 Jianmin Chai
Jianmin Chai Xinting Liu1,2
Xinting Liu1,2 Ying Li
Ying Li Jiangchao Zhao
Jiangchao Zhao- 1Guangdong Provincial Key Laboratory of Animal Molecular Design and Precise Breeding, College of Life Science and Engineering, Foshan University, Foshan, China
- 2School of Life Science and Engineering, Foshan University, Foshan, China
- 3Division of Agriculture, Department of Animal Science, University of Arkansas, Fayetteville, AR, United States
Bovine respiratory disease (BRD), one of the most common and infectious diseases in the beef industry, is associated with the respiratory microbiome and stressors of transportation. The impacts of the bovine respiratory microbiota on health and disease across different geographic locations and sampling niches are poorly understood, resulting in difficult identification of BRD causes. In this study, we explored the effects of geography and niches on the bovine respiratory microbiome and its function by re-analyzing published metagenomic datasets and estimated the main opportunistic pathogens that changed after transportation. The results showed that diversity, composition, structure, and function of the bovine nasopharyngeal microbiota were different across three worldwide geographic locations. The lung microbiota also showed distinct microbial composition and function compared with nasopharyngeal communities from different locations. Although different signature microbiota for each geographic location were identified, a module with co-occurrence of Mycoplasma species was observed in all bovine respiratory communities regardless of geography. Moreover, transportation, especially long-distance shipping, could increase the relative abundance of BRD-associated pathogens. Lung microbiota from BRD calves shaped clusters dominated with different pathogens. In summary, geography, sampling niches, and transportation are important factors impacting the bovine respiratory microbiome and disease, and clusters of lung microbiota by different bacterial species may explain BRD pathogenesis, suggesting the importance of a deeper understanding of bovine respiratory microbiota in health.
Introduction
Bovine respiratory disease (BRD) is one of the most common infectious causes of pneumonia in cattle worldwide and causes morbidity and mortality in newly transported and recently weaned feedlot cattle, resulting in a slower growth rate and economic cost for prevention and treatment (Chai et al., 2022). With developments of sequencing technology, the roles of the respiratory microbiome in maintaining homeostasis of the airway ecosystem and its association with disease are more deeply understood (Dickson et al., 2016; Man et al., 2017; Zeineldin et al., 2019). The most implicated bacterial pathogens in BRD, including Mycoplasma bovis, Mannheimia haemolytica, Histophilus somni, and Pasteurella multocida, have been identified (Nicola et al., 2017; Cirone et al., 2019). Until now, 16S rRNA sequencing has been the most popular technology to investigate the respiratory microbiome of both humans and cattle (Human Microbiome Project, 2012; Zeineldin et al., 2020). Metagenomics that capture sequences from nearly all the organisms inhabiting the respiratory system is more effective and accurate to investigate the composition and functions of the respiratory microbiome (Holman et al., 2017c; Cui et al., 2021). However, fewer studies regarding BRD using metagenomics have been performed.
Geography serves as one of the major factors influencing the respiratory microbiota (Gupta et al., 2017). As cattle are a main source of human protein and nutrition and BRD appears worldwide in cattle, research of the bovine respiratory microbiome at the world level is important and necessary. However, due to feeding strategy, diet, environment, climate, etc., the compositional differences of the bovine airway microbiota are affected by the geographic locations of farms. Using 16S sequencing technology, a study found that, in Canada, the most prominently identified genera in the nasopharynx of calves were Mycoplasma, Lactococcus, Moraxella, Histophilus, and Pasteurella (Mcmullen et al., 2018), while in the United States Mannheimia, Mycoplasma, Moraxella, Psychrobacter, and Pseudomonas were the top five genera in the nasopharynx of calves (Lima et al., 2016). However, to our knowledge, there are fewer studies to specifically investigate how bovine respiratory microbiota and its functions vary geographically.
Other factors, including sampling niche in the respiratory tract, weaning and transportation, time since arrival to the feedlot, and health status, can influence the microbial structure of the bovine respiratory ecosystem and are associated with an increased risk of BRD in recently weaned beef calves (Snowder et al., 2006; Taylor et al., 2010; Zeineldin et al., 2017a; Zeineldin et al., 2020). The biochemical and physiological environments of niches within the respiratory tracts are different, resulting in a different microbial composition (Zeineldin et al., 2019; Chai et al., 2022). A recent study reported that the dominant bacteria were Moraxella and Mycoplasma in the nasopharynx, as well as Mycoplasma in the lungs of healthy calves, and concluded that the nasopharynx could serve as a primary source for lung microbiota (Mcmullen et al., 2020). Although a recent theory that suggests microbiota colonize in the upper respiratory tracts firstly and then disperse to the lungs was reported in humans (Venkataraman et al., 2015), it has not been confirmed in cattle. Furthermore, the nasopharynx has been the most popular niche to determine the relationship between the respiratory microbiota and BRD (Holman et al., 2015a; Timsit et al., 2016a; Holman et al., 2017a; Zeineldin et al., 2017a; Holman et al., 2017d)—for instance, some studies mainly focused on the pathogens in the nasopharynx (Timsit et al., 2016b; Gaeta et al., 2017; Zeineldin et al., 2017a; Holman et al., 2017d; Holman et al., 2019; Mcmullen et al., 2019). In addition, transportation, as an important stressor, is associated with the bovine respiratory microbiota and BRD (Holman et al., 2017d; Mcmullen et al., 2018; Cui et al., 2021). There have been no studies, however, investigating whether consistent microbial changes by stressors (such as transportation) were found in calves from different geographic locations.
Three studies performed metagenomics to determine how transportation or BRD affects the nasopharyngeal microbiome of calves from Canada (Malmuthuge et al., 2021) and China (Cui et al., 2021) and the lung microbiota of calves from Canada (Klima et al., 2019). Their studies provide experimental data that can be analyzed to determine the effects of geography and transportation stress on bovine microbial structure and functional potential. This paper presents the results of our re-analysis of the shotgun metagenomic dataset from these three studies to determine the geographic and transportation effects on the bovine respiratory microbiome.
Materials and methods
Data collection
Our study was based on three public metagenomic datasets published by Malmuthuge et al. (2021); Cui et al. (2021), and Klima et al. (2019). Malmuthuge et al. investigated the effects of weaning and transportation on the temporal dynamics of the nasopharyngeal microbiota in Hereford-crossed calves in Canada, Cui and colleagues measured the longitudinal changes of the nasopharyngeal microbiome before and after long-distance transportation in Simmental calves in China, and Klima et al. collected and characterized the lung microbiota from feedlot calves that died from BRD (Supplementary Table S1). A total of 145 bovine respiratory samples collected from the feedlot calves 5 to 6 months of age were included in this study. In brief, the samples were from three geographic locations [Saskatoon in Canada, two cities (Qiqihaer and Guangan) in China, and Alberta in Canada] and two niches (nasopharynx and lung). Moreover, transportation effects were estimated in the studies of Malmuthuge et al. and Cui et al. Details of the study design and sample collections were described in the original study. Metagenomic sequencing was performed using the Illumina HiSeq platform. Sequences were downloaded from the NCBI SRA database under accession codes PRJNA687519, PRJNA724913, and PRJNA395911.
Metagenomic sequence processing
KneadData (v 0.7.2) was used to remove host contamination and filter reads for the downloaded metagenomic data. Low-quality reads with a Phred score smaller than 15 within a 5-bp sliding window on reads were trimmed using Trimmomatic (v0.39). Clean reads of each sample were acquired, and then all reads were aligned to the Bos taurus reference genome (UMD v3.0) using bmtagger (v.3.102.4) for host contamination read removal. Next, clean reads of each sample were then uploaded into the MG-RAST metagenomic analysis server (v4.0) (Keegan et al., 2016) and against the Kyoto Encyclopedia of Genes and Genomes (KEGG) Orthology database for function analysis and the RefSeq database for microbial taxa classification based on the recommended manual. After obtaining the count table of microbial composition and functional prediction from the MG-RAST server, R software was used to do downstream analyses.
Statistics and bioinformatics
Alpha diversity, including the Shannon index and richness, was calculated to evaluate the corresponding diversities by R (v4.1.2). A two-tailed Wilcoxon signed-rank test was used to test the significance of alpha diversity. Principal coordinate analysis based on Bray–Curtis distance was performed to visualize beta diversity, and dissimilarity among groups was determined using the “adonis2” function on the R “vegan” package. Signature microbiota for geographic locations were identified using the linear discriminant analysis (LDA) effect size (LEfSe) with default settings (e.g., LDA score >2 as a criterion for judging the significant effect size), and the abundances of microbial signatures were visualized using a heat map. The Sankey plot was drawn in R to show the taxonomy of bovine respiratory microbiome in three geographic locations. Other boxplots were made using the “ggplot2” package in R.
To determine the correlation of the bovine respiratory microbiome in the network interface, correlation matrixes by calculating pairwise Spearman’s rank correlations among the top 200 bacteria were constructed. A correlation between two bacteria was considered statistically robust if Spearman’s correlation coefficient (R) was greater than 0.5, and the P-value was less than 0.01. To avoid false positives, all P-values were adjusted with a multiple testing correction using the Benjamini–Hochberg method. The pairwise correlations of the bacteria formed their co-occurrence networks. Network analyses were performed using “VEGAN”, “igraph”, and “Hmisc” packages in R, and the interactive platform of Gephi was employed for network visualization. The size of each node is proportional to the number of connections.
Hierarchical clustering based on the relative abundance of the bronchoalveolar lavage (BAL) microbiome was performed using the Bray–Curtis distance and the “hclust” function in R. Taxa of the top 30 lung microbiota were included in hierarchical clustering. All BAL samples were subjected to unsupervised hierarchical clustering.
Results
Sample characteristics and sequencing analysis
Metagenomic samples (n = 145) during the feedlot period were obtained from three worldwide studies with publicly available datasets (Klima et al., 2019; Cui et al., 2021; Malmuthuge et al., 2021). The characteristics of the samples are summarized in Supplementary Table S1. Three geographic locations, including Saskatoon in Canada, two cities (Qiqihaer and Guangan) in China, and Alberta in Canada, were selected. The respiratory microbial samples were collected from the nasopharynx and lung using nasopharyngeal swabs (NPS; n = 130) and bronchoalveolar lavage (BAL; n = 15). Notably, the effects of short- and long-distance transportation, respectively, were included in the NPS, which were analyzed to estimate the shipping stress on the nasopharyngeal microbiota. After quality control, an average of 3,374,490 clean reads in the respiratory metagenomics were used for downstream analysis, including microbial classification using the RefSeq database and functional prediction by the KEGG Orthology database.
Bovine respiratory microbiome influenced by geographic locations and sampling niches
To analyze the effects of geographic location and sampling niches on the bovine respiratory microbiome, microbial diversities were estimated at the community level. The Shannon index of NPS of Qiqihaer and Guangan was higher compared with the NPS of Saskatoon and the BAL of Alberta (P < 0.05) (Figure 1A). Interestingly, the BAL of Alberta had greater alpha diversity than the NPS of Saskatoon (P < 0.05). Consistently, the NPS of Qiqihaer and Guangan showed distinct clustering compared with the NPS of Saskatoon (analysis of similarity, ANOSIM: R = 0.99, R = 1.0, p = 0.001) and the BAL of Alberta (ANOSIM: R = 0.78, R = 0.49, p = 0.001) based on the Bray–Curtis distance (Figure 1B). At the same time, significant differences between Saskatoon and Alberta (ANOSIM: R = 0.99, p = 0.001) were also observed.
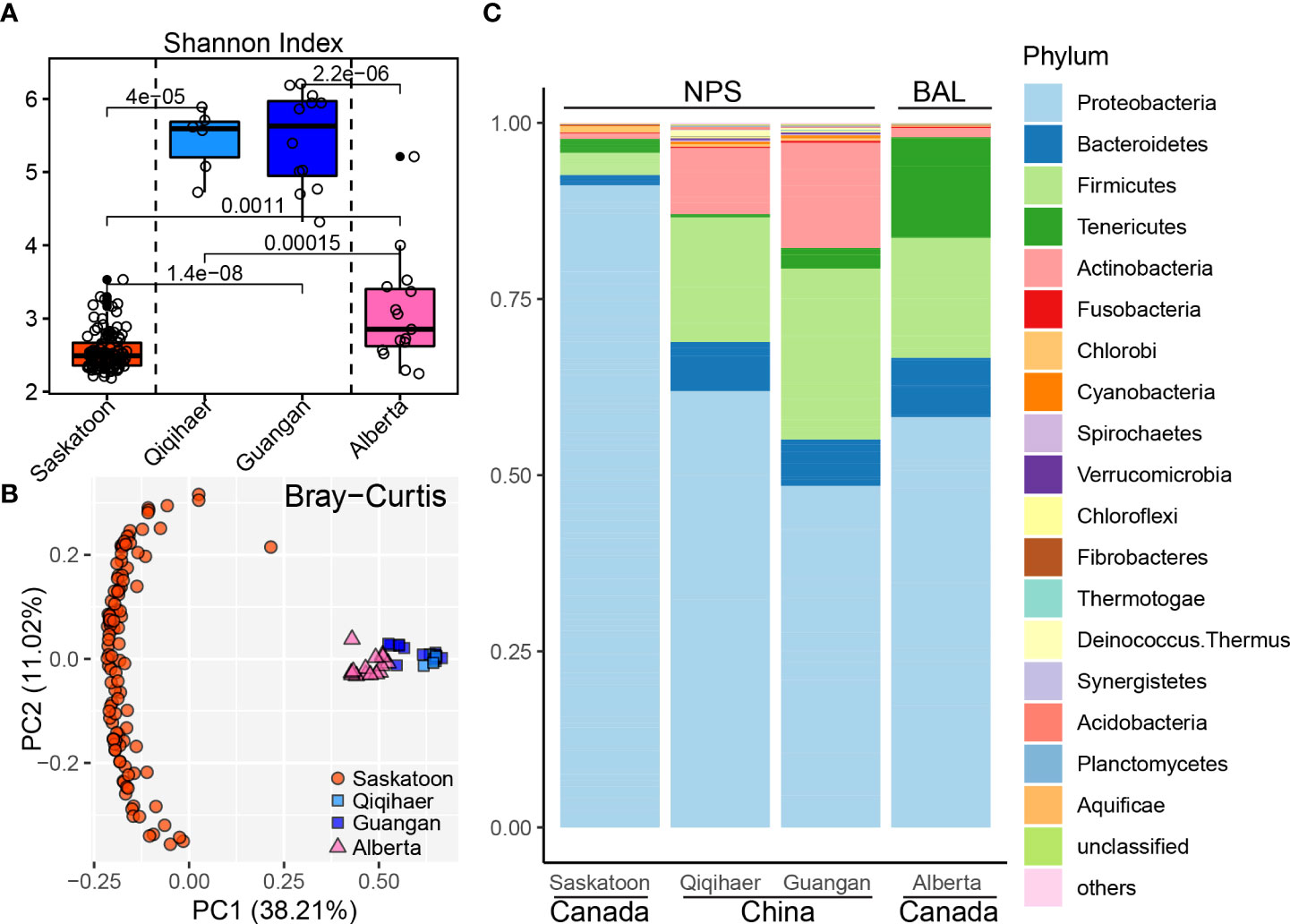
Figure 1 Bovine respiratory microbiome influenced by geographic locations and sampling niches. (A) Shannon index of bovine respiratory microbiota among NPS of Saskatoon (Canada), NPS of Qiqihaer and Guangan (China), and BAL of Alberta (Canada). Alpha diversity was tested using the Kruskal–Wallis test. The line inside the box denotes the median, and the boxes denote the interquartile range between the first and third quartiles (25th and 75th percentiles, respectively). (B) Principal coordinate analysis of the Bray–Curtis distances between microbiota. (C) Bacterial abundances at the phylum level. NPS, nasopharyngeal swab; BAL, bronchoalveolar lavage.
Using the Refseq database, 96.72% high-quality reads were classified as the bacterial kingdom, indicating the bacteria as the main microbiome in the bovine respiratory tracts (Supplementary Figure S1). At the phylum level, a total of 27 bacterial phyla were identified across 145 bovine respiratory samples. The predominant phylum in the NPS of Saskatoon was Proteobacteria (91.19%). In the NPS niche of Qiqihaer and Guangan (China), the most dominant phyla were Proteobacteria (48.49 and 61.94%), Firmicutes (24.26 and 17.68%), Actinobacteria (14.90 and 9.40%), and Bacteroidetes (6.59 and 6.98%) (Figure 1C). The BAL of Alberta was dominated by Proteobacteria (58.27%), Firmicutes (17.03%), Tenericutes (14.27%), and Bacteroidetes (8.41%). At the genus level, Burkholderia (81.71%) was the dominant genus in the NPS of Saskatoon (Supplementary Figure S2). The top genera in the NPS of Qiqihaer and Guangan were Psychrobacter (8.07 and 11.63%), Moraxella (4.94 and 7.13%), and Corynebacterium (4.63 and 2.08%). In the BAL microbiome, the top genera included Mannheimia (16.55%), Mycoplasma (14.11%), Actinobacillus (10.51%), Clostridium (5.73%), Psychrobacter (5.66%), and Haemophilus (5.60%).
The signature microbiota associated with geographic locations and sampling niches
LEfSe was performed to identify the microbial species differentiating both cities and sampling niches. Burkholderia species, including Burkholderia cenocepacia, Burkholderia sp. 383, Burkholderia ambifaria, Burkholderia multivorans, Burkholderia vietnamiensis, Burkholderia xenovorans, Burkholderia dolosa, Burkholderia ubonensis, and Burkholderia phymatum, were enriched in the NPS of Saskatoon (Figure 2), while the NPS of Qiqihaer and Guangan had greater abundances of bacteria such as Moraxella catarrhalis, Psychrobacter sp. PRwf-1, Enhydrobacter aerosaccus, Psychrobacter arcticus, Corynebacterium efficiens, Corynebacterium glutamicum, and Mycoplasma conjunctivae. In the BAL of Alberta (Canada), the abundant bacteria identified by LefSe were Mannheimia haemolytica, Mannheimia succiniciproducens, Mycoplasma species (Mycoplasma agalactiae, Mycoplasma arthritidis, Mycoplasma bovis, Mycoplasma hominis, and Mycoplasma mycoides), Histophilus somni, Haemophilus species (Haemophilus ducreyi, Haemophilus influenzae, and Haemophilus parasuis), Actinobacillus species (Actinobacillus pleuropneumoniae and Actinobacillus succinogenes), Clostridium perfringens, and Prevotella species (Prevotella melaninogenica, Prevotella copri, Prevotella ruminicola, Prevotella buccae, Prevotella oris, and Prevotella bryantii).
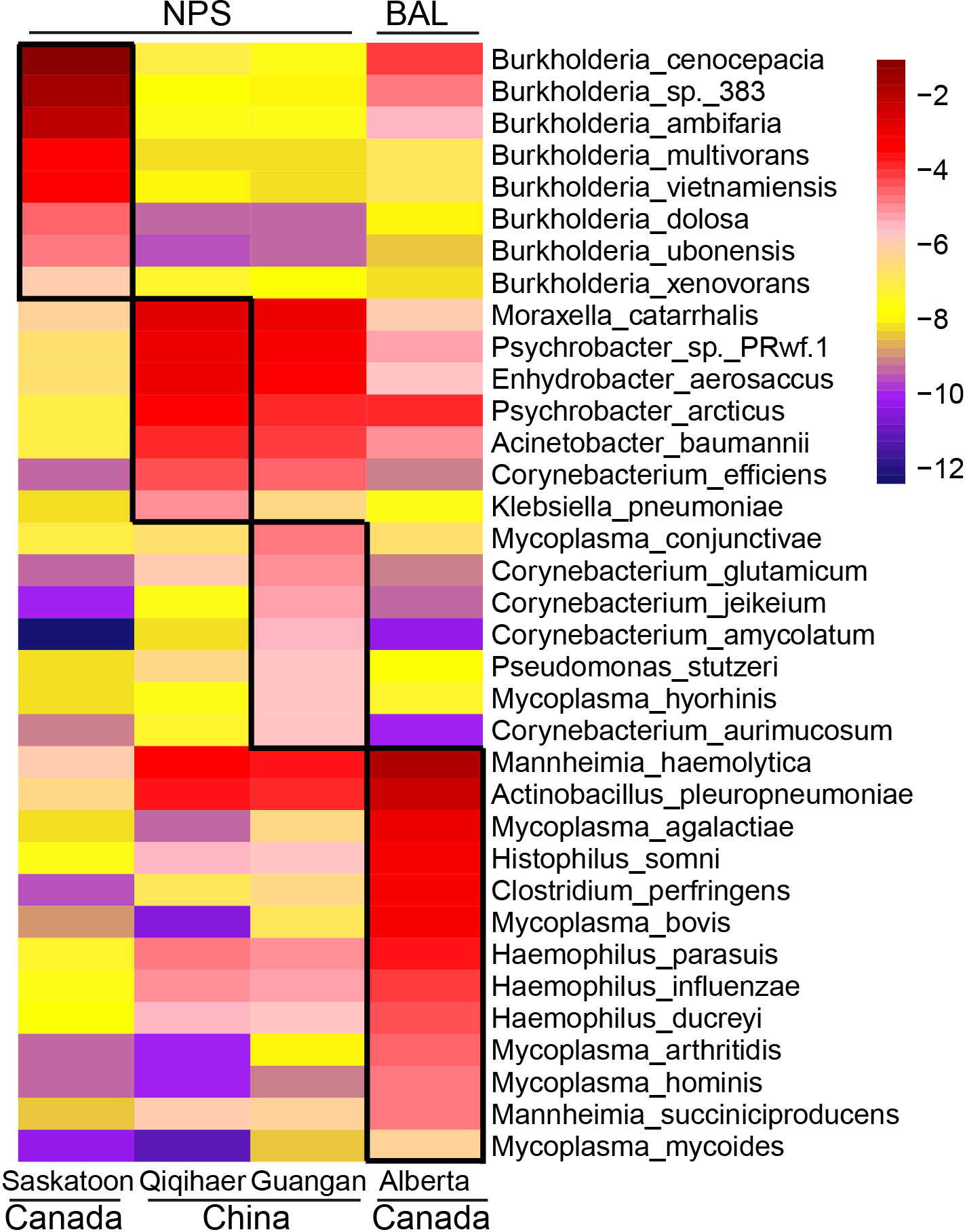
Figure 2 Geographic and niche-associated bacterial signatures that were identified by LEfSe analysis. The heat map shows the average relative abundance of ages on a log scale. The log-scaled relative abundance heat map of city-related bacterial species screened by LEfSe (linear discriminant analysis >2) in the bovine respiratory microbiome. The color of cells from purple to red corresponds to the relative abundance of bacteria from low to high. NPS, nasopharyngeal swab; BAL, bronchoalveolar lavage.
To better visualize the microbial composition of the bovine respiratory microbiome among geographic locations, a Sankey diagram of bacterial taxonomy was drawn (Figure 3). The most abundant bacteria in the NPS of Saskatoon were Burkholderia species, including B. cenocepacia, B. sp. 383, and B. ambifaria (Burkholderiaceae family, Proteobacteria phylum) (Figure 3A). The top bacteria in the NPS of Qiqihaer and Guangan were species under the Moraxellaceae family (Moraxella catarrhalis, Psychrobacter sp. PRwf-1, and Enhydrobacter aerosaccus) and the Pasteurellaceae family (Mannheimia haemolytica and Actinobacillus pleuropneumoniae) belonging to the Proteobacteria phylum, followed by the Streptococcaceae family (Streptococcus salivarius) and the Eubacteriaceae family (Eubacterium hallii) belonging to the Firmicutes phylum (Figure 3B). In the BAL of Alberta (Canada), species members of the Pasteurellaceae family (e.g., Mannheimia haemolytica, Actinobacillus pleuropneumoniae, and Histophilus somni), the Pseudomonadaceae family (e.g., Pseudomonas fluorescens), and the Moraxellaceae family (e.g., Psychrobacter cryohalolentis) belonging to the Proteobacteria phylum, the Clostridiaceae family (e.g., Clostridium perfringens) belonging to the Firmicutes phylum, the Bacteroidaceae family (e.g., Bacteroides sp. 1_1_6 and Bacteroides thetaiotaomicron) belonging to the Bacteroidetes phylum, and the Mycoplasmataceae family (e.g., Mycoplasma bovis and Mycoplasma agalactiae) belonging to the Tenericutes phylum formed the major bacterial composition (Figure 3C).
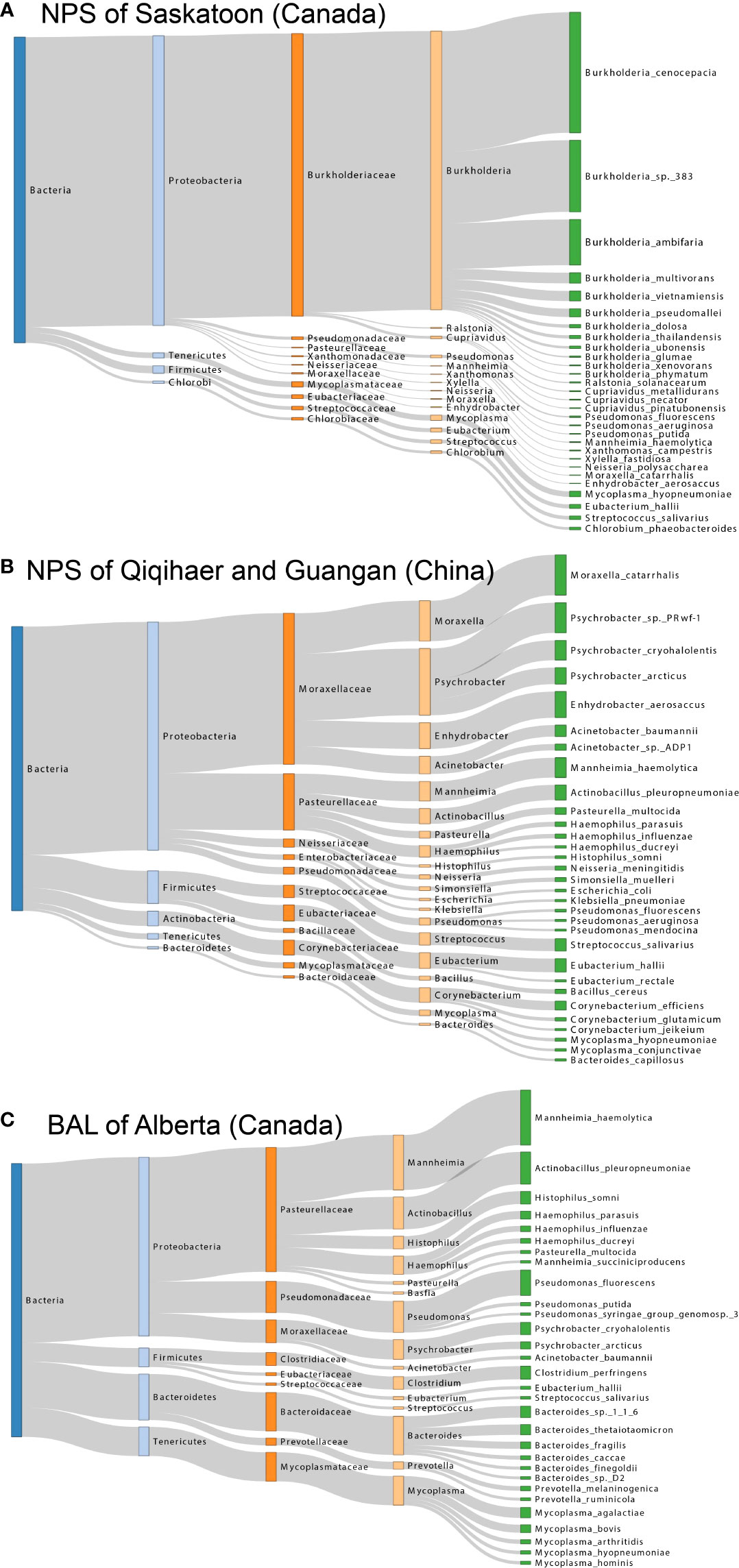
Figure 3 Sankey diagram for the major taxonomy structure of bovine respiratory microbiome. (A, B, and C) Sankey diagrams of bovine airway microbiota among NPS of Saskatoon (Canada), NPS of Qiqihaer and Guangan (China), and BAL of Alberta (Canada), which depict the bacterial flow of taxonomy from the kingdom to species level. NPS, nasopharyngeal swab; BAL, bronchoalveolar lavage.
Co-occurrence analysis of bacterial interconnections within geographic locations and niches
Based on the modularity class, the entire network in the NPS of Saskatoon can be parsed into six major modules (Figure 4A). Burkholderia species, including B. cenocepacia, B. sp. 383, B. ambifaria, B. multivorans and B. pseudomallei, formed a module, while Mycoplasma species (M. agalactiae, Mycoplasma synoviae, Mycoplasma alligatoris, Mycoplasma crocodyli, Mycoplasma hyopneumoniae, and Mycoplasma hyorhinis) co-occurred in another module. Regarding the NPS of Qiqihaer and Guangan (China), six modules with more complex bacterial co-occurrences were found. Interestingly, Mycoplasma species (M. conjunctivae, M. hyopneumoniae, M. hyorhinis, and M. agalactiae) also co-occurred (Figure 4B). In the BAL of Alberta community, Mycoplasma species (M. agalactiae, M. bovis, M. arthritidis, M. hyopneumoniae, and M. hominis), which were inter-connected with each other, were also observed (Figure 4C). Moreover, Mannheimia haemolytica, Histophilus somni, Pasteurella multocida, and Actinobacillus pleuropneumoniae were correlated with Haemophilus species (H. ducreyi, H. influenzae, and H. parasuis).
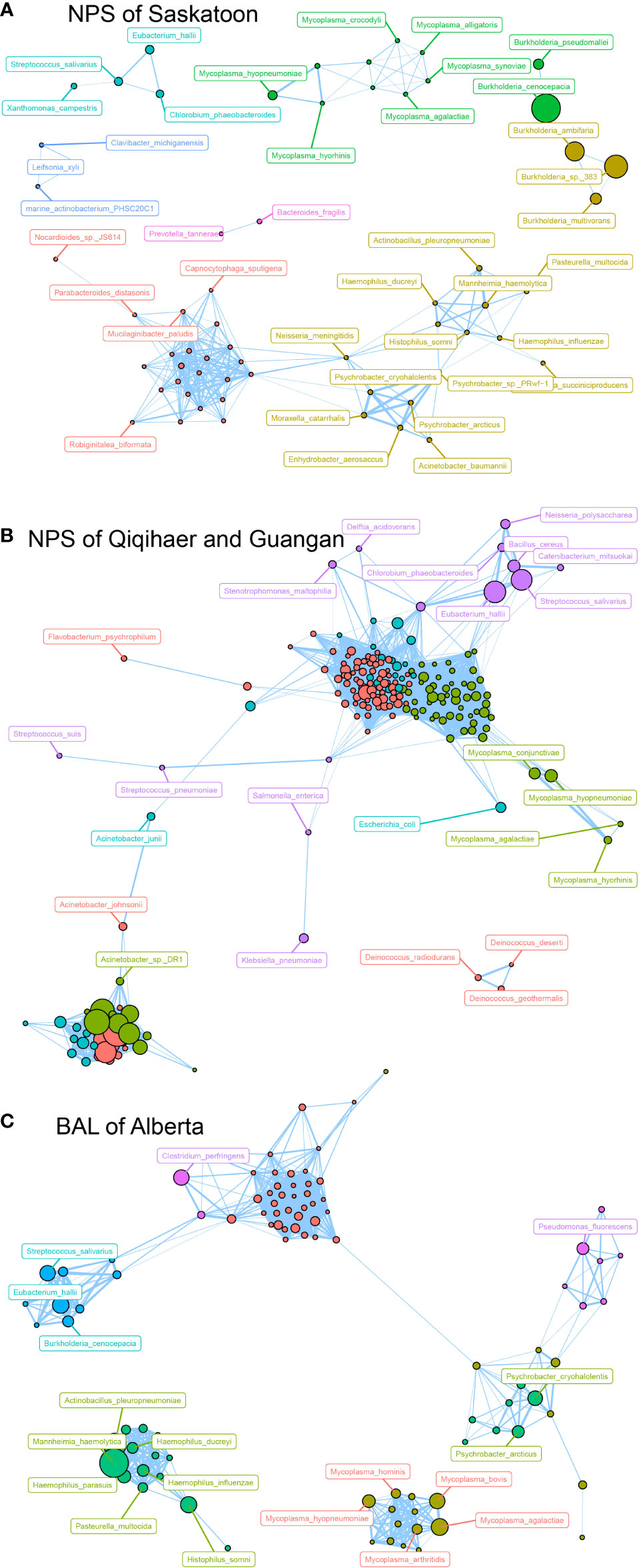
Figure 4 Network analysis revealing the co-occurrence patterns among airway microbial species. The nodes were colored according to modularity class. (A–C) Network analysis of bovine airway microbial species in samples of NPS of Saskatoon (Canada), NPS of Qiqihaer and Guangan (China), and BAL of Alberta (Canada). A connection represents a strong (Spearman’s correlation coefficient R > 0.8) and significant (P < 0.01) correlation. The size of each node is proportional to the number of connections, that is, the degree. NPS, nasopharyngeal swab; BAL, bronchoalveolar lavage.
Geographic locations and sampling niches affect the functions of the bovine respiratory microbiome
Consistent with the bovine microbial structure, the KEGG functional configuration showed significant differences among cities and niches (Figures 5A, B). The Shannon index in the NPS of Qiqihaer and Guangan was higher compared with the NPS of Saskatoon and the BAL of Alberta (P < 0.05) (Figure 5A) and had distinct clustering compared with the NPS of Saskatoon (ANOSIM: R = 0.99, R = 0.98, p = 0.001) and the BAL of Alberta (ANOSIM: R = 0.30, R = 0.66, p = 0.001) based on the Bray–Curtis distance (Figure 5B).
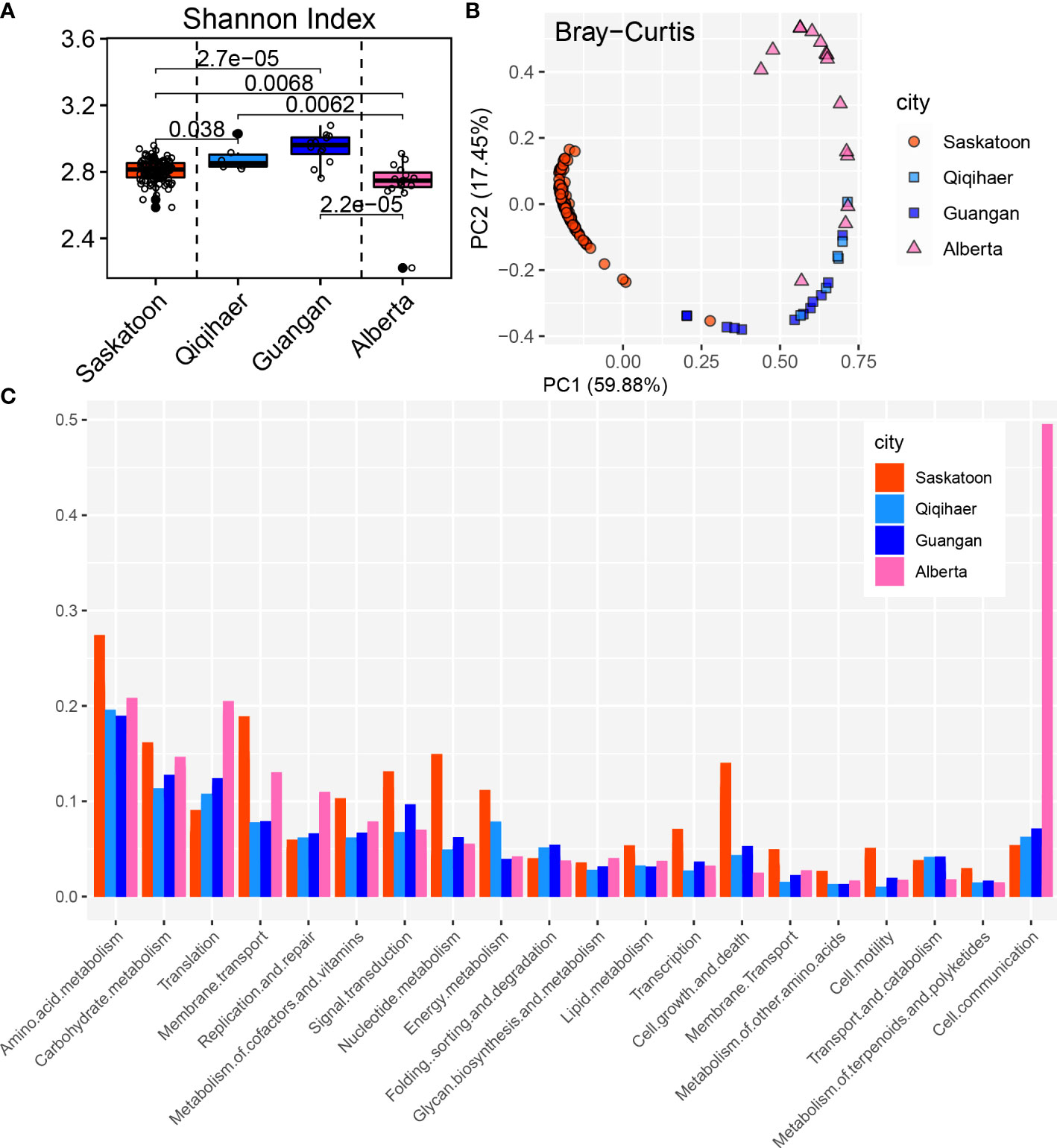
Figure 5 Functional analysis of bovine respiratory microbiome. (A) Shannon index of Kyoto Encyclopedia of Genes and Genomes (KEGG) function. (B) Principal coordinate analysis of the Bray–Curtis distance of the KEGG pathways. NPS, nasopharyngeal swab; BAL, bronchoalveolar lavage. (C) KEGG annotation at level 2.
The NPS of Saskatoon (Canada), the NPS of Qiqihaer and Guangan, and the BAL of Alberta had relative abundances of “cellular processes” (12.00, 8.92, 8.30, and 5.02%), “environmental information processing” (21.24, 14.99, 14.15, and 17.11%), and “genetic information processing” (11.79, 22.49, 22.16, and 23.28%) at level 1 (Supplementary Figure S3). At level 2, the NPS of Saskatoon had higher relative abundance of “membrane transport” (11.92%), “signal transduction” (6.60%), “transcription” (2.41%), “cell growth and death” (6.45%), “cell motility” (2.62%), “amino acid metabolism” (18.57%), “carbohydrate metabolism” (16.53%), “metabolism of cofactors and vitamins” (4.98%), “nucleotide metabolism” (3.06%), and “energy metabolism” (3.22%) (Figure 5C). The NPS of Qiqihaer and Guangan was greater in “folding, sorting, and degradation” (3.98%) and “signal transduction” (5.95%). The BAL of Alberta had higher relative abundances of “translation” (22.34%), “carbohydrate metabolism” (13.53%), “membrane transport” (10.34%), “replication and repair” (6.84%), and “cell communication” (5.51%).
Long-distance transportation changes bovine nasopharyngeal microbiota
Three groups including non-transportation (Control) and short-distance (Short) and long-distance transportation (Long) were selected. Three days was chosen as the long-distance transportation from a study of Cui et al. (2021), and metagenomic data in the Long group at the time before loading to truck (Bfload), unloading (Unload), and 7 days after placement and adaptive feeding (Adfeed) were screened. Correspondingly, we selected data at the same time points from two other groups in the study of Malmuthuge et al. (2021). Interestingly, richness in the Long group at Unload and Adfeed time points significantly decreased (P < 0.05) compared with that at Bfload, while there were no temporal changes of alpha diversity in the Control and Short groups. Beta diversity based on the Bray–Curtis distance had consistent results (Figures 6A, B).
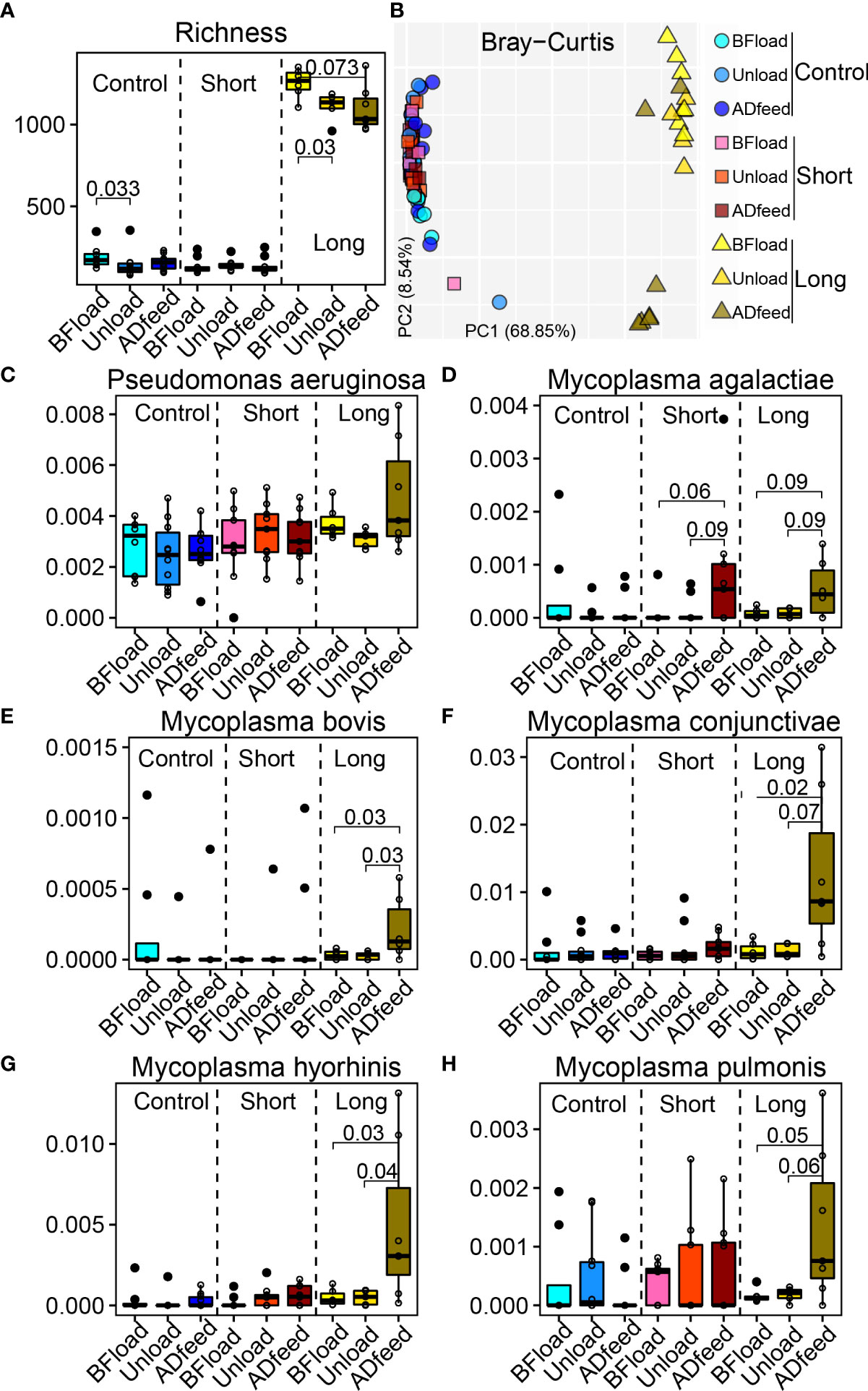
Figure 6 Long-distance transportation changes the bovine nasopharyngeal microbiota. (A) Richness. (B) Principal coordinate analysis plot based on the Bray–Curtis distance. (C–H) Main species associated with transportation. Control, non-transportation; Short, short-distance transportation; Long, long-distance transportation; BFload, time before loading to truck; Unload, unloading; ADfeed, 7 days after placement and adaptive feeding.
Next, LefSe was used to identify the longitudinal changes of microbiota following transportation (Supplementary Figure S4). Due to the variation from different studies and across multiple cities, the signature microbiota after transportation among the three groups were different. Then, we focused on the bacteria among studies to test if they have patterns across transportation distance and time. Although the microbial data were from different studies, shared bacteria, such as Pseudomonas aeruginosa, were not influenced by either short or long transportation, while Mycoplasma agalactiae was influenced by both short- and long-distance transportations such as higher abundances at Adfeed time in the Short and Long groups (Figures 6C, D). Specifically, some bacteria associated with BRD pathogens were influenced by long-distance transportation, such as the greater abundances of Mycoplasma bovis, Mycoplasma conjunctivae, Mycoplasma pulmonis, and Mycoplasma hyorhinis at the ADfeed of the Long group compared with at the BFload and Unload time points (Figures 6E–H).
Colonization of the opportunistic pathogens at the nasopharynx changes with time following weaning and transportation
Measurement of the temporal dynamics of the bovine respiratory microbiome affected by stressors including weaning and short-distance transportation allows us to better understand the changes of the bovine respiratory microbiome at the feedlot, leading to a better understanding of BRD pathogenesis (Chai et al., 2022). Metagenomics of the bovine respiratory microbiome in treatment groups on days 0 (prior to weaning and short-distance transportation, WT), 2, 4, 8, 14, and 28 was used to compare with the control group that stayed with their dams (suckling). Slight differences between suckling and WT were found based on alpha and beta diversities (Supplementary Figure S5), while the temporal dynamics of the bovine respiratory microbiome changed significantly as more statistically significant differences were found among time points (Supplementary Figure S6).
The major bacterial genera associated with BRD were influenced by time and WT (Figure 7). Moraxella was more abundant in the suckling group on day 14 (Figure 7A). Another genus, Pseudomonas, was not influenced by WT since no differences were observed between the two groups during the trial (Figure 7B). Mannheimia was greater in WT at day 14, and Mycoplasma had higher medians than suckling, although there were no statistically significant differences (Figures 7C, D).
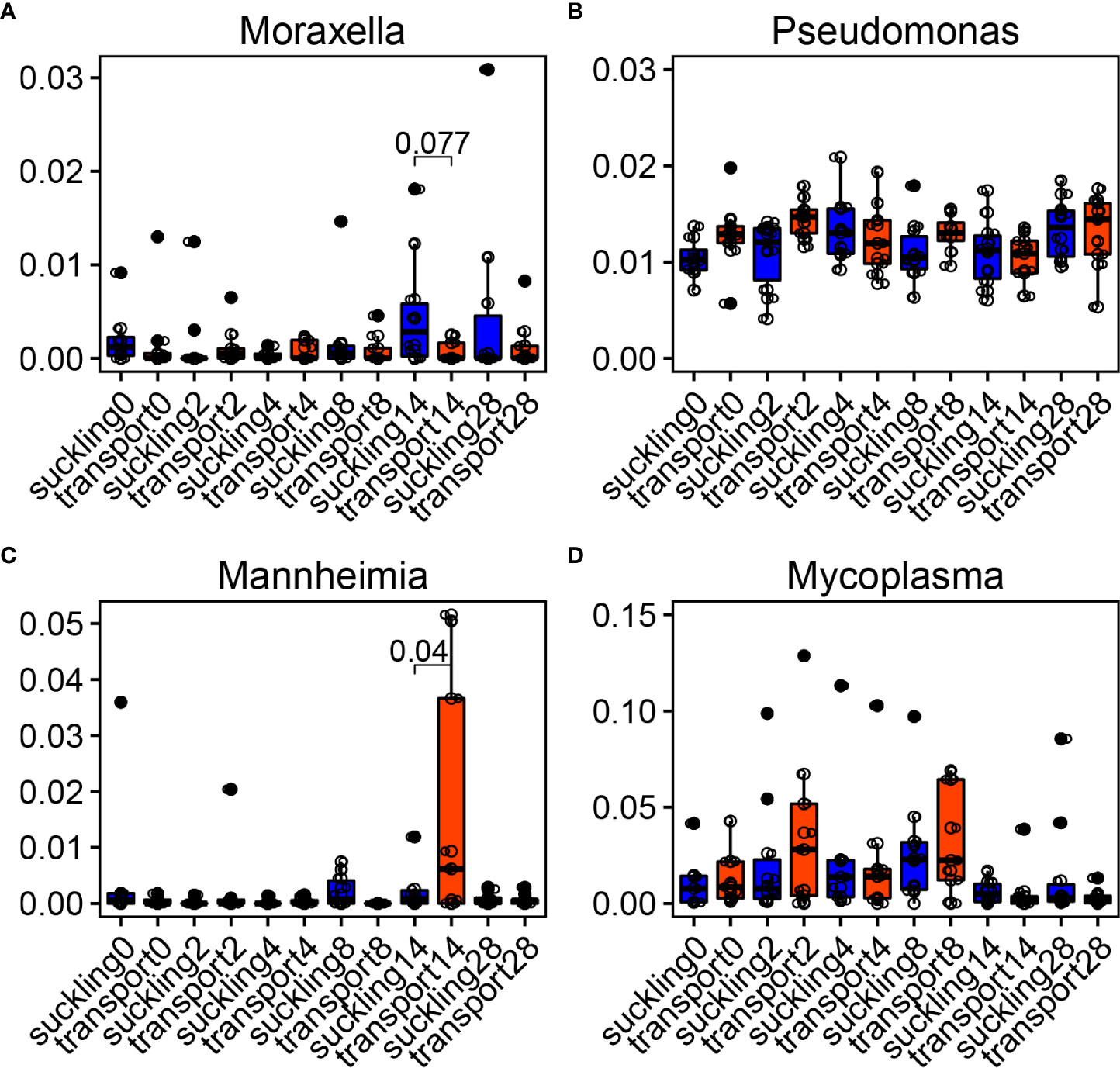
Figure 7 Temporal dynamics of bovine respiratory microbiota after weaning + transportation (WT). (A–D) Main bacterial genera changed after WT compared with the control group that stayed with their dams (suckling). Significances were labeled between suckling and WT at the time when they were statistically different.
Individual variances and clusters of the bovine lung microbiome
In the BAL microbial samples collected from BRD calves, through phylum and genus bar plots (Supplementary Figure S7), we did observe a distinct individual variation of the bovine lung microbiome. Regarding similar microbial composition found in part of the subjects, clustering analysis was performed at the species level (Figure 8A). Four clusters were observed among 15 BAL samples. Cluster 1, including animal IDs Yellow6373, 713_157, and W7839, mainly consisted of Mycoplasma bovis, Histophilus somni, and Mycoplasma agalactiae (Figure 8B). Cluster 2 (animal IDs B2_406LL, Peach2295, Org79, F1_109, 302_62, and 405_202) was abundant with Mannheimia haemolytica and Actinobacillus pleuropneumoniae. Cluster 3 (animal IDs 8577, 302_45, and 511_157WL) was dominated by Mannheimia haemolytica and Psychrobacter cryohalolentis. However, cluster 4 (animal IDs B6_216, G2516, and G3758) did not have a good pattern. The B6_216 had greater abundance of Eubacterium hallii and Streptococcus salivarius, while G3758 was dominated by Clostridium perfringens.
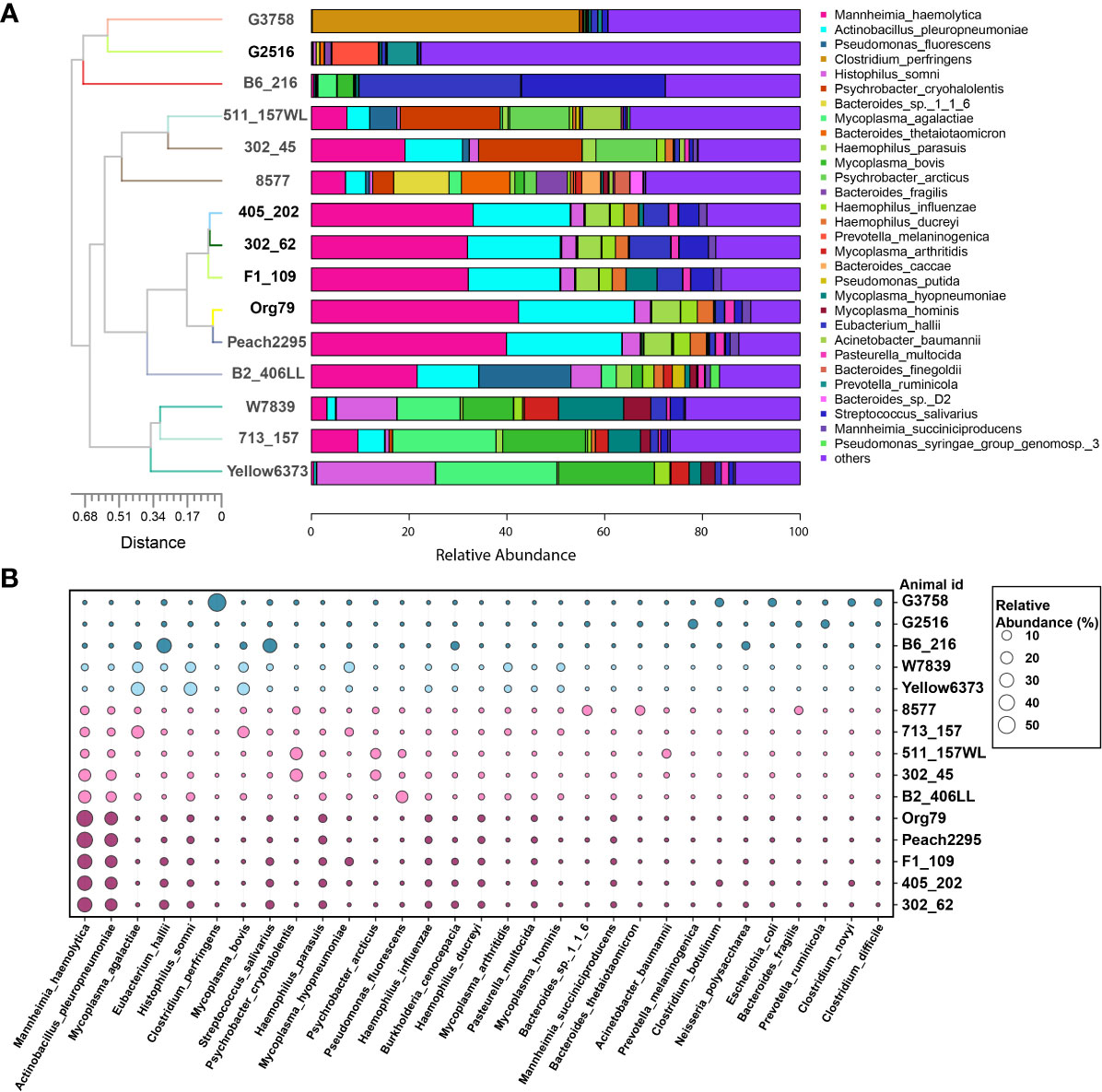
Figure 8 Individual variations and clusters of bovine lung microbiome. (A) Top 30 bacterial species in bronchoalveolar lavage were used for clustering analysis. (B) The major bacterial species distribution in the lungs. Circle size proportionally represents the relative abundance of bacterial species at a certain animal.
Discussion
The investigation of bovine respiratory microbiome and its function benefits us in order to understand microbial functions in health and disease. Although 16S rRNA sequencing technology has been broadly used, less metagenomics of bovine respiratory microbiome is reported. This study characterized microbial communities using metagenomics in the nasopharynx and lungs of calves from worldwide geographic locations, which provides evidence that geography and niches are important factors to impact the bovine respiratory microbiome and its function. Beyond this, long-distance transportation had greater impacts on the nasopharyngeal microbiota, especially BRD pathogens, than short-distance transportation. Consistent changes in microbiota influenced by transportation were found across samples from different countries. Metagenomics results from both healthy and diseased calves suggested the associations between bovine respiratory microbiome and BRD.
Calves in different geographic locations experienced huge variations in living environment, such as breed, gender, feeding strategy, diet, altitude, climate, etc. These factors together result in the variation of the microbial composition in the nasopharynx across different cities or countries. In this study, although the calves were of similar age and maintained healthy throughout the trial, calves in Canada were Hereford-crossed females that had access to water and brome-alfalfa hay, while calves in China were Simmental males that were restricted from eating and drinking. It is not surprising that the diversity, structure, and composition of the nasopharyngeal microbiota in calves from different geographic locations showed distinct variations. A previous report found that weaned calves that consumed selenium-biofortified alfalfa hay for 9 weeks resulted in favorably reformed microbial communities in the nostrils (Hall et al., 2017). Differences of nasopharyngeal microbiota among Charolais and Angus–Hereford calves were also reported (Zeineldin et al., 2017a; Holman et al., 2017d; Zeineldin et al., 2017c). Thus, geography and breed effects resulting in differences of bovine respiratory microbiota increase the difficulties to understand BRD pathogenesis when using calves from different countries. In addition, signature microbiota in the nasopharynx for each geographic location were identified. Burkholderia cenocepacia (Burkholderiaceae family, Proteobacteria phylum) was the dominant species in the NPS of Saskatoon (Canada), while the NPS of Qiqihaer and Guangan had greater abundances of Moraxellaceae family (Proteobacteria phylum). B. cenocepacia is an important opportunistic respiratory pathogen of cystic fibrosis patients (Berlutti et al., 2008). Moraxellaceae is often found to be one of the most abundant genera in the upper respiratory tract of cattle (Mcmullen et al., 2019; Mcmullen et al., 2020), and one previous study found an association between Moraxella and the development of pneumonia and/or otitis in the early life of dairy calves (Lima et al., 2016). Moreover, co-occurrence between the species-associated BRD pathogens was observed in all nasopharyngeal communities. Therefore, although the calves for nasopharyngeal sampling that were from different geographic locations were clinically healthy during the trial, the nasopharynx could be a reservoir of the opportunistic pathogens for BRD. The effect of geographic locations was also a key factor to affect the functions of the bovine respiratory microbiome. As we have observed, the structure and the abundance of the function were different across geographic locations. Regarding the main functions at KEGG level 2, “membrane transport”, “signal transduction”, “transcription”, “cell growth and death”, and “cell motility” were found in the nasopharynx of Saskatoon, indicating the active, healthy, and balanced respiratory ecosystem. The calves in Saskatoon were kept healthy during the trial, which implied that the microbial community had more interactions with the host. It is not surprising that more cell growth and death and nutrient metabolism were found in the calves of Saskatoon when compared with the calves in China that experienced long-distance transportation. Therefore, homeostasis of the respiratory microbiome plays critical roles in bovine airway health, and disease or environmental factors can lead to disequilibrium in the respiratory ecosystem.
The lung microbiome is a critical point for BRD (pneumonia). In this study, the lung microbiome and its functions showed a significantly different composition and structure compared with nasopharyngeal communities regardless of their geographic locations. The BAL samples were collected at necropsy from 15 feedlot cattle that were confirmed to have died of BRD. Higher abundances of Mannheimia haemolytica, Mycoplasma bovis, and Histophilus somni were observed, which is in agreement with previous studies that these bacteria are BRD-associated pathogens (Rice et al., 2007; Singh et al., 2011; Nicola et al., 2017). Notably, Mycoplasma species (M. agalactiae, M. bovis, M. arthritidis, M. hyopneumoniae, and M. hominis) inter-connected with each other were observed in BRD lungs, which reflects the cooperation of pathogens to cause BRD. Based on the hypothesis that the nasopharynx is a reservoir of the opportunistic pathogens for BRD, we assumed that Mycoplasma species may come from the nasopharynx. A study reported that the best recovery condition for Mycoplasma bovis is at 36.91°C (± 0.07) and pH = 7.13 (± 0.05) under an aerobic environment (Parker et al., 2016). Bovine nasopharynx is the niche with pH of about 7 and temperature of about 37°C, which is good for the colonization and growth of Mycoplasma species (Chai et al., 2022). Therefore, the opportunistic pathogens for BRD may colonize the nasopharynx and disperse in and infect the lungs, which provides a new way to explore BRD pathogenesis and prevent BRD. In addition, the interactions of Mycoplasma species might be another reason causing BRD, which need to be investigated in future research.
BRD, also known as “shipping fever”, is caused by various stressors (i.e., weaning, transportation) that also influence the bovine respiratory microbiota (Chai et al., 2022). Understanding the association between transportation and microbial pathogen proliferation may explain BRD pathogenesis. A previous study demonstrated that transportation to a feedlot caused an abrupt shift in the nasopharyngeal microbiota of cattle using 16S rRNA sequencing, and major differences were driven mostly by Mycoplasma (Holman et al., 2017d). A metagenomic study confirmed that stress from long-distance shipping influences the nasopharyngeal microbiota (Cui et al., 2021). Similarly, in our study, long-distance transportation had bigger impacts on the bovine respiratory microbiota, and Mycoplasma species, including M. bovis, M. conjunctivae, M. pulmonis, and M. hyorhinis, increased the abundances after feedlot arrival. Thus, stressors (i.e., long-distance transportation) and the feedlot environment may provide conditions that allow for the proliferation of Mycoplasma in the nasopharynx. In addition, abundance of Mycoplasma agalactiae by both short- and long-distance transportation was observed, which gives more evidence that transportation can cause changes in Mycoplasma species (Stroebel et al., 2018). It was reported that stress can exacerbate respiratory hyperreactivity and inflammation in an animal model of allergic bronchial asthma (Joachim et al., 2003). Newly weaned calves experience huge stress during transportation or shipping, especially for a long distance (Zulkifli et al., 2019; Malmuthuge et al., 2021), which may cause unclear changes in the airway for Mycoplasma proliferation. Keeping microbial balances in bovine airway during transportation might be an alternative strategy to decrease BRD morbidity.
BRD is usually diagnosed in cattle within 4 weeks after having been transported to a feedlot (Currin and Whittier, 2005). Understanding of the temporal dynamics of the bovine respiratory microbiome from the time since arrival to the feedlot can allow us to recognize the association between microbiota and BRD. Here we found that genera associated with BRD, such as Mycoplasma and Mannheimia, were increased at a specific time after weaning and transportation. Since all of the calves were healthy and had never been diagnosed with BRD in this study, these increased pathogens might be due to the time after feedlot arrival or changes of the respiratory health status. Moreover, Moraxella was higher in calves that stayed with their dams. Moraxella is often found to be one of the most abundant genera in the upper respiratory tract of healthy cattle (Zeineldin et al., 2017a; Mcmullen et al., 2019). Therefore, Mycoplasma and Mannheimia may reach a specific level to cause clinical BRD signs, and a greater abundance of Moraxella in the nasopharynx might resist BRD.
The inter-individual variability of the respiratory microbiota was found in cattle and humans (Dickson et al., 2015; Holman et al., 2015b; Zeineldin et al., 2017a), which is not surprising due to individual variation. The bacterial clusters and their association with clinical signs were investigated in human respiratory microbiome studies (Zhou et al., 2019; Widder et al., 2022). However, in cattle, there were no studies to characterize the community type of the respiratory microbiota. In this study, microbial clusters of BRD lungs were found, although there was significant inter-individual variability—for instance, bacterial pathogens, including Mycoplasma bovis, Histophilus somni, and Mycoplasma agalactiae, were enriched in cluster 1, while Mannheimia haemolytica plus different pathogens formed a different cluster in the BRD lung microbiome. Previous studies found that Mycoplasma bovis co-infection with other respiratory bacteria (M. haemolytica and H. somni) can lead to severe pneumonic lesions, and Mannheimia haemolytica might be associated with Actinobacillus pleuropneumoniae in pigs (Gagea et al., 2006; Radaelli et al., 2008). Although the cooperation of different pathogens causing BRD is still unclear, this study provides insights that pathogenic symbiosis is one of the important causes of respiratory diseases. In future BRD studies, large-scale sampling is needed for characterizing the community type of the lung microbiome, which may reveal the interactions of bacterial pathogens and provide an alternative therapeutic strategy for BRD.
A limitation of this study may be the small sample size of lung samples. However, the dominant bacterial pathogens in each cluster could explain the subtypes of BRD pathogenesis since these samples were collected from the lungs of calves that died from BRD. The strengths of this study are the analysis of bovine nasopharyngeal microbiome from different geographic locations using metagenomics and the finding that the general pathogens were influenced by transportation regardless of geography. Future studies to assess more geographic locations of bovine respiratory microbiome and use omics to explore the interactions of the airway microbiome and the host are urgently needed.
Conclusions
Metagenomic sequencing offers the chance to illustrate the species level of community structure and functional profile of the bovine respiratory microbiome. The geographic locations affect the composition and the functions of the bovine respiratory microbiome, and the microbiota from different sampling niches were also distinct. Additionally, transportation is an important factor driving the bovine respiratory microbiome and diseases, and long-distance transportation had a bigger chance to cause increased abundances of pathogens in the nasopharynx. Furthermore, we found that the cluster of lung microbiota from different BRD calves might provide an opportunity to come up with a new guidebook for the category of BRD pathogenesis.
Data availability statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/Supplementary Material.
Ethics statement
All data related to animals were downloaded from the published NCBI database.
Author contributions
JC contributed to data collection, analysis and interpretation, and figure organization and drafted the manuscript. XL and HU contributed to draft proofing. FD contributed to data collection. JZ and YL contributed to conception, manuscript proofing, and project supervision. All authors contributed to the article and approved the submitted version.
Funding
This project was supported by the Agriculture and Food Research Initiative competitive grant no. 20196701629869 from the USDA National Institute of Food and Agriculture, National Natural Science Foundation of China (no. 32170430), Guangdong Provincial Key Laboratory of Animal Molecular Design and Precise Breeding (2019B030301010), and Key Laboratory of Animal Molecular Design and Precise Breeding of Guangdong Higher Education Institutes (2019KSYS011).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fcimb.2022.961644/full#supplementary-material
References
Berlutti, F., Superti, F., Nicoletti, M., Morea, C., Frioni, A., Ammendolia, M. G., et al. (2008). Bovine lactoferrin inhibits the efficiency of invasion of respiratory A549 cells of different iron-regulated morphological forms of pseudomonas aeruginosa and burkholderia cenocepacia. Int. J. Immunopathol. Pharmacol. 21, 51–59. doi: 10.1177/039463200802100107
Chai, J., Capik, S. F., Kegley, B., Richeson, J. T., Powell, J. G., Zhao, J. (2022). Bovine respiratory microbiota of feedlot cattle and its association with disease. Vet. Res. 53, 4. doi: 10.1186/s13567-021-01020-x
Cirone, F., Padalino, B., Tullio, D., Capozza, P., Lo Surdo, M., Lanave, G., et al. (2019). Prevalence of pathogens related to bovine respiratory disease before and after transportation in beef steers: Preliminary results. Anim. an Open Access J. MDPI 9, 1093. doi: 10.3390/ani9121093
Cui, Y., Qi, J., Cai, D., Fang, J., Xie, Y., Guo, H., et al. (2021). Metagenomics reveals that proper placement after long-distance transportation significantly affects calf nasopharyngeal microbiota and is critical for the prevention of respiratory diseases. Front Microbiol. 12, 700704. doi: 10.3389/fmicb.2021.700704.
Currin, J. F., Whittier, W. D. (2005). Recognition and treatment of bovine respiratory disease complex. Vet Med 8, 1–3.
Dickson, R. P., Erb-Downward, J. R., Freeman, C. M., Mccloskey, L., Beck, J. M., Huffnagle, G. B., et al. (2015). Spatial variation in the healthy human lung microbiome and the adapted island model of lung biogeography. Ann. Am. Thorac. Soc. 12, 821–830. doi: 10.1513/AnnalsATS.201501-029OC
Dickson, R. P., Erb-Downward, J. R., Martinez, F. J., Huffnagle, G. B. (2016). The microbiome and the respiratory tract. Annu. Rev. Physiol. 78, 481–504. doi: 10.1146/annurev-physiol-021115-105238
Gaeta, N. C., Lima, S. F., Teixeira, A. G., Ganda, E. K., Oikonomou, G., Gregory, L., et al. (2017). Deciphering upper respiratory tract microbiota complexity in healthy calves and calves that develop respiratory disease using shotgun metagenomics. J. Dairy Sci. 100, 1445–1458. doi: 10.3168/jds.2016-11522
Gagea, M. I., Bateman, K. G., Shanahan, R. A., Van Dreumel, T., Mcewen, B. J., Carman, S., et al. (2006). Naturally occurring mycoplasma bovis-associated pneumonia and polyarthritis in feedlot beef calves. J. Veterinary Diagn. Invest. 18, 29–40. doi: 10.1177/104063870601800105
Gupta, V. K., Paul, S., Dutta, C. (2017). Geography, ethnicity or subsistence-specific variations in human microbiome composition and diversity. Front. Microbiol. 8, 1162. doi: 10.3389/fmicb.2017.01162
Hall, J. A., Isaiah, A., Estill, C. T., Pirelli, G. J., Suchodolski, J. S. (2017). Weaned beef calves fed selenium-biofortified alfalfa hay have an enriched nasal microbiota compared with healthy controls. PloS One 12 (6), e0179215. doi: 10.1371/journal.pone.0179215
Holman, D., Hallewell, J., Alexander, T. W. (2017a). The nasopharyngeal microbiota of beef cattle before and after transport to a feedlot. J. Anim. Sci. 95, 29–29. doi: 10.1186/s12866-017-0978-6
Holman, D. B., Klima, C. L., Ralston, B. J., Niu, Y. D., Stanford, K., Alexander, T. W., et al. (2017c). Metagenomic sequencing of bronchoalveolar lavage samples from feedlot cattle mortalities associated with bovine respiratory disease. Genome Announc 5 (40), e01045-17. doi: 10.1128/genomeA.01045-17
Holman, D., Mcallister, T. A., Topp, E., Wright, A.-D. G., Alexander, T. W. (2015a). The nasopharyngeal microbiota of feedlot cattle that develop bovine respiratory disease. Veterinary Microbiol. 180, 90–95. doi: 10.1016/j.vetmic.2015.07.031
Holman, D., Timsit, E., Alexander, T. (2015b). The nasopharyngeal microbiota of feedlot cattle. Sci. Rep. 5, 15557. doi: 10.1038/srep15557
Holman, D. B., Timsit, E., Amat, S., Abbott, D. W., Buret, A. G., Alexander, T. W. (2017d). The nasopharyngeal microbiota of beef cattle before and after transport to a feedlot. BMC Microbiol. 17, 70. doi: 10.1186/s12866-017-0978-6
Holman, D. B., Yang, W., Alexander, T. W. (2019). Antibiotic treatment in feedlot cattle: a longitudinal study of the effect of oxytetracycline and tulathromycin on the fecal and nasopharyngeal microbiota. Microbiome 7, 86. doi: 10.1186/s40168-019-0696-4
Human Microbiome Project, C. (2012). Structure, function and diversity of the healthy human microbiome. Nature 486, 207–214. doi: 10.1038/nature11234
Joachim, R. A., Quarcoo, D., Arck, P. C., Herz, U., Renz, H., Klapp, B. F. (2003). Stress enhances airway reactivity and airway inflammation in an animal model of allergic bronchial asthma. Psychosom Med. 65, 811–815. doi: 10.1097/01.PSY.0000088582.50468.A3
Keegan, K. P., Glass, E. M., Meyer, F. (2016). MG-RAST, a metagenomics service for analysis of microbial community structure and function. Methods Mol. Biol. 1399, 207–233. doi: 10.1007/978-1-4939-3369-3_13
Klima, C. L., Holman, D. B., Ralston, B. J., Stanford, K., Zaheer, R., Alexander, T. W., et al. (2019). Lower respiratory tract microbiome and resistome of bovine respiratory disease mortalities. Microb. Ecol. 78 (2), 446–456. doi: 10.1007/s00248-019-01361-3
Lima, S. F., Teixeira, A. G., Higgins, C. H., Lima, F. S., Bicalho, R. C. (2016). The upper respiratory tract microbiome and its potential role in bovine respiratory disease and otitis media. Sci. Rep. 6, 29050. doi: 10.1038/srep29050
Malmuthuge, N., Howell, A., Arsic, N., Prysliak, T., Perez-Casal, J., Griebel, P. (2021). Effect of maternal separation and transportation stress on the bovine upper respiratory tract microbiome and the immune response to resident opportunistic pathogens. Anim. Microbiome 3, 62. doi: 10.1186/s42523-021-00123-2
Man, W. H., De Steenhuijsen Piters, W. A., Bogaert, D. (2017). The microbiota of the respiratory tract: gatekeeper to respiratory health. Nat. Rev. Microbiol. 15, 259–270. doi: 10.1038/nrmicro.2017.14
Mcmullen, C., Alexander, T. W., Leguillette, R., Workentine, M., Timsit, E. (2020). Topography of the respiratory tract bacterial microbiota in cattle. Microbiome 8, 91. doi: 10.1186/s40168-020-00869-y
Mcmullen, C., Orsel, K., Alexander, T. W., van der Meer, F., Plastow, G., Timsit, E. (2018). Evolution of the nasopharyngeal bacterial microbiota of beef calves from spring processing to 40 days after feedlot arrival. Veterinary Microbiol. 225, 139–148. doi: 10.1016/j.vetmic.2018.09.019
Mcmullen, C., Orsel, K., Alexander, T. W., van der Meer, F., Plastow, G., Timsit, E. (2019). Comparison of the nasopharyngeal bacterial microbiota of beef calves raised without the use of antimicrobials between healthy calves and those diagnosed with bovine respiratory disease. Veterinary Microbiol. 231, 56–62. doi: 10.1016/j.vetmic.2019.02.030
Nicola, I., Cerutti, F., Grego, E., Bertone, I., Gianella, P., D'angelo, A., et al. (2017). Characterization of the upper and lower respiratory tract microbiota in piedmontese calves. Microbiome 5 (1), 152. doi: 10.1186/s40168-017-0372-5
Parker, A. M., House, J. K., Hazelton, M. S., Bosward, K. L., Mohler, V. L., Maunsell, F. P., et al. (2016). Milk acidification to control the growth of mycoplasma bovis and salmonella Dublin in contaminated milk. J. Dairy Sci. 99, 9875–9884. doi: 10.3168/jds.2016-11537
Radaelli, E., Luini, M., Loria, G. R., Nicholas, R. A., Scanziani, E. (2008). Bacteriological, serological, pathological and immunohistochemical studies of mycoplasma bovis respiratory infection in veal calves and adult cattle at slaughter. Res. Vet. Sci. 85, 282–290. doi: 10.1016/j.rvsc.2007.11.012
Rice, J. A., Carrasco-Medina, L., Hodgins, D. C., Shewen, P. E. (2007). Mannheimia haemolytica and bovine respiratory disease. Anim. Health Res. Rev. 8, 117–128. doi: 10.1017/S1466252307001375
Singh, K., Ritchey, J. W., Confer, A. W. (2011). Mannheimia haemolytica: bacterial-host interactions in bovine pneumonia. Vet. Pathol. 48, 338–348. doi: 10.1177/0300985810377182
Snowder, G. D., Van Vleck, L. D., Cundiff, L. V., Bennett, G. L. (2006). Bovine respiratory disease in feedlot cattle: environmental, genetic, and economic factors. J. Anim. Sci. 84, 1999–2008. doi: 10.2527/jas.2006-046
Stroebel, C., Alexander, T., Workentine, M. L., Timsit, E. (2018). Effects of transportation to and co-mingling at an auction market on nasopharyngeal and tracheal bacterial communities of recently weaned beef cattle. Veterinary Microbiol. 223, 126–133. doi: 10.1016/j.vetmic.2018.08.007
Taylor, J. D., Fulton, R. W., Lehenbauer, T. W., Step, D. L., Confer, A. W. (2010). The epidemiology of bovine respiratory disease: What is the evidence for predisposing factors? Can. Vet. J. 51, 1095–1102.
Timsit, E., Holman, D. B., Hallewell, J., Alexander, T. W. (2016a). The nasopharyngeal microbiota in feedlot cattle and its role in respiratory health. Anim. Front. 6, 44–50. doi: 10.2527/af.2016-0022
Timsit, E., Workentine, M., Schryvers, A. B., Holman, D. B., van der Meer, F., Alexander, T. W. (2016b). Evolution of the nasopharyngeal microbiota of beef cattle from weaning to 40 days after arrival at a feedlot. Veterinary Microbiol. 187, 75–81. doi: 10.1016/j.vetmic.2016.03.020
Venkataraman, A., Bassis, C. M., Beck, J. M., Young, V. B., Curtis, J. L., Huffnagle, G. B., et al. (2015). Application of a neutral community model to assess structuring of the human lung microbiome. MBio 6 (1), e02284-14. doi: 10.1128/mBio.02284-14
Widder, S., Zhao, J., Carmody, L. A., Zhang, Q., Kalikin, L. M., Schloss, P. D., et al. (2022). Association of bacterial community types, functional microbial processes and lung disease in cystic fibrosis airways. ISME J. 16, 905–914. doi: 10.1038/s41396-021-01129-z
Zeineldin, M., Elolimy, A. A., Barakat, R. (2020). Meta-analysis of bovine respiratory microbiota: link between respiratory microbiota and bovine respiratory health. FEMS Microbiol. Ecol. 96(8), fiaa127. doi: 10.1093/femsec/fiaa127
Zeineldin, M., Lowe, J., Aldridge, B. (2019). Contribution of the mucosal microbiota to bovine respiratory health. Trends Microbiol. 27 (9), 753–770. doi: 10.1016/j.tim.2019.04.005
Zeineldin, M., Lowe, J., De Godoy, M., Maradiaga, N., Ramirez, C., Ghanem, M., et al. (2017a). Disparity in the nasopharyngeal microbiota between healthy cattle on feed, at entry processing and with respiratory disease. Veterinary Microbiol. 208, 30–37. doi: 10.1016/j.vetmic.2017.07.006
Zeineldin, M., Lowe, J., Grimmer, E., De Godoy, M., Ghanem, M., Abd El-Raof, Y., et al. (2017c). Relationship between nasopharyngeal and bronchoalveolar microbial communities in clinically healthy feedlot cattle. BMC Microbiol. 17 (1), 138. doi: 10.1186/s12866-017-1042-2
Zhou, Y., Jackson, D., Bacharier, L. B., Mauger, D., Boushey, H., Castro, M., et al. (2019). The upper-airway microbiota and loss of asthma control among asthmatic children. Nat. Commun. 10, 5714. doi: 10.1038/s41467-019-13698-x
Keywords: respiratory microbiota, bovine, metagenomics, geography, bovine respiratory disease, transportation, nasopharynx, lung
Citation: Chai J, Liu X, Usdrowski H, Deng F, Li Y and Zhao J (2022) Geography, niches, and transportation influence bovine respiratory microbiome and health. Front. Cell. Infect. Microbiol. 12:961644. doi: 10.3389/fcimb.2022.961644
Received: 05 June 2022; Accepted: 17 August 2022;
Published: 12 September 2022.
Edited by:
Kristina Marie Feye, National Center for Toxicological Research (FDA), United StatesReviewed by:
Joyce Oh, National Center for Toxicological Research (FDA), United StatesQi Xu, Yangzhou University, China
Copyright © 2022 Chai, Liu, Usdrowski, Deng, Li and Zhao. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jiangchao Zhao, anpoYW83N0B1YXJrLmVkdQ==