 Jose L. Huaman1
Jose L. Huaman1 Carlo Pacioni2,3
Carlo Pacioni2,3 Lily Kenchington-Evans1
Lily Kenchington-Evans1 Mark Doyle4
Mark Doyle4 Karla J. Helbig1
Karla J. Helbig1 Teresa G. Carvalho1*
Teresa G. Carvalho1*- 1Department of Microbiology, Anatomy, Physiology and Pharmacology, School of Agriculture, Biomedicine and Environment, La Trobe University, Melbourne, VIC, Australia
- 2Department of Environment, Land, Water and Planning, Arthur Rylah Institute for Environmental Research, Melbourne, VIC, Australia
- 3Environmental and Conservation Sciences, Murdoch University, Perth, WA, Australia
- 4Far South Coast, South East Local Land Services, Bega, NSW, Australia
Australian wild deer populations have significantly expanded in size and distribution in recent decades. Due to their role in pathogen transmission, these deer populations pose a biosecurity risk to the livestock industry. However, little is known about the infection status of wild deer in Australia. The intestinal parasite Entamoeba bovis has been previously detected in farm and wild ruminants worldwide, but its epidemiology and distribution in wild ruminants remain largely unexplored. To investigate this knowledge gap, faecal samples of wild deer and domestic cattle from south-eastern Australia were collected and analysed for the presence of Entamoeba spp. using PCR and phylogenetic analysis of the conserved 18S rRNA gene. E. bovis parasites were detected at high prevalence in cattle and wild deer hosts, and two distinct Entamoeba ribosomal lineages (RLs), RL1 and RL8, were identified in wild deer. Phylogenetic analysis further revealed the existance of a novel Entamoeba species in sambar deer and a novel Entamoeba RL in fallow deer. While we anticipated cross-species transmission of E. bovis between wild deer and cattle, the data generated in this study demonstrated transmission is yet to occur in Australia. Overall, this study has identified novel variants of Entamoeba and constitutes the first report of Entamoeba in fallow deer and sambar deer, expanding the host range of this parasite. Epidemiological investigations and continued surveillance of Entamoeba parasites in farm ruminants and wild animals will be required to evaluate pathogen emergence and transmission to livestock.
Introduction
Parasites of the genus Entamoeba comprise unicellular anaerobic organism that infect humans (Shirley et al., 2018; Cui et al., 2019), domestic animals (Noble and Noble, 1952; Stensvold et al., 2010; Cui et al., 2019; Ai et al., 2021) and wild animals (Stensvold et al., 2010; Shilton et al., 2018; Cui et al., 2019). Entamoeba parasites develop through a faecal-oral life cycle, and infections with pathogenic species can cause intestinal disease and damage the liver and brain (Ngui et al., 2012). The initial classification of Entamoeba species was established based on the type of host in which the parasites were identified (Noble and Noble, 1952) and on parasite morphological features (Stensvold et al., 2011), such as cyst size and the number of nuclei. Using this approach, Entamoeba species have been classified into four distinct groups, including E. gingivalis-like group (species without cysts), E. bovis-like group (uni-nucleated cysts), E. histolytica-like group (quadri-nucleated cysts), and E. coli-like group (octo-nucleated cysts) (Clark et al., 2006; Stensvold et al., 2011). In recent years, the analysis of Entamoeba 18S ribosomal RNA (18S rRNA) sequences has significantly expanded the repertoire of genetically distinct Entamoeba organisms (Clark et al., 2006; Stensvold et al., 2011; Jacob et al., 2016). Although morphology-based analysis will be required to consolidate such findings, they provide unique insights into variation within species, evolutionary relationships, and host specificity (Clark et al., 2006; Stensvold et al., 2011; Jacob et al., 2016). Moreover, analysis of Entamoeba DNA sequences is an essential tool in endemic countries where microscopy does not allow for the distinction of pathogenic and non-pathogenic Entamoeba species (Nath et al., 2015). The genetic diversity of morphologically identical parasites, and the host promiscuity of Entamoeba organisms, highlights an ongoing need for further characterisation of genetic variants and host range, particularly in pathogenic species and emerging zoonotic species infections.
In animals, ruminants such as cattle and sheep appear to be common hosts of the uni-nucleated cyst Entamoeba species (Noble and Noble, 1952; Clark et al., 2006; Stensvold et al., 2010). Nevertheless, cyst morphology varies greatly within and between uni-nucleated cyst-producing species isolated from different ruminant hosts (Stensvold et al., 2010). The term “ribosomal lineage” (RL) was introduced to name newly discovered Entamoeba 18S rRNA sequences with greater than 5% divergence from known species. These RLs represent organisms not yet described morphologically and not referrable to described species (Jacob et al., 2016). The analysis of Entamoeba 18S rRNA sequences detected in farmed and wild ruminants over the last decade revealed the presence of E. bovis and eight RLs. Of these, four RLs are closely related to E. bovis (Entamoeba RL 1-3 and 8) (Jacob et al., 2016). Besides being detected in cattle (Bos taurus) (Stensvold et al., 2010; Jacob et al., 2016; Nolan et al., 2017; Matsubayashi et al., 2018; Ai et al., 2021) and sheep (Ovis aries) (Stensvold et al., 2010; Jacob et al., 2016; Ai et al., 2021), E. bovis and Entamoeba RLs have also been detected in goats (Capra hircus) (Nolan et al., 2017; Al-Habsi et al., 2017; Ai et al., 2021), horses (Equus ferus) (Ai et al., 2021), camels (Camelus ferus) (Ai et al., 2021), and cervids. Among the studies conducted on cervids, white-tailed deer (Odocoileus virginianus) from the USA (Kingston and Stabler, 1978), fallow deer (Dama dama) from Mauritius (Jacob et al., 2016), and reindeer (Rengifer tarandus) from Iceland (Stensvold et al., 2010) tested positive for E. bovis, while Entamoeba RL 1 was detected in roe deer (Capreolus capreolus) from Sweden (Stensvold et al., 2010). Information about the pathogenicity of E. bovis and Entamoeba RLs in ruminants remains limited; however, their detection in cattle in the absence of clinical symptoms such as diarrhoea, suggests low pathogenicity (Matsubayashi et al., 2018; Ai et al., 2021). E. bovis have a broad ruminant host range and can be transmitted by faecal excretion of cysts followed by oral ingestion of contaminated food or water (Noble and Noble, 1952; Clark et al., 2006; Stensvold et al., 2010). Based on the oral-faecal life cycle of Entamoeba parasites, the transmission of E. bovis between different host taxa that share common land is highly likely.
To date, Entamoeba parasites have only been identified twice in Australian wild animals. E. ranarum was detected and characterised in wild cane toads (Rhinella marina) (Shilton et al., 2018), and E. bovis was detected in feral goats in Western Australia rangeland with a 6.4% prevalence (Al-Habsi et al., 2017). The prevalence and distribution of Entamoeba species in Australian farmed and wild ruminants, such as wild deer, remains yet to be investigated. Wild deer and livestock commonly share grazing areas in agricultural landscapes and are equally susceptible to a wide range of pathogens of agricultural importance (Cripps et al., 2019). Wild deer represent a significant source of pathogen transmission; thus, we hypothesised wild deer to be involved in the transmission of Entamoeba parasites to livestock and vice versa. In recent years, our team has investigated the role of wild deer as carriers of livestock pathogens in Australia (Huaman et al., 2020; Huaman et al., 2021; Huaman et al., 2021a; Huaman et al., 2021b), and here, we report the first detection of Entamoeba parasites in wild deer sampled in Australia. Further, we assess the prevalence, distribution, and characterisation of Entamoeba species and RLs as well as the potential of cross-species transmission.
Materials and Methods
Sample Collection
Faecal samples were collected from Australian wild deer to assess their infection status (Huaman et al., 2021a; Huaman et al., 2021b). Opportunistic sampling during field necropsies was carried out on deer culled with the assistance of recreational and professional hunters as part of control operations in New South Wales and Victoria (Figure 1) between August 2019 and October 2020. All samples were collected from the large intestine and placed in sterile plastic containers (Techno Plas, Australia).
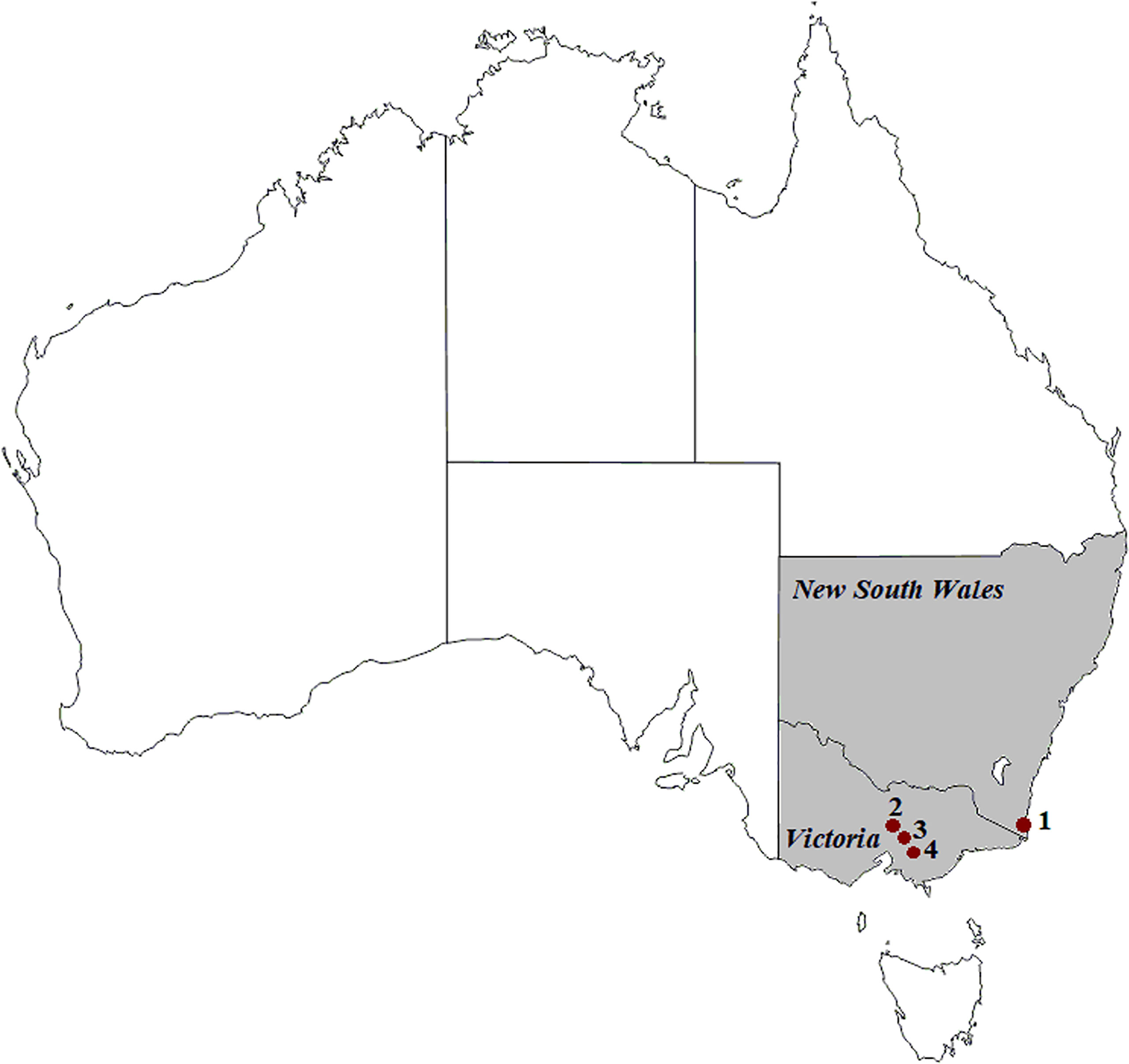
Figure 1 Geographic locations of deer (1 to 4) and cattle (1, 2 and 4) sample collection in south-eastern Australia. (1) Kiah, (2) Outer Melbourne, (3) Yellingbo, (4) Bunyip. ©d-maps.com.
Cattle faecal samples collected for clinical investigations independent from this study were analysed here for the presence of Entamoeba parasites. Cattle samples were collected from beef and dairy farms within a 20 kilometres radius of the deer sampling areas between September 2020 and April 2021. All samples were collected directly from the animals, placed in individual sterile plastic containers and immediately refrigerated. Samples were transported to the Laboratory of Molecular Parasitology within the Department of Microbiology, Anatomy, Physiology and Pharmacology at La Trobe University, and stored at -80° C until further use.
DNA Extraction and PCR Amplification
Frozen faecal samples were aliquoted into 0.25 g frozen portions. Genomic DNA was extracted using a DNeasy® PowerSoil® Kit (Qiagen, Valencia, CA, USA) following the manufacturer’s instructions. PCR was performed with primers EntboF2 and EntboR3 (Matsubayashi et al., 2018; Ai et al., 2021) to amplify an internal fragment of 850 bp of the 18S rRNA Entamoeba gene. In addition, the methodology published by Ali et al. (2005) was employed to detect tRNA-linked short tandem repeats (STRs). Six previously published primer pairs (A-L5/A-L3, D-A5/D-A3, N-K5/N-K3, R-R5/R-R3, STGA-D5/STGA-D3, S-Q5/S-Q3) were selected and tested in all the deer and cattle samples. These primers were originally designed to amplify E. hystolytica t-RNA STRs. PCR amplification was performed in a 25 μL reaction mixture containing 1x Green GoTaq Flexi buffer, 2 mM of MgCl2, 10 mM of dNTPs, 0.2 μM of each primer, 0.625 units of GoTaq G2 DNA polymerase (Promega, Madison, WI, USA), and 1 μL of template DNA. Amplification was carried out in a T100 thermal cycler (BioRad, Hercules, CA, USA), and amplification products were visualised by gel electrophoresis, using a 2% agarose gel stained with RedSafe™ (iNtRON Biotechnology, Gyeonggi-do, Korea), and the high-resolution ChemiDoc™ MP Imaging System (Bio-Rad, Hercules, CA, USA).
DNA Sequencing and Phylogenetic Analysis
PCR products were sequenced by bi-directional Sanger sequencing at the Australian Genome Research Facility (Melbourne, Australia), then analysed and edited using Geneious software 11.1.4 (Biomatters Ltd., Auckland, New Zealand, version 11.1.4). Multiple sequence alignments were performed using Clustal X (Thompson et al., 1997). A phylogenetic tree was built based on Entamoeba 18S rRNA sequences using the substitution model with the lowest BIC scores (Tamura 3-parameter model + G) and the maximum-likelihood method in MEGA 7 (Kumar et al., 2016). Thus, Entamoeba sequences obtained in the present study were aligned with 31 Entamoeba reference sequences deposited in GenBank (Table S1). These reference sequences represented 17 recognised species and 5 published ribosomal lineages. Statistical support for the trees was evaluated by bootstrapping based on 1,000 repetitions. Moreover, the number of nucleotide differences and the mean sequence divergence of Entamoeba clades identified in our sequences were calculated in MEGA 7. The nucleotide 18S rRNA sequences detected in this study were submitted to GenBank under accession number OM415364 - OM415424 (Table S2).
Bayesian Divergence Time Estimates
As deer were introduced in Australia only 200 years ago, estimating the most recent common ancestors (TMRCA) of E. bovis detected in wild deer and cattle can reveal whether parasite transmission occurred between the two hosts in Australia. Therefore, the reported split ages (Romero et al., 2016) between E. nuttalli and E. hystolytica (5.93 ± 0.28Mya), along with E. hystolytica and E. invadens (68.18 ± 16.04 Mya), were used as calibrations for the Bayesian analysis using a lognormal distribution with a mean of 1.78 and 4.5, and a standard deviation of 0.05 and 0.25, respectively. The phylogenetic trees were modelled using a birth-death tree prior, a lognormal relaxed clock in BEAST v2.6.3 (Bouckaert et al., 2019), and a gamma distribution (shape=1, rate=0.00001) for the substitution rate parameter. Two independent runs of 200 million steps were computed, sampling parameters every 10,000 steps and discarding the first 10% of each chain as burn-in. Tracer v1.7.1 (Rambaut et al., 2018) was used to ensure that the length of the burn-in phase was sufficient and guaranteed convergence of the two analyses. Results were obtained after combining the two chains with LogCombiner. The programs TreeAnnotator v2.6.2 (Bouckaert et al., 2019) and FigTree v1.4.4 (http://tree.bio.ed.ac.uk/software/figtree/) were used to summarise the posterior tree distribution and visualise the annotated Maximum Clade Credibility (MCC) tree.
Results
High Prevalence of Entamoeba DNA Found in Wild Deer and Cattle Samples
A total of twenty-three cattle faecal samples were obtained from south-eastern Australia, as well as seventy-one wild deer faecal samples, including sixty samples from fallow deer (Dama dama) and eleven samples from sambar deer (Rusa unicolor) (Table 1). All samples were screened by PCR for the presence of the 18S rRNA Entamoeba gene using primers EntboF2 and EntboR3 (Matsubayashi et al., 2018). The overall prevalence of Entamoeba spp. in wild deer from south-eastern Australia was found to be 81.7% (58/71), ranging from 72.9% to 100% depending on the host species and the sample geographic location (Table 1). In the cattle faecal samples, the prevalence of Entamoeba spp. was 100% (Table 1). All the Entamoeba 18S rRNA PCR amplicons generated (seventy-one from wild deer samples and twenty-three from cattle samples) were analysed by Sanger sequencing. Subsequent analysis of the ninety-four 18S rRNA sequences revealed E. bovis as the dominant species detected with a total prevalence of 74.6% (53/71) in wild deer and 100% (23/23) in cattle.
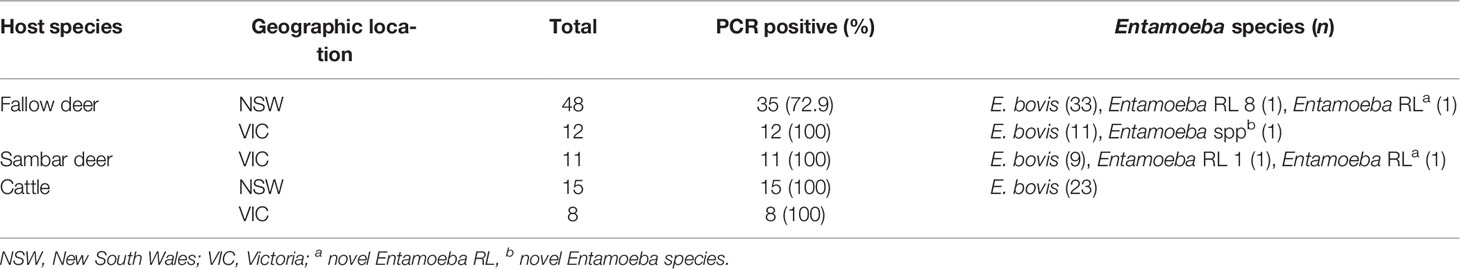
Table 1 Entamoeba species and RLs identified in deer and cattle faecal samples collected across south-eastern Australia.
Phylogenetic Analysis of Entamoeba Sequences Reveals the Existence of RL Variants in Wild Deer Samples
Out of the 850 bp Entamoeba 18S amplicons generated by PCR, a good quality DNA fragment of 778 bp was successfully sequenced for each of the ninety-four deer and cattle samples. This 778 bp fragment covered nearly 50% of the Entamoeba 18S rRNA gene and was therefore used to investigate the phylogenetic relationship and the levels of divergence of the ninety-four Entamoeba sequences. A high proportion of the wild deer-derived sequences fell into the E. bovis clade, which includes isolates from rangeland goats, cattle, sheep, and reindeer (Figure 2). Further, the cattle-derived sequences clustered exclusively within the E. bovis clade. The genetic similarity among all E. bovis sequences obtained in this study (both derived from deer and cattle) ranged from 92.5% to 100%. Moreover, the mean divergence within the cattle-derived sequences is 5 to 7-fold smaller than the divergence observed within the deer-derived sequences (Table 2). Within the 778 bp 18S rRNA gene fragment, a mean of 21 nucleotide differences was found between the two host group sequences (Table 2).

Figure 2 Cladogram of Entamoeba partial 18S rRNA sequences. Deer sequences are indicated in blue and cattle sequences in red. Reference sequences are indicated in black. The tree was constructed using the maximum likelihood method and Tamura 3-parameter + G substitution model. Bootstrap values above 70% are shown at the nodes. Note: substitutions do not scale branches in this tree. The phylogenetic tree with scaled branches and alignment is shown in Figures S2, S3, respectively.
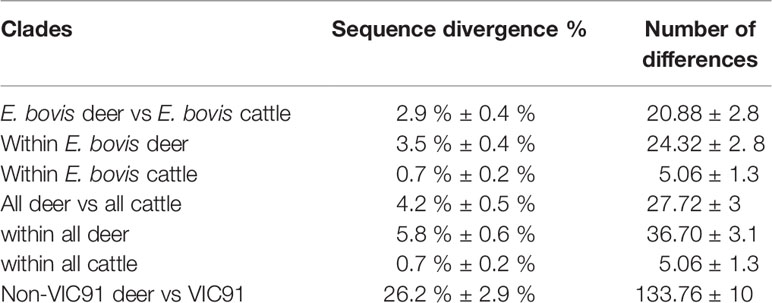
Table 2 Mean sequence divergence and number of differences (nucleotides) between Australian deer and cattle sequences within clades.
Interestingly, five 18S rRNA sequences of deer origin (VIC89, NSW304, NSW332, VIC93 and VIC91) clustered with distinct Entamoeba RLs, while none of the sequences identified in this study clustered with Entamoeba RLs 2, 3 and 4, which were previously reported in ruminants (Figure 2). Sequence VIC89, sourced from sambar deer, clustered with Entamoeba RL1 (FN666253) detected in roe deer from Sweden and shared 98.6% of the nucleotide sequence. Sequence NSW304 sourced from fallow deer, clustered with Entamoeba RL8 (KR025406), detected in cattle from the United Kingdom, with a homology of 99.9%. Comparison of sequence NSW304 with two additional Entamoeba RL8 sequences detected in camel (MN749974) and goat (MN749989) from China revealed a nucleotide identity of 99.9% and 95.1%, respectively. The alignment of these four Entamoeba RL8 sequences (NSW304, FN666253, MN749974, and MN749989) revealed an identity of 100% between sequences NSW304, FN666253, and MN749974; while three insertions (at positions 715, 716 and 750) and one deletion (at position 677) were identified in the strain detected in the goat (MN749989) (Figure S1). Sequences VIC93 and NSW332 sourced from fallow deer fell into the same clade, displaying 99% sequence identity; however, these two sequences did not cluster with any Entamoeba RL reference sequence, therefore emerging as a divergent Entamoeba RL (Figure 2). Sequence VIC91 obtained from sambar deer was genetically distinct from the Entamoeba sequences identified in other deer and cattle samples with high sequence divergence (mean 26.2%) and nucleotide difference (mean 133.76) (Table 2). Nucleotide similarity between sequence VIC91 and the reference Entamoeba sequences ranged from 82% to 86%. Overall, these findings suggest sample VIC91 belongs to a novel Entamoeba species.
STRs Were Amplified in Deer and Cattle Samples But Not Successfully Sequenced
Amplicons were obtained for all the STRs tested except for D-A5/D-A3 (Figure S4), albeit a slight difference in size when compared to amplicons of E. histolytica (Ali et al., 2005). A total of ten samples, including 4 wild deer samples and 6 cattle samples (Figures S4A, B, respectively) were sequenced using primers STGA-D5 and STGA-D3 (Ali et al., 2005). However, DNA sequences of good quality could not obtained, even when cloning the STR amplicons prior to sequencing.
Divergence Time Analysis Suggests Lack of Entamoeba Transmission Between Wild Deer and Cattle
To determine the transmission of Entamoeba parasites between wild deer and cattle in this study, a phylogenetic tree was constructed using a birth-death tree prior under a Bayesian framework and two calibration nodes (Figure 3). This approach aims to reconstruct the speciation process and, by using the time of divergence between two taxa as calibration, it converts the unit of the branches from substitutions to time (years in this case). The trees explored are then annotated in a maximum clade credibility (MCC) tree. The MCC tree revealed a clear species structure with the cattle-derived Entamoeba sequences well separated from the wild deer-derived Entamoeba sequences, like the previously generated maximum likelihood (ML) tree (Figure 2). Sequence NSW340 was, however, an exception to the species separation as it clustered with LV7 and LV14 (Figure 3). Overall, sequences sourced from wild deer clustered within four clades (Figure 3). There was little resolution within the deer group, as reflected in the low node posterior probabilities (< 0.7); however, deer clade 2 and 3 grouped with posterior probabilities > 0.8, and similar strong support was found in the ML tree. The MCC tree confirmed that sequences VIC89 and NSW304 belong to Entamoeba RL1 and RL8, respectively, and corroborated that sequence VIC91 is genetically distinct from the other sequences analysed in this study. In the ML tree sequences, LN1 (cattle origin) and VIC92 (deer origin) clustered within the E. bovis clade, although they fell outside of any other cluster, which is in contrast with the output of the ML tree (Figure 2). The most recent node between sequences obtained from a deer and a cow in Australia was estimated to be 171 million years ago (Mya) (95% HPD: 31.5 – 377.9 Mya). While the most recent common ancestors (TMRCA) between Australian wild deer and cattle clades was estimated at 632 Mya, but with considerable uncertainty (95% HPD: 163 - 1308 Mya) (Figure 3).
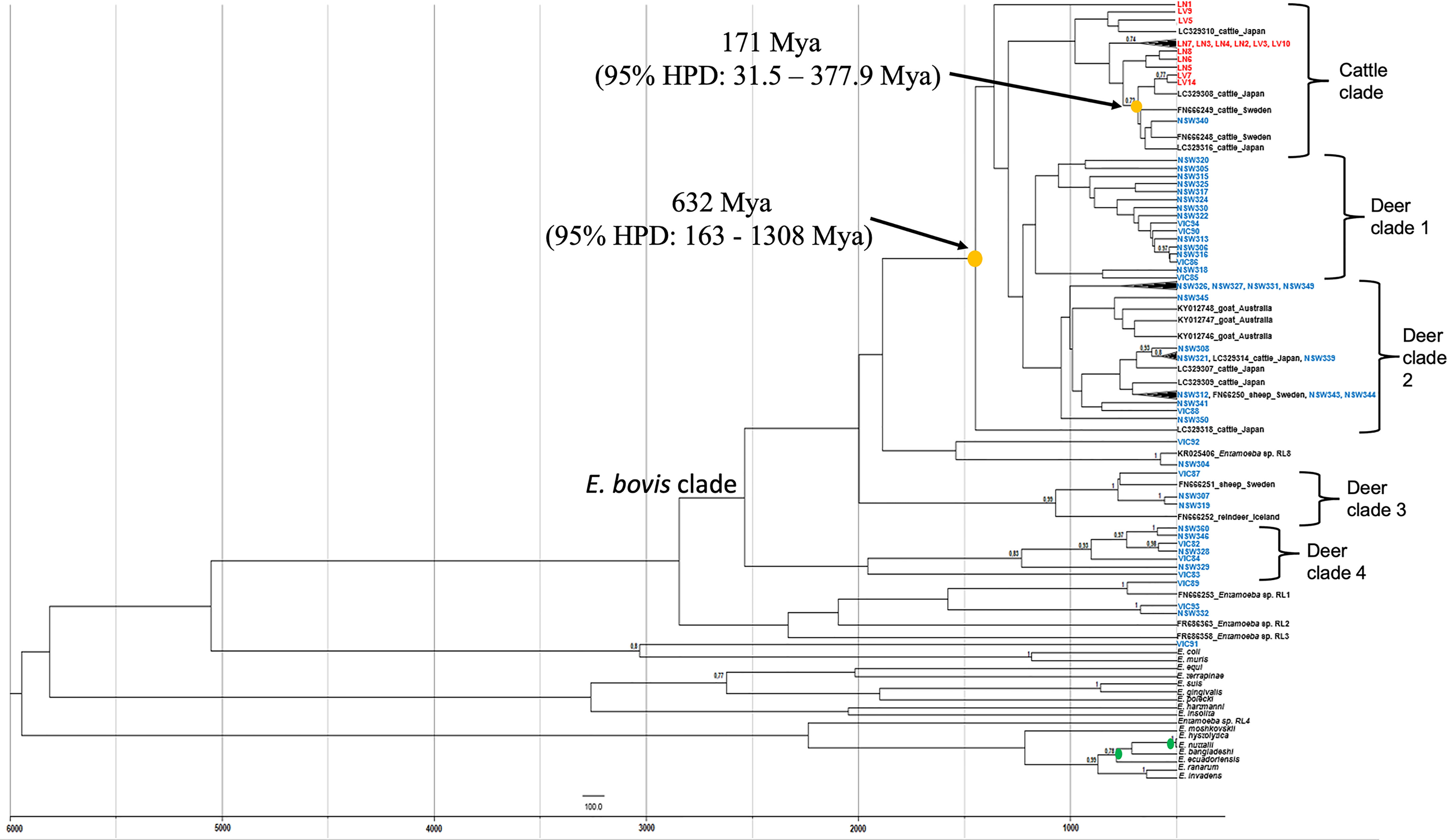
Figure 3 Maximum clade credibility tree of Entamoeba obtained from Bayesian inference using split ages reported previously as calibrations (green dots). Yellow dots indicate the estimated mean ages for the most recent common ancestor (TMRCA) of Entamoeba detected in Australian wild deer and cattle. Deer sequences are indicated in blue and cattle sequences in red. Reference sequences are indicated in black. HPD, highest posterior density, Mya, Million years ago.
Discussion
In recent decades, Australian wild deer populations have significantly increased in abundance and distribution, leading to regular close interactions between deer and livestock, which increases the risk of pathogen transmission (Davis et al., 2016; Cripps et al., 2019). However, little is known about the epidemiology of pathogens that Australian deer may transmit to livestock, other domestic animals, or wildlife. The present study complements our initial work on investigating pathogens in wild deer across multiple geographic locations in Australia (Huaman et al., 2020; Huaman et al., 2021; Huaman et al., 2021a; Huaman et al., 2021b). Here we report the identification of Entamoeba sequences in wild deer and cattle faecal samples collected in south-eastern Australia, with subsequent phylogenetic analysis to evaluate the cross-species transmission of Entamoeba parasites. This baseline information is of value for monitoring the status of parasitic infections in Australian deer and evaluating the risk of disease transmission between wild deer and livestock. Additionally, the data provided by this study increases our knowledge of the host range and distribution of Entamoeba, a group of parasites prevalent in ruminant livestock. Finally, this study represents the first molecular screening and characterisation of Entamoeba in Australian wild deer.
The predominant Entamoeba species identified in the wild deer and cattle samples collected and analysed in this study was Entamoeba bovis, a species recognised to infect ruminants, including livestock animals (Stensvold et al., 2010; Matsubayashi et al., 2018; Ai et al., 2021). The prevalence of Entamoeba infections previously reported in cattle are relatively low [2.5% in Costa Rica (Jimenez et al., 2007), 4.8% in Korea (Ismail et al., 2010)] when detected by microscopic analysis; while higher prevalences have been reported following PCR analysis [72% in Japan (Matsubayashi et al., 2018), 80% in Uganda (Nolan et al., 2017), 100% in China (Ai et al., 2021)]. Similar to these last reports, the present study detected a prevalence of 100% for E. bovis in cattle samples (n=23) using a PCR-based analysis.
E. bovis has also been previously detected in wild cervids, including in wild goats from Western Australia with a prevalence of 6.4% (Al-Habsi et al., 2017). This relatively low E. bovis prevalence contrasts with the much higher prevalence of 74.6% (53/71) reported here in fallow deer and sambar deer from eastern Australia. This difference could be attributed to methodology, as E. bovis was identified in wild goats from Western Australia (Nolan et al., 2017) by microscopy analysis. Indeed, microscopy and morphology-based detection methods are likely to underestimate parasite prevalence, as discussed above for the case of cattle samples, and are less sensitive methods when compared to the molecular detection tools employed in the present study. Although microscopy detection methods might underestimate the number of E. bovis infections, it is not excluded that lower parasite prevalence can exist, for example, due to climatic reasons. The sampling areas of this study are located in south-eastern Australia, where a Mediterranean-like climate prevails with significantly humid winters, which can facilitate the maintenance of parasites in the environment (Shirley et al., 2018). In contrast, the sampling area of Al-Habsi et al. (2017) was the semiarid rangeland area in Western Australia.
Wild deer and cattle-derived Entamoeba 18S rRNA sequences cluster within the E. bovis clade, although different species-specific clades are formed (Figure 3). Pairwise analysis revealed differences within sequences of wild deer origin, indicating high parasite diversity within this host. Polymorphic markers such as serine-rich protein genes and tRNA-linked short tandem repeats (STRs) have been used for the genotyping and correlation with the geographical distribution of other Entamoeba species such as E. histolytica, E. dispar, and E. nuttalli (Tawari et al., 2007; Weedall and Hall, 2011; Feng et al., 2014). However, this approach is yet to be used to identify E. bovis. We employed a methodology previously used in E. histolytica (Ali et al., 2005); however, the sequencing of ten samples (4 wild deer and 6 cattle samples) using primers STGA-D5 and STGA-D3 did not generate good quality DNA sequences, even when STR amplicons were cloned prior to sequencing. The primers used here were originally designed to amplify E. hystolytica t-RNA STRs (Ali et al., 2005). Therefore, the presence of polymorphisms in the t-RNA gene of E. bovis and/or the sensitivity of the primers could account for the low quality of the sequences generated.
In the absence of STR data and to determine whether a potential E. bovis cross-species transmission was possible, the time of the most common ancestor between E. bovis sequences of wild deer and cattle origin was estimated (Figure 3). The most common ancestor of E. bovis identified in wild deer and cattle hosts was estimated to have existed well before 200 years ago (before cattle and deer were introduced in Australia). Both ML and Bayesian phylogenetic analyses grouped the sequences according to their host species with moderate sequence divergence. Therefore, taken together, these results provide no evidence of E. bovis transmission between wild deer and cattle in Australia. This finding was somewhat unexpected, but it is possible that since wild deer populations have only recently increased in density, they did not play an important role in the transmission of these parasites thus far. However, this may change in the future due to deer density expansion, increasing contact rates (direct or indirect) and transmission events with livestock species.
Phylogenetic analysis of all Entamoeba sequences of deer origin identified that two sequences (VIC89 and NSW304) cluster with two distinct Entamoeba RL. The term “ribosomal lineage” (RL) was proposed to name Entamoeba strains with greater than 5% sequence divergence from known species (Jacob et al., 2016). Sequence VIC89 detected in a sambar deer from Victoria clustered with high genetic similarity with an Entamoeba RL1 sequence from a roe deer from Sweden (FN666253). This RL was also detected in one gazelle and one bighorn sheep in the USA; however, their sequences are not available for comparison (Jacob et al., 2016). Sequence NSW304, detected in a fallow deer from NSW, clustered with an Entamoeba RL8 sequence (KR025406). This ribosomal lineage has been previously identified in a variety of hosts, including cow (Jacob et al., 2016), camel (Ai et al., 2021) and goat (Ai et al., 2021). The sequence NSW304 presents high homology with an RL8 sequence of a cow (99.9%; KR025406) and camel (99.9%; MN749974) origin and lower homology with a sequence of goat origin (95.1%; MN749989). Here we present the first identification of Entamoeba RL1 and Entamoeba RL8 sequences in wild deer. These results broaden the host range of both RLs.
Interestingly, Entamoeba sequences (NSW332, VIC93 and VIC91) detected in three wild deer animals did not cluster with any 18S rRNA reference sequence. One sambar deer-sourced sequence (VIC91) was genetically distinct from other Entamoeba species found in ruminants, suggesting a possible novel Entamoeba species. Sequences NSW332 and VIC93 of fallow deer origin displayed 100% identity with each other and clustered as a sister clade with the VIC89/Entamoeba RL1 clade. Jacob et al. (2016) proposed the classification for RL sequences based on two criteria: i) sequences with ≥ 80% coverage of the 18S rRNA region, and ii) ≥ 5% difference with previously known sequences. Our phylogenetic analysis has identified NSW332 and VIC93 as a putative novel RL; however, the sequences generated here are shorter than 80% (48%) of the full Entamoeba 18S rRNA gene. Therefore, future studies to determine the complete 18S rRNA gene sequence of NSW332 and VIC93 are needed to confirm this finding.
In conclusion, here we present evidence of three Entamoeba RLs in Australian ruminants: E. bovis in wild deer and cattle, Entamoeba RL1 in wild sambar deer, and Entamoeba RL8 in wild fallow deer. Our study represents the first identification of Entamoeba parasites in Australian deer, expanding the host range of Entamoeba parasites. Further, we present evidence of a potential novel Entamoeba species (VIC91) of wild deer origin, closely related to Entamoeba RL1. We detected a high prevalence of E. bovis (100%) in cattle in the absence of clinical signs, which aligns with the low pathogenicity of E. bovis and its alleged commensal relationship with its cattle host (Ai et al., 2021). Finally, our study suggests a lack of current E. bovis transmission between wild deer and cattle in Australia. However, considering the ongoing expansion of wild deer populations in Australia, both in size and distribution, this scenario is likely to change in the future.
Data Availability Statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/Supplementary Material.
Ethics Statement
The work presented in this manuscript required no specific ethical approval. Deer culling was carried out as management operations and independently from this research. Similarly, cattle samples were collected as part of clinical investigations independently from this research. In both cases, we accessed the samples opportunistically.
Author Contributions
Conceptualisation, CP and TC. Methodology, JH, CP, and TC. Formal analysis, LK-E and JH. Investigation, JH, CP, and TC. Funding: CP, KH, and TC. Resources, CP, MD and TC. Data curation, JH. Writing—original draft preparation, JH and TC. Writing—review and editing, CP, MD, KH and TC. All authors have read and agreed to the published version of the manuscript.
Funding
This study was funded by the Centre for Invasive Species Solutions (Grant PO1-L-002).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
We thank Jake Haddad (VPAC), Andrew Bengsen, Troy Crittle and Quentin Hart (New South Wales Department of Primary Industries), and the staff from Parks Victoria for assisting with sample collection.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fcimb.2022.883031/full#supplementary-material
Supplementary Figure 1 | Alignment of partial 18S rRNA DNA sequences obtained from sample NSW304 and three Entamoeba sp. RL8 reference strains (KR025406, MN749974, MN749989). Numbers indicate nucleotide positions. ‘*’ denotes an identical nucleotide residue across all four sequences. ‘-’ indicates the absence of a nucleotide residue. Gap positions are highlighted in green.
Supplementary Figure 2 | Phylogenetic analysis of Entamoeba partial 18S rRNA DNA sequences. Deer sequences are highlighted in blue and cattle sequences in red. Reference sequences are indicated in black. The tree was constructed using the maximum likelihood method and Tamura 3-parameter + G substitution model. Only bootstrap values >70% are shown at the nodes. The scale bar indicates nucleotide substitutions per site.
Supplementary Figure 3 | Alignment of 18S rRNA sequences included in our phylogenetic and bayesian analysis. Numbers indicate nucleotide positions. ‘*’ denotes an identical nucleotide across the alignment. ‘-’ indicates the absence of a nucleotide residue.
Supplementary Figure 4 | PCR amplification of the six STRs assessed in this study [based on (21)]. (A) Amplification of STRs D-A5/D-A3 (400/500 bp), STGA-D5/STGA-D3 (150/200 bp), A-L5/A-L3 (500/600 bp), R-R5/R-R3 (700 bp), N-K5/N-K3 (600/800 bp), and SQ5/SQ3 (400/500 bp) was attempted in four deer samples (N301, N304, N306, N308). (B) Amplification of STRs N-K5/N-K3 (600/800 bp), A-L5/A-L3 (500/600 bp), SQ5/SQ3 (400/500 bp), STGA-D5/STGA-D3 (150/200 bp) was attempted in six cattle samples (LN2, LN4, LN6, LV1, LV4, 300).
Supplementary Table 1 | List of 18S rRNA Entamoeba sequences included on the phylogenetic and divergence time analysis.
Supplementary Table 2 | List of accession number of the Entamoeba 18S rRNA DNA sequences obtained in the present study and submitted to GenBank.
References
Ai, S., Zhang, Z., Wang, X., Zhang, Q., Yin, W., Duan, Z. (2021). The First Survey and Molecular Identification of Entamoeba Spp. In Farm Animals on Qinghai-Tibetan Plateau of China. Comp. Immunol. Microbiol. Infect. Dis. 75, 101607. doi: 10.1016/j.cimid.2020.101607
Al-Habsi, K., Yang, R., Ryan, U., Jacobson, C., Miller, D. W. (2017). Morphological and Molecular Characterization of an Uninucleated Cyst-Producing Entamoeba Spp. In Captured Rangeland Goats in Western Australia. Vet. Parasitol 235, 41–46. doi: 10.1016/j.vetpar.2017.01.013
Ali, I. K. M., Zaki, M., Clark, C. G. (2005). Use of PCR Amplification of tRNA Gene-Linked Short Tandem Repeats for Genotyping Entamoeba Histolytica. J. Clin. Microbiol. 43 (12), 5842–5847. doi: 10.1128/JCM.43.12.5842-5847.2005
Bouckaert, R., Vaughan, T. G., Barido-Sottani, J., Duchene, S., Fourment, M., Gavryushkina, A., et al. (2019). BEAST 2.5: An Advanced Software Platform for Bayesian Evolutionary Analysis. PloS Comput. Biol. 15 (4), e1006650. doi: 10.1371/journal.pcbi.1006650
Clark, C. G., Kaffashian, F., Tawari, B., Windsor, J. J., Twigg-Flesner, A., Davies-Morel, M. C. G., et al. (2006). New Insights Into the Phylogeny of Entamoeba Species Provided by Analysis of Four New Small-Subunit rRNA Genes. I J. Syst. Evol. Microbiol. 56 (9), 2235–2239. doi: 10.1099/ijs.0.64208-0
Cripps, J. K., Pacioni, C., Scroggie, M. P., Woolnough, A. P., Ramsey, D. S. L. (2019). Introduced Deer and Their Potential Role in Disease Transmission to Livestock in Australia. Mammal Rev. 49 (1), 60–77. doi: 10.1111/mam.12142
Cui, Z., Li, J., Chen, Y., Zhang, L. (2019). Molecular Epidemiology, Evolution, and Phylogeny of Entamoeba Spp. Infect. Genet. Evol. 75, 104018. doi: 10.1016/j.meegid.2019.104018
Davis, N. E., Bennett, A., Forsyth, D. M., Bowman, D. M. J. S., Lefroy, E. C., Wood, S. W., et al. (2016). A Systematic Review of the Impacts and Management of Introduced Deer (Family Cervidae) in Australia. Wildl. Res. 43 (6), 515–532. doi: 10.1071/WR16148
Feng, M., Komiyama, T., Yanagi, T., Cheng, X., Sherchand, J. B., Tachibana, H. (2014). Correlation Between Genotypes of tRNA-Linked Short Tandem Repeats in Entamoeba Nuttalli Isolates and the Geographical Distribution of Host Rhesus Macaques. Parasitol Res. 113 (1), 367–374. doi: 10.1007/s00436-013-3664-0
Huaman, J. L., Pacioni, C., Forsyth, D. M., Pople, A., Hampton, J. O., Carvalho, T. G., et al. (2020). Serosurveillance and Molecular Investigation of Wild Deer in Australia Reveals Seroprevalence of Pestivirus Infection. Viruses 12 (7), 752–767. doi: 10.3390/v12070752
Huaman, J. L., Pacioni, C., Forsyth, D. M., Pople, A., Hampton, J. O., Helbig, K. J., et al. (2021). Evaluation of Haemoparasite and Sarcocystis Infections in Australian Wild Deer. Int. J. Parasitol. Parasites. Wildl. 15, 262–269. doi: 10.1016/j.ijppaw.2021.06.006
Huaman, J. L., Pacioni, C., Sarker, S., Doyle, M., Forsyth, D. M., Pople, A., et al. (2021a). Molecular Epidemiology and Characterization of Picobirnavirus in Wild Deer and Cattle From Australia: Evidence of Genogroup I and II in the Upper Respiratory Tract. Viruses 13 (8), 1492. doi: 10.3390/v13081492
Huaman, J. L., Pacioni, C., Sarker, S., Doyle, M., Forsyth, D. M., Pople, A., et al. (2021b). Novel Picornavirus Detected in Wild Deer: Identification, Genomic Characterisation, and Prevalence in Australia. Viruses 13 (12), 2412. doi: 10.3390/v13122412
Ismail, H. A., Jeon, H. K., Yu, Y. M., Do, C., Lee, Y. H. (2010). Intestinal Parasite Infections in Pigs and Beef Cattle in Rural Areas of Chungcheongnam-Do, Korea. Korean J. Parasitol. 48 (4), 347–349. doi: 10.3347/kjp.2010.48.4.347
Jacob, A. S., Busby, E. J., Levy, A. D., Komm, N., Clark, C. G. (2016). Expanding the Entamoeba Universe: New Hosts Yield Novel Ribosomal Lineages. J. Eukaryot. Microbiol. 63 (1), 69–78. doi: 10.1111/jeu.12249
Jimenez, A. E., Montenegro, V. M., Hernandez, J., Dolz, G., Maranda, L., Galindo, J., et al. (2007). Dynamics of Infections With Gastrointestinal Parasites and Dictyocaulus Viviparus in Dairy and Beef Cattle From Costa Rica. Vet. Parasitol 148 (3-4), 262–271. doi: 10.1016/j.vetpar.2007.06.015
Kingston, N., Stabler, R. M. (1978). Two Species of Entamoeba From White-Tailed Deer, Odocoileus Virginianus, From Georgia. J. Parasitol. 64 (1), 14–16. doi: 10.2307/3279600
Kumar, S., Stecher, G., Tamura, K. (2016). MEGA7: Molecular Evolutionary Genetics Analysis Version 7.0 for Bigger Datasets. Mol. Biol. Evol. 33 (7), 1870–1874. doi: 10.1093/molbev/msw054
Matsubayashi, M., Matsuura, Y., Nukata, S., Daizi, Y., Shibahara, T., Teramoto, I., et al. (2018). First Detection and Molecular Identification of Entamoeba Bovis From Japanese Cattle. Parasitol. Res. 117 (1), 339–342. doi: 10.1007/s00436-017-5689-2
Nath, J., Ghosh, S. K., Singha, B., Paul, J. (2015). Molecular Epidemiology of Amoebiasis: A Cross-Sectional Study Among North East Indian Population. PloS Negl. Trop. Dis. 9 (12), e0004225. doi: 10.1371/journal.pntd.0004225
Ngui, R., Angal, L., Fakhrurrazi, S. A., Lian, Y. L., Ling, L. Y., Ibrahim, J., et al. (2012). Differentiating Entamoeba Histolytica, Entamoeba Dispar and Entamoeba Moshkovskii Using Nested Polymerase Chain Reaction (PCR) in Rural Communities in Malaysia. Parasit. Vectors 5, 187. doi: 10.1186/1756-3305-5-187
Noble, G. A., Noble, E. R. (1952). Entamoebae in Farm Mammals. J. Parasitol. 38 (6), 571–595. doi: 10.2307/3273985
Nolan, M. J., Unger, M., Yeap, Y. T., Rogers, E., Millet, I., Harman, K., et al. (2017). Molecular Characterisation of Protist Parasites in Human-Habituated Mountain Gorillas (Gorilla Beringei Beringei), Humans and Livestock, From Bwindi Impenetrable National Park, Uganda. Parasit. Vectors 10 (1), 340. doi: 10.1186/s13071-017-2283-5
Rambaut, A., Drummond, A. J., Xie, D., Baele, G., Suchard, M. A. (2018). Posterior Summarization in Bayesian Phylogenetics Using Tracer 1.7. Syst. Biol. 67 (5), 901–904. doi: 10.1093/sysbio/syy032
Romero, M., Cerritos, R., Ximenez, C. (2016). Horizontal Gene Transfers From Bacteria to Entamoeba Complex: A Strategy for Dating Events Along Species Divergence. J. Parasitol. Res. 2016, 3241027. doi: 10.1155/2016/3241027
Shilton, C. M., Slapeta, J., Shine, R., Brown, G. P. (2018). Invasive Colonic Entamoebiasis in Wild Cane Toads, Australia. Emerg. Infect. Dis. 24 (8), 1541–1543. doi: 10.3201/eid2408.180101
Shirley, D. T., Farr, L., Watanabe, K., Moonah, S. (2018). A Review of the Global Burden, New Diagnostics, and Current Therapeutics for Amebiasis. Open Forum Infect. Dis. 5 (7), 161. doi: 10.1093/ofid/ofy161
Stensvold, C. R., Lebbad, M., Clark, C. G. (2010). Genetic Characterisation of Uninucleated Cyst-Producing Entamoeba Spp. From Ruminants. Int. J. Parasitol. 40 (7), 775–778. doi: 10.1016/j.ijpara.2010.03.003
Stensvold, C. R., Lebbad, M., Victory, E. L., Verweij, J. J., Tannich, E., Alfellani, M., et al. (2011). Increased Sampling Reveals Novel Lineages of Entamoeba: Consequences of Genetic Diversity and Host Specificity for Taxonomy and Molecular Detection. Protist 162 (3), 525–541. doi: 10.1016/j.protis.2010.11.002
Tawari, B., Ali, I. K. M., Scott, C., Quail, M. A., Berriman, M., Hall, N., et al. (2007). Patterns of Evolution in the Unique tRNA Gene Arrays of the Genus Entamoeba. Mol. Biol. Evol. 25 (1), 187–198. doi: 10.1093/molbev/msm238
Thompson, J. D., Gibson, T. J., Plewniak, F., Jeanmougin, F., Higgins, D. G. (1997). The CLUSTAL_X Windows Interface: Flexible Strategies for Multiple Sequence Alignment Aided by Quality Analysis Tools. Nucleic Acids Res. 25 (24), 4876–4882. doi: 10.1093/nar/25.24.4876
Keywords: Entamoeba bovis, wild deer, cattle, cross-species infection, Australia, ribosomal lineages, 18S rRNA
Citation: Huaman JL, Pacioni C, Kenchington-Evans L, Doyle M, Helbig KJ and Carvalho TG (2022) First Evidence of Entamoeba Parasites in Australian Wild Deer and Assessment of Transmission to Cattle. Front. Cell. Infect. Microbiol. 12:883031. doi: 10.3389/fcimb.2022.883031
Received: 24 February 2022; Accepted: 10 May 2022;
Published: 10 June 2022.
Edited by:
Maria Carolina Touz, Medical Research Institute Mercedes and Martín Ferreyra (INIMEC), ArgentinaReviewed by:
Manigandan Lejeune, Cornell University, United StatesKaren Luisa Haag, Federal University of Rio Grande do Sul, Brazil
Copyright © 2022 Huaman, Pacioni, Kenchington-Evans, Doyle, Helbig and Carvalho. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Teresa G. Carvalho, dC5jYXJ2YWxob0BsYXRyb2JlLmVkdS5hdQ==