
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Cell. Infect. Microbiol. , 02 September 2022
Sec. Bacteria and Host
Volume 12 - 2022 | https://doi.org/10.3389/fcimb.2022.931635
This article is part of the Research Topic Peptidyl-Prolyl Cis/Trans Isomerases (PPIases) in Host-Pathogen Interactions View all 5 articles
 Sreekanth Rajan1
Sreekanth Rajan1 Ho Sup Yoon1,2,3*
Ho Sup Yoon1,2,3*Malaria is one of the most prevalent infectious diseases posing a serious challenge over the years, mainly owing to the emergence of drug-resistant strains, sparking a need to explore and identify novel protein targets. It is a well-known practice to adopt a chemo-genomics approach towards identifying targets for known drugs, which can unravel a novel mechanism of action to aid in better drug targeting proficiency. Immunosuppressive drugs cyclosporin A, FK506 and rapamycin, were demonstrated to inhibit the growth of the malarial parasite, Plasmodium falciparum. Peptidyl prolyl cis/trans isomerases (PPIases), comprising cylcophilins and FK506-binding proteins (FKBPs), the specific target of these drugs, were identified in the Plasmodium parasite and proposed as an antimalarial drug target. We previously attempted to decipher the structure of these proteins and target them with non-immunosuppressive drugs, predominantly on FKBP35. This review summarizes the structural insights on Plasmodium PPIases, their inhibitor complexes and perspectives on drug discovery.
Malaria is one of the world’s most dreadful parasitic diseases affecting millions each year, prevalently in tropical and subtropical regions (Liu et al., 2021; White et al., 2022). This vector-borne disease is caused by a single-celled parasite belonging to the genus Plasmodium, transmitted and spread by the infected female Anopheles mosquitos. Among the many Plasmodium species, five have been identified as responsible for transmitting malaria in humans. Plasmodium falciparum (Pf) is the most infectious, followed by P. vivax (Pv), apart from P. malariae, P. ovale, and P. knowlesi. Vector control (Van De Straat et al., 2022), chemoprophylaxis (Plowe, 2022), antimalarial drugs (Tse et al., 2019), and, recently, a protein-based vaccine (Laurens, 2020) are the presently adopted methods for treating malaria. The complex life cycle of the Plasmodium (Supplementary Figure 1) involving the pathogen (parasite), carrier (mosquito), and the host (human) compounded by the emergence of drug-resistant strains (White, 2004; Balikagala et al., 2021), makes it a challenging organism for drug targeting. The most common malaria drug targets are the parasite proteins such as lactate dehydrogenase, plasmepsin2, dihydrofolate reductase, and others (Mehlin, 2005; Yang et al., 2021). Drug repurposing is an efficient starting point for identifying and characterizing novel targets (Pushpakom et al., 2019). In one such chemo-genomics effort immunosuppressive drugs, cyclosporin A (CsA), FK506 and rapamycin were shown to possess antimalarial properties (Bell et al., 1994), leading to the identification of their target PPIases in the parasite.
CsA, FK506 and rapamycin are well-known high-affinity/specific inhibitors of a family of proteins, peptidyl-prolyl cis/trans isomerases (PPIases) comprising of (a) cyclophilins (CyPs), which bind CsA, (b) FK506-binding proteins (FKBPs), which bind FK506 and rapamycin, and (c) parvulins (Galat, 2003; Harikishore and Yoon, 2015). As the name indicates, PPIases primarily function as chaperone proteins, enabling the conversion of cis to trans configuration of proline, a rate-limiting step in protein folding. Apart from inhibiting the PPIase activity, CsA/FK506 acts as an intermolecular glue between CyP/FKBP and calcineurin (CaN), a Ca2+/calmodulin (CaM)-dependent protein serine/threonine phosphatase, thereby inhibiting CaN activity. This signaling leads to a cascade of downstream events resulting in immunosuppression. On the other hand, rapamycin binds to FKBPs to bridge its interaction with the mammalian target of rapamycin (mTOR), also resulting in immunosuppression. Therefore, PPIases are also called immunophilins, due to the immunosuppressive action exerted by these molecules. While these ligands have been primarily used to treat rejection during organ transplant, they have also been repurposed for eczema, cancer (Hasskarl, 2018) and other clinical applications.
Angus Bell and coworkers showed that CsA, FK506 and rapamycin (Bell et al., 1994) could be repurposed for malaria, as they exhibited in vitro P. falciparum inhibition. Further they also showed that the non-immunosuppressive analogs of FK506 (Monaghan et al., 2005) and CsA (Bell et al., 1996) also have the similar effects, which was considered a promising sign toward drug discovery.
A search of Plasmodium PPIases in the PlasmoDB (Amos et al., 2022) revealed the presence of 2 FKBPs and 11 CyPs (Supplementary Table 1). All of them possess a PPIase catalytic domain, apart from other domains such as Tetratricopeptide repeat (TPR), CaM, indicating that these PPIases may act as chaperones or be part of a multi-protein complex (Supplementary Figure 2). Both PPIases were characterized for their (a) PPIase, (b) CaN inhibition, and (c) chaperone activities to decipher their probable function (Gavigan et al., 2003; Monaghan and Bell, 2005; Bell et al., 2006; Yoon et al., 2007; Marin-Menendez et al., 2012).
CsA and its non-immunosuppressive analogs inhibit the intraerythrocytic growth of P. facliparum. An affinity pulldown on P. facliparum proteins using CsA identified two cyclophilins, PfCyP19A and PfCyP19B, constituting ~1.2% and ~0.5% of the parasite protein, respectively. The mRNA levels of PfCyP19A, PfCyP19B, and PfCyP24, revealed that they are abundant in the intraerythrocytic cytosol (Hirtzlin et al., 1995; Reddy, 1995; Le Roch et al., 2003), predominantly in the middle of the erythrocytic cycle, late trophozoite/schizont and immature (ring) stages (Gavigan et al., 2003; Bell et al., 2006), respectively. Apart from the above, a few other PfCyPs were also identified (Marin-Menendez and Bell, 2011; Marin-Menendez et al., 2012). Sequence comparison of the CsA-interacting residues of human (hs)CyPs with PfCyPs revealed minor variation (Marin-Menendez et al., 2012). The highly conserved Trp121 (hsCyPA-numbering) is found in six of 11 Plasmodium CyPs. Most of the other residues within 4Å from CsA are also mostly conserved among PfCyPs, with exceptions in pCyP72 and pCyP81 (Supplementary Table 2). It was shown that only PfCyP19A and PfCyP19B possessed PPIase activity which was inhibited by CsA with an IC50 of 10nM and 15nM, respectively (Marin-Menendez et al., 2012). Nonetheless, all PfCyPs exhibited chaperone activity, by preventing the aggregation of the model substrate, rhodanese and a few against citrate synthase (Marin-Menendez et al., 2012). The CaN inhibition of PfCyP19 was observed in the presence of CsA (Bell et al., 1994; Gavigan et al., 2003), similar to the hsCyP. Thus, one can conclude that PfCyP19 could be a good target for drug discovery. To this end, the crystal structure of PfCyP19A in complex with CsA (Supplementary Figure 3A), adopts a canonical CyP-fold mainly made up of eight β-strands and two α-helices (Peterson et al., 2000), and comparison revealed negligible differences with its human counterpart (Supplementary Figure 3B). The CsA-interacting residues in PfCyPA are identical to those in hsCyPs, and so are their interactions. Owing to the highly hydrophobic active site, a few non-classical C-H … O hydrogen bonds (Rajan et al., 2013), apart from the hydrogen-bonded and non-polar interactions, were observed (Peterson et al., 2000), which are also conserved in the human counterpart (Supplementary Table 3). In addition, a serine protease-like catalytic triad was identified near the active site, but their mutation did not alter the enzyme’s catalytic properties. Thus, it was concluded that generating specific inhibitors for PfCyPA would be difficult, owing to the indistinguishable active site residues and interactions. We tried to compare the CsA-interacting residues within 4-5Å to identify any residue differences and observed that Thr63 (hsCyPA numbering) mutated to Ser; this could be considered a starting point toward specificity (Supplementary Table 2). Outside the active site region, additional six residues are inserted in the loop between α1 and β3, observed in PfCyp19A and PyCyP24 but not in PyCyP23 and PfCyP87 (Supplementary Figure 3C). The functional relevance, if any, of this insertion still needs to be deciphered and could play a role in their localization or chaperone function.
Extensive studies were done on PfFKBPs compared to PfCyPs for reasons mentioned herein. The 35-kDa FKBP (Braun et al., 2003) was the first identified FKBP in P. falciparum (PfFKBP35), and later highly similar homologues in the other malarial parasites, P. vivax and P. knowlesi, were also observed (Cui et al., 2005; Hall et al., 2005) (Supplementary Table 1). PfFKBP35 is expressed throughout the intraerythrocytic life cycle (Kumar et al., 2005a), predominantly in the cytosol during the ring stage. However, it can translocate into the nucleus in the trophozoites and schizonts stages. PfFKBP35’s localization in the nucleus could be due to the presence of the TPR domain (Alag et al., 2009a) (Supplementary Figure 2), which interacts with PfHsp90 (Kumar et al., 2005a; Kumar et al., 2005b). Functionally, FK506 showed an IC50 of 260nM and 160nM against the PPIase activity of Pf FK506 binding domain35 (PfFKBD35) and PvFKBD35, respectively (Alag et al., 2010), while rapamycin’s IC50 is 480nM against PfFKBD35 (Monaghan and Bell, 2005). In addition, PfFKBP35 also exhibited chaperone activity against model substrates, rhodanese, and citrate synthase. Although FK506 did not inhibit the activity of full-length PfFKBP35 or TPR domain alone, it affected PfFKBD35’s PPIase activity, suggesting that PfFKBD35 has a role in PfFKBP35’s chaperone activity (Monaghan and Bell, 2005). Earlier studies reported that FKBP35 binds to CaN in the absence of FK506 (Kumar et al., 2005b; Monaghan and Bell, 2005; Monaghan et al., 2017). On the other hand, our studies showed that this molecular interaction is negligible in the absence of FK506, which was enhanced when FK506 was added (Yoon et al., 2007). Future studies may help validate the discrepancy.
While the above observations promised to coin PfFKBP35 as a probable drug target, it is the two unique active site residues identified from the sequence-structure comparison that made it an attractive target. The Pf/PvFKBD35 structures in apo and FK506-bound forms were determined using NMR and crystallography (Kang et al., 2008; Kotaka et al., 2008; Alag et al., 2009b; Alag et al., 2010), respectively. The structure adopts a typical FKBP fold, consisting of a six stranded β-sheet and a short α-helix, with an additional β-strand at the N-terminus (Figure 1A). We also successfully determined the substrate-bound structure of FKBP, the first of its kind to our knowledge, in which the critical proline residue of the substrate (ALPF) adopted a cis configuration, overlaying on the pipecolinyl moiety of FK506 (Alag et al., 2013). A comparison of the FK506 interacting residues in hsFKBP12 with Plasmodium FKBD35 indicated that most of them are identical, except for Q53G, H87C, and I90S (hsFKBP12 numbering) (Supplementary Table 4). Of these, the sidechain atoms of Cys106 and Ser109 in the β5-β6 loop of PfFKBD35 were located at a distance of 3.9Å and 5.4Å from the pyranose group of FK506 (Figures 1B, D). A similar observation was also made on the PvFKBD35-FK506 complex, with residues Cys105 and Ser108 (Figures 1C, D). Sequence comparison with all hsFKBPs also revealed that these two residues are unique to Plasmodium FKBP35s (Supplementary Table 4), which is promising as it helps to address the specificity towards the parasite inhibition. To support this, the estimated lower physiological levels of PfFKBP35 (0.05-0.1μM) in the parasite in comparison to hsFKBP12 (3-5μM) (Braun et al., 2003) also aid in achieving this, provided we design non-immunosuppressive inhibitors with higher affinity towards Cys106 and Ser109. With this aim, an in silico pharmacophore approach was adopted to identify small molecules targeting these two residues (Harikishore et al., 2013b). This resulted in a molecule containing purine-like and phenyl-ethyl ring systems linked by a thioacetamide linker, labeled as D44 (Supplementary Table 5). The IC50 of D44 against the PPIase activity of PfFKBD35 and PvFKBD35 was shown to be 132nM and 125nM, respectively. D44 also exhibited in vitro inhibition of P. falciparum 3D7 intraerythrocytic growth with an IC50 of 234nM. It arrested the parasite growth at the schizont stage, similar to FK506 (Harikishore et al., 2013b). D44 is a non-immunosuppressive FKBP35 inhibitor, as observed from the calcineurin phosphatase assay. Based on the above results, we studied the molecular binding of D44 with Pf/PvFKBD35 using NMR titration and X-ray crystallography. NMR revealed that D44 perturbs the essential active site residues, while the co-crystal structures revealed the precise mode of interactions. A few critical active site residues, especially Trp78, swings inwards in the D44-bound structure to compensate the bulkier pipecolinyl moiety in FK506 (Supplementary Figure 4). More importantly, the purine-like ring in D44 can be seen oriented towards Cys106/105 and Ser109/108 in Pf/PvFKBD35, at a distance of 3.6/3.8Å and 4.3/3.7Å, respectively, compared to 3.9/4.0Å and 5.4/8.1Å in the FK506-bound form (Figure 1D). The hydroxyl sidechain group of Ser108 flips towards the purine-like ring of D44 in the PvFKBD35-D44 structure, probably owing to its better resolution. Alternately, we also identified another inhibitor using a ligand-based approach in which adamantyl and pyridine rings were connected by a carbonyl hydrazide linker, which we labelled SRA (Harikishore et al., 2013a) (Supplementary Table 5). This small molecule inhibited the PPIase activity of PfFKBD35 and PvFKBD35, with an IC50 of 83nM and 75nM, respectively, and did not impinge on calcineurin, similar to D44. in vitro parasite inhibition revealed that SRA acts on the trophozoite stage of parasite growth, with an IC50 of 250nM. The crystal structure of PvFKBD35 bound to SRA revealed that the adamantyl and pyridine rings of SRA overlay on to the pipecolinyl and cyclohexyl rings of FK506, respectively. The four-atom linker is stabilized by hydrogen bonds to orient the pyridine ring. The key here is the anchor-like interaction made by the bulky adamantyl moiety, which occupies the base of the active site pocket. It was evident that both these small molecules inhibited the parasite’s growth in the intraerythrocytic stages, where FKBP35 expresses abundantly (Supplementary Figure 1). Recently, another small molecule, [4.3.1]-aza-bicyclic sulfonamide ([4.3.1]-ABS) (Supplementary Table 5), possessed an enzymatic IC50 of 98nM and inhibited the P. falciparum growth with an IC50 of 2.3μM (Pomplun et al., 2018) (Table 1).
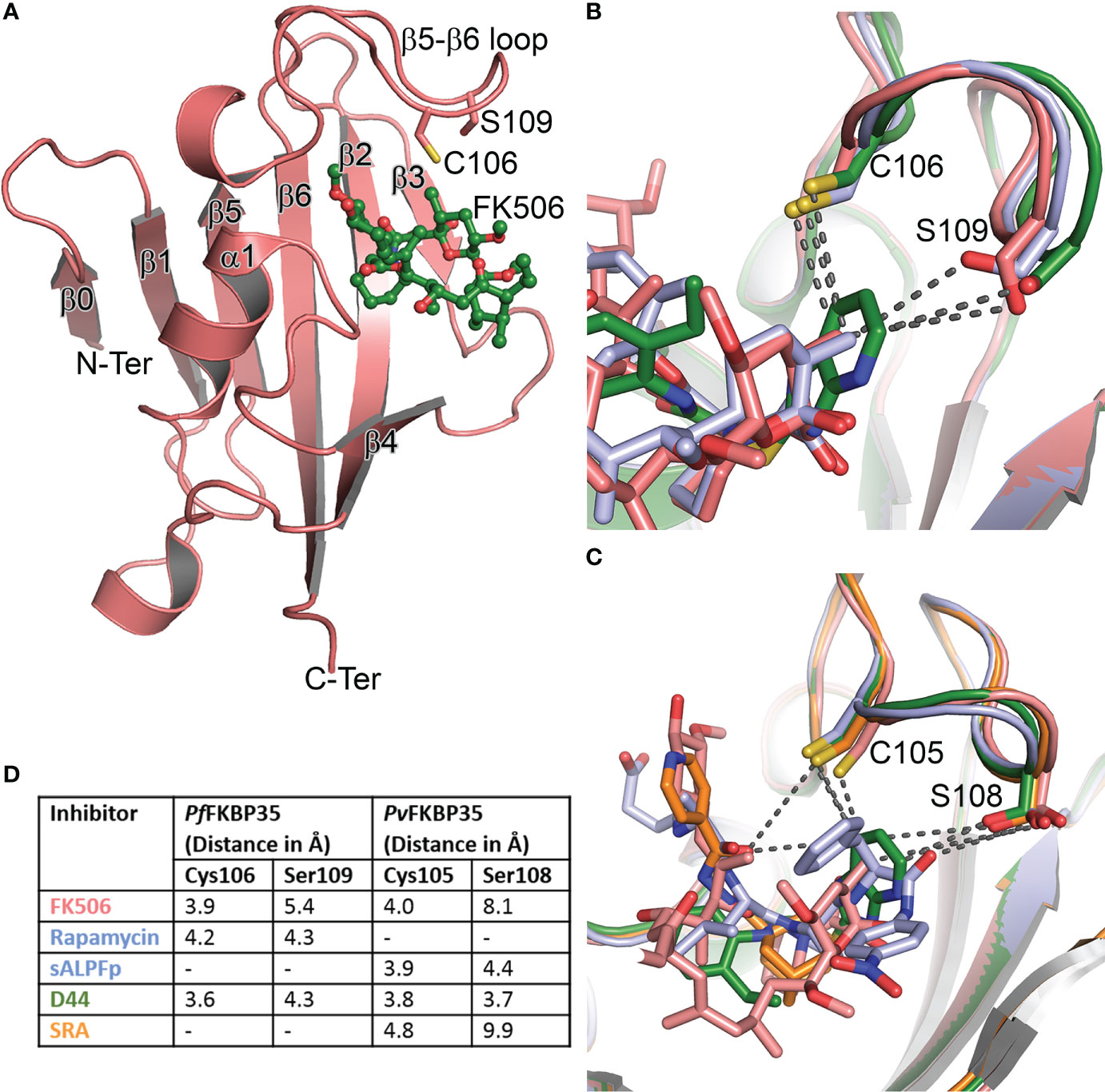
Figure 1 Structure and comparison of Plasmodium FKBPs. (A) The cartoon representation of PfFKBD35 in complex with FK506 (in ball and stick mode). The β0 observed here is not present in hsFKBPs. Similarly, the two unique residues pertaining to Plasmodium FKBP35’s Cys106 and Ser109 in the β5-β6 loop are shown in stick mode. (B) The overlay of FK506 (salmon), rapamycin (pale blue) and D44 (green) bound structures of PfFKBD35, with the nearest atom from Cys106 and Ser109 shown in broken lines. (C) Similarly, the overlay of FK506 (salmon), substrate-ALPF (pale blue), D44 (green) and SRA (orange) bound structures of PvFKBD35. (D) The nearest distance between the ligands and the unique Cys and Ser residues are listed for comparison, indicating the D44 is oriented closer to these residues.
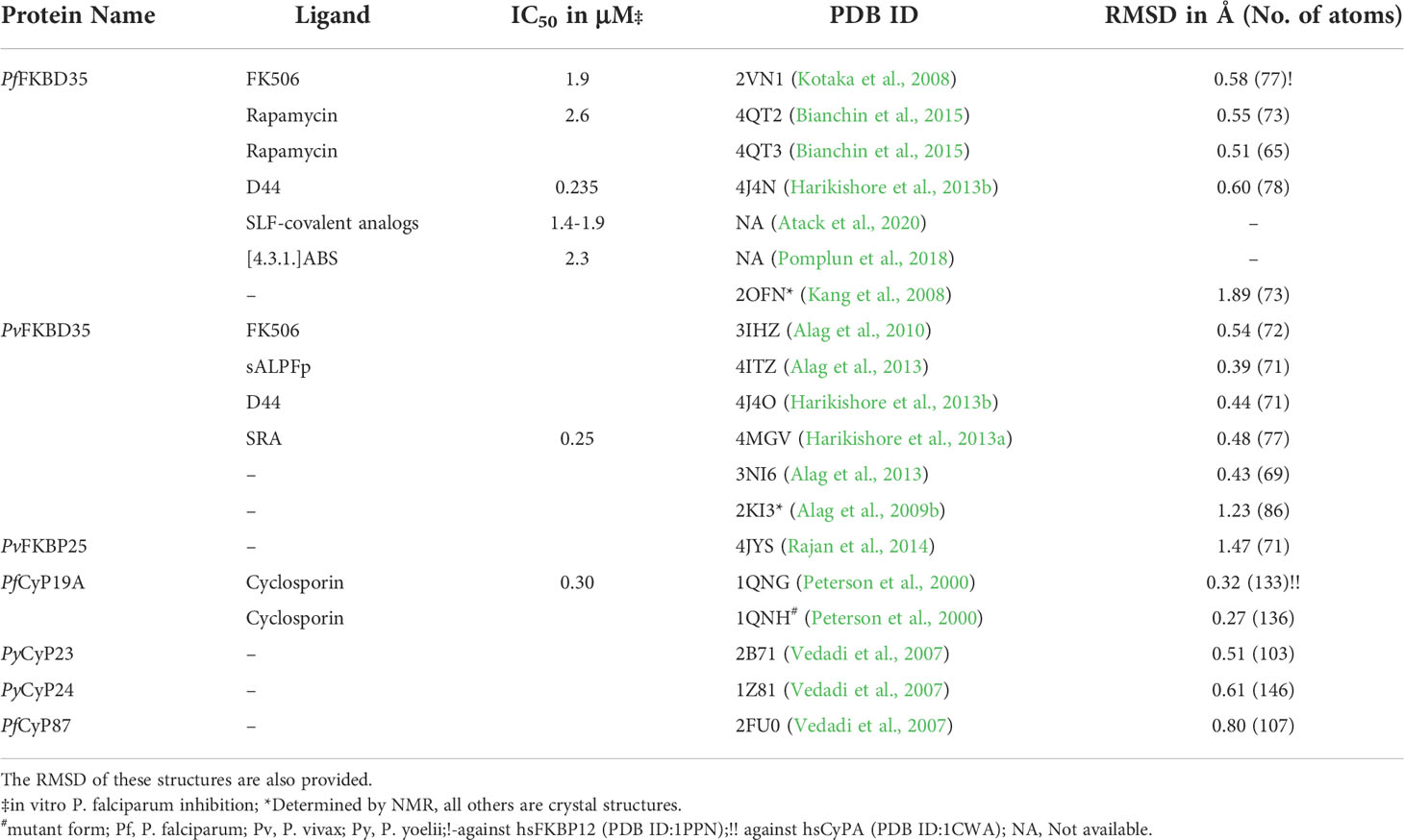
Table 1 List of Plasmodium PPIases, their inhibitors, IC50, and PDB ID, if available.
In all these crystal structures (Table 1), the FKBDs form a crystallographic dimer, either in the asymmetric unit or by crystal packing, primarily owing to the Cys105/106, which forms a disulfide bond with the same cysteine from the neighboring chain, except in PfFKBD35-FK506 complex, with the active site from each chain facing each other. Interestingly, the FKBD35 remains a monomer in solution, whereas the full-length FKBP35 is present as a dimer (Yoon et al., 2007), indicating that the dimer formation is due to TPR domain and not FKBD. Therefore, the observed dimer and disulfide bond could have resulted from the crystallization conditions and incubation time for crystal formation. The crystal structures showed that this is a reactive cysteine and being a unique residue of the Plasmodium FKBPs, it was proposed to be an attractive site for designing covalent inhibitors which can enhance the specificity of Plasmodium FKBP35 (Kotaka et al., 2008). Toward this end, the rapamycin-bound PfFKBD35 was determined in the oxidized and reduced forms (Bianchin et al., 2015). Monomeric units were captured in the crystal. In the oxidized form the Cys106 could be seen covalently attached to dithiothreitol and did not affect the rapamycin interactions. Moreover, the nearest distance from the sidechains of Cys106 and Ser109 to rapamycin is 4.2Å and 4.3Å, respectively (Figure 1D).
Recently, another study showed that the modification of D44 terminal phenyl ethyl with covalent warheads were not promising. In contrast, synthetic ligand for FKBP (SLF) analogs (Supplementary Table 5) were shown to covalently bind to FKBP35 (Atack et al., 2020), possessing an in vitro IC50 of 1.4 – 1.9μM against P. falciparum (Table 1). It was observed that the covalent bond formation is a slow reaction and requires stronger electrophilic warheads, corroborating with our solution studies (Yoon et al., 2007) where it remained a monomer and disulfide bond formation was observed only under the crystal conditions, probably owing to the longer time required for crystal formation. Thus, targeted covalent inhibition of FKBP35 has been considered the way forward (Macdonald and Boyd, 2015).
Another annotated Plasmodium FKBP, a 25kDa PvFKBP25, was also characterized by our group (Rajan et al., 2014). The domain architecture indicated the presence of an N-terminal Helix-Loop-Helix domain, followed by a FKBD and a C-terminal CaM-binding domain (Supplementary Figure 2), similar to the hsFKBP25 (Prakash et al., 2016). HsFKBP25 is a unique member of the FKBP family possessing multiple binding partners. Biochemical characterization of PvFKBP25 exposed its noncanonical nature lacking catalytic activity, owing to the active site mutations (Supplementary Table 4). Its structure unveiled the concealed active site, which is unconducive for ligand binding to validate the noncanonical function of this protein (Supplementary Figure 5). Thus, it would be interesting to decipher the role of this protein in the parasite life cycle.
11 Plasmodium CyPs and 2 Plasmodium FKBPs have been identified, while only one in each, CyP19 and FKBP35, possess catalytic activity. The question remains as to the function of other PPIases and why the parasite needs multiple PPIases. The possible role of these PPIases in liver stage (Derbyshire et al., 2012; Fontinha et al., 2020) can be explored, though most of them are abundantly produced in the intraerythrocytic stage. Their inhibitors also act on stages which correlate with this (Supplementary Figure 1). PfCyP19, only when in complex with CsA, can inhibit CaN, unlike the conundrum revolving the role of FKBP35/FK506 and CaN. The parasite possesses a CaN but lacks an mTOR, the targets of FK506 and rapamycin, respectively. Both inhibitors affect the parasite growth, similarly, indicating that FKBP35 is the target of interest. Although these inhibitors have an affinity for Plasmodium FKBP35, they bind stronger with host FKBPs which remains to be a challenge for targeting. On the other hand, if it is true that PlasmodiumFKBP35 can bind CaN sans FK506, it could be one of the parasite’s arsenal to enhance immunosuppression to hijack the host during infection (Calle et al., 2021). Studies have shown that Plasmodium CaN is involved in the erythrocyte invasion by the parasite and regulated host-cell interaction (Paul et al., 2015; Philip and Waters, 2015), which is also intriguing. Thus, a few open questions still lure around FKBP35’s precise function in the parasite and their relationship with CaN, warranting further investigation. Identifying Plasmodium PPIase’s natural substrates and characterizing the binding proteins (Leneghan and Bell, 2015) may also help the cause to understand their functional relevance as a chaperone. From a structural perspective, the role of an additional β-strand (β0) (Figure 1A) at the N-terminal of PlasmodiumFKBP35, and insertion in the loop between α1 and β3 of PfCyP19A/PyCyP24 (Supplementary Figure 3C) is also inquisitive. In future, the structures (Table 1) and the interactions (Supplementary Tables 3, 6) made by these inhibitors (Supplementary Table 5) can aid the design of more specific inhibitors. In addition, the D44’s purine-like ring can be linked with the adamantyl ring of SRA, which could improve its affinity for FKBP35 or covalent warheads can be attached to the purine-like moiety closer to Cys106. The promising [4.3.1.]ABS analogs can also be modified as potential covalent inhibitors. Nonetheless, its inhibition still would be beneficial, especially with the help of non-immunosuppressive covalent inhibitors.
SR and HY wrote the manuscript and approved the submitted version.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fcimb.2022.931635/full#supplementary-material
Alag, R., Balakrishna, A. M., Rajan, S., Qureshi, I. A., Shin, J., Lescar, J., et al. (2013). Structural insights into substrate binding by PvFKBP35, a peptidylprolyl cis-trans isomerase from the human malarial parasite plasmodium vivax. Eukaryot. Cell 12 (4), 627–634. doi: 10.1128/EC.00016-13
Alag, R., Bharatham, N., Dong, A., Hills, T., Harikishore, A., Widjaja, A. A., et al. (2009a). Crystallographic structure of the tetratricopeptide repeat domain of plasmodium falciparum FKBP35 and its molecular interaction with Hsp90 c-terminal pentapeptide. Protein Sci. 18 (10), 2115–2124. doi: 10.1002/pro.226
Alag, R., Qureshi, I. A., Bharatham, N., Shin, J., Lescar, J., Yoon, H. S. (2010). NMR and crystallographic structures of the FK506 binding domain of human malarial parasite plasmodium vivax FKBP35. Protein Sci. 19 (8), 1577–1586. doi: 10.1002/pro.438
Alag, R., Shin, J., Yoon, H. S. (2009b). NMR assignments of the FK506-binding domain of FK506-binding protein 35 from plasmodium vivax. Biomol. NMR Assign. 3 (2), 243–245. doi: 10.1007/s12104-009-9185-1
Amos, B., Aurrecoechea, C., Barba, M., Barreto, A., Basenko, E. Y., Bazant, W., et al. (2022). VEuPathDB: the eukaryotic pathogen, vector and host bioinformatics resource center. Nucleic Acids Res. 50 (D1), D898–D911. doi: 10.1093/nar/gkab929
Atack, T. C., Raymond, D. D., Blomquist, C. A., Pasaje, C. F., Mccarren, P. R., Moroco, J., et al. (2020). Targeted covalent inhibition of plasmodium FK506 binding protein 35. ACS Med. Chem. Lett. 11 (11), 2131–2138. doi: 10.1021/acsmedchemlett.0c00272
Balikagala, B., Fukuda, N., Ikeda, M., Katuro, O. T., Tachibana, S. I., Yamauchi, M., et al. (2021). Evidence of artemisinin-resistant malaria in Africa. N Engl. J. Med. 385 (13), 1163–1171. doi: 10.1056/NEJMoa2101746
Bell, A., Monaghan, P., Page, A. P. (2006). Peptidyl-prolyl cis-trans isomerases (immunophilins) and their roles in parasite biochemistry, host-parasite interaction and antiparasitic drug action. Int. J. Parasitol. 36 (3), 261–276. doi: 10.1016/j.ijpara.2005.11.003
Bell, A., Roberts, H. C., Chappell, L. H. (1996). The antiparasite effects of cyclosporin a: possible drug targets and clinical applications. Gen. Pharmacol. 27 (6), 963–971. doi: 10.1016/0306-3623(95)02148-5
Bell, A., Wernli, B., Franklin, R. M. (1994). Roles of peptidyl-prolyl cis-trans isomerase and calcineurin in the mechanisms of antimalarial action of cyclosporin a, FK506, and rapamycin. Biochem. Pharmacol. 48 (3), 495–503. doi: 10.1016/0006-2952(94)90279-8
Bianchin, A., Allemand, F., Bell, A., Chubb, A. J., Guichou, J. F. (2015). Two crystal structures of the FK506-binding domain of plasmodium falciparum FKBP35 in complex with rapamycin at high resolution. Acta Crystallogr. D Biol. Crystallogr. 71 (Pt 6), 1319–1327. doi: 10.1107/S1399004715006239
Braun, P. D., Barglow, K. T., Lin, Y. M., Akompong, T., Briesewitz, R., Ray, G. T., et al. (2003). A bifunctional molecule that displays context-dependent cellular activity. J. Am. Chem. Soc. 125 (25), 7575–7580. doi: 10.1021/ja035176q
Calle, C. L., Mordmuller, B., Singh, A. (2021). Immunosuppression in malaria: Do plasmodium falciparum parasites hijack the host? Pathogens 10 (10):1277. doi: 10.3390/pathogens10101277
Cui, L., Fan, Q., Hu, Y., Karamycheva, S. A., Quackenbush, J., Khuntirat, B., et al. (2005). Gene discovery in plasmodium vivax through sequencing of ESTs from mixed blood stages. Mol. Biochem. Parasitol. 144 (1), 1–9. doi: 10.1016/j.molbiopara.2005.05.016
Derbyshire, E. R., Prudencio, M., Mota, M. M., Clardy, J. (2012). Liver-stage malaria parasites vulnerable to diverse chemical scaffolds. Proc. Natl. Acad. Sci. U.S.A. 109 (22), 8511–8516. doi: 10.1073/pnas.1118370109
Fontinha, D., Moules, I., Prudencio, M. (2020). Repurposing drugs to fight hepatic malaria parasites. Molecules 25 (15):3409. doi: 10.3390/molecules25153409
Galat, A. (2003). Peptidylprolyl cis/trans isomerases (immunophilins): biological diversity–targets–functions. Curr. Top. Med. Chem. 3 (12), 1315–1347. doi: 10.2174/1568026033451862
Gavigan, C. S., Kiely, S. P., Hirtzlin, J., Bell, A. (2003). Cyclosporin-binding proteins of plasmodium falciparum. Int. J. Parasitol. 33 (9), 987–996. doi: 10.1016/s0020-7519(03)00125-5
Hall, N., Karras, M., Raine, J. D., Carlton, J. M., Kooij, T. W., Berriman, M., et al. (2005). A comprehensive survey of the plasmodium life cycle by genomic, transcriptomic, and proteomic analyses. Science 307 (5706), 82–86. doi: 10.1126/science.1103717
Harikishore, A., Leow, M. L., Niang, M., Rajan, S., Pasunooti, K. K., Preiser, P. R., et al. (2013a). Adamantyl derivative as a potent inhibitor of plasmodium FK506 binding protein 35. ACS Med. Chem. Lett. 4 (11), 1097–1101. doi: 10.1021/ml400306r
Harikishore, A., Niang, M., Rajan, S., Preiser, P. R., Yoon, H. S. (2013b). Small molecule plasmodium FKBP35 inhibitor as a potential antimalaria agent. Sci. Rep. 3:2501. doi: 10.1038/srep02501
Harikishore, A., Yoon, H. S. (2015). Immunophilins: Structures, mechanisms and ligands. Curr. Mol. Pharmacol. 9 (1), 37–47. doi: 10.2174/1874467208666150519113427
Hasskarl, J. (2018). Everolimus. Recent Results Cancer Res. 211, 101–123. doi: 10.1007/978-3-319-91442-8_8
Hirtzlin, J., Farber, P. M., Franklin, R. M., Bell, A. (1995). Molecular and biochemical characterization of a plasmodium falciparum cyclophilin containing a cleavable signal sequence. Eur. J. Biochem. 232 (3), 765–772. doi: 10.1111/j.1432-1033.1995.0765a.x
Kang, C. B., Ye, H., Yoon, H. R., Yoon, H. S. (2008). Solution structure of FK506 binding domain (FKBD) of plasmodium falciparum FK506 binding protein 35 (PfFKBP35). Proteins 70 (1), 300–302. doi: 10.1002/prot.21623
Kotaka, M., Ye, H., Alag, R., Hu, G., Bozdech, Z., Preiser, P. R., et al. (2008). Crystal structure of the FK506 binding domain of plasmodium falciparum FKBP35 in complex with FK506. Biochemistry 47 (22), 5951–5961. doi: 10.1021/bi800004u
Kumar, R., Adams, B., Musiyenko, A., Shulyayeva, O., Barik, S. (2005a). The FK506-binding protein of the malaria parasite, plasmodium falciparum, is a FK506-sensitive chaperone with FK506-independent calcineurin-inhibitory activity. Mol. Biochem. Parasitol. 141 (2), 163–173. doi: 10.1016/j.molbiopara.2005.02.007
Kumar, R., Musiyenko, A., Barik, S. (2005b). Plasmodium falciparum calcineurin and its association with heat shock protein 90: mechanisms for the antimalarial activity of cyclosporin a and synergism with geldanamycin. Mol. Biochem. Parasitol. 141 (1), 29–37. doi: 10.1016/j.molbiopara.2005.01.012
Laurens, M. B. (2020). RTS,S/AS01 vaccine (Mosquirix): an overview. Hum. Vaccin. Immunother. 16 (3), 480–489. doi: 10.1080/21645515.2019.1669415
Leneghan, D., Bell, A. (2015). Immunophilin-protein interactions in plasmodium falciparum. Parasitology 142 (11), 1404–1414. doi: 10.1017/S0031182015000803
Le Roch, K. G., Zhou, Y., Blair, P. L., Grainger, M., Moch, J. K., Haynes, J. D., et al. (2003). Discovery of gene function by expression profiling of the malaria parasite life cycle. Science 301 (5639), 1503–1508. doi: 10.1126/science.1087025
Liu, Q., Jing, W., Kang, L., Liu, J., Liu, M. (2021). Trends of the global, regional and national incidence of malaria in 204 countries from 1990 to 2019 and implications for malaria prevention. J. Travel Med. 28 (5):taab046. doi: 10.1093/jtm/taab046
Macdonald, C. A., Boyd, R. J. (2015). Computational insights into the suicide inhibition of plasmodium falciparum Fk506-binding protein 35. Bioorg. Med. Chem. Lett. 25 (16), 3221–3225. doi: 10.1016/j.bmcl.2015.05.079
Marin-Menendez, A., Bell, A. (2011). Overexpression, purification and assessment of cyclosporin binding of a family of cyclophilins and cyclophilin-like proteins of the human malarial parasite plasmodium falciparum. Protein Expr. Purif. 78 (2), 225–234. doi: 10.1016/j.pep.2011.04.012
Marin-Menendez, A., Monaghan, P., Bell, A. (2012). A family of cyclophilin-like molecular chaperones in plasmodium falciparum. Mol. Biochem. Parasitol. 184 (1), 44–47. doi: 10.1016/j.molbiopara.2012.04.006
Mehlin, C. (2005). Structure-based drug discovery for plasmodium falciparum. Comb. Chem. High Throughput Screen 8 (1), 5–14. doi: 10.2174/1386207053328093
Monaghan, P., Bell, A. (2005). A plasmodium falciparum FK506-binding protein (FKBP) with peptidyl-prolyl cis-trans isomerase and chaperone activities. Mol. Biochem. Parasitol. 139 (2), 185–195. doi: 10.1016/j.molbiopara.2004.10.007
Monaghan, P., Fardis, M., Revill, W. P., Bell, A. (2005). Antimalarial effects of macrolactones related to FK520 (ascomycin) are independent of the immunosuppressive properties of the compounds. J. Infect. Dis. 191 (8), 1342–1349. doi: 10.1086/428454
Monaghan, P., Leneghan, D. B., Shaw, W., Bell, A. (2017). The antimalarial action of FK506 and rapamycin: evidence for a direct effect on FK506-binding protein PfFKBP35. Parasitology 144 (7), 869–876. doi: 10.1017/S0031182017000245
Paul, A. S., Saha, S., Engelberg, K., Jiang, R. H., Coleman, B. I., Kosber, A. L., et al. (2015). Parasite calcineurin regulates host cell recognition and attachment by apicomplexans. Cell Host Microbe 18 (1), 49–60. doi: 10.1016/j.chom.2015.06.003
Peterson, M. R., Hall, D. R., Berriman, M., Nunes, J. A., Leonard, G. A., Fairlamb, A. H., et al. (2000). The three-dimensional structure of a plasmodium falciparum cyclophilin in complex with the potent anti-malarial cyclosporin a. J. Mol. Biol. 298 (1), 123–133. doi: 10.1006/jmbi.2000.3633
Philip, N., Waters, A. P. (2015). Conditional degradation of plasmodium calcineurin reveals functions in parasite colonization of both host and vector. Cell Host Microbe 18 (1), 122–131. doi: 10.1016/j.chom.2015.05.018
Plowe, C. V. (2022). Malaria chemoprevention and drug resistance: a review of the literature and policy implications. Malar. J. 21 (1), 104. doi: 10.1186/s12936-022-04115-8
Pomplun, S., Sippel, C., Hahle, A., Tay, D., Shima, K., Klages, A., et al. (2018). Chemogenomic profiling of human and microbial FK506-binding proteins. J. Med. Chem. 61 (8), 3660–3673. doi: 10.1021/acs.jmedchem.8b00137
Prakash, A., Shin, J., Rajan, S., Yoon, H. S. (2016). Structural basis of nucleic acid recognition by FK506-binding protein 25 (FKBP25), a nuclear immunophilin. Nucleic Acids Res. 44 (6), 2909–2925. doi: 10.1093/nar/gkw001
Pushpakom, S., Iorio, F., Eyers, P. A., Escott, K. J., Hopper, S., Wells, A., et al. (2019). Drug repurposing: progress, challenges and recommendations. Nat. Rev. Drug Discovery 18 (1), 41–58. doi: 10.1038/nrd.2018.168
Rajan, S., Austin, D., Harikishore, A., Nguyen, Q. T., Baek, K., Yoon, H. S. (2014). Crystal structure of plasmodium vivax FK506-binding protein 25 reveals conformational changes responsible for its noncanonical activity. Proteins 82 (7), 1235–1244. doi: 10.1002/prot.24487
Rajan, S., Baek, K., Yoon, H. S. (2013). C-h O hydrogen bonds in FK506-binding protein-ligand interactions. J. Mol. Recognit. 26 (11), 550–555. doi: 10.1002/jmr.2299
Reddy, G. R. (1995). Cloning and characterization of a plasmodium falciparum cyclophilin gene that is stage-specifically expressed. Mol. Biochem. Parasitol. 73 (1-2), 111–121. doi: 10.1016/0166-6851(95)00103-8
Tse, E. G., Korsik, M., Todd, M. H. (2019). The past, present and future of anti-malarial medicines. Malar. J. 18 (1), 93. doi: 10.1186/s12936-019-2724-z
Van De Straat, B., Sebayang, B., Grigg, M. J., Staunton, K., Garjito, T. A., Vythilingam, I., et al. (2022). Zoonotic malaria transmission and land use change in southeast Asia: what is known about the vectors. Malar. J. 21 (1), 109. doi: 10.1186/s12936-022-04129-2
Vedadi, M., Lew, J., Artz, J., Amani, M., Zhao, Y., Dong, A., et al. (2007). Genome-scale protein expression and structural biology of plasmodium falciparum and related apicomplexan organisms. Mol. Biochem. Parasitol. 151 (1), 100–110. doi: 10.1016/j.molbiopara.2006.10.011
White, N. J. (2004). Antimalarial drug resistance. J. Clin. Invest. 113 (8), 1084–1092. doi: 10.1172/JCI21682
White, N. J., Day, N. P. J., Ashley, E. A., Smithuis, F. M., Nosten, F. H. (2022). Have we really failed to roll back malaria? Lancet 399 (10327), 799–800. doi: 10.1016/S0140-6736(22)00175-1
Yang, T., Ottilie, S., Istvan, E. S., Godinez-Macias, K. P., Lukens, A. K., Baragana, B., et al. (2021). MalDA, accelerating malaria drug discovery. Trends Parasitol. 37 (6), 493–507. doi: 10.1016/j.pt.2021.01.009
Keywords: malaria, plasmodium, PPIase, FKBP, cyclophilin, FK506, cyclosporin
Citation: Rajan S and Yoon HS (2022) Structural insights into Plasmodium PPIases. Front. Cell. Infect. Microbiol. 12:931635. doi: 10.3389/fcimb.2022.931635
Received: 29 April 2022; Accepted: 16 August 2022;
Published: 02 September 2022.
Edited by:
Felix Hausch, Darmstadt University of Technology, GermanyReviewed by:
Thomas Atack, Broad Institute, United StatesCopyright © 2022 Rajan and Yoon. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ho Sup Yoon, aHN5b29uQG50dS5lZHUuc2c=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.