 Amanda N. Samuels
Amanda N. Samuels Erin R. Wang
Erin R. Wang Gregory A. Harrison
Gregory A. Harrison Joy C. Valenta
Joy C. Valenta Christina L. Stallings
Christina L. Stallings
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Cell. Infect. Microbiol., 22 August 2022
Sec. Molecular Bacterial Pathogenesis
Volume 12 - 2022 | https://doi.org/10.3389/fcimb.2022.958555
This article is part of the Research TopicDrug-resistant Mycobacterium tuberculosisView all 21 articles
Treatment of Mycobacterium tuberculosis (Mtb) infections is particularly arduous. One challenge to effectively treating tuberculosis is that drug efficacy in vivo often fails to match drug efficacy in vitro. This is due to multiple reasons, including inadequate drug concentrations reaching Mtb at the site of infection and physiological changes of Mtb in response to host derived stresses that render the bacteria more tolerant to antibiotics. To more effectively and efficiently treat tuberculosis, it is necessary to better understand the physiologic state of Mtb that promotes drug tolerance in the host. Towards this end, multiple studies have converged on bacterial central carbon metabolism as a critical contributor to Mtb drug tolerance. In this review, we present the evidence that changes in central carbon metabolism can promote drug tolerance, depending on the environment surrounding Mtb. We posit that these metabolic pathways could be potential drug targets to stymie the development of drug tolerance and enhance the efficacy of current antimicrobial therapy.
Tuberculosis, an infection caused by the pathogen Mycobacterium tuberculosis (Mtb) is one of the leading causes of death world-wide by an infectious agent (WHO, 2021). Standard of care treatment for drug sensitive Mtb infections requires at least 6 months of antibiotic therapy with 4 or more antibiotics (WHO, 2017). Infection with Mtb mutants that are resistant to the frontline antibiotics isoniazid and rifampicin constituted approximately half a million tuberculosis cases in 2019 (WHO, 2021) and contributes to treatment failure (Chen et al., 2020). The treatment regimen for patients harboring drug resistant Mtb is even longer and more expensive than drug sensitive cases and has an increased risk of adverse side effects (Nahid et al., 2019; WHO, 2021; Ghazy et al., 2022). Overall, the emergence and prevalence of Mtb drug resistance threatens treatment efficacy globally.
In addition, treatment failure and relapse can occur even in the absence of drug resistance. Dating as far back as the 1950’s, it is documented that Mtb can be recovered from some patients after antibiotic treatment, with a fraction of these isolates remaining drug sensitive in vitro (Hobby, 1955; Wallace and Sutherland, 1955). In a 2014 study, 8% of patients that were treated with the standard of care isoniazid, rifampin, pyrazinamide, and ethambutol for 8 weeks followed by 18 weeks of isoniazid and rifampicin had an unfavorable outcome (Gillespie et al., 2014). The most common unfavorable outcome was relapse, which was differentiated from patients re-infected with another strain by using 24-locus mycobacterial-interspersed-repetitive-unit analysis to confirm that the strains isolated during relapse were the same as the primary infection (Gillespie et al., 2014). In this study, only 25% of the patients receiving the standard of care that relapsed after conversion to culture-negative status were suspected to have acquired drug resistance (Gillespie et al., 2014). Shortening the antibiotic regimen results in even further increased rates of treatment failure and relapse (Gillespie et al., 2014; Jindani et al., 2014). Another study collected serial Mtb isolates from tuberculosis patients that had relapsed infection after antibiotic treatment, where relapse was defined by paired isolates exhibiting 0-6 single nucleotide polymorphisms by whole genome sequencing (Bryant et al., 2013). In this study, all the relapsed Mtb isolates were drug sensitive in vitro (Bryant et al., 2013). Collectively, this data support that a reservoir of drug sensitive Mtb can persist in the host despite antibiotic therapy, contributing to treatment failure in some patients.
Multiple factors have been identified that enable Mtb to persist in the host during antibiotic treatment without acquiring a drug resistance mutation. One factor is the pathology that develops within the lung during Mtb infection. During infection, the interaction between Mtb and the host immune response can result in the development of a granuloma, which is made up of host immune cells, Mtb, and tissue debris (Ehlers and Schaible, 2013). Antibiotic penetration into the granuloma can be limited based on the chemical properties of the antibiotic, which creates a challenge for efficient delivery of the antibiotic to the various sites where Mtb resides (Kjellsson et al., 2012; Prideaux et al., 2015; Sarathy et al., 2018). In addition, Mtb can reside within various compartments inside innate immune cells, which can impact antibiotic efficacy. For example, pyrazinamide preferentially accumulates and is maximally active against Mtb in acidified compartments within the macrophage (Santucci et al., 2021; Santucci et al., 2022).
In addition to the host response impacting antibiotic accessibility to Mtb, the pathogen itself changes its physiology in response to the host environment, resulting in phenotypic drug tolerance. Importantly, drug tolerance is different from drug resistance in that a drug tolerant population can survive in the presence of an antibiotic but cannot grow until the antibiotic pressure is removed, whereas a drug resistant population can both survive and replicate in the presence of an antibiotic. In unstressed axenic culture conditions, Mtb populations display a basal level of heterogeneity such that a subpopulation of bacteria is transiently tolerant to antibiotics (Aldridge et al., 2012; Manina et al., 2015; Rego et al., 2017). Because of this drug tolerant subpopulation, treatment with a bactericidal antibiotic, such as isoniazid or rifampicin, leads to a significant decrease in viable bacteria, but fails to sterilize the culture (Jain et al., 2016; Sukheja et al., 2017; Vilcheze et al., 2017). Some of the drug susceptibility heterogeneity results from Mtb’s asymmetric cell division (Aldridge et al., 2012; Rego et al., 2017). Deletion of the gene lamA/mmpS3 leads to a loss of asymmetric cell elongation and cell size heterogeneity in Mycobacterium smegmatis, and an Mtb lamA/mmpS3 mutant is more susceptible to killing by rifampicin and vancomycin, suggesting that asymmetric cell elongation and cell size heterogeneity contributes to the emergence of drug tolerant subpopulations (Rego et al., 2017). In addition, there are stochastic differences in gene expression within mycobacterial cultures that can affect antibiotic susceptibility. For example, mycobacteria exhibit stochastic variation in the expression of katG, which is required to activate the pro-drug isoniazid, leading to a small population of bacteria with transiently low katG expression that can survive exposure to isoniazid (Wakamoto et al., 2013).
The proportion of drug tolerant Mtb is higher in vivo when compared to the small population that exists at basal levels in unstressed axenic cultures. Mtb directly isolated from patient sputum samples exhibited a nearly 10-fold reduction in killing by streptomycin, isoniazid, ethambutol, or rifampicin in comparison to when those same isolates were passaged through normal culture conditions (Turapov et al., 2016). Mtb in caseum isolated from infected rabbit granulomas also exhibited a >100-fold increase in the minimum bactericidal concentration for rifampicin and isoniazid compared to Mtb growing in vitro (Sarathy et al., 2018). Therefore, the Mtb population at the site of infection is enriched for drug tolerant cells, indicating that the host environment causes the Mtb population to shift towards a more drug tolerant state. Understanding the mechanistic basis for this enhanced drug tolerance is essential for developing therapies that target the Mtb population that is recalcitrant to treatment.
During infection of macrophages, Mtb may be exposed to low pH, nitrosative stress, oxidative stress, osmotic changes, carbohydrate limitation, and cell envelope damage (Schnappinger et al., 2003; Tan et al., 2013; Larrouy-Maumus et al., 2016; Pisu et al., 2020). The environment within granulomas also poses additional stresses on Mtb, where granulomas can be hypoxic (Via et al., 2008), contain host factors that sequester iron (Kurthkoti et al., 2017), and harbor host enzymes that produce reactive oxygen species (Marakalala et al., 2016). Despite this harsh host environment, Mtb can survive due to its robust stress response capabilities. Mtb responds transcriptionally and metabolically to survive exposure to hypoxia (Wayne and Hayes, 1996), nitric oxide (Voskuil et al., 2003), reactive oxygen species (Voskuil et al., 2011), carbon limitation (Loebel et al., 1933; Betts et al., 2002; Gengenbacher et al., 2010), iron limitation (Kurthkoti et al., 2017), and low pH (Baker et al., 2014). Importantly, when exposed to stress in vitro, such as hypoxia, low pH, changes in osmolarity, or nutrient limitation, the proportion of drug tolerant Mtb increases, leading to higher minimal inhibitory concentrations or minimum bactericidal concentrations for several antibiotics (Wayne and Hayes, 1996; Deb et al., 2009; Gengenbacher et al., 2010; Larrouy-Maumus et al., 2016; Sarathy et al., 2018; Baker and Abramovitch, 2018; Xie et al., 2005). These data support that exposure to host derived stresses contributes to the increased Mtb antibiotic tolerance observed during infection.
Mtb stress responses are complex and involve multiple transcriptional, proteomic, and metabolic changes aimed at promoting pathogen survival. The resulting increase in drug tolerance that emerges in these conditions is indisputably multifactorial. Recent reviews have focused on the role of transcriptional adaptation (Kundu and Basu, 2021), the stringent response (Sharma et al., 2021), bacterial respiration (Hasenoehrl et al., 2021), and drug efflux in Mtb drug tolerance (Remm et al., 2021). In this review, we will focus on the role of fluctuations in central carbon metabolism in promoting drug tolerance of Mtb and discuss how continued dissection of the link between central carbon metabolism and drug tolerance will provide novel therapeutic approaches to target drug tolerant Mtb.
Mtb grown in vitro can metabolize multiple carbon sources, even simultaneously (de Carvalho et al., 2010). Some of the most common carbon sources used to culture Mtb include glucose, glycerol, and oleic acid (Larsen et al., 2007). Glucose and other sugars are metabolized primarily through glycolysis and the pentose phosphate pathway to generate ATP and reducing equivalents (Figure 1). Glycerol is also used to generate ATP and reducing equivalents through glycolysis, or it can be anabolized via gluconeogenesis to synthesize sugars. To assimilate into these pathways, glycerol must first be converted to glycerol-3-phosphate by GlpK and then oxidized to dihydroxyacetone phosphate (DHAP)(Figure 1). Oleic acid and other even-chain fatty acids are catabolized to acetyl-CoA, which enters the tricarboxylic acid (TCA) cycle (Figure 1). The TCA cycle is critical for the generation of the reducing equivalents NADH and NADPH, as well as biosynthetic precursors for multiple other pathways, including synthesis of several amino acids. In particular, α-ketoglutarate can be converted to glutamate, which is a precursor for glutamine, arginine, and proline synthesis, and oxaloacetate can be converted to aspartate, which serves as a precursor for the synthesis of several amino acids including asparagine, methionine, lysine, threonine, and isoleucine. Mutants that are auxotrophic for one or more of these amino acids, including glutamine (Lee et al., 2006), arginine (Gordhan et al., 2002), aspartate (Jansen et al., 2020), methionine (Berney et al., 2015; Hasenoehrl et al., 2019), lysine (Pavelka et al., 2003), and threonine (Hasenoehrl et al., 2019) are severely attenuated during infection, demonstrating that the ability to synthesize these amino acids from TCA cycle intermediates is critical for Mtb to establish and maintain infection in the host. The essentiality of de novo amino acid biosynthesis during infection is particularly surprising because Mtb can assimilate nitrogen from asparagine, aspartate, glutamate, glutamine, leucine, alanine, and glycine during growth in macrophages in vitro (Gouzy et al., 2014; Borah et al., 2019). Mtb can also divert carbon from the CO2-generating steps of the TCA cycle via the glyoxylate shunt pathway (Muñoz-Elías and Mckinney, 2005). The glyoxylate shunt enables growth on fatty acids as a sole carbon source because it prevents loss of carbon via CO2, allowing for net gain of carbon from acetyl-CoA. This carbon can then be routed to other essential biosynthetic pathways such as amino acid synthesis or gluconeogenesis to generate cell wall precursors. In contrast, carbon sources that feed into glycolysis can be used to re-generate TCA cycle intermediates, allowing for carbon to leave the TCA cycle for biosynthesis and also be replenished independent of the glyoxylate shunt.
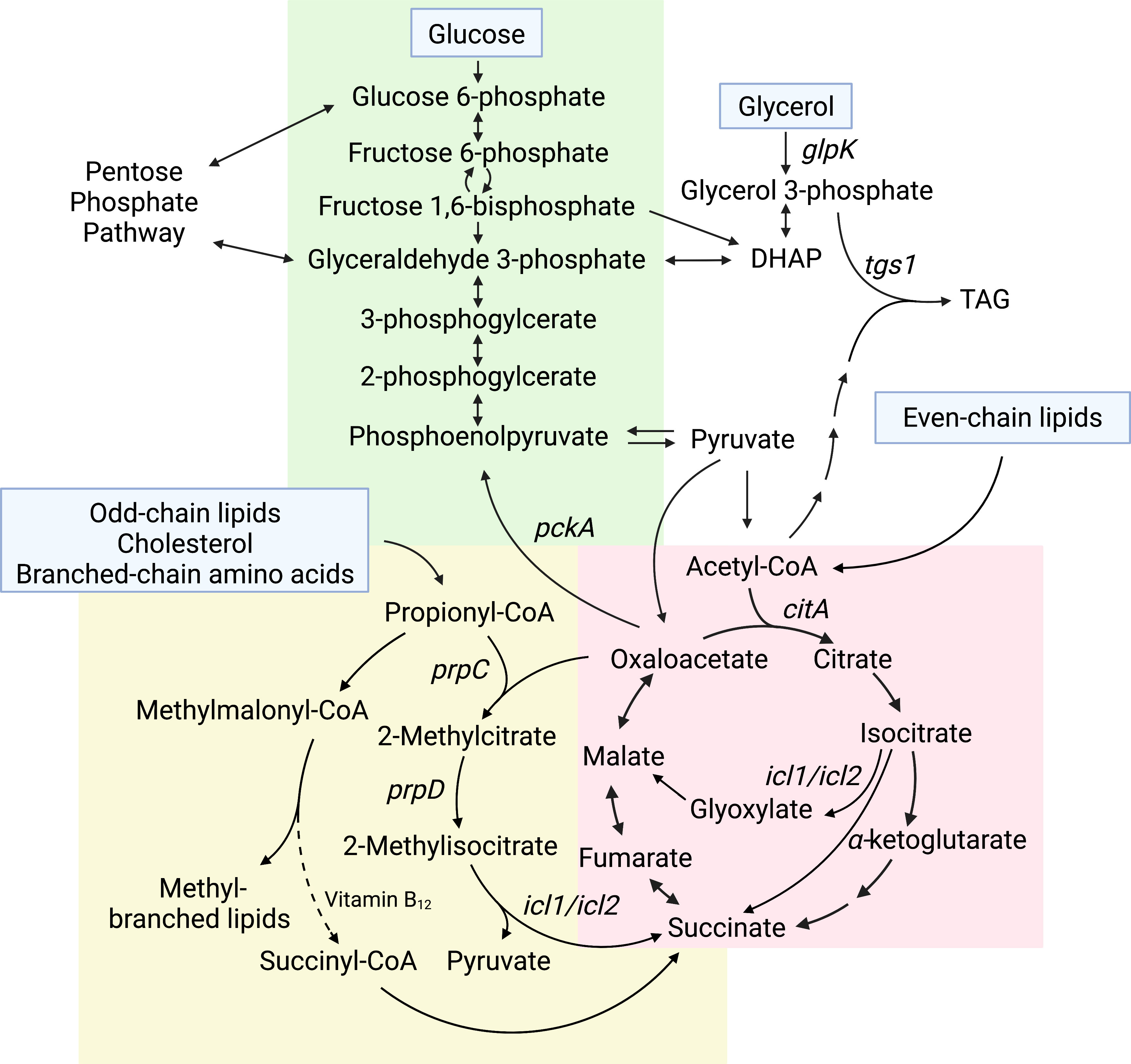
Figure 1 Core central carbon metabolism pathways that impact Mtb drug sensitivity. Carbon sources that feed into Mtb central carbon metabolism are listed in blue boxes. Carbon flowing down the pathway from glucose toward pyruvate, indicated by the downward pointing arrows, is glycolysis, whereas the reverse pathway, indicated by upward pointing arrows, is gluconeogenesis (green background). Even-chain lipids feed into the TCA cycle (red background). Odd-chain lipids, cholesterol, and branched-chain amino acids feed into the MCC and methylmalonyl-CoA pathways (yellow background). Genes discussed in the text that impact antibiotic sensitivity are indicated on the pathway.
As opposed to in vitro cultures where Mtb can utilize multiple different carbon sources, Mtb isolated directly from infected mouse lungs was found to preferentially metabolize fatty acids over other carbon sources such as glucose or glycerol (Segal and Bloch, 1956). In humans, direct RNA-sequencing of Mtb from patient sputum revealed up-regulation of transcripts encoding enzymes required for cholesterol degradation (Lai et al., 2021). Furthermore, the Mtb-specific cholesterol byproduct 4-cholesten-3-one is increased in patients with active tuberculosis, suggesting that Mtb actively metabolizes cholesterol during infection (Chandra et al., 2022). These data indicate that Mtb carbon metabolism is shifted in the host to preferentially rely on lipids over carbohydrate carbon sources.
The preferential use of lipids by Mtb during infection is further supported by experiments using Mtb mutants in metabolic pathways, which demonstrate that Mtb requires the glyoxylate shunt to colonize mice and requires cholesterol uptake and catabolism to maintain infection (Muñoz-Elías and Mckinney, 2005; Pandey and Sassetti, 2008; Nesbitt et al., 2010). This is consistent with data showing that gluconeogenesis, which allows TCA cycle intermediates to be used to generate essential cell wall precursors, is more important than glycolysis for Mtb growth in the host. While a mutant that lacks hexose kinase activity, the first step of glycolysis, is only slightly attenuated later during infection (Marrero et al., 2013), mutants lacking enzymes required for gluconeogenesis are unable to grow in mice at all (Marrero et al., 2013; Puckett et al., 2014; Trujillo et al., 2014; Ganapathy et al., 2015). These findings demonstrate that Mtb relies on gluconeogenic substrates, such as lipids, for growth during infection, rather than sugars or glycerol. Therefore, the host environment, which induces a higher proportion of drug tolerant Mtb, also leads to a shift in Mtb metabolic requirements compared to unstressed in vitro culturing conditions.
Triacylglycerols (TAG) and cholesterol are abundant lipid carbon sources available to Mtb during infection (Kim et al., 2010). Mtb liberates free fatty acids from TAG Deb et al.,2006, which are oxidized to acetyl-CoA, and degrades cholesterol through a series of reactions to pyruvate, acetyl-CoA, succinyl-CoA, and propionyl-CoA (Wilburn et al., 2018). The majority of cholesterol degradation products can directly feed into the TCA cycle or serve as substrates for gluconeogenesis. The exception is propionyl-CoA, which is toxic to the bacteria if it is not metabolized further (Munoz-Elias et al., 2006; Eoh and Rhee, 2014). Propionyl-CoA can be coupled with oxaloacetate through the methylcitrate cycle (MCC) to be detoxified to succinate and pyruvate (Figure 1) (Munoz-Elias et al., 2006; Eoh and Rhee, 2014). However, the MCC is dispensable for infection (Munoz-Elias et al., 2006), which may be because the environment encountered in the host enables propionyl-CoA detoxification through two alternative pathways. Specifically, the presence of exogenous even-chain fatty acids would enable Mtb to detoxify propionyl-CoA through incorporation into methyl-branched lipids, and access to vitamin B12 would enable detoxification of propionyl-CoA to succinyl-CoA (Jain et al., 2007; Savvi et al., 2008; Lee et al., 2013). Therefore, access to lipids or to vitamin B12 may obviate the need for the MCC during growth in the host even though the bacteria are catabolizing cholesterol.
Metabolism of cholesterol and the production of propionyl-CoA are associated with increased Mtb drug tolerance (Figure 2). Mtb grown in media containing cholesterol as a sole carbon source or containing mixed carbon sources including propionate exhibits decreased sensitivity to rifampicin (Koh et al., 2022). Exposure to propionate also activates PrpR, a regulator that induces expression of the prpDC operon, which encodes MCC enzymes PrpD and PrpC (Figure 1) (Masiewicz et al., 2012). Mutants with reduced or no PrpR activity, which are presumed to accumulate propionyl-CoA due to decreased expression of prpDC, exhibit slower growth in media containing propionate and increased tolerance to isoniazid, rifampicin, and ofloxacin (Hicks et al., 2018). Supplementing the prpR mutants with vitamin B12 enables shunting of propionyl-CoA to succinyl-CoA via methylmalonyl-CoA and is sufficient to rescue the growth defect in propionate media and reverse the drug tolerance of the mutants, supporting that accumulation of MCC intermediates contributes to drug tolerance (Hicks et al., 2018). The prpR mutants are similarly less sensitive to killing by antibiotics during in vitro infection of human macrophages (Hicks et al., 2018). Consistent with a role for PrpR-mediated regulation of the MCC in drug tolerance, mutations in prpR were enriched in drug resistant clinical isolates (Hicks et al., 2018). Since prpR mutations are associated with but do not confer drug resistance, it is possible these mutations promote a drug tolerance phenotype during infection, allowing the bacteria to survive and subsequently acquire drug resistance mutations. Consistent with decreased MCC activity promoting drug tolerance, knocking down expression of Icl1, which performs the final enzymatic step in the MCC, leads to accumulation of MCC intermediates in Mtb cultured in propionate media and causes nearly 10-fold less killing by isoniazid (Quinonez et al., 2022). In addition, exposure of Mtb to exogenous methylisocitrate, an MCC intermediate, is sufficient to decrease killing by isoniazid (Quinonez et al., 2022). Collectively, these studies suggest that accumulation of propionyl-CoA or MCC intermediates leads to antibiotic tolerance.
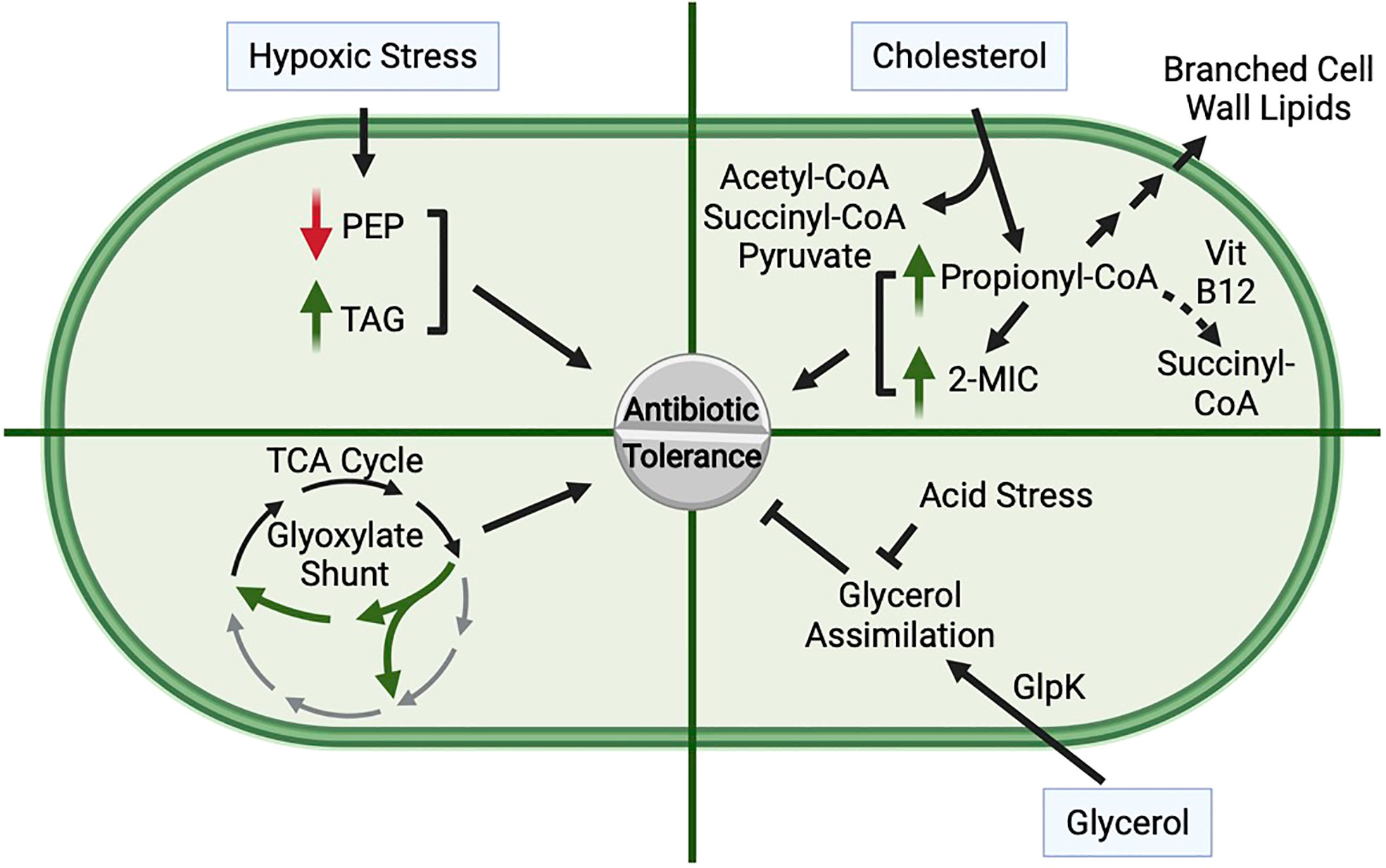
Figure 2 Role of Central Carbon Metabolism in Promoting Antibiotic Tolerance. Hypoxic stress, cholesterol metabolism, glycerol assimilation, low pH, and shunting of the TCA cycle via the glyoxylate shunt can each impact antibiotic tolerance of Mtb. Hypoxia: Exposure of Mtb to hypoxia leads to decreased levels of phosphoenolpyruvate (PEP) and an accumulation of triacylglycerol (TAG), both of which lead to an increase in drug tolerance. Cholesterol: Cholesterol is catabolized to acetyl-CoA, succinyl-CoA, pyruvate, and propionyl-CoA. Propionyl-CoA is detoxified through multiple pathways, including assimilation into branched chain lipids, conversion to succinyl-CoA through a vitamin B12-dependent pathway, or through the methylcitrate cycle (MCC) in which methylisocitrate (2-MIC) is an intermediate. Supplementation with cholesterol, propionate, or 2-MIC promotes antibiotic tolerance, and mutant strains that accumulate elevated levels of propionyl-CoA or 2-MIC are more tolerant to antibiotics. Glycerol Assimilation: Glycerol is assimilated into glycolysis and gluconeogenesis through phosphorylation by GlpK. Loss of glycerol catabolism leads to increased drug tolerance, suggesting that glycerol assimilation antagonizes antibiotic tolerance. Furthermore, Mtb in low pH is unable to efficiently catabolize glycerol, likely due to defects in glycolysis, resulting in increased antibiotic tolerance. Glyoxylate Shunt Activity: Mutants that lack the glyoxylate shunt are more sensitive to antibiotics, suggesting that rerouting carbon through the glyoxylate shunt promotes antibiotic tolerance.
Propionyl-CoA accumulation slows Mtb growth and slow growth rate has been associated with drug tolerance (Wayne and Hayes, 1996; Baek et al., 2011). Therefore, reduced growth rate could explain how cholesterol metabolism decreases antibiotic sensitivity. Another possible contributor to cholesterol-induced drug tolerance could be the changes to cell wall lipids that occur during propionyl-CoA metabolism. Growth on propionyl-CoA-generating carbon sources causes Mtb to synthesize branched lipids such as phthiocerol dimycocerosate (PDIM) and sulfolipid-1 with increased chain lengths (Jain et al., 2007; Yang et al., 2009; Griffin et al., 2012; Borah et al., 2021; Koh et al., 2022). PDIM is a major structural lipid intercalated in the outer Mtb envelope and has been shown to create a barrier that is particularly impenetrable to polar molecules (Wang et al., 2020). Thus, it is possible that alterations to PDIM chain length during growth on cholesterol or propionate may impact the permeability of the Mtb cell envelope, which could explain the altered antibiotic sensitivity.
During exposure to hypoxia, Mtb exhibits decreased levels of phosphoenolpyruvate (PEP) (Figure 2), an intermediate in glycolysis and gluconeogenesis, which is likely caused by decreased synthesis of PEP from oxaloacetate (Lim et al., 2021) (Figure 1). Supplementing hypoxic Mtb with exogenous PEP enhances killing by isoniazid (Lim et al., 2021), suggesting that the decrease in PEP during hypoxia contributes to hypoxia-induced drug tolerance. Notably, supplementation with pyruvate does not have the same effect, suggesting that this effect is specific for PEP, and access to additional carbon alone is not sufficient to sensitize Mtb to isoniazid. PEP supplementation also promotes Mtb sensitivity to D-cycloserine, a cell wall biosynthesis inhibitor, in aerated conditions, suggesting that the effect of PEP on drug tolerance is not specific for hypoxia (Lim et al., 2021). In addition to feeding into glycolysis and/or gluconeogenesis, PEP can also feed into the TCA cycle by conversion into oxaloacetate, can serve as a substrate for synthesis of the peptidoglycan precursor N-acetylmuramic acid, and is a substrate for the shikimate pathway (Lim et al., 2021). Which of these pathways contributes to the PEP-dependent drug sensitivity is still unknown.
During hypoxia, there is also decreased flux through several NAD(P)H-generating steps of the TCA cycle, likely to prevent production of NAD(P)H in conditions that these cofactors cannot be re-oxidized. This altered flux is caused by re-routing of acetyl-CoA to fatty acid and subsequent TAG biosynthesis (Baek et al., 2011), increased glyoxylate shunt activation (Eoh and Rhee, 2013), and reversal of several steps in the TCA cycle to generate succinate from oxaloacetate (Watanabe et al., 2011; Zimmermann et al., 2015). Deletion of the TAG biosynthesis gene tgs1 or overexpression of the citrate synthase gene citA prevented re-rerouting of acetyl-CoA to fatty acid and TAG biosynthesis during hypoxia and iron starvation (Baek et al., 2011). These mutants failed to arrest growth and exhibited enhanced sensitivity to isoniazid, streptomycin, ciprofloxacin, and ethambutol in hypoxic and iron starvation conditions (Baek et al., 2011). The Δtgs1 and citA-overexpressing strains were also significantly more sensitive to killing by isoniazid in a mouse model of infection, supporting that the re-routing of acetyl-CoA to fatty acid biosynthesis promotes drug tolerance (Figure 2) (Baek et al., 2011). Redirecting carbon away from the TCA cycle can also promote drug tolerance in aerated conditions. The glyoxylate shunt enables bypassing of two NAD(P)H- and CO2-generating steps of the TCA cycle. In addition to promoting growth on lipids by conserving carbon, the glyoxylate shunt may also decrease the generation of NAD(P)H during growth on glucose, preventing oxidative stress caused by electron transport chain activity (Nandakumar et al., 2014). A Δicl1/icl2 double mutant, which lacks the first step of the glyoxylate shunt, exhibits >10-fold enhanced killing by isoniazid, rifampicin, or streptomycin compared to the wild-type strain during aerobic growth on glucose (Nandakumar et al., 2014). Therefore, diverting carbon through the glyoxylate shunt can promote Mtb drug tolerance (Figure 2), likely by alleviating oxidative stress caused by TCA cycle and downstream electron transport chain activity. These studies demonstrate that by re-routing carbon away from the TCA cycle, Mtb becomes more tolerant to antibiotics.
Mtb is unable to grow on glycerol as the sole carbon source in low pH (Baker et al., 2014). This is likely due to inefficient assimilation of glycerol into lower glycolysis caused by decreased glyceraldehyde-3-phosphate dehydrogenase activity in low pH (Gouzy et al., 2021) (Figure 1). This nonpermissive growth condition results in acidic pH-induced drug tolerance (Figure 2), whereas growth on pyruvate, which enables Mtb growth in low pH, prevents the pH-induced drug tolerance (Baker and Abramovitch, 2018). Glycerol is assimilated into glycolysis and gluconeogenesis through phosphorylation by GlpK and subsequent conversion to DHAP (Figure 1). In media containing glycerol and other carbon sources, a ΔglpK mutant exhibited decreased sensitivity to isoniazid and rifampicin compared to wild-type Mtb, further supporting that decreasing glycerol metabolism increases drug tolerance (Bellerose et al., 2019; Safi et al., 2019). glpK is dispensable in the mouse model of Mtb infection, suggesting that glycerol is not a primary carbon source in mice (Pethe et al., 2010). However, free glycerol is detectable in infected mouse lungs, suggesting Mtb would have access to glycerol in the host (Safi et al., 2019). Furthermore, a ΔglpK mutant survived better than wild-type Mtb during treatment with pyrazinamide or any drug combination involving pyrazinamide, but not during isoniazid or rifampicin monotherapy, in a mouse model of infection (Bellerose et al., 2019). Therefore, the inability to metabolize glycerol during infection in mice promotes Mtb drug tolerance specifically to pyrazinamide. Since the glpk mutant does not exhibit increased tolerance to pyrazinamide in vitro, the pyrazinamide-specific tolerance in the glpK mutant is dependent upon the microenvironment within the host (Bellerose et al., 2019). Multiple groups have also identified glpK mutations in Mtb clinical isolates (Bellerose et al., 2019; Safi et al., 2019; Vargas and Farhat, 2020), and in some datasets these mutants are more commonly found in drug resistant isolates than in drug sensitive isolates (Bellerose et al., 2019; Safi et al., 2019). The glpK mutations are associated with but do not confer drug resistance themselves. However, if these mutations promote drug tolerance, they may enable Mtb to survive during antibiotic therapy, extending the time wherein Mtb may acquire a drug resistance mutation.
In this review, we have highlighted studies demonstrating that Mtb undergoes changes in carbon metabolism in response to host derived stresses that render Mtb more tolerant to antibiotics. However, the observed drug tolerance is rarely pan-antibiotic. For example, in hypoxic conditions, while Mtb becomes extremely tolerant to some antibiotics, including isoniazid, rifampicin, and streptomycin, it remains susceptible to antibiotics that target ATP synthase, and in some cases becomes more sensitive to killing by ATP synthase inhibitors and other antibiotics that target the electron transport chain (Koul et al., 2008; Rao et al., 2008; Gengenbacher et al., 2010; Sarathy et al., 2018; Lee et al., 2021). Furthermore, in an experiment to identify Mtb mutants with altered sensitivity to either isoniazid, rifampicin, pyrazinamide, or ethambutol during mouse infection, the majority of mutants identified only exhibited significantly altered susceptibility to a single antibiotic (Bellerose et al., 2020). Therefore, antibiotic tolerance can be conditional and specific to individual antibiotics.
Metabolic changes in Mtb that result in increased drug tolerance are often correlated with growth arrest, including toxicity mediated by propionyl-CoA or MCC intermediate accumulation (Hicks et al., 2018; Quinonez et al., 2022), hypoxia-induced TAG accumulation (Baek et al., 2011), and decreased glycerol metabolism in low pH conditions (Baker et al., 2014). However, there is also data, particularly in infection models, that suggests growth arrest is not universally associated with antibiotic tolerance (Raffetseder et al., 2014; Bellerose et al., 2019, Bellerose et al., 2020). Specifically, the ΔglpK mutant had no fitness defect in mice yet had altered antibiotic susceptibility to pyrazinamide (Bellerose et al., 2019). In addition, although there is a correlation between mutants that were less sensitive to isoniazid and mutants that had a fitness defect in mice, this association was not observed with mutants that were less sensitive to the other antibiotics (Bellerose et al., 2020).
Understanding the mechanisms by which Mtb metabolism impacts tolerance to specific antibiotics, particularly in the host environment, could lead to novel therapeutic approaches. We have highlighted several studies that demonstrate it is possible to manipulate metabolic pathways to reverse tolerance to a number of frontline antibiotics. For example, the Mtb Δtgs1 mutant is more susceptible to killing by isoniazid, rifampicin, or streptomycin during stress and in mice (Baek et al., 2011). This suggests that developing inhibitors of TAG biosynthesis could be a viable approach to enhance the efficacy of these antibiotics in the clinic. Additionally, supplementation with exogenous PEP was sufficient to enhance killing of hypoxic Mtb by isoniazid (Lim et al., 2021). Thus, changing the metabolic state of Mtb can potentiate killing by frontline antibiotics. Our review focused on central carbon metabolism, however these pathways are intricately connected to other metabolic networks, such as amino acid biosynthesis. De novo biosynthesis of amino acids from the TCA cycle and other pathways is essential for Mtb virulence and recent studies have shown that inhibiting amino acid biosynthesis is a promising approach for therapeutic development (Wellington et al., 2017). Although some metabolic enzymes may not be druggable targets due to shared structural homology with the mammalian homolog or difficulty in identifiying small molecules that effectively inhibit the enzyme, further elucidation of which metabolic pathways are essential during infection and how specific pathways contribute to drug tolerance will provide new opportunities for exploration.
All authors participated in the conceptualization and writing of this mini-review.
CS is supported by NIH grant AI134847 as well as a Burroughs Wellcome Fund Investigators in the Pathogenesis of Infectious Disease Award. GH is supported by National Science Foundation graduate research fellowship DGE-1745038 and NIGMS cell and molecular biology training grant GM007067. EW is supported by NIAID award T32AI007172. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
We are grateful to H. Eoh for kindly sharing his unpublished manuscript for this issue while we prepared our final versions.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Aldridge, B. B., Fernandez-Suarez, M., Heller, D., Ambravaneswaran, V., Irimia, D., Toner, M., et al. (2012). Asymmetry and aging of mycobacterial cells lead to variable growth and antibiotic susceptibility. Science 335, 100–104. doi: 10.1126/science.1216166
Baek, S.-H., Li, A. H., Sassetti, C. M. (2011). Metabolic regulation of mycobacterial growth and antibiotic sensitivity. PloS Biol. 9, 1–10. doi: 10.1371/journal.pbio.1001065
Baker, J. J., Abramovitch, R. B. (2018). Genetic and metabolic regulation of mycobacterium tuberculosis acid growth arrest. Sci. Rep. 8, 1–16. doi: 10.1038/s41598-018-22343-4
Baker, J. J., Johnson, B. K., Abramovitch, R. B. (2014). Slow growth of mycobacterium tuberculosis at acidic pH is regulated by phoPR and host-associated carbon sources. Mol. Microbiol. 94, 56–69. doi: 10.1111/mmi.12688
Bellerose, M. M., Baek, S.-H., Huang, C.-C., Moss, C. E., Koh, E.-I., Proulx, M. K., et al. (2019). Common variants in the glycerol kinase gene reduce tuberculosis drug efficacy. mBio 10, 1–15. doi: 10.1128/mBio.00663-19
Bellerose, M. M., Proulx, M. K., Smith, C. M., Baker, R. E., Ioerger, T. R., Sassetti, C. M. (2020). Distinct bacterial pathways influence the efficacy of antibiotics against mycobacterium tuberculosis. mSystems 5, 1–18. doi: 10.1128/mSystems00396-20
Berney, M., Berney-Meyer, L., Wong, K. W., Chen, B., Chen, M., Kim, J., et al. (2015). Essential roles of methionine and s-adenosylmethionine in the autarkic lifestyle of mycobacterium tuberculosis. PNAS 112, 10008–10013. doi: 10.1073/pnas.1513033112
Betts, J. C., Lukey, P. T., Robb, L. C., McAdam, R. A., Duncan, K. (2002). Evaluation of a nutrient starvation model of mycobacterium tuberculosis persistence by gene and protein expression profiling. Mol. Microbiol. 43, 717–731. doi: 10.1046/j.1365-2958.2002.02779.x
Borah, K., Beyß, M., Theorell, A., Wu, H., Basu, P., Mendum, T. A., et al. (2019). Intracellular mycobacterium tuberculosis exploits multiple host nitrogen sources during growth in human macrophages. Cell Rep. 29, 3580–3591. doi: 10.1016/j.celrep.2019.11.037
Borah, K., Mendem, T. A., Hawkins, D. D., Ward, J. L., Beale, M. H., Larrouy-Maumus, G., et al. (2021). Metabolic fluxes for nutritional flexibility of mycobacterium tuberculosis. Mol. Syst. Biol. 17, 1–17. doi: 10.15252/msb.202110280
Bryant, J. M., Harris, S. R., Parkhill, J., Dawson, R., Diacon, A. H., van Helden, P., et al. (2013). Whole-genome sequencing to establish relapse or re-infection with mycobacterium tuberculosis: a retrospective observational study. Lancet Respir. Med. 1, 786–792. doi: 10.1016/S2213-2600(13)70231-5
Chandra, P., Coullon, H., Agarwal, M., Goss, C. W., Philips, J. A. (2022). Macrophage global metabolomics identifies cholestenone as host/pathogen cometabolite present in humn mycobacterium tuberculosis infection. J. Clin. Invest. 132, 1–15. doi: 10.1172/JCI152509
Chen, X., He, G., Lin, S., Wang, S., Sun, F., Chen, J., et al. (2020). Analysis of serial multidrug-resistant tuberculosis strains causing treatment failure and within-host evolution by whole-genome sequencing. mSphere 5, e00884–e00820. doi: 10.1128/mSphere
Deb, C., Daniel, J., Sirokova, T. D., Abomoelak, B., Dubey, V. S., Kolattukudy, P. E. A novel lipase belonging to the hormon-sensitive lipase family induced under starvation to utilize stored triacylglycerol in mycobacterium tuberculosis. (2006) 7, 3866–3875. doi: 10.1074/jbc.M505556200
Deb, C., Lee, C.-M., Dubey, V. S., Daniel, J., Abomoelak, B., Sirakova, T. D., et al. (2009). A novel In vitro multiple-stress dormancy model for mycobacterium tuberculosis generates a lipid-loaded, drug-tolerant, dormant pathogen. PloS One 4, 1–15. doi: 10.1371/journal.pone.0006077
de Carvalho, L. P. S., Fischer, S. M., Marrero, J., Nathan, C., Ehrt, S., Rhee, K. Y. (2010). Metabolomics of mycobacterium tuberculosis reveals compartmentalized co-catabolism of carbon substrates. Chem. Biol. 17, 1122–1131. doi: 10.1016/j.chembiol.2010.08.009
Ehlers, S., Schaible, U. E. (2013). The granuloma in tuberculosis: dynamics of a host–pathogen collusion. Front. Immunol. 3. doi: 10.3389/fimmu.2012.00411
Eoh, H., Rhee, K. Y. (2013). Multifunctional essentiality of succinate metabolism in adaptation to hypoxia in mycobacterium tuberculosis. PNAS 110, 6554–6559. doi: 10.1073/pnas.1219375110
Eoh, H., Rhee, K. Y. (2014). Methylcitrate cycle defines the bactericidal essentiality of isocitrate lyase for survival of mycobacterium tuberculosis on fatty acids. PNAS 111, 4976–4981. doi: 10.1073/pnas.1400390111
Ganapathy, U., Marrero, J., Calhoun, S., Eoh, H., Pedro Sorio de Carvalho, L., Rhee, K., et al. (2015). Two enzymes with redundant fructose bisphosphatase activity sustain gluconeogenesis and virulence in mycobacterium tuberculosis. Nat. Commun. 6, 1–12. doi: 10.1038/ncomms8912
Gengenbacher, M., Rao, S. P. S., Pethe, K., Dick, T. (2010). Nutrient-starved, non-replicating mycobacterium tuberculosis requires respiration, ATP synthase and isocitrate lyase for maintenance of ATP homeostasis and viability. Microbiol. (N Y) 156, 81–87. doi: 10.1099/mic.0.033084-0
Ghazy, R. M., el Saeh, H. M., Abdulaziz, S., Hammouda, E. A., Elzorkany, A. M., Khidr, H., et al. (2022). A systematic review and meta-analysis of the catastrophic costs incurred by tuberculosis patients. Sci. Rep. 12, 558–574. doi: 10.1038/s41598-021-04345-x
Gillespie, S. H., Crook, A. M., McHugh, T. D., Mendel, C. M., Meredith, S. K., Murray, S. R., et al. (2014). Four-month moxifloxacin-based regimens for drug-sensitive tuberculosis. New Engl. J. Med. 371, 1577–1587. doi: 10.1056/nejmoa1407426
Gordhan, B. G., Smith, D. A., Alderton, H., McAdam, R. A., Bancroft, G. J., Mizrahi, V. (2002). Construction and phenotypic characterization of an auxotrophic mutant of mycobacterium tuberculosis defective in l-arginine biosynthesis. Infection Immun. 70, 3080–3084. doi: 10.1128/IAI.70.6.3080-3084.2002
Gouzy, A., Healy, C., Black, K. A., Rhee, K. Y., Ehrt, S. (2021). Growth of mycobacterium tuberculosis at acidic pH depends on lipid assimilation and is accompanied by reduced GAPDH activity. PNAS 118, 1–9. doi: 10.1073/pnas.2024571118/-/DCSupplemental
Gouzy, A., Larrouy-Maumus, G., Bottai, D., Levillain, F., Dumas, A., Wallach, J. B., et al. (2014). Mycobacterium tuberculosis exploits asparagine to assimilate nitrogen and resist acid stress during infection. PloS Pathog. 10, 1–14. doi: 10.1371/journal.ppat.1003928
Griffin, J. E., Pandey, A. K., Gilmore, S. A., Mizrahi, V., McKinney, J. D., Bertozzi, C. R., et al. (2012). Cholesterol catabolism by mycobacterium tuberculosis requires transcriptional and metabolic adaptations. Chem. Biol. 19, 218–227. doi: 10.1016/j.chembiol.2011.12.016
Hasenoehrl, E. J., Sajorda, D. R., Berney-Meyer, L., Johnson, S., Tufariello, J. M., Fuhrer, T., et al. (2019). Derailing the aspartate pathway of mycobacterium tuberculosis to eradicate persistent infection. Nat. Commun. 10, 1–12. doi: 10.1038/s41467-019-12224-3
Hasenoehrl, E. J., Wiggins, T. J., Berney, M. (2021). Bioenergetic inhibitors: Antibiotic efficacy and mechanisms of action in mycobacterium tuberculosis. Front. Cell. Infection Microbiol. 10. doi: 10.3389/fcimb.2020.611683
Hesper Rego, E., Audette, R. E., Rubin, E. J. (2017). Deletion of a mycobacterial divisome factor collapses single-cell phenotypic heterogeneity. Nature 546, 153–157. doi: 10.1038/nature22361
Hicks, N. D., Yang, J., Zhang, X., Zhao, B., Grad, Y. H., Liu, L., et al. (2018). Clinically prevalent mutations in mycobacterium tuberculosis alter propionate metabolism and mediate multidrug tolerance. Nat. Microbiol. 3, 1032–1042. doi: 10.1038/s41564-018-0218-3
Hobby, G. L. (1955). The fate of tubercle bacilli In vitro. Am. J. Med. 18, 753–763. doi: 10.1016/0002-9343(55)90189-3
Jain, M., Petzold, C., Schelle, M. W., Leavell, M. D., Mougous Joseph, D., Bertozzi, C. R., et al. (2007). Lipidomics reveals control of mycobacterium tuberculosis virulence lipids via metabolic coupling. PNAS 104, 5133–5138. doi: 10.1073/pnas.0610634104
Jain, P., Weinrick, B. C., Kalivoda, E. J., Yang, H., Munsamy, V., Vilcheze, C., et al. (2016). Dual-reporter mycobacteriophages (Φ2DRMS) reveal preexisting mycobacterium tuberculosis persistent cells in human sputum. mBio 7, 1–22. doi: 10.1128/mBio.01023-16
Jansen, R. S., Mandyoli, L., Hughes, R., Wakabayashi, S., Pinkham, J. T., Selbach, B., et al. (2020). Aspartate aminotransferase Rv3722c governs aspartate-dependent nitrogen metabolism in mycobacterium tuberculosis. Nat. Commun. 11, 1–13. doi: 10.1038/s41467-020-15876-8
Jindani, A., Harrison, T. S., Nunn, A. J., Phillips, P. P. J., Churchyard, G. J., Charalambous, S., et al. (2014). High-dose rifapentine with moxifloxacin for pulmonary tuberculosis. New Engl. J. Med. 371, 1599–1608. doi: 10.1056/nejmoa1314210
Kim, M. J., Wainwright, H. C., Locketz, M., Bekker, L. G., Walther, G. B., Dittrich, C., et al. (2010). Caseation of human tuberculosis granulomas correlates with elevated host lipid metabolism. EMBO Mol. Med. 2, 258–274. doi: 10.1002/emmm.201000079
Kjellsson, M. C., Via, L. E., Goh, A., Weiner, D., Low, K. M., Kern, S., et al. (2012). Pharmacokinetic evaluation of the penetration of antituberculosis agents in rabbit pulmonary lesions. Antimicrob Agents Chemother 56, 446–457. doi: 10.1128/AAC.05208-11
Koh, E.-I., Oluoch, P. O., Ruecker, N., Proulx, M. K., Soni, V., Murphy, K. C., et al. (2022). Chemical-genetic interaction mapping links carbon metabolism and cell wall structure to tuberculosis drug efficacy. PNAS 119, 1–12. doi: 10.1073/pnas.2201632119
Koul, A., Vranckx, L., Dendouga, N., Balemans, W., van den Wyngaert, I., Vergauwen, K., et al. (2008). Diarylquinolines are bactericidal for dormant mycobacteria as a result of disturbed ATP homeostasis. J. Biol. Chem. 283, 25273–25280. doi: 10.1074/jbc.M803899200
Kundu, M., Basu, J. (2021). Applications of transcriptomics and proteomics for understanding dormancy and resuscitation in mycobacterium tuberculosis. Front. Microbiol. 12. doi: 10.3389/fmicb.2021.642487
Kurthkoti, K., Amin, H., Marakalala, M. J., Ghanny, S., Subbian, S., Sakatos, A., et al. (2017). The capacity of mycobacterium tuberculosis to survive iron starvation might enable it to persist in iron- deprived microenvironments of human granulomas. mBio 8, 1–17. doi: 10.1128/mBio.01092-17
Lai, R. P. J., Cortes, T., Marais, S., Rockwood, N., Burke, M. L., Garza-Garcia, A., et al. (2021). Transcriptomic characterization of tuberculous sputum reveals a host warburg effect and microbial cholesterol catabolism. mBio 12, 1–13. doi: 10.1128/mBio.01766-21
Larrouy-Maumus, G., Marino, L. B., Madduri, A. V. R., Ragan, T. J., Hunt, D. M., Bassano, L., et al. (2016). Cell-envelope remodeling as a determinant of phenotypic antibacterial tolerance in mycobacterium tuberculosis. ACS Infect. Dis. 2, 352–360. doi: 10.1021/acsinfecdis.5b00148
Larsen, M. H., Biermann, K., Tandberg, S., Hsu, T., Jacobs, W. R. (2007). Genetic manipulation of mycobacterium tuberculosis. Curr. Protoc. Microbiol. 6, 1–21. doi: 10.1002/9780471729259.mc10a02s6
Lee, B. S., Hards, K., Engelhart, C. A., Hasenoehrl, E. J., Kalia, N. P., Mackenzie, J. S., et al. (2021). Dual inhibition of the terminal oxidases eradicates antibiotic-tolerant mycobacterium tuberculosis. EMBO Mol. Med. 13, 1–16. doi: 10.15252/emmm.202013207
Lee, S., Jeon, B. Y., Bardarov, S., Chen, M., Morris, S. L., Jacobs, W. R. (2006). Protection elicited by two glutamine auxotrophs of mycobacterium tuberculosis and in vivo growth phenotypes of the four unique glutamine synthetase mutants in a murine model. Infection Immun. 74, 6491–6495. doi: 10.1128/IAI.00531-06
Lee, W., VanderVen, B. C., Fahey, R. J., Russell, D. G. (2013). Intracellular mycobacterium tuberculosis exploits host-derived fatty acids to limit metabolic stress. J. Biol. Chem. 288, 6788–6800. doi: 10.1074/jbc.M112.445056
Lim, J., Lee, J. J., Lee, S., Kim, S., Eum, S.-Y., Eoh, H. (2021). Phosphoenolpyruvate depletion mediates both growth arrest and drug tolerance of mycobacterium tuberculosis in hypoxia. PNAS 118, 1–12. doi: 10.1073/pnas.2105800118
Loebel, R. O., Shorr, E., Richardson, H. B. (1933). The influence of foodstuffs upon the respiratory metabolism and growth of human tubercle bacilli. J. Bacteriol 2, 139–166. doi: 10.1128/jb.26.2.139-166
Manina, G., Neeraj, D., McKinney, J. D. (2015). Stress and host immunity amplify mycobacterium tuberculosis phenotypic heterogeneity and induce nongrowing metabolically active forms. Cell Host Microbe 17, 32–46. doi: 10.1016/j.chom.2014.11.016
Marakalala, M. J., Raju, R. M., Sharma, K., Zhang, Y. J., Eugenin, E. A., Prideaux, B., et al. (2016). Inflammatory signaling in human tuberculosis granulomas is spatially organized. Nat. Med. 22, 531–538. doi: 10.1038/nm.4073
Marrero, J., Trujillo, C., Rhee, K. Y., Ehrt, S. (2013). Glucose phosphorylation is required for mycobacterium tuberculosis persistence in mice. PloS Pathog. 9, 1–11. doi: 10.1371/journal.ppat.1003116
Masiewicz, P., Brzostek, A., Wolanski, M., Dziadek, J., Zakrzewska-Czerwinska, J. (2012). A novel role of the PrpR as a transcription factor involved in the regulation of methylcitrate pathway in mycobacterium tuberculosis. PloS One 7, 1–14. doi: 10.1371/journal.pone.0043651
Muñoz-Elías, E. J., Mckinney, J. D. (2005). Mycobacterium tuberculosis isocitrate lyases 1 and 2 are jointly required for in vivo growth and virulence. Nat. Med. 11, 638–644. doi: 10.1038/nm1252
Munoz-Elias, E. J., Upton, A. M., Cherian, J., McKinney, J. D. (2006). Role of the methylcitrate cycle in mycobacterium tuberculosis metabolism, intracellular growth, and virulence. Mol. Microbiol. 60, 1109–1122. doi: 10.1111/j.1365-2958.2006.05155.x
Nahid, P., Mase, S. R., Migliori, G. B., Sotgiu, G., Bothamley, G. H., Brozek, J. L., et al. (2019). Treatment of drug-resistant tuberculosis an official ATS/CDC/ERS/IDSA clinical practice guideline. Am. J. Respir. Crit. Care Med. 200, E93–E142. doi: 10.1164/rccm.201909-1874ST
Nandakumar, M., Nathan, C., Rhee, K. Y. (2014). Isocitrate lyase mediates broad antibiotic tolerance in mycobacterium tuberculosis. Nat. Commun. 5, 1–10. doi: 10.1038/ncomms5306
Nesbitt, N. M., Yang, X., Fontán, P., Kolesnikova, I., Smith, I., Sampson, N. S., et al. (2010). A thiolase of mycobacterium tuberculosis is required for virulence and production of androstenedione and androstadienedione from cholesterol. Infection Immun. 78, 275–282. doi: 10.1128/IAI.00893-09
Pandey, A. K., Sassetti, C. M. (2008). Mycobacterial persistence requires the utilization of host cholesterol. PNAS 105, 4376–4380. doi: 10.1073/pnas.0711159105
Pavelka, M. S., Chen, B., Kelley, C. L., Collins, F. M., Jacobs, W. R. (2003). Vaccine efficacy of a lysine auxotroph of mycobacterium tuberculosis. Infection Immun. 71, 4190–4192. doi: 10.1128/IAI.71.7.4190-4192.2003
Pethe, K., Sequeira, P. C., Agarwalla, S., Rhee, K., Kuhen, K., Phong, W. Y., et al. (2010). A chemical genetic screen in mycobacterium tuberculosis identifies carbon-source-dependent growth inhibitors devoid of in vivo efficacy. Nat. Commun. 1, 1–8. doi: 10.1038/ncomms1060
Pisu, D., Huang, L., Grenier, J. K., Russell, D. G. (2020). Dual RNA-seq of mtb-infected macrophages In vivo reveals ontologically distinct host-pathogen interactions. Cell Rep. 30, 335–350. doi: 10.1016/j.celrep.2019.12.033
Prideaux, B., Via, L. E., Zimmerman, M. D., Eum, S., Sarathy, J., O’Brien, P., et al. (2015). The association between sterilizing activity and drug distribution into tuberculosis lesions. Nat. Med. 21, 1223–1227. doi: 10.1038/nm.3937
Puckett, S., Trujillo, C., Eoh, H., Marrero, J., Spencer, J., Jackson, M., et al. (2014). Inactivation of fructose-1,6-Bisphosphate aldolase prevents optimal Co-catabolism of glycolytic and gluconeogenic carbon substrates in mycobacterium tuberculosis. PLosPathogens 10, 1–11. doi: 10.1371/journal.ppat.1004144
Quinonez, C. G., Jin Lee, J., Lim, J., Odell, M., Lawson, C. P., Anyogu, A., et al. (2022). The role of fatty acid metabolism in drug tolerance of mycobacterium tuberculosis. mBio 13, 1–16. doi: 10.1128/mbio.03559-21
Raffetseder, J., Pienaar, E., Blomgran, R., Eklund, D., Brodin, V. P., Andersson, H., et al. (2014). Replication rates of mycobacterium tuberculosis in human macrophages do not correlate with mycobacterial antibiotic susceptibility. PloS One 9, e112426. doi: 10.1371/JOURNAL.PONE.0112426
Rao, S. P. S., Alonso, S., Rand, L., Dick, T., Pethe, K. (2008). The proton motive force is required for maintaining ATP homeostasis and viability of hypoxic, nonreplicating mycobacterium tuberculosis. PNAS 105, 11945–11950. doi: 10.1073/pnas.0711697105
Remm, S., Earp, J. C., Dick, T., Dartois, V., Seeger, M. A. (2021). Critical discussion on drug efflux in mycobacterium tuberculosis. FEMS Microbiol. Rev. 46, 1–15. doi: 10.1093/femsre/fuab050
Safi, H., Gopal, P., Lingaraju, S., Ma, S., Levine, C., Dartois, V., et al. (2019). Phase variation in mycobacterium tuberculosis glpK produces transiently heritable drug tolerance. PNAS 116, 19665–19674. doi: 10.1073/pnas.1907631116
Santucci, P., Aylan, B., Botella, L., Bernard, E. M., Bussi, C., Pellegrino, E., et al. (2022). Visualizing pyrazinamide action by live single-cell imaging of phagosome acidification and mycobacterium tuberculosis pH homeostasis. mBio 13, 1–21. doi: 10.1128/mbio.00117-22
Santucci, P., Greenwood, D. J., Fearns, A., Chen, K., Jiang, H., Gutierrez, M. G. (2021). Intracellular localisation of mycobacterium tuberculosis affects efficacy of the antibiotic pyrazinamide. Nat. Commun. 12, 1–16. doi: 10.1038/s41467-021-24127-3
Sarathy, J. P., Zuccotto, F., Hsinpin, H., Sandberg, L., Via, L. E., Marriner, G. A., et al. (2018). Prediction of drug penetration in tuberculosis lesions. ACS Infect. Dis. 2, 552–563. doi: 10.1021/acsinfecdis.6b00051
Savvi, S., Warner, D. F., Kana, B. D., McKinney, J. D., Mizrahi, V., Dawes, S. S. (2008). Functional characterization of a vitamin B12-dependent methylmalonyl pathway in mycobacterium tuberculosis: Implications for propionate metabolism during growth on fatty acids. J. Bacteriol 190, 3886–3895. doi: 10.1128/JB.01767-07
Schnappinger, D., Ehrt, S., Voskuil, M. I., Liu, Y., Mangan, J. A., Monahan, I. M., et al. (2003). Transcriptional adaptation of mycobacterium tuberculosis within macrophages: Insights into the phagosomal environment. J. Exp. Med. 198, 693–704. doi: 10.1084/jem.20030846
Segal, W., Bloch, H. (1956). BIOCHEMICAL DIFFERENTIATION OF MYCOBACTERIUM TUBERCULOSIS GROWN IN VIVO AND IN VITRO. J. Biochem. 72, 132–141. doi: 10.1128/jb.72.2.132-141
Sharma, S., Briffotaux, J., Khan, A., Karakousis, P. C., Danchik, C., Wang, S. (2021). Targeting the mycobacterium tuberculosis stringent response as a strategy for shortening tuberculosis treatment. Front. Microbiol. 12. doi: 10.3389/fmicb.2021.744167
Sukheja, P., Kumar, P., Mittal, N., Li, S. G., Singleton, E., Russo, R., et al. (2017). A novel small-molecule inhibitor of the mycobacterium tuberculosis demethylmenaquinone methyltransferase meng is bactericidal to both growing and nutritionally deprived persister cells. mBio 8, e02022–16. doi: 10.1128/mBio.02022-16
Tan, S., Sukumar, N., Abramovitch, R. B., Parish, T., Russell, D. G. (2013). Mycobacterium tuberculosis responds to chloride and pH as synergistic cues to the immune status of its host cell. PloS Pathog. 9, e1003282. doi: 10.1371/journal.ppat.1003282
Trujillo, C., Blumenthal, A., Marrero, J., Rhee, K. Y., Schnappinger, D., Ehrt, S. (2014). Triosephosphate isomerase is dispensable in vitro yet essential for mycobacterium tuberculosis to establish infection. mBio 5, e00085. doi: 10.1128/mBio.00085-14
Turapov, O., O’Connor, B. D., Sarybaeva, A. A., Williams, C., Patel, H., Kadyrov, A. S., et al. (2016). Phenotypically adapted mycobacterium tuberculosis populations from sputum are tolerant to first-line drugs. Antimicrob Agents Chemother 60, 2476–2483. doi: 10.1128/AAC.01380-15
Vargas, R., Farhat, M. R. (2020). Antibiotic treatment and selection for glpK mutations in patients with active tuberculosis disease. PNAS 117, 3910–3912. doi: 10.1073/pnas.1920788117
Via, L. E., Lin, P. L., Ray, S. M., Carrillo, J., Allen, S. S., Seok, Y. E., et al. (2008). Tuberculous granulomas are hypoxic in guinea pigs, rabbits, and nonhuman primates. Infection Immun. 76, 2333–2340. doi: 10.1128/IAI.01515-07
Vilcheze, C., Hartman, T., Weinrick, B., Jain, P., Weisbrod, T. R., Leung, L. W., et al. (2017). Enhanced respiration prevents drug tolerance and drug resistance in mycobacterium tuberculosis. PNAS 114, 4495–4500. doi: 10.1073/pnas.1704376114
Voskuil, M. I., Bartek, I. L., Visconti, K., Schoolnik, G. K. (2011). The response of mycobacterium tuberculosis to reactive oxygen and nitrogen species. Front. Microbiol. 2. doi: 10.3389/fmicb.2011.00105
Voskuil, M. I., Schnappinger, D., Visconti, K. C., Harrell, M. I., Dolganov, G. M., Sherman, D. R., et al. (2003). Inhibition of respiration by nitric oxide induces a mycobacterium tuberculosis dormancy program. J. Exp. Med. 198, 705–713. doi: 10.1084/jem.20030205
Wakamoto, Y., Dhar, N., Chait, R., Schneider, K., Signorino-Gelo, F., Leibler, S., et al. (2013). Dynamic persistence of antibiotic-stressed mycobacteria. Science 339, 91–95. doi: 10.1126/science.1229858
Wallace, F., Sutherland, I. (1955). THE CLINICAL SIGNIFICANCE OF POSITIVE CULTURES AND OF ISONIAZID-RESISTANT TUBERCLE BACILLI DURING THE TREATMENT OF PULMONARY TUBERCULOSIS. Thorax 10, 85–98. doi: 10.1136/thx.10.2.85
Wang, Q., Boshoff, H. I. M., Harrison, J. R., Ray, P. C., Green, S. R., Wyatt, P. G., et al. (2020). PE/PPE proteins mediate nutrient transport across the outer membrane of mycobacterium tuberculosis. Science 367, 1147–1151. doi: 10.1126/science.aav5912
Watanabe, S., Zimmermann, M., Goodwin, M. B., Sauer, U., Barry, C. E., Boshoff, H. I. (2011). Fumarate reductase activity maintains an energized membrane in anaerobic mycobacterium tuberculosis. PloS Pathog. 7, 1002287. doi: 10.1371/journal.ppat.1002287
Wayne, L. G., Hayes, L. G. (1996). An In vitro model for sequential study of shiftdown of mycobacterium tuberculosis through two stages of nonreplicating persistence. Infection Immun. 64, 2062–2069. doi: 10.1128/iai.64.6.2062-2069
Wellington, S., Nag, P. P., Michalska, K., Johnston, S., Jedrzejczak, R., Kaushik, V. K., et al. (2017). A small-molecule allosteric inhibitor of mycobacterium tuberculosis tryptophan synthase. Nat. Chem. Biol. 13, 943–955. doi: 10.1038/nCHeMBIO.2420
WHO (2017) Guidelines for treatment of drug-susceptible tuberculosis and patient care. Available at: https://www.who.int/publications/i/item/9789241550000.
WHO (2021) Global tuberculosis report 2021. Available at: https://www.who.int/publications/i/item/9789240037021.
Wilburn, K. M., Fieweger, R. A., VanderVen, B. C. (2018). Cholesterol and fatty acids grease the wheels of mycobacterium tuberculosis pathogenesis. Pathog. Dis. 76, 1–14. doi: 10.1093/femspd/fty021
Xie, Z., Siddiqi, N., Rubin, E. J. (2005). Differential antibiotic susceptibilities of starved mycobacterium tuberculosis isolates. Antimicrob. Agents Chemother. 49, 4778–4780. doi: 10.1128/AAC.49.11.4778-4780.2005
Yang, X., Nesbitt, N. M., Dubnau, E., Smith, I., Sampson, N. S. (2009). Cholesterol metabolism increases the metabolic pool of propionate in mycobacterium tuberculosis. Biochemistry 48, 3819–3821. doi: 10.1021/bi9005418
Keywords: tuberculosis, antibiotics, tolerance, metabolism, granuloma, cholesterol, hypoxia
Citation: Samuels AN, Wang ER, Harrison GA, Valenta JC and Stallings CL (2022) Understanding the contribution of metabolism to Mycobacterium tuberculosis drug tolerance. Front. Cell. Infect. Microbiol. 12:958555. doi: 10.3389/fcimb.2022.958555
Received: 31 May 2022; Accepted: 29 July 2022;
Published: 22 August 2022.
Edited by:
Helena Ingrid Boshoff, National Institutes of Health (NIH), United StatesReviewed by:
Khushboo Borah, University of Surrey, United KingdomCopyright © 2022 Samuels, Wang, Harrison, Valenta and Stallings. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Christina L. Stallings, c3RhbGxpbmdzQHd1c3RsLmVkdQ==
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.