 Kweku S. Enninful1
Kweku S. Enninful1 Samuel K. Kwofie2,3
Samuel K. Kwofie2,3 Mark Tetteh-Tsifoanya1Amanda N. L. Lamptey1Georgina Djameh1Samuel Nyarko1
Mark Tetteh-Tsifoanya1Amanda N. L. Lamptey1Georgina Djameh1Samuel Nyarko1 Anita Ghansah1
Anita Ghansah1 Michael D. Wilson1,4*
Michael D. Wilson1,4*- 1Department of Parasitology, Noguchi Memorial Institute for Medical Research, University of Ghana, Accra, Ghana
- 2Department of Biomedical Engineering, School of Engineering Sciences, University of Ghana, Accra, Ghana
- 3West African Centre for Cell Biology of Infectious Pathogens, College of Basic and Applied Sciences, University of Ghana, Accra, Ghana
- 4Stritch School of Medicine, Loyola University of Chicago, Maywood, IL, United States
Recent reports of resistance to artemisinin-based combination drugs necessitate the need to discover novel antimalarial compounds. The present study was aimed at identifying novel antimalarial compounds from natural product libraries using computational methods. Plasmodium falciparum is highly dependent on the pyrimidine biosynthetic pathway, a de novo pathway responsible for the production of pyrimidines, and the parasite lacks the pyrimidine salvage enzymes. The P. falciparum thymidylate monophosphate kinase (PfTMPK) is an important protein necessary for rapid DNA replication; however, due to its broad substrate specificity, the protein is distinguished from its homologs, making it a suitable drug target. Compounds from AfroDB, a database of natural products originating from Africa, were screened virtually against PfTMPK after filtering the compounds for absorption, distribution, metabolism, excretion, and toxicity (ADMET)-acceptable compounds with FAF-Drugs4. Thirteen hits with lower binding energies than thymidine monophosphate were selected after docking. Among the thirteen compounds, ZINC13374323 and ZINC13365918 with binding energies of −9.4 and −8.9 kcal/mol, respectively, were selected as plausible lead compounds because they exhibited structural properties that ensure proper binding at the active site and inhibitory effect against PfTMPK. ZINC13374323 (also called aurantiamide acetate) is known to exhibit anti-inflammatory and antiviral activities, and ZINC13365918 exhibits antileishmanial activity. Furthermore, aurantiamide acetate, which is commercially available, is a constituent of Artemisia annua, the herb from which artemisinin was derived. The compound also shares interactions with several residues with a potent thymidine analog inhibitor of PfTMPK. The anti-plasmodial activity of aurantiamide acetate was evaluated in vitro, and the mean half-maximal inhibitory concentration (IC50) was 69.33 μM when synchronized P. falciparum 3D7 culture was used as compared to IC50 > 100 μM with asynchronized culture. The significance of our findings within the context of malaria treatment strategies and challenges is discussed.
Introduction
Plasmodium falciparum is among the five Plasmodium parasites that cause human malaria and is also responsible for the most severe form of the disease (Tuteja, 2007; Ludin et al., 2012; Spitzmüller and Mestres, 2013; Bhatt et al., 2015). Recent studies have reported P. falciparum to be developing resistance to current major antimalarial drugs (Egwu et al., 2022), which warrants the identification and development of new antimalarials as a necessity. Throughout the history of malaria chemotherapy, the most successful antimalarials have been natural products. Antimalarials such as chloroquine were developed from quinine, which was extracted from the bark of the Cinchona tree from South America. Artemisinin was also obtained from Artemisia annua originating from China (Tajuddeen and Van Heerden, 2019). Medicinal herbs have generally proven to be very effective drugs against parasitic diseases (Siddiqui et al., 2014). Even though both chloroquine and artemisinin have been plagued with resistance to the parasite, research into natural product antimalarials remains a priority. Out of about 1,524 compounds with anti-plasmodial activity reported between 2010 and 2017, 39% were natural products, with 29% of the compounds having half-maximal inhibitory concentration (IC50) ≤ 3.0 µM against at least one Plasmodium strain (Tajuddeen and Van Heerden, 2019). This reinforces the urgent need to exploit natural products to unravel future potent biotherapeutic molecules.
Research efforts at developing new antimalarials have uncovered diverse pathways and protein targets of which some are novel (Belete, 2020; Agamah et al., 2021). It is worthwhile to mention that new antimalarials, currently in the clinical trial phase, have been identified against critical pathways and targets (Kumar et al., 2018; Belete, 2020). However, a good number remain unexploited, while others have not yielded any potent drugs. Identification of new antimalarial targets from novel pathways not associated with resistance must be explored by screening diverse compound libraries (Ludin et al., 2012; Spitzmüller and Mestres, 2013).
Thymidylate monophosphate kinase catalyzes the reversible phosphorylation of dTMP to deoxythymidine diphosphate (dTDP), which is an important step for cellular DNA synthesis (Cassera et al., 2011; Krungkrai and Krungkrai, 2016; Vanoevelen et al., 2022). It has a broad substrate specificity, which distinguishes it from other homologs, making it a suitable target (Reyes et al., 1982; Cassera et al., 2011; Krungkrai and Krungkrai, 2016). P. falciparum thymidylate monophosphate kinase (PfTMPK) is involved in the pyrimidine biosynthetic pathway, a de novo pathway responsible for the production of pyrimidines, which are necessary for rapid DNA replication (Reyes et al., 1982; Cassera et al., 2011; Krungkrai and Krungkrai, 2016). The pathway is preferred because the parasite is highly dependent on it since it lacks pyrimidine salvage enzymes (Cassera et al., 2011; Krungkrai and Krungkrai, 2016).
So far, thiourea has been shown to exhibit inhibitory activity against PfTMPK, the compound that was first discovered to inhibit the TMPK of Mycobacterium tuberculosis (MtTMPK) (Cui et al., 2012; Patrick and Turner, 2020). The inhibitory effect of thiourea on PfTMPK is however weaker than that of MtTMPK. Several analogs containing thiourea or urea have since been tested for antimalarial activity through the inhibition of PfTMPK (Cui et al., 2012). However, the analogs have mostly exhibited weak inhibition against PfTMPK. The most active analog, phenylurea, exhibited an EC50 of 28 nM but was reported to be a weak inhibitor of PfTMPK with a binding affinity of 200 µM (Cui et al., 2012; Chen et al., 2018). It was also observed that ureas showed more antimalarial activities than thioureas (Cui et al., 2012; Patrick and Turner, 2020).
In this study, new antimalarials are predicted for P. falciparum by virtually screening an African natural product library against PfTMPK. Recently, the development of quantitative structure–activity relationship (QSAR) models has contributed significantly to antimalarial discovery (Neves et al., 2020). As such, this study explores the structural insights for PfTMPK inhibition using QSAR modeling, pharmacophore mapping, and docking studies (Ojha and Roy, 2013) to select the most plausible antimalarial lead compounds with good pharmacological profiles from compounds in the AfroDB database. Molecular dynamic simulations were also undertaken to provide insights into the binding mechanisms of PfTMPK.
Methodology
In-Silico Absorption, Distribution, Metabolism, Excretion, and Toxicity Filtering of the Natural Product Database
A total of 885 African natural products were retrieved from AfroDB (Ntie-Kang et al., 2013) in “.sdf” format for absorption, distribution, metabolism, excretion, and toxicity (ADMET) analysis. The compounds were screened via FAF-Drugs4 (Lagorce et al., 2017) to elucidate their pharmacokinetic, structural, and physicochemical properties. The physicochemical filter used for the compounds was “Drug likeness”. The compounds were categorized as “rejected”, “intermediate”, or “accepted” by FAF-Drugs4. “Accepted” compounds were then chosen for molecular docking.
Virtual Screening
The “accepted” compounds from the FAF-Drugs4 ADMETox prediction were docked against PfTMPK, using AutoDock Vina (Trott and Olson, 2010) integrated with PyRx (Dallakyan and Olson, 2015). The substrate of PfTMPK (TMP) was also extracted from the complex (Protein Data Bank (PDB) ID: 2wwf) and added to the library of “accepted” compounds for docking. Before the docking, the compounds were energy minimized with OpenBabel using the universal force field (uff) before converting to “.pdbqt” formats (Kwofie et al., 2021). A grid box size of 17.3, 9.7, and 11.2 Å and center dimensions of 42.5, 46.7, and 47.6 Å all in the x, y, and z coordinate axes were used, respectively.
Docking Method Validation
For validation of the performance of the docking technique, the SMILES of five potent inhibitors (compounds 28, 30, 53, 54, and 55) against PfTMPK (Cui et al., 2012) were used to generate their decoys via RApid DEcoy Retriever (RADER) (Wang et al., 2017). A total of 243 decoys were obtained and screened together with the five inhibitors against PfTMPK using AutoDock Vina. The docking results were used to generate a receiver operating characteristic (ROC) curve, and the area under the curve (AUC) was computed using easy ROC Ver. 1.3 (Goksuluk et al., 2016). Parameters used for the ROC generation and calculation of AUC were a non-parametric method for curve fitting (DeLong et al., 1988) for SE estimation and CI as well as a Type I error of 0.05.
The co-crystallized ligands of PfTMPK were removed from their binding site and re-docked against PfTMPK. The best-predicted docking pose of each ligand was superimposed with its respective experimental co-crystallized ligand pose, and their root mean square deviation (RMSD) was calculated using LigAlign (Heifets and Lilien, 2010).
Molecular Dynamic Simulations of Complexes
Molecular dynamics simulations were performed using the PfTMPK in complex with TMP and the respective chosen potential lead compounds. GROningen MAchine for Chemical Simulation (GROMACS) version 5.1.4 (Van Der Spoel et al., 2005) was used to perform the molecular dynamics simulations using the GROMOS96 43A1 force field. The topology of the compounds was generated using PRODRG (Schüttelkopf and Van Aalten, 2004). As part of the preparation before the simulation, the complexes were first solvated in a 1-nm dodecahedron water box and neutralized by adding one positive ion. The complexes were relaxed through energy minimization to remove any steric clashes or bad geometry and equilibrated to the required temperature (300K) and density (1,020 kg/m3). After the respective systems were equilibrated and set in the desired temperature and density, a 100-ns production run was performed, and the results of the simulations were analyzed using Xmgrace version 5.1.25 (Vaught, 1996).
In-Vitro Parasite Growth Inhibition Assay
One of the potential lead compounds, aurantiamide acetate, was tested for anti-plasmodial activity using the SYBR Green I fluorescence assay as described previously (Smilkstein et al., 2004). Stock concentrations of 100 mM [100% dimethyl sulfoxide (DMSO)] of compounds were diluted with culture media to a working concentration of 100 µM (0.1% DMSO). A serial dilution of 1:2 concentrations of the compound (10 to 0.781 µM) was prepared for the assay. Artesunate was the reference drug diluted from 100 to 3.125 nM. Test wells were initially seeded with 90 µl of ring-stage (synchronized) parasitized red blood cells (pRBCs), culture media, and pRBCs at 2% hematocrit and 1% parasitemia. An aliquot of 10 µl of each concentration was dispensed into each well in triplicates. The wells containing RBCs (2% hematocrit), pRBCs, and cultured protoplast washing (CPW) media served as negative and blank controls and were then incubated for 48 h. An aliquot of 100 µl of 4× buffered SYBR Green I (0.20 µl of 10,000× SYBR Green I/ml of 1× phosphate-buffered saline) was then added for a further 30 min at 37°C. The presence and amount of pRBCs were detected by fluorescence using the Guava EasyCyte HT FACS machine (Millipore, Billerica, MA, USA), and parasitemia was recorded in percentages. The IC50 was extrapolated from non-linear regression curves of percentage inhibition versus log-concentration curves from GraphPad Prism (Graph Pad Software, San Diego, CA, USA) using algorithms obtained from flow cytometry (fluorescence-activated cell sorting (FACS)) data. The experiment was repeated using an asynchronized parasite culture using the same protocol.
Results and Discussion
Target Structure
The three-dimensional structure of PfTMPK retrieved from the PDB (PDB ID: 2wwf) was solved using X-ray crystallography at a resolution of 1.89 Å (Whittingham et al., 2010). The 2wwf is a homo-oligomer (homodimer) consisting of three chains of PfTMPK in complex adenosine diphosphate (ADP) and thymidine monophosphate (TMP), which is a natural substrate. Other ligands like sodium-ion and glycerol are also present. Figure 1 shows chain A of 2wwf with the active site, and TMP is highlighted. The protein has a sequence length of 212 and is significantly different from the human TMPK with a sequence identity of 36.9%.
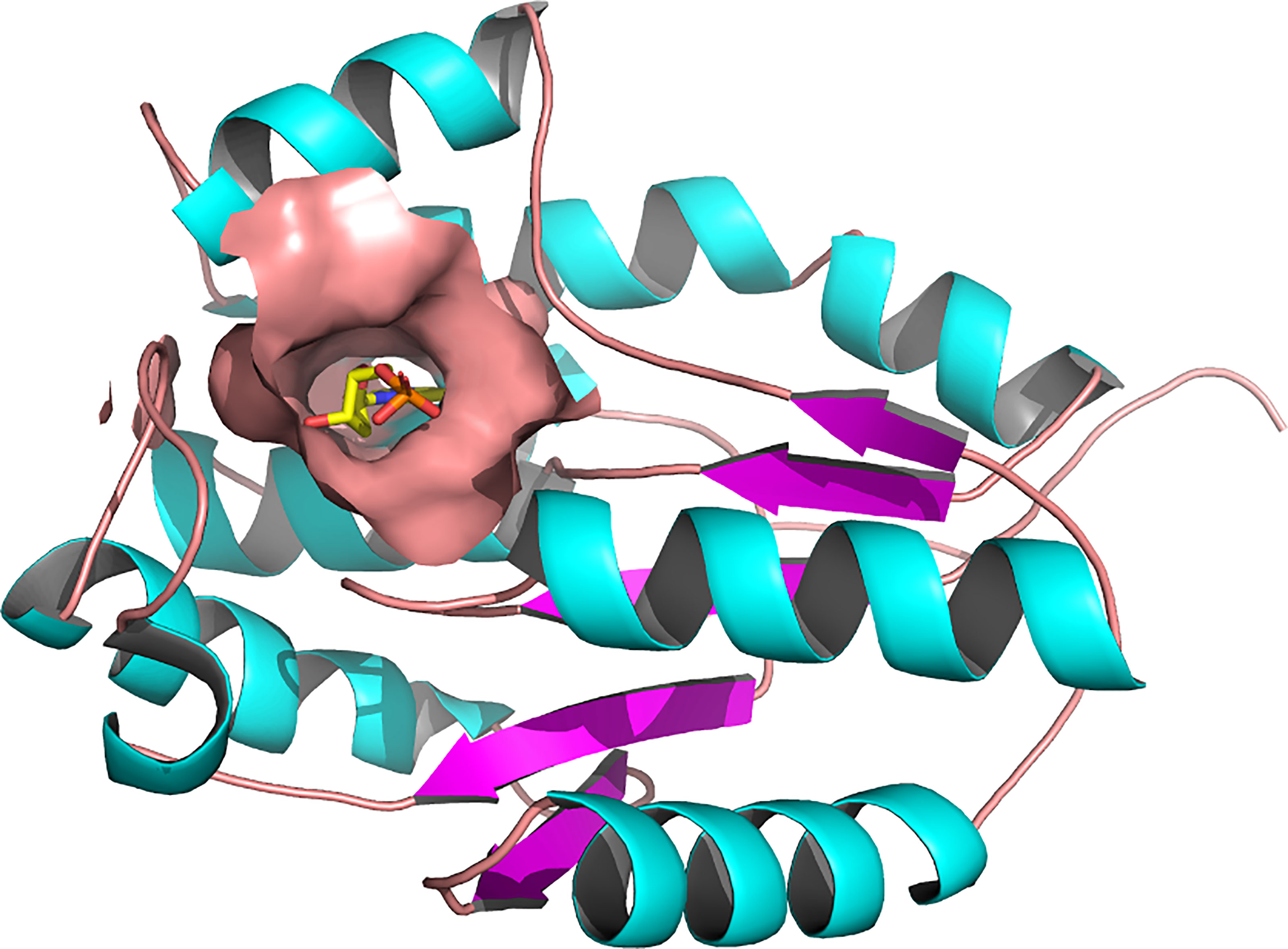
Figure 1 Three-dimensional structure of Plasmodium falciparum thymidylate monophosphate kinase (PDB ID: 2WWF) and its active site (Whittingham et al., 2010). A cartoon representation of the 3D structure of PfTMPK. The surface representation shows the active site of PfTMPK and binding in its natural substrate, thymidine monophosphate (TMP), represented by yellow sticks. Active site residues include Asp17, Lys21, Leu59, Phe44, Arg47, Pro45, Phe74, Arg78, Arg99, Tyr100, Ser103, Gly104, Tyr107, and Tyr153 (Kandeel et al., 2009). The protein structure was retrieved from the Protein Data Bank, and image was generated with PyMOL.
Absorption, Distribution, Metabolism, Excretion, and Toxicity-Acceptable Compounds
ADMET tests are performed to eliminate compounds that may be weak drug candidates so that potential drug-like compounds are prioritized (Sliwoski et al., 2013). FAF drug server uses quantitative QSAR models to predict specific properties or toxicological endpoints of compounds (Lagorce et al., 2017). The predicted properties were compared with the standard range of accepted values concerning the chosen physicochemical filter, which was “drug-likeness”. The filtered compounds were categorized as “rejected”, “intermediate”, or “accepted”. The “accepted” compounds are those with no structural alerts and satisfy the physicochemical filter. Intermediate and rejected compounds show some structural alerts and do not satisfy completely the physicochemical filter. Structural alerts are substructures that are related to mutagenic and carcinogenic properties, which are undesirable for drug-likeness (Benigni and Bossa, 2006). After the compounds were screened using the FAF drug, 91 compounds categorized as “accepted” were used for docking against PfTMPK.
Docking Protocol Validation
To validate AutoDock Vina’s ability to distinguish between active and inactive compounds concerning PfTMPK, a ROC curve (Figure 2) was generated after virtually screening five potent inhibitors of PfTMPK (Cui et al., 2012) (Table 1) and their decoys against the receptor. The AUC value was calculated to assess the docking performance. AUC value less than 0.5 is considered a poor discrimination ability, from 0.5 to 0.7 is considered moderate, and greater than 0.7 is acceptable (Mandrekar, 2010). AUC value very close to 1 indicates an excellent discriminatory ability of the docking model for the receptor (Mandrekar, 2010). The AUC obtained was 0.95 with a p-value of 9.776159e−78, indicating an excellent discriminatory ability of AutoDock Vina to distinguish between active compounds and decoys of PfTMPK.
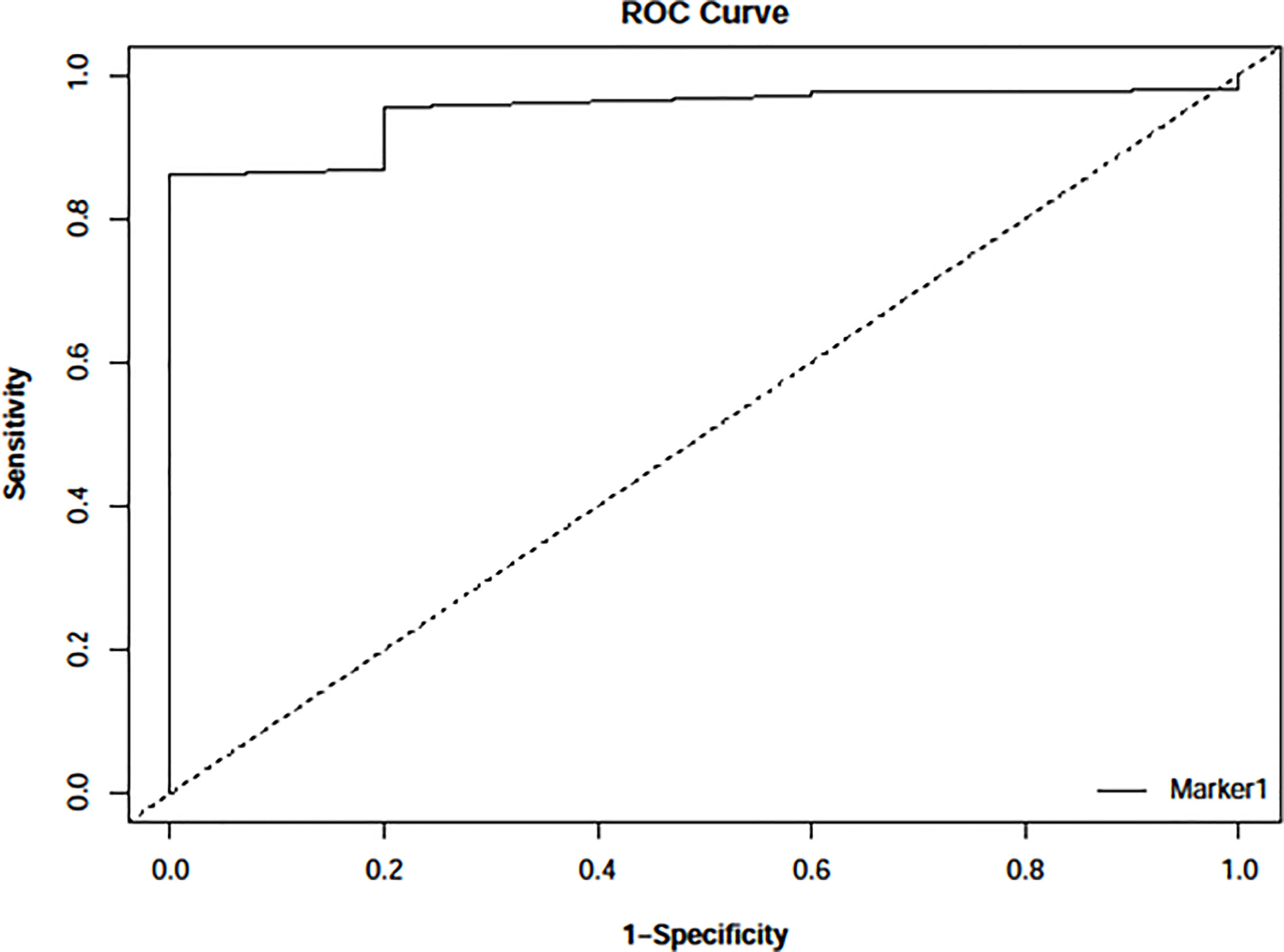
Figure 2 Receiver operating characteristic (ROC) curve generated with easyROC (Goksuluk et al., 2016) after docking 243 decoys and 5 potent inhibitors against PfTMPK using AutoDock Vina via PyRx version 0.8. The area under the curve (AUC) obtained was 0.94897 with a p-value of 9.776159e−78.

Table 1 List of potent inhibitors and co-crystallized ligands showing the Ki values (Cui et al., 2012), the respective PDB IDs, and RMSD values of the aligned co-crystallized and re-docked ligands.
To validate further the docking approach used, four co-crystallized PfTMPK ligands (three of which are complexed with inhibitors and a substrate) were retrieved from their PDB crystal structures and re-docked against the PfTMPK protein structures. The co-crystallized binding poses of the ligands were superimposed to their respective predicted docking poses, and RMSDs less than 2 Å were obtained for all of them. This suggests that AutoDock Vina reasonably distinguished between active and inactive compounds for PfTMPK and accurately predicted binding poses.
Virtual Screening Analysis and Lead Identification
Ninety-one pre-filtered drug-like compounds were screened against PfTMPK together with its natural substrate and known potent inhibitors. After docking, thirteen of the drug-like compounds had binding affinities greater than the protein’s substrate and were selected as hits (Table 2). The binding energies of three potent inhibitors were −8.9, −9.3, and −9.7 kcal/mol for compounds 28, 53, and 55, respectively, which fell within the same range as the hits. The lowest energies among them were −10.5 and −10.2 kcal/mol for compounds 54 and 30, respectively, which were not far from the lowest among the hits (−9.9 kcal/mol). This is a good indication of the potential antimalarial activity of the hits. Lead compounds were then selected among the hits. Four structural properties of a molecule that ensure proper binding at the active site and inhibitory effect against PfTMPK, developed using QSAR analysis, pharmacophore modeling, and docking studies with a set of thymidine analogs that have well-defined PfTMPK inhibitory activity, were considered in selecting possible lead compounds (Ojha and Roy, 2013). These properties are the presence of –NH fragment, –OH group, urea moiety, and a considerable amount of oxygen atoms (Ojha and Roy, 2013).
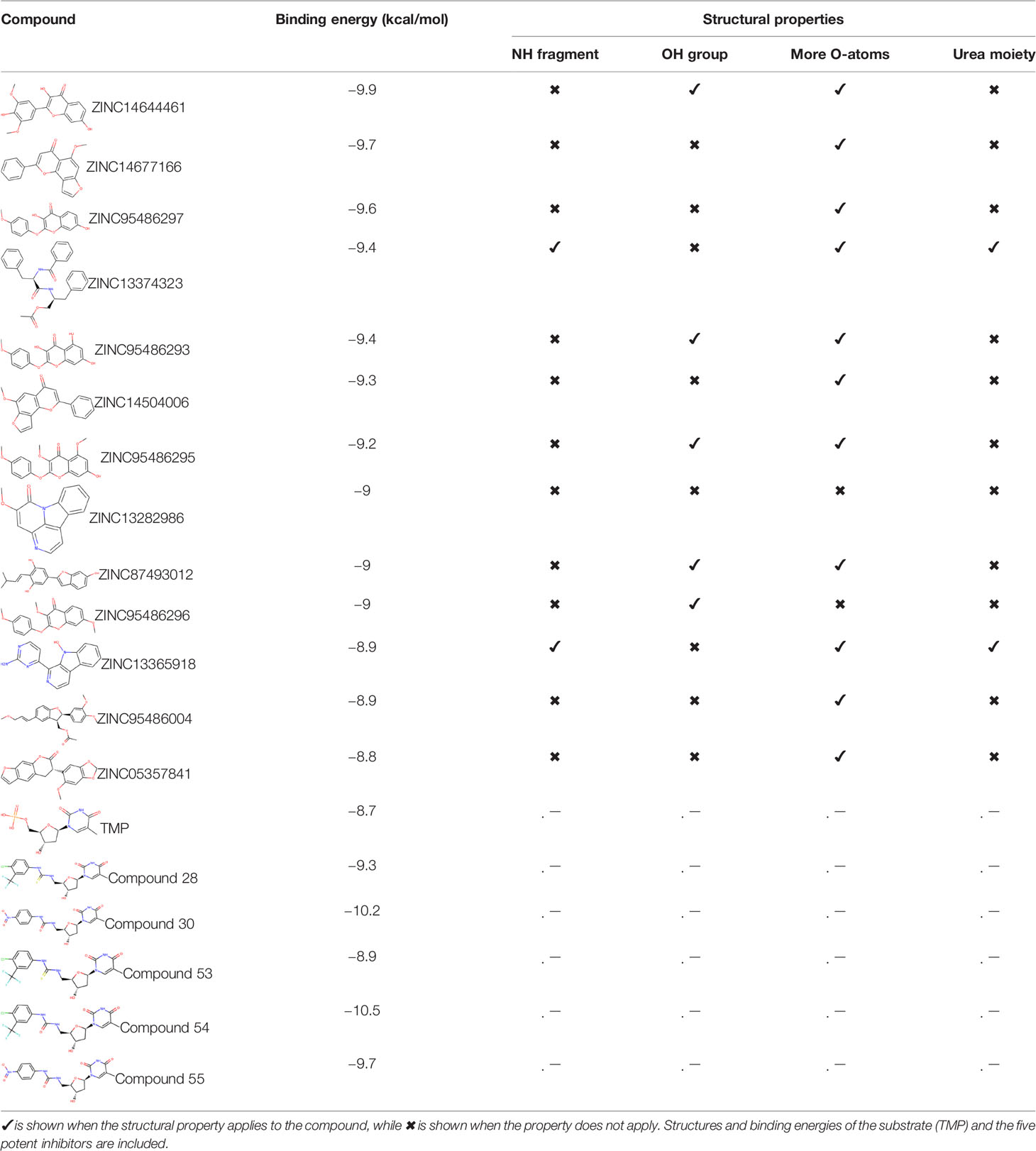
Table 2 List of hits showing their structures, binding energies, and structural properties.
The molecular structures of the hit compounds were analyzed to identify any of the aforementioned structural properties. ZINC13374323 possesses –NH group, urea moiety, and a considerable amount of oxygen atoms, while ZINC13365918 also has all the properties but lacks oxygen atoms. ZINC14644461, ZINC95486293, ZINC95486295, and ZINC87493012 all have –OH groups and considerable amounts of oxygen atoms, while ZINC13282986 possesses none of the structural properties. The rest of the six compounds only possess oxygen atoms among the aforementioned structural properties. Most of the compounds seem to possess considerable amounts of O-atoms, which is an essential requirement for high binding affinity toward PfTMPK. The most distinctive structural properties were the presence of NH fragment and urea moiety, which were exhibited only by ZINC13374323 and ZINC13365918. Although all four properties are essential for good inhibitory activity against PfTMPK (Ojha and Roy, 2013), a ligand with at least three of the properties, placing priority on the distinctive properties (presence of -NH fragment and urea moiety), may exhibit a good inhibitory effect against the compound with no or little modifications. As such, ZINC13374323 and ZINC13365918 were considered the potential lead compounds among the 13 hits.
Protein–Ligand Interactions
Protein–ligand molecular interactions of ZINC13374323 and ZINC13365918 were further studied and compared to the protein–ligand molecular interaction of compound 25, a PfTMPK inhibitor thymidine analog, which has been shown to exhibit very high activity against the protein (Ojha and Roy, 2013). Compound 25 has been reported to share molecular interactions with protein residues Arg78, Arg99, Arg47, Asp17, Ser22, Phe74, and Tyr43 (Ojha and Roy, 2013). In Figure 3A, we show the substrate to share interactions with similar residues such as Arg78, Arg99, Arg47, and Phe74. From Figure 3C, ZINC13374323 is also shown to have interactions with common residues such as compound 25. ZINC13365918, on the other hand, exhibited interactions with Asp17 and Phe74 among the listed residues (Figure 3B). This warranted the experimental characterization of ZINC13374323 to corroborate potential inhibitory activity against PfTMPK. H-bond interactions with Asp17 and Pi-Pi stacked interactions with Tyr107 and Phe74 are common among both ligands. This may indicate the importance of the residues for effective binding. One can suggest that the lack of more O-atoms in ZINC13365918 significantly affects its ability to form H-bonds with important residues such as Arg47 and Arg99. TMP and ZINC13374323 were able to form H-bonds with both Arg47 and Arg99 using their O-atoms. However, a lack of an OH-group did not seem to affect the ligands’ interactions, since none of them formed any bonds with it.
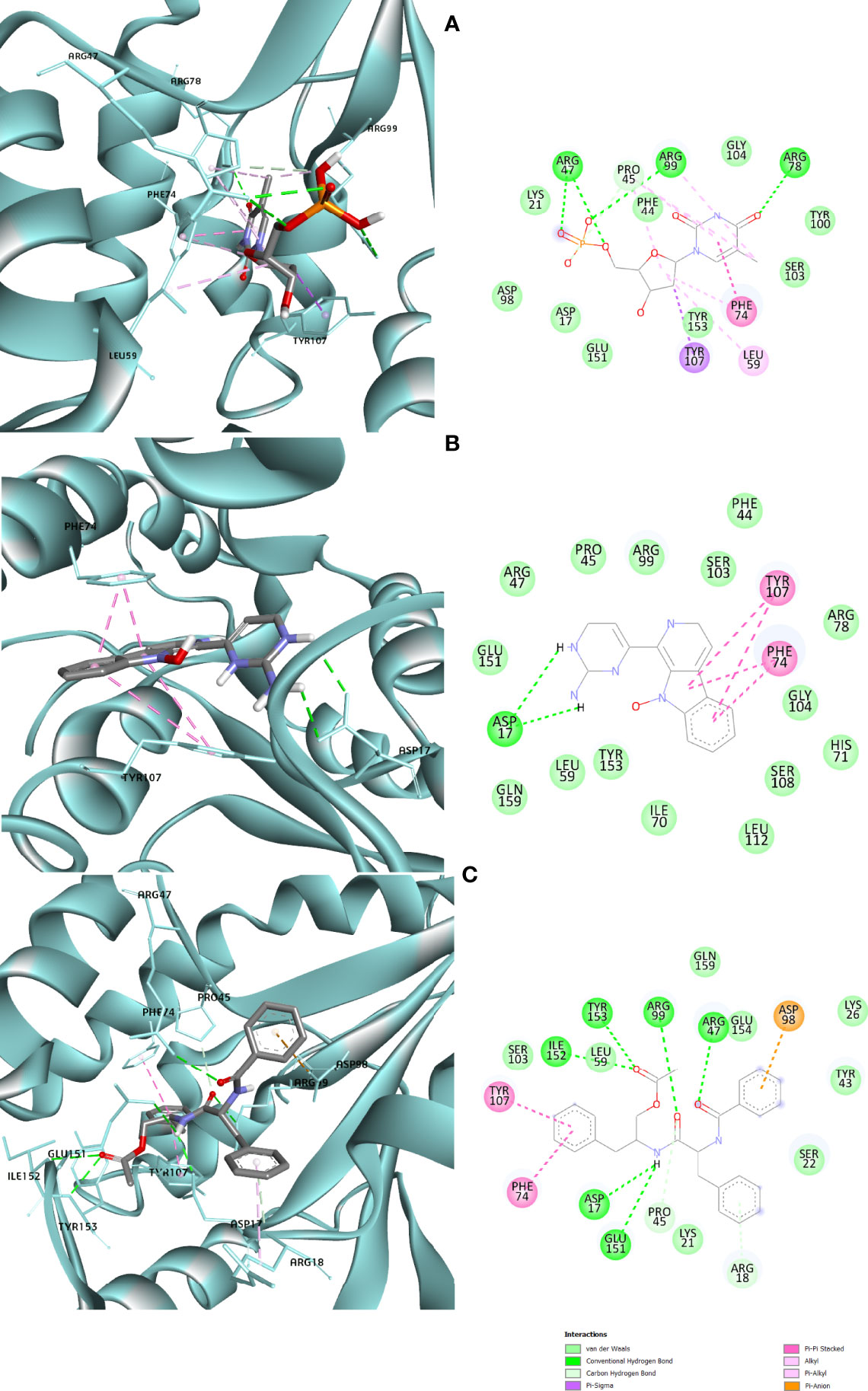
Figure 3 3D and 2D images of the protein–ligand interactions between PfTMPK and (A) TMP, (B) ZINC13365918, and (C) ZINC13374323. TMP forms H-bonds with Arg47, Arg99, and Arg78; ZINC13374323 is shown to have H-bond interactions with Arg99, Arg47, Asp17, Glu151, Ile152, and Tyr153; ZINC13365918 forms H-bonds with only Asp17.
Molecular Dynamics Simulations
Stability of Protein–Ligand Complexes
Molecular dynamics simulations were performed to compare the stability of the distinctive protein–ligand complexes of PfTMPK with TMP, ZINC13374323, and ZINC13365918. To accomplish that, the RMSDs of the complexes were generated after the simulation (Figure 4). The results showed that PfTMPK–ZINC13374323 behaved very similarly to PfTMPK–TMP in terms of stability. Complex PfTMPK–ZINC13365918 shows a higher RMSD but has much fewer fluctuations than PfTMPK–TMP and PfTMPK–ZINC13374323, maintaining an RMSD of 0.34 nm from 15- to 90-ns simulation time. Complexes PfTMPK–TMP and PfTMPK–ZINC13374323 exhibited RMSD from 0.25 nm at the first 20 ns of the simulation to 0.26 nm between 20 ns and 60 ns and 0.27 nm in the final 10 ns of the simulation.
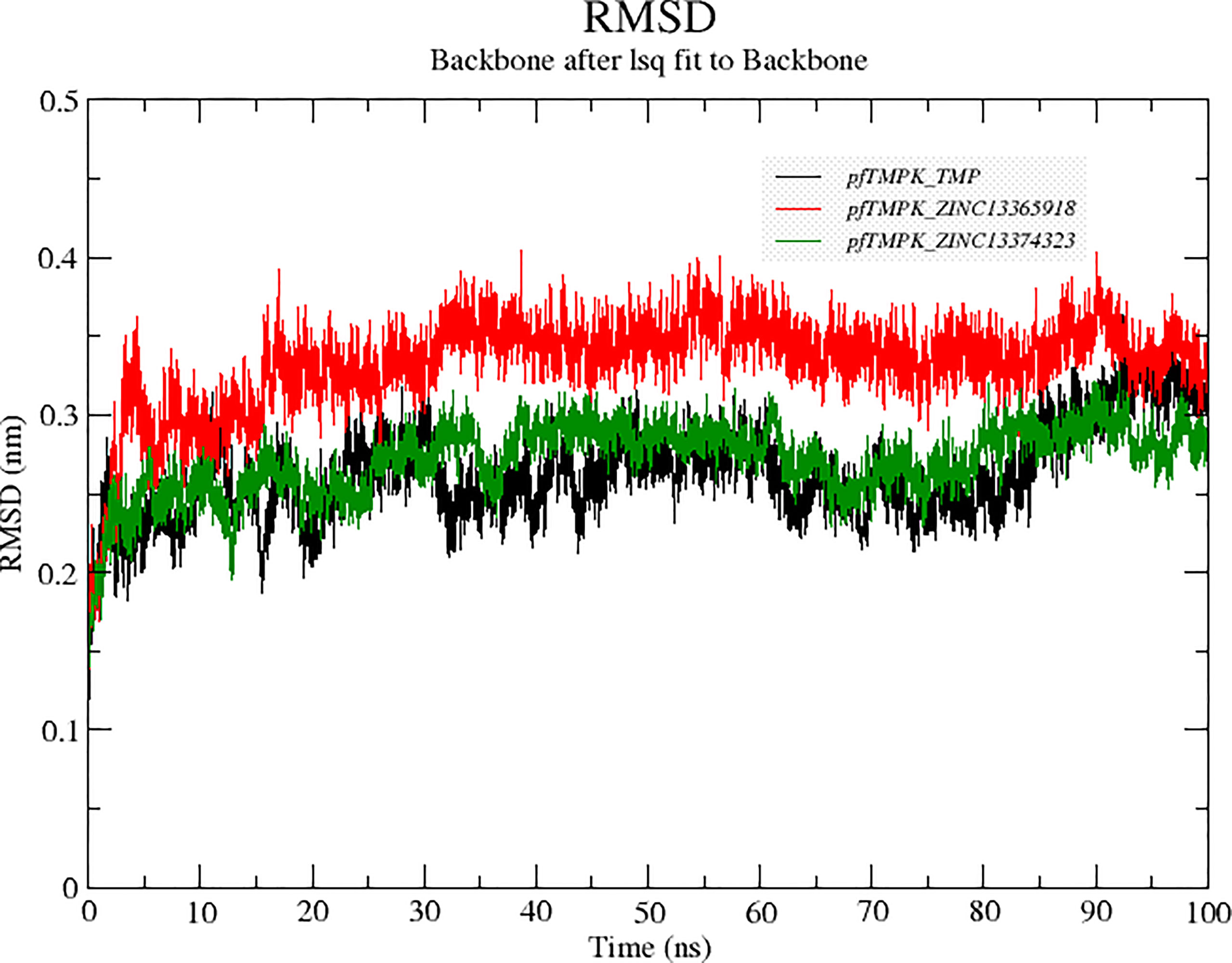
Figure 4 Root mean square deviation (RMSD) graphs comparing the RMSDs of PfTMPK in complex with its natural substrate (TMP) and the respective lead compounds. The RMSD graph for PfTMPK–TMP is colored black, for PfTMPK–ZINC13365918 red, and for PfTMPK–ZINC13374323 green.
Influence of Ligand Binding on the Flexibility of PfTMPK
A comparison of the behavior of the protein residues in each complex was also done using the root mean square fluctuations (RMSFs) (Figure 5). It can be observed from the graph that the protein residues experienced higher fluctuations in PfTMPK–ZINC13365918 than in the other complexes and the unbounded PfTMPK. The fluctuations of PfTMPK–ZINC13374323 and PfTMPK–TMP seem to be very close, indicating a similar margin of flexibility within the protein when complexed with either ZINC13374323 or TMP. A similar observation was made with their RMSDs. However, all the complexes generally experienced more fluctuations than the unbound protein throughout the simulation. The fluctuations were most probably induced by the effect of the binding of the ligands.
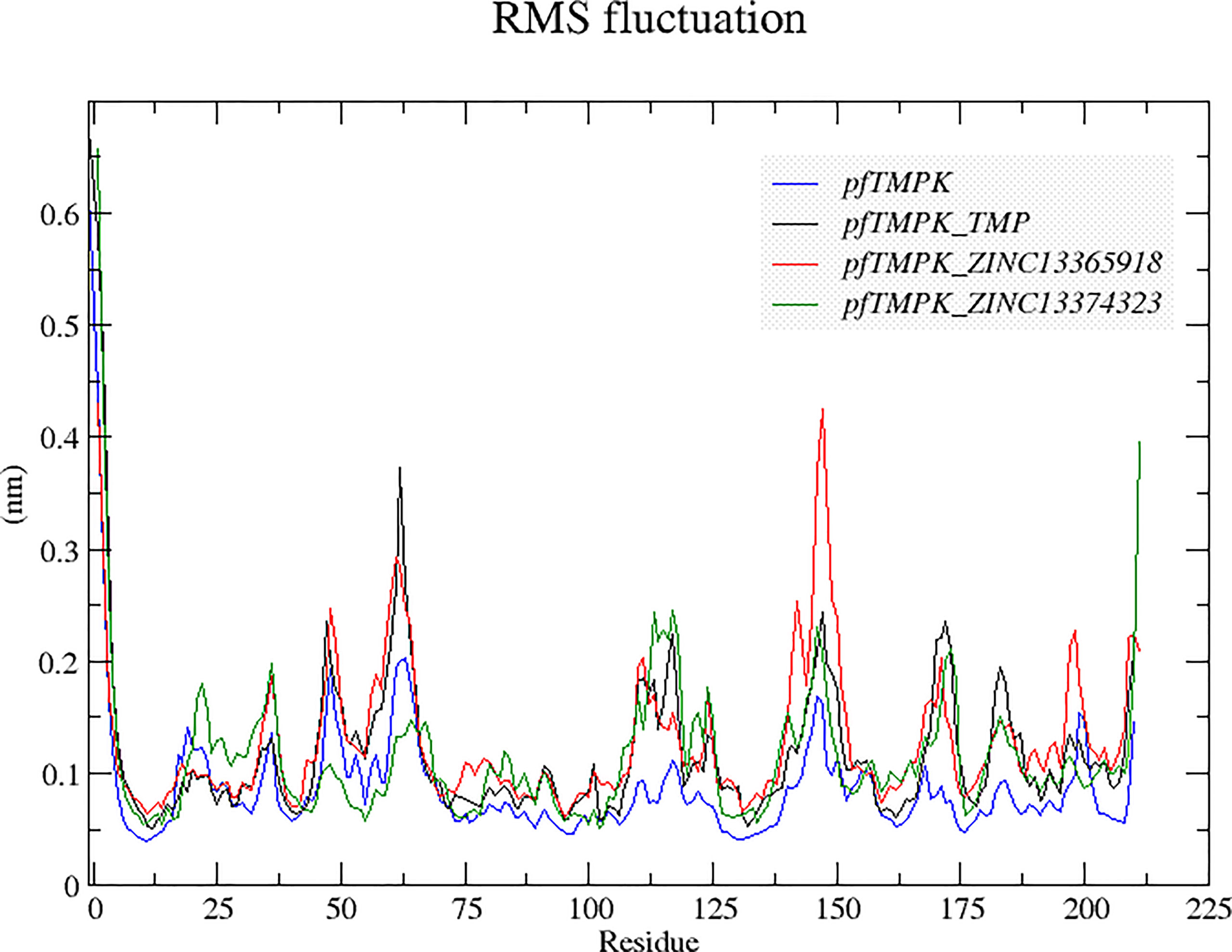
Figure 5 Root mean square fluctuation (RMSF) graphs comparing the behaviors of the residues of PfTMPK in complex with its natural substrate (TMP) and the respective compounds with those of the unbound protein. The RMSF graph for the unbound PfTMPK is shown in blue, for PfTMPK–TMP black, for PfTMPK–ZINC13365918 red, and green for PfTMPK–ZINC13374323.
Binding Interactions During Molecular Dynamics Simulations
To determine if ZINC13374323 and ZINC13365918 can maintain strong interactions with PfTMPK, the numbers of hydrogen bonds formed between the respective compounds and PfTMPK throughout the simulation were analyzed. This suggests a continuous bind of the ligand in the binding site under harsh dynamic conditions. The average number of hydrogen bonds (Figure 6) shared between PfTMPK–ZINC13365918 and PfTMPK–ZINC13374323 complexes during the entire simulation decreased to between 1 and 2. Complex PfTMPK–ZINC13374323 formed 3 and 4 hydrogen bonds more frequently during the simulation as compared to PfTMPK–ZINC13365918. Complex PfTMPK–ZINC13365918 reached as high as 5 and 6 hydrogen bonds, but this happened very few times in the simulation. The average hydrogen bonds shown were consistent with the H-bond interactions obtained after molecular docking (Figures 3B, C).
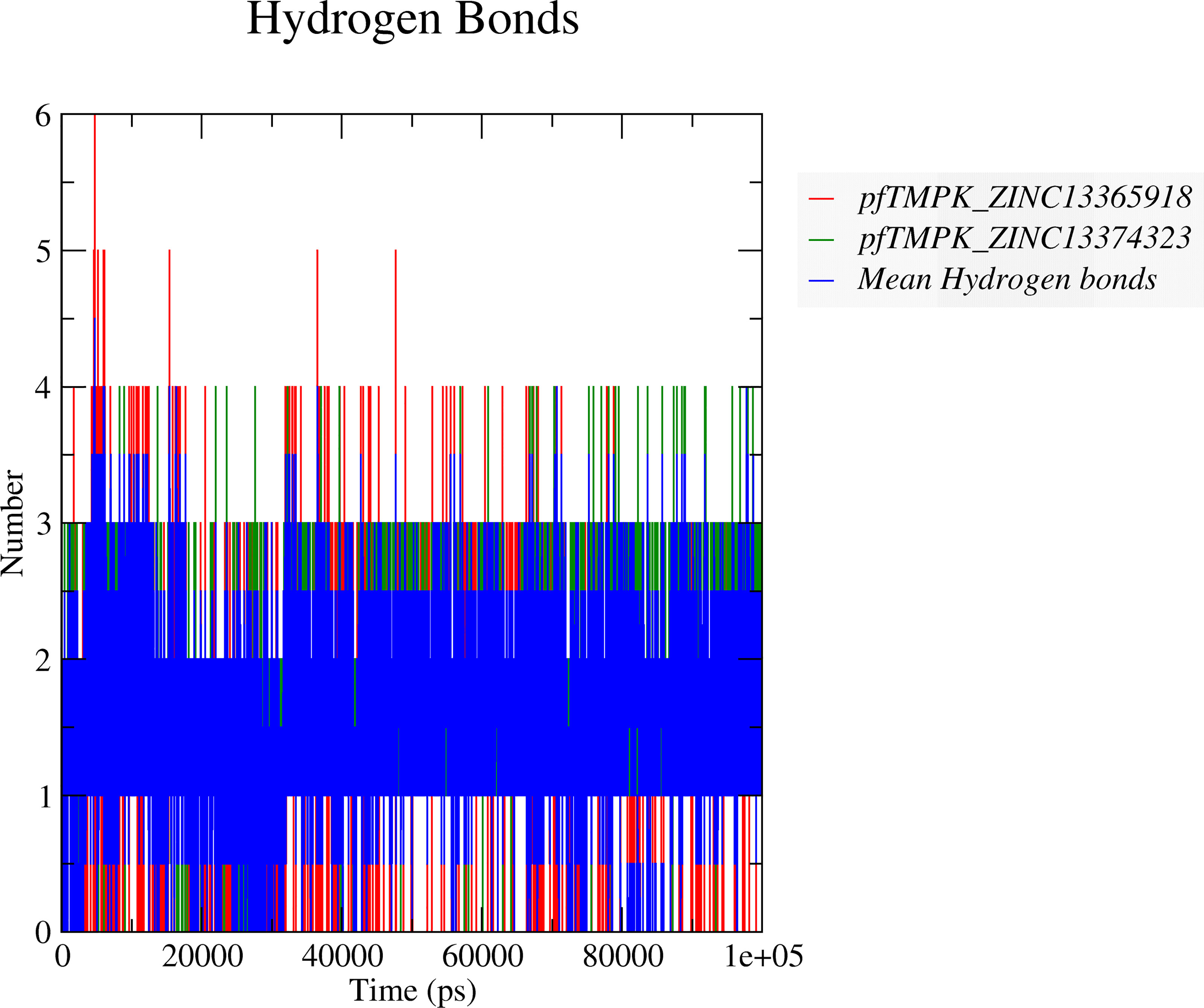
Figure 6 Hydrogen bond predictions between the complexes of PfTMPK and the respective potential lead compounds. The hydrogen bonds formed between PfTMPK and ZINC13365918 are shown as red lines, while those of the protein and ZINC13374323 are shown as green lines. The mean hydrogen bonds formed throughout the simulation in both complexes are highlighted in blue.
Exploring Biological Activities of Potential Lead Compounds
The common name of ZINC13374323 is aurantiamide acetate, which has been shown to exhibit anti-inflammatory and antiviral activities in influenza-infected cells (Zhou et al., 2017). The compound is a constituent of herbs such as the bark of Albizia adianthifolia and Brillantaisia lamium, where it has been shown to exhibit antimicrobial activity (Tamokou et al., 2011; Tamokou et al., 2012). Interestingly, aurantiamide acetate is also an ingredient of A. annua, the Chinese herb from which artemisinin was discovered (Milne et al., 2018). ZINC13365918 is a pyrimidine analog with the name N-hydroxyannomontine. N-Hydroxyannomontine has been shown to exhibit antileishmanial activity against Leishmania braziliensis and Leishmania guyanensis (Costa et al., 2006). The toxicity profiles of both compounds as shown in Table 3 indicate the compounds to be generally safe, with aurantiamide acetate tending to cause some irritation. The toxicity profile was generated with OSIRIS DataWarrior version 5.5.0 (Sander et al., 2015).

Table 3 Toxicity profile of potential lead compounds.
We explored the anti-plasmodial activity of aurantiamide acetate further since it is commercially available. N-Hydroxyannomontine is currently not commercially available; as such, in vitro experimentation for this analog was not carried out. An in vitro parasite growth inhibition assay was prepared for the P. falciparum 3D7 strain using the SYBR Green I fluorescence assay (Smilkstein et al., 2004). Artesunate was used as the positive control. The compounds were added to synchronized cultures of the ring stages, and inhibition of the parasite’s growth was determined through IC50, with the experiment conducted in triplicates. The IC50 values obtained for artesunate were comparable to those reported previously with the average being 23.22 nM (Touré et al., 2008; Quashie et al., 2013). The average IC50 value obtained for aurantiamide acetate was 69.33 μM (Table 4). The inhibition curves of both compounds are shown in Figure 7. A single screen of aurantiamide acetate was also performed on asynchronized cultures of P. falciparum 3D7 strain. The IC50 value achieved for the asynchronized culture was >100 μM (Supplementary Figure 1), which indicates an increase in the potency of the compound when screened against the ring stages of the parasite. This could imply a clue into the target specificity of the compound but requires further investigation; therefore, a stage-of-action study to assess the potency of the compound against different asexual stages is recommended. The activity of aurantiamide acetate, despite being appreciable for a natural compound, must be further improved through optimization to cater to its lack of O-atoms, which is necessary for PfTMPK inhibition.
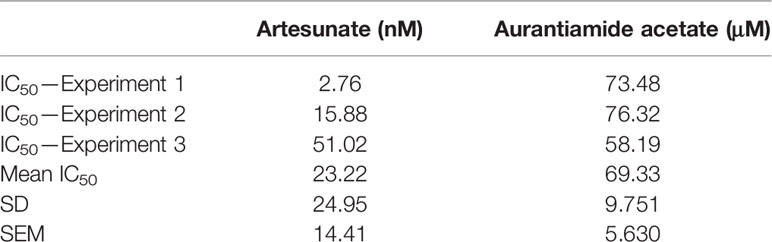
Table 4 IC50 values of artesunate and aurantiamide acetate for the synchronized culture of Plasmodium falciparum 3D7 ring stages.
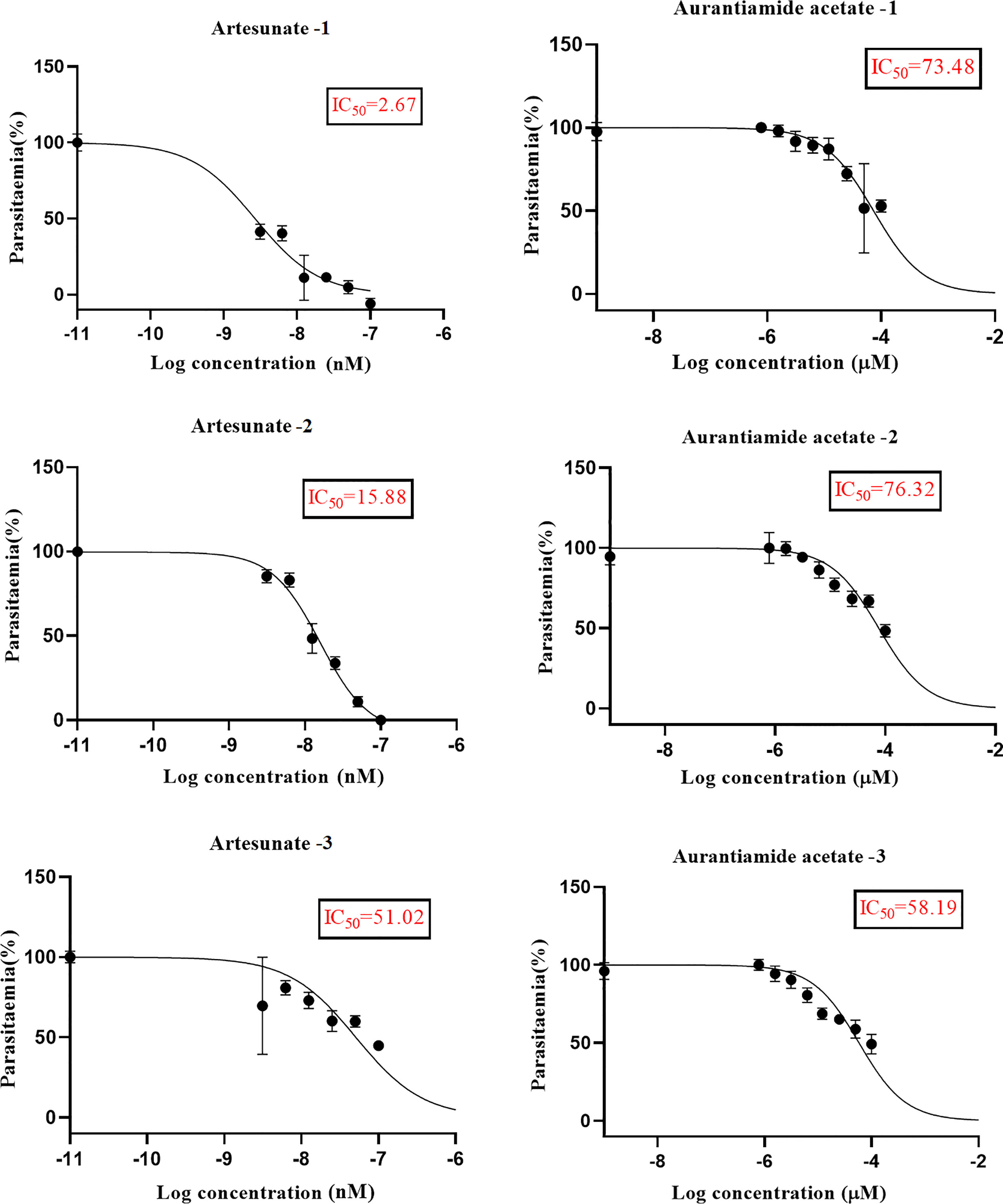
Figure 7 The inhibition curves of artesunate and aurantiamide acetate for the synchronized culture of Plasmodium falciparum 3D7 ring stages.
Significance of the Study
In recent years, a minimal number of natural products have made it to antimalarial lead optimization projects (Guantai and Chibale, 2011). Natural products have proven to be a rich source of antimalarials throughout the history of malaria drug discovery, with breakthroughs like quinine and artemisinin being prime examples (Saxena et al., 2003; Itokawa et al., 2008). Most other drugs like clindamycin and azithromycin have also had their foundation in natural products (Guantai and Chibale, 2011). This shows that developing new drug candidates from natural product sources remains a favorable line of research for antimalarial drug development. It is evident, however, that most compounds developed from natural products show moderate inhibition activity (Saxena et al., 2003; Guantai and Chibale, 2011). As such, through the incorporation of in silico techniques, we can focus on compounds with desirable properties with the potential to become lead compounds and later become drug candidates through further optimization. The thirteen hits obtained after molecular docking represent compounds that can compete with the natural substrate of PfTMPK based on their binding affinity. By screening those compounds against important structural properties obtained through extensive in silico work (Ojha and Roy, 2013), we have been able to highlight two natural products, namely, aurantiamide acetate and N-hydroxyannomontine, as potential lead compounds. The binding affinities of the compounds as compared to known active inhibitors of PfTMPK corroborate this claim. Also, further experimental in vitro studies of the biological activity of aurantiamide acetate against the P. falciparum 3D7 strain show the compound to express appreciable antimalarial activity (IC50 of 69.33). In vitro experimentation could not be done for N-hydroxyannomontine because it is not commercially available. It will be necessary to synthesize and test the compound in future studies and also develop cytotoxicity assays for both compounds. To develop these compounds into lead compounds, it is crucial to optimize them, taking into account the structural properties needed for effective binding as earlier described (Ojha and Roy, 2013).
Resistance to chloroquine, a cheap and efficacious antimalarial drug, has led to the change to artemisinin-based combination therapy (ACT) as the recommended first-line treatment option. Chloroquine resistance was first observed in Southeast Asia (in Thailand in 1957), which spread globally (Packard, 2014) and rendered chloroquine useless. However, resistance to ACT has been detected again in Southeast Asia with hints of delayed parasite clearance in Africa (Global Malaria Program, 2015). Delayed parasite clearance, however, may not necessarily lead to treatment failure, but in the Greater Mekong Subregion, treatment failures following treatment with an ACT have only been observed where there is resistance to the partner drug (World Health Organization, 2018). The drugs used in the ACT are few, necessitating the need to have additional compounds, thus the significance of our finding of aurantiamide acetate as a potential antimalarial compound. Moreover, it is known to possess anti-inflammatory properties (Zhou et al., 2017), which is good. Although its anti-plasmodial activity in vitro is not comparable to that of artesunate, we believe that the addition of oxygen atoms to the molecule will significantly increase its performance. Studies have reported that dried leaves of A. annua used as a tea infusion in treating malaria have a higher potency than pure artemisinin (Weathers, 2014). Using the dried-leaf treatment has also been shown to overcome resistance to artemisinin. These effects may be due to the other chemical components of A. annua, which enhance the bioavailability and efficacy of artemisinin even though they have significantly less potent anti-plasmodial activity than artemisinin (Weathers, 2014). Since aurantiamide acetate is a constituent of A. annua, we recommend further studies to determine its ability to reduce resistance to artemisinin and/or enhance anti-plasmodial activity when used as a partner drug in ACT.
Conclusion
A total of 885 compounds retrieved from AfroDB were screened in FAF-Drugs4 server, which produced 91 ADMET-acceptable compounds. Thirteen compounds out of the 91, which scored higher binding energy than the substrate of PfTMPK, were selected as hits. Four structural properties of a molecule that ensures proper binding at the active site and inhibitory effect against PfTMPK were considered in selecting lead compounds. ZINC13374323 and ZINC13365918 were selected as plausible lead compounds since they exhibited three of the structural properties. ZINC13374323 shares interactions with many similar residues as a PfTMPK inhibitor thymidine analog, which has been shown to exhibit high activity against the protein. ZINC13374323, also known as aurantiamide acetate, is an ingredient of A. annua and exhibits anti-inflammatory, antiviral, and antimicrobial activities. ZINC13365918, on the other hand, is a pyrimidine analog that has been shown to exhibit antileishmanial activity. Analysis of the molecular dynamics simulations of the lead compounds complexed with the protein showed the complex of PfTMPK and ZINC13374323 to have similar RMSD and RMSF as that of the protein in complex with its natural substrate, TMP. In vitro testing of aurantiamide acetate for anti-plasmodial activity resulted in an IC50 of 69.33 μM. The compounds could be used as scaffolds for lead optimization.
Data Availability Statement
The original contributions presented in the study are included in the article/Supplementary Material. Further inquiries can be directed to the corresponding author.
Author Contributions
AG, MW, and SK developed the concept and designed the study. KE conducted the computational aspects of the study and drafted the manuscript. MT-T, AL, SN, and GD performed the in vitro experiments of the study. AG, MW, and SK edited and proofread the manuscript. All authors contributed to the manuscript.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
We are grateful to Dr. Rita Afriyie Boateng for her support in the molecular dynamics simulations and the director of Noguchi Memorial Institute for Medical Research for allowing us to publish this paper.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fcimb.2022.868529/full#supplementary-material.
References
Agamah, F. E., Damena, D., Skelton, M., Ghansah, A., Mazandu, G. K., Chimusa, E. R. (2021). Network-Driven Analysis of Human–Plasmodium falciparum Interactome: Processes for Malaria Drug Discovery and Extracting In Silico Targets. Malar. J. 20, 421. doi: 10.1186/s12936-021-03955-0
Belete, T. M. (2020). Recent Progress in the Development of New Antimalarial Drugs With Novel Targets. Drug Des. Devel. Ther. 14, 3875–3889. doi: 10.2147/DDDT.S265602
Benigni, R., Bossa, C. (2006). Structural Alerts of Mutagens and Carcinogens. Curr. Comput. Aided-Drug Des. 2, 169–176. doi: 10.2174/157340906777441663
Bhatt, S., Weiss, D. J., Cameron, E., Bisanzio, D., Mappin, B., Dalrymple, U., et al. (2015). The Effect of Malaria Control on Plasmodium falciparum in Africa Between 2000 and 2015. Nature 526, 207–211. doi: 10.1038/nature15535
Cassera, M. B., Zhang, Y., Hazleton, K. Z., Schramm, V. L. (2011). Purine and Pyrimidine Pathways as Targets in Plasmodium falciparum. Curr. Top. Med. Chem. 11, 2103–2115. doi: 10.2174/156802611796575948
Chen, M.D., Sinha, K., Rule, G. S., Ly, D. H. (2018). Interaction of α-Thymidine Inhibitors With Thymidylate Kinase From Plasmodium falciparum. Biochemistry 57, 2868–2875. doi: 10.1021/acs.biochem.8b00162
Costa, E. V., Pinheiro, M. L. B., Xavier, C. M., Silva, J. R. A., Amaral, A. C. F., Souza, A. D. L., et al. (2006). A Pyrimidine-β-Carboline and Other Alkaloids From Annona F Oetida With Antileishmanial Activity. J. Nat. Prod. 69, 292–294. doi: 10.1021/np050422s
Cui, H., Carrero-Lérida, J., Silva, A. P. G., Whittingham, J. L., Brannigan, J. A., Ruiz-Pérez, L. M., et al. (2012). Synthesis and Evaluation of α-Thymidine Analogues as Novel Antimalarials. J. Med. Chem. 55, 10948–10957. doi: 10.1021/jm301328h
Dallakyan, S., Olson, A. J. (2015). Small-Molecule Library Screening by Docking With Pyrx. Methods Mol. Biol. 1263, 243–250. doi: 10.1007/978-1-4939-2269-7_19
DeLong, E. R., DeLong, D. M., Clarke-Pearson, D. L. (1988). Comparing the Areas Under Two or More Correlated Receiver Operating Characteristic Curves: A Nonparametric Approach. Biometrics 44, 837. doi: 10.2307/2531595
Egwu, C. O., Pério, P., Augereau, J.-M., Tsamesidis, I., Benoit-Vical, F., Reybier, K. (2022). Resistance to Artemisinin in Falciparum Malaria Parasites: A Redox-Mediated Phenomenon. Free Radic. Biol. Med. 179, 317–327. doi: 10.1016/j.freeradbiomed.2021.08.016
Global Malaria Program. (2015). Status Report on Artemisinin and ACT Resistance. Available at: https://www.who.int/malaria/publications/atoz/status-rep-artemisinin-act-resistance-sept2015.pdf?ua=1 (Accessed February 2, 2022).
Goksuluk, D., Korkmaz, S., Zararsiz, G., Karaagaoglu, A. E. (2016). Easyroc: An Interactive Web-Tool for Roc Curve Analysis Using R Language Environment. R J. 8, 213–230. doi: 10.32614/RJ-2016-042
Guantai, E., Chibale, K. (2011). How can Natural Products Serve as a Viable Source of Lead Compounds for the Development of New/Novel Anti-Malarials? Malar. J. 10, S2. doi: 10.1186/1475-2875-10-S1-S2
Heifets, A., Lilien, R. H. (2010). Ligalign: Flexible Ligand-Based Active Site Alignment and Analysis. J. Mol. Graph. Model. 29, 93–101. doi: 10.1016/j.jmgm.2010.05.005
Itokawa, H., Morris-Natschke, S. L., Akiyama, T., Lee, K. H. (2008). Plant-Derived Natural Product Research Aimed at New Drug Discovery. J. Nat. Med. 62, 263–280. doi: 10.1007/s11418-008-0246-z
Kandeel, M., Ando, T., Kitamura, Y., Abdel-Aziz, M., Kitade, Y. (2009). Mutational, Inhibitory and Microcalorimetric Analyses of Plasmodium falciparum TMP Kinase. Implications for Drug Discovery. Parasitology 136, 11. doi: 10.1017/S0031182008005301
Krungkrai, S. R., Krungkrai, J. (2016). Insights Into the Pyrimidine Biosynthetic Pathway of Human Malaria Parasite Plasmodium falciparum as Chemotherapeutic Target. Asian Pac. J. Trop. Med. 9, 525–534. doi: 10.1016/j.apjtm.2016.04.012
Kumar, S., Bhardwaj, T. R., Prasad, D. N., Singh, R. K. (2018). Drug Targets for Resistant Malaria: Historic to Future Perspectives. Biomed. Pharmacother. 104, 8–27. doi: 10.1016/j.biopha.2018.05.009
Kwofie, S. K., Broni, E., Asiedu, S. O., Kwarko, G. B., Dankwa, B., Enninful, K. S., et al. (2021). Cheminformatics-Based Identification of Potential Novel Anti-SARS-CoV-2 Natural Compounds of African Origin. Molecules 26, 406. doi: 10.3390/molecules26020406
Lagorce, D., Bouslama, L., Becot, J., Miteva, M. A., Villoutreix, B. O. (2017). Faf-Drugs4: Free ADME-Tox Filtering Computations for Chemical Biology and Early Stages Drug Discovery. Bioinformatics 33, 3658–3660. doi: 10.1093/bioinformatics/btx491
Ludin, P., Woodcroft, B., Ralph, S. A., Mäser, P. (2012). In Silico Prediction of Antimalarial Drug Target Candidates. Int. J. Parasitol. Drugs Drug Resist. 2, 191–199. doi: 10.1016/j.ijpddr.2012.07.002
Mandrekar, J. N. (2010). Receiver Operating Characteristic Curve in Diagnostic Test Assessment. J. Thorac. Oncol. 5, 1315–1316. doi: 10.1097/JTO.0b013e3181ec173d
Milne, G. W. A., Zhou, J., Xie, G., Yan, X. (2018). Traditional Chinese Medicines: Molecular Structures, Natural Sources and Applications. Eds. Yan, X., Xie, G., Zhou, J., Milne Routledge, G. W. A.. (London: Routledge). doi: 10.4324/9781315193441
Neves, B. J., Braga, R. C., Alves, V. M., Lima, M. N. N., Cassiano, G. C., Muratov, E. N., et al. (2020). Deep Learning-driven Research for Drug Discovery: Tackling Malaria. PLoS Comput. Biol. 16, e1007025. doi: 10.1371/journal.pcbi.1007025
Ntie-Kang, F., Zofou, D., Babiaka, S. B., Meudom, R., Scharfe, M., Lifongo, L. L., et al. (2013). Afrodb: A Select Highly Potent and Diverse Natural Product Library From African Medicinal Plants. PLoS One 8, e78085. doi: 10.1371/journal.pone.0078085
Ojha, P. K., Roy, K. (2013). First Report on Exploring Structural Requirements of Alpha and Beta Thymidine Analogs for PfTMPK Inhibitory Activity Using Iin Silico Studies. BioSystems 113, 177–195. doi: 10.1016/j.biosystems.2013.07.005
Packard, R. M. (2014). The Origins of Antimalarial-Drug Resistance. N. Engl. J. Med. 371, 397–399. doi: 10.1056/NEJMp1403340
Patrick, G. L., Turner, H. (2020). “Kinases and Kinase Inhibitors,” in Antimalarial Agents (Amsterdam: Elsevier). doi: 10.1016/B978-0-08-101210-9.00013-5
Quashie, N. B., Duah, N. O., Abuaku, B., Quaye, L., Ayanful-Torgby, R., Akwoviah, G. A., et al. (2013). A SYBR Green 1-Based In Vitro Test of Susceptibility of Ghanaian Plasmodium falciparum Clinical Isolates to a Panel of Anti-Malarial Drugs. Malar. J. 12, 450. doi: 10.1186/1475-2875-12-450
Reyes, P., Rathod, P. K., Sanchez, D. J., Mrema, J. E., Rieckmann, K. H., Heidrich, H. G. (1982). Enzymes of Purine and Pyrimidine Metabolism From the Human Malaria Parasite, Plasmodium Falciparum. Mol. Biochem. Parasitol. 5, 275–290. doi: 10.1016/0166-6851(82)90035-4
Sander, T., Freyss, J., von Korff, M., Rufener, C. (2015). Datawarrior: An Open-Source Program for Chemistry Aware Data Visualization And Analysis. J. Chem. Inf. Model. 55, 460–473. doi: 10.1021/ci500588j
Saxena, S., Pant, N., Jain, D. C., Bhakuni, R. S. (2003). Antimalarial Agents From Plant Sources. Curr. Sci. 85, 1314–1329.
Schüttelkopf, A. W., Van Aalten, D. M. F. (2004). Prodrg: A Tool for High-Throughput Crystallography of Protein-Ligand Complexes. Acta Crystallogr. Sect. D Biol. Crystallogr. 60, 1355–1363. doi: 10.1107/S0907444904011679
Siddiqui, A. A., Iram, F., Siddiqui, S., Sahu, K. (2014). Role of Natural Products in Drug Discovery Process. Int. J. Drug Dev. Res. 6, 172–204.
Sliwoski, G., Kothiwale, S., Meiler, J., Lowe, E. W. (2013). Computational Methods in Drug Discovery. Pharmacol. Rev. 66, 334–395. doi: 10.1124/pr.112.007336
Smilkstein, M., Sriwilaijaroen, N., Kelly, J. X., Wilairat, P., Riscoe, M. (2004). Simple and Inexpensive Fluorescence-Based Technique for High-Throughput Antimalarial Drug Screening. Antimicrob. Agents Chemother. 48, 1803–1806. doi: 10.1128/AAC.48.5.1803-1806.2004
Spitzmüller, A., Mestres, J. (2013). Prediction of the P. falciparum Target Space Relevant to Malaria Drug Discovery. PLoS Comput. Biol. 9, e1003257. doi: 10.1371/journal.pcbi.1003257
Tajuddeen, N., Van Heerden, F. R. (2019). Antiplasmodial Natural Products: An Update. Malar. J. 18, 404. doi: 10.1186/s12936-019-3026-1
Tamokou, J., de, D., Kuiate, J. R., Tene, M., Kenla Nwemeguela, T. J., Tane, P. (2011). The Antimicrobial Activities of Extract and Compounds Isolated From Brillantaisia Lamium. Iran. J. Med. Sci. 36, 24–31.
Tamokou, J., de, D., Simo Mpetga, D. J., Keilah Lunga, P., Tene, M., Tane, P., et al. (2012). Antioxidant and Antimicrobial Activities of Ethyl Acetate Extract, Fractions and Compounds From Stem Bark of Albizia Adianthifolia (Mimosoideae). BMC Complement. Altern. Med. 12, 1074. doi: 10.1186/1472-6882-12-99
Touré, A. O., Koné, L. P., Jambou, R., Konan, T. D., Demba, S., Beugre, G. E., et al. (2008). Sensibilité In Vitro De P. falciparum À La Quinine, L’artésunate Et La Chloroquine À Abidjan (Côte d’ivoire). Cah. Santé 18, 043–047. doi: 10.1684/san.2008.0103
Trott, O., Olson, A. J. (2010). Software News and Update AutoDock Vina: Improving the Speed and Accuracy of Docking With a New Scoring Function, Efficient Optimization, and Multithreading. J. Comput. Chem. 31, 455–461. doi: 10.1002/jcc.21334
Tuteja, R. (2007). Helicases - Feasible Antimalarial Drug Target for Plasmodium falciparum. FEBS J. 274, 4699–4704. doi: 10.1111/j.1742-4658.2007.06000.x
Van Der Spoel, D., Lindahl, E., Hess, B., Groenhof, G., Mark, A. E., Berendsen, H. J. C. (2005). Gromacs: Fast, Flexible, and Free. J. Comput. Chem. 26, 1701–1718. doi: 10.1002/jcc.20291
Vanoevelen, J. M., Bierau, J., Grashorn, J. C., Lambrichs, E., Kamsteeg, E.-J., Bok, L. A., et al. (2022). DTYMK is Essential for Genome Integrity and Neuronal Survival. Acta Neuropathol. 143, 245–262. doi: 10.1007/s00401-021-02394-0
Vaught, A. (1996). Graphing With Gnuplot and Xmgr: Two Graphing Packages Available Under Linux. Linux J. 1996, 7.
Wang, L., Pang, X., Li, Y., Zhang, Z., Tan, W. (2017). Rader: A Rapid DEcoy Retriever to Facilitate Decoy Based Assessment of Virtual Screening. Bioinformatics 33, 1235–1237. doi: 10.1093/bioinformatics/btw783
Weathers, P. J. (2014). Dried-Leaf Artemisia Annua: A Practical Malaria Therapeutic for Developing Countries? World J. Pharmacol. 3, 39. doi: 10.5497/wjp.v3.i4.39
Whittingham, J. L., Carrero-Lerida, J., Brannigan, J. A., Ruiz-Perez, L. M., Silva, A. P. G., Fogg, M. J., et al. (2010). Structural Basis for the Efficient Phosphorylation of AZT-MP (3′-azido-3′-deoxythymidine Monophosphate) and dGMP by Plasmodium falciparum Type I Thymidylate Kinase. Biochem. J. 428, 499–509. doi: 10.1042/BJ20091880
World Health Organization (2018) Status Report on Artemisinin Resistance and ACT Efficacy (August 2018) (World Heal. Organ). Available at: https://www.who.int/publications/i/item/WHO-CDS-GMP-2019.17 (Accessed February 2, 2022).
Zhou, B., Yang, Z., Feng, Q., Liang, X., Li, J., Zanin, M., et al. (2017). Aurantiamide Acetate From Baphicacanthus cusia Root Exhibits Anti-Inflammatory and Anti-Viral Effects via Inhibition of the NF-κB Signaling Pathway in Influenza A virus-Infected Cells. J. Ethnopharmacol. 199, 60–67. doi: 10.1016/j.jep.2017.01.038
Keywords: Plasmodium falciparum, aurantiamide acetate, PfTMPK, Artemisia annua, natural compounds
Citation: Enninful KS, Kwofie SK, Tetteh-Tsifoanya M, Lamptey ANL, Djameh G, Nyarko S, Ghansah A and Wilson MD (2022) Targeting the Plasmodium falciparum’s Thymidylate Monophosphate Kinase for the Identification of Novel Antimalarial Natural Compounds. Front. Cell. Infect. Microbiol. 12:868529. doi: 10.3389/fcimb.2022.868529
Received: 02 February 2022; Accepted: 25 April 2022;
Published: 25 May 2022.
Edited by:
Vipan Kumar, Guru Nanak Dev University, IndiaReviewed by:
Conrad Simoben Veranso, University of Buea, CameroonTingxiang Yan, Shanghai Jiao Tong University, China
Copyright © 2022 Enninful, Kwofie, Tetteh-Tsifoanya, Lamptey, Djameh, Nyarko, Ghansah and Wilson. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Michael D. Wilson, mwilson@noguchi.ug.edu.gh