 Kalen M. Hall
Kalen M. Hall Zachary F. Pursell
Zachary F. Pursell Lisa A. Morici
Lisa A. Morici- 1Department of Microbiology and Immunology, Tulane University School of Medicine, New Orleans, LA, United States
- 2Department of Biochemistry and Molecular Biology, Tulane University School of Medicine, New Orleans, LA, United States
Chronic respiratory infection (CRI) with Pseudomonas aeruginosa (Pa) presents many unique challenges that complicate treatment. One notable challenge is the hypermutator phenotype which is present in up to 60% of sampled CRI patient isolates. Hypermutation can be caused by deactivating mutations in DNA mismatch repair (MMR) genes including mutS, mutL, and uvrD. In vitro and in vivo studies have demonstrated hypermutator strains to be less virulent than wild-type Pa. However, patients colonized with hypermutators display poorer lung function and a higher incidence of treatment failure. Hypermutation and MMR-deficiency create increased genetic diversity and population heterogeneity due to elevated mutation rates. MMR-deficient strains demonstrate higher rates of mucoidy, a hallmark virulence determinant of Pa during CRI in cystic fibrosis patients. The mucoid phenotype results from simple sequence repeat mutations in the mucA gene made in the absence of functional MMR. Mutations in Pa are further increased in the absence of MMR, leading to microcolony biofilm formation, further lineage diversification, and population heterogeneity which enhance bacterial persistence and host immune evasion. Hypermutation facilitates the adaptation to the lung microenvironment, enabling survival among nutritional complexity and microaerobic or anaerobic conditions. Mutations in key acute-to-chronic virulence “switch” genes, such as retS, bfmS, and ampR, are also catalyzed by hypermutation. Consequently, strong positive selection for many loss-of-function pathoadaptive mutations is seen in hypermutators and enriched in genes such as lasR. This results in the characteristic loss of Pa acute infection virulence factors, including quorum sensing, flagellar motility, and type III secretion. Further study of the role of hypermutation on Pa chronic infection is needed to better inform treatment regimens against CRI with hypermutator strains.
Introduction
Pseudomonas aeruginosa (Pa) is a ubiquitous Gram-negative bacterium known to cause a wide range of opportunistic infections, including respiratory, wound, urinary tract, surgical site, and bloodstream infections. Pa has been designated a global threat due to increasing rates of multidrug resistant nosocomial infections, but it has long been known as the primary cause of morbidity and mortality in cystic fibrosis (CF) patients (Rajan, 2002). CF is an autosomal recessive disorder caused by over 2,000 documented variants in the CF transmembrane conductance regulator (CFTR) gene, leading to multisystem pathology (Bergeron and Cantin, 2019). The disease affects over 30,000 people in the United States and has a poor prognosis with a median age of death of 34 (Cystic Fibrosis Foundation, 2021). In the lungs, altered CFTR function leads to thick mucosal secretions which create a unique hospitable environment for microbes (Ciofu et al., 2013). Patients become colonized in the respiratory tract in ages as early as 1 with bacteria such as Pa, Staphylococcus aureus, Haemophilus influenzae, and Stenotrophomonas maltophilia, but Pa predominates by age 18 (Rajan, 2002; Cystic Fibrosis Foundation, 2021). Chronic respiratory infection (CRI) with Pa leads to excessive inflammation and eventual tissue necrosis and lung failure (Ciofu et al., 2013).
CRI with Pa in the context of the CF lung poses many unique challenges for treatment. The infection is characterized by the Pa mucoid phenotype, high levels of drug resistance, persistence, and a shift from an acute to a chronic virulence state resulting in treatment failure. During CRI, Pa downregulates the virulence factors needed for establishment of acute infection, including LasR-mediated quorum sensing, type III secretion, twitching motility and adhesion mediated by flagella and pili. Instead, Pa expresses factors that favor persistence in the CF lung such as alginate overproduction, biofilm formation, and alternate metabolic pathways (Smith et al., 2006; Bragonzi et al., 2009). In the chronic virulence state, alginate overproduction, or “mucoidy”, in particular leads to greater regional lung inflammation and impairs both host immune defenses and therapeutic treatments (Malhotra et al., 2019).
Another challenge is the high prevalence (up to 60% of sampled patients) of hypermutator (defined as having 20-fold higher mutants per total viable cells than wild-type) strains in chronic Pa CF lung infections, which are overwhelmingly caused by deactivating mutations in the DNA mismatch repair (MMR) genes such as mutS, mutL, and uvrD (Table 1) (Oliver et al., 2000; Oliver et al., 2002a; Hogardt et al., 2006). Hypermutator isolates deficient in the GO system are very rarely observed, but most isolates to be complemented with GO system genes such as mutT or mutM (Oliver et al., 2002b) The bacterial DNA MMR system is responsible for repairing replicative insertion or slippage errors that were not corrected by DNA polymerase proof-reading activity (Kunkel and Erie, 2005; Iyer et al., 2006). In the absence of MMR, mutations are biased towards T>C and C>T transitions and insertion-deletions (indels) in homopolymers, implicating genes containing these sequences as mutational hotspots in hypermutators (Lee et al., 2012; Marvig et al., 2013). Hypermutators are thought to be so prevalent in CRI because of the positive co-selection of the resulting adaptive mutations consequent of MMR-deficiency (Gutiérrez et al., 2004). Induction of the hypermutator phenotype has been associated with chronic oxidative stress and with chronic antibiotic treatment (Ciofu et al., 2005; Dößelmann et al., 2017). Hypermutators appear to be associated with chronicity of infection, as none were found until 5 years after the onset of infection in a sample of CF isolates and are incredibly rare in acute infection isolates (<1%) (Gutiérrez et al., 2004; Ciofu et al., 2005). In addition, patients colonized with hypermutators are reported to have poorer patient lung function measured via percent forced expiratory volume and mean forced expiratory volume per forced vital capacity (Waine et al., 2008; Ferroni et al., 2009). Colonization with hypermutators is also associated with greater instance of multidrug resistance and treatment failure (Macía et al., 2005).

Table 1 Pa MMR genes and their respective functions, along with common point mutation positions resulting in protein inactivation (Oliver et al., 2002a; Hogardt et al., 2006; On and Welch, 2021).
Despite these trends in clinical data, mutS-knockout strains of Pa are outcompeted by wild-type in vitro and in vivo murine models. Strains deficient in MutS also display attenuated virulence and reduced capacity for colonization (Mena et al., 2007; Montanari et al., 2007). Clinical data has long suggested the independent emergence of hypermutators in CF patients, but recent phylogenetic analyses of widespread clonal lineages of Pa CF isolates demonstrated evidence for interpatient transmission (Oliver and Mena, 2010; López-Causapé et al., 2017). The discrepancy between clinical and experimental observations suggests that the hypermutator phenotype may be critical for bacterial adaption or survival during CRI. High mutation rates have been shown to be beneficial in early colonization and mutator alleles can become fixed in a fraction of the population due to their evolutionary advantage, even though randomly occurring deleterious mutations at secondary sites can be disadvantageous (Taddei et al., 1997; Giraud et al., 2001). Hypermutation has also been observed in S. aureus, H. influenzae, Escherichia coli, Salmonella enterica and Neisseria meningitidis, possibly implicating it as a conserved mechanism for rapid adaptation (LeClerc et al., 1996; Matic et al., 1997; Sniegowski et al., 1997; Richardson et al., 2002; Prunier et al., 2003; Román et al., 2004).
In this review, we examine the role of the Pa hypermutator phenotype (caused by MMR-deficiency) found in CRI in the shift from the acute to chronic virulence state (summarized in Figure 1). We will describe how hypermutation allows for genetic population heterogeneity and phenotypic diversity and how MMR-deficiency catalyzes the establishment of the most common mutant mucA22 allele causing mucoidy (Moyano et al., 2007). Hypermutation allows for the rapid adaptation to the CF lung microenvironment via mutations in metabolic pathways allowing for survival in the high amino acid content and microaerobic conditions. In addition, mutations in master transcriptional regulators act as ‘switches’ that shift Pa from the acute to the chronic virulence state. Hypermutators are strongly associated with pathoadaptive loss-of-function mutations in acute virulence genes that contribute to the transition of virulence state as well.
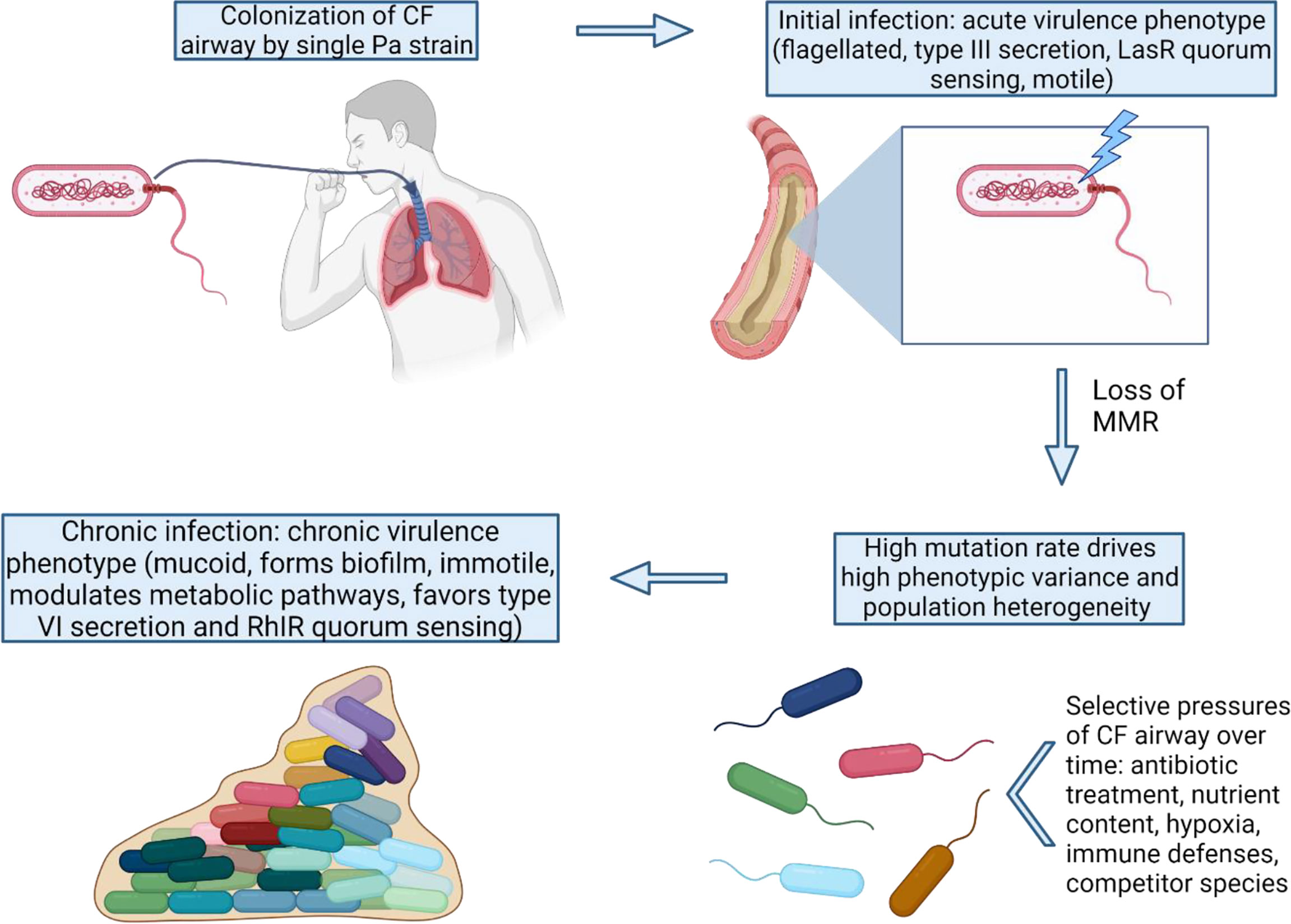
Figure 1 MMR-deficiency catalyzes the shift from an acute to chronic P. aeruginosa virulence state. Once MMR is lost in the initial colonizing strain, many adaptive pathways can be exploited by Pa via high mutation rates. The variants with mutations favoring the chronic virulence state confer advantages for long-term survival and persistence under the strong selective pressures of the CF lung.
Hypermutation creating population heterogeneity and phenotypic diversity
A hallmark of Pa CRI is a phenomenon known as adaptive radiation that contributes to Pa persistence in the CF lung. Due to its large genome size (>6 Mb) and sophisticated networks of transcriptional regulation, a single parent Pa strain has the capacity to occupy specific environmental niches via divergence into adapted sublineages (Stover et al., 2000; Klockgether et al., 2011; Markussen et al., 2014). These sublineages differ at the genomic and phenotypic levels but coexist creating a heterogenous population (Chung et al., 2012; la Rosa et al., 2018). Nutritional complexity and high mucin levels in CF airways alone are sufficient to drive Pa’s divergence into sublineages and phenotypic diversity, but this diversification is even further enhanced by host immune pressure and competitor microbial species (Schick and Kassen, 2018; la Rosa et al., 2019). Whole genome sequencing of isolates derived from CF patients has revealed that an initial colonizing strain of Pa undergoes a period of rapid adaptation followed by a long period of genetic drift with minor changes. This is seen in non-mutator Pa populations in CF airways with a reported mutation rate of 7.2 x 10-11 single nucleotide polymorphisms (SNPs) per base pair (bp) per generation (Yang et al., 2011).
Hypermutation creates greater genetic diversity in a Pa population, allowing for further phenotypic diversity and driving evolution (Mena et al., 2008). Laboratory evolution experiments with a MutS-deficient Pa strain showed significantly greater diversification of colony morphology that demonstrate increased antibiotic resistance and decreased cytotoxicity similar to CF isolates (Smania et al., 2004). Hypermutation affecting genetic and phenotypic diversity is observed extensively in clinical isolates. MutS-deficient paired isolates differed by 344 SNPs and 93 indels, compared to 54 SNPs/38 indels and 1 SNP/8 indels of two pairs of wild-type isolates from different patients (Chung et al., 2012). A longitudinal genetic analysis of 13 isolates from an Argentinian patient and 14 isolates from a Danish patient revealed sublineages with extensive intra-patient genomic diversity (Feliziani et al., 2014). Hypermutators comprised 90% of the heterogenous population of isolates from both the Argentinian and Danish patient, indicating that they dominate and outcompete non-mutator isolates under CF airway selective pressures. The reported in vivo mutation rate of the populations was 100 SNPs/year, which is 40-fold higher than non-mutator isolates. Characteristic of adaptive radiation, genomic variation showed coexistence of equally fit subpopulations that arose from multiple evolutionary events. Parallel convergent evolution across sublineages and patient populations indicate hypermutation can target genes that optimize fitness in the CF airway independent of geography (Feliziani et al., 2014). A retrospective study of the DK2 clone type (a lineage of Pa strains causing chronic infection in Danish CF patients) and its transmission events and subsequent divergence into intra-patient sublineages also demonstrated parallel evolution in genes related to antibiotic resistance, regulatory functions, and the cell envelope, which is thought to play a role in immune evasion. Many of these genes contained homopolymers, which are known MMR-deficiency mutation hotspots, and the number of mutations accumulated correlated with homopolymer run length (Marvig et al., 2013). This has been expanded upon with another analysis of longitudinally collected CF isolates containing many hypermutators that showed parallel convergent evolution in genes involved in central metabolism and virulence factors (Marvig et al., 2015).
The shift to mucoidy and biofilm development
Mucoidy is a unique characteristic of CRIs with Pa and is caused by the overproduction of the exopolysaccharide alginate. Pa’s conversion to mucoidy during CRI is mostly caused by inactivating mutations in mucA (~85%), a negative regulator of sigma factor algU. Disruption of MucA leads to constitutive expression of AlgU and alginate biosynthesis (Martin et al., 1993; Boucher et al., 1997). The mucoid phenotype is highly virulent and is associated with chronic infections, increased inflammation, and increased patient mortality (Malhotra et al., 2019). Alginate overproduction aids Pa in evading macrophage killing via scavenging of free radicals (Simpson et al., 1989). Alginate also interferes with antibody-independent opsonic killing and Th1-mediated killing (Pier et al., 2001). Alginate expression is associated with increased pro-inflammatory cytokines that exacerbate tissue damage and pathology (Song et al., 2003).
Pa mucoid isolates are more likely to be hypermutators than non-mucoid isolates, demonstrating an association between the two phenotypes (Waine et al., 2008). This could be because both phenotypes are associated with chronic infection. However, in vitro data suggests that hypermutation could be driving mucoid conversion. A MutS-deficient Pa strain showed significantly increased emergence of mucoid mutants when cultured in vitro compared to wild-type. This was associated with a single base deletion in a run of five consecutive guanines (G5-SSR426). This deletion causes a frameshift and results in an inactivated mutant allele (mucA22), which is observed in up to 40% of mucoid CF isolates (Bragonzi et al., 2006; Moyano et al., 2007). A forward mutation model of mucA demonstrated a critical role of G5-SSR426 in mucoid conversion in MMR-deficient cells and emphasized homopolymers as a main target for hypermutators (Moyano and Smania, 2009). It is important to note that mucA mutations have been found to occur prior to MMR-inactivating mutations, and that no statistically significant link could be established between hypermutability and mucA mutations in two studies: one assessing 70 samples from 10 CF patients and another with 38 isolates from 26 CF patients (Ciofu et al., 2010; Feliziani et al., 2010). Together, these data suggest that hypermutation is not a prerequisite for mucoidy but may drive conversion when present.
Alginate overproduction also plays a key role in Pa biofilm formation and architecture (Nivens et al., 2001; Ghafoor et al., 2011). In addition to mucoid conversion, hypermutators also show high rates of missense mutations in bfmS, a sensor histidine kinase that negatively regulates bfmR which is responsible for biofilm maturation (Cao et al., 2020). Biofilms display increased resistance to antibiotics and phagocytosis and worsen inflammation during CRI. Biofilms also display increased mutagenesis, promoting adaptation to the CF lung (Høiby et al., 2010). However, sequencing of 12 isolates of the DK2 lineage, all deemed to be non-mutators, showed no increase in mutation rate in biofilms (Yang et al., 2011). Mutagenesis data obtained in situ with biofilms implicates the importance of microcolonies. MMR-deficient Pa showed enhanced microcolony formation and growth, and the mutation rates in the microcolonies were elevated compared to planktonic forms (Conibear et al., 2009). This implicates hypermutability in Pa’s exceptional capacity to adapt as a biofilm.
The adaptation to the CF lung microenvironment
Hypermutation aids in survival in complex nutritional environment
The CF lung poses a unique and complex environment in terms of bacterial nutrient uptake and survival as it is comprised of high amounts of mucin, lipids, and amino acids (Thomas et al., 2000). Thick mucosal secretions also create pockets of hypoxia within the airways, creating unusual microaerobic to anaerobic bacterial growth conditions (Worlitzsch et al., 2002). A longitudinal study of sequential isolates of the DK2 clone family revealed oxygen metabolism as a hotspot for adaptive evolution (la Rosa et al., 2018). A transcriptomic and proteomic analysis of 13 sequential isolates including both MMR-deficient hypermutators and MMR-intact non-mutators revealed key metabolic adaptation catalyzed by hypermutation. Transcripts of genes involved in the anaerobic arginine deaminase pathway, such as oprF, azu, ccpR, aotJ, and braC, were increased in mutators. This pathway allows for production of adenine triphosphate using amino acid arginine under low oxygen conditions, suggesting the hypermutators were well adapted to the rich amino acid content and hypoxia in the CF lung. Interestingly, OprF has been implicated in biofilm viability in anaerobic environments, and Azu and CcpR protect against reactive nitrogen species released as byproducts of anaerobic respiration (Foote et al., 1992; Hassett et al., 2002; Yoon et al., 2002). Expression of genes in the arginine succinyltransferase pathway, which converts arginine into TCA cycle intermediates, was also increased in hypermutators. Genes needed for lipid metabolism (PA2886-93, foaAB, acpP, accB, and fabI) were also highly up-regulated in hypermutators. Gene expression profiles differed between hypermutators but showed a signature of convergent parallel evolution on these gene sets, suggesting adaptive evolution (Hoboth et al., 2009). Adaptive laboratory evolution of a hypermutator CF isolate showed overexpression of nos, nor, and nir operons due to RpoN overexpression which can mitigate toxic effects of anaerobic respiration as a vital adaptive event. Reversion to acute phenotype during laboratory evolution also showed upregulation in cioA and cioB needed for aerobic respiration (la Rosa et al., 2021). Genomic analysis of longitudinal hypermutator isolates showed parallel reductive evolution in catabolism pathways, resulting in a high number of auxotrophies, thought to serve as an energy conservation mechanism due to the rich amino acid environment in the CF airway (Feliziani et al., 2014).
Hypermutation catalyzes mutagenesis in master transcriptional regulators
Pa employs a large arsenal of virulence factors that are tightly controlled by a complex network of transcription factors to minimize unnecessary energy expenditure (Balasubramanian et al., 2013). Transcriptional plasticity has been implicated in the flexibility of Pa to occupy many environmental niches and to persist in chronic CF lung infections (Rossi et al., 2018). Hypermutator strains have been documented to have many nonsynonymous mutations in master transcription regulators that mediate the switch from an acute to chronic virulence state. Inactivating mutations in lasR (a master quorum sensing regulator) are highly correlated with hypermutability in CF isolates (Whiteley et al., 1999; Bjarnsholt et al., 2010). LasR mutants are associated with chronic infection and poorer patient outcome similar to hypermutability (Hoffman et al., 2009). MutS-deficient Pa displayed significant emergence of LasR-mutants caused by indel frameshifts compared to wild-type in vitro (Luján et al., 2007). It is important to note that, like MucA, LasR mutants have been observed before MMR-deficiency, so it is not a requirement (Ciofu et al., 2010).
Additional common loci for mutation in hypermutator strains are gacS and retS that regulate the Gac/Rsm signaling pathway. GacS negatively regulates the pathway and promotes expression of type VI secretion and Pel polysaccharides. Through upregulation of small regulatory RNAs RsmY and RsmZ, GacS activity also downregulates type III secretion (Sall et al., 2014; Valentini et al., 2018). Through these pathways, GacS promotes characteristics of the chronic virulence state. RetS activity attenuates GacS signaling and therefore promotes expression of factors of the acute virulence state (Francis et al., 2018). RetS has been found to be a hotspot for loss-of-function frameshifts in hypermutators. Interestingly, GacS and GacA (the other member of the GacS/GacA two-component system controlling RsmY and RsmZ expression) have also been identified as hotspots for convergent evolution in hypermutators (Feliziani et al., 2014; Marvig et al., 2015; la Rosa et al., 2021). GacA mutants have been associated with nitrogen metabolism upregulation, type VI secretion, and reduced motility (Wei et al., 2013; Huang et al., 2019). GacS mutants appear to confer an evolutionary advantage in the CF airway through formation of small colony variants in biofilms with increased resistance to immune defense and antibiotics (Davies et al., 2007; Nelson et al., 2010). Large numbers of AmpR and ExsA mutants have also been observed in hypermutators with adaptive consequences in type III secretion system functions, quorum sensing, immune evasion, and nitrogen metabolism (Balasubramanian et al., 2014; Marvig et al., 2015; Huang et al., 2019; Tian et al., 2019; la Rosa et al., 2021).
Pathoadaptive loss of function mutations in key acute virulence genes
Numerous genetic analyses of longitudinal CF isolates have revealed overwhelmingly large numbers of mutations in key Pa virulence genes and have consistently showed a strong signature of purifying selection for these loss-of-function pathoadaptive mutations (Smith et al., 2006; Chung et al., 2012; Feliziani et al., 2014; Marvig et al., 2015; Wee et al., 2018). One example is the loss of quorum sensing in CRI with Pa due to LasR loss-of-function. However, recent evidence has shown that quorum sensing may not be lost, but hypermutation helps rewire it to favor the LasR-independent RhII-RhIR alternate pathway (Feltner et al., 2016; Chen et al., 2019; Kostylev et al., 2019). RhIR mutants are highly correlated with hypermutability in CF isolates (Bjarnsholt et al., 2010). In vitro, RhIR mutants were not found with evolution of a MutS-deficient Pa strain, whereas LasR mutants did emerge, suggesting differing selective pressures on the two pathways (Luján et al., 2007). In the context of Pa CRI, RhII has also been associated with anaerobic biofilm viability, which quorum sensing plays an especially important role in due to close spatial arrangement and population heterogeneity (Hassett et al., 2002; Darch et al., 2018).
Another common pathoadaptive mutation in CF isolates is the loss of type III secretion. This usually results from mutations in transcription regulators such as retS and exsA, as mentioned previously. Hypermutators show significantly more downregulation of genes involved in type III secretion compared to non-mutators, and they do not produce the major type III secretion product ExoS (Hoboth et al., 2009). Instead, adaptive laboratory evolution with a hypermutator CF isolate shows that shift in expression of type III to type VI secretion is a key adaptive event (Moscoso et al., 2011; la Rosa et al., 2021). Hypermutators also show significantly more downregulation in flagellar proteins compared to non-mutators, and show convergent evolution in flgG and fliD, associated with the chronic virulence state of loss of motility (Hoboth et al., 2009; la Rosa et al., 2021). This is recapitulated in vitro, as MutS-deficient Pa shows impaired swimming and twitching motility, suggesting the switch to favor biofilm formation (Smania et al., 2004). The genes targeted for pathoadaptive mutations in hypermutators are summarized in Table 2.

Table 2 Summary of genes targeted for convergent evolution in the CF lung during CRI with Pa, catalyzed by hypermutation.
Conclusions and perspectives
Hypermutators play a vital role in the survival and persistence of Pa in CRI by allowing rapid diversification and adaptation to the CF lung environment. This results in the shift from the acute virulence phenotypes (type III secretion, motility, toxin production) to the chronic virulence phenotypes. Isolates having undergone the shift to a chronic virulence state display mucoidy, biofilm formation, modulation of metabolic pathways, alteration of quorum sensing, type VI secretion, and loss of motility. The high prevalence of hypermutators arising in the CF lung underscores the need for the adaptability afforded by genetically diverse isolates. This mutagenesis is preferred despite the simultaneous increased probability of accumulating deleterious mutations and potential reduced virulence.
This review reveals many discrepancies between longitudinal genetic analyses and in vitro adaptive evolution, notably in data concerning the effect of hypermutability on MucA and LasR. Although mutation rate and accumulation of mutations is higher in hypermutators, numbers of variants in target genes are usually not significantly different between hypermutator and non-mutator CF isolates (Mena et al., 2008). This suggests that hypermutation may not affect a specific adaptive trait significantly over the course of CRI, but rather has a generalized effect of facilitating adaptive evolution. As the selective pressures of the CF lung are the same on both hypermutators and non-mutators, it is reasonable for both to undergo similar genetic and phenotypic changes just at different rates. In fact, the only trait significantly linked to hypermutation is antibiotic resistance (Macía et al., 2005; Feliziani et al., 2010). This could be due to the large bottlenecking effect of antibiotic treatment on a population (Windels et al., 2021).
Although this review mainly addresses the role of hypermutation in the adaption of Pa to the CF lung environment, hypermutation has also been shown to play a role in adaptation in other disease states as well. PAHM4 (a bronchiectasis Pa isolate) displays mutS inactivating alleles similar to CF isolates (Warren et al., 2011). It also contains mucA22 causing mucoidy, likely caused by MutS-deficiency as discussed above. The isolate shows similar characteristics of loss of motility and type III secretion and demonstrates high levels of antibiotic resistance (Varga et al., 2015). Similar to CF, bronchiectasis airways are known for having high mucin levels, altered nutritional complexity, and a widely diverse diseased-state lung microbiome, so perhaps similar selective pressures are driving convergence on these highlighted similarities (H. Richardson et al., 2019).
The prevalence of the hypermutator phenotype in Pa CRI and its prominent role in adaptation challenges the common assumption in microbiology that strains and isolates are clonal and can be treated as such. Population heterogeneity is overwhelmingly seen in CF isolates. With the occurrence of hypermutation in other diseases and even other species, it is possible that many other bacterial isolates also display high levels of population heterogeneity. Hypermutator S. aureus and H. influenzae isolates have been documented in CF patients and are associated with higher rates of antibiotic resistance, similar to Pa (Prunier et al., 2003; Román et al., 2004). How the hypermutator phenotype affects interspecies competition in the context of the CF lung is yet to be understood. As mentioned previously, hypermutability also occurs in pathogenic E. coli, S. enterica, and N. meningitidis (LeClerc et al., 1996; Matic et al., 1997; A. R. Richardson et al., 2002). It would be interesting to explore the role of hypermutation in different species and disease contexts and to see if it similarly drives adaptation to the host environment fostering survival. Hypermutation and its effect on bacterial pathogenesis poses many interesting questions for future study.
Author contributions
KH performed the literature review and drafted the manuscript. LM and ZP revised the manuscript and provided supervision. All authors contributed to the article and approved the submitted version.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Balasubramanian, D., Kumari, H., Mathee, K. (2014). Pseudomonas aeruginosa AmpR: an acute-chronic switch regulator. Pathog. Dis 73 (2, 1–14. doi: 10.1111/2049-632X.12208
Balasubramanian, D., Schneper, L., Kumari, H., Mathee, K. (2013). A dynamic and intricate regulatory network determines pseudomonas aeruginosa virulence. Nucleic Acids Res. 41, 1–20). doi: 10.1093/nar/gks1039
Bergeron, C., Cantin, A. M. (2019). Cystic fibrosis: Pathophysiology of lung disease. Semin. Respir. Crit. Care Med. 40 (06), 715–726. doi: 10.1055/s-0039-1694021
Bjarnsholt, T., Jensen, P. Ø., Jakobsen, T. H., Phipps, R., Nielsen, A. K., Rybtke, M. T., et al. (2010). Quorum sensing and virulence of pseudomonas aeruginosa during lung infection of cystic fibrosis patients. PLoS One 5 (4). doi: 10.1371/journal.pone.0010115
Boucher, J. C., Yu, H., Mudd, M. H., Deretic, V. (1997). Mucoid pseudomonas aeruginosa in cystic fibrosis: characterization of muc mutations in clinical isolates and analysis of clearance in a mouse model of respiratory infection. Infect. Immun. 65 (9), 3838–3846. doi: 10.1128/iai.65.9.3838-3846.1997
Bragonzi, A., Paroni, M., Nonis, A., Cramer, N., Montanari, S., Rejman, J., et al. (2009). Pseudomonas aeruginosa microevolution during cystic fibrosis lung infection establishes clones with adapted virulence. Am. J. Respir. Crit. Care Med. 180 (2), 138–145. doi: 10.1164/rccm.200812-1943OC
Bragonzi, A., Wiehlmann, L., Klockgether, J., Cramer, N., Worlitzsch, D., Döring, G., et al. (2006). Sequence diversity of the mucABD locus in pseudomonas aeruginosa isolates from patients with cystic fibrosis. Microbiology 152 (11), 3261–3269. doi: 10.1099/mic.0.29175-0
Cao, Q., Yang, N., Wang, Y., Xu, C., Zhang, X., Fan, K., et al. (2020). Mutation-induced remodeling of the BfmRS two-component system in pseudomonas aeruginosa clinical isolates. Sci. Signal 13, eaaz1529. doi: 10.1126/scisignal.aaz1529
Chen, R., Déziel, E., Groleau, M.-C., Schaefer, A., Peter Greenberg, E. (2019). Social cheating in a pseudomonas aeruginosa quorum-sensing variant. Natl. Acad. Sci. 116 (14), 7021–7026. doi: 10.1073/pnas.1819801116ï
Chung, J. C. S., Becq, J., Fraser, L., Schulz-Trieglaff, O., Bond, N. J., Foweraker, J., et al. (2012). Genomic variation among contemporary pseudomonas aeruginosa isolates from chronically infected cystic fibrosis patients. J. Bacteriol. 194 (18), 4857–4866. doi: 10.1128/JB.01050-12
Ciofu, O., Hansen, C. R., Høiby, N. (2013). Respiratory bacterial infections in cystic fibrosis. Curr. Opin. Pulm. Med. 19 (3), 251–258. doi: 10.1097/MCP.0b013e32835f1afc
Ciofu, O., Mandsberg, L. F., Bjarnsholt, T., Wassermann, T., Høiby, N. (2010). Genetic adaptation of pseudomonas aeruginosa during chronic lung infection of patients with cystic fibrosis: Strong and weak mutators with heterogeneous genetic backgrounds emerge in mucA and/or lasR mutants. Microbiology 156 (4), 1108–1119. doi: 10.1099/mic.0.033993-0
Ciofu, O., Riis, B., Pressler, T., Poulsen, H. E., Høiby, N. (2005). Occurrence of hypermutable pseudomonas aeruginosa in cystic fibrosis patients is associated with the oxidative stress caused by chronic lung inflammation. Antimicrobial. Agents Chemother. 49 (6), 2276–2282. doi: 10.1128/AAC.49.6.2276-2282.2005
Conibear, T. C. R., Collins, S. L., Webb, J. S. (2009). Role of mutation in pseudomonas aeruginosa biofilm development. PLoS One 4 (7), e6289. doi: 10.1371/journal.pone.0006289
Cystic Fibrosis Foundation (2021). Cystic fibrosis foundation 2020 annual data report (Bethesda, Maryland: ©2021 Cystic Fibrosis Foundation
Darch, S. E., Simoska, O., Fitzpatrick, M., Barraza, J. P., Stevenson, K. J., Bonnecaze, R. T., et al. (2018). Spatial determinants of quorum signaling in a pseudomonas aeruginosa infection model. Proc. Natl. Acad. Sci. U.S.A 115 (18), 4779–4784. doi: 10.1073/pnas.1719317115
Davies, J. A., Harrison, J. J., Marques, L. L. R., Foglia, G. R., Stremick, C. A., Storey, D. G., et al. (2007). The GacS sensor kinase controls phenotypic reversion of small colony variants isolated from biofilms of pseudomonas aeruginosa PA14. FEMS Microbiol. Ecol. 59 (1), 32–46. doi: 10.1111/j.1574-6941.2006.00196.x
Dößelmann, B., Willmann, M., Steglich, M., Bunk, B., Nübel, U., Peter, S., et al. (2017). Rapid and consistent evolution of colistin resistance in extensively drug-resistant pseudomonas aeruginosa during morbidostat culture. Antimicrobial. Agents Chemother. 61 (9), e00043–17. doi: 10.1128/AAC.00043-17
Feliziani, S., Luján, A. M., Moyano, A. J., Sola, C., Bocco, J. L., Montanaro, P., et al. (2010). Mucoidy, quorum sensing, mismatch repair and antibiotic resistance in pseudomonas aeruginosa from cystic fibrosis chronic airways infections. PLoS One 5 (9), 1–12. doi: 10.1371/journal.pone.0012669
Feliziani, S., Marvig, R. L., Luján, A. M., Moyano, A. J., di Rienzo, J. A., Krogh Johansen, H., et al. (2014). Coexistence and within-host evolution of diversified lineages of hypermutable pseudomonas aeruginosa in long-term cystic fibrosis infections. PLoS Genet. 10 (10), e1004651. doi: 10.1371/journal.pgen.1004651
Feltner, J. B., Wolter, D. J., Pope, C. E., Groleau, M. C., Smalley, N. E., Greenberg, E. P., et al. (2016). LasR variant cystic fibrosis isolates reveal an adaptable quorum-sensing hierarchy in pseudomonas aeruginosa. MBio 7 (5), e01513–16. doi: 10.1128/mBio.01513-16
Ferroni, A., Guillemot, D., Moumile, K., Bernede, C., le Bourgeois, M., Waernessyckle, S., et al. (2009). Effect of mutator p. aeruginosa on antibiotic resistance acquisition and respiratory function in cystic fibrosis. Pediatr. Pulmonol. 44 (8), 820–825. doi: 10.1002/ppul.21076
Foote, N., Turner, R., Brittain, T., Greenwood, C. (1992). A quantitative model for the mechanism of action of the cytochrome c peroxidase of pseudomonas aeruginosa. Biochem. J. 283 (3), 839–843. doi: 10.1042/bj2830839
Francis, V. I., Waters, E. M., Finton-James, S. E., Gori, A., Kadioglu, A., Brown, A. R., et al. (2018). Multiple communication mechanisms between sensor kinases are crucial for virulence in pseudomonas aeruginosa. Nat. Commun. 9 (1), 2219. doi: 10.1038/s41467-018-04640-8
Ghafoor, A., Hay, I. D., Rehm, B. H. A. (2011). Role of exopolysaccharides in pseudomonas aeruginosa biofilm formation and architecture. Appl. Environ. Microbiol. 77 (15), 5238–5246. doi: 10.1128/AEM.00637-11
Giraud, A., Matic, I., Tenaillon, O., Clara, A., Radman, M., Fons, M., et al. (2001). Costs and benefits of high mutation rates: Adaptive evolution of bacteria in the mouse gut. Science 291 (5513), 2606–2608. doi: 10.1126/science.1056421
Gutiérrez, O., Juan, C., Pérez, J. L., Oliver, A. (2004). Lack of association between hypermutation and antibiotic resistance development in pseudomonas aeruginosa isolates from intensive care unit patients. Antimicrobial. Agents Chemother. 48 (9), 3573–3575. doi: 10.1128/AAC.48.9.3573-3575.2004
Høiby, N., Ciofu, O., Bjarnsholt, T. (2010). Pseudomonas aeruginosa biofilms in cystic fibrosis. Future Microbiol. 5 (11), 1663–1674. doi: 10.2217/fmb.10.125
Hassett, D. J., Cuppoletti, J., Trapnell, B., Lymar, S., Rowe, J. J., Yoon, S. S., et al. (2002). Anaerobic metabolism and quorum sensing by pseudomonas aeruginosa biofilms in chronically infected cystic fibrosis airways: rethinking antibiotic treatment strategies and drug targets. Adv. Drug Deliv. Rev. 54 (11), 1425–1443. doi: 10.1016/S0169-409X(02)00152-7
Hoboth, C., Hoffmann, R., Eichner, A., Henke, C., Schmoldt, S., Imhof, A., et al. (2009). Dynamics of adaptive microevolution of hypermutable pseudomonas aeruginosa during chronic pulmonary infection in patients with cystic fibrosis. J. Infect. Dis. 200 (1), 118–130. doi: 10.1086/599360
Hoffman, L. R., Kulasekara, H. D., Emerson, J., Houston, L. S., Burns, J. L., Ramsey, B. W., et al. (2009). Pseudomonas aeruginosa lasR mutants are associated with cystic fibrosis lung disease progression. J. Cyst. Fibrosis 8 (1), 66–70. doi: 10.1016/j.jcf.2008.09.006
Hogardt, M., Schubert, S., Adler, K., Götzfried, M., Heesemann, J. (2006). Sequence variability and functional analysis of MutS of hypermutable pseudomonas aeruginosa cystic fibrosis isolates. Int. J. Med. Microbiol. 296 (4–5), 313–320. doi: 10.1016/j.ijmm.2005.12.018
Huang, H., Shao, X., Xie, Y., Wang, T., Zhang, Y., Wang, X., et al. (2019). An integrated genomic regulatory network of virulence-related transcriptional factors in pseudomonas aeruginosa. Nat. Commun. 10 (1), 2931. doi: 10.1038/s41467-019-10778-w
Iyer, R. R., Pluciennik, A., Burdett, V., Modrich, P. L. (2006). DNA Mismatch repair: Functions and mechanisms. Chem. Rev. 106 (2), 302–323. doi: 10.1021/cr0404794
Klockgether, J., Cramer, N., Wiehlmann, L., Davenport, C. F., Tümmler, B. (2011). Pseudomonas aeruginosa genomic structure and diversity. Front. Microbiol. 2. doi: 10.3389/fmicb.2011.00150
Kostylev, M., Kim, D. Y., Smalley, N. E., Salukhe, I., Peter Greenberg, E., Dandekar, A. A. (2019). Evolution of the pseudomonas aeruginosa quorum-sensing hierarchy. Proc. Natl. Acad. Sci. U.S.A 116 (14), 7027–7032. doi: 10.1073/pnas.1819796116
Kunkel, T. A., Erie, D. A. (2005). DNA Mismatch repair. Annu. Rev. Biochem. 74, 681–710). doi: 10.1146/annurev.biochem.74.082803.133243
la Rosa, R., Johansen, H. K., Molin, S. (2018). Convergent metabolic specialization through distinct evolutionary paths in pseudomonas aeruginosa. MBio 9 (2), e00269–18. doi: 10.1128/mBio.00269-18
la Rosa, R., Johansen, H. K., Molin, S. (2019). Adapting to the airways: Metabolic requirements of pseudomonas aeruginosa during the infection of cystic fibrosis patients. Metabolites 9 (10), 234. doi: 10.3390/metabo9100234
la Rosa, R., Rossi, E., Feist, A. M., Johansen, H. K., Molin, S. (2021). Compensatory evolution of pseudomonas aeruginosa’s slow growth phenotype suggests mechanisms of adaptation in cystic fibrosis. Nat. Commun. 12 (1), 3186. doi: 10.1038/s41467-021-23451-y
LeClerc, J. E., Li, B., Payne, W. L., Cebula, T. A. (1996). High mutation frequencies among escherichia coli and salmonella pathogens. Science 274 (5290), 1208–1211. doi: 10.1126/science.274.5290.1208
Lee, H., Popodi, E., Tang, H., Foster, P. L. (2012). Rate and molecular spectrum of spontaneous mutations in the bacterium escherichia coli as determined by whole-genome sequencing. Proc. Natl. Acad. Sci. U.S.A 109 (41), E2774–E2783. doi: 10.1073/pnas.1210309109
López-Causapé, C., Sommer, L. M., Cabot, G., Rubio, R., Ocampo-Sosa, A. A., Johansen, H. K., et al. (2017). Evolution of the pseudomonas aeruginosa mutational resistome in an international cystic fibrosis clone. Sci. Rep. 7 (1), 5555. doi: 10.1038/s41598-017-05621-5
Luján, A. M., Moyano, A. J., Segura, I., Argaraña, C. E., Smania, A. M. (2007). Quorum-sensing-deficient (lasR) mutants emerge at high frequency from a pseudomonas aeruginosa mutS strain. Microbiology 153 (1), 225–237. doi: 10.1099/mic.0.29021-0
Macía, M. D., Blanquer, D., Togores, B., Sauleda, J., Pérez, J. L., Oliver, A. (2005). Hypermutation is a key factor in development of multiple-antimicrobial resistance in pseudomonas aeruginosa strains causing chronic lung infections. Antimicrobial. Agents Chemother. 49 (8), 3382–3386. doi: 10.1128/AAC.49.8.3382-3386.2005
Malhotra, S., Hayes, D., Wozniak, D. J. (2019). Mucoid pseudomonas aeruginosa and regional inflammation in the cystic fibrosis lung. J. Cyst. Fibrosis 18 (6), 796–803. doi: 10.1016/j.jcf.2019.04.009
Markussen, T., Marvig, R. L., Gómez-Lozano, M., Aanæs, K., Burleigh, A. E., Høiby, N., et al. (2014). Environmental heterogeneity drives within-host diversification and evolution of pseudomonas aeruginosa. MBio 5 (5), e01592–14. doi: 10.1128/mBio.01592-14
Martin, D. W., Schurr, M. J., Mudd, M. H., Govant, J. R. W., Hollowayi, B. W., Deretic, V. (1993). Mechanism of conversion to mucoidy in pseudomonas aeruginosa infecting cystic fibrosis patients. Proc. Natl. Acad. Sci. U.S.A. 90, e1003741. doi: 10.1073/pnas.90.18.8377
Marvig, R. L., Johansen, H. K., Molin, S., Jelsbak, L. (2013). Genome analysis of a transmissible lineage of pseudomonas aeruginosa reveals pathoadaptive mutations and distinct evolutionary paths of hypermutators. PLoS Genet. 9 (9), 8377–81. doi: 10.1371/journal.pgen.1003741
Marvig, R. L., Sommer, L. M., Molin, S., Johansen, H. K. (2015). Convergent evolution and adaptation of pseudomonas aeruginosa within patients with cystic fibrosis. Nat. Genet. 47 (1), 57–64. doi: 10.1038/ng.3148
Matic, I., Radman, M., Taddei, F., Picard, B., Doit, C., Bingen, E., et al. (1997). Highly variable mutation rates in commensal and pathogenic escherichia coli. Science 277 (5333), 1833–1834. doi: 10.1126/science.277.5333.1833
Mena, A., Macía, M. D., Borrell, N., Moya, B., de Francisco, T., Pérez, J. L., et al. (2007). Inactivation of the mismatch repair system in pseudomonas aeruginosa attenuates virulence but favors persistence of oropharyngeal colonization in cystic fibrosis mice. J. Bacteriol. 189 (9), 3665–3668. doi: 10.1128/JB.00120-07
Mena, A., Smith, E. E., Burns, J. L., Speert, D. P., Moskowitz, S. M., Perez, J. L., et al. (2008). Genetic adaptation of pseudomonas aeruginosa to the airways of cystic fibrosis patients is catalyzed by hypermutation. J. Bacteriol. 190 (24), 7910–7917. doi: 10.1128/JB.01147-08
Montanari, S., Oliver, A., Salerno, P., Mena, A., Bertoni, G., Tümmler, B., et al. (2007). Biological cost of hypermutation in pseudomonas aeruginosa strains from patients with cystic fibrosis. Microbiology 153 (5), 1445–1454. doi: 10.1099/mic.0.2006/003400-0
Moscoso, J. A., Mikkelsen, H., Heeb, S., Williams, P., Filloux, A. (2011). The pseudomonas aeruginosa sensor RetS switches type III and type VI secretion via c-di-GMP signalling. Environ. Microbiol. 13 (12), 3128–3138. doi: 10.1111/j.1462-2920.2011.02595.x
Moyano, A. J., Luján, A. M., Argaraña, C. E., Smania, A. M. (2007). MutS deficiency and activity of the error-prone DNA polymerase IV are crucial for determining mucA as the main target for mucoid conversion in pseudomonas aeruginosa. Mol. Microbiol. 64 (2), 547–559. doi: 10.1111/j.1365-2958.2007.05675.x
Moyano, A. J., Smania, A. M. (2009). Simple sequence repeats and mucoid conversion: Biased muca mutagenesis in mismatch repair-deficient pseudomonas aeruginosa. PLoS One 4 (12), e8203. doi: 10.1371/journal.pone.0008203
Nelson, L. K., Stanton, M. M., Elphinstone, R. E. A., Helwerda, J., Turner, R. J., Ceri, H. (2010). Phenotypic diversification in vivo: Pseudomonas aeruginosa gacS- strains generate small colony variants in vivo that are distinct from in vitro variants. Microbiology 156 (12), 3699–3709. doi: 10.1099/mic.0.040824-0
Nivens, D. E., Ohman, D. E., Williams, J., Franklin, M. J. (2001). Role of alginate and its O acetylation in formation of pseudomonas aeruginosa microcolonies and biofilms. J. Bacteriol. 183 (3), 1047–1057. doi: 10.1128/JB.183.3.1047-1057.2001
Oliver, A., Baquero, F., Blázquez, J. (2002a). The mismatch repair system (mutS, mutL and uvrD genes) in pseudomonas aeruginosa: Molecular characterization of naturally occurring mutants. Mol. Microbiol. 43 (6), 1641–1650. doi: 10.1046/j.1365-2958.2002.02855.x
Oliver, A., Cantón, R., Campo, P., Baquero, F., Blázquez, J. (2000). High frequency of hypermutable pseudomonas aeruginosa in cystic fibrosis lung infection. Science 288 (5469), 1251–1253. doi: 10.1126/science.288.5469.1251
Oliver, A., Mena, A. (2010). Bacterial hypermutation in cystic fibrosis, not only for antibiotic resistance. Clin. Microbiol. Infect. 16, 798–808). doi: 10.1111/j.1469-0691.2010.03250.x
Oliver, A., Sánchez, J. M., Blázquez, J. (2002b). Characterization of the GO system of pseudomonas aeruginosa. FEMS Microbiol. Lett. 217 (1), 31–35. doi: 10.1111/j.1574-6968.2002.tb11452.x
On, Y. Y., Welch, M. (2021). The methylation-independent mismatch repair machinery in pseudomonas aeruginosa. Microbiology 167 (12), 001120. doi: 10.1099/mic.0.001120
Pier, G. B., Coleman, F., Grout, M., Franklin, M., Ohman, D. E. (2001). Role of alginate O acetylation in resistance of mucoid pseudomonas aeruginosa to opsonic phagocytosis. Infect. Immun. 69 (3), 1895–1901. doi: 10.1128/IAI.69.3.1895-1901.2001
Prunier, A., Malbruny, B., Laurans, M., Brouard, J., Duhamel, J., Leclercq, R. (2003). High rate of macrolide resistance in staphylococcus aureus strains from patients with cystic fibrosis reveals high proportions of hypermutable strains. J. Infect. Dis. 187 (11), 1709–1716. doi: 10.1086/374937
Rajan, S. (2002). Pulmonary infections in patients with cystic fibrosis. Semin. Respir. Infect. 17 (1), 47–56. doi: 10.1053/srin.2002.31690
Richardson, H., Dicker, A. J., Barclay, H., Chalmers, J. D. (2019). The microbiome in bronchiectasis. Eur. Respir. Rev. 28 (153), 190048. doi: 10.1183/16000617.0048-2019
Richardson, A. R., Yu, Z., Popovic, T., Stojiljkovic, I. (2002). Mutator clones of neisseria meningitidis in epidemic serogroup a disease. Proc. Natl. Acad. Sci. 99 (9), 6103–6107. doi: 10.1073/pnas.092568699
Román, F., Cantón, R., Pérez-Vázquez, M., Baquero, F., Campos, J. (2004). Dynamics of long-term colonization of respiratory tract by haemophilus influenzae in cystic fibrosis patients shows a marked increase in hypermutable strains. J. Clin. Microbiol. 42 (4), 1450–1459. doi: 10.1128/JCM.42.4.1450-1459.2004
Rossi, E., Falcone, M., Molin, S., Johansen, H. K. (2018). High-resolution in situ transcriptomics of pseudomonas aeruginosa unveils genotype independent patho-phenotypes in cystic fibrosis lungs. Nat. Commun. 9 (1), 3459. doi: 10.1038/s41467-018-05944-5
Sall, K. M., Casabona, M. G., Bordi, C., Huber, P., de Bentzmann, S., Attrée, I., et al. (2014). A gacS deletion in pseudomonas aeruginosa cystic fibrosis isolate CHA shapes its virulence. PLoS One 9 (4), e95936. doi: 10.1371/journal.pone.0095936
Schick, A., Kassen, R. (2018). Rapid diversification of pseudomonas aeruginosa in cystic fibrosis lung-like conditions. Proc. Natl. Acad. Sci. 115 (42), 10714–10719. doi: 10.1073/pnas.1721270115
Simpson, J. A., Smith, S. E., Dean, R. T. (1989). Scavenging by alginate of free radicals released by macrophages. Free Radical Biol. Med. 6 (4), 347–353. doi: 10.1016/0891-5849(89)90078-6
Smania, A. M., Segura, I., Pezza, R. J., Becerra, C., Albesa, I., Argaraña, C. E. (2004). Emergence of phenotypic variants upon mismatch repair disruption in pseudomonas aeruginosa. Microbiology 150 (5), 1327–1338. doi: 10.1099/mic.0.26751-0
Smith, E. E., Buckley, D. G., Wu, Z., Saenphimmachak, C., Hoffman, L. R., D’Argenio, D., et al. (2006). Genetic adaptation by pseudomonas aeruginosa to the airways of cystic fibrosis patients Proc. Natl. Acad. Sci. U.S.A. 103 (22), 8487–8492.
Sniegowski, P. D., Gerrish, P. J., Lenski, R. E. (1997). Evolution of high mutation rates in experimental populations of e. Coli Nat. 387 (6634), 703–705. doi: 10.1038/42701
Song, Z., Wu, H., Ciofu, O., Kong, K.-F., Høiby, N., Rygaard, J., et al. (2003). Pseudomonas aeruginosa alginate is refractory to Th1 immune response and impedes host immune clearance in a mouse model of acute lung infection. J. Med. Microbiol. 52 (9), 731–740. doi: 10.1099/jmm.0.05122-0
Stover, C. K., Pham, X. Q., Erwin, A. L., Mizoguchi, S. D., Warrener, P., Hickey, M. J., et al. (2000). Complete genome sequence of pseudomonas aeruginosa PAO1, an opportunistic pathogen. Nature 406, 959–64. doi: 10.1073/pnas.0602138103
Taddei, F., Radman, M., Maynard-Smith, J., Toupance, B., Gouyon, P. H., Godelle, B. (1997). Role of mutator alleles in adaptive evolution. Nature 387 (6634), 700–702. doi: 10.1038/42696
Thomas, S. R., Anjana, R., Hodson, M. E., Pitt, T. L. (2000). Increased sputum amino acid concentrations and auxotrophy of pseudomonas aeruginosa in severe cystic fibrosis lung disease. Thorax 55 (9), 795–797. doi: 10.1136/thorax.55.9.795
Tian, Z., Cheng, S., Xia, B., Jin, Y., Bai, F., Cheng, Z., et al. (2019). Pseudomonas aeruginosa ExsA regulates a metalloprotease, ImpA, that inhibits phagocytosis of macrophages. Infect. Immun. 87 (12), e00695–19. doi: 10.1128/IAI.00695-19
Valentini, M., Gonzalez, D., Mavridou, D. A., Filloux, A. (2018). Lifestyle transitions and adaptive pathogenesis of pseudomonas aeruginosa. Curr. Opin. Microbiol. 41, 15–20). doi: 10.1016/j.mib.2017.11.006
Varga, J. J., Barbier, M., Mulet, X., Bielecki, P., Bartell, J. A., Owings, J. P., et al. (2015). Genotypic and phenotypic analyses of a pseudomonas aeruginosa chronic bronchiectasis isolate reveal differences from cystic fibrosis and laboratory strains. BMC Genomics 16 (1), 883. doi: 10.1186/s12864-015-2069-0
Waine, D. J., Honeybourne, D., Smith, E. G., Whitehouse, J. L., Dowson, C. G. (2008). Association between hypermutator phenotype, clinical variables, mucoid phenotype, and antimicrobial resistance in pseudomonas aeruginosa. J. Clin. Microbiol. 46 (10), 3491–3493. doi: 10.1128/JCM.00357-08
Warren, A. E., Boulianne-Larsen, C. M., Chandler, C. B., Chiotti, K., Kroll, E., Miller, S. R., et al. (2011). Genotypic and phenotypic variation in pseudomonas aeruginosa reveals signatures of secondary infection and mutator activity in certain cystic fibrosis patients with chronic lung infections. Infect. Immun. 79 (12), 4802–4818. doi: 10.1128/IAI.05282-11
Wee, B. A., Tai, A. S., Sherrard, L. J., ben Zakour, N. L., Hanks, K. R., Kidd, T. J., et al. (2018). Whole genome sequencing reveals the emergence of a pseudomonas aeruginosa shared strain sub-lineage among patients treated within a single cystic fibrosis centre. BMC Genomics 19 (1), 644. doi: 10.1186/s12864-018-5018-x
Wei, X., Huang, X., Tang, L., Wu, D., Xu, Y. (2013). Global control of GacA in secondary metabolism, primary metabolism, secretion systems, and motility in the rhizobacterium pseudomonas aeruginosa M18. J. Bacteriol. 195 (15), 3387–3400. doi: 10.1128/JB.00214-13
Whiteley, M., Lee, K. M., Greenberg, E. P. (1999). Identification of genes controlled by quorum sensing in pseudomonas aeruginosa. Proc. Natl. Acad. Sci. 96 (24), 13904–13909. doi: 10.1073/pnas.96.24.13904
Windels, E. M., Fox, R., Yerramsetty, K., Krouse, K., Wenseleers, T., Swinnen, J., et al. (2021). Population bottlenecks strongly affect the evolutionary dynamics of antibiotic persistence. Mol. Biol. Evol. 38 (8), 3345–3357. doi: 10.1093/molbev/msab107
Worlitzsch, D., Tarran, R., Ulrich, M., Schwab, U., Cekici, A., Meyer, K. C., et al. (2002). Effects of reduced mucus oxygen concentration in airway pseudomonas infections of cystic fibrosis patients. J. Clin. Invest. 109 (3), 317–325. doi: 10.1172/JCI13870
Yang, L., Jelsbak, L., Marvig, R. L., Damkiær, S., Workman, C. T., Rau, M. H., et al. (2011). Evolutionary dynamics of bacteria in a human host environment. Proc. Natl. Acad. Sci. U.S.A. 108 (18), 7481–7486. doi: 10.1073/pnas.1018249108
Keywords: Pseudomonas, hypermutation, mismatch repair, microevolution, chronic respiratoryinfection, virulence
Citation: Hall KM, Pursell ZF and Morici LA (2022) The role of the Pseudomonas aeruginosa hypermutator phenotype on the shift from acute to chronic virulence during respiratory infection. Front. Cell. Infect. Microbiol. 12:943346. doi: 10.3389/fcimb.2022.943346
Received: 13 May 2022; Accepted: 30 June 2022;
Published: 22 July 2022.
Edited by:
William D Picking, University of Kansas, United StatesReviewed by:
Zhenyu Cheng, Dalhousie University, CanadaDebaki R Howlader, University of Kansas, United States
Copyright © 2022 Hall, Pursell and Morici. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Lisa A. Morici, bG1vcmljaUB0dWxhbmUuZWR1