
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Cell. Infect. Microbiol., 26 August 2022
Sec. Microbiome in Health and Disease
Volume 12 - 2022 | https://doi.org/10.3389/fcimb.2022.903828
This article is part of the Research TopicMicrobiota and Cardiovascular DiseasesView all 4 articles
 Hao Yu1†
Hao Yu1† Le Li1†Yu Deng1Guolan Zhang1Mimi Jiang1He Huang1
Le Li1†Yu Deng1Guolan Zhang1Mimi Jiang1He Huang1 Cheng Li2Zhiyu Lv3
Cheng Li2Zhiyu Lv3 Yingshun Zhou4*
Yingshun Zhou4* Xing Liu1,5*
Xing Liu1,5*An increasing number of studies have shown that the gut microbiome plays an important role in the development of coronary heart disease (CHD). However, there are no clear studies on the relationship between the gut microbiome and the number of stenotic coronary arteries. To clarify whether the gut microbiome is associated with the number of stenotic coronary arteries in CHD, we performed the 16S rRNA gene sequencing for the V3-V4 region in the gut microbiota from 9 healthy controls (C) and 36 CHD patients, which including 25 CHD patients with multivessel (MV) lesion and 11 CHD patients with single-vessel (SV) lesion. It showed that the abundance of the genus Escherichia-Shigella was significantly increased in the MV and SV groups compared with C group, while the abundance of the genera Subdoligranulum and Collinsella was significantly decreased. Biomarkers based on three gut microbiotas (Escherichia-Shigella, Subdoligranulum, and Collinsella) and three plasma metabolites(left atrial diameter (LA), low density lipoprotein (LDL), and total bile acids (TBA)) were able to distinguish CHD patients with different numbers of stenotic coronary arteries. Functional prediction of the gut microbiome was performed based on the Kyoto Encyclopedia of Genes and Genomes (KEGG) database. The results showed that the gut microbial function of MV and SV group patients was richer than C group in betaine biosynthesis and unsaturated fatty acid biosynthesis, in the contrast less than C group in sphingolipid metabolism and primary bile acid biosynthesis. In summary, our study showed that the composition and function of the gut microbiome changed significantly from healthy controls to CHD patients with different numbers of coronary lesions.
Coronary heart disease (CHD) is defined as cardiac dysfunction and/or organic disease caused by insufficient myocardial blood supply due to coronary artery stenosis. Atherosclerosis (AS) is the most common cause of coronary artery stenosis. More diseased coronary arteries caused worse outcome in patients. Cohort studies have shown that the number of coronary with ≥75% lumen obstruction is independently associated with an increased risk of 1-year mortality and major adverse cardiovascular events (Ndrepepa et al., 2012). An increasing number of studies have shown significant changes in the composition and function of the gut microbiome in patients with CHD. Koren et al. identified Chryseomonas, Veillonella, and Streptococcus in AS plaque samples, and some gut microbiome are common to atherosclerotic plaques and are correlated with cholesterol levels (Koren et al., 2011). A metagenome-wide association study showed that the atherosclerotic cardiovascular disease (ACVD) gut microbiome deviates from the healthy status by increased abundance of Enterobacteriaceae and Streptococcus spp. (Jie et al., 2017). Liu et al. found that the bacterial co-abundance group (CAG), represented by Roseburia, Klebsiella, Clostridium IV, and Ruminococcaceae, was enriched in the gut microbiota of samples with CHD and had characteristic changes at different stages of CHD (Liu et al., 2019). CHD can be categorized as either acute coronary syndromes (ACS) or chronic coronary syndromes (CCS) according to the severity of clinical symptoms (Knuuti et al., 2020). Previous study showed that that the CCS group experienced a significantly higher ratio of Firmicutes/Bacteroidetes compared with the control group (Sawicka-Smiarowska et al., 2021).A new study shows that ACS patients had distinct serum metabolome and gut microbial signatures as compared with control individuals, and were depleted in a previously unknown bacterial species of the Clostridiaceae family (Talmor-Barkan et al., 2022). The relationship between the alteration of microbiota and patients with CHD is not only correlation but also causality (Kwon et al., 2022). Several studies have shown that the occurrence and clinical classification of CHD are related to the changes of gut microbiome, but it remains to be determined whether these changes can affect the number of stenotic coronary arteries.
To address the above questions, we analyzed the gut microbial characteristics of 45 hospitalized patients who had undergone coronary angiography through high-throughput sequencing, including 36 CHD patients (including multivessel (MV) group N=25, single-vessel (SV) group N=11) and 9 healthy controls. Based on 16S rRNA V3-V4 region sequencing and statistical analysis of clinical features, we identified the gut microbiota and clinical features associated with an increased number of stenotic coronary arteries in CHD and further established relationships. This information may contribute to construct a disease classifier to distinguish healthy controls from patients with CHD with different numbers of coronary stenotic lesions.
The studies involving human participants were reviewed and approved by The Ethics Committee of The Affiliated Hospital of Southwest Medical University. (Approval no. KY2022104). All subjects were voluntarily recruited and informed of the nature of the study before sample collection. Written informed consent was obtained from all study subjects.
We recruited 45 patients who had undergone coronary angiography at the Affiliated Hospital of Southwest Medical University. We took 9 people with no coronary plaques and smooth intima as the control (C) group; subject with more than 75% stenosis of any of coronary arteries or branches was regarded as the CHD group: 1) 11 people with only one coronary artery stenosis degree ≥75% and other coronary intima completely smooth were regarded as the single-vessel (SV) disease group; 2) 25 people with 2 or more coronary arteries with a stenosis degree ≥75% were regarded as the multivessel (MV) disease group. Patients were excluded if they had any gastrointestinal disease, had a history of gastrointestinal surgery in one year, or had used gut microbiome preparations or antibiotics in the past 1 month. All patients’ feces were collected at the time of their initial appointment ensuring they haven’t eaten lipid-lowering and antiplatelet drugs, which may prevent coronary atherosclerotic. Fresh feces were collected from each subject on the day following coronary angiography, and all collected samples were transported immediately to the laboratory and stored at -80°C.
Bacterial DNA was isolated from fecal samples by bead milling for DNA extraction (Godon et al., 1997) and sequenced of the V3-V4 region of the 16S rRNA gene. Deoxyribonucleic acid extracted from each sample was used as a template to amplify the V3-V4 region of the 16S rRNA gene with PCR. Polymerase chain reaction amplification, polymerase chain reaction amplicon sequencing, and quality control of the raw data were performed (Zhang et al., 2010). Sequencing libraries of the V3-V4 region of the 16S rRNA gene were prepared by mixing the purified products in equal proportions for sequencing using the Illumina MiSeq system (Illumina, USA) to generate 100 bp paired-end reads in the forwards and reverse directions (Zhang et al., 2016).
Operational taxonomic units (OTUs) were clustered at the cutoff of 97% by using USEARCH v.8.0 (Edgar, 2013). The protocol can be found on the website (http://drive5.com/usearch/manual/uparse_pipeline.html). By comparison with the Silva database (Release138, http://www.arb-silva.de), the RDP classifier (RDP database version 11.5, http://rdp.cme.msu.edu/classifier/classifier.jsp) Bayesian algorithm was used to taxonomically analyze the OTU representative sequences at a 97% similarity level, and the community species composition of each sample was counted at each taxonomic level: domain, kingdom, phylum, class, order, family, genus, and species. The taxonomic composition of each group was visualized as a stacked bar plot at the phylum level and genus level with the ggplot2 package. The QIIME platform (http://qiime.org/scripts/assign_taxonomy.html) was used for alpha and beta diversity analysis. The Shannon index, observed OTUs, and Simpson index were evaluated. Beta diversity analysis was performed using standardized OTU abundance tables, including principal component analysis (PCA) and principal coordinate analysis (PCoA) based on Bray-Curtis distance. Partial least-squares discrimination analysis (PLS-DA) and analysis of similarities (ANOSIM) were used to test for statistical significance among the three groups. Wherever mentioned, the Benjamini-Hochberg method was used to control the false discovery rate (FDR). Data visualization was achieved using the vegan package and the mixOmics package of the R language. One-way analysis of variance (ANOVA) and Kruskal-Wallis test were used to find differential species between groups. Linear discriminant analysis Effect Size (LEfSe, http://huttenhower.sph.harvard.edu/galaxy/root?tool_id=lefse_upload) was used to find communities or species that had significant differential effects on grouping. Data visualization was achieved using the stats package for R. DESeq2 was utilized to identify significantly differential features, and the Benjamini-Hochberg method was used to control the FDR. Phylogenetic Investigation of Communities by Reconstruction of Unobserved States (PICRUSt2) was utilized to predict the functional compositions. Pathways that were different in abundance among the C, SV and MV groups were obtained using Welch’s t-test by STAMP software (v2.1.3). The visualization of the identified pathways was obtained by using the pheatmap package. To obtain functional predictions based on the 16S rRNA sequences, the taxonomic classification of sequences was performed based on the Kyoto Encyclopedia of Genes and Genomes (KEGG,http://www.genome.jp/kegg/).
Spearman correlations among important bacterial taxa, clinical features and metabolic pathway were calculated by using SPSS 23.0. The correlations between features were visualized using the pheatmap package.
According to the relative abundance of bacteria at the genus level, the Bray-Curtis distance was calculated, and PAM (Partitioning Around Medoids) clustering was performed. Then, the optimal cluster K value was calculated by the Calinski-Harabasz (CH) index. Finally, principal coordinates analysis (PCoA, K ≥ 2) was used for visualization. Data analysis and visualization were performed using the R package ade4 package, cluster package, and clusterSim. At the same time, based on the species abundance information and the results of enterotype analysis, species with significant differences among enterotypes were identified by statistical testing. Species with significant differences and the highest relative abundance among enterotypes were defined as the name of the enterotype.
To identify the relationship between the number of stenotic coronary arteries and gut microbiota, we collected feces from 45 patients who underwent coronary angiography at the Affiliated Hospital of Southwest Medical University (detailed in the “Materials and methods” section) and divided them into a control group (C) (n = 9), a MV group (n = 25), and a SV group (n = 11). The characteristics and traditional cardiovascular risk factors for the participants were summarized in Table 1. Left ventricular ejection fraction (LVEF) and left ventricular end diastolic diameter (LVDd) are the most important indicators to evaluate left ventricular function. Right ventricular diameter (RV) can be used to assess right ventricular function. N-terminal pro-B type natriuretic peptide (NT-proBNP) is an index for the overall evaluation of cardiac function. We also counted NT-proBNP, LVEF, LVDd, RV and other indicators in Table 1 to analyze the differences in cardiac function of patients in each group as a whole. Except for the significant differences in the triglyceride (TG), high density lipoprotein (HDL) and LVDd levels between C vs. SV and C vs. MV (P < 0.05), other clinical features showed no significant difference between the C and CHD subgroups. However, the LVDd of patients in MV group and SV group is basically within the normal range. Therefore, we supposed that the number of coronary stenotic vessels in patients with CHD is only affected by the differences in the composition and structure of the gut microbiota.
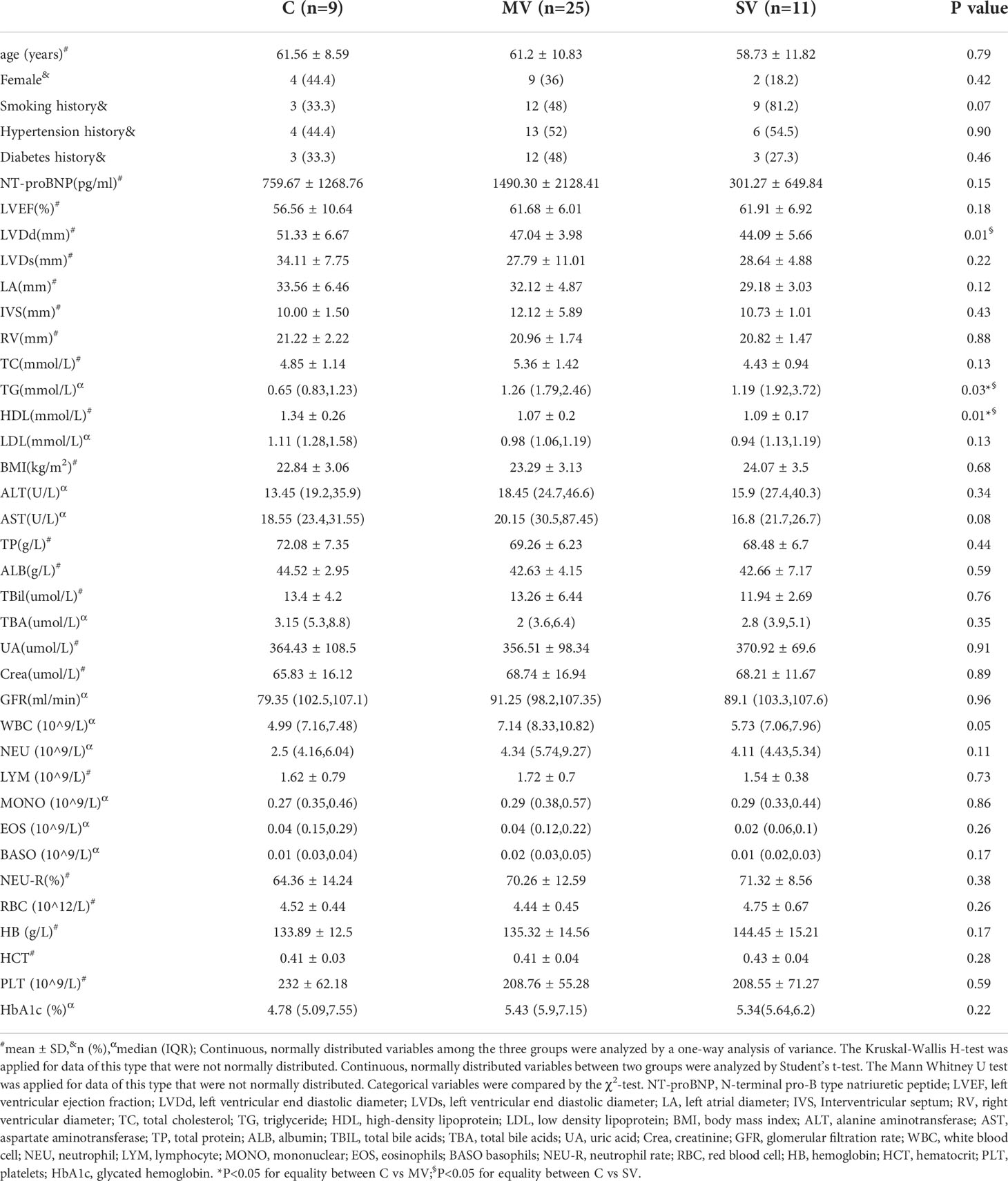
Table 1 Clinical characteristics of study participants.
We sequenced the V3-V4 region of the gut microbiome 16S rRNA gene in 9 healthy controls (C), 25 patients with multivessel coronary artery disease (MV) CHD, and 11 patients with single-vessel coronary artery disease (SV) CHD (sequences were detailed in Supplementary Data 1). After quality control and removal of human DNA, a total of 5,396,535 sequences were obtained, with an average of 119,923 sequences per sample and an average length of 417 bp per sample sequence. Using the Silva database (Release138http://www.arb-silva.de), reads from all samples were species annotated, and species clustering was performed according to 97% similarity, resulting in a total of 1002 OTUs. Then, we clustered the OTUs at the genus level, resulting in 306 gut microbiota genera. After excluding the genera with an average abundance contributing with less than 0.01% to the total, 211 genera were used for the subsequent analyses (Supplementary Table 1). Rarefaction analysis shows that observed-genera numbers tend to stabilize when the sample size of each group reaches 10 (Figure 1A). The revealed that the gut microbiota in our population capture most gut microbiota members of human. At the same time, rarefaction analysis also shows that the number of OTUs observed also tends to be stable when the sequence reads reaches 60000, indicating the sequencing depth is sufficient (Figure 1B). At the phylum level, the gut microbiota of all subjects was mainly classified into five phyla: Firmicutes, Bacteroides, Proteobacteria, Actinobacteriota, and Verrucomicrobiota. At the genus level, the gut microbiota was mainly composed of Bacteroides, Blautia, Escherichia-Shigella, Faecalibacterium, and Streptococcus (Supplementary Figure S1).
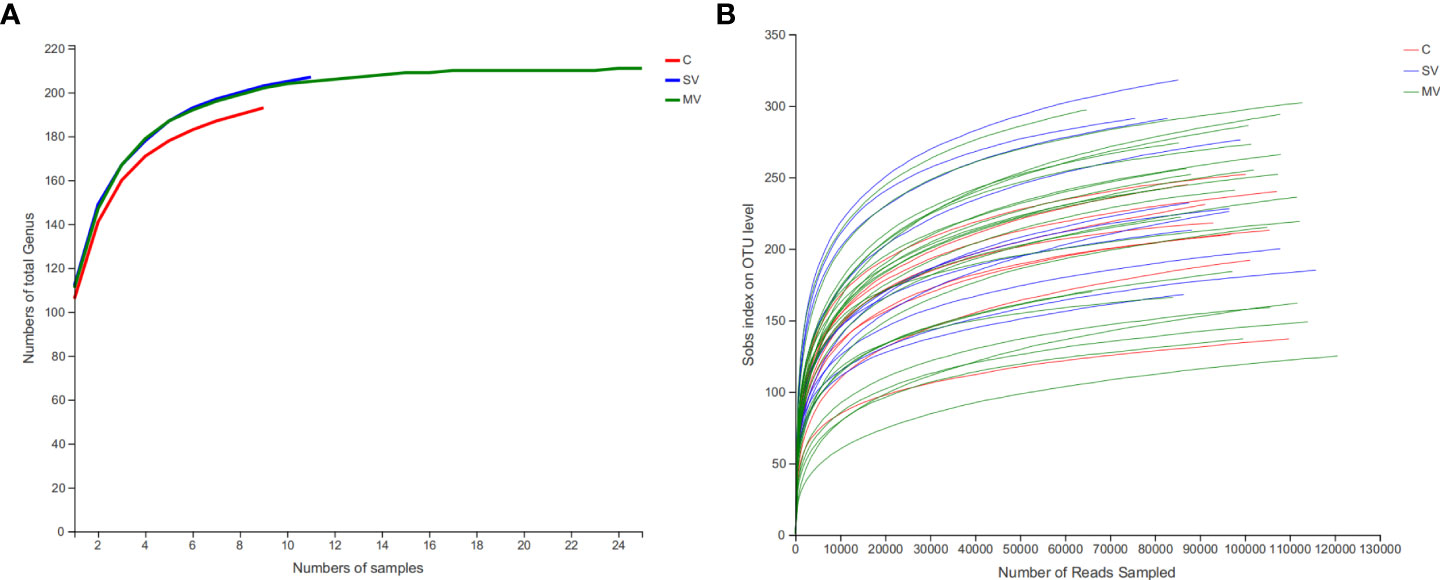
Figure 1 Coverage of members in the gut microbiota of each group subject. (A) Rarefaction curves of detected gut microbiota genus of patient in each group reach the saturation stage with increasing numbers of samples, indicating that the gut microbiota in our population capture most gut microbiota members of human. (B) Rarefaction curves of detected bacterial OTUs of the gut microbiota from each group subject reach saturation stage with increasing sequencing depth.
Alpha diversity analysis showed no significant difference among three groups in either gene richness or diversity (Supplementary Figure S2). As seen by principal component analysis (PCA) and principal coordinates analysis (PCoA), the distribution of gut microbiota among the three groups showed a tendency to separate (Supplementary Figure S3), indicating that the flora among the three groups was evidently different. We further showed differences in microbiota among the three groups by PLS-DA (Figure 2A). The Kruskal-Wallis test was performed to identify differential species of each group. The results showed that the Subdoligranulum and Collinsella genera were significantly more abundant in group C subjects than in the SV and MV groups (MV vs. C, P < 0.05; MV vs. SV, P < 0.01). At the same time, the abundance of the Escherichia-Shigella genus was significantly higher in subjects in the MV group than in those in the C and SV groups (Figure 2B) (C vs. MV, P < 0.05; C vs. SV, P < 0.01).
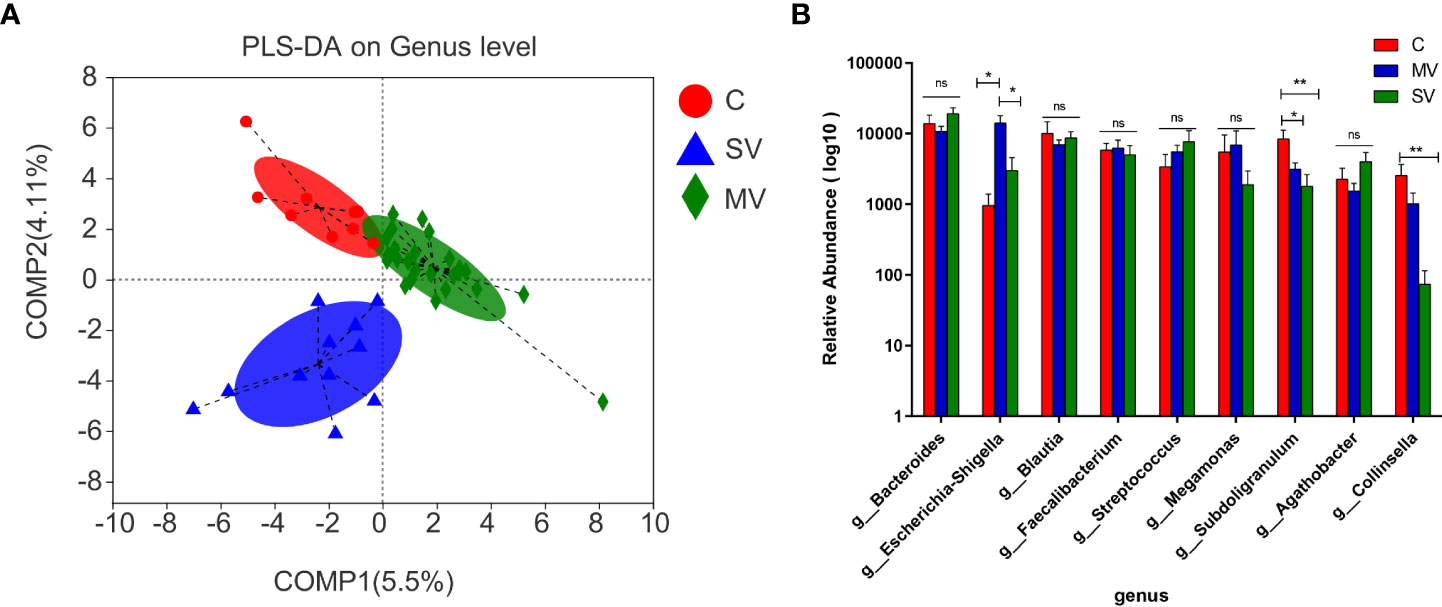
Figure 2 Difference test and significant different genera among groups microbiome. (A) Partial least-square discrimination analysis (PLS-DA). (B) The Kruskal-Wallis test was applied for the difference of gut microbiota among three groups. Tukey test was performed followup tests between any two groups. *P values < 0.05, **P values < 0.01, ns, no significance.
Next, we used linear discriminant analysis (LDA) to identify the gut microbiota that affected grouping differences (Figure 3). The results showed that Escherichia-Shigella and Subdoligranulum contributed the most to the difference in C vs. MV, Collinsella, and Subdoligranulum contributed the most to the difference in C vs. SV, and Geobacillus and Escherichia-Shigella contributed the most to the difference in SV vs. MV. At the bacterial phylum level, Proteobacteria was the main phylum distinguishing the C vs. MV group, Bacteroides was the main phylum distinguishing the SV vs. MV group, while the C vs. SV group had no significant difference at the phylum level. Our study showed that enrichment of Escherichia-Shigella was associated with multivessel coronary stenosis. In contrast, Subdoligranulum and Collinsella may be associated with normal coronary arteries.
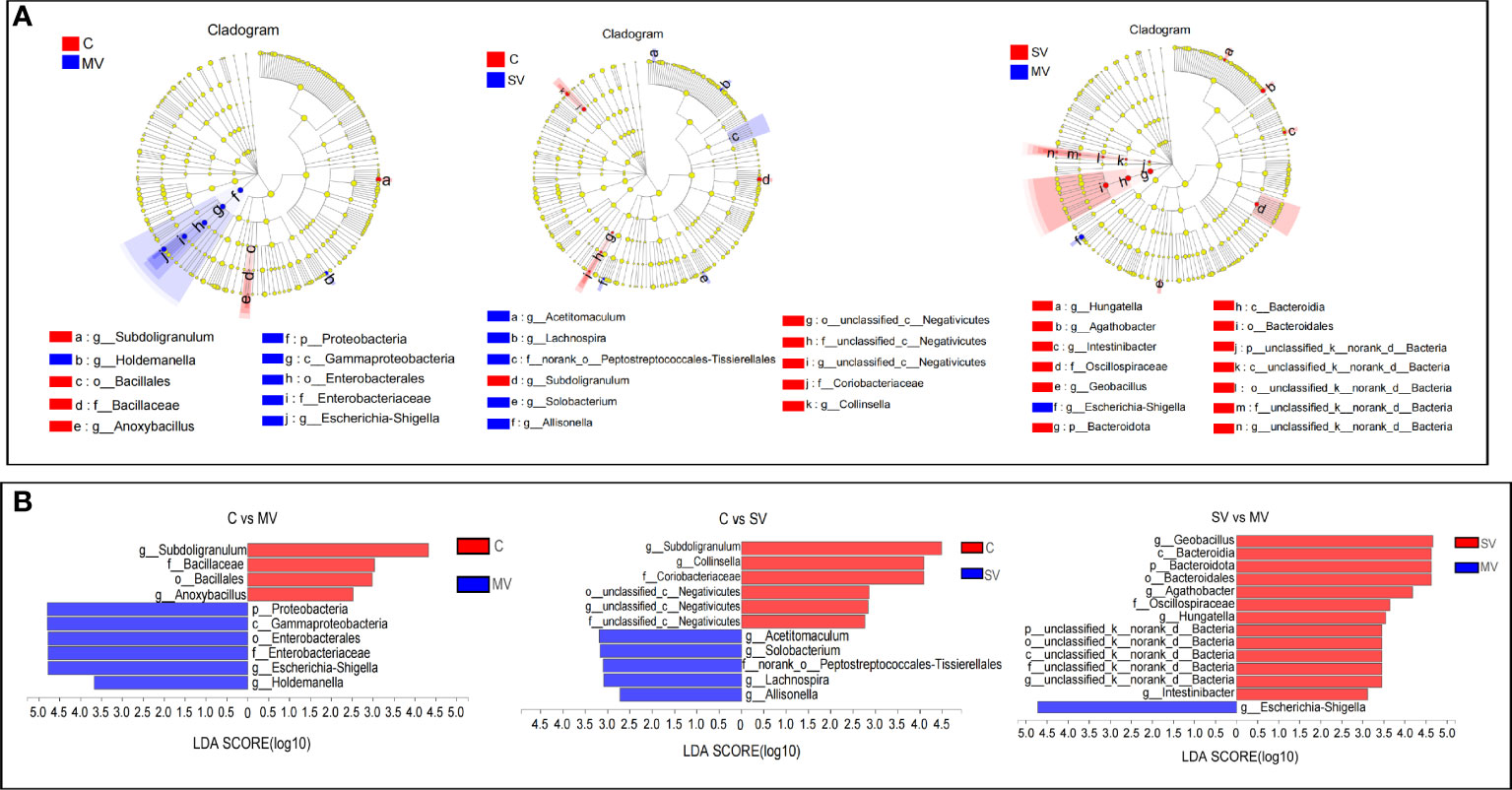
Figure 3 Linear discriminant analysis effect size (LEfSe) analysis of species differences. The non-parametricfactor Kruskal-Wallis (K-W) sum-rank test was used to detect characteristics of significant abundance differences and to find classes that were significantly different from abundance. Linear discriminant analysis (LDA) was employed to estimate the magnitude of the effect of each component (species) abundance on the differential effect. (A) Hierarchical dendrogram of multilevel species. (B) Linear discriminant analysis (LDA).
It has been shown that normal human gut microbiota can be divided into three types: Bacteroides enrichment leads to enterotype 1, Prevotella enrichment leads to enterotype 2, and Ruminococcus enrichment leads to enterotype 3 (Arumugam et al., 2011). We calculated the Bray-Curtis distance of genus abundances to cluster the samples, and the Calinski-Harabasz index indicated that the optimal number of clusters was five (Supplementary Figure S4). The five enterotypes we observed had different contributors at the genus level: Bacteroides resulted in enterotype 1 and enterotype 2, Megamonas resulted in enterotype 3, Lactobacillus resulted in enterotype 4, and Escherichia-Shigella resulted in enterotype 5 (Supplementary Figure S4). At the same time, we found that enterotype 5 was composed of only SV and MV group samples, so we speculated that enterotype 5, characterized by enrichment of Escherichia-Shigella, may be associated with the occurrence of CHD.
We calculated the Spearman correlation coefficient between a range of clinical indicators that may be associated with the onset of CHD (Table 1) and gut microbiota in the top 10 most abundant genera and Collinsella genus (Figure 4). We found that the genus Escherichia-Shigella was positively correlated with plasma low density lipoprotein (LDL) and left atrial diameter (LA), and negatively correlated with total bile acids (TBA); The genus Collinsella was negatively correlated with neutrophil ratio (NEU-R) and TG, and the genus Subdoligranulum was negatively correlated with alanine aminotransferase (ALT) and aspartate aminotransferase (AST). In addition, the genus Megamonas was positively correlated with LDL and total cholesterol (TC).
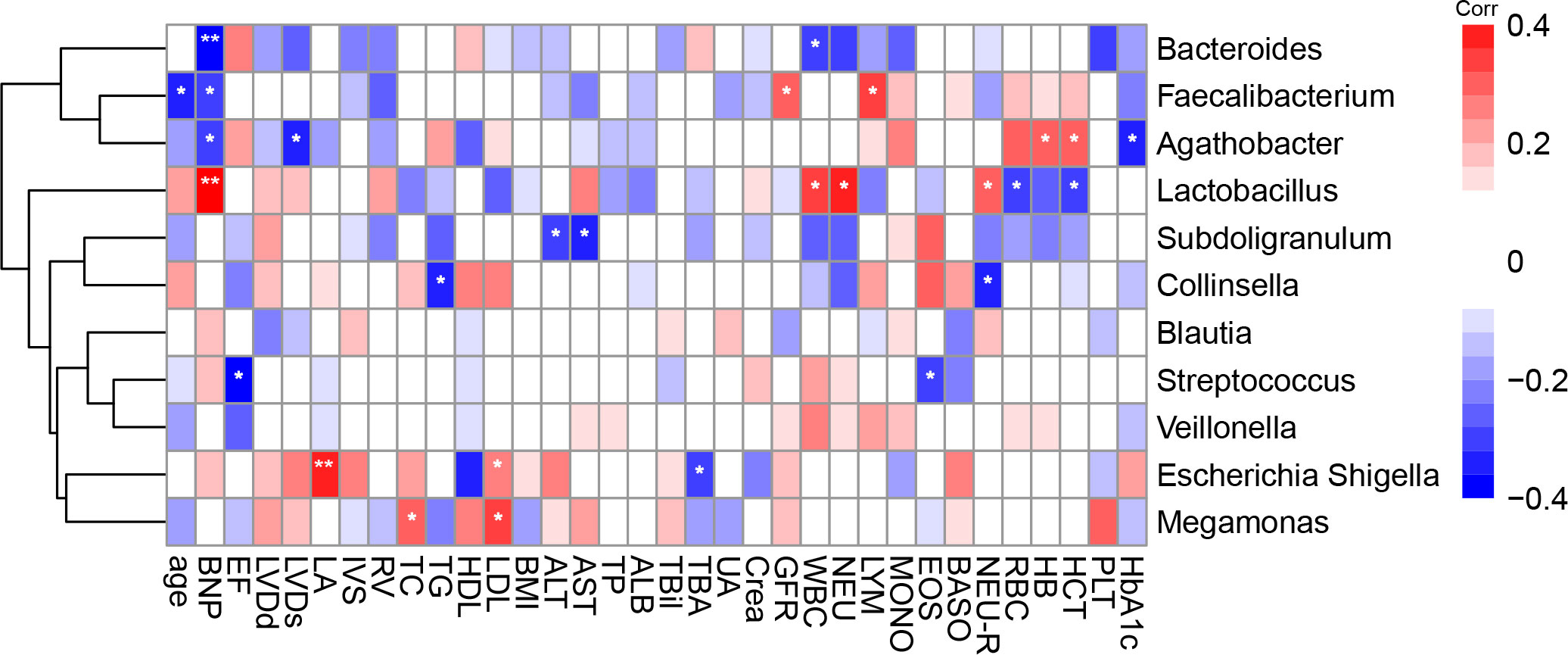
Figure 4 Spearman correlations between gut microbiota genus and clinical indicators. The colour represents positive (red) or negative (blue) correlations, and FDRs are denoted as follows: *FDR < 0.05, **FDR < 0.01. NT –proBNP, N-terminal pro-B type natriuretic peptide; LVEF, left ventricular ejection fraction; LVDd; left ventricular end diastolic diameter; LVDS, left ventricular end diastolic diameter; LA, left atrial diameter; IVS, Interventricular septum; RV, right ventricular diameter; TC, total cholesterol; TG, triglyceride; HDL, high-density lipoprotein; LDL, low density lipoprotein; BMI, body mass index; ALT, alanine aminotransferase; AST, aspartate aminotransferase; TP, total protein; ALB, albumin; TBIL, total bile acids; TBA, total bile acids; UA, uric acid; Crea, creatinine; GFR, glomerular filtration rate; WBC, white blood cell; NEU, neutrophil; LYM, lymphocyte; MONO, mononuclear; EOS, eosinophils; BASO, basophils; NEU-R, neutrophil rate; RBC, red blood cell; HB, haemoglobin; HCT, haematocrit; PLT, platelets; HbA1c, glycated haemoglobin.
To determine whether gut microbiota and clinical indicators can be regarded as biomarkers to distinguish the number of stenotic coronary arteries in patients with CHD, we constructed a few prediction models based on gut microbiota and clinical features as mentioned above. The Kruskal-Wallis test showed that the Subdoligranulum and Collinsella genera were significantly more abundant in group C subjects than in the SV and MV groups, and the abundance of the Escherichia-Shigella genus was significantly higher in the MV group subjects than in the C and SV groups. We finally selected Subdoligranulum and Collinsella genera as a gut bacterial biomarker set for controls. Receiver operating characteristic (ROC) analysis revealed that this gut bacterial set could distinguish C from CHD, SV, and MV with area under the curve (AUC) values of 0.9, 0.98, and 0.87, respectively (Figure 5A). However, the predictive potential of this biomarker set for SV and MV groups is low. Spearman correlation coefficient analysis showed that the genus Escherichia-Shigella was positively correlated with LDL and LA, and negatively correlated with TBA. Therefore, we added Escherichia-Shigella and three clinical features (LDL, LA, and TBA) to construct a new prediction model to distinguish SV and MV groups. The result revealed the new biomarker set exhibited a higher predicting potential to distinguish SV from MV patients (AUC 0.66 vs 0.80) (Figure 5B).
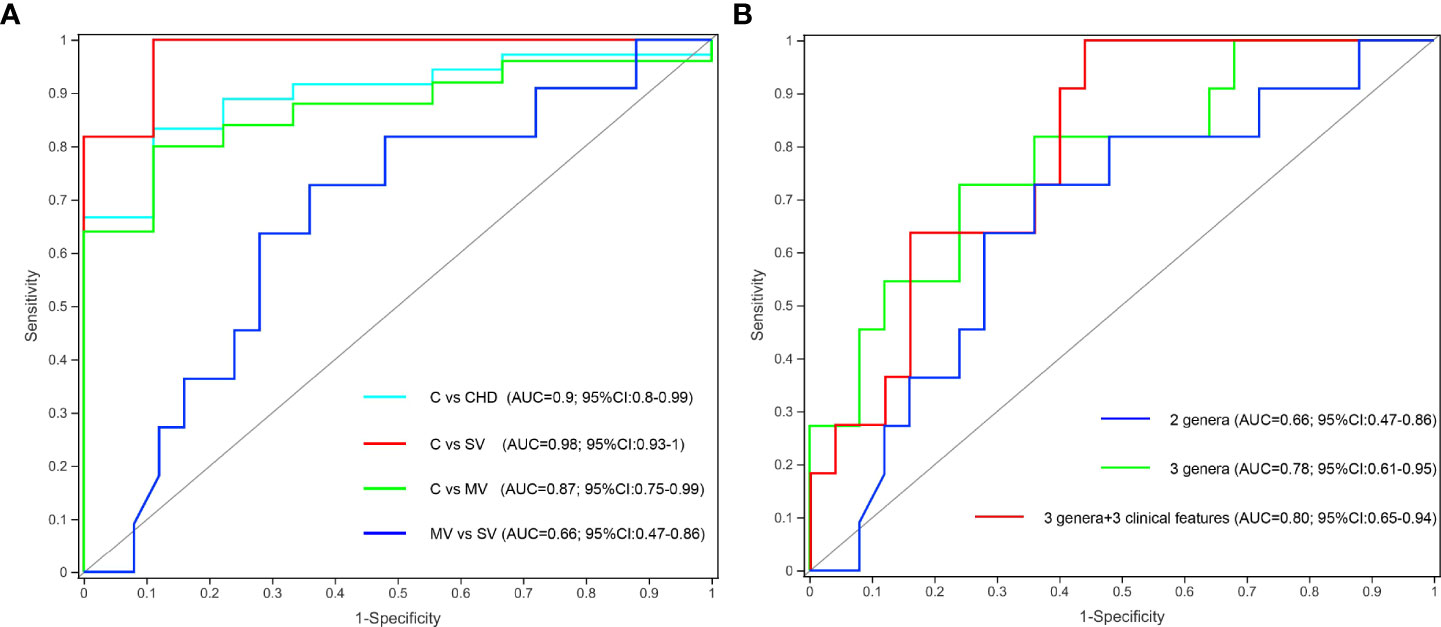
Figure 5 Gut microbiota and clinical features could effectively distinguish C from CHD, SV, and MV group subjects. (A) Two specific genera to build the prediction model yielded an AUC based on ROC analysis. (B) Gut microbiota clinical features to build the prediction model yielded an AUC based on ROC analysis.
To identify the gut microbiome functional changes in CHD patients with different numbers of coronary lesions, we first functionally annotated all sample sequences using the KEGG database (Supplementary Table 2). We focused on the metabolic pathways of gut microbiota and focused on lipid, carbohydrate, and glycan metabolism. We normalized the resulting functional annotation table for abundance. The results showed that the enrichment of metabolic pathways such as biosynthesis of unsaturated fatty acids, α-linolenic acid metabolism, betaine biosynthesis, and linoleic acid metabolism showed a trend of MV > SV > C group. Enrichment of metabolic pathways such as sphingolipid metabolism, glycosphingolipid biosynthesis, primary bile acid biosynthesis, and secondary bile acid biosynthesis showed the C > SV > MV group (Figure 6A). In addition, there were also many metabolic pathways with reduced enrichment in the CHD group, such as butyrate metabolism, fatty acid degradation, glyoxylate, and dicarboxylic acid metabolism, propionate metabolism, C5 - branched-chain dibasic acid metabolism, peptidoglycan biosynthesis, fatty acid biosynthesis, carotenoid biosynthesis, glyceride metabolism, synthesis and degradation of ketones, and steroid biosynthesis etc.
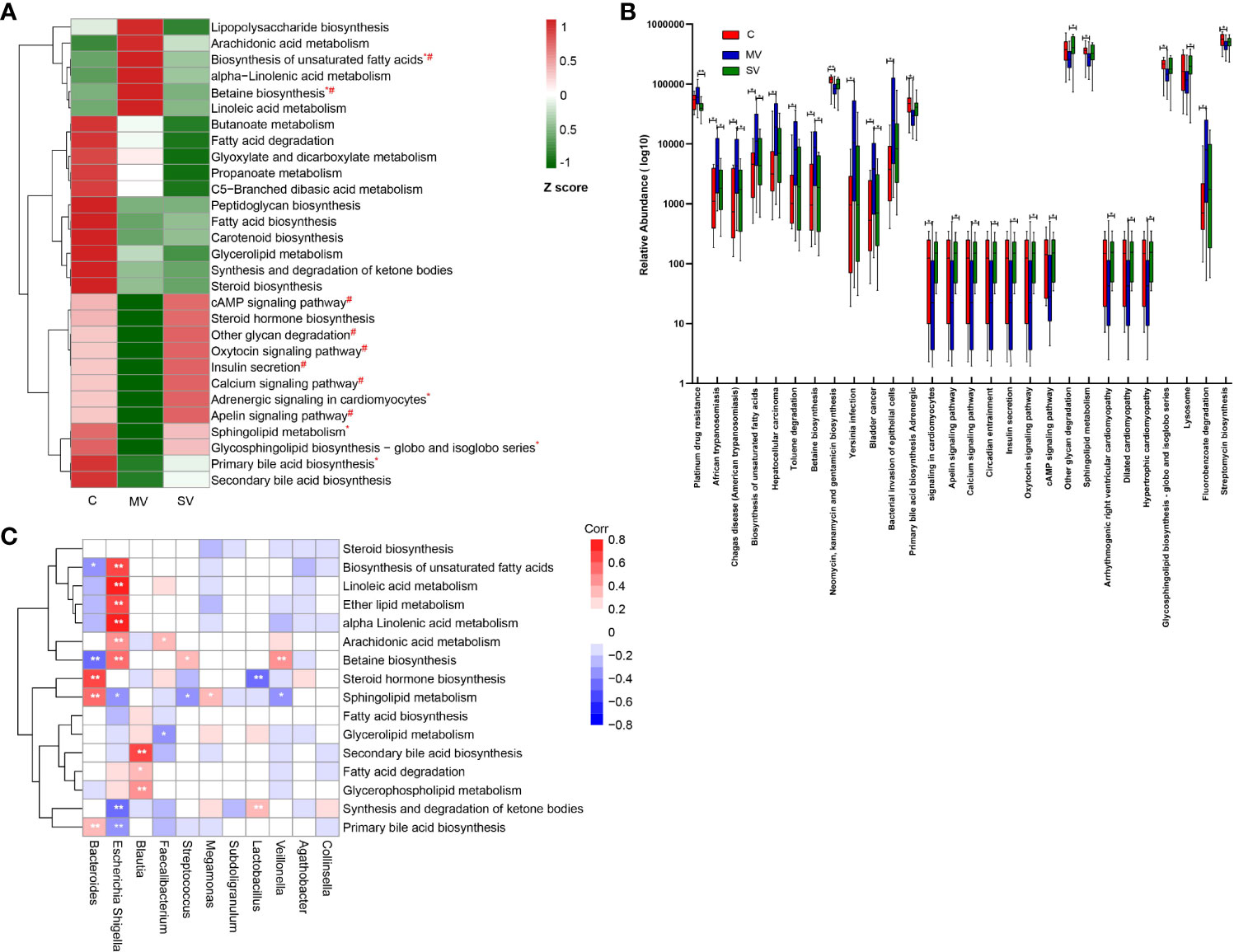
Figure 6 Changes in gut microbiome function in CHD patients. (A) Relative abundance of part of bacterial metabolic pathways in different groups. The abundance profiles were transformed into Z scores by substracting the average abundances and dividing the standard deviations of all the samples. The Z score was negative (shown in green) when the row abundance was lower than the mean. Statistical analysis of metabolic pathways was performed using the Kruskal-Wallis test. *P<0.05 for equality between C vs MV: #P<0.05 for equality between MV vs SV. (B) The box plot shows that metabolism pathway (KEGG pathway level 3) significantly changed between different groups by Kruskal-Wallis test. *, Kruskal-Wallis test P-values<0.05; **, Kruskal-Wallis test P-values<0.01, boxes represent the inter-quartile ranges, and lines inside the boxes denote medians. (C) Spearman correlations between gut microbiota genera and functions. The colour represents positive (red) or negative (blue) correlations, and FDRs are denoted as follows: *FDR < 0.05, **FDR < 0.01.
In order to further clarify which gut microbiota function may be related to the occurrence of CHD and lead to different numbers of coronary artery stenoses, we performed Kruskal-Wallis tests of the functional annotation table. We found that the group C gut microbiota was significantly superior to the SV and MV groups in sphingolipid metabolism and primary bile acid biosynthesis (Figure 6B) (P < 0.05). In contrast, the gut microbiota was clearly dominant in MV group patients in terms of betaine biosynthesis and biosynthesis of unsaturated fatty acids (Figure 6B) (P < 0.05). In addition, spearman correlation between gut microbiota and function indicated that Escherichia-Shigella was negatively correlated with sphingolipid metabolism and primary bile acid biosynthesis, and positively correlated with betaine biosynthesis and biosynthesis of unsaturated fatty acids (Figure 6C). The result indicates that Escherichia-Shigella may affect the metabolism of host to lead to the occurrence and development of coronary heart disease. In conclusion, our study showed that gut microbial function was markedly altered in patients with CHD compared to controls. At the same time, the functional changes in the gut microbiota were more obvious in patients in MV group.
In the current study, we prove that CHD patients had significant differences in the composition and function of the gut microbiota compared with healthy group and may further change with the number of stenotic coronary arteries. Through 16S rDNA sequencing and determination of clinical indicators, we found that gut microbiota and clinical indicators that exhibited significant changes with an increasing number of stenotic coronary arteries were significantly correlated and might be used independently as biomarkers to distinguish the number of stenotic coronary arteries in CHD patients. In addition, we also found that Escherichia-Shigella may affect the biosynthesis of betaine and thus lead to the occurrence and development of coronary heart disease.
Our data indicated that the abundance of Escherichia-Shigella was significantly increased in the gut microbiota of patients in the MV group compared with groups C and SV. This indicates that the high abundance of Escherichia-Shigella is associated with multivessel coronary artery lesions. Interestingly, enterotype analysis also showed that Escherichia-Shigella enterotype was enrichment in CHD patients. Shigella genus is defined clinically, and Escherichia is defined phylogenetically. Escherichia-Shigella is actually a collective name for pathogenic species in the Shigella genus. Studies had shown that Escherichia-Shigella can cause endotoxemia and systemic inflammation (Lang et al., 2020), and the inflammatory response has been proven to be an important intermediate link in the onset of cardiovascular disease (Durpes et al., 2015; Gao et al., 2016; Herrero-Fernandez et al., 2019; Marchio et al., 2019). These showed that Escherichia-Shigella may cause CHD through inflammation, and as the abundance of Escherichia-Shigella increases, the number of coronary stenosis lesions also increases. In addition, our research also found that compared with the control group, the abundance of Collinsella and Subdoligranulum in the gut microbiota of subjects in the MV and SV groups was significantly reduced. This showed that the high abundance of the genera Collinsella and Subdoligranulum may be associated with milder coronary atherosclerotic lesions. Related studies have shown that prebiotic oligofructose can increase the content of Collinsella in the gut microbiota of rats, thereby reducing the content of triglycerides in the plasma to inhibit weight gain in rats (Klancic et al., 2020). A series of studies had shown that an increase in the abundance of Subdoligranulum can significantly reduce subjects’ fasting blood glucose, plasma triglyceride content and body fat rate, and reduce the incidence of hyperlipidemia, hyperglycemia, and obesity (Everard et al., 2011; Bajaj et al., 2012; Leclercq et al., 2014; Dao et al., 2016; Louis et al., 2016). However, an animal experiment showed that treating diabetic mice with Subdoligranulum can increase the abundance of the gut microbiota Subdoligranulum, but there was no significant improvement in the characteristics of diabetes,for instance weight gain, increased fat mass, glucose tolerance, blood lipids, etc. (Van Hul et al., 2020). In other words, although studies had shown that increasing Subdoligranulum can improve metabolic diseases such as obesity, hyperlipidemia, and hyperglycemia, the exact physiological role of Subdoligranulum in animals or humans is still unclear. By observing that Collinsella and Subdoligranulum were simultaneously enriched in the C group samples, we boldly hypothesized that Subdoligranulum can have beneficial effects on the body when it coexists in large numbers with Collinsella. Further animal experiments are required to verify our hypothesizes. In conclusion, our research showed that the composition of the gut microbiota and clinical indicators change with the increase in the number of stenotic coronary arteries in patients with CHD.
One-way ANOVA of clinical characteristics showed that except for the significant differences in the TG, HDL, and LVDd levels between C vs. SV and C vs. MV, other clinical features showed no significant difference between the C and CHD subgroups. It indicated that the number of coronary stenotic vessels in patients with CHD is only affected by the differences in the composition and structure of the gut microbiota. In addition, spearman correlation coefficient analysis showed that the genus Escherichia-Shigella was positively correlated with LDL and LA, and negatively correlated with TBA. A new study showed that bile acid could promote intestinal cholesterol absorption and suppress hepatic cholesterol synthesis to prevent hypercholesterolemia (Semova et al., 2022). High levels of LDL are recognized as one of the risk factors leading to the occurrence and development of coronary heart disease. These results suggest that the cause of coronary heart disease caused by Escherichia-Shigella may be closely related to host metabolism. We selected Subdoligranulum and Collinsella genera as a gut bacterial biomarker set to distinguish C from CHD, SV, and MV with AUC values of 0.9, 0.98, and 0.87, respectively, which display a promising prediction utility. Meanwhile, the prediction model constructed by three genera (Subdoligranulum, Collinsella, and Escherichia-Shigella) and three clinical features (LDL, LA, and TBA) distinguished effectively SV and MV groups with AUC 0.80.
The human gut microbiota interacts extensively with the host through metabolic exchange and substrate co-metabolism. Human metabolites are composed of endogenous metabolites, exogenous metabolites, metabolites from the gut microbiota, and cometabolites between bacteria and the host. Changes in the metabolic function of the gut microbiota can predict changes in the metabolic function and metabolites of the host to a certain extent (Kaye et al., 2020). We observed that the gut microbiota of group C was significantly higher than that of the SV group and the MV group in terms of sphingolipid metabolism and primary bile acid biosynthesis, trending to the C>SV>MV group. Sphingolipids are the basic components of cell membranes and organelles and include ceramide, sphingosine 1-phosphate (S1P), sphingosine, sphingomyelin, glycosphingolipids, and other molecules (Maceyka and Spiegel, 2014). Research showed that genes functioning in sphingosine metabolism and the S1P pathway showed drastically increased expression in the germ-free mice (Formes et al., 2021). Ceramide, the core product of sphingolipid metabolism, has long been considered a substance that causes atherosclerosis (Maceyka and Spiegel, 2014). In mouse, rabbit, and rat cardiac ischemia/reperfusion injury (IRI) models, it was found that the infarct site and blood ceramide increased, while S1P decreased (Cui et al., 2004; Knapp et al., 2013; Pan et al., 2014). Studies had shown that ceramide produces oxidative stress in human endothelial cells, thereby reducing biologically active NO. The decrease in the bioavailability of NO aggravates the proatherogenic effect of ceramide (Li et al., 2002). At the same time, many studies have shown that S1P has a significant cardioprotective effect (Keul et al., 2016; Alessenko et al., 2018; Kurano and Yatomi, 2018). The sphingosine 1-phosphate receptor-1 (S1PR1)-specific agonist can alleviate the hypoxic damage of mouse cardiomyocytes. Animal experiments have shown that S1P can induce significant recovery of heart function and reduction of infarction in patients with IRI (Karliner et al., 2001; Vessey et al., 2006; Jin et al., 2007). In addition, an animal experiment showed that pretreatment of animals with sphingosine before ischemia or perfusion can cause a significant reduction in infarct size. This indicates that sphingosine is also a cardioprotective agent in cardiovascular disease (Vessey et al., 2009). However, it is not clear which sphingolipid molecules increase in the host plasma caused by the gut microbiota. Through bile salt hydrolysis and bile acid 7a-dehydroxylation, the gut microbiota affects the host’s physiology and produces a pool of unbound hydrophobic bile acids (secondary bile acids) (Everard et al., 2011). Secondary bile acids (such as deoxycholic acid) can be used as direct antibacterial agents to reduce bacterial translocation and the systemic inflammatory response (Begley et al., 2005; Ubeda et al., 2016). Relevant studies have shown that damage to the vascular endothelial barrier and inflammation are important factors in the formation of coronary atherosclerosis (Durpes et al., 2015; Gao et al., 2016; Herrero-Fernandez et al., 2019; Marchio et al., 2019). Meanwhile, spearman correlation coefficient analysis showed that the genus Escherichia-Shigella was negatively correlated with sphingolipid metabolism and primary bile acid biosynthesis. These results indicates that the genus Escherichia-Shigella may inhibite sphingolipid metabolism and primary bile acid biosynthesis to exacerbate vascular inflammation and vascular endothelial dysfunction, thus leading to coronary endothelial injury and atherosclerosis. Our research provided basic support for further exploring the influence of gut microbiota on human sphingolipid and bile acid metabolism and its mechanism.
In contrast, the betaine biosynthesis pathway was significantly higher in the MV group than in the C group and SV group. Spearman correlation coefficient analysis showed that the genus Escherichia-Shigella was positively correlated with betaine biosynthesis. Betaine is a raw material for the synthesis of trimethylamine oxide (TMAO), which can be converted to TMAO under the action of gut microbiota (Wang et al., 2014). Specifically, endogenous or exogenous betaine produces trimethylamine (TMA) in response to gut microbes, and then host liver flavin monooxygenase (FMO) catalyzes the conversion of TMA to TMAO (Koeth et al., 2013). A large number of studies had shown that TMAO can promote the occurrence and development of atherosclerosis in animal models (Wang et al., 2011; Koeth et al., 2013; Tang et al., 2013). Multiple cohort studies had shown that blood TMAO levels are associated with the risk of coronary atherosclerotic heart disease and major adverse cardiac events (Lever et al., 2014; Mente et al., 2015; Heianza et al., 2020). In addition, an animal study has shown that nonlethal inhibition of the production of trimethylamine by intestinal microbes can treat atherosclerosis (Wang et al., 2015). Taken together, we speculated that Escherichia-Shigella may promote plasma TMAO content by promoting betaine biosynthesis, which leads to coronary atherosclerosis.
In conclusion, this study suggests that the composition and diversity of the gut microbiota change significantly from healthy controls to CHD subgroups with different numbers of coronary lesions. At the same time, we also found several gut microbiotas associated with leading to CHD and affecting the number of coronary lesions. We constructed a disease classifier based on these related gut microbiota and plasma metabolites to distinguish the control group from CHD, providing a new direction for the diagnosis and prognosis of CHD. In addition, our results predict several functional pathways based on gut microbiome information in patients with CHD, which may enhance our understanding of the pathogenesis of CHD. In summary, the changes in gut microbiota structure and function are closely related to the occurrence and development of CHD, and changing the structure and function of gut microbiota may become a new measure to prevent and treat CHD.
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found below: https://www.ncbi.nlm.nih.gov/, PRJNA810651.
The studies involving human participants were reviewed and approved by The Ethics Committee of The Affiliated Hospital of Southwest Medical University. The patients/participants provided their written informed consent to participate in this study. Written informed consent was obtained from the individual(s) for the publication of any potentially identifiable images or data included in this article.
HY, YZ and XL: designed the experiments. YD and HH: collected samples. HY, YZ, and LL: analyzed the data. HY, GZ, and MJ: visualization. HY: writing - original draft. LL, XL, ZL, CL, and YZ: writing-review and editing. XL and YZ: project administration. All the authors read and approved the final manuscript. All authors contributed to the article and approved the submitted version.
We thank Dr Mingzhi Liu(Karolinska Institutet) for proofreading. This study was supported by the Collaborative Innovation Center for Prevention and Treatment of Cardiovascular Disease of Sichuan Province (xtcx2016-17). Sichuan Province Science and Technology project (2020YJ0338), Southwest Medical University Foundation (21YYJC0529). Doctoral Research Initiation Fund of Affiliated Hospital of Southwest Medical University, China (Grant No.20118).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fcimb.2022.903828/full#supplementary-material
Alessenko, A. V., Lebedev, А.Т., Kurochkin, I. N. (2018). The role of sphingolipids in cardiovascular pathologies. Biomeditsinskaia khimiia 64, 487–495. doi: 10.18097/PBMC20186406487
Arumugam, M., Raes, J., Pelletier, E., Le paslier, D., Yamada, T., Mende, D. R., et al. (2011). Enterotypes of the human gut microbiome. Nature 473, 174–180. doi: 10.1038/nature09944
Bajaj, J. S., Hylemon, P. B., Ridlon, J. M., Heuman, D. M., Daita, K., White, M. B., et al. (2012). Colonic mucosal microbiome differs from stool microbiome in cirrhosis and hepatic encephalopathy and is linked to cognition and inflammation. Am. J. Physiol-Gastrointest. Liver Physiol. 303, G675–G685. doi: 10.1152/ajpgi.00152.2012
Begley, M., Gahan, C. G. M., Hill, C. (2005). The interaction between bacteria and bile. FEMS Microbiol. Rev. 29, 625–651. doi: 10.1016/j.femsre.2004.09.003
Cui, J., Engelman, R. M., Maulik, N., Das, D. K. (2004). Role of ceramide in ischemic preconditioning. J. Am. Coll. Surgeons 198, 770–777. doi: 10.1016/j.jamcollsurg.2003.12.016
Dao, M. C., Everard, A., Aron-wisnewsky, J., Sokolovska, N., Prifti, E., Verger, E. O., et al. (2016). Akkermansia muciniphila and improved metabolic health during a dietary intervention in obesity: relationship with gut microbiome richness and ecology. Gut 65, 426–436. Consortium, M. I.-O. doi: 10.1136/gutjnl-2014-308778
Durpes, M.-C., Morin, C., Paquin-veillet, J., Beland, R., Pare, M., Guimond, M.-O., et al. (2015). PKC-beta activation inhibits IL-18-binding protein causing endothelial dysfunction and diabetic atherosclerosis. Cardiovasc. Res. 106, 303–313. doi: 10.1093/cvr/cvv107
Edgar, R. C. (2013). UPARSE: highly accurate OTU sequences from microbial amplicon reads. Nat. Methods 10, 996–99+. doi: 10.1038/nmeth.2604
Everard, A., Lazarevic, V., Derrien, M., Girard, M., Muccioli, G. M., Neyrinck, A. M., et al. (2011). Responses of gut microbiota and glucose and lipid metabolism to prebiotics in genetic obese and diet-induced leptin-resistant mice. Diabetes 60, 2775–2786. doi: 10.2337/db11-0227
Formes, H., Bernardes, J. P., Mann, A., Bayer, F., Pontarollo, G., Kiouptsi, K., et al. (2021). The gut microbiota instructs the hepatic endothelial cell transcriptome. Iscience 24(10):103092. doi: 10.1016/j.isci.2021.103092
Gao, W., Liu, H., Yuan, J., Wu, C., Huang, D., Ma, Y., et al. (2016). Exosomes derived from mature dendritic cells increase endothelial inflammation and atherosclerosis via membrane TNF-alpha mediated NF-kappa b pathway. J. Cell. Mol. Med. 20, 2318–2327. doi: 10.1111/jcmm.12923
Godon, J. J., Zumstein, E., Dabert, P., Habouzit, F., Moletta, R. (1997). Molecular microbial diversity of an anaerobic digestor as determined by small-subunit rDNA sequence analysis. Appl. Environ. Microbiol. 63, 2802–2813. doi: 10.1128/aem.63.7.2802-2813.1997
Heianza, Y., Ma, W., Didonato, J. A., Sun, Q., Rimm, E. B., Hu, F. B., et al. (2020). Long-term changes in gut microbial metabolite trimethylamine n-oxide and coronary heart disease risk. J. Am. Coll. Cardiol. 75, 763–772. doi: 10.1016/j.jacc.2019.11.060
Herrero-Fernandez, B., Gomez-bris, R., Somovilla-crespo, B., Gonzalez-granado, J. M. (2019). Immunobiology of atherosclerosis: A complex net of interactions. Int. J. Mol. Sci. 20, 204–212. doi: 10.1093/he/9780198748830.001.0001
Jie, Z., Xia, H., Zhong, S.-L., Feng, Q., Li, S., Liang, S., et al. (2017). The gut microbiome in atherosclerotic cardiovascular disease. Nat. Commun. 8, 845. doi: 10.1038/s41467-017-00900-1
Jin, Z.-Q., Zhang, J., Huang, Y., Hoover, H. E., Vessey, D. A., Karliner, J. S. (2007). A sphingosine kinase 1 mutation sensitizes the myocardium to ischemia/reperfusion injury. Cardiovasc. Res. 76, 41–50. doi: 10.1016/j.cardiores.2007.05.029
Karliner, J. S., Honbo, N., Summers, K., Gray, M. O., Goetzl, E. J. (2001). The lysophospholipids sphingosine-1-phosphate and lysophosphatidic acid enhance survival during hypoxia in neonatal rat cardiac myocytes. J. Mol. Cell. Cardiol. 33, 1713–1717. doi: 10.1006/jmcc.2001.1429
Kaye, D. M., Shihata, W. A., Jama, H. A., Tsyganov, K., Ziemann, M., Kiriazis, H., et al. (2020). Deficiency of prebiotic fiber and insufficient signaling through gut metabolite-sensing receptors leads to cardiovascular disease. Circulation 141, 1393–1403. doi: 10.1161/CIRCULATIONAHA.119.043081
Keul, P., Van borren, M. M. G. J., Ghanem, A., Mueller, F. U., Baartscheer, A., verkerk, A. O., et al. (2016). Sphingosine-1-Phosphate receptor 1 regulates cardiac function by modulating Ca2+ sensitivity and Na+/H+ exchange and mediates protection by ischemic preconditioning. J. Am. Heart Assoc. 5(5):e003393. doi: 10.1161/JAHA.116.003393
Klancic, T., Laforest-lapointe, I., Choo, A., Nettleton, J. E., Chleilat, F., Noye tuplin, E. W., et al. (2020). Prebiotic oligofructose prevents antibiotic-induced obesity risk and improves metabolic and gut microbiota profiles in rat dams and offspring. Mol. Nutr. Food Res. 64(16):e2000288. doi: 10.1002/mnfr.202000288
Knapp, M., Lisowska, A., Zabielski, P., Musial, W., Baranowski, M. (2013). Sustained decrease in plasma sphingosine-1-phosphate concentration and its accumulation in blood cells in acute myocardial infarction. Prostaglandins Other Lipid Mediators 106, 53–61. doi: 10.1016/j.prostaglandins.2013.10.001
Knuuti, J., WIJNS, W., SARASTE, A., Capodanno, D., Barbato, E., Funck-brentano, C., et al. (2020). 2019 ESC guidelines for the diagnosis and management of chronic coronary syndromes the task force for the diagnosis and management of chronic coronary syndromes of the European society of cardiology (ESC). Eur. Heart J. 41, 407–477. C. doi: 10.1093/eurheartj/ehz425
Koeth, R. A., Wang, Z., Levison, B. S., Buffa, J. A., ORG, E., Sheehy, B. T., et al. (2013). Intestinal microbiota metabolism of l-carnitine, a nutrient in red meat, promotes atherosclerosis. Nat. Med. 19, 576–585. doi: 10.1038/nm.3145
Koren, O., Spor, A., Felin, J., Fak, F., Stombaugh, J., Tremaroli, V., et al. (2011). Human oral, gut, and plaque microbiota in patients with atherosclerosis. Proc. Natl. Acad. Sci. United States America 108, 4592–4598. doi: 10.1073/pnas.1011383107
Kurano, M., Yatomi, Y. (2018). Sphingosine 1-phosphate and atherosclerosis. J. Atheroscl. Thromb. 25, 16–26. doi: 10.5551/jat.RV17010
Kwon, S.-K., Park, J. C., Kim, K. H., YOON, J., CHO, Y., LEE, B., et al. (2022). Human gastric microbiota transplantation recapitulates premalignant lesions in germ-free mice. Gut 71, 1266–126+. doi: 10.1136/gutjnl-2021-324489
Lang, M., Baumgartner, M., Rozalska, A., Frick, A., Riva, A., Jarek, M., et al. (2020). Crypt residing bacteria and proximal colonic carcinogenesis in a mouse model of lynch syndrome. Int. J. Cancer 147, 2316–2326. doi: 10.1002/ijc.33028
Leclercq, S., Matamoros, S., Cani, P. D., Neyrinck, A. M., Jamar, F., Staerkel, P., et al. (2014). Intestinal permeability, gut-bacterial dysbiosis, and behavioral markers of alcohol-dependence severity. Proc. Natl. Acad. Sci. United States America 111, E4485–E4493. doi: 10.1073/pnas.1415174111
Lever, M., George, P. M., Slow, S., Bellamy, D., Young, J. M., Ho, M., et al. (2014). Betaine and trimethylamine-N-Oxide as predictors of cardiovascular outcomes show different patterns in diabetes mellitus: An observational study. PloS One 9(12):e114969. doi: 10.1371/journal.pone.0114969
Li, H., Junk, P., Huwiler, A., Burkhardt, C., Wallerath, T., Pfeilschifter, J., et al. (2002). Dual effect of ceramide on human endothelial cells: induction of oxidative stress and transcriptional upregulation of endothelial nitric oxide synthase. Circulation 106, 2250–2256. doi: 10.1161/01.CIR.0000035650.05921.50
Liu, H., Chen, X., Hu, X., Niu, H., Tian, R., Wang, H., et al. (2019). Alterations in the gut microbiome and metabolism with coronary artery disease severity. Microbiome 7, 68. doi: 10.1186/s40168-019-0683-9
Louis, S., Tappu, R.-M., Damms-machado, A., Huson, D. H., Bischoff, S. C. (2016). Characterization of the gut microbial community of obese patients following a weight-loss intervention using whole metagenome shotgun sequencing. PloS One 11(2):e0149564. doi: 10.1371/journal.pone.0149564
Maceyka, M., Spiegel, S. (2014). Sphingolipid metabolites in inflammatory disease. Nature 510, 58–67. doi: 10.1038/nature13475
Marchio, P., Guerra-ojeda, S., Vila, J. M., Aldasoro, M., Victor, V. M., Mauricio, M. D. (2019). Targeting early atherosclerosis: A focus on oxidative stress and inflammation. Oxid. Med. Cell. Longevity 2019, 1–32. doi: 10.1155/2019/8563845
Mente, A., Chalcraft, K., Ak, H., Davis, A. D., Lonn, E., Miller, R., et al. (2015). The relationship between trimethylamine-N-Oxide and prevalent cardiovascular disease in a multiethnic population living in Canada. Can. J. Cardiol. 31, 1189–1194. doi: 10.1016/j.cjca.2015.06.016
Ndrepepa, G., Tada, T., Fusaro, M., Cassese, S., King, L., Hadamitzky, M., et al. (2012). Association of coronary atherosclerotic burden with clinical presentation and prognosis in patients with stable and unstable coronary artery disease. Clin. Res. Cardiol. 101, 1003–1011. doi: 10.1007/s00392-012-0490-9
Pan, W., Yu, J., Shi, R., Yan, L., Yang, T., Li, Y., et al. (2014). Elevation of ceramide and activation of secretory acid sphingomyelinase in patients with acute coronary syndromes. Coronary Artery Dis. 25, 230–235. doi: 10.1097/MCA.0000000000000079
Sawicka-Smiarowska, E., Bondarczuk, K., Bauer, W., Niemira, M., Szalkowska, A., Raczkowska, J., et al. (2021). Gut microbiome in chronic coronary syndrome patients. J. Clin. Med. 10, 5074. doi: 10.3390/jcm10215074
Semova, I., Levenson, A. E., Krawczyk, J., Bullock, K., Gearing, M. E., Ling, A. V., et al. (2022). Insulin prevents hypercholesterolemia by suppressing 12 alpha-hydroxylated bile acids. Circulation 145, 969–982. doi: 10.1161/CIRCULATIONAHA.120.045373
Talmor-Barkan, Y., Bar, N., Shaul, A. A., Shahaf, N., Godneva, A., Bussi, Y., et al. (2022). Metabolomic and microbiome profiling reveals personalized risk factors for coronary artery disease. Nat. Med. 28, 295–302. doi: 10.1038/s41591-022-01686-6
Tang, W. H. W., Wang, Z., Levison, B. S., Koeth, R. A., Britt, E. B., Fu, X., et al. (2013). Intestinal microbial metabolism of phosphatidylcholine and cardiovascular risk. New Engl. J. Med. 368, 1575–1584. doi: 10.1056/NEJMoa1109400
Ubeda, M., Lario, M., Munoz, L., Borrero, M.-J., Rodriguez-serrano, M., Sanchez-diaz, A.-M., et al. (2016). Obeticholic acid reduces bacterial translocation and inhibits intestinal inflammation in cirrhotic rats. J. Hepatol. 64, 1049–1057. doi: 10.1016/j.jhep.2015.12.010
Van Hul, M., Le Roy, T., Prifti, E., Dao, M. C., Paquot, A., Zucker, J.-D., et al. (2020). From correlation to causality: the case of subdoligranulum. Gut. Microbes 12, 1–13. doi: 10.1080/19490976.2020.1849998
Vessey, D. A., Kelley, M., Li, L., Huang, Y. (2009). Sphingosine protects aging hearts from ischemia/reperfusion injury superiority to sphingosine 1-phosphate and ischemic pre- and post-conditioning. Oxid. Med. Cell. Longevity 2, 146–151. doi: 10.4161/oxim.2.3.8622
Vessey, D. A., Kelley, M., Li, L., Huang, Y., Zhou, H.-Z., Zhu, B. Q., et al. (2006). Role of sphingosine kinase activity in protection of heart against ischemia reperfusion injury. Med. Sci. monitor Int. Med. J. Exp. Clin. Res. 12, BR318–BR324.
Wang, Z., Klipfell, E., Bennett, B. J., Koeth, R., Levison, B. S., Dugar, B., et al. (2011). Gut flora metabolism of phosphatidylcholine promotes cardiovascular disease. Nature 472, 57–U82. doi: 10.1038/nature09922
Wang, Z., Roberts, A. B., Buffa, J. A., Levison, B. S., Zhu, W., Org, E., et al. (2015). Non-lethal inhibition of gut microbial trimethylamine production for the treatment of atherosclerosis. Cell 163, 1585–1595. doi: 10.1016/j.cell.2015.11.055
Wang, Z., Tang, W. H. W., Buffa, J. A., Fu, X., Britt, E. B., Koeth, R. A., et al. (2014). Prognostic value of choline and betaine depends on intestinal microbiota-generated metabolite trimethylamine-n-oxide. Eur. Heart J. 35, 904–910. doi: 10.1093/eurheartj/ehu002
Zhang, Q., Wu, Y., Wang, J., Wu, G., Long, W., Xue, Z., et al. (2016). Accelerated dysbiosis of gut microbiota during aggravation of DSS-induced colitis by a butyrate-producing bacterium. Sci. Rep. 6(1):27572. doi: 10.1038/srep27572
Keywords: coronary heart disease, stenotic coronary arteries, gut microbiome, 16S rRNA sequencing, metabolites, functional prediction
Citation: Yu H, Li L, Deng Y, Zhang G, Jiang M, Huang H, Li C, Lv Z, Zhou Y and Liu X (2022) The relationship between the number of stenotic coronary arteries and the gut microbiome in coronary heart disease patients. Front. Cell. Infect. Microbiol. 12:903828. doi: 10.3389/fcimb.2022.903828
Received: 24 March 2022; Accepted: 09 August 2022;
Published: 26 August 2022.
Edited by:
Lei Li, Wenzhou Medical University, ChinaReviewed by:
Almagul Kushugulova, Nazarbayev University, KazakhstanCopyright © 2022 Yu, Li, Deng, Zhang, Jiang, Huang, Li, Lv, Zhou and Liu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Xing Liu, bGl1eGluZ2NhcmRpb0Bzd211LmVkdS5jbg==; Yingshun Zhou, eWluZ3NodW56aG91QHN3bXUuZWR1LmNu
†These authors have contributed equally to this work and share first authorship
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.