 Breno A. B. Salgado‡
Breno A. B. Salgado‡ Josephine C. Moran
Josephine C. Moran Malcolm J. Horsburgh
Malcolm J. Horsburgh
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Cell. Infect. Microbiol. , 04 August 2022
Sec. Molecular Bacterial Pathogenesis
Volume 12 - 2022 | https://doi.org/10.3389/fcimb.2022.874138
This article is part of the Research Topic “Omics”- Revolution in Elucidating the Virulence and Resistance in Staphylococcus aureus View all 6 articles
Staphylococcus aureus nasal colonization is a risk factor for infection. A large proportion of the population are identified as potential S. aureus carriers yet we only partially understand the repertoire of genetic factors that promote long-term nasal colonization. Here we present a murine model of nasopharyngeal colonization that requires a low S. aureus inoculum and is amenable to experimental evolution approaches. We used this model to experimentally evolve S. aureus using successive passages in the nasopharynx to identify those genetic loci under selection. After 3 cycles of colonization, mutations were identified in mannitol, sorbitol, arginine, nitrite and lactate metabolism genes promoting key pathways in nasal colonization. Stress responses were identified as being under selective pressure, with mutations in DNA repair genes including dnaJ and recF and key stress response genes clpL, rpoB and ahpF. Peptidoglycan synthesis pathway genes also revealed mutations indicating potential selection for alteration of the cell surface. The murine model used here is versatile to question colonization, persistence and evolution studies.
We studied the human pathogen Staphylococcus aureus in our search to determine factors that contribute to its ability to live in the human nose and throat. The anterior nares and nasopharynx are considered primary habitats but we do not understand how the pathogen adapts as it moves from one person to the next. We first determined sustained survival of the pathogen over multiple days in the nasopharynx that might act as a good model for human persistence due to the low numbers of bacteria needed for it to establish. By using successive rounds of colonization of the nasopharynx across different mice we revealed that multiple genetic changes in the S. aureus occurred. These changes were found in genes associated with the cell surface and metabolism and might indicate adaptation to the niche. One gene showed an accumulation of multiple mutations supporting a key contribution in adaptation but the role of the protein it encodes is not yet known. The contribution of these genes and genetic changes are unclear but indicate an area for future research to better understand how this common human pathogen is so successful at human colonization and survival.
S. aureus is an opportunistic pathogen commonly found in both the anterior and inner nasal cavity. While S. aureus carriage is typically asymptomatic, it is a risk factor for infection. Since the eradication of carriage prevents both infection and transmission of S. aureus (Grothe et al., 2014; Von Eiff et al., 2014; Liu et al., 2017), knowledge of the determinants that enable S. aureus to colonize the human nasopharynx are critical in developing strategies to control the spread of S. aureus.
The major components contributing to S. aureus nasal colonization depends on the composition of the niche (Leonard et al., 2019). The anterior nares are lined with keratinized epithelium identical to skin, with loricrin, K1 and K10 cytokeratin molecules present in high numbers. S. aureus cell wall-anchored (CWA) proteins, such as clumping factor B (ClfB) and iron-regulated surface determinant A (IsdA) are major adhesins contributing to successful colonization of the anterior nasal cavity (Hanssen et al., 2017). Other CWA proteins, such as SdrC, SdrD and SasG, are also associated with S. aureus nasal colonization (Corrigan et al., 2009). The epithelium of the inner nasal sites where S. aureus is commonly found is characterized by a ciliated and pseudostratified morphology. The cells in this niche express SREC-I, a member of the F-type scavenger receptor family, a key component in S. aureus colonization recognized by S. aureus glycopolymer wall teichoic acid (WTA) (Baur et al., 2014; Schade and Weidenmaier, 2016). While the interaction between S. aureus adhesins and its ligands in the anterior nares are implicated in persistent colonization, the WTA/SREC-I interface is proposed to be important in the early stages of nasal colonization (Leonard et al., 2019).
During long-term colonization, S. aureus is exposed to diverse selective pressures that include competing microbiota, the host immune system and nutrient limitation (Coates et al., 2014; Krismer et al., 2017). S. aureus must therefore be versatile in order to persist, and is known for its high evolutionary rate, resulting in large genomic and phenotypic diversity (Dean et al., 2012; Szafrańska et al., 2019). Genetic variation from mutations in the form of single nucleotide polymorphisms (SNPs) or short insertion/deletions (INDELs) can be tracked by whole-genome sequencing; a method previously used to identify contributory genes involved in S. aureus adaptation to specific selective pressures over an evolutionary time-frame (Kennedy et al., 2008; Lopez-Collazo et al., 2015). For example, comparative genomics was used to detect S. aureus genes and alleles with a role in bacteraemia (Young et al., 2012) and infections during cystic fibrosis (Lopez-Collazo et al., 2015). However, detailed temporal mechanisms of nasal colonization remain unclear with several determinants of S. aureus carriage yet to be characterized, highlighting the need for robust models that mirror more reliably in vivo conditions (Sollid et al., 2014).
Experimental evolution of microbes selects for adaptation to applied environmental pressure and the use of whole-genome sequencing simplifies the identification of genomic adaptations acquired by this directed evolution. Several studies described S. aureus evolution in vitro by serially passaging isolates over days or even weeks under selective pressure from antimicrobials to reveal adaptation and survival mechanisms (Johnston et al., 2016; Kubicek-Sutherland et al., 2017; Coates-Brown et al., 2018), however there are no reports of equivalent studies using in vivo models.
Here we developed a murine asymptomatic nasopharyngeal colonization model for the experimental evolution of S. aureus to investigate temporal colonization of the mouse nasopharynx. The isolates generated using this approach were sequenced to identify genetic changes that might reveal key components, pathways and mechanisms that enable S. aureus to colonize and persist in the nasal cavity.
A murine S. aureus nasopharyngeal colonization model was established by inoculating groups of mice intranasally with ~ 5 x 104 CFU S. aureus USA300 LAC JE2 strain. The mice were maintained for a period of 14 days post-inoculation. At chosen time points (1, 3-, 5-, 7- and 14-days post-inoculation), the nasopharyngeal CFU load in each mouse was determined (Figure 1).
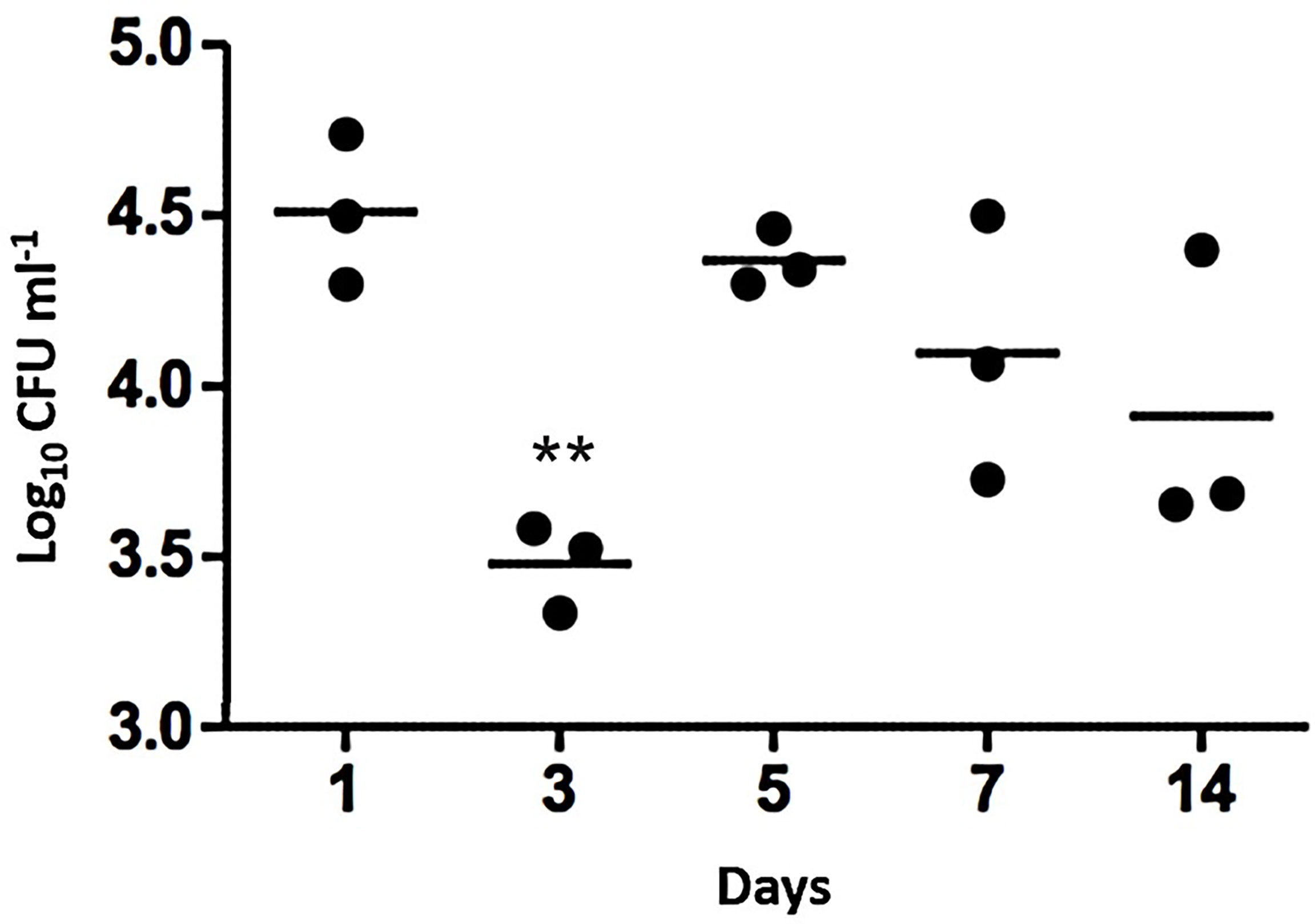
Figure 1 Nasopharyngeal colonization of mice by S. aureus USA300 LAC JE2. Intranasal inoculation with ~5 x 104 CFU. At each time point post-inoculation, the colonizing bacteria were recovered and cultured to identify S. aureus. Bars represent mean values of the mice in each time group. 2-way ANOVA was used for statistics with ** indicating P<0.01.
Across the 14 days of nasopharyngeal colonization, numbers of viable S. aureus CFU decreased by ~1 log by day 3 compared to day 1 (P<0.01), followed by an increase in CFU from day 5 onwards, with the population remaining constant thereafter until day 14 when the experiment was ended (Figure 1). The model was therefore determined to be sufficiently robust to design an experimental evolution using serial passages.
Based upon the established colonization model, 3 serial passages of S. aureus were performed in total with each passage lasting for 7 days (Figure 2). Isolated bacteria from the first passage were used as inoculum for the second passage. The process was repeated using the bacteria from the second passage as inoculum for the third passage. Mice were sacrificed to recover the colonizing S. aureus after each passage with no deaths recorded otherwise. After completion of the serial passages, spa typing of the bacteria was performed by DNA sequencing of the amplified gene to confirm nasopharyngeal isolates matched the input strain.
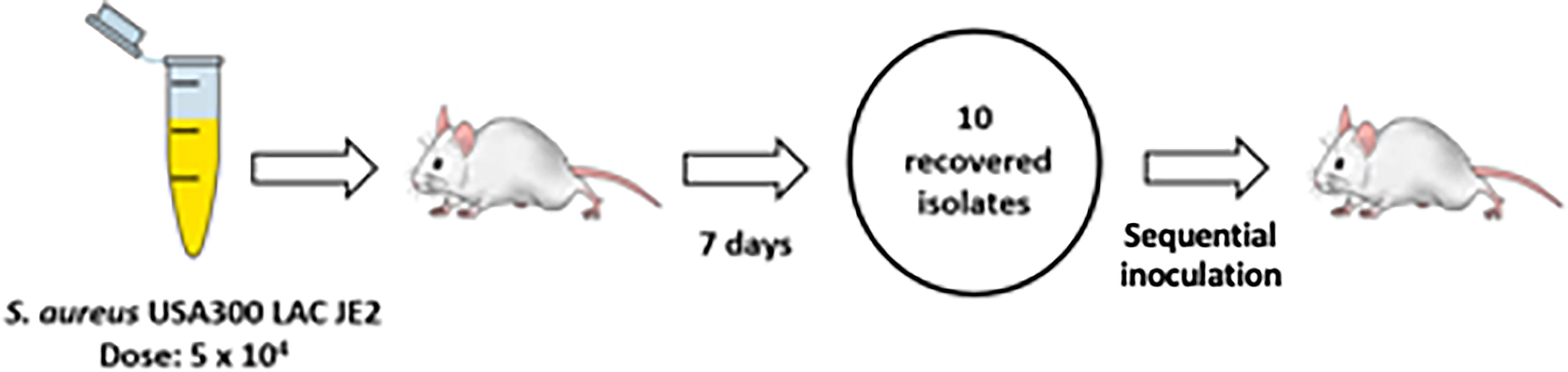
Figure 2 Serial passage of S. aureus in a murine nasopharyngeal colonization model. Schematic demonstration of sequential colonization of mouse nasopharynx by S. aureus USA300 LAC JE2. Three passages were performed with each lasting 7 d. An inoculum of ~5x104 CFU was used for the first passage. For the second and third passages 10 isolates randomly collected from the previous passage were pooled and adjusted to ~5x104 CFU to serve as inoculum. Three mice were used per round of colonization.
Whole genome sequencing of 10 pooled isolates from each mouse in the three passages was used to determine whether repeated S. aureus colonization of the nasopharynx selected for genetic changes. The isolate inputs for pooled for DNA sequencing are shown in Table 1.

Table 1 List of strains used and generated in this study.
After the first passage, there was determined to be limited intragenic and intergenic sequence variation (Figure 3) based upon a threshold imposed of variants being represented at >20% i.e., in at least two isolates. Seven single nucleotide polymorphisms (SNPs) in total were identified from the 3 mice, with only one SNP being observed in more than one pool of isolates (S1 and S2 Tables). Identified SNPs showed low frequency of around 20% of total reads, indicating that at least two of the ten isolates forming the pooled genomic DNA carried the identified mutation. One sequenced isolate DNA pool (mouse B1) did not bear any SNPs. Overall, the low abundance and frequency of SNPs suggests there was not a pronounced or uniform selection on S. aureus during the first seven days of nasopharyngeal colonization as evidenced by the lack of a dominant variant causing population change.
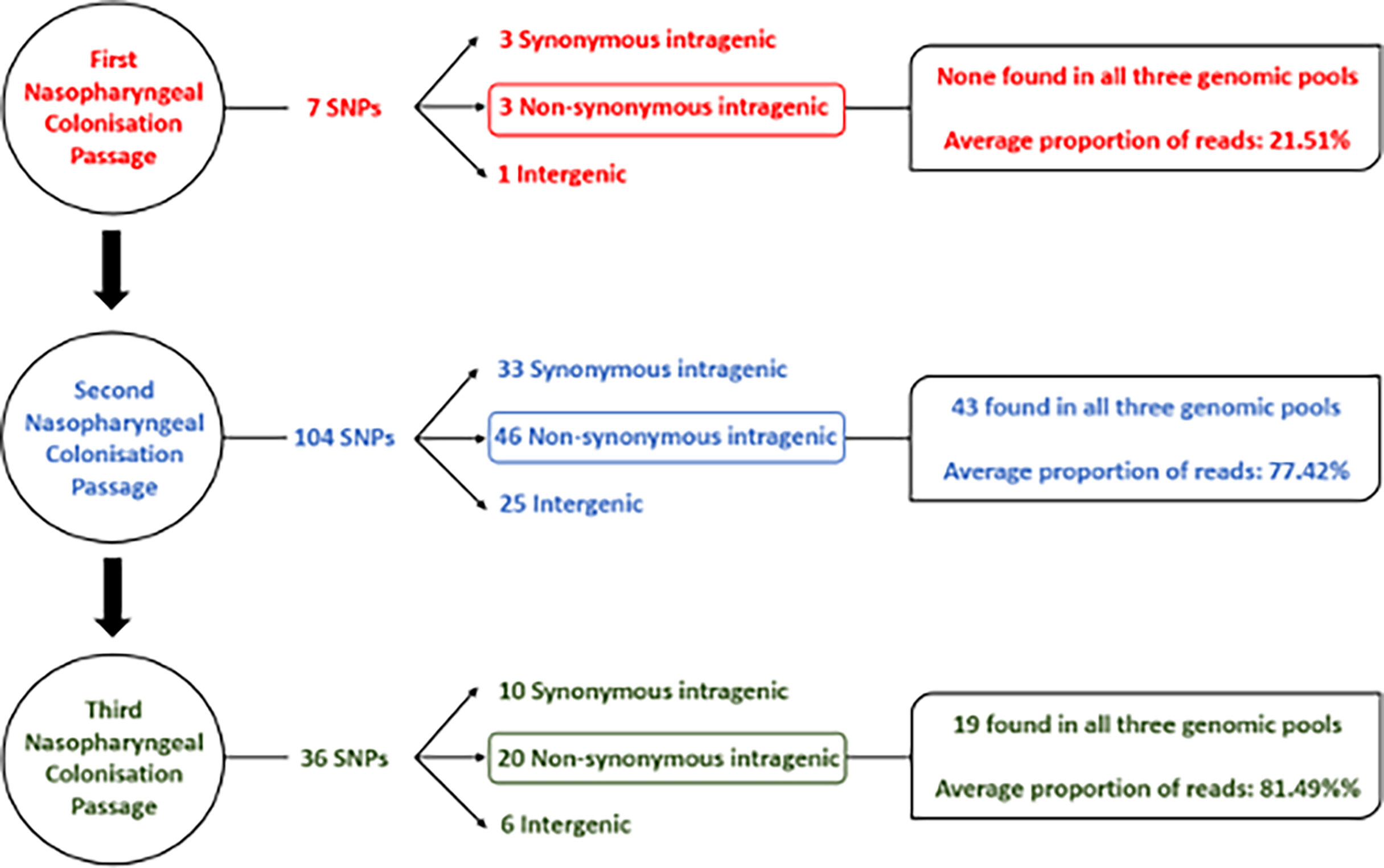
Figure 3 Flow chart detailing sequence variants identified in S. aureus isolates after each nasopharyngeal passage.
DNA sequencing of isolates after the second nasopharyngeal passage revealed a greater number of variants in the S. aureus clones, with 104 SNPs found across the three genomic DNA pools (Figure 3) based on the same >20% threshold imposed in the first round of sequence analysis. From these variants, 43 out of 46 non-synonymous intragenic SNPs and 29 out of 30 intergenic SNPs were observed in all three genomic pools (S3 and S4 Tables). A further difference between the profiles of sequence variants from the first and second passages is the higher proportion of reads for each variant in the second round. The DNA sequence pools from the second passage (B2 and C2) (Figure 2; S3 Table) revealed 37 intragenic SNPs that were present in at least 90% of the reads, (i.e., in 9/10 of the recovered S. aureus isolates). The pool of isolates from A2 had a lower proportion of reads for sequence variants, where most of the SNPs (n=36; 78.2%) were detected in a lower proportion (60%) of the reads, though still substantially higher than the variant frequencies of the first passage. The increased number of sequence variants identified after the second passage was suggestive of selection S. aureus during nasopharynx colonization leading to a third passage.
A reduced range of sequence variants was identified across the isolate pools from the three mice after the third 7-day colonization of the nasopharynx passage. A total of 36 SNPs were detected from the genome DNA pools (Figure 3). Above the threshold, sequence variants were present at ~90% of reads shared across the three sequence pools, with the exception of those detected in the hypothetical protein SAUSA300_RS09205. Among the 20 non-synonymous SNPs detected in the sequence pools of the third passage, 16 persisted from the first and/or second passage. In addition, three out of six intergenic SNPs also persisted across nasopharyngeal passages (Table 2). Culture of isolates on agar was minimized though the possibility of additional selection between nasal passages cannot be discounted.
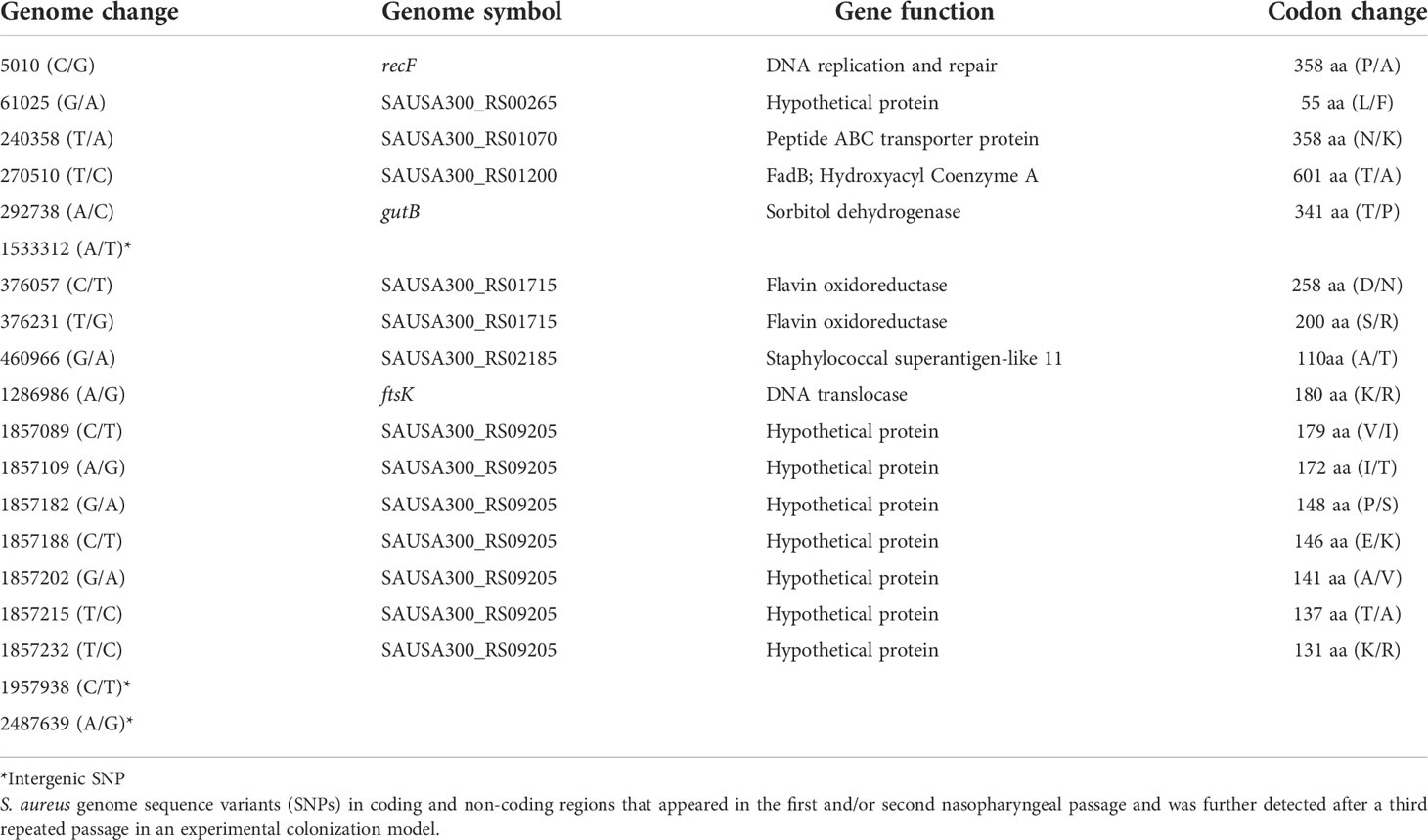
Table 2 SNPs of nasopharyngeal colonization isolates that persisted across repeated passages.
The functional category of genes containing sequence variants after each of the three 7-day nasopharyngeal colonization was identified. The variants included non-synonymous intragenic SNPs, and intergenic sequence variants determined to have the potential to alter transcription of downstream genes. The majority of the functional categories belong to genes encoding proteins involved in cell metabolism (amino acid and co-factors biosynthesis; energy and fatty acid metabolism) or DNA metabolism (replication, recombination and repair) (Figure 4).
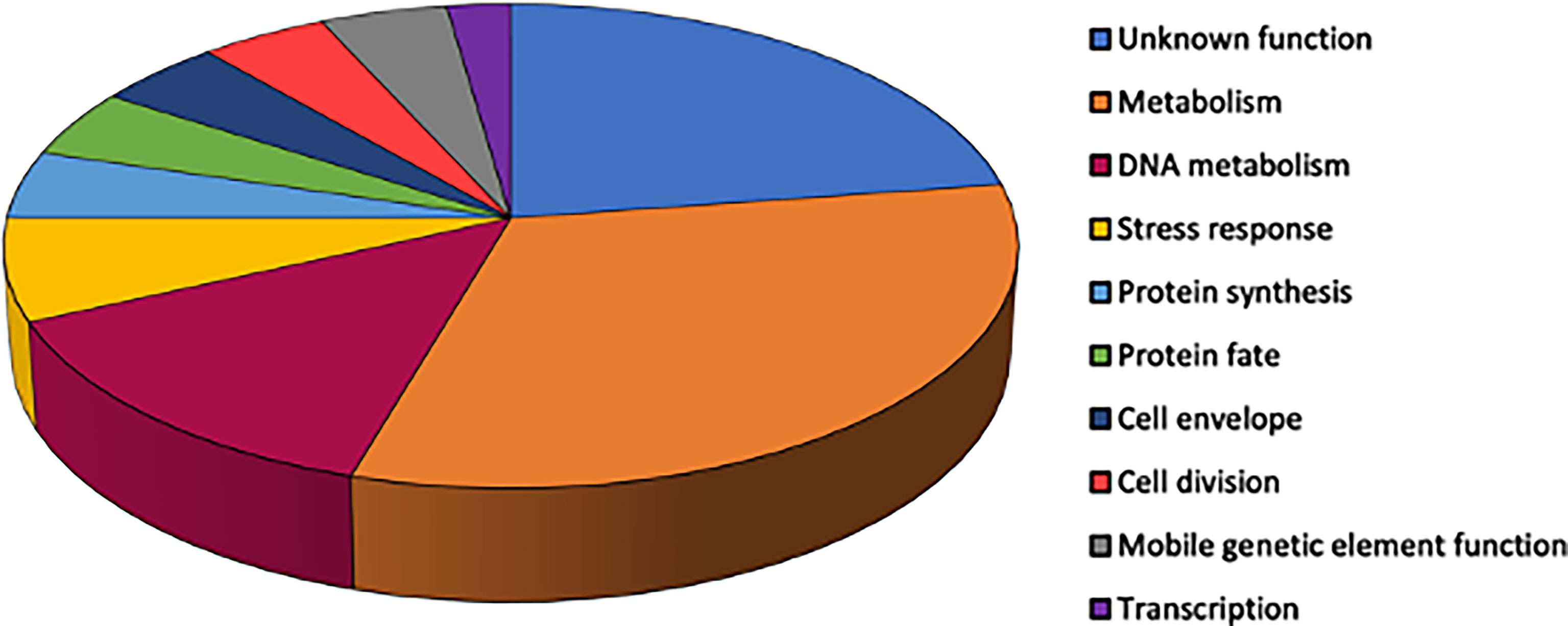
Figure 4 Relative proportions of functional groups ascribed to S. aureus genes with identified sequence variants across three serial nasopharyngeal passages. Variants were included for non-synonymous intragenic SNPs (n=44).
Proteins in pathways of DNA replication, recombination and repair with intergenic SNPs included: topoisomerases (GyrA, GyrB, GyrC); translocases (FtsK); nuclease (SbcC), and the DNA replication and repair protein RecF. The relative proportion of genes associated with these DNA pathways harboring SNPs could reflect selection for genome adaptation. Increased mutability by S. aureus was reported as an adaptation mechanism (Baharoglu and Mazel, 2014), whereby weakened DNA replication control and repair promotes allelic variability, benefiting S. aureus in its niche.
Variants associated with transcriptional regulation were observed for genes encoding proteins involved in transcription (RpoB), RNA modification (Sun, SAUSA300_RS04890 and SAUSA300_RS09885) and RNA decay (SAUSA300_RS06375) (S3 and S5 Tables). These genes play a role in cellular pathways that modulate ribosomal stability and the synthesis of proteins (Decatur and Fournier, 2002) as part of mechanisms the bacteria use to changes in the microenvironment (Khemici et al., 2015).
Sequence variants were identified in genes encoding metabolism proteins including mannitol (MtlA) and sorbitol fermentation (GutB), fatty acid metabolism (FadB; SAUSA300_RS01200 and SAUSA300_RS07925) and peptide metabolism (AmpA aminopeptidase). Further sequence variants were identified in genes encoding proteins associated with the biosynthesis of co-factors biotin (BioA) and thiamine (SAUSA300_RS11285), haem (SAUSA300_RS08410), and amino acids valine (ValS) and histidine (HisG) following S. aureus nasopharyngeal colonization. Genes encoding proteins within arginine metabolism revealed variants: ArcA and ArgF. Both enzymes catalyze formation of citrulline, the former as part of the arginine deiminase pathway that serves as energy source for S. aureus in anaerobic conditions, and the latter within arginine synthesis from glutamate (Diep et al., 2006; Pi et al., 2009). Pathways for responses to a variety of stresses revealed sequence variants within genes encoding ClpL (general stress tolerance), AhpF (oxidative stress) and DnaJ (hyperosmotic and heat shock stresses) together with variants observed in genes associated with anaerobic metabolism (NarK) and lactate (LctP2).
Those sequence variants that were identified within the pathways described during nasopharyngeal colonization revealed a smaller subset that persisted across the successive rounds of colonization (Table 2). One gene was identified as the target for multiple mutations across its length with varied individual frequencies of the variants from the three mice in the final passage. The gene (SAUSA300_RS09205) encodes a 399 amino acid hypothetical membrane protein described within the TIGRFAM: transport proteins and PFAM: metallopeptidase family M24. The collective sequence variants identified reveal this gene as showing selection most clearly across the sequential passages. Conversely, from the experimental evolution sequence data, there were sequence variants not maintained temporally, indicating potential refinement of selection during the repeated nasopharyngeal colonization passages of S. aureus. The vast majority of selected variants that persisted across repeated nasopharyngeal passages of S. aureus are genes involved in metabolism and DNA replication (Table 2).
Experiments to perform the third nasopharyngeal passage repeatedly returned S. aureus with a different spa type from the USA300 LAC species inoculum indicating that sets of mice were colonized by S. aureus upon arrival. Recent studies reported there is frequent colonization of laboratory mice by S. aureus (Schulz et al., 2017) in contrast to earlier studies that S. aureus are not natural colonizers of mice (McCarthy et al., 2012; Mulcahy et al., 2012). Others have determined that colonization by S. aureus prevents the invasion of distinct S. aureus strains (Margolis et al., 2010). To gain knowledge of the resident strain in the purchased mice and enable a simple comparative interrogation, we performed whole-genome sequencing and variant analysis on the mouse resident S. aureus (named SA_MOU). We sought insights of genome sequence polymorphism that converged with the experimental evolution selection from repeated colonization of the nasopharynx.
Multilocus sequence typing technique (MLST) identified SA_MOU as ST8, the same sequence type of USA300. A recent study demonstrated that the genetic variation within ST8 clones is rather substantial temporally and globally (Strauß et al., 2017) and that ST8 possesses clonal heterogeneity in Europe, where it co-exists with the dominant ST80, and others such as ST22 (Otter and French, 2010). The sequenced and assembled SA_MOU genome was aligned with the USA300 reference genome plus representatives of dominant STs in Europe: ST22 and ST80. Alignment showed fewest gaps with the SA_MOU assembled contig against the USA300 reference genome, supported by a pairwise identify of 99.5% across 94.5% coverage as the highest values among the three genome alignments (data not shown). Alignment of SA_MOU against ST22 and ST80 genomes revealed high pairwise identify (97.9% and 99.4%, respectively) but with lower coverage (88.0% and 90.9%, respectively).
Because we identified that SA_MOU was resident in mouse nasopharynx and was the same ST as USA300 LAC JE2, we hypothesized there was concordance between sequence variants that occurred in passaged isolates and the corresponding SA_MOU allele. Matches could provide support for key loci undergoing in vivo selection. The genes containing 13 non-synonymous intragenic SNPs from the third experimental nasopharyngeal passage were compared with those of the SA_MOU annotated genome. Sequences for 9 of the 13 genes were identified in the SA_MOU genome (Table 3) with four loci not located in the assembled genome. The cognate protein sequences detected in SA_MOU were compared with the translated USA300 reference genome (NC_007793.1) using BLASTp, which identified two proteins with matched sequence variation: RecF and FtsK.
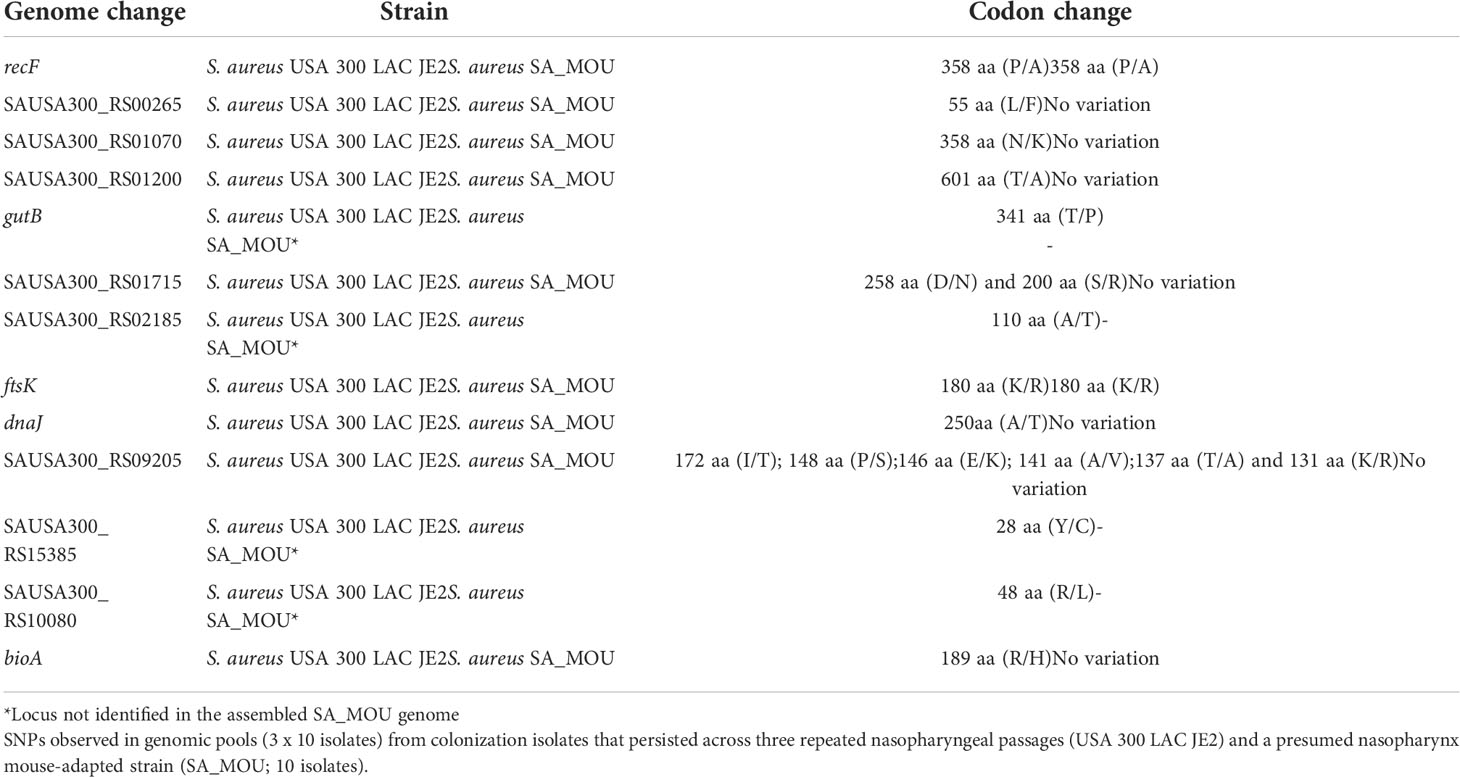
Table 3 Non-synonymous intragenic SNPs comparison between two ST8 S. aureus nasopharyngeal strains.
In this study, a murine S. aureus nasopharyngeal colonization model with a low inoculum was established to enable experimental evolutionary approaches to reveal contributory genetic factors. The colonization model used S. aureus USA300 LAC JE2 and showed relatively stable colonization of the bacteria in the nasopharynx over a 14-day post-inoculation period. Based on these data, day 7 was selected as a timepoint for the design of sequential colonization that would allow stable carriage and isolation of S. aureus.
The colonization data that was determined here was broadly similar to that of a previously developed cotton rat nasal colonization model (Kokai-Kun, 2008). A range of different animal models exist for the study of S. aureus nasal carriage where an inoculum of >107 CFU are typically used for prolonged colonization (Sanford and Ramsay, 1987; Sanford et al., 1989; Kiser et al., 1999; Kokai-kun et al., 2003; Sun et al., 2018). Several models use antibiotic pretreatment to promote stable nasal carriage, showing colonization >7 d being achieved with streptomycin-treated mice (Kiser et al., 1999; Mulcahy et al., 2012). In all of these models though, the infecting inoculum is high (>107 CFU) which is distinctly dissimilar to natural human colonization. Furthermore, such high infective doses can disrupt the host barrier, significantly altering host-pathogen interactions, while the use of antibiotics can disrupt the native flora potentially altering the host niche. The novel model described here uses a relatively low inoculum of S. aureus (5 x 104 CFU), more in keeping with natural colonization, and which resulted in long term stable carriage without antibiotic pretreatment.
After a first round of colonization lasting 7 d in our study, DNA sequencing of 10 pooled isolates revealed a low level of genetic variance with only 3 SNPs identified above a set threshold of 20% of sequencing reads. Two subsequent passages of S. aureus in the nasopharynx resulted in considerably greater genetic variation. The basis for the expansion of genetic variants by selection in these passages was not identified but could arise from the genetic variants present below the 20% threshold set for calling SNPs after the first round of colonization or the SNPs present above the threshold increasing mutation rate. Across the three rounds of colonization, there was evidence of genetic variants predominantly being within the DNA repair, recombination and replication category that might signify pathways through which selection has occurred sequentially. The nasopharyngeal colonization model demonstrates a platform to design experimental evolution over a short evolutionary timescale and could be modified to remove the population bottleneck that was imposed with each passage by choosing a limited number of isolates as the input inoculum. Future study of the model will also require control experimental evolution experiments designed to remove caution of the outcomes presented here and help interpret niche-specific drivers of selection. The inclusion of comparator S. aureus strains will also aid discrimination and help identify sequence changes resulting from strain or host-based factors.
A diverse set of loci and gene functions showed sequence variation as a result of the experimental evolution and reveal determinants that could contribute to the establishment and maintenance of nasopharyngeal colonization by S. aureus. Notably, there was low maintenance of SNPs across passages, potentially indicating that selective pressures of each passage were distinct as the genetic variants exerted their phenotypes. Multiple contributing effects could also alter maintenance of sequence variants, including competitor microbiota, differences in host genetics of each host producing variations in the immune response or variation in the S. aureus community composition as the inoculated isolate pool replicates. The experimental design here, with sequential in vivo and in vitro steps could also contribute to the conservation of phenotype expression. These factors would account for both different mutations emerging, purifying selection and reversion of mutations. Alternatively, changes could indicate there was refinement in the repertoire of S. aureus genome variants during nasopharyngeal colonization. Observed selection outcomes would require that some mutations were dominant in their effects or could alternatively overcome reproduction limitation. There may also be multiple routes through selection of mutations to achieve the same broad aims. Determining the cause for the low SNP maintenance would require extensive assessment of the functional outcome of the mutations and mutation combinations.
The finding in this study that several polymorphisms led to amino acid changes identical with those identified in a previous S. aureus epidemiology study of epidemic community-associated methicillin-resistant S. aureus highlights there could be relevant functional outcomes of SNPs (Kennedy et al., 2008). Moreover, the overlap supports both the methodology used here to investigate relevant features of S. aureus nasopharyngeal colonization and further study of the shared polymorphisms.
Various genetic changes detected in our colonization study are relevant to metabolism and nucleic acid functions rather than virulence factors. Krismer et al. (2014) determined that key metabolic pathways are essential during S. aureus nasal colonization owing to the low nutrient levels. Mutations located in genes related to carbohydrate biosynthesis and transport (mannitol, sorbitol and fructose-6-P metabolism) were identified in our study that could impact upon the acquisition and use of alternative carbon sources during nasopharynx colonization. Further genetic variation was detected in genes for co-factors biotin and thiamine, linked to amino acid and carbohydrate metabolism. Chaffin et al. (2012) reported increased in vivo transcription of co-factor genes associated with changes in the energy source and higher growth rate during lung infection. Young et al. (2012) investigated S. aureus genetic changes from carriage to disease and found the majority of mutations were associated with regulation of persistence components instead of virulence factors. Mutations found in stress resistance genes in this study (hemN, ahpF and clpL) which are possibly linked to altered expression for S. aureus components involved in protection from oxidative stress in the anterior nares (Chaves-Moreno et al., 2016). The absence of genetic variance selection in our study within virulence factors could reflect a predominant adaptive selection of metabolic pathways although the clear interplay in S. aureus between metabolism and virulence factor expression must not be overlooked (Somerville and Proctor, 2009).
Our identification of batches of mice pre-colonized with S. aureus proved to be a hindrance and future studies require that this effect is mitigated in the experimental plan. High rates of colonization are reported in these rodents from commercial vendors (Schulz et al., 2017) and this appears to be the problem in our study, as none of the experimental S. aureus USA300 LAC JE2 inoculum was recovered from mice naturally pre-colonized with S. aureus SAU_MOU. Both strains belonged to ST8 with different spa types (USA300 and SAU_MOU are t008 and t024, respectively). ST8 is an important clonal group, which includes the American epidemic CA-MRSA USA300 clone; this genotype is characterized by the unique linkage between SCCmecIVa and arginine catabolic mobile element (ACME), which provides both drug resistance and pronounced virulence factor expression during colonization/infection (Diep et al., 2008). While pre-colonization of mice complicated completion of the experimental plan in our study, the outcomes support the view that mice are better models for both colonization and infection than often argued (Mrochen et al., 2018).
Although USA300 LAC JE2 and SA_MOU share identical ST, genetic differences between these strains might account for the inability to identify that USA300 LAC JE2 had invaded the resident SA_MOU community colonized nasopharyngeal tissue to establish colonization, at least as judged by the lack of its retrieval. Genetic diversity within ST8 is known to be relatively high (Kawaguchiya et al., 2013; Glaser et al., 2016; Von Dach et al., 2016; Strauß et al., 2017) and intraspecies genetic variation is known to prevent colonization by incoming S. aureus (Margolis et al., 2010). Votintseva et al. (2014) used spa typing to detect multiple-strain S. aureus colonization and found that cocolonization was only 3.4 to 5.8% in samples pooled from healthy individuals.
In conclusion, our study highlights S. aureus genomic variants are selected using a model of nasopharynx colonization that incorporates successive inoculation cycles. The sequence variants were mostly within metabolism and stress pathways suggesting the associated gene functions could contribute to S. aureus nasal carriage. The individual understanding of their relevance will serve as basis for future studies focused on genetic factors that contribute to S. aureus nasal colonization and persistence.
The clinically-derived S. aureus strain USA300 LAC JE2 (Fey et al., 2013) was chosen as the strain to study in the model. This strain was used in the murine model described below which generated 90 nasopharyngeal colonization clones. Cultures were grown for 18 h at 37°C with shaking unless stated otherwise. Brain Heart Infusion (BHI) broth or agar was the medium used unless otherwise stated.
A nasopharyngeal model for S. aureus colonization was established to enable experimental evolution approaches. Mouse experiments were approved by the United Kingdom Home Office (Home Office Project License Number 40/3602) and the University of Liverpool Animal Welfare and Ethics Committee.
Groups of three mice (female BALB/c mice of 6-8-week-old, Charles River, UK) were assigned to a cage and the small group size was based on the subsequent genetic analysis. Mice were maintained with food and water ad libitum.
Animals were anaesthetized with a mixture of oxygen and isofluorane and 5 x 104 cfu of USA300 LAC JE2 in a volume of 15 μl were horizontally instilled into the nares. Inocula were adjusted to their cfu by dilution in phosphate-buffered saline (PBS) and confirmed by viable counts. Mice were monitored over 14 days post-inoculation and did not show any visible signs of disease. At five time points over a period of 14 days, mice were culled by cervical dislocation followed by aseptic removal of nasopharynx to determine CFU load. No CFUs were recovered from the lungs or blood over this period. Nasopharyngeal tissue was mixed with 3 ml of PBS and mechanically disrupted using an IKA T10 handled tissue homogenizer. Bacteria were enumerated from viable counts of serially diluted nasopharyngeal homogenates with growth and species confirmation on mannitol salt agar after incubation for up to 48 h at 37°C.
Staphylococcus aureus USA300 LAC JE2 was passaged three times by sequential colonization of the nasopharynx, with three mice per passage. Animal handling and bacterial inoculum preparation was performed as described above. An inoculum of USA300 LAC JE2 culture was prepared for the first nasopharyngeal passage and instilled in each of three mice. After the 7 d incubation period, mice were euthanized, each nasopharynx was aseptically removed and homogenized and the resulting tissue homogenates grown on separate mannitol salt agar plates. Ten clones were randomly selected from each replicate and subculture. Equal volumes (1 ml) of the ten cultures were mixed to generate a new inoculum followed by dilution in PBS to obtain a bacterial load of ~ 5 x 104 cfu. This inoculum was used for a second passage of murine nasopharyngeal colonization. A third and final serial passage was achieved by repeating this approach. Both control and resulting clones from each of the three nasopharyngeal passages were stored alongside nasal homogenates at -80°C with 15% glycerol (v/v).
DNA was purified from ten isolates randomly picked after plating nasal homogenates for each mouse from the three nasopharyngeal passages; purification methods were as described elsewhere (Coates-Brown et al., 2018) with the addition of an RNase step. The 9 genomic DNA pools for sequencing were prepared by mixing equimolar concentrations of DNA from which 9 libraries were prepared with an insert size of 350 bp using a TruSeq PCR-free sample prep kit (Illumina), according to the manufacturer’s instructions. DNA libraries were sequenced as barcoded pools on the Illumina MiSeq using V2 reagents generating paired sequencing reads of 350 bases in length. Reads were quality-filtered and trimmed for Nextera adaptor sequences using Trimmomatic (Bolger et al., 2014). Reads were aligned to parental S. aureus USA300 LAC JE2 that was sequenced in this study as a control. DNA library preparation and samples sequencing were done by the CGR (Centre for Genomic Research), University of Liverpool. Variant analysis was performed as described in Coates-Brown et al. (2018) with a threshold of 20% of reads.
S. aureus strain SA_MOU isolated from mouse nasopharynx in this study was assembled then visualized using Geneious software (version R11.1) where the resulting assembly was aligned by BLASTZ software (version 7.0.1) to the USA300 reference genome (USA300_FPR3757; NCBI Ref Seq: NC_007793.1) and two additional reference genomes representing dominant STs in Europe: ST22 (HO 5096 0412; NCBI Ref Seq: NC_017763.1) and ST80 (NCTC 8325; NCBI Ref Seq: NC_007795.1).
Mouse nasopharynx isolates were screened by PCR amplification of the staphylococcal protein A (spa) gene to confirm that the S. aureus found in the nasopharynx was the same spa type as the inoculum strain. The reactions were undertaken using Mastercycler pro S (Eppendorf) and each contained 0.5 μM PA 1095F forward PCR primer, 0.5 μM PA 1517R reverse PCR primer (Shopsin et al., 1999), 80 ng DNA, 25 μL of BioMix Red (Bioline) and DEPC-treated water up to a total reaction size of 50 μL mixed into 0.2 mL PCR tubes. The run cycle parameters were an initial 10 min at 95°C and then 30 cycles of 30 s at 95°C, 30 s at 60°C and 45 s at °C with a final 10 min at 72°C. PCR products were purified by using the Isolate II PCR and gel kit (Bioline), analyzed by agarose gel electrophoresis and then sequenced (GATC Biotech) with samples prepared according to the company requirements. The spa sequence was analyzed by using BLASTn (Madden, 2002) with default settings. Matched PCR fragment sizes and BLASTn output data for both control and passaged clones were used to indicate that they belonged to the same strain.
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found below: https://www.ebi.ac.uk/ena, PRJEB53542.
Mouse experiments were approved by the United Kingdom Home Office (Home Office Project License Number 40/3602) and the University of Liverpool Animal Welfare and Ethics Committee.
BS and EW performed the research. BS, EW and JM analyzed the data. AK and MH conceived and designed the study. BS, JM, AK and MH wrote the manuscript. All authors contributed to the article and approved the submitted version
BS was funded by the CAPES Foundation, Ministry of Education, Brazil. EW and AK were funded by the Joint Programming Initiative on Antimicrobial Resistance (JPI-AMR).
DNA sequencing was performed by the Centre for Genomic Research, Liverpool. We thank Charlotte Chong for technical assistance.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fcimb.2022.874138/full#supplementary-material
Baharoglu, Z., Mazel, D. (2014). SOS, The formidable strategy of bacteria against aggressions. FEMS Microbiol. Rev. 38 (6), 1126–1145. doi: 10.1111/1574-6976.12077
Baur, S., Rautenberg, M., Faulstich, M., Grau, T., Severin, Y., Unger, C., et al. (2014). A nasal epithelial receptor for staphylococcus aureus WTA governs adhesion to epithelial cells and modulates nasal colonization. PloS Pathog. 10 (5), e1004089. doi: 10.1371/journal.ppat.1004089
Bolger, A. M., Lohse, M., Usadel, B. (2014). Trimmomatic: a flexible trimmer for illumina sequence data. Bioinformatics 30 (15), 2114–2120. doi: 10.1093/bioinformatics/btu170
Chaffin, D. O., Taylor, D., Skerrett, S. J., Rubens, C. E. (2012). Changes in the staphylococcus aureus transcriptome during early adaptation to the lung. PloS One 7 (8), e41329. doi: 10.1371/journal.pone.0041329
Chaves-Moreno, D., Wos-Oxley, M. L., Jáuregui, R., Medina, E., Oxley, A. P., Pieper, D. H. (2016). Exploring the transcriptome of staphylococcus aureus in its natural niche. Sci. Rep. 6, 33174. doi: 10.1038/srep33174
Coates-Brown, R., Moran, J. C., Pongchaikul, P., Darby, A. C., Horsburgh, M. J. (2018). Comparative genomics of staphylococcus reveals determinants of speciation and diversification of antimicrobial defense. Front. Microbiol. 9. doi: 10.3389/fmicb.2018.02753
Coates, R., Moran, J. C., Horsburgh, M. J. (2014). Staphylococci: colonizers and pathogens of human skin. Future Microbiol. 9 (1), 75–91. doi: 10.2217/fmb.13.145
Corrigan, R. M., Miajlovic, H., Foster, T. J. (2009). Surface proteins that promote adherence of staphylococcus aureus to human desquamated nasal epithelial cells. BMC Microbiol. 9, 22. doi: 10.1186/1471-2180-9-22
Dean, R., Van Kan, J. A. L., Pretorius, Z. A., Hammond-Kosack, K. E., Di Pietro, A., Spanu, P. D., et al. (2012). The top 10 fungal pathogens in molecular plant pathology. Mol. Plant Pathol. 13 (4), 414–430. doi: 10.1111/j.1364-3703.2011.00783.x
Decatur, W. A., Fournier, M. J. (2002). rRNA modifications and ribosome function. Trends Biochem. Sci. 27 (7), 344–351. doi: 10.1016/S0968-0004(02)02109-6
Diep, B. A., Gill, S. R., Chang, R. F., Phan, T. H., Chen, J. H., Davidson, M. G., et al. (2006). Complete genome sequence of USA300, an epidemic clone of community-acquired methicillin-resistant staphylococcus aureus. Lancet 367, 731–739. doi: 10.1016/S0140-6736(06)68231-7
Diep, B. A., Stone, G. G., Basuino, L., Graber, C. J., Miller, A., Etages, S., et al. (2008). The arginine catabolic mobile element and staphylococcal chromosomal cassette mec linkage: convergence of virulence and resistance in the USA300 clone of methicillin-resistant staphylococcus aureus. J. Infect. Dis. 197 (11), 1523–1530. doi: 10.1086/587907
Fey, P. D., Endres, J. L., Yajjala, V. K., Widhelm, T. J., Boissy, R. J., Bose, J. L., et al. (2013). A genetic resource for rapid and comprehensive phenotype screening of nonessential staphylococcus aureus genes. mBio 4 (1), e00537–e00512. doi: 10.1128/mBio.00537-12
Glaser, P., Martins-Simões, P., Villain, A., Barbier, M., Tristan, A., Bouchier, C., et al. (2016). Demography and intercontinental spread of the USA300 community-acquired methicillin-resistant staphylococcus aureus lineage. mBio 7 (1), e02183. doi: 10.1128/mBio.02183-15
Grothe, C., Taminato, M., Belasco, A., Sesso, R., Barbosa, D. (2014). Screening and treatment for staphylococcus aureus in patients undergoing hemodialysis: a systematic review and meta-analysis. BMC Nephrolog 15, 202. doi: 10.1186/1471-2369-15-202
Hanssen, A. M., Kindlund, B., Stenklev, N. C., Furberg, A. S., Fismen, S., Olsen, R. S., et al. (2017). Localization of staphylococcus aureus in tissue from the nasal vestibule in healthy carriers. BMC Microbiol. 17, 89. doi: 10.1186/s12866-017-0997-3
Johnston, P. R., Dobson, A. J., Roff, J. (2016). Genomic signatures of experimental adaptation to antimicrobial peptides in staphylococcus aureus. G3-Genes Genom Genet. 6 (6), 1535–1539. doi: 10.1534/g3.115.023622
Kawaguchiya, M., Urushibara, N., Ghosh, S., Kuwahara, O., Morimoto, S., Ito, M., et al. (2013). Genetic diversity of emerging panton-valentine leukocidine/arginine catabolic mobile element (ACME)-positive ST8 SSCmec-iva methicillin-resistant staphylococcus aureus (MRSA) strains and ACME-positive CC% (ST5/ST764) MRSA strains in northern Japan. J. Med. Microbiol. 62, 1852–1863. doi: 10.1099/jmm.0.062125-0
Kennedy, A. D., Otto, M., Braughton, K. R., Whitney, A. R., Chen, L., Mathema, B., et al. (2008). Epidemic community-associated methicillin-resistant staphylococcus aureus: Recent clonal expansion and diversification. PNAS 105 (4), 1327–1332. doi: 10.1073/pnas.0710217105
Khemici, V., Prados, J., Linder, P., Redder, P. (2015). Decay-initiating endoribonucleolytic cleavage by RNase y is kept under tight control via sequence preference and sub-cellular localisation. PloS Genet. 11 (10), e1005577. doi: 10.1371/journal.pgen.1005577
Kiser, K. B., Cantey-Kiser, J. M., Lee, J. C. (1999). Development and characterization of a staphylococcus aureus nasal colonization model in mice. Infect. Immun. 67 (10), 5001–5006. doi: 10.1128/iai.67.10.5001-5006.1999
Kokai-Kun, J. F. (2008). The cotton rat as a model for staphylococcus aureus nasal colonization in humans: cotton rat s. aureus nasal colonization model. Methods Mol. Bio 431, 241–254. doi: 10.1007/978-1-60327-032-8_19
Kokai-kun, J. F., Walsh, S. M., Chanturiya, T., Mond, J. J. (2003). Lysostaphin cream eradicates staphylococcus aureus nasal colonization. Antimicrob. Agents Chemother. 47 (5), 1589–1597. doi: 10.1128/AAC.47.5.1589-1597.2003
Krismer, B., Liebeke, M., Janek, D., Nega, M., Rautenberg, M., Hornig, G., et al. (2014). Nutrient limitation governs staphylococcus aureus metabolism and niche adaptation in the human nose. PloS Pathog. 10 (1), e1003862. doi: 10.1371/journal.ppat.1003862
Krismer, B., Weidenmaier, C., Zipperer, A., Peschel, A. (2017). The commensal lifestyle of staphylococcus aureus and its interactions with the nasal microbiota. Nat. Rev. Microbiol. 15, 675–687. doi: 10.1038/nrmicro.2017.104
Kubicek-Sutherland, J. Z., Lofton, H., Vestergaard, M., Hjort, K., Ingmer, H., Andersson, D. I. (2017). Antimicrobial peptide exposure selects for staphylococcus aureus resistance to human defence peptides. J. Antimicrob. Chemother. 72 (1), 115–127. doi: 10.1093/jac/dkw381
Leonard, A. C., Petrie, L. E., Cox, G. (2019). Bacterial anti-adhesives: Inhibition of staphylococcus aureus nasal colonization. ACS Infect. Dis. 5 (10), 1668–1681. doi: 10.1021/acsinfecdis.9b00193
Liu, Z., Norman, G., Iheozor-Ejiofor, Z., Wong, J. K., Crosbie, E. J., Wilson, P. (2017). Nasal decontamination for the prevention of surgical site infection in staphylococcus aureus carriers. Cochrane Database Syst. Rev. 18 (5), CD012462. doi: 10.1002/14651858.CD012462.pub2
Lopez-Collazo, E., Jurado, T., De Dios Caballero, J., Pérez-Vázquez, M., Vindel, A., Hernández-Jiménez, E., et al. (2015). In vivo attenuation and genetic evolution of a ST247-SCCmecI MRSA clone after 13 years of pathogenic bronchopulmonary colonization in a patient with cystic fibrosis: Implications of the innate immune response. Mucosal Immunol. 8 (2), 362–371. doi: 10.1038/mi.2014.73
Madden, T. (2002). “The BLAST sequence analysis tool,” in The NCBI handbook. Eds. McEntyre, J., Ostell, J. (Bethesda, MD: National Center for Biotechnology Information (US). Available at: http://www.ncbi.nlm.nih.gov/books/NBK21097/.
Margolis, E., Yates, A., Levin, B. R. (2010). The ecology of nasal colonization of streptococcus pneumoniae, haemophilus influenzae and staphylococcus aureus: The role of competition and interactions with host’s immune response. BMC Microbiol. 10, 59. doi: 10.1186/1471-2180-10-59
McCarthy, A. J., Lindsay, J. A., Loeffler, A. (2012). Are all methicillin-resistant staphylococcus aureus (MRSA) equal in all hosts? epidemiological and genetic comparison between animal and human MRSA. Vet. Dermatol. 23 (4), 267–275. doi: 10.1111/j.1365-3164.2012.01072.x
Mrochen, D. M., Grumann, D., Schulz, D., Gumz, J., Trube, P., Pritchett-Corning, K., et al. (2018). Global spread of mouse-adapted staphylococcus aureus lineages cc1, cc15, and cc88 among mouse breeding facilities. Int. J. Med. Microbiol. 308 (6), 598–606. doi: 10.1016/j.ijmm.2017.11.006
Mulcahy, M. E., Geoghegan, J. A., Monk, I. R., O’Keeffe, K. M., Walsh, E. J., Foster, T. J., et al. (2012). Nasal colonisation by staphylococcus aureus depends upon clumping factor b binding to the squamous epithelial cell envelope protein loricrin. PloS Pathogens 8 (12), e1003092. doi: 10.1371/journal.ppat.1003092
Otter, J. A., French, G. L. (2010). Molecular epidemiology of community-associated meticillin-resistant staphylococcus aureus in Europe. Lancet Infect. Dis. 10 (4), 227–239. doi: 10.1016/S1473-3099(10)70053-0
Pi, B., Yu, M., Chen, Y., Yu, Y., Li, L. (2009). Distribution of the ACME-arcA gene among meticillin-resistant staphylococcus haemolyticus and identification of a novel ccr allotype in ACME-arcA-positive isolates. J. Med. Microbiol. 58 (Pt6), 731–736. doi: 10.1099/jmm.0.007351-0
Sanford, B. A., Ramsay, M. A. (1987). Bacterial adherence to the upper respiratory tract of ferrets infected with influenza a virus. Proc. Soc. Exp. Biol. Med. 185 (2), 120–128. doi: 10.3181/00379727-185-42525
Sanford, B. A., Thomas, V. L., Ramsay, M. A. (1989). Binding of staphylococci to mucus in vivo and in vitro. Infect. Immun. 57 (12), 3735–3742. doi: 10.1128/iai.57.12.3735-3742.1989
Schade, J., Weidenmaier, C. (2016). Cell wall glycopolymers of firmicutes and their role as nonprotein adhesins. FEBS Lett. 590 (21), 3758–3771. doi: 10.1002/1873-3468.12288
Schulz, D., Grumann, D., Trübe, P., Pritchett-Corning, K., Johnson, S., Reppschläger, K., et al. (2017). Laboratory mice are frequently colonized with staphylococcus aureus and mount a systemic immune response - note of caution for in vivo infection experiments. Front. Cell Infect. Microbiol. 7. doi: 10.3389/fcimb.2017.00152
Shopsin, B., Gomez, M., Montgomery, S. O., Smith, D. H., Waddington, M., Dodge, D. E. (1999). Evaluation of protein a gene polyphormic region DNA sequencing typing of staphylococcus aureus strains. J. Clin. Microbiol. 37 (11), 3556–3563. doi: 10.1128/JCM.37.11.3556-3563.1999
Sollid, J. U. E., Furberg, A. S., Hansen, A. M., Johannessen, M. (2014). Staphylococcus aureus. determinants of human carriage. Infect. Genet. Evol. 21, 531–541. doi: 10.1016/j.meegid.2013.03.020
Somerville, G. A., Proctor, R. A. (2009). At The crossroads of bacterial metabolism and virulence factor synthesis in staphylococci. Microbiol. Mol. Biol. Rev. 73 (2), 233–248. doi: 10.1128/MMBR.00005-09
Strauß, L., Stegger, M., Akpaka, P. E., Alabi, A., Breurec, S., Coombs, G., et al. (2017). Origin, evolution, and global transmission of community-acquired staphylococcus aureus ST8. Proc. Natl. Acad. Sci. U S A 114 (49), E10596–E10604. doi: 10.1073/pnas.1702472114
Sun, Y., Emolo, C., Holtfreter, S., Wiles, S., Kreiswirth, B., Missiakas, D., et al. (2018). Staphylococcal protein a contributes to persistent colonization of mice with staphylococcus aureus. J. Bacteriol. 200, e00735–e00 17. doi: 10.1128/JB.00735-17
Szafrańska, A. K., Junker, V., Steglich, M., Nubel, U. (2019). Rapid cell division of staphylococcus aureus during colonization of the human nose. BMC Genomics 20, 229. doi: 10.1186/s12864-019-5604-6
Von Dach, E., Diene, S. M., Fankhause, C., Schrenzel, J., Harbarth, S., François, P. (2016). Comparative genomics of community-associated methicillin-resistant staphylococcus aureus shows the emergence of clone ST8-USA300 in Geneva, Switzerland. J. Infect. Dis. 213 (9), 1370–1379. doi: 10.1093/infdis/jiv489
Von Eiff, V., Becker, K., Machka, K., Stammer, H., Peters, G. (2014). Nasal carriage as a source of staphylococcus aureus bacteremia. N Engl. J. Med. 344 (1), 11–16. doi: 10.1056/NEJM200101043440102
Votintseva, A. A., Miller, R. R., Fung, R., Knox, K., Godwin, H., Peto, T. E., et al. (2014). Multiple-strain colonization in nasal carriers of staphylococcus aureus. J. Clin. Microbiol. 52 (4), 1192–1200. doi: 10.1128/JCM.03254-13
Keywords: Staphylococcus aureus, nasopharyngeal colonization, evolution, genomics, adaptation
Citation: Salgado BAB, Waters EM, Moran JC, Kadioglu A and Horsburgh MJ (2022) Selection of Staphylococcus aureus in a murine nasopharyngeal colonization model. Front. Cell. Infect. Microbiol. 12:874138. doi: 10.3389/fcimb.2022.874138
Received: 11 February 2022; Accepted: 15 July 2022;
Published: 04 August 2022.
Edited by:
Francois Vandenesch, Université de Lyon, FranceReviewed by:
Frieder Schaumburg, University of Münster, GermanyCopyright © 2022 Salgado, Waters, Moran, Kadioglu and Horsburgh. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Malcolm J. Horsburgh, TS5KLkhvcnNidXJnaEBsaXZlcnBvb2wuYWMudWs=; Aras Kadioglu, QS5LYWRpb2dsdUBsaXZlcnBvb2wuYWMudWs=
†Present address: Elaine M. Waters, School of Natural Sciences, National University of Ireland (NUI) Galway, Ireland
Josephine C. Moran, Division of Infection, Immunity and Respiratory Medicine, Faculty of Biology, Medicine and Health, University of Manchester, United Kingdom
‡These authors have contributed equally to this work and share senior authorship
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.