 Eliza Gil
Eliza Gil Mahdad Noursadeghi
Mahdad Noursadeghi Jeremy S. Brown
Jeremy S. Brown
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Cell. Infect. Microbiol., 28 July 2022
Sec. Molecular Bacterial Pathogenesis
Volume 12 - 2022 | https://doi.org/10.3389/fcimb.2022.929483
This article is part of the Research TopicDeceiving the Host: Mechanisms of Immune Evasion and Survival By Pneumococcal BacteriaView all 5 articles
Host innate and adaptive immunity to infection with Streptococcus pneumoniae is critically dependent on the complement system, demonstrated by the high incidence of invasive S. pneumoniae infection in people with inherited deficiency of complement components. The complement system is activated by S. pneumoniae through multiple mechanisms. The classical complement pathway is activated by recognition of S. pneumoniae by C-reactive protein, serum amyloid P, C1q, SIGN-R1, or natural or acquired antibody. Some S. pneumoniae strains are also recognised by ficolins to activate the mannose binding lectin (MBL) activation pathway. Complement activation is then amplified by the alternative complement pathway, which can also be activated by S. pneumoniae directly. Complement activation results in covalent linkage of the opsonic complement factors C3b and iC3b to the S. pneumoniae surface which promote phagocytic clearance, along with complement-mediated immune adherence to erythrocytes, thereby protecting against septicaemia. The role of complement for mucosal immunity to S. pneumoniae is less clear. Given the major role of complement in controlling infection with S. pneumoniae, it is perhaps unsurprising that S. pneumoniae has evolved multiple mechanisms of complement evasion, including the capsule, multiple surface proteins, and the toxin pneumolysin. There is considerable variation between S. pneumoniae capsular serotypes and genotypes with regards to sensitivity to complement which correlates with ability to cause invasive infections. However, at present we only have a limited understanding of the main mechanisms causing variations in complement sensitivity between S. pneumoniae strains and to non-pathogenic streptococci.
Streptococcus pneumoniae colonises the nasopharynx of a high proportion of infants and up to 10% of adults. S. pneumoniae can spread from the nasopharynx to the lungs to cause pneumonia, the blood to cause septicaemia (often associated with pneumonia), or to the meninges to cause meningitis and is a common cause of severe infections worldwide (Troeger et al., 2018). Similar to other mainly extracellular bacterial pathogens, host immunity to S. pneumoniae is critically dependent on phagocytosis and therefore opsonisation by circulating soluble host immune mediators, particularly complement. The importance of complement for prevention of S. pneumoniae infection is demonstrated by the high incidence of invasive S. pneumoniae infections people with complement deficiencies (Sjöholm et al., 2006; Skattum et al., 2011; Woehrl et al., 2011). For example, around 50% of subjects with inherited deficiency of the classical complement pathway component C2 have had an episode of pneumonia, septicaemia, or meningitis, the majority of which were caused by S. pneumoniae (Jönsson et al., 2005). Genome wide association studies showing an association between genotypic complement variants and poor outcomes in S. pneumoniae meningitis provide additional evidence of the importance of complement for immunity to S. pneumoniae (Weiss et al., 1998; Biesma et al., 2001). Furthermore, human infections with S. pneumoniae are associated with significant reductions in circulating levels of complement factors (Coonrod and Rylko-Bauer, 1977) suggesting complement activation and consumption.
The complement system is activated by three mechanisms: the classical, alternative and lectin pathways (Walport, 2001). Data obtained using genetically modified mice or serum from human subjects with complement deficiencies have demonstrated a key role for the classical pathway for initiation of complement deposition on S. pneumoniae. Classical pathway deficiency reduced opsonisation with complement and complement-mediated phagocytosis of S. pneumoniae by neutrophils, and increased target organ CFU and disease severity in mouse models of pneumonia, septicaemia and meningitis (Brown et al., 2002; Rupprecht et al., 2007; Yuste et al., 2008). Although the classical pathway was previously considered to be activated by antibody and therefore mediate adaptive immunity, mouse data have demonstrated an essential role for the classical pathway for innate complement-mediated immunity to S. pneumoniae (Brown et al., 2002). Further work has demonstrated that multiple pathways activate classical pathway recognition of S. pneumoniae independent of acquired antibody, including direct binding of C1q (the first complement of the classical pathway), natural antibody recognition (mainly of cell wall phosphocholine), binding of the serum components C-reactive protein (CRP) and serum amyloid P (SAP) protein, and activation on splenic macrophage cell surfaces through SIGN-R1 recognition of the S. pneumoniae capsule (Figure 1) (Brown et al., 2002; Kang et al., 2004; Rupprecht et al., 2007; Suresh et al., 2007; Yuste et al., 2007; Agrawal et al., 2008)
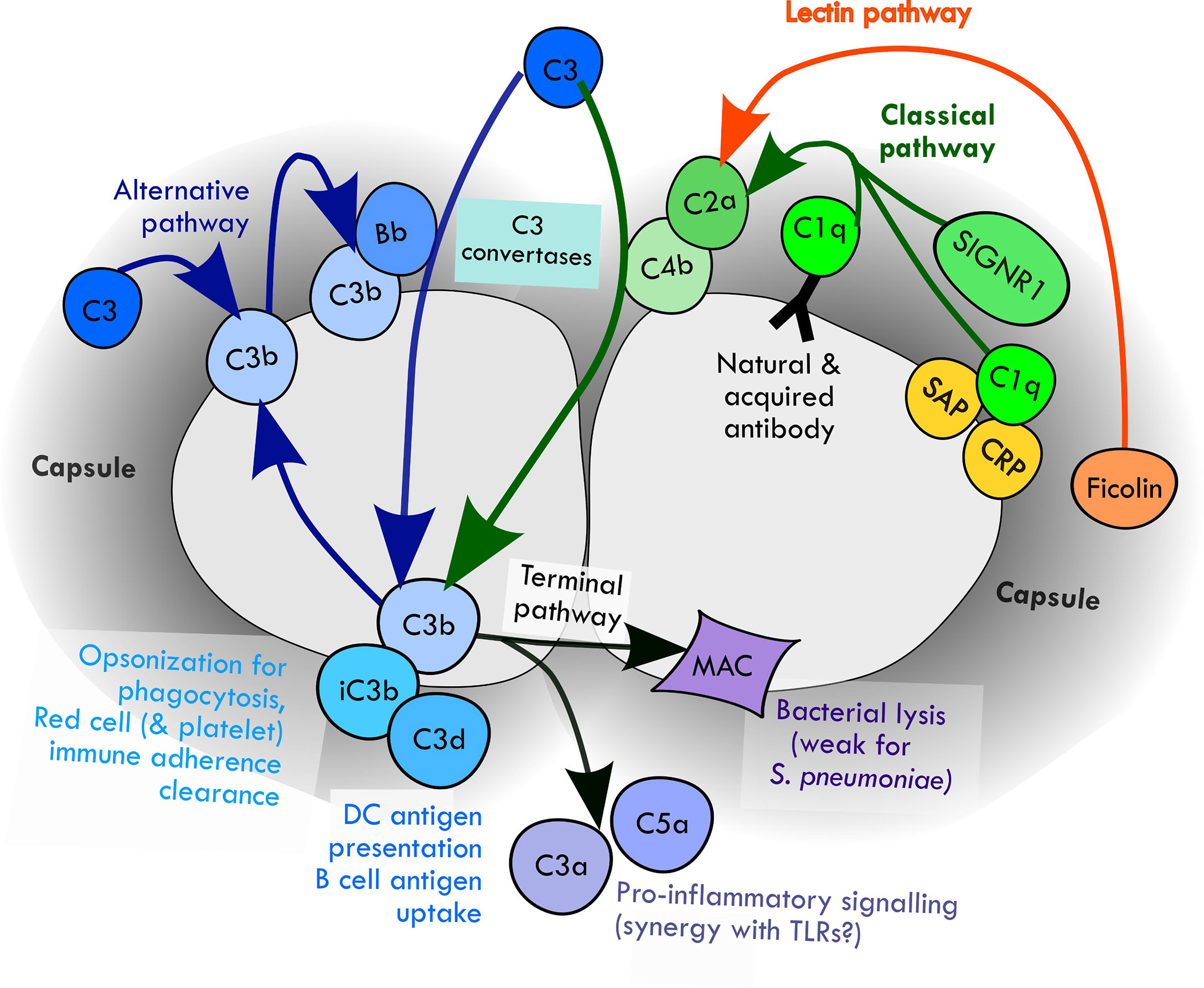
Figure 1 Mechanisms of activation of complement pathways by S. pneumoniae and the downstream potential effects on the immune response.
The alternative pathway amplifies complement activation by S. pneumoniae initiated by the classical and lectin pathways (Xu et al., 2001; Brown et al., 2002) and is also activated directly through recognition of cell wall phosphocholine (Winkelstein and Tomasz, 1978; Hummell et al., 1981; Brown et al., 1983b; Hummell et al., 1985; Hyams et al., 2010). Mice with deficiencies in components of the alternative pathway show impaired opsonophagocytosis of S. pneumoniae and increased susceptibility to infection (Xu et al., 2001; Brown et al., 2002; Li et al., 2011). In humans, hereditary deficiency of the alternative pathway component factor D is associated with severe pneumococcal disease (Weiss et al., 1998; Biesma et al., 2001). The role of the lectin complement pathway for protection against S. pneumoniae infection is less clear. The lectin pathway is activated by the binding to microbial carbohydrates of mannose-binding lectin (MBL) and the ficolins (Krarup et al., 2005). Meta-analysis has suggested MBL deficiency in humans is associated with an increased susceptibility to invasive S. pneumoniae infections (Eisen et al., 2008; Brouwer et al., 2009) and L-ficolin binds to at least some capsular serotypes of S. pneumoniae (Krarup et al., 2005). However, data from genetically modified mice is contradictory with some data showing the lectin pathway only has a small role in complement recognition of S. pneumoniae (Neth et al., 2000; Brown et al., 2002; Krarup et al., 2005; Brady et al., 2014) but other data demonstrating deficiency of MASP-2, the effector enzyme of the lectin pathway, increases susceptible to S. pneumoniae (Ali et al., 2012).
Recognition of S. pneumoniae by the classical, alternative and lectin pathways results in activation of protease cascades that converge on the central complement component C3 (Figure 1). Cleavage of C3 by C3 convertases results in formation of the opsonins C3b and iC3b, along with activation of the terminal complement pathway. This results in the three main mechanisms by which complement directly prevents infection: (1) opsonisation and increased phagocytosis of the pathogen by covalent linkage of C3b and iC3b to bacterial surface molecules; (2) pro-inflammatory signaling by the complement components C3a and C5a; and (3) direct killing of the pathogen by formation of the membrane attack complex, a lipophilic complex of the terminal complement components which forms a pore in the pathogen phospholipid bilayer leading to bacterial lysis (Walport, 2001). Opsonisation with C3b/iC3b strongly promotes both innate and adaptive (mediated by acquired antibody) clearance of S. pneumoniae by phagocytosis (Gordon et al., 1986; van Lookeren Campagne et al., 2007; Hyams et al., 2010; Hyams et al., 2013; Wilson et al., 2017). C5a production in response to S. pneumoniae increases cytokine production by human white cells, possibly involving cross-talk with toll like receptor-mediated inflammatory responses (van der Maten et al., 2016). Some data also suggest complement-mediated inflammation is in fact detrimental in meningitis through promoting host neurotoxicity (Kasanmoentalib et al., 2017). S. pneumoniae is generally resistant to MAC-mediated killing (Joiner et al., 1983).
Along with direct effects on innate immunity, additional mechanisms by which complement promotes immunity to bacteria have been described. Red cells express complement receptor 1 (CR1), and thereby bind S. pneumoniae opsonised with C3b and transport them to the liver and spleen to be phagocytosed by tissue macrophages (Li et al., 2009; van Lookeren Campagne and Verschoor, 2018). In transgenic mice expressing human CR1 on red cells this mechanism promotes clearance of S. pneumoniae from the blood (Li et al., 2009). A similar mechanism of bacterial clearance involving bacterial adhesion to platelets via CR1 has also been described for other pathogens (Broadley et al., 2016), but it is not known whether this is relevant for S. pneumoniae. Recent data has also shown that complement-opsonised S. pneumoniae captured by splenic macrophages can then be killed by splenic neutrophils (Deniset et al., 2017). In addition, opsonisation with C3b also promotes adaptive antibody responses to S. pneumoniae by improving antigen presentation by dendritic cells and antigen uptake by B cells (Griffioen et al., 1991; Peset Llopis et al., 1996; Haas et al., 2002; Donius et al., 2013; Heesters and Carroll, 2016). Other complement dependent mechanisms of immunity that have recently been described include intracellular activation of pro-inflammatory pathways, bacterial killing via autophagy, and improved activation of T cells (Reis et al., 2019), but there are only limited or no clear data for their relevance for S. pneumoniae.
There are also likely to be differences in the role or significance of complement-mediated immunity to S. pneumoniae in different anatomical sites. The high incidence of septicaemia, meningitis, pneumoniae and otitis media in humans with inherited complement deficiencies (Skattum et al., 2011) suggest complement is required for prevention of both systemic and respiratory tract infections. Mouse infection models data convincingly show complement has a very powerful role in preventing septicaemia, with a less important but significant role for protecting against lung infection and otitis media (Brown et al., 2002; Ren et al., 2004b; Yuste et al., 2005; Bogaert et al., 2009; Sabharwal et al., 2009). Experiments using complement deficient mice suggest complement has a limited role in promoting clearance of S. pneumoniae in simple models of nasopharyngeal colonisation (van Rossum et al., 2005; Bogaert et al., 2010). However, in a dual infection model Haemophilus influenzae stimulated S. pneumoniae clearance from the nasopharynx and this required complement, suggesting that complement may help limit S. pneumoniae colonisation when the nasopharynx is inflamed (Lysenko et al., 2005). How important complement-mediated inflammation is for the pathogenesis S. pneumoniae infections at different anatomical sites is not clear and requires further investigation.
The importance of complement for immunity to S. pneumoniae is also demonstrated by the multiple mechanisms S. pneumoniae has evolved to evade complement mediated immunity, several of which are important for S. pneumoniae to establish invasive infection. The key mechanisms used by S. pneumoniae to inhibit complement-mediated immunity are described below and summarised in Table 1.
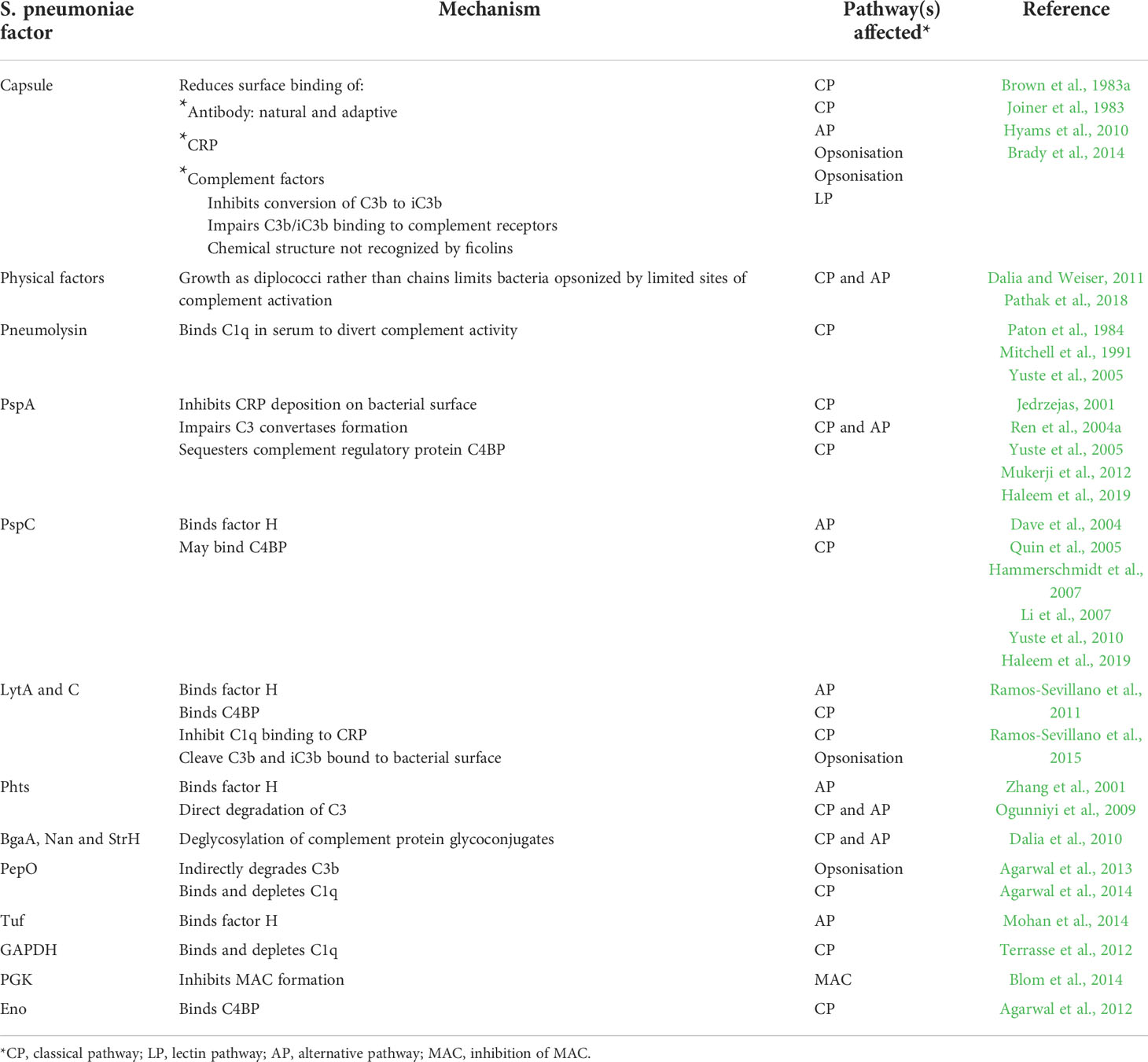
Table 1 Mechanisms of inhibition of complement activity by S. pneumoniae.
Virulent strains of S. pneumoniae capable of causing invasive infections have an external capsular layer made of repeating chains of polysaccharide anchored to the peptidoglycan cell wall (Kadioglu et al., 2008). The chemical composition of the polysaccharide chains varies, resulting in at least 100 different S. pneumoniae serotypes. The capsule is a key virulence factor, in part due to its role in complement evasion; unencapsulated S. pneumoniae have low virulence and are rapidly removed from the circulation (Watson and Musher, 1990; Watson et al., 1995; Casal and Tarragó, 2003). The capsule blocks S. pneumoniae activation of the classical pathway by reducing bacterial recognition by natural and adaptive antibody, as well as direct binding of C1q and C reactive protein (Hyams et al., 2010). Furthermore the capsule inhibits activation of the alternative pathway and conversion of C3b to iC3b, and physically masks complement on the bacterial surface from binding to phagocyte complement receptors (Brown et al., 1983a; Joiner et al., 1983; Hyams et al., 2010). The result is that encapsulated S. pneumoniae are relatively resistant to complement-dependent phagocytosis (Hyams et al., 2010).
A perhaps surprising but important concept about complement recognition of S. pneumoniae is the relative paucity of activation sites per bacterium. Instead of complement activation occurring evenly across the whole bacterial surface, there is focal formation of C3b/iC3b on the S. pneumoniae surface with a predilection for the site of divisional septa where the capsule layer is thinnest (Dalia and Weiser, 2011; Pathak et al., 2018). These foci of activated complement allow the spread of complement activation across the S. pneumoniae surface. As a consequence longer chains of S. pneumoniae are more susceptible to complement than short chains (Dalia and Weiser, 2011), providing one potential reason why S. pneumoniae is usually found as diplococci rather than in chains. Conversely, S. pneumoniae agglutination by antibody improves complement recognition as it allows the initial focal sites of complement activation to spread across multiple bacteria (Dalia and Weiser, 2011).
The S. pneumoniae pore-forming toxin pneumolysin is released during bacterial growth and also found attached to the cell wall in some strains (Johnson, 1977; Balachandran et al., 2001; Price and Camilli, 2009). Extra-cellular pneumolysin activates the classical complement pathway by binding the Fc portion of IgG, diverting complement activation away from S. pneumoniae and increasing resistance to complement-mediated immunity (Paton et al., 1984; Mitchell et al., 1991; Yuste et al., 2005). In a mouse model the importance of pneumolysin for S. pneumoniae virulence was largely dependent on complement, and inhibition of both the alternative and classical complement pathways by a combination of pneumolysin and PspA respectively has a synergistic effect on S. pneumoniae complement evasion and virulence (Yuste et al., 2005).
An increasing number of S. pneumoniae surface proteins have been described to inhibit complement activity against S. pneumoniae (Andre et al., 2017) (Table 1). The most notable are the cell wall choline binding proteins PspA and PspC which both have multiple roles for S. pneumoniae virulence, including complement evasion.
PspA is expressed by all clinically important serotypes of S. pneumoniae and appears to inhibit both the classical and alternative complement pathways (Yuste et al., 2005), preventing complement-mediated clearance and phagocytosis of S. pneumoniae (Briles et al., 1997; Ren et al., 2004a; Quin et al., 2007; Ochs et al., 2008). In mouse infection models, PspA-dependent promotion of S. pneumoniae virulence is largely (but not completely) lost in mice deficient in C3, and both alternative and classical pathways contribute to this effect (Yuste et al., 2005). PspA is highly polar, with a strong electronegative charge on the region protruding through the cell wall that may directly impair complement deposition (Jedrzejas, 2001; Ren et al., 2004a). PspA also blocks classical pathway activity by preventing C-reactive protein binding to S. pneumoniae (Mukerji et al., 2012), and (in common with PspC), PspA can sequester the complement regulatory protein C4 binding protein (C4BP) from host serum (Haleem et al., 2019).
PspC binds factor H, a negative regulator of complement, thereby inhibiting factor B binding to C3b and accelerating the breakdown of the alternative pathway C3 convertase resulting in the inhibition of complement-dependent S. pneumoniae phagocytosis (Dave et al., 2004; Quin et al., 2005; Hammerschmidt et al., 2007; Li et al., 2007; Yuste et al., 2010). Assessing the relevance of PspC/factor H interactions in infection models has been impaired by the lack of PspC binding to rodent factor H (Lu et al., 2008). As discussed above, there is also evidence that PspC can also bind the classical pathway regulatory factor C4BP (Dieudonné-Vatran et al., 2009; Haleem et al., 2019).
Additional S. pneumoniae choline binding proteins may have a role in complement resistance, including murein hydrolases LytA and C, which are involved in remodelling of the cell wall and cell division. LytA binds both C4BP and factor H, inhibits C1q binding to CRP, and cleaves C3b and iC3b on the bacterial surface thereby inhibiting complement deposition by both the classical and alternative pathways (Ramos-Sevillano et al., 2015). LytC also seems to inhibit C3b binding to S. pneumoniae (Ramos-Sevillano et al., 2011).
There are several other S. pneumoniae surface proteins that might be involved in complement evasion (Table 1). These include the histidine triad proteins PhtA, PhtB, PhtD and PhtE, with deletion of all four Pht proteins increasing C3 deposition on S. pneumoniae serotype 4 although not other strains (Melin et al., 2010a). The proposed mechanisms include increased factor H binding (Ogunniyi et al., 2009; André et al., 2021) and/or direct degradation of C3 by PhtA (Angel et al., 1994; Hostetter, 1999; Zhang et al., 2001). Various S. pneumoniae biochemical enzymes affect complement activity including the exoglycosidases BgaA, Nan and StrH, which are thought to impair complement activity by deglycosylation of complement protein glycoconjugates (Dalia et al., 2010). Phosphoglycerate kinase can inhibit MAC formation (Blom et al., 2014), and the metallopeptidase endopeptidase O (PepO) cleaves plasminogen to plasmin which in turn cleaves C3b, thereby reducing C3b deposition on the S. pneumoniae surface (Agarwal et al., 2013; Agarwal et al., 2014). PepO has also been shown to interact with complement C1q, inhibiting the classical pathway (Agarwal et al., 2014).
In vitro studies of complement activation of genetically engineered S. pneumoniae have demonstrated that capsular serotype causes very large differences in complement sensitivity (Hyams et al., 2010; Melin et al., 2010b). Opaque phase variants with a thicker capsule layer are more resistant to complement compared to transparent phase variants. However, capsular serotype effects on complement sensitivity are partially independent of capsule layer thickness (Hyams et al., 2013), implying the capsular carbohydrate structure is also important. This has been demonstrated for some specific serotypes: the related serotype 11A and 11E capsules only differ by O-acetylation of the galactose residue in the 11A capsule, but this results in ficolin-2 binding to the 11A capsule and activation of the lectin pathway whereas serotype 11E remains relatively complement resistant (Brady et al., 2014). Atomic force microscopy demonstrates that identical capsular serotypes expressed by S. pneumoniae and Streptococcus mitis have similar biomechanical properties, suggesting a relationship between capsule biochemistry and mechanical properties independent of bacterial strain. However, other factors must influence complement resistance as S. mitis strains were significantly more sensitive to complement than S. pneumoniae strains expressing the same capsular serotype (Marshall et al., 2020).
Along with capsular serotype, non-capsular variation in genetic background also determines differences between S. pneumoniae in their ability to resist complement (Hyams et al., 2011). Both pspA and pspC have marked allelic variation between S. pneumoniae strains (Croucher et al., 2017), and this is likely to cause at least some of the capsule-independent variation in complement sensitivity between strains. Indeed, the degree of factor H binding to S. pneumoniae dependent on PspC varies between strains (Yuste et al., 2010), with PspC subgroup 1 binding significantly more factor H than subgroup 2 (van der Maten et al., 2018). PspC subgroup 1 is the dominant subgroup in human cases of invasive S. pneumoniae infections (van der Maten et al., 2018) suggesting factor H binding to S. pneumoniae is potentially important for virulence. This is supported by data showing that PspC-dependent factor H binding variation between S. pneumoniae strains correlates with strain and capsular serotype ability to cause invasive infections in humans (Hyams et al., 2013). The association of complement-mediated phagocytosis with invasiveness within a pathogenic species suggests that complement resistance may be one important factor differentiating virulent from less virulent pathogens. This hypothesis is supported by data showing S. mitis strains were highly complement sensitive compared to S. pneumoniae (Marshall et al., 2020). However, increasing S. mitis resistance to complement by expression of a S. pneumoniae capsule or PspC (Rukke et al., 2014; Marshall et al., 2021) did not overcome the low virulence of S. mitis in mouse models of infection, indicating other factors are also important.
Characterization of S. pneumoniae protein interactions with complement have often relied on mutational analysis to demonstrate a role for a particular protein for complement evasion. However, care is necessary in interpreting these data - mutation of one gene can have direct and indirect effects on other genes or proteins, altering the complement resistance phenotype indirectly. Furthermore, the large degree of genetic variation between S. pneumoniae strains suggests the relative contributions of different complement evasion proteins will differ between S. pneumoniae strains. Lastly, even if a specific interaction between a S. pneumoniae protein and complement has been described it may not be functionally relevant; for example although other S. pneumoniae proteins have been shown to bind to factor H deletion of pspC results in complete loss of detectable factor H binding to multiple S. pneumoniae strains (Yuste et al., 2010; Hyams et al., 2013). There remain significant gaps in our understanding of the relative contributions of pneumococcal proteins for complement evasion during invasive disease in humans, partly reflecting the limitations in studying virulence factors using animal models, the complexity of the interplay between virulence factors, and the additional effects on pathogenesis of individual proteins beyond those related to complement.
The complement system is central to the protective immune response to S. pneumoniae, with the balance between complement activation by host recognition of S. pneumoniae and complement evasion by multiple bacterial mechanisms significantly affecting the outcome of infection. Many questions remain about the interactions between S. pneumoniae and the complement system, including how capsule structure alters complement recognition, the exact roles during infection of the multiple non-capsular factors thought to inhibit complement activity, and the importance of complement for inflammatory and other non-phagocytic mechanisms of immunity to S. pneumoniae. A greater understanding of the mechanisms by which complement mediates immunity against S. pneumoniae and how S. pneumoniae evades these will provide a clearer understanding of disease pathogenesis, and more broadly potentially clarify why some bacterial commensal species are common pathogens and others rarely cause severe infections.
EG, JB and MN conceived the manuscript. EG and JB wrote the text and figures, which was critically revised by MN. All have provided approval for publication.
This work was supported by Wellcome Trust awards to EG (107311/Z/15/Z), MN (207511/Z/17/Z), and JB (221803/Z/20/Z). Meningitis Now and MRC awards (MR/S004394/1) to JB, and the National Institute for Health Research University College London Hospitals Biomedical Research Centre awards to MN and JB (IS-BRC-1215-20016 NIHR University College London Hospitals Biomedical Research Centre).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Agarwal, V., Hammerschmidt, S., Malm, S., Bergmann, S., Riesbeck, K., Blom, A. M., et al (2012). Enolase of Streptococcus pneumoniae binds human complement inhibitor C4b-binding protein and contributes to complement evasion. J Immunol 189(7):3575–3584. doi: 10.4049/jimmunol.1102934
Agarwal, V., Kuchipudi, A., Fulde, M., Riesbeck, K., Bergmann, S., Blom, A. M. (2013). Streptococcus pneumoniae endopeptidase O (PepO) is a multifunctional plasminogen- and fibronectin-binding protein, facilitating evasion of innate immunity and invasion of host cells. J. Biol. Chem. 288, 6849–6863. doi: 10.1074/jbc.M112.405530
Agarwal, V., Sroka, M., Fulde, M., Bergmann, S., Riesbeck, K., Blom, A. M. (2014). Binding of streptococcus pneumoniae endopeptidase O (PepO) to complement component C1q modulates the complement attack and promotes host cell adherence. J. Biol. Chem. 289, 15833–15844. doi: 10.1074/jbc.M113.530212
Agrawal, A., Suresh, M. V., Singh, S. K., Ferguson, D. A. J. R. (2008). The protective function of human c-reactive protein in mouse models of streptococcus pneumoniae infection. Endocr. Metab. Immune Disord. Drug Targets 8, 231–237. doi: 10.2174/187153008786848321
Ali, Y. M., Lynch, N. J., Haleem, K. S., Fujita, T., Endo, Y., Hansen, S., et al. (2012). The lectin pathway of complement activation is a critical component of the innate immune response to pneumococcal infection. PloS Pathog. 8, e1002793. doi: 10.1371/journal.ppat.1002793
André, G. O., Borges, M. T., Assoni, L., Ferraz, L. F. C., Sakshi, P., Adamson, P., et al. (2021). Protective role of PhtD and its amino and carboxyl fragments against pneumococcal sepsis. Vaccine 39, 3626–3632. doi: 10.1016/j.vaccine.2021.04.068
Andre, G. O., Converso, T. R., Politano, W. R., Ferraz, L. F. C., Ribeiro, M. L., Leite, L. C. C., et al. (2017). Role of streptococcus pneumoniae proteins in evasion of complement-mediated immunity. Front. Microbiol. 8. doi: 10.3389/fmicb.2017.00224
Angel, C. S., Ruzek, M., Hostetter, M. K. (1994). Degradation of C3 by streptococcus pneumoniae. J. Infect. Dis. 170, 600–608. doi: 10.1093/infdis/170.3.600
Balachandran, P., Hollingshead, S. K., Paton, J. C., Briles, D. E. (2001). The autolytic enzyme LytA of streptococcus pneumoniae is not responsible for releasing pneumolysin. J. Bacteriol. 183, 3108–3116. doi: 10.1128/JB.183.10.3108-3116.2001
Biesma, D. H., Hannema, A. J., van Velzen-Blad, H., Mulder, L., van Zwieten, R., Kluijt, I., et al. (2001). A family with complement factor d deficiency. J. Clin. Invest. 108, 233–240. doi: 10.1172/JCI12023
Blom, A. M., Bergmann, S., Fulde, M., Riesbeck, K., Agarwal, V. (2014). Streptococcus pneumoniae phosphoglycerate kinase is a novel complement inhibitor affecting the membrane attack complex formation. J. Biol. Chem. 289, 32499–32511. doi: 10.1074/jbc.M114.610212
Bogaert, D., Thompson, C. M., Trzcinski, K., Malley, R., Lipsitch, M. (2010). The role of complement in innate and adaptive immunity to pneumococcal colonization and sepsis in a murine model. Vaccine 28, 681–685. doi: 10.1016/j.vaccine.2009.10.085
Bogaert, D., Weinberger, D., Thompson, C., Lipsitch, M., Malley, R. (2009). Impaired innate and adaptive immunity to streptococcus pneumoniae and its effect on colonization in an infant mouse model. Infect. Immun. 77, 1613–1622. doi: 10.1128/IAI.00871-08
Brady, A. M., Calix, J. J., Yu, J., Geno, K. A., Cutter, G. R., Nahm, M. H. (2014). Low invasiveness of pneumococcal serotype 11A is linked to ficolin-2 recognition of O-acetylated capsule epitopes and lectin complement pathway activation. J. Infect. Dis. 210, 1155–1165. doi: 10.1093/infdis/jiu195
Briles, D. E., Hollingshead, S. K., Swiatlo, E., Brooks-Walter, A., Szalai, A., Virolainen, A., et al. (1997). PspA and PspC: their potential for use as pneumococcal vaccines. Microb. Drug Resist. 3, 401–408. doi: 10.1089/mdr.1997.3.401
Broadley, S. P., Plaumann, A., Coletti, R., Lehmann, C., Wanisch, A., Seidlmeier, A., et al. (2016). Dual-track clearance of circulating bacteria balances rapid restoration of blood sterility with induction of adaptive immunity. Cell Host Microbe 20, 36–48. doi: 10.1016/j.chom.2016.05.023
Brouwer, M. C., de Gans, J., Heckenberg, S. G. B., Zwinderman, A. H., van der Poll, T., van de Beek, D. (2009). Host genetic susceptibility to pneumococcal and meningococcal disease: a systematic review and meta-analysis. Lancet Infect. Dis. 9, 31–44. doi: 10.1016/S1473-3099(08)70261-5
Brown, J. S., Hussell, T., Gilliland, S. M., Holden, D. W., Paton, J. C., Ehrenstein, M. R., et al. (2002). The classical pathway is the dominant complement pathway required for innate immunity to streptococcus pneumoniae infection in mice. Proc. Natl. Acad. Sci. U.S.A. 99, 16969–16974. doi: 10.1073/pnas.012669199
Brown, E. J., Joiner, K. A., Cole, R. M., Berger, M. (1983a). Localization of complement component 3 on streptococcus pneumoniae: anti-capsular antibody causes complement deposition on the pneumococcal capsule. Infect. Immun. 39, 403–409. doi: 10.1128/iai.39.1.403-409.1983
Brown, E. J., Joiner, K. A., Gaither, T. A., Hammer, C. H., Frank, M. M. (1983b). The interaction of C3b bound to pneumococci with factor h (beta 1H globulin), factor I (C3b/C4b inactivator), and properdin factor b of the human complement system. J. Immunol. 131, 409–415.
Casal, J., Tarragó, D. (2003). Immunity to streptococcus pneumoniae: Factors affecting production and efficacy. Curr. Opin. Infect. Dis. 16, 219–224. doi: 10.1097/01.qco.0000073770.11390.3c
Coonrod, J. D., Rylko-Bauer, B. (1977). Complement levels in pneumococcal pneumonia. Infect. Immun. 18, 14–22. doi: 10.1128/iai.18.1.14-22.1977
Croucher, N. J., Campo, J. J., Le, T. Q., Liang, X., Bentley, S. D., Hanage, W. P., et al. (2017). Diverse evolutionary patterns of pneumococcal antigens identified by pangenome-wide immunological screening. Proc. Natl. Acad. Sci. U.S.A. 114, E357–E366. doi: 10.1073/pnas.1613937114
Dalia, A. B., Standish, A. J., Weiser, J. N. (2010). Three surface exoglycosidases from streptococcus pneumoniae, NanA, BgaA, and StrH, promote resistance to opsonophagocytic killing by human neutrophils. Infect. Immun. 78, 2108–2116. doi: 10.1128/IAI.01125-09
Dalia, A. B., Weiser, J. N. (2011). Minimization of bacterial size allows for complement evasion and is overcome by the agglutinating effect of antibody. Cell Host Microbe 10, 486–496. doi: 10.1016/j.chom.2011.09.009
Dave, S., Carmicle, S., Hammerschmidt, S., Pangburn, M. K., McDaniel, L. S. (2004). Dual roles of PspC, a surface protein of streptococcus pneumoniae, in binding human secretory IgA and factor h. J. Immunol. 173, 471–477. doi: 10.4049/jimmunol.173.1.471
Deniset, J. F., Surewaard, B. G., Lee, W.-Y., Kubes, P. (2017). Splenic Ly6Ghigh mature and Ly6Gint immature neutrophils contribute to eradication of s. pneumoniae. J. Exp. Med. 214, 1333–1350. doi: 10.1084/jem.20161621
Dieudonné-Vatran, A., Krentz, S., Blom, A. M., Meri, S., Henriques-Normark, B., Riesbeck, K., et al. (2009). Clinical isolates of streptococcus pneumoniae bind the complement inhibitor C4b-binding protein in a PspC allele-dependent fashion. J. Immunol. 182, 7865–7877. doi: 10.4049/jimmunol.0802376
Donius, L. R., Handy, J. M., Weis, J. J., Weis, J. H. (2013). Optimal germinal center b cell activation and T-dependent antibody responses require expression of the mouse complement receptor Cr1. J. Immunol. 191, 434–447. doi: 10.4049/jimmunol.1203176
Eisen, D. P., Dean, M. M., Boermeester, M. A., Fidler, K. J., Gordon, A. C., Kronborg, G., et al. (2008). Low serum mannose-binding lectin level increases the risk of death due to pneumococcal infection. Clin. Infect. Dis. 47, 510–516. doi: 10.1086/590006
Gordon, D. L., Johnson, G. M., Hostetter, M. K. (1986). Ligand-receptor interactions in the phagocytosis of virulent streptococcus pneumoniae by polymorphonuclear leukocytes. J. Infect. Dis. 154, 619–626. doi: 10.1093/infdis/154.4.619
Griffioen, A. W., Rijkers, G. T., Janssens-Korpela, P., Zegers, B. J. (1991). Pneumococcal polysaccharides complexed with C3d bind to human b lymphocytes via complement receptor type 2. Infect. Immun. 59, 1839–1845. doi: 10.1128/iai.59.5.1839-1845.1991
Haas, K. M., Hasegawa, M., Steeber, D. A., Poe, J. C., Zabel, M. D., Bock, C. B., et al. (2002). Complement receptors CD21/35 link innate and protective immunity during streptococcus pneumoniae infection by regulating IgG3 antibody responses. Immunity 17, 713–723. doi: 10.1016/S1074-7613(02)00483-1
Haleem, K. S., Ali, Y. M., Yesilkaya, H., Kohler, T., Hammerschmidt, S., Andrew, P. W., et al. (2019). The pneumococcal surface proteins PspA and PspC sequester host C4-binding protein to inactivate complement C4b on the bacterial surface. Infect. Immun. 87, e00742–e00718. doi: 10.1128/IAI.00742-18
Hammerschmidt, S., Agarwal, V., Kunert, A., Haelbich, S., Skerka, C., Zipfel, P. F. (2007). The host immune regulator factor h interacts via two contact sites with the PspC protein of streptococcus pneumoniae and mediates adhesion to host epithelial cells. J. Immunol. 178, 5848–5858. doi: 10.4049/jimmunol.178.9.5848
Heesters, B. A., Carroll, M. C. (2016). The role of dendritic cells in s. pneumoniae transport to follicular dendritic cells. Cell Rep. 16, 3130–3137. doi: 10.1016/j.celrep.2016.08.049
Hostetter, M. K. (1999). Opsonic and nonopsonic interactions of C3 with streptococcus pneumoniae. Microb. Drug Resist. 5, 85–89. doi: 10.1089/mdr.1999.5.85
Hummell, D. S., Berninger, R. W., Tomasz, A., Winkelstein, J. A. (1981). The fixation of C3b to pneumococcal cell wall polymers as a result of activation of the alternative complement pathway. J. Immunol. 127, 1287–1289.
Hummell, D. S., Swift, A. J., Tomasz, A., Winkelstein, J. A. (1985). Activation of the alternative complement pathway by pneumococcal lipoteichoic acid. Infect. Immun. 47, 384–387. doi: 10.1128/iai.47.2.384-387.1985
Hyams, C., Camberlein, E., Cohen, J. M., Bax, K., Brown, J. S. (2010). The streptococcus pneumoniae capsule inhibits complement activity and neutrophil phagocytosis by multiple mechanisms. Infect. Immun. 78, 704–715. doi: 10.1128/IAI.00881-09
Hyams, C., Opel, S., Hanage, W., Yuste, J., Bax, K., Henriques-Normark, B., et al. (2011). Effects of streptococcus pneumoniae strain background on complement resistance. PloS One 6, e24581. doi: 10.1371/journal.pone.0024581
Hyams, C., Trzcinski, K., Camberlein, E., Weinberger, D. M., Chimalapati, S., Noursadeghi, M., et al. (2013). Streptococcus pneumoniae capsular serotype invasiveness correlates with the degree of factor h binding and opsonization with C3b/iC3b. Infect. Immun. 81, 354–363. doi: 10.1128/IAI.00862-12
Jedrzejas, M. J. (2001). Pneumococcal virulence factors: structure and function. Microbiol. Mol. Biol. Rev. 65, 187–207. doi: 10.1128/MMBR.65.2.187-207.2001
Johnson, M. K. (1977). Cellular location of pneumolysin. FEMS Microbiol. Lett. 2, 243–245. doi: 10.1111/j.1574-6968.1977.tb00951.x
Joiner, K., Brown, E., Hammer, C., Warren, K., Frank, M. (1983). Studies on the mechanism of bacterial resistance to complement-mediated killing. III. C5b-9 deposits stably on rough and type 7S, Immunology (1983) 130 (2), 845–849.
Jönsson, G., Truedsson, L., Sturfelt, G., Oxelius, V.-A., Braconier, J. H., Sjöholm, A. G. (2005). Hereditary C2 deficiency in Sweden: frequent occurrence of invasive infection, atherosclerosis, and rheumatic disease. Med. (Baltimore). 84, 23–34. doi: 10.1097/01.md.0000152371.22747.1e
Kadioglu, A., Weiser, J. N., Paton, J. C., Andrew, P. W. (2008). The role of streptococcus pneumoniae virulence factors in host respiratory colonization and disease. Nat. Rev. Microbiol. 6, 288–301. doi: 10.1038/nrmicro1871
Kang, Y.-S., Kim, J. Y., Bruening, S. A., Pack, M., Charalambous, A., Pritsker, A., et al. (2004). The c-type lectin SIGN-R1 mediates uptake of the capsular polysaccharide of streptococcus pneumoniae in the marginal zone of mouse spleen. Proc. Natl. Acad. Sci. U.S.A. 101, 215–220. doi: 10.1073/pnas.0307124101
Kasanmoentalib, E. S., Valls Seron, M., Ferwerda, B., Tanck, M. W., Zwinderman, A. H., Baas, F., et al. (2017). Mannose-binding lectin-associated serine protease 2 (MASP-2) contributes to poor disease outcome in humans and mice with pneumococcal meningitis. J. Neuroinflamm. 14, 2. doi: 10.1186/s12974-016-0770-9
Krarup, A., Sørensen, U. B. S., Matsushita, M., Jensenius, J. C., Thiel, S. (2005). Effect of capsulation of opportunistic pathogenic bacteria on binding of the pattern recognition molecules mannan-binding lectin, l-ficolin, and h-ficolin. Infect. Immun. 73, 1052–1060. doi: 10.1128/IAI.73.2.1052-1060.2005
Li, J., Glover, D. T., Szalai, A. J., Hollingshead, S. K., Briles, D. E. (2007). PspA and PspC minimize immune adherence and transfer of pneumococci from erythrocytes to macrophages through their effects on complement activation. Infect. Immun. 75, 5877–5885. doi: 10.1128/IAI.00839-07
Li, Q., Li, Y. X., Stahl, G. L., Thurman, J. M., He, Y., Tong, H. H. (2011). Essential role of factor b of the alternative complement pathway in complement activation and opsonophagocytosis during acute pneumococcal otitis media in mice. Infect. Immun. 79, 2578–2585. doi: 10.1128/IAI.00168-11
Li, J., Szalai, A. J., Hollingshead, S. K., Nahm, M. H., Briles, D. E. (2009). Antibody to the type 3 capsule facilitates immune adherence of pneumococci to erythrocytes and augments their transfer to macrophages. Infect. Immun. 77, 464–471. doi: 10.1128/IAI.00892-08
Lu, L., Ma, Z., Jokiranta, T. S., Whitney, A. R., DeLeo, F. R., Zhang, J.-R. (2008). Species-specific interaction of streptococcus pneumoniae with human complement factor h. J. Immunol. 181, 7138–7146. doi: 10.4049/jimmunol.181.10.7138
Lysenko, E. S., Ratner, A. J., Nelson, A. L., Weiser, J. N. (2005). The role of innate immune responses in the outcome of interspecies competition for colonization of mucosal surfaces. PloS Pathog. 1, e1. doi: 10.1371/journal.ppat.0010001
Marshall, H., Aguayo, S., Kilian, M., Petersen, F., Bozec, L., Brown, J. (2020). In vivo relationship between the nano-biomechanical properties of streptococcal polysaccharide capsules and virulence phenotype. ACS Nano. 14, 1070–1083. doi: 10.1021/acsnano.9b08631
Marshall, H., José, R. J., Kilian, M., Petersen, F. C., Brown, J. S. (2021). Effects of expression of streptococcus pneumoniae PspC on the ability of streptococcus mitis to evade complement-mediated immunity. Front. Microbiol. 12. doi: 10.3389/fmicb.2021.773877
Melin, M., Di Paolo, E., Tikkanen, L., Jarva, H., Neyt, C., Käyhty, H., et al. (2010a). Interaction of pneumococcal histidine triad proteins with human complement. Infect. Immun. 78, 2089–2098. doi: 10.1128/IAI.00811-09
Melin, M., Trzciński, K., Meri, S., Käyhty, H., Väkeväinen, M. (2010b). The capsular serotype of streptococcus pneumoniae is more important than the genetic background for resistance to complement. Infect. Immun. 78, 5262–5270. doi: 10.1128/IAI.00740-10
Mitchell, T. J., Andrew, P. W., Saunders, F. K., Smith, A. N., Boulnois, G. J. (1991). Complement activation and antibody binding by pneumolysin via a region of the toxin homologous to a human acute-phase protein. Mol. Microbiol. 5, 1883–1888. doi: 10.1111/j.1365-2958.1991.tb00812.x
Mohan, S., Hertweck, C., Dudda, A., Hammerschmidt, S., Skerka, C., Hallström, T., et al (2012). Tuf of Streptococcus pneumoniae is a surface displayed human complement regulator binding protein. Mol. Immunol 62(1):249–264. doi: 10.1016/j.molimm.2014.06.029
Mukerji, R., Mirza, S., Roche, A. M., Widener, R. W., Croney, C. M., Rhee, D.-K., et al. (2012). Pneumococcal surface protein a inhibits complement deposition on the pneumococcal surface by competing with the binding of c-reactive protein to cell-surface phosphocholine. J. Immunol. 189, 5327–5335. doi: 10.4049/jimmunol.1201967
Neth, O., Jack, D. L., Dodds, A. W., Holzel, H., Klein, N. J., Turner, M. W. (2000). Mannose-binding lectin binds to a range of clinically relevant microorganisms and promotes complement deposition. Infect. Immun. 68, 688–693. doi: 10.1128/IAI.68.2.688-693.2000
Ochs, M. M., Bartlett, W., Briles, D. E., Hicks, B., Jurkuvenas, A., Lau, P., et al. (2008). Vaccine-induced human antibodies to PspA augment complement C3 deposition on streptococcus pneumoniae. Microb. Pathog. 44, 204–214. doi: 10.1016/j.micpath.2007.09.007
Ogunniyi, A. D., Grabowicz, M., Mahdi, L. K., Cook, J., Gordon, D. L., Sadlon, T. A., et al. (2009). Pneumococcal histidine triad proteins are regulated by the Zn2+-dependent repressor AdcR and inhibit complement deposition through the recruitment of complement factor h. FASEB J. 23, 731–738. doi: 10.1096/fj.08-119537
Pathak, A., Bergstrand, J., Sender, V., Spelmink, L., Aschtgen, M.-S., Muschiol, S., et al. (2018)3398. Factor h binding proteins protect division septa on encapsulated streptococcus pneumoniae against complement C3b deposition and amplification. Nat. Commun. 9. doi: 10.1038/s41467-018-05494-w
Paton, J. C., Rowan-Kelly, B., Ferrante, A. (1984). Activation of human complement by the pneumococcal toxin pneumolysin. Infect. Immun. 43, 1085–1087. doi: 10.1128/iai.43.3.1085-1087.1984
Peset Llopis, M. J., Harms, G., Hardonk, M. J., Timens, W. (1996). Human immune response to pneumococcal polysaccharides: complement-mediated localization preferentially on CD21-positive splenic marginal zone b cells and follicular dendritic cells. J. Allergy Clin. Immunol. 97, 1015–1024. doi: 10.1016/S0091-6749(96)80078-9
Price, K. E., Camilli, A. (2009). Pneumolysin localizes to the cell wall of streptococcus pneumoniae. J. Bacteriol. 191, 2163–2168. doi: 10.1128/JB.01489-08
Quin, L. R., Carmicle, S., Dave, S., Pangburn, M. K., Evenhuis, J. P., McDaniel, L. S. (2005). In vivo binding of complement regulator factor h by streptococcus pneumoniae. J. Infect. Dis. 192, 1996–2003. doi: 10.1086/497605
Quin, L. R., Moore, Q. C., McDaniel, L. S. (2007). Pneumolysin, PspA, and PspC contribute to pneumococcal evasion of early innate immune responses during bacteremia in mice. Infect. Immun. 75, 2067–2070. doi: 10.1128/IAI.01727-06
Ramos-Sevillano, E., Moscoso, M., García, P., García, E., Yuste, J. (2011). Nasopharyngeal colonization and invasive disease are enhanced by the cell wall hydrolases LytB and LytC of streptococcus pneumoniae. PloS One 6, e23626. doi: 10.1371/journal.pone.0023626
Ramos-Sevillano, E., Urzainqui, A., Campuzano, S., Moscoso, M., González-Camacho, F., Domenech, M., et al. (2015). Pleiotropic effects of cell wall amidase LytA on streptococcus pneumoniae sensitivity to the host immune response. Infect. Immun. 83, 591–603. doi: 10.1128/IAI.02811-14
Reis, E. S., Mastellos, D. C., Hajishengallis, G., Lambris, J. D. (2019). New insights into the immune functions of complement. Nat. Rev. Immunol. 19, 503–516. doi: 10.1038/s41577-019-0168-x
Ren, B., McCrory, M. A., Pass, C., Bullard, D. C., Ballantyne, C. M., Xu, Y., et al. (2004a). The virulence function of streptococcus pneumoniae surface protein a involves inhibition of complement activation and impairment of complement receptor-mediated protection. J. Immunol. 173, 7506–7512. doi: 10.4049/jimmunol.173.12.7506
Ren, B., Szalai, A. J., Hollingshead, S. K., Briles, D. E. (2004b). Effects of PspA and antibodies to PspA on activation and deposition of complement on the pneumococcal surface. Infect. Immun. 72, 114–122. doi: 10.1128/IAI.72.1.114-122.2004
Rukke, H. V., Kalluru, R. S., Repnik, U., Gerlini, A., José, R. J., Periselneris, J., et al. (2014). Protective role of the capsule and impact of serotype 4 switching on streptococcus mitis. Infection. Immun. 82, 3790–3801. doi: 10.1128/IAI.01840-14
Rupprecht, T. A., Angele, B., Klein, M., Heesemann, J., Pfister, H.-W., Botto, M., et al. (2007). Complement C1q and C3 are critical for the innate immune response to streptococcus pneumoniae in the central nervous system. J. Immunol. 178, 1861–1869. doi: 10.4049/jimmunol.178.3.1861
Sabharwal, V., Ram, S., Figueira, M., Park, I. H., Pelton, S. I. (2009). Role of complement in host defense against pneumococcal otitis media. Infect. Immun. 77, 1121–1127. doi: 10.1128/IAI.01148-08
Sjöholm, A. G., Jönsson, G., Braconier, J. H., Sturfelt, G., Truedsson, L. (2006). Complement deficiency and disease: an update. Mol. Immunol. 43, 78–85. doi: 10.1016/j.molimm.2005.06.025
Skattum, L., van Deuren, M., van der Poll, T., Truedsson, L. (2011). Complement deficiency states and associated infections. Mol. Immunol. 48, 1643–1655. doi: 10.1016/j.molimm.2011.05.001
Suresh, M. V., Singh, S. K., Ferguson, D. A. J. R., Agrawal, A. (2007). Human c-reactive protein protects mice from streptococcus pneumoniae infection without binding to pneumococcal c-polysaccharide. J. Immunol. 178, 1158–1163. doi: 10.4049/jimmunol.178.2.1158
Terrasse, R., Tacnet-Delorme, P., Moriscot, C., Pérard, J., Schoehn, G., Vernet, T., et al (2012). Human and pneumococcal cell surface glyceraldehyde-3-phosphate dehydrogenase (GAPDH) proteins are both ligands of human C1q protein. J. Biol. Cheml 287(51):42620–42633. doi: 10.1074/jbc.M112.423731
Troeger, C., Blacker, B., Khalil, I. A., Rao, P. C., Cao, J., Zimsen, S. R. M., et al. (2018). Estimates of the global, regional, and national morbidity, mortality, and aetiologies of lower respiratory infections in 195 countrie 1990–2016: A systematic analysis for the global burden of disease study 2016. Lancet Infect. Dis. 18, 1191–1210. doi: 10.1016/S1473-3099(18)30310-4
van der Maten, E., de Bont, C. M., de Groot, R., de Jonge, M. I., Langereis, J. D., van der Flier, M. (2016). Alternative pathway regulation by factor h modulates streptococcus pneumoniae induced proinflammatory cytokine responses by decreasing C5a receptor crosstalk. Cytokine 88, 281–286. doi: 10.1016/j.cyto.2016.09.025
van der Maten, E., van den Broek, B., de Jonge, M. I., Rensen, K. J. W., Eleveld, M. J., Zomer, A. L., et al. (2018). Streptococcus pneumoniae PspC subgroup prevalence in invasive disease and differences in contribution to complement evasion. Infect. Immun. 86, e00010–e00018. doi: 10.1128/IAI.00010-18
van Lookeren Campagne, M., Verschoor, A. (2018). Pathogen clearance and immune adherence “revisited”: Immuno-regulatory roles for CRIg. Semin. Immunol. 37, 4–11. doi: 10.1016/j.smim.2018.02.007
van Lookeren Campagne, M., Wiesmann, C., Brown, E. J. (2007). Macrophage complement receptors and pathogen clearance. Cell. Microbiol. 9, 2095–2102. doi: 10.1111/j.1462-5822.2007.00981.x
van Rossum, A. M. C., Lysenko, E. S., Weiser, J. N. (2005). Host and bacterial factors contributing to the clearance of colonization by streptococcus pneumoniae in a murine model. Infect. Immun. 73, 7718–7726. doi: 10.1128/IAI.73.11.7718-7726.2005
Walport, M. J. (2001). Complement. first of two parts. N. Engl. J. Med. 344, 1058–1066. doi: 10.1056/NEJM200104053441406
Watson, D. A., Musher, D. M. (1990). Interruption of capsule production in streptococcus pneumonia serotype 3 by insertion of transposon Tn916. Infect. Immun. 58, 3135–3138. doi: 10.1128/iai.58.9.3135-3138.1990
Watson, D. A., Musher, D. M., Verhoef, J. (1995). Pneumococcal virulence factors and host immune responses to them. Eur. J. Clin. Microbiol. Infect. Dis. 14, 479–490. doi: 10.1007/BF02113425
Weiss, S. J., Ahmed, A. E., Bonagura, V. R. (1998). Complement factor d deficiency in an infant first seen with pneumococcal neonatal sepsis. J. Allergy Clin. Immunol. 102, 1043–1044. doi: 10.1016/S0091-6749(98)70346-X
Wilson, R., Cohen, J. M., Reglinski, M., Jose, R. J., Chan, W. Y., Marshall, H., et al. (2017). Naturally acquired human immunity to pneumococcus is dependent on antibody to protein antigens. PloS Pathog. 13, e1006137. doi: 10.1371/journal.ppat.1006137
Winkelstein, J. A., Tomasz, A. (1978). Activation of the alternative complement pathway by pneumococcal cell wall teichoic acid. J. Immunol. 120, 174–178.
Woehrl, B., Brouwer, M. C., Murr, C., Heckenberg, S. G. B., Baas, F., Pfister, H. W., et al. (2011). Complement component 5 contributes to poor disease outcome in humans and mice with pneumococcal meningitis. J. Clin. Invest. 121, 3943–3953. doi: 10.1172/JCI57522
Xu, Y., Ma, M., Ippolito, G. C., Schroeder, H. W. J. R., Carroll, M. C., Volanakis, J. E. (2001). Complement activation in factor d-deficient mice. Proc. Natl. Acad. Sci. U.S.A. 98, 14577–14582. doi: 10.1073/pnas.261428398
Yuste, J., Botto, M., Bottoms, S. E., Brown, J. S. (2007). Serum amyloid p aids complement-mediated immunity to streptococcus pneumoniae. PloS Pathog. 3, e120. doi: 10.1371/journal.ppat.0030120
Yuste, J., Botto, M., Paton, J. C., Holden, D. W., Brown, J. S. (2005). Additive inhibition of complement deposition by pneumolysin and PspA facilitates streptococcus pneumoniae septicemia. J. Immunol. 175, 1813–1819. doi: 10.4049/jimmunol.175.3.1813
Yuste, J., Khandavilli, S., Ansari, N., Muttardi, K., Ismail, L., Hyams, C., et al. (2010). The effects of PspC on complement-mediated immunity to streptococcus pneumoniae vary with strain background and capsular serotype. Infect. Immun. 78, 283–292. doi: 10.1128/IAI.00541-09
Yuste, J., Sen, A., Truedsson, L., Jönsson, G., Tay, L.-S., Hyams, C., et al. (2008). Impaired opsonization with C3b and phagocytosis of streptococcus pneumoniae in sera from subjects with defects in the classical complement pathway. Infect. Immun. 76, 3761–3770. doi: 10.1128/IAI.00291-08
Zhang, Y., Masi, A. W., Barniak, V., Mountzouros, K., Hostetter, M. K., Green, B. A. (2001). Recombinant PhpA protein, a unique histidine motif-containing protein from streptococcus pneumoniae, protects mice against intranasal pneumococcal challenge. Infect. Immun. 69, 3827–3836. doi: 10.1128/IAI.69.6.3827-3836.2001
Keywords: Streptococcus pneumoniae, streptococcal infections, complement - immunological terms, immune evasion, innate immunity
Citation: Gil E, Noursadeghi M and Brown JS (2022) Streptococcus pneumoniae interactions with the complement system. Front. Cell. Infect. Microbiol. 12:929483. doi: 10.3389/fcimb.2022.929483
Received: 26 April 2022; Accepted: 08 July 2022;
Published: 28 July 2022.
Edited by:
Karthik Subramanian, Rajiv Gandhi Centre for Biotechnology, IndiaReviewed by:
Michelle Darrieux, Sao Francisco University, BrazilCopyright © 2022 Gil, Noursadeghi and Brown. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Eliza Gil, ZWxpemEuZ2lsQG5ocy5uZXQ=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.