 Michael W. Shultis
Michael W. Shultis Claire V. Mulholland
Claire V. Mulholland Michael Berney
Michael Berney
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Cell. Infect. Microbiol., 17 August 2022
Sec. Molecular Bacterial Pathogenesis
Volume 12 - 2022 | https://doi.org/10.3389/fcimb.2022.933458
This article is part of the Research TopicDrug-resistant Mycobacterium tuberculosisView all 21 articles
Antibiotic persisters are a sub-population of bacteria able to survive in the presence of bactericidal antibiotic despite the lack of heritable drug resistance mechanisms. This phenomenon exists across many bacterial species and is observed for many different antibiotics. Though these bacteria are often described as “multidrug persisters” very few experiments have been carried out to determine the homogeneity of a persister population to different drugs. Further, there is much debate in the field as to the origins of a persister cell. Is it formed spontaneously? Does it form in response to stress? These questions are particularly pressing in the field of Mycobacterium tuberculosis, where persisters may play a crucial role in the required length of treatment and the development of multidrug resistant organisms. Here we aim to interpret the known mechanisms of antibiotic persistence and how they may relate to improving treatments for M. tuberculosis, exposing the gaps in knowledge that prevent us from answering the question: Are all antibiotic persisters created equal?
In 2020 the WHO reported an estimated 10 million people contracted tuberculosis and 1.5 million died from the disease, making it the second most deadly infectious disease behind COVID-19 worldwide (WHO 2021). The agent responsible for this previously mysterious disease was first identified in 1882 when Robert Koch discovered the bacterium Mycobacterium tuberculosis as its cause (Murray et al., 2015). It wasn’t until the 1950s that reliable chemotherapy was developed (Murray et al., 2015). This began with streptomycin and para-aminosalycylic acid in 1944, but monotherapy treatments resulted in drug resistance, emphasizing the need for combination therapies. Isoniazid was introduced in 1951, leading to a combined “triple therapy”. This therapy required treatment times lasting up to 24 months long (Murray, 2004). Continued emergence of drug resistant populations led to the development of ethambutol in 1961 (Murray et al., 2015). It wasn’t until 5 years later that the introduction of rifampin combined with isoniazid was able to shorten treatment times to 9 months. Finally, the introduction of pyrazinamide further reduced treatment times to 6 months, bringing us to the combination therapies that remain in use today (Murray et al., 2015). The most common regimen includes the combination of isoniazid (INH), ethambutol (EMB), rifampicin (RIF), and pyrazinamide (PZA) for a period of 6 to 9 months (Nahid et al., 2016). Curiously, the two drugs that enabled greatly shortened treatment times, RIF and PZA, share a common property: the ability to kill persistent bacteria (Zhang et al., 2014; Hu et al., 2015).
Persistent bacteria are a sub-population of bacteria that demonstrate slower killing kinetics in response to a stress, yielding a bimodal kill curve (Figure 1, blue line) (Boldrin et al., 2020). Persistence is distinct from antibiotic resistance because the state of persistence is non-heritable (Balaban et al., 2019). If persistent bacteria are regrown and exposed to the same stressor, they will again exhibit a heterogeneous response with a bimodal kill curve. Conversely, if a small subpopulation of resistant bacteria is isolated, regrown, and retreated with the same drug, growth would be observed instead of bimodal killing. Persistent bacteria exist across bacterial species, though are commonly referred to by the umbrella term ‘persisters’.
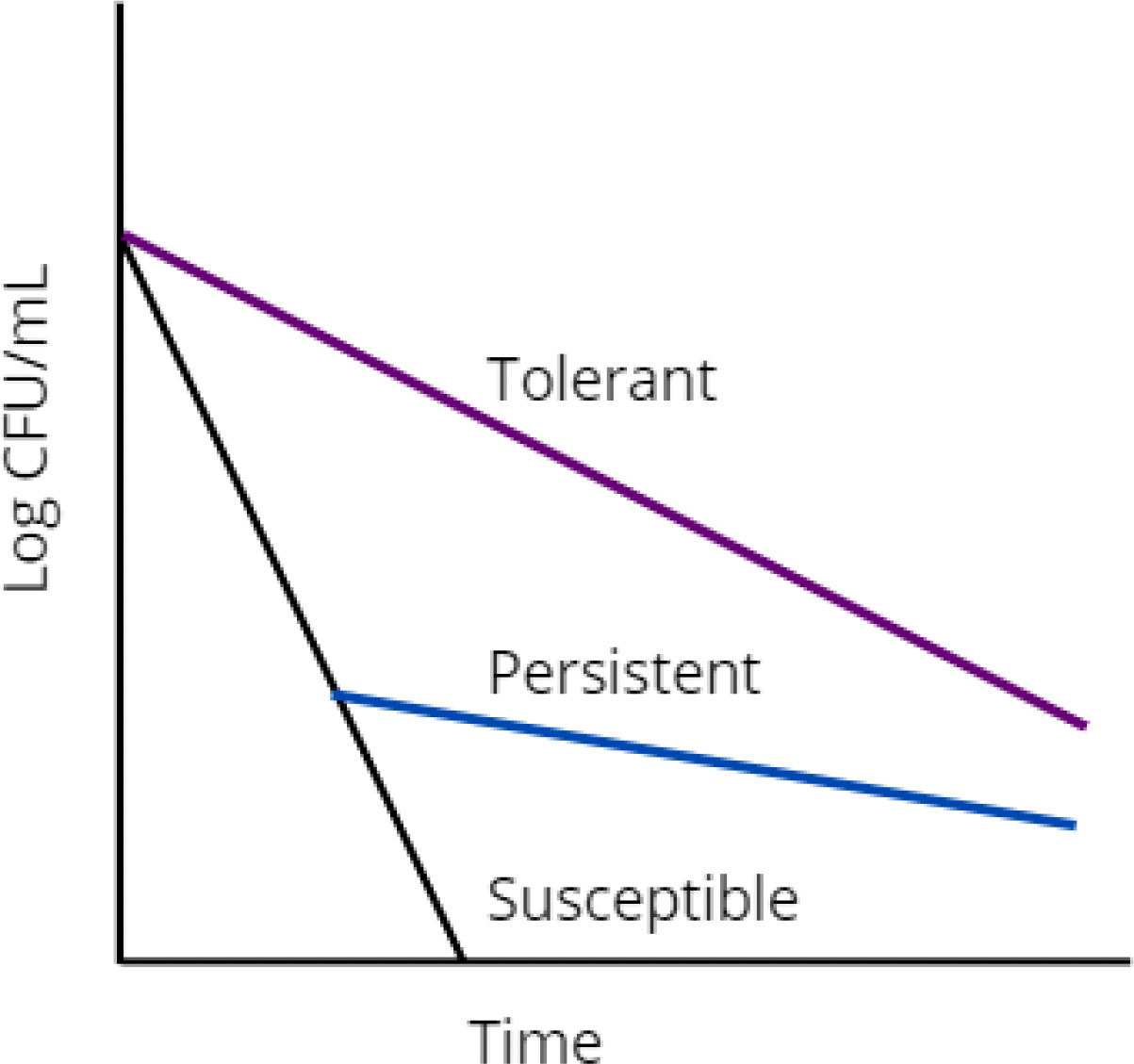
Figure 1 Graphical representation of drug susceptible (black), persistent (blue), and tolerant (purple) bacterial populations.
The first observation of antibiotic persistence was made in 1944 when Joseph Bigger demonstrated penicillin was incapable of sterilizing a culture of staphylococci (Bigger, 1944). In 1964 physicians with Cornell medical school made observations of “disappearing” M. tuberculosis bacilli in mice (McCune et al., 1966; McCune et al., 1966). Treatment of mice with a specific regimen of INH and PZA could push M. tuberculosis into an undetectable state by microscopy, culture, or reinfection. When treatment was removed for 90 days, M. tuberculosis became detectable in 1/3 mice (McCune et al., 1966). The undetectable state was only achievable when the mice were treated in order first with INH for a period of 2-4 weeks, followed by PZA for 8 weeks (McCune et al., 1966). If the duration of therapy was extended to 26 weeks no bacterial regrowth was observed up to 6 months after treatment cessation, suggesting these bacteria were persistent to sequential INH then PZA treatment after 12 weeks, but were killed with prolonged 26 week treatment (McCune et al., 1966). In these discussed conditions, treatment was generally carried out immediately following inoculation of mice. When mice were left untreated for 15 weeks, the undetectable state was not achievable with 12- or 26-week therapy. Among the bacilli that were detectable in the 26 week treated condition, only INH and PZA dual resistant mutant strains were identified, indicating that after 15 weeks enough pre-existing INH+PZA resistant mutants were generated, preventing the study of persistent bacteria (McCune et al., 1966). Although persistence is a common phenomenon in bacteria, a mechanism ubiquitously required for antibiotic persistence has yet to be identified, specifically in M. tuberculosis. The results described in the above Cornell study indicate that revealing the dynamics of persister cell formation are critical to accelerating the sterilization of M. tuberculosis infections.
Comprehensive and systematic reviews of persister phenomena have been presented elsewhere e.g (Boldrin et al., 2020). In the present review we focus on offering interpretations of the available data to illustrate the gaps in knowledge that prevent us from drawing conclusions about the heterogeneity of M. tuberculosis persisters to multidrug therapies.
The terms persistence and tolerance are often used interchangeably. Some prefer to draw a line between the two, describing them as similar but different phenomena (Brauner et al., 2016). The label of ‘persister’ appears largely to be definitionally confined. Under the current definition, an antibiotic persister must be a part of a small subpopulation, non-growing in the presence of the drug, genetically identical to the population that was killed by the stressor, vary only slightly with drug concentration at high concentrations of antibiotic, the list goes on (Balaban et al., 2019). By this definition, a bacterium could be labelled tolerant or persistent depending only on the characteristics of surrounding bacteria. If alone, this bacterium is a persister (Figure 1, blue line). If part of a larger population of bacteria exhibiting the same survival advantage, it is tolerant (Figure 1, purple line). There is no evidence to support the mechanistic distinction of persistent and tolerant bacteria, which is ultimately why many use these terms interchangeably. The mechanisms of M. tuberculosis tolerance are described in striking similarity to those of persistence. Metabolic slowdown, transcriptional and translational responses to stress, toxin-antitoxin system utilization, and efflux pumps are used to describe the mechanisms of both persistence and tolerance, [reviewed elsewhere in (Boldrin et al., 2020; Goossens et al., 2020)]. Comparatively it is very easy to draw the line between resistant bacteria and persistent bacteria. Resistance is a heritable adaptation that enables bacteria to grow in the presence of antibiotic (Balaban et al., 2019).
In summary, persistent bacteria are a drug tolerant sub-population that endures bactericidal antibiotic treatment through non-heritable mechanisms. Therefore, is it natural to wonder: If these survival advantages are non-heritable, where do persisters come from?
The exact origins of persistent bacteria remain shrouded in mystery. Persisters have been described as belonging to two categories - type 1 and type 2 (Balaban et al., 2004). These types will be referred to here as triggered and stochastic persisters respectively. The distinction between these two categories can generally be understood as a description of when a persister is formed. Does a persister already exist in a population before a stress is introduced? Does a normally growing bacterium respond to stress in its environment by becoming a persister cell? Triggered persisters form in response to a trigger in their environment, whereas stochastic persisters form in the absence of external triggers (Balaban et al., 2004; Balaban et al., 2019). This distinction can be difficult to make when persisters make up 1% or less of a bacterial population (Lewis, 2007) and can only be definitively identified by their survival in response to a stress.
In Escherichia coli some studies have reported persistence arising in a stochastic pattern consistent with the decreased availability of nutrients that enhance ATP production in the cell (Shan et al., 2017; Manuse et al., 2021). This increase in persistence to ciprofloxacin and ampicillin is suggested to be due to decreased ATP generation, leading to lower activity of antibiotic targets, resulting in drug tolerance (Shan et al., 2017). This result was interrogated following single cells in solution, revealing 15 out of 16 ampicillin persister cells were not growing prior to ampicillin treatment, supporting their identification as stochastic persisters (Manuse et al., 2021). Other groups have identified E. coli persisters that originate from metabolically active cells (Dorr et al., 2010; Goormaghtigh and Van Melderen, 2019). In a single-cell experiment, microfluidics were used to image cells in a culture every 15 minutes, enabling retroactive observation of persister cell growth once they were identified by ofloxacin treatment (Goormaghtigh and Van Melderen, 2019). The authors tracked the cell area of persistent cells and were able to identify a rate of elongation immediately before ofloxacin treatment. Though a small significant decrease in growth rate was identified in persisters compared to the total population, the authors attributed this to a high statistical power as they were unable to find a significant difference when they randomly sampled a smaller subset of the non-persistent population (Goormaghtigh and Van Melderen, 2019). Another group found that ciprofloxacin, also a fluoroquinolone, was able to induce E. coli persister cell formation via the SOS response triggered by DNA damage (Dorr et al., 2010). In Pseudomonas aeruginosa, cells have been observed to upregulate persistence in response to quorum-sensing signals secreted into their media (Moker et al., 2010).
These points together indicate that the mechanisms for persister cell formation can vary between bacterial species and can vary depending on the stress applied to bacteria within the same species. When ampicillin was selected to reveal the E. coli persister population in the first experiment endorsing stochastic persistence, bacteria that may have been persistent to another drug were killed, preventing their characterization (Manuse et al., 2021). When ofloxacin or ciprofloxacin were used to reveal the E. coli persister population instead, a different group of bacteria capable of triggered persistence may have ended up being analyzed (Dorr et al., 2010; Goormaghtigh and Van Melderen, 2019). Both populations seem to exist, meaning the origins of persister populations can be varied. It is likely that stochastic persistence remains present at a certain level and that triggered persistence can occur with different intensities to different stressors.
Mycobacteria also demonstrate characteristics consistent both with stochastic and triggered persistence. The growth of mycobacteria are inherently heterogenous. The bacilli elongate asymmetrically on one of their two poles (Aldridge et al., 2012). The growing pole deemed the accelerator pole and the nongrowing pole deemed the alternator pole (Aldridge et al., 2012). During each division one daughter cell inherits the accelerator pole while the other inherits the alternator pole (Aldridge et al., 2012). Cell growth continues along the older of the two poles, requiring the alternator pole be converted to an accelerator pole (Aldridge et al., 2012). It was found that daughter cells inheriting the accelerator pole elongated faster than daughter cells inheriting the alternator pole (Aldridge et al., 2012). Further, in Mycobacterium smegmatis, it was found that accelerator cells were generally more susceptible to cell wall antibiotics like INH and alternator cells were generally more susceptible to RIF (Aldridge et al., 2012). This result is consistent with the observation that most antimycobacterial drugs have poor activity on slow growing cells, with RIF and PZA being the exceptions (Xie et al., 2005; Pullan et al., 2016). Deletion of lamA, the gene responsible for the inhibition of growth at new growth poles, results in a more symmetrical growth from each pole. Of note, these more uniform cells demonstrate a faster killing rate in response to RIF in M. tuberculosis (Rego et al., 2017). In M. smegmatis these cells were killed faster in response to RIF as well as cell wall targeting drugs (Rego et al., 2017). When considering this slow, asymmetric growth pattern of M. tuberculosis it isn’t difficult to rationalize that this heterogeneity could lead to stochastic persisters. Indeed, authors Jain et al. demonstrated using a dual reporter mycobacteriophage that genes linked to persistence in M. tuberculosis were upregulated prior to INH treatment, and that the bacteria expressing these genes were enriched in the persistent population (Jain et al., 2016).
In another vein, M. tuberculosis excels at responding to stressors in the host environment. Signal transduction systems have been shown to be essential for M. tuberculosis to establish latent infection in lung tissue and play a role in the response of M. tuberculosis to environmental stressors (Zahrt and Deretic, 2001; Bretl et al., 2011). Mistranslation has been shown to be more prevalent in stressed M. tuberculosis, which has led to increased bacterial survival to RIF (Javid et al., 2014). When sequenced, the surviving bacteria contained no mutations in the RIF resistance determining region (RRDR), suggesting these bacteria were demonstrating triggered persistence (Javid et al., 2014). Collectively these observations indicate that persistent M. tuberculosis is composed of a mixed population of pre-existing and triggered persistent bacteria. This heterogeneity is likely further exacerbated in the host environment where M. tuberculosis encounters a variety of stressors (Warner and Mizrahi, 2006; Adams et al., 2011; Liu et al., 2016).
Regardless of its origin, it is important to consider the nature of a persister when attempting to design therapies to sterilize these bacteria. As discussed above, many drugs that impact the bacterial cell wall require actively growing cells to impact their targets (Xie et al., 2005). This fact has led to the belief that all persisters are non-growing cells, but is this truly the case?
Perhaps the most accepted characteristic of a persister is the characteristic of dormancy (Wood et al., 2013). Persisters are generally thought of as metabolically stunted bacteria that are inaccessible by antibiotics because the systems the antibiotics impact are inactive. It has been suggested that vitamin C and cysteine can prevent this metabolic shutdown by stimulating respiration, leading to sterilization of M. tuberculosis in vitro (Vilcheze et al., 2013; Vilcheze et al., 2017). In starved or stationary phase E. coli and P. aeruginosa, where bacteria metabolize more slowly, populations seem to be enriched for persistence (Keren et al., 2004a; Volzing and Brynildsen, 2015). This effect is observed in starved M. tuberculosis, where activation of the stringent response mediates persister formation, and deletion of a stringent response enzyme reduces persistence (Dutta et al., 2019). This stringent response enzyme, Rel, initiates metabolic arrest in M. tuberculosis (Dahl et al., 2003). The enrichment of persisters in stationary phase has been theorized to be a result of ATP-depletion (Manuse et al., 2021).
Contrarily, some groups presented evidence that persister cells can be metabolically active, and even actively dividing, in E. coli and M. smegmatis (Orman and Brynildsen, 2013; Wakamoto et al., 2013). In the case of E. coli 20/100 persisters were identified to be metabolically active by fluorescence-activated cell sorting (Orman and Brynildsen, 2013). This result comes with scrutiny as the experimental design is accused of carrying over persistent bacteria in the inoculum of the assay (Wood et al., 2013). M. smegmatis cells were observed to grow and divide in the presence of INH so long as expression of KatG, the activator of the prodrug INH, was suppressed (Wakamoto et al., 2013). Proponents of dormancy believe this to be an outlier, stating these cells are not persisters but instead normally growing cells that haven’t expressed KatG (Wood et al., 2013). This interaction is seen as unique because the cells aren’t under any stress from the antibiotic until converted to the active form, making this case inapplicable to persisters as a whole (Wood et al., 2013). Though model M. smegmatis is often used in experiments due to its faster growth and reduced biosafety requirements, it is important to be cautious when making direct comparisons to M. tuberculosis given the inherent differences of these two bacteria. However, we believe that when considering persistence in M. tuberculosis this result is important to consider given it is a direct study of mycobacterial persistence to a clinically relevant antimycobacterial drug. Furthermore, even E. coli persisters have been reported as having a reduced, but nonzero growth rate prior to antibiotic treatment (Balaban et al., 2004).
Briefly mentioned above, work in M. smegmatis has demonstrated bacterial growth in the presence of RIF, despite cells remaining genotypically sensitive to the drug (Javid et al., 2014). In this study, investigators generated a strain of M. smegmatis that resulted in higher rates of protein mistranslation, and these bacteria demonstrated 1000-fold more colonies on RIF containing agar, despite containing no mutations in the RRDR (Javid et al., 2014). The authors then utilized a bacterial strain with a higher fidelity ribosome, resulting in less mistranslation. This strain resulted in a decrease of bacterial survival to RIF compared to wild-type, suggesting that bacterial mistranslation is a unique way mycobacteria can survive antibiotic stress, though it is unknown if a resistance conferring mutation existed beyond the RRDR in the high protein mistranslation strain (Javid et al., 2014).
Another study was carried out in M. smegmatis and M. tuberculosis that demonstrated “semi-heritable” growth in the presence of RIF (Zhu et al., 2018). The cell wall of M. smegmatis was fluorescently labelled and then exposed to increasing concentrations of RIF. Cells that can grow will become less fluorescent as they “dilute” the fluorescent labels in their cell walls. It was found that unique to RIF, a small subset of cells was able to grow in the presence of RIF, up to 36 µg/mL (Zhu et al., 2018). The peak serum concentration for RIF appears to reach 3 to 5 µg/mL in humans (Seth et al., 1993; Lei et al., 2019). When M. tuberculosis or M. smegmatis exposed to RIF were plated on RIF-containing agar, a significant sub-population of colonies were observed (Zhu et al., 2018). When sequenced, all 30 of the present M. smegmatis colonies were found to be wild-type in the RRDR of the rpoB gene, the gene controlling the target of RIF. When these colonies were picked and re-plated onto RIF containing agar, there was a 10-fold increase in survival compared to the first exposure to the drug. When the same procedure was performed plating RIF-sensitive clinical isolates from active M. tuberculosis infections, a larger surviving sub-population was observed the longer the patient was on RIF therapy (Zhu et al., 2018). When these colonies were picked and regrown in non-selective media for 16 hours, no survival advantage was observed when compared to cells never exposed to RIF, making this effect “semi-heritable” (Zhu et al., 2018). This effect was correlated with increased transcription of rpoB.
Another study assessed the impact of asymmetric mycobacterial growth on RIF tolerance, and it was found that RIF tolerance was correlated with large cell size and older inherited growth poles (accelerator poles) (Richardson et al., 2016). This is consistent with the previously mentioned study demonstrating that alternator cells are more susceptible to RIF, since alternator cells will tend to be smaller on average, given the need to convert the nongrowing alternator pole to a growing pole (Aldridge et al., 2012). A study of persistent M. tuberculosis revealed persisters to PZA or RIF continue to engage in active transcription despite the apparent growth arrest associated with persistence (Hu et al., 2000). Given that mycobacteria seem to operate on a time based replication schedule, rather than a size based replication schedule (Aldridge et al., 2012), it becomes unclear if a persistent population is truly nongrowing or if this population is in a state of dynamic equilibrium between life and death, similar to the M. smegmatis cells observed by Wakamoto et al. (Aldridge et al., 2012; Wakamoto et al., 2013).
In this section we have discussed three instances of mycobacteria growing in the presence of antimycobacterial drugs, RIF and INH, despite remaining genotypically sensitive (Wakamoto et al., 2013; Javid et al., 2014; Zhu et al., 2018). These observations lend themselves to the conclusion that while most persisters are ‘dormant’, some persistent bacteria can grow in the presence of antimycobacterial drugs when treated with monotherapy.
Now, with the arguments for non-dormant persister cells established, we introduce with this section the phenomenon of phenotypic resistance and discuss the roles this phenomenon may play alongside persistence and tolerance in complicating the treatment of M. tuberculosis.
As already discussed in the above section on tolerance and persistence, the line between tolerance and persistence is very thin and often crossed in discussions of both topics. In contrast, drug resistance has been clearly differentiated based on two factors, heritability and growth. However, discussion of metabolically active bacteria that survive antibiotic treatment begs the question: Are both parameters, heritability and growth, necessary to exclude the persister label or is heritability alone sufficient?
Phenotypic resistance describes a phenomenon where bacteria can grow in the presence of antibiotics, but the mechanism that enables their growth is non-heritable. This phenomenon was discussed above in the context of protein mistranslation (Javid et al., 2014), and is reviewed in greater depth elsewhere (Corona and Martinez, 2013). The bacteria mentioned previously did not contain mutations in the RRDR, indicating that if these bacteria were regrown and exposed to RIF under conditions that did not promote mistranslation, they would remain drug sensitive unless a resistance mutation was present outside the RRDR (Javid et al., 2014). To some the growth of these bacteria excludes the persister label, but why? As discussed previously, we highlight here three instances of mycobacteria that survive treatment with antimycobacterial drugs through a non-heritable mechanism (Wakamoto et al., 2013; Javid et al., 2014; Zhu et al., 2018). Each author used a different descriptor for their population of cells, dynamic persistence (Wakamoto et al., 2013), phenotypic resistance (Javid et al., 2014; Zhu et al., 2018), and tolerance (Zhu et al., 2018). These studies all focus on a sub-population of bacteria undergoing non-heritable (or semi-heritable), heterogenous mechanisms of survival in response to an applied antibiotic stress. If any of these mechanisms arise during an infection, they would likely prolong treatment times and warrant a solution. Though these mechanisms seem to be drug specific, there is evidence, expanded on in the next section, that patient noncompliance and drug pharmacokinetics can impact the effective concentrations of certain drugs in the lesions where M. tuberculosis is present (Kimerling et al., 1998; Tostmann et al., 2013; Prideaux et al., 2015). In these cases, bacteria exhibiting drug-specific persistence mechanisms that we would otherwise expect combination therapies to kill, may be of greater clinical concern.
The important take away from these experiments is the identification of the mechanisms that lead to bacterial survival. If these mechanisms, transcriptional downregulation and protein mistranslation, impacted more general pathways, the resulting bacteria may exhibit survival to a wider array of drugs, making them of even greater clinical concern. Therefore, it is important to consider these as persister mechanisms, rather than being concerned whether the resulting phenotype is labelled tolerant, phenotypically resistant, or ‘persistent’. These definitional limitations introduce obstacles in communication making it difficult to solve the underlying issue that motivates all this research, how do we improve treatment of M. tuberculosis?
As discussed above, M. tuberculosis is capable of adapting to changes to its environment, including entering states of metabolic inactivity, rendering most antimycobacterials ineffective until the bacteria reactivate (Connolly et al., 2007). Although, if indeed some bacteria exist in a state of ‘dynamic persistence’ (Wakamoto et al., 2013) or ‘phenotypic resistance’ (Javid et al., 2014; Zhu et al., 2018), there is some plausibility that these growing persistent populations may undergo spontaneous mutations that drive them towards resistance and out of persistence. Though there is little evidence demonstrating that resistant populations arise directly from persistent populations (Cohen et al., 2013; Sebastian et al., 2017), any bacteria that survive antibiotic stress can reactivate and grow (Hu et al., 2000; Wood et al., 2013; Boldrin et al., 2020). This cycle of reactivation in the event of improper treatment or poor adherence enables the rise of drug resistant populations (Zhang et al., 2012). This concern is amplified when considering the evidence that mycobacteria can develop phenotypic resistance due to errors in transcription or translation triggered by various stressors (Hu et al., 2000; Wakamoto et al., 2013; Zhu et al., 2018). Though these results were only demonstrated with monotherapy, the spectrum of drug-noncompliant patients is vast. Non-compliant patients range from those that take no drugs to those that miss some of their doses (Munro et al., 2007). In a study of M. tuberculosis infected patients in New York City in 1997, 48% were found to be nonadherent. Non-adherent patients took longer to recover and were more likely to develop drug resistant tuberculosis (Pablos-Mendez et al., 1997).
Even if patients remain adherent to therapy, there have been observations of patients with serum drug concentrations below the therapeutic range for these drugs (Kimerling et al., 1998; Tostmann et al., 2013). When drug concentrations fall below the level required to inhibit bacterial growth, drug resistant populations can arise. Furthermore, even when patients maintain therapeutic serum concentrations, authors Prideaux et al. demonstrated that different drugs INH, RIF, PZA, and moxifloxacin (MXF) have “different spatial and temporal patterns of distribution across TB lesion types and compartments” (Prideaux et al., 2015). Though it appears that INH demonstrated good penetration into critical compartments, INH never reached its minimum anaerobic cidal concentration (MAC), as the drug has poor activity on non-replicating bacteria. Inversely, MXF, a drug that has demonstrated promising activity against non-replicating bacteria in vitro but failed to shorten treatments in clinical trials (Li et al., 2015) demonstrated sub-cidal concentrations in regions of cavities containing non-replicating bacteria (Prideaux et al., 2015). The two drugs most active against persisters in this study, RIF and PZA, achieved cidal concentrations within relevant compartments of the studied lesions (Prideaux et al., 2015). These results indicate that bacteria exhibiting drug-specific persistence mechanisms may still contribute to the rise of drug resistant bacteria in compartments of relative monotherapy. Of particular concern are persisters to RIF and PZA or the persisters generated to second line therapies used to treat RIF and PZA drug resistant strains, discussed later this section.
The development of drug resistant M. tuberculosis can be considered a stepwise process (Allue-Guardia et al., 2021). Before acquiring “high-level” resistance mutations typically associated with clinical strains of M. tuberculosis, strains may first accrue “low-level” resistance mutations that enable the bacteria to survive higher concentrations of antibiotics before undergoing cell death (Safi et al., 2013). The importance of low-level resistance to the pathogenesis of M. tuberculosis, has been discussed well in a review on the evolution of antibiotic resistance in M. tuberculosis (Fonseca et al., 2015). Scientists studying the development of EMB resistance in clinical isolates suggest that these low-level resistant mutants are preferentially selected in patients exposed to sub-therapeutic drug concentrations (Safi et al., 2013). These low-level resistance mutations are typically thought to be associated with efflux pump systems (Machado et al., 2012). As the strains accumulate low-level resistance mutations, they provide a background for high-level resistant mutants to arise from (Martins et al., 2009). Aside from the upregulation of efflux pumps, other mutations that increase antibiotic tolerance have been implicated in the development of high-level drug resistant M. tuberculosis (Allue-Guardia et al., 2021). Two such mutations include transcription factor prpR and the gene encoding glycerol-3-kinase, glpK, which have been demonstrated to promote drug tolerance to clinically relevant antimycobacterial drugs (Hicks et al., 2018; Bellerose et al., 2019). These examples demonstrate the role that persistence could be playing in the development of drug resistant M. tuberculosis.
Among persister populations may exist low-level resistant mutations that would otherwise not survive if not for the protection offered by persistence. As time goes on, in some patient’s compliance decreases (Jin et al., 2008), in others their metabolism induces sub-therapeutic drug levels (Kimerling et al., 1998), or in some lesions drug concentrations poorly penetrate compartments of lesions (Prideaux et al., 2015) providing the opportunity for either dynamic persistence (Wakamoto et al., 2013; Javid et al., 2014; Zhu et al., 2018) or low-level resistance mutations to exert their survival benefit. Subsequent growth leads to accumulation of low-level resistance or drug tolerance mutations until eventually strains are fully drug resistant. Once fully drug resistant, strains begin to undergo compensatory mutations to reduce the fitness cost of preliminary resistance mutations, resulting in clinically relevant drug resistant strains (Comas et al., 2012). The concept of compensatory mutations is reviewed well elsewhere (Castro et al., 2020). The bacterial mechanisms discussed in previous sections result in greater bacterial survival to monotherapy and therefore increased risk of spontaneous drug resistance, further underscoring the need for multidrug therapy when treating M. tuberculosis.
Once drug resistant organisms develop, the most concerning strains are those resistant to RIF and PZA. So long as strains are susceptible to these two drugs, treatment times remain between 6-9 months (Mase and Chorba, 2019). PZA resistant but RIF susceptible strains require 9-month treatment, RIF resistant but PZA susceptible strains require 12–18-month treatment, and strains resistant to both of these drugs require 18-month treatment, the same time required prior to the discovery of these two drugs (Mase and Chorba, 2019). Development of new drugs for M. tuberculosis has been slow, with only 3 new drugs, pretomanid, delamanid, and bedaquiline being approved in the last 40 years (Murray et al., 2015). Two of these drugs, bedaquiline and delamanid, have demonstrated activity against dormant bacteria (Koul et al., 2008; Chen et al., 2017). These drugs are typically reserved for multidrug resistant cases, but as evidenced by the treatment times of multidrug resistant bacteria, neither of these have demonstrated the same impact on treatment times as RIF and PZA (Mase and Chorba, 2019).
While we are unaware of studies comparing treatment outcomes of INH monoresistant (INHR) M. tuberculosis directly to RIF monoresistant (RIFR) M. tuberculosis in the same patient populations, studies of these monoresistant strains have been carried out in separate patient populations. In the following paragraph we discuss the general trend that patient outcomes for RIFR M. tuberculosis are worse than those of INHR M. tuberculosis. Given the knowledge that RIF has greater activity on persistent M. tuberculosis than INH, we suggest that the reason for these worse outcomes can be attributed to the reduced killing of persistent bacteria that RIF otherwise provides.
In 2019 a meta-analysis found the success rate for drug susceptible M. tuberculosis infections was 80.1%, multidrug resistant M. tuberculosis was 58.4%, and extensively drug resistant M. tuberculosis was 27.1% (Chaves Torres et al., 2019). Success was defined as patients that fit the criteria for “cure” or “treatment completion”. In one retrospective cohort analysis RIF monoresistance was found to occur less than INH monoresistance with 178 cases compared to 3469 (Prach et al., 2013). In this study it was concluded that compared to drug susceptible strains, patients with RIF resistant M. tuberculosis were twice as likely to die (Prach et al., 2013). Another study of 39 patients with RIF resistant M. tuberculosis identified only 20 patients that were cured. Of the 39 patients only 30 could be assessed for outcome as the other 9 had either died or been lost to follow up (Meyssonnier et al., 2014). In a study of 165 patients with INH resistant M. tuberculosis 140 had treatment success, while 12 had an unsuccessful outcome (Romanowski et al., 2017). The issue of RIF monoresistance was reviewed well by Malenfant and Brewer in 2021 (Malenfant and Brewer, 2021).
Given the heterogeneity of patient populations across these studies, it is difficult to draw conclusions about the outcomes of RIF monoresistant M. tuberculosis compared directly to other monoresistant strains. However, from the studies presented here it appears that patients with INHR M. tuberculosis experience more positive outcomes than those with RIFR M. tuberculosis. If further analyses were carried out that validated this trend that RIF resistance leads to worse outcomes than other resistances, this could illustrate further the importance of persisters and RIF’s role in killing them to patient outcomes. Persistent bacteria that would otherwise be killed by RIF may serve as a reservoir for the rise of multidrug resistant strains in compliant patients, as discussed above. In lieu of this line of experimentation, it is evident that rifampicin resistant strains require the longest treatment times (Mase and Chorba, 2019). It has been documented that longer treatment regimens have a lower compliance rate than shorter treatment regimens, and as discussed low adherence can lead to drug resistant populations (Jin et al., 2008).
It is common for persisters to be deemed or implied to be ‘multidrug tolerant’ (Keren et al., 2004b; Willenborg et al., 2014). In this context, multidrug tolerance means that when bacteria are grown, split into different aliquots, and treated with a range of different drugs, each aliquot demonstrates a persister population. Though persister populations are more and more frequently referred to as heterogenous, descriptions such as these paint a less clear picture. In two such studies a range of drugs were administered to Streptococcus suis and E. coli (Keren et al., 2004b; Willenborg et al., 2014). Analysis of kill curves performed in these two studies reveal persister populations that vary depending on the drug used in the experiment. As discussed earlier in this review, these populations are likely a mixture of pre-formed stochastic persisters and triggered persisters formed in response to the applied antibiotic stress. However, since this is still up for debate its informative to analyze these results under both paradigms.
Going stepwise, the first analysis comes through the lens of stochastic persistence. Since these bacteria are obtained from the same culture prior to antibiotic treatment, it is safe to assume the level of persistent bacteria should be the same prior to treatment. In the case of the E. coli experiment (Keren et al., 2004b), the clearest difference is between ofloxacin, a DNA gyrase inhibitor, and tobramycin, a bacterial ribosome inhibitor. Treatment with ofloxacin revealed a persister population approximately 2-3x in size to the tobramycin treated population (Keren et al., 2004b). Assuming pre-existing persister populations of the same size, this indicates that the true persister population size is closer to that revealed by ofloxacin and that the population observed after tobramycin treatment is a smaller portion of that larger population. This begs the question, is something about these bacteria different? Why did these bacteria persist to this point in the presence of ofloxacin, but not in the presence of tobramycin? This could be attributed to the efficacy of the drug. For example, RIF has a greater capacity to kill M. tuberculosis persister cells, but has very little impact on persister cells of S. suis or Borrelia burgdorferi (Keren et al., 2004b; Hu et al., 2015; Feng et al., 2015).
To illustrate the concepts discussed here, we generated the schematic seen in Figure 2. This schematic illustrates a susceptible population compared to persistent populations observed to two different drugs, x and y (Figure 2A). In this case, assuming the persistent bacteria are the same or persistent via the same mechanism, tobramycin may be demonstrating a higher efficiency in killing these persistent bacteria than ofloxacin (Figure 2B1). The alternative explanation is that these bacteria are different, and that the populations revealed are engaging in two distinct mechanisms that confer persistence to one drug or the other (Figure 2B3), possibly with some overlap (Figure 2B2).
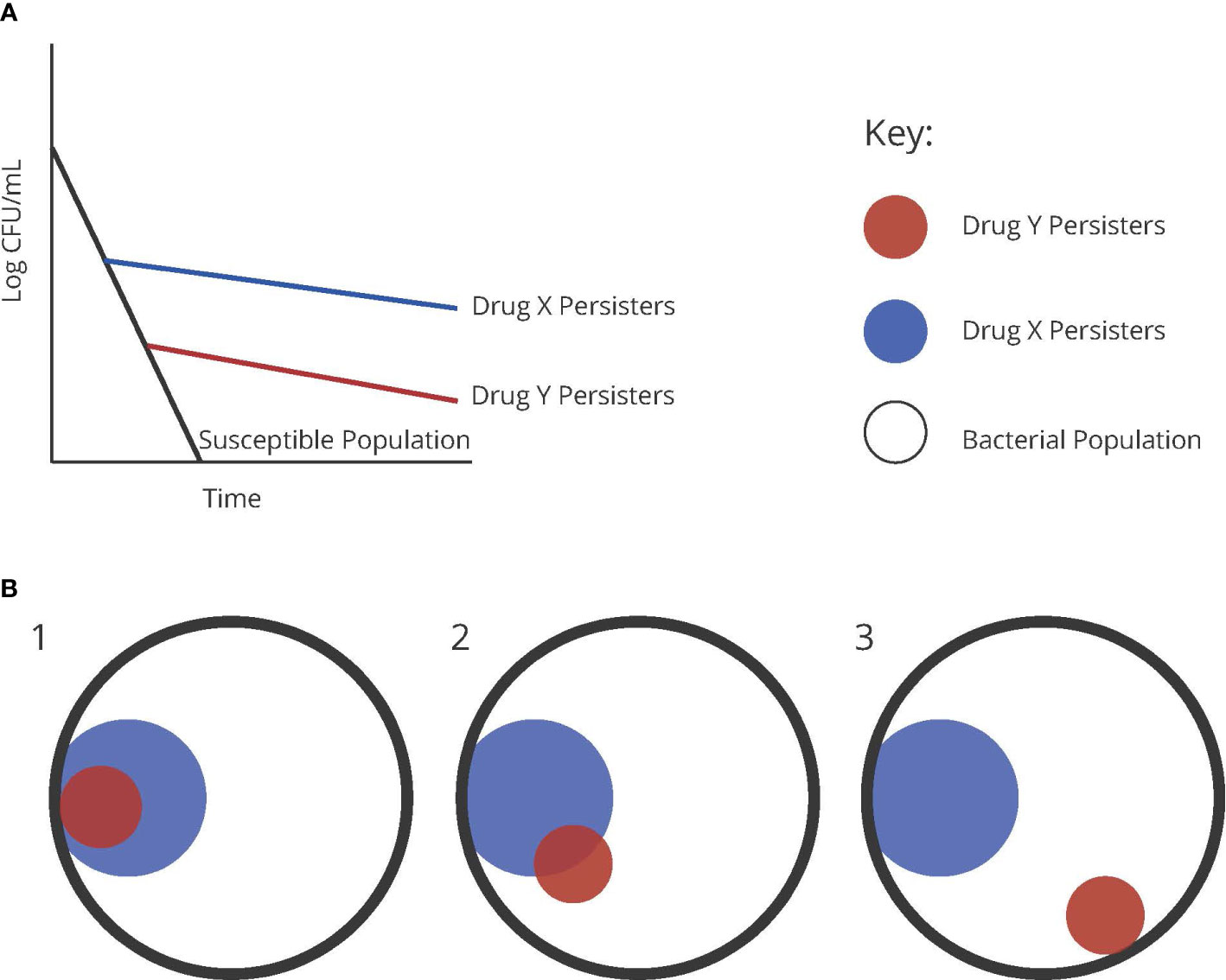
Figure 2 (A) Graphical representation of persister populations to two theoretical drugs, x and y, compared to a susceptible population of bacteria. (B) Diagrams representing the possible distributions of heterogenous persister populations of two different antibiotics.
Through another lens, this higher amount of persisters in the ofloxacin group could be a result of triggered persistence, since ofloxacin and the similar drug ciprofloxacin have both been shown to induce persistence in metabolically active cells (Dorr et al., 2010; Goormaghtigh and Van Melderen, 2019). This explanation continues to beg the question whether triggered persisters are the same as the pre-existing persisters. The only way to discern this concept of cross-persistence is to sequentially treat a population with one drug and then the other, assessing what is commonly referred to as ‘cross-tolerance’.
This line of experimentation was carried out on E. coli with the drugs ciprofloxacin, ampicillin, rifampin, streptomycin, tetracycline, and levofloxacin (Wiuff et al., 2005; Singh et al., 2010). The results revealed that while some drugs conferred cross tolerance, this cross tolerance was not observed for all drugs even among those that did exhibit cross tolerance in certain combinations. For example, streptomycin persisters demonstrated cross tolerance to ampicillin, but not ciprofloxacin. Ciprofloxacin persisters demonstrated cross tolerance to RIF, but not streptomycin. Rifampin persisters did not exhibit cross tolerance to any drugs (Wiuff et al., 2005). However, even when cross-tolerance was not observed, sterilization did not occur, indicating possible sub-populations within persister populations that were cross-tolerant (Wiuff et al., 2005). Another experiment showed that E. coli persisters to streptomycin, ampicillin, and levofloxacin seem to all exhibit cross tolerance (Singh et al., 2010). These results suggest that persisters to one drug are not the same as persisters to another drug and may arrive in the persister state via unique mechanisms. In fact, RelE homologues, a toxin module found in E. coli and M. tuberculosis have demonstrated differential impacts on persistence to different antimycobacterial drugs in M. tuberculosis (Singh et al., 2010).
Toxin-antitoxin systems are used by many bacterial species. In these systems, bacteria synthesize both a toxin capable of suppressing cellular processes, as well as an anti-toxin capable of binding the toxin and preventing its effects (Boldrin et al., 2020). The anti-toxin tends to degrade much more rapidly than the toxin, requiring active upkeep to maintain antitoxin levels. DNA toxins can impact DNA gyrase, polymerase, or even cleave DNA directly (Bernard and Couturier, 1992; Critchlow et al., 1997). RNA toxins can degrade tRNAs and mRNAs or impair their activity through chemical modifications such as acetylation and phosphorylation (Mets et al., 2017; Culviner and Laub, 2018; Mets et al., 2019). These systems are covered well in a review on toxin-antitoxin systems (Jurėnas et al., 2022).
RelE is an mRNA toxin that cleaves mRNA entering the ribosomal translation site (Pedersen et al., 2003). In the case of RelE homologues in M. tuberculosis, RelE2 overexpression increased bacterial persistence to RIF, while its deletion decreased persistence to RIF. RelE3 overexpression increased persistence to INH, while its deletion decreased persistence to INH, but not EMB (Singh et al., 2010). When the authors assessed M. tuberculosis for cross-tolerance they found no cross-tolerance between INH, RIF, and PZA (Singh et al., 2010).
Other investigators Grant et al. observed persister populations in M. smegmatis and M. tuberculosis to drug combinations INH+RIF, Ciprofloxacin (CIP) +RIF, and OFX+INH, suggesting the presence of cross-persistence to these combinations. Interestingly, the persister population generated by 7 days of treatment with a combination of CIP and INH demonstrated cross-tolerance to RIF (Grant et al., 2012). The authors found accelerated killing of these persistent populations when cultures were maintained at a high level of dissolved oxygen (Grant et al., 2012). Though this condition would be difficult to maintain in hypoxic granulomas, this result indicates that an underlying process can be activated by the presence of oxygen in these cross-tolerant populations, either awakening them from the persister state or promoting death of persistent bacteria.
Though most of these studies of persistence have been carried out in vitro, experiments performed by Bellerose et al. have shown signs of multidrug-persistence in vivo (Bellerose et al., 2019). These authors inoculated mice with wild-type M. tuberculosis as well as a glpK mutant. As mentioned in the previous section, this glpK mutant is unable to phosphorylate glycerol, making the mutant incapable of utilizing glycerol metabolism in the host. The authors demonstrated that this mutant does not exhibit a growth disadvantage in the lungs of mice up to 40 days post infection but when exposed to antimycobacterial PZA, glpK mutants in the lungs demonstrated a significant survival advantage compared to the wild-type. Further, when mice were infected with M. tuberculosis and treated with INH, EMB, RIF, PZA, the authors identified varying amounts of surviving bacteria. While these surviving bacteria could be interpreted to be persistent, it is difficult to make that claim given bacterial counts were only obtained at a single time point after treatment, so no biphasic killing can be observed (Bellerose et al., 2019). More interestingly, when all four drugs were given in combination, the amount of surviving bacteria most closely resembled the amount surviving after PZA monotherapy (Bellerose et al., 2019). This may be indicative that the mechanism of persistence to PZA that glpK mutants undergo in vivo demonstrate cross-persistence to other drugs in the standard M. tuberculosis drug regimen. One limitation to this interpretation is the fact that drugs were administered as a combination therapy, making the potential mechanisms of persister cell formation much more complex than if drugs were administered in sequence (Bellerose et al., 2019).
It is logical to think that drug-specific INH persisters that are actively growing so long as they downregulate KatG would remain susceptible to other antimycobacterials (Wakamoto et al., 2013) and that those with phenotypic resistance to RIF due to alterations in rpoB transcription would remain susceptible to other antimycobacterials (Zhu et al., 2018). It is unclear from any of these studies if the persisters generated will persist when exposed to other antimycobacterials with different mechanisms. Though some investigations begin to show avenues for killing multidrug persistent M. tuberculosis, an additional universal mechanism that is more general to all antibiotics may be occurring in the background of these experiments. It would be very illuminating to identify the presence of one of these general mechanisms, as they may lie at the heart of improving treatment of M. tuberculosis infections.
So far, we discussed that persistent bacteria are relevant for their potential role in M. tuberculosis infection severity, treatment times, and development of drug resistant populations. With this importance in mind, we set out to establish the origin, nature, and heterogeneity of persistent M. tuberculosis.
The origin of persisters is important to consider in the context of an infected patient. Which persisters are already present in a patient because of the various host environments M. tuberculosis finds itself in (Adams et al., 2011; Liu et al., 2016)? Which persisters do we induce when we give treatment? Answering these questions should shape how we configure and administer drug combinations.
The nature of persistent bacteria is important to consider in the evolution of drug resistance. History has already taught us the lesson that M. tuberculosis requires a multidrug chemotherapy and never to add a single drug to a failing regimen. Evidence discussed here reveals that dynamic persistence may play a role in this process of single drug resistance.
The heterogeneity of persistent M. tuberculosis is important to consider in improving treatment of both drug susceptible and drug resistant M. tuberculosis. Given the lack of study on cross-tolerance, it is unclear whether dynamic persistence exists to the multidrug regimens administered to patients. Patient non-compliance aside, it is also of concern whether drugs remain within the therapeutic range for all compliant patients (Kimerling et al., 1998; Pasipanodya et al., 2012; Tostmann et al., 2013). Drug concentrations outside the therapeutic range may enable dynamic persistence or regrowth despite the administration of combination therapy. The possibilities are endless, but the data are shallow. If persister populations vary in the presence of different drugs as argued here, then what are the next steps?
As illustrated in numerous examples discussed in this review, RIF appears to be key in the killing of persistent bacteria, with pyrazinamide a few steps behind. In the short term, we suggest that it is imperative to find new compounds that replicate RIF’s success in killing persistent bacteria in order to shorten treatment times and improve outcomes for RIF resistant M. tuberculosis. Salvage of efficacious antibiotics has previously been achieved using adjuvants such as beta-lactamase inhibitors or drug-drug conjugates such as tobramycin-ciprofloxacin (Maiti et al., 1998; Gorityala et al., 2016). One such attempt at a drug-drug conjugate was attempted for RIF by linking it to clofazimine (Saravanan et al., 2021). Though this conjugation did not result in a compound effective against RIF resistant M. tuberculosis, it did show activity at lower concentrations than either of the individual compounds. Whether through an adjuvant, RIF analogues that can impact clinical resister mutations, or new drugs that can kill the same persistent bacteria that RIF can, avenues like these could provide a short-term improvement to RIF resistant M. tuberculosis.
In the longer term it is imperative to identify general mechanisms of persistence in M. tuberculosis, particularly those mechanisms of persistence present in RIF persisters. Any new treatment capable of killing RIF persisters may hold promise to shorten the current treatment of drug susceptible M. tuberculosis and provide alternatives in the case of RIF resistance. Before these treatments can be developed, mechanisms of persistence must be further identified and currently known mechanisms of persister enrichment need to be screened for multidrug tolerance to ensure redundancy is avoided.
Finally, we return to our initial question. Current knowledge on this topic paints a very multifaceted picture of persistence. Convincing data exist to support the existence of some general mechanism of persistence that leads to a basal level of stochastic persisters in a population (Shan et al., 2017; Manuse et al., 2021). Convincing data exist that support the process of triggered persistence, suggesting a more adaptive mechanism to arrive in the persister state (Dorr et al., 2010; Goormaghtigh and Van Melderen, 2019). As argued here, some experiments suggest that persistent bacteria engage in different mechanisms of persistence to survive the presence of different drugs (Singh et al., 2010). Of the data that exist, it appears persistent bacteria to one drug do not necessarily persist in the presence of another (Wiuff et al., 2005; Singh et al., 2010). To our knowledge, neither data exist to suggest stochastic and triggered persisters are composed of the same subpopulation of bacteria, nor does data exist to suggest INH and RIF persisters are composed of the same subpopulation of bacteria. Therefore, based on the current information available, we must assert that no, not all antibiotic persisters are created equal.
MWS, CVM, and MB conceptualized and designed the review. MS wrote the first draft of the manuscript. All authors contributed to manuscript revision, read, and approved the submitted version.
This work was supported by Potts Memorial Foundation and NIH grants T32AI007501 (supporting MWS) and R01AI139465 (supporting CVM and MB).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Adams, K. N., Takaki, K., Connolly, L. E., Wiedenhoft, H., Winglee, K., Humbert, O., et al. (2011). Drug tolerance in replicating mycobacteria mediated by a macrophage-induced efflux mechanism. Cell 145 (1), 39–53. doi: 10.1016/j.cell.2011.02.022
Aldridge, B. B., Fernandez-Suarez, M., Heller, D., Ambravaneswaran, V., Irimia, D., Toner, M., et al. (2012). Asymmetry and aging of mycobacterial cells lead to variable growth and antibiotic susceptibility. Science 335 (6064), 100–104. doi: 10.1128/mSystems.00396-20
Allue-Guardia, A., Garcia, J. I., Torrelles, J. B. (2021). Evolution of drug-resistant mycobacterium tuberculosis strains and their adaptation to the human lung environment. Front. Microbiol. 12, 612675. doi: 10.3389/fmicb.2021.612675
Balaban, N.Q., Merrin, J., Chait, R., Kowalik, L., Leibler, S. (2004). Bacterial persistence as a phenotypic switch. Science 305 (5690), 1622–1625. doi: 10.1126/science.1099390
Balaban, N.Q., Helaine, S., Lewis, K., Ackermann, M., Aldridge, B., Andersson, D.I., et al. (2019). Definitions and guidelines for research on antibiotic persistence. Nat. Rev. Microbiol. 17 (7), 441–448. doi: 10.1038/s41579-019-0196-3
Bellerose, M. M., Baek, S.H., Huang, C.C., Moss, C. E., Koh, E. I., Proulx, M. K., et al. (2019). Common variants in the glycerol kinase gene reduce tuberculosis drug efficacy. mBio 10 (4), 1–8. doi: 10.1128/mBio.00663-19
Bernard, P., Couturier, M. (1992). Cell killing by the f plasmid CcdB protein involves poisoning of DNA-topoisomerase II complexes. J. Mol. Biol. 226 (3), 735–745. doi: 10.1016/0022-2836(92)90629-X
Bigger, J. (1944). Treatment of staphylococcal infections with penicillin by intermittent sterilisation. Lancet 244 (6320), 497–500. doi: 10.1016/S0140-6736(00)74210-3
Boldrin, F., Provvedi, R., Cioetto Mazzabo, L., Segafreddo, G., Manganelli, R. (2020). Tolerance and persistence to drugs: A main challenge in the fight against mycobacterium tuberculosis. Front. Microbiol. 11, 1924. doi: 10.3389/fmicb.2020.01924
Brauner, A., Fridman, O., Gefen, O., Balaban, N. Q. (2016). Distinguishing between resistance, tolerance and persistence to antibiotic treatment. Nat. Rev. Microbiol. 14 (5), 320–330. doi: 10.1038/nrmicro.2016.34
Bretl, D. J., Demetriadou, C., Zahrt, T. C. (2011). Adaptation to environmental stimuli within the host: two-component signal transduction systems of mycobacterium tuberculosis. Microbiol. Mol. Biol. Rev. 75 (4), 566–582. doi: 10.1128/MMBR.05004-11
Castro, R. A. D., Borrell, S., Gagneux, S. (2020). The within-host evolution of antimicrobial resistance in mycobacterium tuberculosis. FEMS Microbiol. Rev. 45 (4), 1–24. doi: 10.1093/femsre/fuaa071
Chaves Torres, N. M., Quijano Rodriguez, J. J., Porras Andrade, P.S., Arriaga, M. B., Netto, E. M. (2019). Factors predictive of the success of tuberculosis treatment: A systematic review with meta-analysis. PloS One 14 (12), e0226507. doi: 10.1371/journal.pone.0226507
Chen, X., Hashizume, H., Tomishige, T., Nakamura, I., Matsuba, M., Fujiwara, M., et al. (2017). Delamanid kills dormant mycobacteria in vitro and in a Guinea pig model of tuberculosis. Antimicrob. Agents Chemother. 61 (6), 1–11. doi: 10.1128/AAC.02402-16
Cohen, N. R., Lobritz, M. A., Collins, J. J. (2013). Microbial persistence and the road to drug resistance. Cell Host Microbe 13 (6), 632–642. doi: 10.1016/j.chom.2013.05.009
Comas, I., Borrell, S., Roetzer, A., Rose, G., Malla, B., Kato-Maeda, M., et al. (2012). Whole-genome sequencing of RIF-resistant mycobacterium tuberculosis strains identifies compensatory mutations in RNA polymerase genes. Nat. Genet. 44 (1), 106–110. doi: 10.1038/ng.1038
Connolly, L. E., Edelstein, P. H., Ramakrishnan, L. (2007). Why is long-term therapy required to cure tuberculosis? PloS Med. 4 (3), e120. doi: 10.1371/journal.pmed.0040120
Corona, F., Martinez, J. L. (2013). Phenotypic resistance to antibiotics. Antibiot (Basel) 2 (2), 237–255. doi: 10.3390/antibiotics2020237
Critchlow, S. E., O'dea, M. H., Howells, A. J., Couturier, M., Gellert, M., Maxwell, A., et al. (1997). The interaction of the f plasmid killer protein, CcdB, with DNA gyrase: induction of DNA cleavage and blocking of transcription. J. Mol. Biol. 273 (4), 826–839. doi: 10.1006/jmbi.1997.1357
Culviner, P. H., Laub, M. T. (2018). Global analysis of the e. coli toxin MazF reveals widespread cleavage of mRNA and the inhibition of rRNA maturation and ribosome biogenesis. Mol. Cell 70 (5), 868–880.e10. doi: 10.1016/j.molcel.2018.04.026
Dahl, J. L., Kraus, C. N., Boshoff, H. I., Doan, B., Foley, K., Avarbock, D., et al. (2003). The role of RelMtb-mediated adaptation to stationary phase in long-term persistence of mycobacterium tuberculosis in mice. Proc. Natl. Acad. Sci. U.S.A. 100 (17), 10026–10031. doi: 10.1073/pnas.1631248100
Dorr, T., Vulic, M., Lewis, K. (2010). Ciprofloxacin causes persister formation by inducing the TisB toxin in escherichia coli. PloS Biol. 8 (2), e1000317. doi: 10.1371/journal.pbio.1000317
Dutta, N. K., Klinkenberg, L.G., Vazquez, M. J., Segura-Carro, D., Colmenarejo, G., Ramon, F., et al. (2019). Inhibiting the stringent response blocks mycobacterium tuberculosis entry into quiescence and reduces persistence. Sci. Adv., 5(3) eaav2104. doi: 10.1126/sciadv.aav2104
Feng, J., Auwaerter, P. G., Zhang, Y. (2015). Drug combinations against borrelia burgdorferi persisters in vitro: eradication achieved by using daptomycin, cefoperazone and doxycycline. PloS One 10 (3), e0117207. doi: 10.1371/journal.pone.0117207
Fonseca, J. D., Knight, G. M., McHugh, T. D. (2015). The complex evolution of antibiotic resistance in mycobacterium tuberculosis. Int. J. Infect. Dis. 32, 94–100. doi: 10.1016/j.ijid.2015.01.014
Goormaghtigh, F., Van Melderen, L. (2019). Single-cell imaging and characterization of escherichia coli persister cells to ofloxacin in exponential cultures. Sci. Adv. 5 (6), eaav9462. doi: 10.1126/sciadv.aav9462
Goossens, S. N., Sampson, S. L., Van Rie, A. (2020). Mechanisms of drug-induced tolerance in mycobacterium tuberculosis. Clin. Microbiol. Rev. 34 (1), 1–21. doi: 10.1128/CMR.00141-20
Gorityala, B. K., Guchhait, G., Fernando, D. M., Deo, S., Mckenna, S.A., Zhanel, G. G, et al. (2016). Adjuvants based on hybrid antibiotics overcome resistance in pseudomonas aeruginosa and enhance fluoroquinolone efficacy. Angew Chem. Int. Ed Engl. 55 (2), 555–559. doi: 10.1002/anie.201508330
Grant, S. S., Kaufmann, B B., Chand, N. S., Haseley, N., Hung, D. T. (2012). Eradication of bacterial persisters with antibiotic-generated hydroxyl radicals. Proc. Natl. Acad. Sci. U.S.A. 109 (30), 12147–12152. doi: 10.1073/pnas.1203735109
Hu, Y., Mangan, J. A., Dhillon, J., Sole, K. M., Mitchison, D. A., Butcher, P.D., et al. (2018). Clinically prevalent mutations in mycobacterium tuberculosis alter propionate metabolism and mediate multidrug tolerance. Nat. Microbiol. 3 (9), 1032–1042. doi: 10.1038/s41564-018-0218-3
Hu, Y., Liu, A., Ortega-Muro, F., Alameda-Martin, L., Mitchison, D., Coates, A. (2000). Detection of mRNA transcripts and active transcription in persistent mycobacterium tuberculosis induced by exposure to rifampin or Pyrazinamide. J. Bacteriol 182 (22), 6358–6365. doi: 10.1128/JB.182.22.6358-6365.2000
Hu, Y., Liu, A., Ortega-Muro, F., Alameda-Martin, L., Mitchison, D., Coates, A, et al. (2015). High-dose RIF kills persisters, shortens treatment duration, and reduces relapse rate in vitro and in vivo. Front. Microbiol. 6, 641. doi: 10.3389/fmicb.2015.00641
Jain, P., Weinrick, B. C., Kalivoda, E. J., Yang, H., Munsamy, V., Vilcheze, C, et al. (2016). Dual-reporter mycobacteriophages (Phi2DRMs) reveal preexisting mycobacterium tuberculosis persistent cells in human sputum. mBio 7 (5), 1–13. doi: 10.1128/mBio.01023-16
Javid, B., Sorrentino, F., Toosky, M., Zheng, W., Pinkham, J. T., Jain, N., et al. (2014). Mycobacterial mistranslation is necessary and sufficient for RIF phenotypic resistance. Proc. Natl. Acad. Sci. U.S.A. 111 (3), 1132–1137. doi: 10.1073/pnas.1317580111
Jin, J., Sklar, G. E., Min Sen Oh, V., Chuen Li, S. (2008). Factors affecting therapeutic compliance: A review from the patient's perspective. Ther. Clin. Risk Manag 4 (1), 269–286. doi: 10.2147/tcrm.s1458
Jurėnas, D., Fraikin, N., Goormaghtigh, F., Van Melderen, L.. (2022). Biology and evolution of bacterial toxin–antitoxin systems. Nat. Rev. Microbiol 20, 335–350. doi: 10.1038/s41579-021-00661-1
Keren, I., Kaldalu, N., Spoering, A., Wang, Y., Lewis, K. (2004a). Persister cells and tolerance to antimicrobials. FEMS Microbiol. Lett. 230 (1), 13–18. doi: 10.1016/S0378-1097(03)00856-5
Keren, I., Shah, D., Spoering, A., Kaldalu, N., Lewis, K. (2004b). Specialized persister cells and the mechanism of multidrug tolerance in escherichia coli. J. Bacteriol 186 (24), 8172–8180. doi: 10.1128/JB.186.24.8172-8180.2004
Kimerling, M. E., Phillips, P., Patterson, P., Hall, M., Robinson, C. A., Dunlap, N. E. (1998). Low serum antimycobacterial drug levels in non-HIV-infected tuberculosis patients. Chest 113 (5), 1178–1183. doi: 10.1378/chest.113.5.1178
Koul, A., Vranckx, L., Dendouga, N., Balemans, W., Van Den Wyngaert, I., Vergauwen, K., et al. (2008). Diarylquinolines are bactericidal for dormant mycobacteria as a result of disturbed ATP homeostasis. J. Biol. Chem. 283 (37), 25273–25280. doi: 10.1074/jbc.M803899200
Lei, Q., Wang, H., Zhao, Y., Dang, L., Zhu, C., Lv, X., et al. (2019). Determinants of serum concentration of first-line anti-tuberculosis drugs from China. Medicine 98 (41), e17523. doi: 10.1097/MD.0000000000017523
Lewis, K. (2007). Persister cells, dormancy and infectious disease. Nat. Rev. Microbiol. 5 (1), 48–56. doi: 10.1038/nrmicro1557
Li, S. Y., Irwin, S. M., Converse, P.J., Mdluli, K. E., Lenaerts, A. J., Nuermberger, E. L. (2015). Evaluation of moxifloxacin-containing regimens in pathologically distinct murine tuberculosis models. Antimicrob. Agents Chemother. 59 (7), 4026–4030. doi: 10.1128/AAC.00105-15
Liu, Y., Tan, S., Huang, L., Abramovitch, R. B., Rohde, K. H., Zimmerman, M. D., et al. (2016). Immune activation of the host cell induces drug tolerance in mycobacterium tuberculosis both in vitro and in vivo. J. Exp. Med. 213 (5), 809–825. doi: 10.1084/jem.20151248
Machado, D., Couto, I., Perdigao, J., Rodrigues, L., Portugal, I., Baptista, P., et al. (2012). Contribution of efflux to the emergence of INH and multidrug resistance in mycobacterium tuberculosis. PloS One 7 (4), e34538. doi: 10.1371/journal.pone.0034538
Maiti, S. N., Phillips, O. A., Micetich, R. G., Livermore, D. M. (1998). Beta-lactamase inhibitors: agents to overcome bacterial resistance. Curr. Med. Chem. 5 (6), 441–456. doi: 10.2174/0929867305666220319110127
Malenfant, J. H., Brewer, T. F. (2021). RIF mono-resistant tuberculosis–a review of an uncommon but growing challenge for global tuberculosis control. Open Forum Infect. Dis. 8 (2), 1–6. doi: 10.1093/ofid/ofab018
Manuse, S., Shan, Y., Canas-Duarte, S. J., Bakshi, S., Sun, W. S., Mori, H., et al. (2021). Bacterial persisters are a stochastically formed subpopulation of low-energy cells. PloS Biol. 19 (4), e3001194. doi: 10.1371/journal.pbio.3001194
Martins, A., Iversen, C., Rodrigues, L., Spengler, G., Ramos, J., Kern, W. V., et al. (2009). An AcrAB-mediated multidrug-resistant phenotype is maintained following restoration of wild-type activities by efflux pump genes and their regulators. Int. J. Antimicrob. Agents 34 (6), 602–604. doi: 10.1016/j.ijantimicag.2009.06.029
Mase, S. R., Chorba, T. (2019). Treatment of drug-resistant tuberculosis. Clin. Chest Med. 40 (4), 775–795. doi: 10.1016/j.ccm.2019.08.002
McCune, R. M., Feldmann, F. M., Lambert, H. P., Mcdermott, W. (1966). Microbial persistence. i. the capacity of tubercle bacilli to survive sterilization in mouse tissues. J. Exp. Med. 123 (3), 445–468. doi: 10.1084/jem.123.3.445
McCune, R. M., Feldmann, F. M., McDermott, W. (1966). Microbial persistence. II. characteristics of the sterile state of tubercle bacilli. J. Exp. Med. 123 (3), 469–486. doi: 10.1084/jem.123.3.469
Mets, T., Lippus, M., Schryer, D., Liiv, A., Kasari, V., Paier, A., et al. (2017). Toxins MazF and MqsR cleave escherichia coli rRNA precursors at multiple sites. RNA Biol. 14 (1), 124–135. doi: 10.1080/15476286.2016.1259784
Mets, T., Kasvandik, S., Saarma, M., Maivali, U., Tenson, T., Kaldalu, N, et al. (2019). Fragmentation of escherichia coli mRNA by MazF and MqsR. Biochimie 156, 79–91. doi: 10.1016/j.biochi.2018.10.004
Meyssonnier, V., Bui, T.V., Veziris, N., Jarlier, V., Robert, J. (2014). RIF mono-resistant tuberculosis in France: a 2005-2010 retrospective cohort analysis. BMC Infect. Dis. 14, 18. doi: 10.1186/1471-2334-14-18
Moker, N., Dean, C. R., Tao, J. (2010). Pseudomonas aeruginosa increases formation of multidrug-tolerant persister cells in response to quorum-sensing signaling molecules. J. Bacteriol 192 (7), 1946–1955. doi: 10.1128/JB.01231-09
Munro, S. A., Lewin, S.A., Smith, H. J., Engel, M. E., Fretheim, A., Volmink, J. (2007). Patient adherence to tuberculosis treatment: a systematic review of qualitative research. PloS Med. 4 (7), e238. doi: 10.1371/journal.pmed.0040238
Murray, J. F. (2004). A century of tuberculosis. Am. J. Respir. Crit. Care Med. 169 (11), 1181–1186. doi: 10.1164/rccm.200402-140OE
Murray, J. F., Schraufnagel, D. E., Hopewell, P. C. (2015). Treatment of tuberculosis. a historical perspective. Ann. Am. Thorac. Soc. 12 (12), 1749–1759. doi: 10.1513/AnnalsATS.201509-632PS
Nahid, P., Dorman, S. E., Alipanah, N., Barry, P. M., Brozek, J. L., Cattamanchi, A., et al. (2016). Executive summary: Official American thoracic Society/Centers for disease control and Prevention/Infectious diseases society of America clinical practice guidelines: Treatment of drug-susceptible tuberculosis. Clin. Infect. Dis. 63 (7), 853–867. doi: 10.1093/cid/ciw566
Orman, M. A., Brynildsen, M. P. (2013). Dormancy is not necessary or sufficient for bacterial persistence. Antimicrob. Agents Chemother. 57 (7), 3230–3239. doi: 10.1128/AAC.00243-13
Pablos-Mendez, A., Knirsch, C. A., Barr, R. G., Lerner, B. H., Frieden, T. R. (1997). Nonadherence in tuberculosis treatment: predictors and consequences in new York city. Am. J. Med. 102 (2), 164–170. doi: 10.1016/S0002-9343(96)00402-0
Pasipanodya, J. G., Srivastava, S., Gumbo, T. (2012). Meta-analysis of clinical studies supports the pharmacokinetic variability hypothesis for acquired drug resistance and failure of antituberculosis therapy. Clin. Infect. Dis. 55 (2), 169–177. doi: 10.1093/cid/cis353
Pedersen, K., Zavialov, A. V., Pavlov, M. Y., Elf, J., Gerdes, K., Ehrenberg, M. (2003). The bacterial toxin RelE displays codon-specific cleavage of mRNAs in the ribosomal a site. Cell 112 (1), 131–140. doi: 10.1016/S0092-8674(02)01248-5
Prach, L. M., Pascopella, L., Barry, P. M., Flood, J., Porco, T. C., Hopewell, P. C., et al. (2013). Rifampin monoresistant tuberculosis and HIV comorbidity in California, 1993-2008: a retrospective cohort study. AIDS 27 (16), 2615–2622. doi: 10.1097/01.aids.0000432445.07437.07
Prideaux, B., Via, L. E., Zimmerman, M. D., Eum, S., Sarathy, J., O'brien, P., et al. (2015). The association between sterilizing activity and drug distribution into tuberculosis lesions. Nat. Med. 21 (10), 1223–1227. doi: 10.1038/nm.3937
Pullan, S. T., Allnutt, J. C., Devine, R., Hatch, K. A., Jeeves, R. E., Hendon-Dunn, C. L., et al. (2016). The effect of growth rate on PZA activity in mycobacterium tuberculosis - insights for early bactericidal activity? BMC Infect. Dis. 16, 205. doi: 10.1186/s12879-016-1533-z
Rego, E. H., Audette, R. E., Rubin, E. J. (2017). Deletion of a mycobacterial divisome factor collapses single-cell phenotypic heterogeneity. Nature 546 (7656), 153–157. doi: 10.1038/nature22361
Richardson, K., Bennion, O. T., Tan, S., Hoang, A. N., Cokol, M., Aldridge, B. B. (2016). Temporal and intrinsic factors of RIF tolerance in mycobacteria. Proc. Natl. Acad. Sci. 113 (29), 8302–8307. doi: 10.1073/pnas.1600372113
Romanowski, K., Chiang, L. Y., Roth, D. Z., Krajden, M., Tang, P., Cook, V. J., et al. (2017). Treatment outcomes for isoniazid-resistant tuberculosis under program conditions in British Columbia, Canada. BMC Infect. Dis. 17 (1), 604. doi: 10.1186/s12879-017-2706-0
Safi, H., Lingaraju, S., Amin, A., Kim, S., Jones, M., Holmes, M., et al. (2013). Evolution of high-level EMB-resistant tuberculosis through interacting mutations in decaprenylphosphoryl-beta-D-arabinose biosynthetic and utilization pathway genes. Nat. Genet. 45 (10), 1190–1197. doi: 10.1038/ng.2743
Saravanan, P., Dusthackeer, V. N. A., Rajmani, R. S., Mahizhaveni, B., Nirmal, C. R., Rajadas, S. E., et al. (2021). Discovery of a highly potent novel RIF analog by preparing a hybrid of the precursors of the antibiotic drugs RIF and clofazimine. Sci. Rep. 11 (1), 1029. doi: 10.1038/s41598-020-80439-2
Sebastian, J., Swaminath, S., Nair, R. R., Jakkala, K., Pradhan, A., Ajitkumar, P. (2017). De Novo emergence of genetically resistant mutants of mycobacterium tuberculosis from the persistence phase cells formed against antituberculosis drugs in vitro. Antimicrob. Agents Chemother. 61 (2), 1–25. doi: 10.1128/AAC.01343-16
Seth, V., Beotra, A., Seth, S. D., Semwal, O. P., Kabra, S., Jain, Y., et al. (1993). Serum concentrations of RIF and isoniazid in tuberculosis. Indian Pediatr. 30 (9), 1091–1098. doi: 10.4269/ajtmh.2009.81.322
Shan, Y., Brown Gandt, A., Rowe, S. E., Deisinger, J. P., Conlon, B.P., Lewis, K. (2017). ATP-dependent persister formation in escherichia coli. mBio 8 (1), 1–14. doi: 10.1128/mBio.02267-16
Singh, R., Barry, C. E., 3rd, Boshoff, H. I. (2010). The three RelE homologs of mycobacterium tuberculosis have individual, drug-specific effects on bacterial antibiotic tolerance. J. Bacteriol 192 (5), 1279–1291. doi: 10.1128/JB.01285-09
Tostmann, A., Mtabho, C. M., Semvua, H. H., Van Den Boogaard, J., Kibiki, G.S., Boeree, M. J., et al. (2013). Pharmacokinetics of first-line tuberculosis drugs in Tanzanian patients. Antimicrob. Agents Chemother. 57 (7), 3208–3213. doi: 10.1128/AAC.02599-12
Vilcheze, C., Hartman, T., Weinrick, B., Jacobs, W. R., Jr. (2013). Mycobacterium tuberculosis is extraordinarily sensitive to killing by a vitamin c-induced fenton reaction. Nat. Commun. 4, 1881. doi: 10.1038/ncomms2898
Vilcheze, C., Hartman, T., Weinrick, B., Jain, P., Weisbrod, T. R., Leung, L. W., et al. (2017). Enhanced respiration prevents drug tolerance and drug resistance in mycobacterium tuberculosis. Proc. Natl. Acad. Sci. U.S.A. 114 (17), 4495–4500. doi: 10.1073/pnas.1704376114
Volzing, K. G., Brynildsen, M. P. (2015). Stationary-phase persisters to ofloxacin sustain DNA damage and require repair systems only during recovery. mBio 6 (5), e00731–e00715. doi: 10.1128/mBio.00731-15
Wakamoto, Y., Dhar, N., Chait, R., Schneider, K., Signorino-Gelo, F., Leibler, S., et al. (2013). Dynamic persistence of antibiotic-stressed mycobacteria. Science 339 (6115), 91–95. doi: 10.1126/science.1229858.
Warner, D. F., Mizrahi, V. (2006). Tuberculosis chemotherapy: the influence of bacillary stress and damage response pathways on drug efficacy. Clin. Microbiol. Rev. 19 (3), 558–570. doi: 10.1128/CMR.00060-05
Willenborg, J., Willms, D., Bertram, R., Goethe, R., Valentin-Weigand, P. (2014). Characterization of multi-drug tolerant persister cells in streptococcus suis. BMC Microbiol. 14 (1), 120. doi: 10.1186/1471-2180-14-120
Wiuff, C., Zappala, R. M., Regoes, R. R., Garner, K. N., Baquero, F., Levin, B. R. (2005). Phenotypic tolerance: antibiotic enrichment of noninherited resistance in bacterial populations. Antimicrob. Agents Chemother. 49 (4), 1483–1494. doi: 10.1128/AAC.49.4.1483-1494.2005
Wood, T. K., Knabel, S. J., Kwan, B. W. (2013). Bacterial persister cell formation and dormancy. Appl. Environ. Microbiol. 79 (23), 7116–7121. doi: 10.1128/AEM.02636-13
Xie, Z., Siddiqi, N., Rubin, E. J. (2005). Differential antibiotic susceptibilities of starved mycobacterium tuberculosis isolates. Antimicrob. Agents Chemother. 49 (11), 4778–4780. doi: 10.1128/AAC.49.11.4778-4780.2005
Zahrt, T. C., Deretic, V. (2001). Mycobacterium tuberculosis signal transduction system required for persistent infections. Proc. Natl. Acad. Sci. U.S.A. 98 (22), 12706–12711. doi: 10.1073/pnas.221272198
Zhang, Y., Shi, W., Zhang, W., Mitchison, D. (2014). Mechanisms of PZA action and resistance. Microbiol. Spectr. 2 (4), MGM2–0023-2013. doi: 10.1128/9781555818845.ch24
Zhang, Y., Yew, W. W., Barer, M. R. (2012). Targeting persisters for tuberculosis control. Antimicrob Agents Chemother 56 (5), 2223–2230. doi: 10.1128/AAC.06288-11
Keywords: mycobacteria, tuberculosis, multidrug, persister, persistence, tolerance, resistance
Citation: Shultis MW, Mulholland CV and Berney M (2022) Are all antibiotic persisters created equal? Front. Cell. Infect. Microbiol. 12:933458. doi: 10.3389/fcimb.2022.933458
Received: 30 April 2022; Accepted: 25 July 2022;
Published: 17 August 2022.
Edited by:
Digby Warner, University of Cape Town, South AfricaReviewed by:
Anh K. Lam, Indiana University, United StatesCopyright © 2022 Shultis, Mulholland and Berney. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Michael Berney, bWljaGFlbC5iZXJuZXlAZWluc3RlaW5tZWQuZWR1
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.