Rezaie, A. R., Yang, L., Manithody, C. (2004). Mutagenesis studies toward understanding the mechanism of differential reactivity of factor Xa with the native and heparin-activated antithrombin. Biochemistry 43 (10), 2898–2905. doi: 10.1021/bi036145a
Rittirsch, D., Flierl, M. A., Nadeau, B. A., Day, D. E., Huber-Lang, M., Mackay, C. R., et al. (2008). Functional roles for C5a receptors in sepsis. Nat. Med. 14 (5), 551–557. doi: 10.1038/nm1753
Rouhiainen, A., Tumova, S., Valmu, L., Kalkkinen, N., Rauvala, H. (2007). Pivotal advance: analysis of proinflammatory activity of highly purified eukaryotic recombinant HMGB1 (amphoterin). J. Leukoc. Biol. 81 (1), 49–58. doi: 10.1189/jlb.0306200
Sauer, A., Peukert, K., Putensen, C., Bode, C. (2021). Antibiotics as immunomodulators: a potential pharmacologic approach for ARDS treatment. Eur. Respir. Rev. 30 (162), 210093. doi: 10.1183/16000617.0093-2021
Sborgi, L., Rühl, S., Mulvihill, E., Pipercevic, J., Heilig, R., Stahlberg, H., et al. (2016). GSDMD membrane pore formation constitutes the mechanism of pyroptotic cell death. EMBO J. 35 (16), 1766–1778. doi: 10.15252/embj.201694696
Sha, Y., Zmijewski, J., Xu, Z., Abraham, E. (2008). HMGB1 develops enhanced proinflammatory activity by binding to cytokines. J. Immunol. 180 (4), 2531–2537. doi: 10.4049/jimmunol.180.4.2531
Shi, J., Zhao, Y., Wang, Y., Gao, W., Ding, J., Li, P., et al. (2014). Inflammatory caspases are innate immune receptors for intracellular LPS. Nature 514 (7521), 187–192. doi: 10.1038/nature13683
Shi, J., Zhao, Y., Wang, K., Shi, X., Wang, Y., Huang, H., et al. (2015). Cleavage of GSDMD by inflammatory caspases determines pyroptotic cell death. Nature 526 (7575), 660–665. doi: 10.1038/nature15514
Tang, Y., Wang, X., Li, Z., He, Z., Yang, X., Cheng, X., et al. (2021). Heparin prevents caspase-11-dependent septic lethality independent of anticoagulant properties. Immunity 54 (3), 454–467.e6. doi: 10.1016/j.immuni.2021.01.007
Travers, A. A. (2003). Priming the nucleosome: a role for HMGB proteins? EMBO Rep. 4 (2), 131–136. doi: 10.1038/sj.embor.embor741
Vanaja, S. K., Russo, A. J., Behl, B., Banerjee, I., Yankova, M., Deshmukh, S. D., et al. (2016). Bacterial Outer Membrane Vesicles Mediate Cytosolic Localization of LPS and Caspase-11 Activation. Cell. 165 (5), 1106–1119. doi: 10.1016/j.cell.2016.04.015
Vanden Berghe, T., Linkermann, A., Jouan-Lanhouet, S., Walczak, H., Vandenabeele, P. (2014). Regulated necrosis: the expanding network of non-apoptotic cell death pathways. Nat. Rev. Mol. Cell Biol. 15 (2), 135–147. doi: 10.1038/nrm3737
Vande Walle, L., Lamkanfi, M. (2016). Pyroptosis. Curr. Biol. 26 (13), R568–R572. doi: 10.1016/j.cub.2016.02.019
Vercammen, D., Beyaert, R., Denecker, G., Goossens, V., Van Loo, G., Declercq, W., et al. (1998). Inhibition of caspases increases the sensitivity of L929 cells to necrosis mediated by tumor necrosis factor. J. Exp. Med. 187 (9), 1477–1485. doi: 10.1084/jem.187.9.1477
Wallach, D., Kang, T. B., Dillon, C. P., Green, D. R. (2016). Programmed necrosis in inflammation: Toward identification of the effector molecules. Science 352 (6281), aaf2154. doi: 10.1126/science.aaf2154
Wang, H., Bloom, O., Zhang, M., Vishnubhakat, J. M., Ombrellino, M., Che, J., et al. (1999). HMG-1 as a late mediator of endotoxin lethality in mice. Science 285 (5425), 248–251. doi: 10.1126/science.285.5425.248
Wang, C., Chi, C., Guo, L., Wang, X., Guo, L., Sun, J., et al. (2014). Heparin therapy reduces 28-day mortality in adult severe sepsis patients: a systematic review and meta-analysis. Crit. Care 18 (5), 563. doi: 10.1186/s13054-014-0563-4
Wang, H., Liao, H., Ochani, M., Justiniani, M., Lin, X., Yang, L., et al. (2004). Cholinergic agonists inhibit HMGB1 release and improve survival in experimental sepsis. Nat. Med. 10 (11), 1216–1221. doi: 10.1038/nm1124
Wang, S., Miura, M., Jung, Y. K., Zhu, H., Li, E., Yuan, J. (1998). Murine caspase-11, an ICE-interacting protease, is essential for the activation of ICE. Cell 92 (4), 501–509. doi: 10.1016/s0092-8674(00)80943-5
Weinlich, R., Oberst, A., Beere, H. M., Green, D. R. (2017). Necroptosis in development, inflammation and disease. Nat. Rev. Mol. Cell Biol. 18 (2), 127–136. doi: 10.1038/nrm.2016.149
Wildhagen, K. C., García de Frutos, P., Reutelingsperger, C. P., Schrijver, R., Aresté, C., Ortega-Gómez, A., et al. (2014). Nonanticoagulant heparin prevents histone-mediated cytotoxicity in vitro and improves survival in sepsis. Blood 123 (7), 1098–1101. doi: 10.1182/blood-2013-07-514984
Wolfson, R. K., Chiang, E. T., Garcia, J. G. (2011). HMGB1 induces human lung endothelial cell cytoskeletal rearrangement and barrier disruption. Microvasc Res. 81 (2), 189–197. doi: 10.1016/j.mvr.2010.11.010
Xu, J., Jiang, Y., Wang, J., Shi, X., Liu, Q., Liu, Z., et al. (2014). Macrophage endocytosis of high-mobility group box 1 triggers pyroptosis. Cell Death Differ. 21 (8), 1229–1239. doi: 10.1038/cdd.2014.40
Xu, D., Young, J., Song, D., Esko, J. D. (2011). Heparan sulfate is essential for high mobility group protein 1 (HMGB1) signaling by the receptor for advanced glycation end products (RAGE). J. Biol. Chem. 286 (48), 41736–41744. doi: 10.1074/jbc.M111.299685
Yang, H., Hreggvidsdottir, H. S., Palmblad, K., Wang, H., Ochani, M., Li, J., et al. (2010). A critical cysteine is required for HMGB1 binding to Toll-like receptor 4 and activation of macrophage cytokine release. Proc. Natl. Acad. Sci. U S A. 107 (26), 11942–11947. doi: 10.1073/pnas.1003893107
Yang, L., Manithody, C., Rezaie, A. R. (2004). Heparin-activated antithrombin interacts with the autolysis loop of target coagulation proteases. Blood 104 (6), 1753–1759. doi: 10.1182/blood-2004-03-1092
Yang, H., Wang, H., Ju, Z., Ragab, A. A., Lundbäck, P., Long, W., et al. (2015). MD-2 is required for disulfide HMGB1-dependent TLR4 signaling. J. Exp. Med. 212 (1), 5–14. doi: 10.1084/jem.20141318
Yan, S. F., Ramasamy, R., Schmidt, A. M. (2010). The RAGE axis: a fundamental mechanism signaling danger to the vulnerable vasculature [published correction appears in Circ Res. 2010 Apr 30;106(8):e6]. Circ. Res. 106 (5), 842–853. doi: 10.1161/CIRCRESAHA.109.212217
Youn, J. H., Oh, Y. J., Kim, E. S., Choi, J. E., Shin, J. S. (2008). High mobility group box 1 protein binding to lipopolysaccharide facilitates transfer of lipopolysaccharide to CD14 and enhances lipopolysaccharide-mediated TNF-alpha production in human monocytes. J. Immunol. 180 (7), 5067–5074. doi: 10.4049/jimmunol.180.7.5067
Yuan, X., Bhat, O. M., Lohner, H., Zhang, Y., Li, P. L. (2020). Downregulation of Lysosomal Acid Ceramidase Mediates HMGB1-Induced Migration and Proliferation of Mouse Coronary Arterial Myocytes. Front. Cell Dev. Biol. 8. doi: 10.3389/fcell.2020.00111
Yuan, J., Najafov, A., Py, B. F. (2016). Roles of Caspases in Necrotic Cell Death. Cell 167 (7), 1693–1704. doi: 10.1016/j.cell.2016.11.047
Zanoni, I., Tan, Y., Di Gioia, M., Broggi, A., Ruan, J., Shi, J., et al. (2016). An endogenous caspase-11 ligand elicits interleukin-1 release from living dendritic cells. Science 352 (6290), 1232–1236. doi: 10.1126/science.aaf3036
Keywords: acute lung injury, pyroptosis, endothelial cells, high mobility group 1, caspase-11, heparin
Citation: Yang R and Zhang X (2022) A potential new pathway for heparin treatment of sepsis-induced lung injury: inhibition of pulmonary endothelial cell pyroptosis by blocking hMGB1-LPS-induced caspase-11 activation. Front. Cell. Infect. Microbiol. 12:984835. doi: 10.3389/fcimb.2022.984835
Received: 02 July 2022; Accepted: 29 August 2022;
Published: 15 September 2022.
Edited by:
Mathieu Coureuil, Institut National de la Santé et de la Recherche Médicale (INSERM), FranceReviewed by:
Mahmoud El-Daly, Minia University, EgyptCopyright © 2022 Yang and Zhang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Xiaojuan Zhang, enhqOTUwMTIyQHNpbmEuY29t
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.