 Inaya Hayek
Inaya Hayek Anja Lührmann
Anja Lührmann
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Cell. Infect. Microbiol., 09 June 2022
Sec. Bacteria and Host
Volume 12 - 2022 | https://doi.org/10.3389/fcimb.2022.867689
This article is part of the Research TopicObligate Intracellular Bacteria: Evasion and Adaptative Tactics Shaping the Host-Pathogen InterfaceView all 11 articles
HIF1α is an important transcription factor regulating not only cellular responses to hypoxia, but also anti-infective defense responses. We recently showed that HIF1α hampers replication of the obligate intracellular pathogen Coxiella burnetii which causes the zoonotic disease Q fever. Prior to development of chronic Q fever, it is assumed that the bacteria enter a persistent state. As HIF1α and/or hypoxia might be involved in the induction of C. burnetii persistence, we analyzed the role of HIF1α and hypoxia in the interaction of macrophages with C. burnetii to understand how the bacteria manipulate HIF1α stability and activity. We demonstrate that a C. burnetii-infection initially induces HIF1α stabilization, which decreases then over the course of an infection. This reduction depends on bacterial viability and a functional type IV secretion system (T4SS). While neither the responsible T4SS effector protein(s) nor the molecular mechanism leading to this partial HIF1α destabilization have been identified, our results demonstrate that C. burnetii influences the expression of HIF1α target genes in multiple ways. Therefore, a C. burnetii infection promotes HIF1α-mediated upregulation of several metabolic target genes; affects apoptosis-regulators towards a more pro-apoptotic signature; and under hypoxic conditions, shifts the ratio of the inflammatory genes analyzed towards a pro-inflammatory profile. Taken together, C. burnetii modulates HIF1α in a still elusive manner and alters the expression of multiple HIF1α target genes.
Hypoxia-inducible factor (HIF)-1 was first recognized as an essential regulator of cellular responses to limited oxygen availability (Majmundar et al., 2010). Recent research has shown that HIF1 activity is also critical for shifting cellular metabolism, regulating immune cell activity, and mounting anti-infective defense responses (Cramer et al., 2003; Knight and Stanley, 2019). HIF1 is a heterodimer, consisting of HIF1α and HIF1β (Wang et al., 1995). The activity of the complex is controlled by proteasomal degradation of the α-subunit. Thus, prolyl hydroxylases (PHDs) hydroxylate HIF1α, which mediates binding to the von Hippel-Lindau (VHL) E3 ubiquitin ligase and leads to proteasomal degradation of HIF1α (Maxwell et al., 1999; Ohh et al., 2000; Jaakkola et al., 2001). Importantly, PHDs require oxygen, Fe2+ and 2-oxoglutarate for HIF1α hydroxylation [reviewed in: (Greer et al., 2012; Hayek et al., 2021)]. Therefore, in the absence of oxygen, its co-factors or co-substrates, HIF1α is stabilized. However, HIF1α stabilization can also occur under normoxic conditions (in the presence of oxygen) in response to increased levels of the TCA cycle intermediates succinate or fumarate, or in the presence of nitric oxide (NO) (Hewitson et al., 2007; Tannahill et al., 2013; Mills et al., 2016). In addition, bacterial, viral, fungal, and parasitic infections might also induce HIF1α stabilization (Devraj et al., 2017; Knight and Stanley, 2019). Once the heterodimer is formed, it attaches to the promoter region of genes containing the hypoxia response element (HRE) and induces their transcription. In addition, HIF1 interacts with other signaling pathways (including Notch, Wnt and Myc) in an HRE-independent manner (Koshiji et al., 2004; Gustafsson et al., 2005; Kaidi et al., 2007; Semenza, 2014; Strowitzki et al., 2019). Thereby, HIF1 regulates transcription of genes involved in metabolic reprogramming, immune responses, and anti-infectious activity (Obach et al., 2004; Kelly and O'Neill, 2015; Devraj et al., 2017).
Hypoxia, a state of insufficient oxygen availability, impairs several important antimicrobial defense mechanisms. To control bacterial infections under hypoxia, myeloid cells induce the production of anti-microbial peptides and pro-inflammatory cytokines, deplete essential metabolites, and modulate their phagocytic capacity and phagosome maturation (Hayek et al., 2021). Under these conditions, some bacteria are controlled under hypoxia and/or HIF1α, while other pathogens survive or even replicate.
We recently showed that in hypoxic murine macrophages, HIF1α or HIF1α -mediated signaling impedes C. burnetii replication (Hayek et al., 2019). This obligate intracellular bacterium is a zoonotic pathogen. Its primary reservoir are domestic ruminants such as cattle, sheep and goats ( Maurin and Raoult, 1999). Although infected ruminants are mainly asymptomatic, in pregnant animals the infection might lead to abortion, premature delivery or stillbirth. Infected animals shed the pathogen through birthing products, feces or milk which are the main source for human infection (Van den Brom et al., 2015). Although often asymptomatic, Q fever may manifest in humans as an acute disease (mainly as a self-limited febrile illness, pneumonia, or hepatitis) or as a chronic disease (mainly endocarditis). Importantly, chronic Q fever develops several months or years after the primary infection (Anderson et al., 2013). A short-term treatment with doxycycline is still considered the mainstay of antibiotic therapy of acute Q fever, whereas chronic Q fever patients have to be treated with doxycycline in combination with hydroxyl chloroquine for at least 18 months. Thus, a more efficient therapy to treat chronic Q fever has to be developed. In addition, it is crucial to increase our knowledge of chronic Q fever development, especially since it develops months or years after the primary infection, during which the patient does not show any symptoms, suggesting a prolonged state of bacterial persistence (Harris et al., 2000; Sukocheva et al., 2016). Our previous results suggest that in macrophages, HIF1α is required for impeding C. burnetii replication by impairing STAT3 activation, which results in reduced levels of the TCA intermediate citrate (Hayek et al., 2019). Importantly, bacterial viability was maintained allowing bacterial persistence. Thus, HIF1α might play an important role in the induction of C. burnetii persistence, and consequently, the development of chronic Q fever. Therefore, we aim to analyze the roles of HIF1α and hypoxia for the interaction of macrophages with C. burnetii in more detail.
Bone marrow derived macrophages from C57BL/6 “J” male mice (Charles River; Strain Code: 027) were prepared as described (Hayek et al., 2019). Briefly, bone marrow cells were extracted from femur and tibia of at least 6 weeks old mice and propagated in sterile Teflon bags (Angst+Pfister) containing DMEM + GlutaMax (Thermo Fisher), 10% Fetal Calf Serum (FCS) (Biochrom), 5% Horse Serum (Cell Concepts), 1% MEM Non-Essential Amino Acids Solution (Life Technologies), 0.5% HEPES (AppliChem) and 20% supernatant of L929 cells for 7-10 days at 37°C, 10% CO2 and 21% O2. Macrophages were cultured for infection experiments in CMoAB medium, consisting of RPMI 1640 medium (Thermo Fisher) supplemented with 10% FCS, 1% HEPES and 0.5% β-mercaptoethanol (Sigma Aldrich). Murine macrophages were seeded and left to adhere for 1 to 2 h at 37°C, 5% CO2, 21% O2 (normoxia) prior to infection.
All C. burnetii strains used in this study were inoculated at a concentration of 1 x 106 C. burnetii/ml in ACCM-2 (Sunrise Science Products, Cat#4700-300) medium and cultivated for 5 days at 37°C, 5% CO2, and 2.5% O2. The C. burnetii Nine Mile phase II (NMII) clone 4 (RSA439) served as wild type (wt) strain in this study. When growing C. burnetii ΔdotA (Schäfer et al., 2020) or the ΔdotA C. burnetii transposon mutant (kindly provided by Matteo Bonazzi (Martinez et al., 2014)), 3 μg/ml chloramphenicol was added to the axenic medium. C. burnetii NMII was heat-killed (Hk wt) at 70°C for 30 min under shaking at 500 rpm.
E. coli DH5α were plated on a Luria broth (LB) agar plate and placed overnight at 37°C. A single colony was picked to inoculate 3 ml LB medium, which was left to rotate for 5 h at 37°C. Then, 50 µl of the liquid culture were transferred into 3 ml of fresh LB medium and rotated overnight at 37°C.
To adjust C. burnetii infection concentrations, the optical density at OD600 was measured, with an OD600 of 1 equaling 1 x 109 C. burnetii/ml. To adjust E. coli infection concentrations, the optical density at OD600 was measured, where an OD600 of 1 equals 8 x 108 E. coli/ml. Unless otherwise mentioned, macrophages were infected with C. burnetii or E. coli at an MOI (multiplicity of infection) of 10. After macrophage seeding, the cells were infected with the bacteria and placed under normoxia or 0.5% O2 (hypoxia) for 4 h at 37°C, 5% CO2. At the 4h time point, the cells were either harvested or the medium was discarded and replaced with fresh CMoAB for the later time points.
The concentration of E. coli LPS (Sigma, L4391) was adjusted in CMoAB at 100 ng/ml. After macrophage seeding, the cells were treated with LPS and placed under normoxia or hypoxia. After 4 h, the medium was discarded and replaced with fresh CMoAB. Samples were harvested 24 h post-infection.
Chloramphenicol (Roth) was adjusted to a concentration of 25 μg/ml in CMoAB and then applied to the infected macrophages to induce bacterial growth arrest along the course of infection.
Hypoxic conditions were set to 0.5% O2 and 5% CO2 at 37°C in an InvivO2 hypoxic chamber (Baker Ruskinn). Media and buffers were equilibrated at least 4 h in the hypoxic chamber before starting an experiment.
For HIF1α and actin immunoblot samples, uninfected, infected or LPS-treated macrophages were lysed with 10 mM Tris-HCl pH 6.8, 6.65 M Urea, 10% Glycerol, 1% SDS with freshly added 1 mM DTT and cOmplete Mini Protease Inhibitor Cocktail (Roche, Cat#04693124001). Hypoxic samples were harvested in the hypoxic chamber to prevent HIF1α destabilization. The samples were mixed for 30 s with the homogenizer unit (VWR) and corresponding pestles (VWR). Finally, 20 μL 4x Laemmli SDS buffer was added to the samples, which were then heated at 85°C for 8 min, shaking at 450 rpm.
Proteins were separated by SDS-PAGE using 4-12% Bis-Tris Gels (Thermo Fischer Scientific) and transferred to a PVDF membrane (Merck Millipore). The membranes were probed with primary antibodies directed against HIF1α (Cayman 10006421/Biomol) or actin (Sigma-Aldrich A2066). The proteins were visualized by using the secondary antibody α - Rabbit IgG (H+L)-HRP (Jackson ImmunoResearch Labs, Cat#111-035-045) and a chemiluminescence detection system (Thermo Fisher). Densitometric analysis was performed using ImageJ (NIH).
The experimental steps of immunofluorescence staining were described in detail elsewhere (Hayek et al., 2019). Briefly, macrophages were seeded on 10 mm sterile coverslips in 24-well plates. After infection and incubation, the cells were fixed with 4% paraformaldehyde (PFA) and permeabilized with ice-cold methanol. The cells were then quenched with 50 mM NH4Cl in PBS/5% goat serum (GS) followed by incubation with the primary antibody against C. burnetii NMII (Davids Biotechnology). Alexa Fluor 594 (Jackson ImmunoResearch Labs) was used as the secondary antibody. Finally, the slides were mounted with ProLong Diamond containing DAPI (Invitrogen). Immunofluorescent images were taken using the Carl Zeiss LSM 700 Laser Scan Confocal Microscope and the ZEN2009 software.
RNA samples were harvested with peqGOLD TriFast (Peqlab VWR, Cat#30-2010) or the RNeasy Plus Kit (Qiagen) and isolated according to manufacturer’s protocol. Isolated RNA was treated with DNase and RDD buffer (QIAGEN, Cat#79254) for 10 min at 37°C, followed by DNase inactivation at 75°C for 5 min. The successful removal of any DNA contamination was confirmed by PCR analysis. Next, first strand cDNA was synthesized from the DNase-treated RNA with SuperScript II Reverse Transcriptase (Invitrogen by Life Technologies, Cat#18064-022) according to manufacturer’s protocol. The resulting cDNA was diluted 5-fold (final concentration of about 100 ng) and served as template in qPCR using the QuantiFast SYBR Green PCR Kit (QIAGEN, Cat#204054), along with a final concentration of 100 nM of each primer in a final volume of 10 µL per reaction. Murine hypoxanthine guanine phosphoribosyl transferase (mHprt1) was the housekeeping gene. The sequence of the primer pairs used to investigate gene expression (HIF1α, PHD1, PHD2, PHD3, VHL, IL1β, Nos2, IL10, IL6, PKM2, LDHA, Glut1, PDK1, Bcl2, Bax, Trp53, Becn1, Bnip3, Bnip3l, P300, FIH, and CBP) are listed in Table 1. The expression levels of these genes were quantified by referencing to mHPRT1 and normalizing to the uninfected or wt-infected normoxic sample. To calculate the fold change, the 2^-(ΔΔCT) method was applied.

Table 1 Primers used.
The primers used are listed in Table 1.
Using GraphPad Prism 5, statistical analysis of the presented data was performed. As mentioned in the individual figure legends, a one sample t-test or an unpaired two-tailed Student’s t test was used. The one-sample t-test was used when comparing datasets to normalized values. A value of p < 0.05 was considered significant.
Previous experiments suggested that C. burnetii increases the HIF1α protein level under hypoxic conditions (Hayek et al., 2019). As HIF1α accumulation is responsible for inhibiting C. burnetii replication, we aimed to determine whether C. burnetii is capable of modulating the HIF1α protein level. Thus, we infected bone marrow derived macrophages (BMDM) with C. burnetii at an MOI of 10 for 4 or 24 hours under normoxic (21% O2, 5% CO2) or under reduced oxygen (0.5% O2, 5% CO2) conditions. In the following, we refer to this reduced oxygen condition as hypoxia. We analyzed the HIF1α protein level of the infected cells, kept under different oxygen conditions, by immunoblot analysis. Under normoxia, HIF1α is constantly degraded. However, the infection with C. burnetii for 4 h resulted in transient stabilization of HIF1α, which was almost absent at 24 h post-infection (Figures 1A, B). Under hypoxia, we observed HIF1α protein accumulation in uninfected cells at 4 and 24 h, which was further augmented by infection with C. burnetii. Importantly, lipopolysaccharide (LPS) stimulation also led to HIF1α stabilization under normoxia and, more pronounced, under hypoxia (Figures 1C, D). Thus, the C. burnetii-mediated HIF1α stabilization might be partially due to the recognition of LPS. However, we used LPS from E. coli. Thus, the comparison has to be taken with caution, as the lipid A of C. burnetii LPS differs significantly from enterobacterial lipid A and fails to signal via toll-like receptor (TLR) 2 and 4 (Zamboni et al., 2004; Abnave et al., 2017; Beare et al., 2018). The fact, that C. burnetii infection increased HIF1α stabilization, prompted us to analyze the impact of bacterial load on HIF1α stabilization. Therefore, we infected BMDM at an MOI of 10, 50 or 100 under hypoxia and analyzed the infection by immunofluorescence and the HIF1α protein level by immunoblot. As shown in Figure 2A, the increased MOI led to a higher bacterial load. Importantly, the increase in bacterial load did not result in increased HIF1α stabilization at 4 h post-infection (Figure 2B). However, at 24 h and 48 h post-infection, the increasing infection dose seemed to result in higher HIF1α protein levels (Figures 2B, C). Moreover, at an MOI of 10, HIF1α stabilization decreases during the course of the infection. Thus, not only the oxygen level influences HIF1α stabilization, but also the pathogen seems to modulate this important transcription factor. This hypothesis prompted us to determine whether bacterial viability is required for affecting HIF1α stabilization.
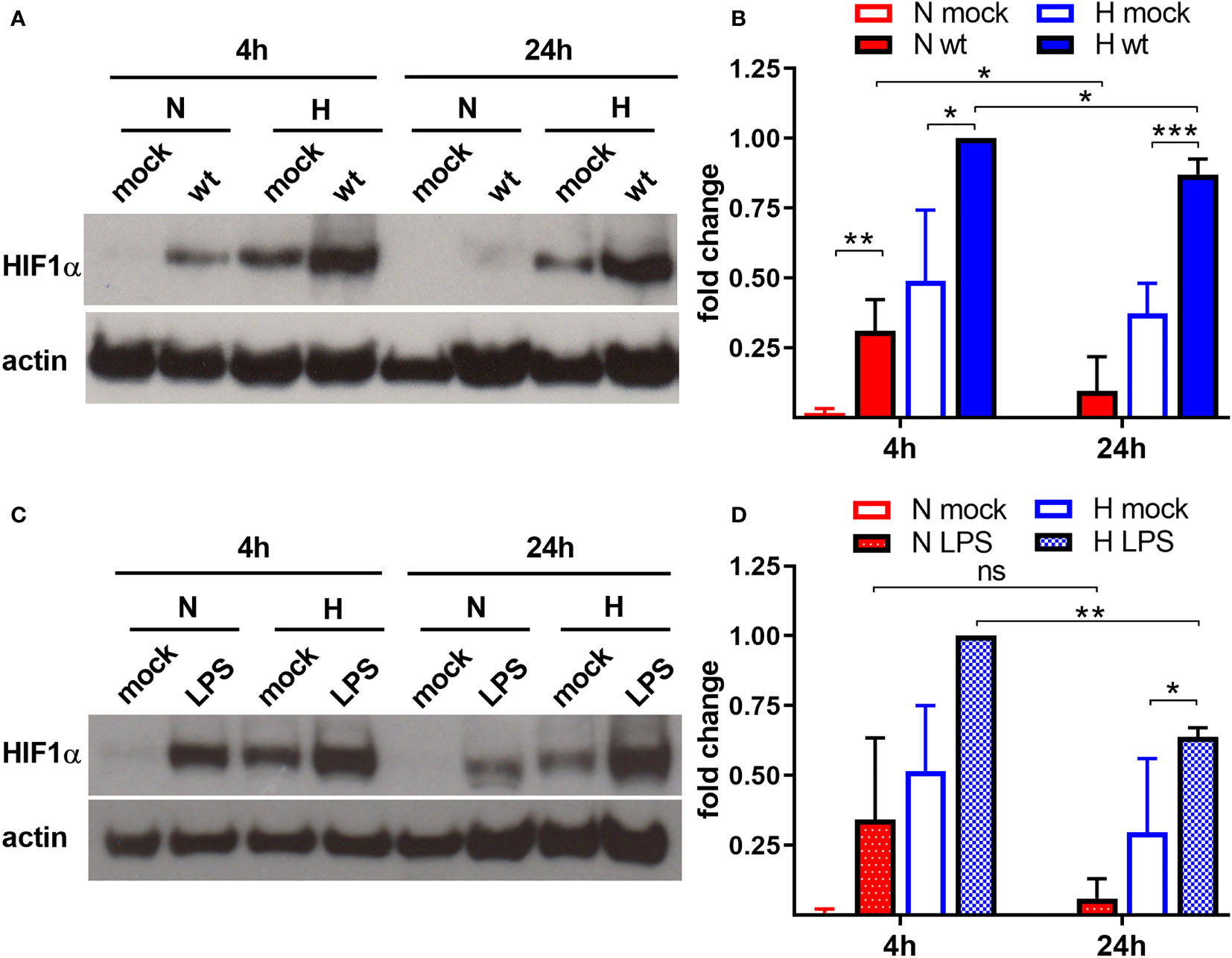
Figure 1 C. burnetii and LPS boost HIF1α stabilization. (A, B) Murine BMDM either uninfected (mock) or infected with C. burnetii (wt) for 4 and 24 h under normoxia (N) or hypoxia (H) were analyzed by immunoblot analysis using antibodies against HIF1α and actin as loading control. (A) One representative experiment out of four independent experiments is shown. (B) Densitometric analysis of the HIF1α/actin ratio was performed using ImageJ. Fold changes are shown relative to cells infected for 4 hours under (H) Mean ± SD, n = 4, one-sample t test or t test. ***p < 0.001, **p < 0.01, *p < 0.05. (C, D) Murine BMDM either untreated (mock) or treated with LPS (100 ng/ml) for 4 and 24 h under N or H were analyzed by immunoblot analysis using antibodies against HIF1α and actin as loading control. (C) One representative experiment out of three independent experiments is shown. (D) Densitometric analysis of the HIF1α/actin ratio was performed using ImageJ. Fold changes are shown relative to cells treated with LPS for 4 hours under (H) Mean ± SD, n = 3, one-sample t test or t test. **p < 0.01, *p < 0.05, ns = non-significant.
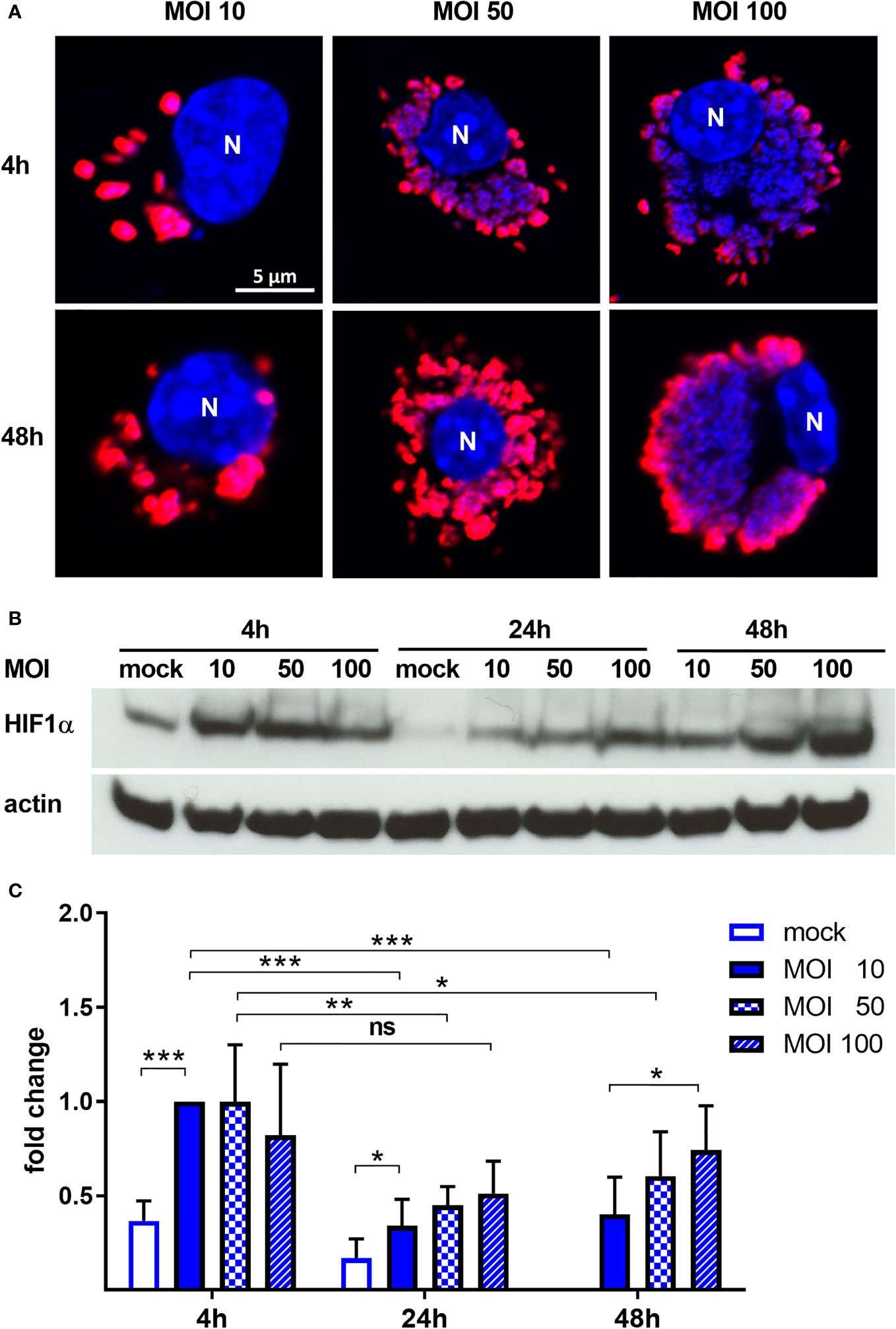
Figure 2 C. burnetii intensifies HIF1α stabilization under hypoxia in a dose-dependent manner. (A) Representative immunofluorescence micrographs of murine BMDM infected with C. burnetii for 4 and 48 h under hypoxia at MOI 10, 50 or 100. The cells were fixed, permeabilized and stained with DAPI (blue) and anti-C. burnetii (red). N = nucleus. (B, C) BMDM infected with C. burnetii for 4, 24 and 48 h under hypoxia at MOI 10, 50 or 100 were analyzed by immunoblot using antibodies against HIF1α and actin as loading control. Importantly, uninfected BMDMs were only cultivated for 24 h under hypoxia, as cell viability was significantly reduced at later time points. (B) One representative immunoblot from 4 independent experiments is shown. (C) Densitometric analysis of the HIF1α/actin ratio was performed using ImageJ. Fold changes are shown relative to cells infected with MOI of 10 for 4 hours. Mean ± SD, n=6, one-sample t-test or t-test. ***p < 0.001, **p < 0.01, *p < 0.05, ns = p > 0.05.
To analyze the role of C. burnetii in HIF1α stabilization, we infected BMDM at an MOI of 10 with either untreated C. burnetii, heat-killed C. burnetii, C. burnetii treated with chloramphenicol to inhibit bacterial protein synthesis or E. coli at an MOI of 10. In normoxic BMDM, C. burnetii led to HIF1α stabilization only at 4 h post-infection (Figure 3A). As this was observed regardless of the viability or physiological state of the pathogen, we assumed that this might be the reaction of the host cell to a pathogen associated molecular pattern (PAMP). This is in line with observations that microbial products, such as LPS, induce HIF1α accumulation also in the presence of O2 (Blouin et al., 2004; Werth et al., 2010). The fact that HIF1α is degraded in infected normoxic macrophages at later time points of infection might be due to the intracellular lifestyle of C. burnetii, which hides in an intracellular vacuole (Pechstein et al., 2017). Importantly, under hypoxic conditions the infection with E. coli induced a higher HIF1α protein level at 4 and 24 h post-infection compared to the infection with viable C. burnetii, indicating that C. burnetii might be able to restrict HIF1α accumulation. Similarly, at 48 h post-infection, the HIF1α protein level was increased in hypoxic BMDM infected with heat-killed C. burnetii compared to BMDM infected with viable C. burnetii (Figures 3A, B). We hypothesized that C. burnetii might be able to actively curtail HIF1α accumulation under hypoxic conditions. The observation that hypoxic BMDM infected with chloramphenicol-treated bacteria showed an increased HIF1α level too, suggests that bacterial protein synthesis is important for C. burnetii-mediated restriction of HIF1α.
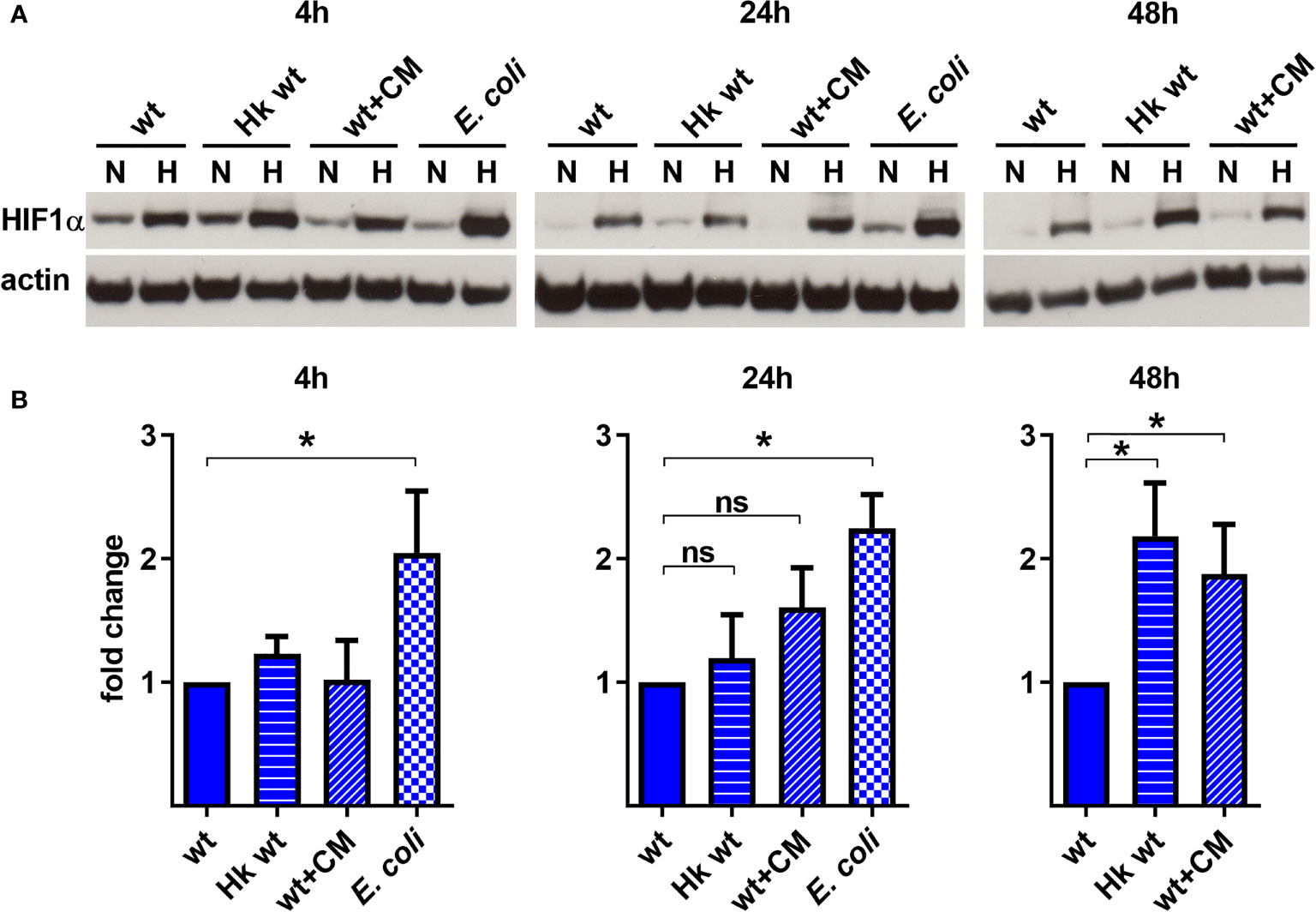
Figure 3 C. burnetii-mediated reduction of infection-induced HIF1α depends on bacterial viability. (A, B) Murine BMDM infected with C. burnetii (wt), heat-killed C. burnetii (Hk), chloramphenicol-treated C. burnetii (CM, 25 µg/ml) or E. coli for 4, 24 and 48 h under normoxia (N) or hypoxia (H) were analyzed by immunoblot using antibodies against HIF1α and actin as loading control. Importantly, E. coli infected BMDMs were only cultivated for 24 h under hypoxia, as cell viability was significantly reduced at later time points. (A) One representative immunoblot from 4 independent experiments is shown. (B) Densitometric analysis of the HIF1α/actin ratio was performed using ImageJ. Fold changes under hypoxia (H) are shown relative to cells infected with viable bacteria. Mean ± SD, n=4, one-sample t-test. *p < 0.05, ns=p > 0.05.
C. burnetii utilizes a type IV secretion system (T4SS) to inject bacterial effector proteins into the host cell to modify host cell pathways for the benefit of the pathogen (Lührmann et al., 2017). Bacteria lacking a functional T4SS are unable to replicate intracellularly, confirming that T4SS-driven modulation of host cell pathways is essential (Beare et al., 2011; Carey et al., 2011). Importantly, inhibition of bacterial protein synthesis by chloramphenicol-treatment also impairs T4SS function (Pan et al., 2008). Therefore, we asked whether the ability of C. burnetii to reduce HIF1α protein level under hypoxic conditions depends on the T4SS. We focused on hypoxic conditions, as, under normoxia, HIF1α is degraded starting at 24 h post-infection regardless of the pathogen viability (Figure 3A). Four hours of infection with the wild-type and the T4SS mutant (ΔdotA) similarly augmented hypoxia-induced HIF1α stabilization. Starting from 24 h post-infection, we detected increased HIF1α stabilization in hypoxic BMDM infected with the ΔdotA mutant (Figures 4A, B). Importantly, this was not mediated by differences in replication ability, as we did not observe any bacterial replication during the course of infection (Figure 4C). This is in line with our previous results, showing that C. burnetii is unable to replicate in hypoxic BMDM (Hayek et al., 2019). Taken together, our results suggest that C. burnetii infection results in HIF1α stabilization under hypoxia. However, viable C. burnetii are able to control HIF1α level in a T4SS-dependent manner.
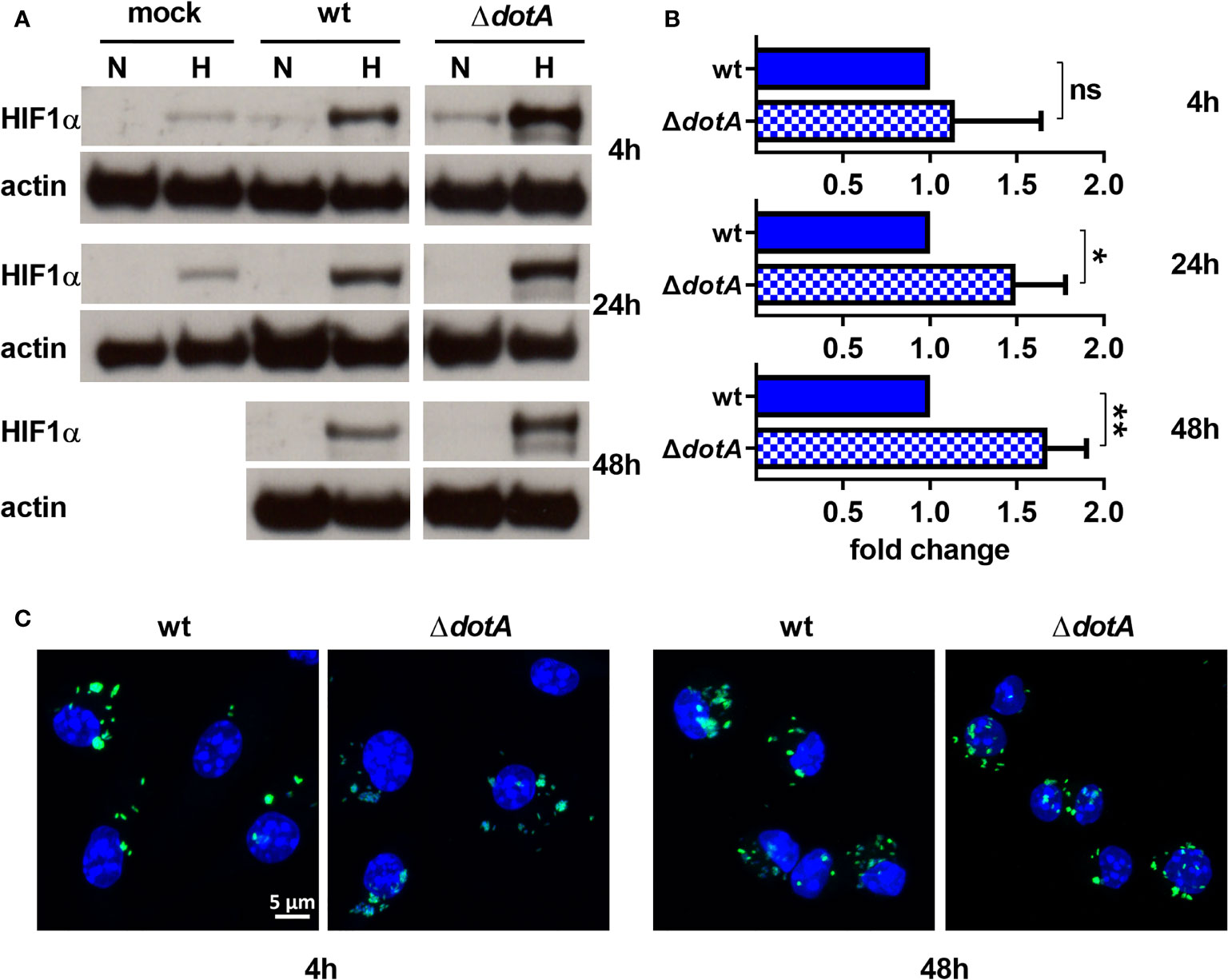
Figure 4 C. burnetii reduces HIF1α levels in a T4SS-dependent manner. (A, B) Murine BMDM either uninfected (mock) or infected with C. burnetii (wt) or C. burnetii lacking a functional T4SS (ΔdotA) for 4, 24 and 48 h under normoxia (N) or hypoxia (H) were analyzed by immunoblot using antibodies against HIF1α and actin as loading control. Importantly, uninfected BMDMs were only cultivated for 24 h under hypoxia, as cell viability was significantly reduced at later time points. (A) One representative Western blot from 4 independent experiments is shown. (B) Densitometric analysis of the HIF1α/actin ratio was performed using ImageJ. Fold changes under hypoxia (H) are shown relative to cells infected with wt. Mean ± SD, n=4, one-sample t-test. **p < 0.01, *p < 0.05, ns=p > 0.05. (C) Representative immunofluorescence micrographs of murine BMDM infected with C. burnetii (wt) (green) or the T4SS transposon mutant (ΔdotA) (green) for 4 and 48 h under hypoxia are shown. The cells were fixed, permeabilized and stained with DAPI (blue).
To determine how C. burnetii might be able to manipulate HIF1α stabilization, we first analyzed the mRNA levels of HIF1α, factors influencing HIF1α degradation and factors important for HIF1α transcription activation by qRT-PCR. The degradation of HIF1α is controlled by prolyl hydroxylases (PHDs), which hydroxylate HIF1α, leading to the recruitment of the von Hippel-Lindau (VHL) E3 ubiquitin ligase, that ubiquitinates HIF1α targeting it for proteasomal degradation (Maxwell et al., 1999; Ohh et al., 2000; Jaakkola et al., 2001). Other factors influence HIF1α transcriptional activity: Factor Inhibiting HIF (FIH) hydroxylates HIF, preventing recruitment of p300 and CREB-binding protein (CBP) (Mahon et al., 2001), which are important for maximal transcriptional activation of HIF (Arany et al., 1996; Dyson and Wright, 2016; Pugh, 2016). A C. burnetii infection, but not LPS stimulation, led to upregulation of HIF1α expression under hypoxia regardless of the pathogen´s genotype (wild-type or ΔdotA) (Figure 5). The expression of the PHDs was affected differently by hypoxia. While the PHD1 expression level was slightly increased by hypoxia, the levels of PHD2 and PHD3 were strongly increased, with a particularly striking induction of PHD3. These results are in line with previous publications (Appelhoff et al., 2004; Marxsen et al., 2004). Importantly, the infection with wild-type or ΔdotA C. burnetii did not alter the PHD2 and PHD3 expression level. The infection with both C. burnetii strains, but not LPS, resulted in PHD1 upregulation under normoxic conditions (Figure 5). The mRNA level of VHL was increased by hypoxia, which was augmented by LPS and by infection with wild-type or ΔdotA C. burnetii. While neither the oxygen level nor the infection state influenced FIH and p300 expression, we observed that LPS resulted in reduced expression of FIH under normoxia and hypoxia and of p300 under normoxia (Figure 5). In addition, CBP expression was reduced in hypoxic conditions and under normoxia when infected with ΔdotA C. burnetii (Figure 5). These data demonstrate that a C. burnetii infection influenced HIF1α (H) and PHD1 (N) mRNA levels, regardless of the T4SS and in a different manner than LPS. Furthermore, under normoxia, the infection with ΔdotA C. burnetii, but not with the wild-type, reduces CBP expression.
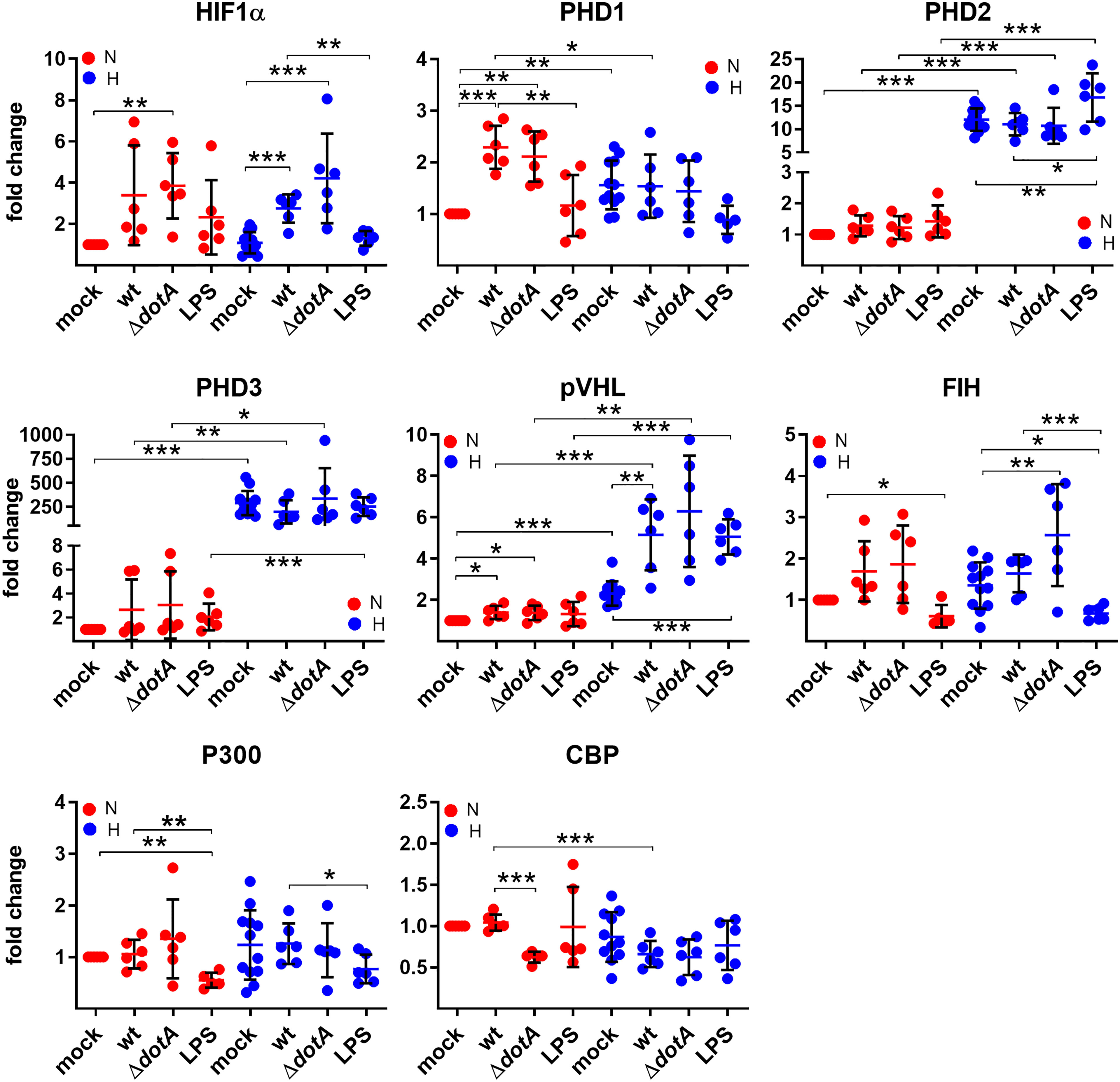
Figure 5 C. burnetii only marginally influences the expression of genes regulating HIF1α stability. BMDM were either uninfected (mock), infected with C. burnetii (wt), the T4SS transposon mutant (ΔdotA), or treated with LPS (100 ng/ml) for 24 h under normoxia (N) and hypoxia (H). Using qRT-PCR, the gene expression of murine HIF1α, PHD1, PHD2, PHD3, VHL, FIH, p300 and CBP was analyzed. The data are displayed as Mean ± SD of 2^-ΔΔCT values (using murine HPRT1 as a calibrator). Fold changes are shown relative to uninfected cells under N. The data shown for each of the C. burnetii (wt and ΔdotA) infection experiment and the LPS treatment experiment represent 3 independent experiments with biological duplicates. One sample t test or t test, n=5-6. ***p < 0.001, **p < 0.01, *p < 0.05.
HIF1α is an important transcription factor critical for cellular metabolism, for regulation of apoptosis and autophagy and for immune responses (Cramer et al., 2003; Corcoran and O'Neill, 2016; Knight and Stanley, 2019). Therefore, we analyzed the role of oxygen availability in combination with a C. burnetii infection or with LPS stimulation as a control on HIF1α target gene expression. First, we concentrated on metabolic genes. As shown in Figure 6, oxygen limitation resulted in upregulation of PKM2, LDHA, Glut1 and PDK1. These factors are involved in glucose uptake (Glut1), generation of pyruvate (PKM2), conversion of pyruvate to lactate (LDHA), and inhibition of the conversion of pyruvate to acetyl-CoA (PDK1), which indirectly increases the conversion of pyruvate to lactate. These data are in line with previous findings showing that HIF1α is essential for the switch to glycolysis in macrophages (Cramer et al., 2003). The infection with wild-type and ΔdotA C. burnetii increases the expression of PKM2 and LDHA in normoxic and hypoxic BMDM, and the expression of Glut1 and PDK1 only in normoxic BMDM (Figure 6). Importantly, treatment with LPS resulted in a similar modulation of the expression of the metabolic genes analyzed. There were only two exceptions: 1) the infection with C. burnetii induced a significantly higher expression of PKM2 under hypoxia than LPS; 2) the infection with C. burnetii induced a significantly higher expression of PDK1 under normoxia than LPS. These data suggest that an infection partially promotes upregulation of the metabolic target genes analyzed. How C. burnetii supports the switch to glycolysis mechanistically is unknown. LPS might play a role in this shift (Figure 6), which is in line with previous reports (Rodriguez-Prados et al., 2010).
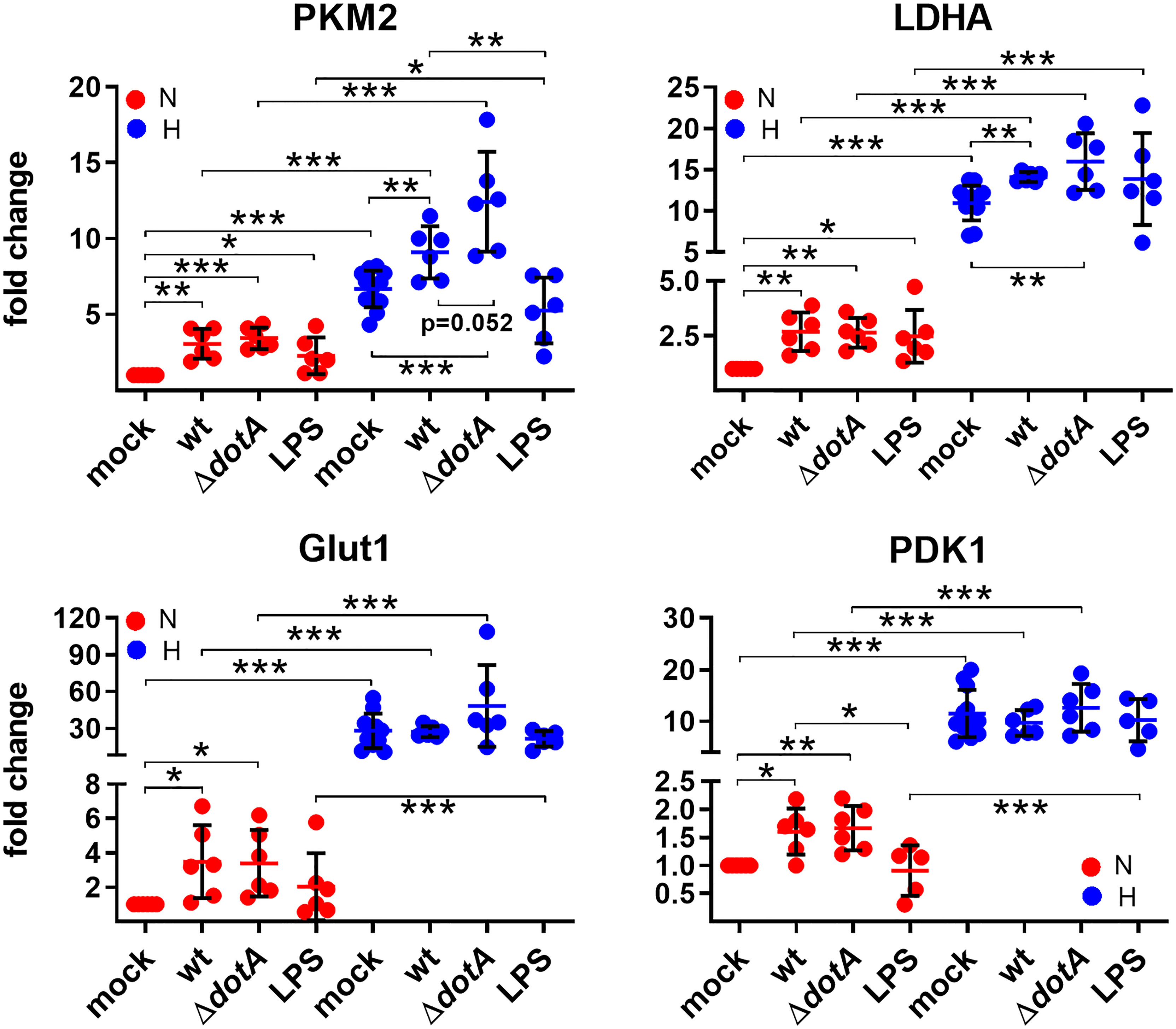
Figure 6 C. burnetii-infected macrophages reveal a shift to glycolysis. BMDM were either uninfected (mock), infected with C. burnetii (wt), the T4SS transposon mutant (ΔdotA), or treated with LPS (100 ng/ml) for 24 h under normoxia (N) and hypoxia (H). Using qRT-PCR, the gene expression of murine PKM2, LDHA, Glut1, and PDK1 was analyzed. The data are shown as Mean ± SD of 2^-ΔΔCT values (using murine HPRT as a calibrator). Fold changes are shown relative to uninfected cells under N. The data shown for each of the C. burnetii (wt and ΔdotA) infection experiments and the LPS treatment experiment represent 3 independent experiments with biological duplicates. One sample t test or t test, n=5-6. ***p < 0.001, **p < 0.01, *p < 0.05.
Next, we analyzed HIF1α target genes involved in regulating apoptotic and autophagic cell death. We then analyzed the mRNA levels of anti-apoptotic Bcl-2, pro-apoptotic Bax and p53, which regulates ~500 target genes, thereby influencing DNA repair, cell cycle arrest, metabolism and cell death (Aubrey et al., 2018). While hypoxia decreased the expression of anti-apoptotic Bcl-2, it increased the expression of Bax. The infection with wild-type C. burnetii did not alter the transcription levels of Bcl-2 and Bax under hypoxia. However, infection with ΔdotA C. burnetii resulted in down-regulation of Bcl-2 and upregulation of Bax under hypoxia. Under normoxia, the infection resulted in down-regulation of Bcl-2 and up-regulation of Bax regardless of the genotype of the pathogen (Figure 7). This result was unexpected, as C. burnetii displays anti-apoptotic activity, and no influence on Bcl-2 and Bax protein levels was determined (Lührmann and Roy, 2007; Voth et al., 2007; Cordsmeier et al., 2019). However, this might be due to different cell types, primary versus cell lines, used.
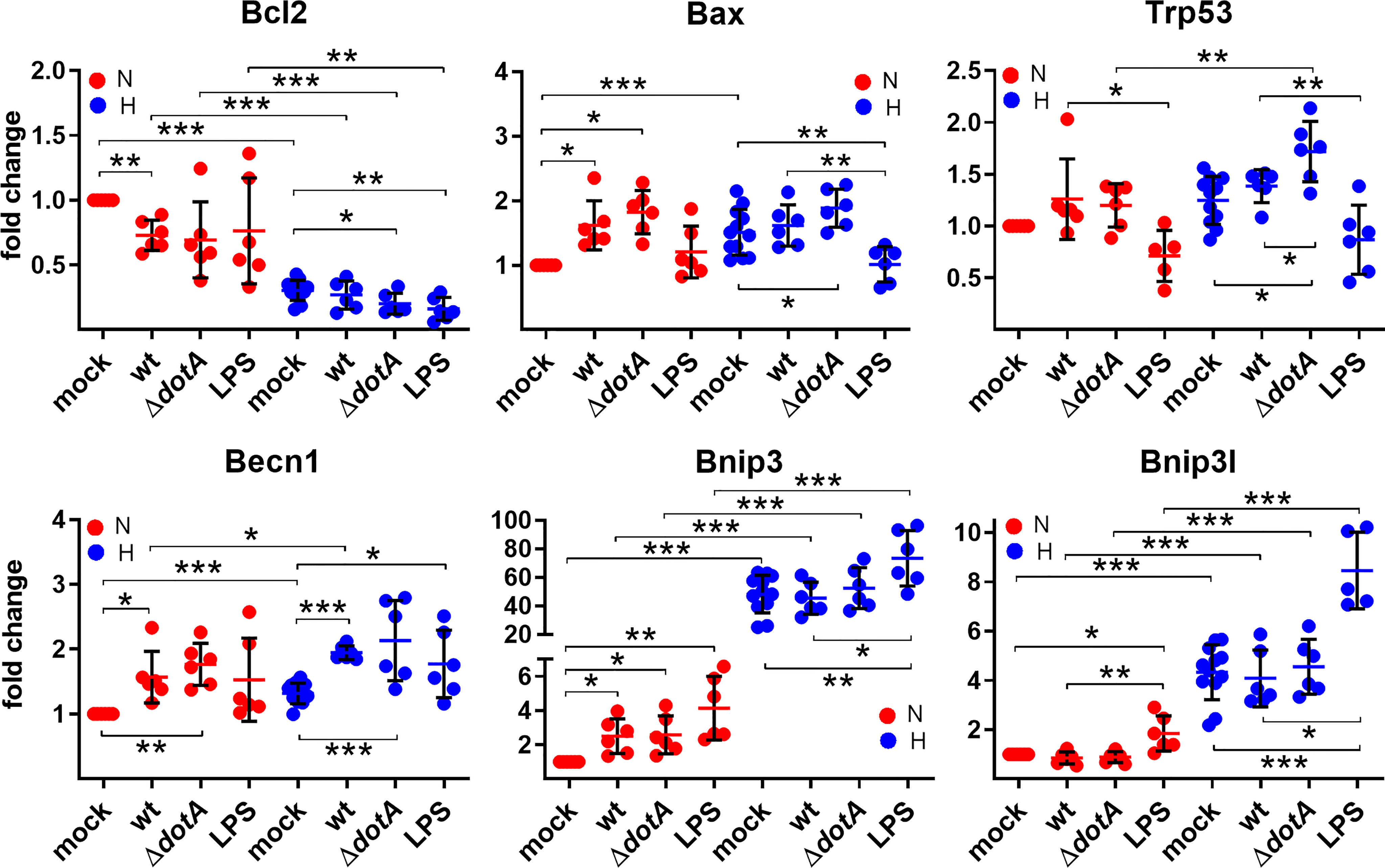
Figure 7 A pro-apoptotic expression signature is more prominent in C. burnetii-infected or hypoxic macrophages. BMDM were either uninfected (mock), infected with C. burnetii (wt), the T4SS transposon mutant (ΔdotA), or treated with LPS (100 ng/ml) for 24 h under normoxia (N) and hypoxia (H). Using qRT-PCR, the gene expression of murine Bcl2, Bax, Trp53, Becn1, Bnip3, and Bnip3l was analyzed. The data are depicted as Mean ± SD of 2^-ΔΔCT values (using murine HPRT as a calibrator). Fold changes are shown relative to uninfected cells under N. The data shown for each of the C. burnetii (wt and ΔdotA) infection experiments and the LPS treatment experiment represent 3 independent experiments with biological duplicates. One sample t test or t test, n=5-6. ***p < 0.001, **p < 0.01, *p < 0.05.
Hypoxia and HIF1α regulate p53 in several ways and vice versa (Zhang et al., 2021). We did not find an influence of hypoxia on p53 transcription level, but the infection under hypoxia resulted in an increased p53 expression (Figure 7). Importantly, cells infected with the ΔdotA mutant showed a significant higher expression of p53 compared to cells infected with wild-type C. burnetii (Figure 7).
This might be due to an increased HIF1α level in cells infected with the ΔdotA mutant (Figures 4A, B), but independent of LPS signaling, as LPS resulted in downregulation of p53 expression under normoxia and hypoxia.
While analyzing genes involved in autophagic cell death induction, we observed an upregulation of Beclin 1, Bnip3 and Bnip3l by hypoxia. The infection influenced the expression level of Beclin 1, both under normoxia and hypoxia, similarly as did LPS.
Bnip3 expression was only upregulated by a C. burnetii infection under normoxia, but not under hypoxia, while Bnip3l expression was not modulated by the infection at all. Importantly, LPS treatment resulted in significant upregulation of Bnip3 and Bnip3l under hypoxia, demonstrating that C. burnetii-induced expression modulation of the genes analyzed was partially independent of LPS signaling (Figure 7). These data suggest that hypoxia and the infection with C. burnetii affect the apoptosis-regulators analyzed independently towards a more pro-apoptotic signature.
Next, we analyzed the role of hypoxia and/or a C. burnetii infection on the transcription of inflammatory genes. We analyzed the pro-inflammatory HIF1α target genes IL1β, IL6, Nos2 and the anti-inflammatory gene IL10. In the absence of infection, we detected an increase of IL1β and a decrease in IL10, when comparing normoxia versus hypoxia (Figure 8), which is in line with the observation that the HIF pathway regulates cytokine production in multiple cell types (Malkov et al., 2021). In contrast, the infection increased the expression of all genes analyzed. While the expression of the pro-inflammatory genes was increased under hypoxia compared to under normoxia, this was the opposite for the anti-inflammatory gene IL10 (Figure 8). Importantly, the C. burnetii infection resulted in significantly stronger induction of IL10 under normoxia than the LPS treatment. In contrast, the combination of hypoxia and LPS treatment resulted in an upregulation of IL6 expression by ~9 fold, while the combination of hypoxia and C. burnetii infection only led to a ~3 fold upregulation. However, we did not detect a difference between BMDM infected with the wild-type or the T4SS mutant, indicating that the effect of C. burnetii on inflammatory HIF1α-target genes is independent of the T4SS. Thus, our data indicates that the C. burnetii infection results in upregulation of pro- and anti-inflammatory genes. Importantly, under hypoxia, the expression profile of the genes analyzed shifts towards a pronounced pro-inflammatory signature.
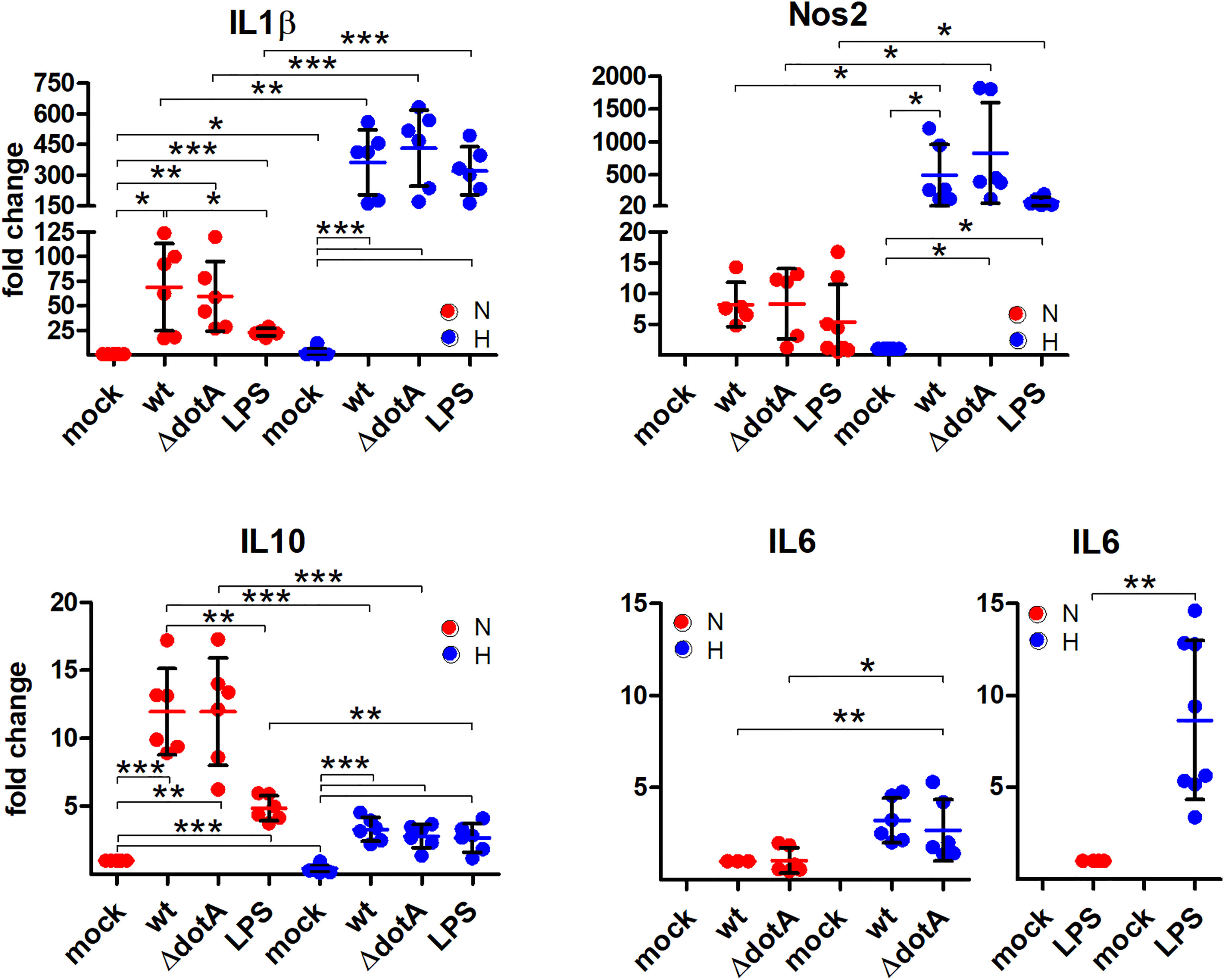
Figure 8 A pro-inflammatory profile is observed through C. burnetii infection under hypoxia. BMDM were either uninfected (mock), infected with C. burnetii (wt), the T4SS transposon mutant (ΔdotA), or treated with LPS (100 ng/ml) for 24 h under normoxia (N) and hypoxia (H). Using qRT-PCR, the gene expression of murine IL1β, Nos2, IL10, and IL6 was analyzed. The data are plotted as Mean ± SD of 2^-ΔΔCT values (using murine HPRT as a calibrator). In case of IL1β and IL10, fold changes are shown relative to uninfected cells under N, while for Nos2 they are represented relative to uninfected cells under H. For IL6, fold changes are shown relative to wt-infected cells or LPS-treated cells under N. The data shown for each of the C. burnetii (wt and ΔdotA) infection experiments and the LPS treatment experiment represent 3 independent experiments with biological duplicates. One sample t test or t test, n=5-6. ***p < 0.001, **p < 0.01, *p < 0.05.
While HIF1α was first identified as an essential regulator of hypoxia (Majmundar et al., 2010), it is now clear that this transcription factor is also activated by several human pathogens even under normoxia (Werth et al., 2010). As HIF1α regulates cellular metabolism, immune cell activity, and inflammatory responses (Knight and Stanley, 2019), it is a central player during host-pathogen interaction.
Thus, it is not surprising that several pathogens have evolved proteins that modulate HIF1α activity (Knight and Stanley, 2019). For example, the Salmonella enterica siderophore Sal activates HIF1 (Hartmann et al., 2008), as does BadA from Bartonella henselae (Riess et al., 2004). In contrast, the AQ signaling molecule from Pseudomonas aeruginosa leads to proteasomal degradation of HIF1 (Legendre et al., 2012). These examples demonstrate that dependent on the nature and requirements of the respective pathogen, the ability to interfere with HIF1 is distinct.
We recently showed that HIF1α is responsible for controlling C. burnetii infection in an in vitro infection model using primary murine and human macrophages (Hayek et al., 2019). However, although HIF1α was proven beneficial for limiting bacterial replication (Hayek et al., 2021), it did not affect the cell´s ability to clear C. burnetii (Hayek et al., 2019). This is in line with previous observations that hypoxia and/or HIF1α induce a state of bacterial persistence and dormancy, which might impair bacterial clearance and allow the emergence of reoccurring or chronic infections (Sershen et al., 2016; Hayek et al., 2019; Hayek et al., 2021).
Here, we show that C. burnetii is able to curtail HIF1α, which depends on bacterial viability and protein synthesis (Figures 3A, B). The data suggests that the T4SS is involved (Figures 4A, B) indicating that a bacterial factor is required for this activity. The T4SS, an essential virulence factor of C. burnetii, injects over 150 effector proteins into the host cell to manipulate several host cell pathways enabling the pathogen to survive and replicate intracellularly (Lührmann et al., 2017). Only a few of these effector proteins have been functionally characterized. They interfere with host cell transcription, apoptosis, pyroptosis, ER stress, autophagy, and vesicular trafficking (Cordsmeier et al., 2019; Burette and Bonazzi, 2020b; Thomas et al., 2020; Dragan and Voth, 2020). The effector protein(s) involved in destabilizing HIF1α is currently unknown. The reason why increasing infection rates, which most likely result in increased numbers of secreted effector proteins, did not result in increased HIF1α degradation (Figures 2B, C), is currently unknown. It might be the balance between activation by PAMPs and dampening by effector proteins. As we could not show the biological consequence of the T4SS-dependent HIF1α destabilization (Figures 5 – 8), we hypothesize that HIF1α destabilization might be a side effect and not the primary function of a so far unknown effector protein. Thus, an effector protein interfering with the NF-κB signaling pathway might be involved, as NF-κB regulates HIF1α (Rius et al., 2008). Importantly, NF-κB modulation by the C. burnetii T4SS has been described (Mahapatra et al., 2016) and recently the C. burnetii T4SS effector protein NopA was identified to perturb NF-κB activation (Burette et al., 2020a). Thus, it can be speculated that NopA or a so far unknown effector protein might be indirectly involved in HIF1α activation. The increased level of HIF1α in cells infected with the T4SS mutant (ΔdotA) in comparison to cells infected with wild-type C. burnetii did not correlate with differences in the expression levels of HIF1α modulators (Figure 5), suggesting that a so far unknown effector protein might not interfere with the expression of HIF1α modulators. It might be possible that the effector protein interferes with the enzymatic activity of the PHDs or the availability of PHD co-factors (Siegert et al., 2015). Further research will be necessary to determine the molecular mechanisms leading to T4SS-dependent reduction of C. burnetii-induced HIF1α stabilization.
Nevertheless, we did not detect a difference in the expression of most of the HIF1α target genes analyzed in BMDM infected with either wild-type C. burnetii or the ΔdotA mutant (Figures 6 – 8), suggesting that the HIF1α protein level does not correlate with the level of HIF1α target gene expression. This was an unexpected finding, as correlation between HIF1α protein level and HIF1α target gene expression has been reported (Lee and Thorgeirsson, 2004; Lv et al., 2021). However, those reports analyzed the role of HIF1α in cancer or in cell lines, while we analyzed the role of HIF1α in primary cells during infection. Infected tissue is commonly found to be hypoxic, which triggers HIF1α stabilization (Jantsch and Schödel, 2015), and pathogens or their products are known to trigger HIF1α accumulation also under normoxia. In addition, bacterial products also activate transcription factors that might act synergistically or antagonistically with HIF1α (Hayek et al., 2021). Importantly, our data clearly demonstrates that the C. burnetii infection under hypoxia leads to upregulation of the pro-inflammatory genes IL1β, IL6 and Nos2, and to downregulation of the anti-inflammatory gene IL10 (Figure 8). This is in line with reports that HIF1α is required for mounting a pro-inflammatory response to bacterial and fungal pathogens (Peyssonnaux et al., 2005; Tannahill et al., 2013; Mills et al., 2016; Li et al., 2018). Especially the increased expression of Nos2 and IL1β might be of biological consequence for the C. burnetii infection. The homodimeric enzyme NOS2 converts L-arginine and oxygen into L-citrulline and nitric oxide (NO) (Bogdan, 2015). The latter is important for controlling bacterial infections (Nathan and Shiloh, 2000), including a C. burnetii infection (Howe et al., 2002; Zamboni and Rabinovitch, 2003; Brennan et al., 2004). IL1β is produced as an inactive pro-form, which has to be cleaved to its active form following inflammasome activation (Dinarello, 2018). C. burnetii avoids activation of the inflammasome, and thus, pyroptosis (Cunha et al., 2015; Delaney et al., 2021). However, whether C. burnetii is able to prevent IL1β secretion induced by potent inflammasome stimuli has to be clarified, as conflicting reports exist (Cunha et al., 2015; Delaney et al., 2021). Of note, NO was found to inhibit the NLRP3 inflammasome-dependent processing of IL1β (Mishra et al., 2013). Thus, it will be of importance to analyze whether not only the expression of IL1β is increased, but also its secretion. In addition, we have to elucidate whether the increased levels of NO in hypoxic C. burnetii infected BMDM might inhibit IL1β processing and secretion.
In summary, our data demonstrate that C. burnetii influences HIF1α stability and activity. As HIF1α is important for mounting anti-bacterial responses, this might have consequences for the host-pathogen interaction and, thus, disease outcome.
The original contributions presented in the study are included in the article. Further inquiries can be directed to the corresponding author.
IH and MS performed the experiments and analyzed the data. AL and IH conceived the study. AL obtained funding, supervised the study and drafted the manuscript. All authors contributed to the writing of the manuscript.
This work was supported by the Deutsche Forschungsgemeinschaft (DFG) through the Collaborative Research Initiative 1181 (CRC1181) project A06 to AL. IH was partially funded by the Bavarian Equal Opportunities Sponsorship – Realisierung von Chancengleichheit von Frauen in Forschung und Lehre (FFL) – Realization Equal Opportunities for Women in Research and Teaching.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
This work was supported by the Deutsche Forschungsgemeinschaft (DFG) through the Collaborative Research Initiative 1181 (CRC1181) project A06 to AL. We thank all members of the Lührmann, Soulat and Petter labs for discussions, Christian Berens for critically reading the manuscript, Matteo Bonazzi (IRIM, CNRS, Université de Montpellier, France) for providing C. burnetii transposon mutants, and Philipp Tripal (Optical Imaging Center Erlangen (OICE)) for support with confocal microscopy.
Abnave, P., Muracciole, X., Ghigo, E. (2017). Coxiella burnetii Lipopolysaccharide: What Do We Know? Int. J. Mol. Sci. 18, 2509. doi: 10.3390/ijms18122509
Anderson, A., Bijlmer, H., Fournier, P. E., Graves, S., Hartzell, J., Kersh, G. J., et al. (2013). ). Diagnosis and Management of Q Fever–United States 2013: Recommendations From CDC and the Q Fever Working Group. MMWR. Recommend. Rep. 62, 1–30.
Appelhoff, R. J., Tian, Y. M., Raval, R. R., Turley, H., Harris, A. L., Pugh, C. W., et al. (2004). Differential Function of the Prolyl Hydroxylases PHD1, PHD2, and PHD3 in the Regulation of Hypoxia-Inducible Factor. J. Biol. Chem. 279, 38458–38465. doi: 10.1074/jbc.M406026200
Arany, Z., Huang, L. E., Eckner, R., Bhattacharya, S., Jiang, C., Goldberg, M. A., et al. (1996). An Essential Role for P300/CBP in the Cellular Response to Hypoxia. Proc. Natl. Acad. Sci. U. S. A. 93, 12969–12973. doi: 10.1073/pnas.93.23.12969
Aubrey, B. J., Kelly, G. L., Janic, A., Herold, M. J., Strasser, A. (2018). How Does P53 Induce Apoptosis and How Does This Relate to P53-Mediated Tumour Suppression? Cell Death Differ. 25, 104–113. doi: 10.1038/cdd.2017.169
Beare, P. A., Gilk, S. D., Larson, C. L., Hill, J., Stead, C. M., Omsland, A., et al. (2011). Dot/Icm Type IVB Secretion System Requirements for Coxiella Burnetii Growth in Human Macrophages. MBio 2, e00175–e00111. doi: 10.1128/mBio.00175-11
Beare, P. A., Jeffrey, B. M., Long, C. M., Martens, C. M., Heinzen, R. A. (2018). Genetic Mechanisms of Coxiella burnetii Lipopolysaccharide Phase Variation. PLoS Pathog. 14, e1006922. doi: 10.1371/journal.ppat.1006922
Blouin, C. C., Page, E. L., Soucy, G. M., Richard, D. E. (2004). Hypoxic Gene Activation by Lipopolysaccharide in Macrophages: Implication of Hypoxia-Inducible Factor 1alpha. Blood 103, 1124–1130. doi: 10.1182/blood-2003-07-2427
Bogdan, C. (2015). Nitric Oxide Synthase in Innate and Adaptive Immunity: An Update. Trends Immunol. 36, 161–178. doi: 10.1016/j.it.2015.01.003
Brennan, R. E., Russell, K., Zhang, G., Samuel, J. E. (2004). Both Inducible Nitric Oxide Synthase and NADPH Oxidase Contribute to the Control of Virulent Phase I Coxiella Burnetii Infections. Infect. Immun. 72, 6666–6675. doi: 10.1128/IAI.72.11.6666-6675.2004
Burette, M., Allombert, J., Lambou, K., Maarifi, G., Nisole, S., Di Russo Case, E., et al. (2020a). Modulation of Innate Immune Signaling by a Coxiella Burnetii Eukaryotic-Like Effector Protein. Proc. Natl. Acad. Sci. U. S. A. 117, 13708–13718. doi: 10.1073/pnas.1914892117
Burette, M., Bonazzi, M. (2020b). From Neglected to Dissected: How Technological Advances are Leading the Way to the Study of Coxiella Burnetii Pathogenesis. Cell Microbiol. 22, e13180. doi: 10.1111/cmi.13180
Carey, K. L., Newton, H. J., Lührmann, A., Roy, C. R. (2011). The Coxiella Burnetii Dot/Icm System Delivers a Unique Repertoire of Type IV Effectors Into Host Cells and is Required for Intracellular Replication. PLoS Pathog. 7, e1002056. doi: 10.1371/journal.ppat.1002056
Corcoran, S. E., O'Neill, L. A. (2016). HIF1alpha and Metabolic Reprogramming in Inflammation. J. Clin. Invest. 126, 3699–3707. doi: 10.1172/JCI84431
Cordsmeier, A., Wagner, N., Lührmann, A., Berens, C. (2019). Defying Death - How Coxiella Burnetii Copes With Intentional Host Cell Suicide. Yale J. Biol. Med. 92, 619–628. doi: 10.1038/cdd.2017.169
Cramer, T., Yamanishi, Y., Clausen, B. E., Forster, I., Pawlinski, R., Mackman, N., et al. (2003). HIF-1alpha is Essential for Myeloid Cell-Mediated Inflammation. Cell 112, 645–657. doi: 10.1016/S0092-8674(03)00154-5
Cunha, L. D., Ribeiro, J. M., Fernandes, T. D., Massis, L. M., Khoo, C. A., Moffatt, J. H., et al. (2015). Inhibition of Inflammasome Activation by Coxiella Burnetii Type IV Secretion System Effector IcaA. Nat. Commun. 6, 10205. doi: 10.1038/ncomms10205
Delaney, M. A., Hartigh, A. D., Carpentier, S. J., Birkland, T. P., Knowles, D. P., Cookson, B. T., et al. (2021). Avoidance of the NLRP3 Inflammasome by the Stealth Pathogen, Coxiella Burnetii. Vet. Pathol. 58, 624–642. doi: 10.1177/0300985820981369
Devraj, G., Beerlage, C., Brune, B., Kempf, V. A. (2017). Hypoxia and HIF-1 Activation in Bacterial Infections. Microbes Infect. 19, 144–156. doi: 10.1016/j.micinf.2016.11.003
Dinarello, C. A. (2018). Overview of the IL-1 Family in Innate Inflammation and Acquired Immunity. Immunol. Rev. 281, 8–27. doi: 10.1111/imr.12621
Dragan, A. L., Voth, D. E. (2020). Coxiella Burnetii: International Pathogen of Mystery. Microbes Infect. 22, 100–110. doi: 10.1016/j.micinf.2019.09.001
Dyson, H. J., Wright, P. E. (2016). Role of Intrinsic Protein Disorder in the Function and Interactions of the Transcriptional Coactivators CREB-Binding Protein (CBP) and P300. J. Biol. Chem. 291, 6714–6722. doi: 10.1074/jbc.R115.692020
Greer, S. N., Metcalf, J. L., Wang, Y., Ohh, M. (2012). The Updated Biology of Hypoxia-Inducible Factor. EMBO J. 31, 2448–2460. doi: 10.1038/emboj.2012.125
Gustafsson, M. V., Zheng, X., Pereira, T., Gradin, K., Jin, S., Lundkvist, J., et al. (2005). Hypoxia Requires Notch Signaling to Maintain the Undifferentiated Cell State. Dev. Cell 9, 617–628. doi: 10.1016/j.devcel.2005.09.010
Harris, R. J., Storm, P. A., Lloyd, A., Arens, M., Marmion, B. P. (2000). Long-Term Persistence of Coxiella Burnetii in the Host After Primary Q Fever. Epidemiol. Infect. 124, 543–549. doi: 10.1017/S0950268899003763
Hartmann, H., Eltzschig, H. K., Wurz, H., Hantke, K., Rakin, A., Yazdi, A. S., et al. (2008). Hypoxia-Independent Activation of HIF-1 by Enterobacteriaceae and Their Siderophores. Gastroenterology 134, 756–767. doi: 10.1053/j.gastro.2007.12.008
Hayek, I., Fischer, F., Schulze-Luehrmann, J., Dettmer, K., Sobotta, K., Schatz, V., et al. (2019). Limitation of TCA Cycle Intermediates Represents an Oxygen-Independent Nutritional Antibacterial Effector Mechanism of Macrophages. Cell Rep. 26, 3502–3510, e3506. doi: 10.1016/j.celrep.2019.02.103
Hayek, I., Schatz, V., Bogdan, C., Jantsch, J., Lührmann, A. (2021). Mechanisms Controlling Bacterial Infection in Myeloid Cells Under Hypoxic Conditions. Cell Mol. Life Sci. 78, 1887–1907. doi: 10.1007/s00018-020-03684-8
Hewitson, K. S., Lienard, B. M., McDonough, M. A., Clifton, I. J., Butler, D., Soares, A. S., et al. (2007). Structural and Mechanistic Studies on the Inhibition of the Hypoxia-Inducible Transcription Factor Hydroxylases by Tricarboxylic Acid Cycle Intermediates. J. Biol. Chem. 282, 3293–3301. doi: 10.1074/jbc.M608337200
Howe, D., Barrows, L. F., Lindstrom, N. M., Heinzen, R. A. (2002). Nitric Oxide Inhibits Coxiella Burnetii Replication and Parasitophorous Vacuole Maturation. Infect. Immun. 70, 5140–5147. doi: 10.1128/IAI.70.9.5140-5147.2002
Jaakkola, P., Mole, D. R., Tian, Y. M., Wilson, M. I., Gielbert, J., Gaskell, S. J., et al. (2001). Targeting of HIF-Alpha to the Von Hippel-Lindau Ubiquitylation Complex by O2-Regulated Prolyl Hydroxylation. Science 292, 468–472. doi: 10.1126/science.1059796
Jantsch, J., Schödel, J. (2015). Hypoxia and Hypoxia-Inducible Factors in Myeloid Cell-Driven Host Defense and Tissue Homeostasis. Immunobiology 220, 305–314. doi: 10.1016/j.imbio.2014.09.009
Kaidi, A., Williams, A. C., Paraskeva, C. (2007). Interaction Between Beta-Catenin and HIF-1 Promotes Cellular Adaptation to Hypoxia. Nat. Cell Biol. 9, 210–217. doi: 10.1038/ncb1534
Kelly, B., O'Neill, L. A. J. (2015). Metabolic Reprogramming in Macrophages and Dendritic Cells in Innate Immunity. Cell Res. 25, 771–784. doi: 10.1038/cr.2015.68
Knight, M., Stanley, S. (2019). HIF-1alpha as a Central Mediator of Cellular Resistance to Intracellular Pathogens. Curr. Opin. Immunol. 60, 111–116. doi: 10.1016/j.coi.2019.05.005
Koshiji, M., Kageyama, Y., Pete, E. A., Horikawa, I., Barrett, J. C., Huang, L. E. (2004). HIF-1alpha Induces Cell Cycle Arrest by Functionally Counteracting Myc. EMBO J. 23, 1949–1956. doi: 10.1038/sj.emboj.7600196
Lee, J. S., Thorgeirsson, S. S. (2004). Genome-Scale Profiling of Gene Expression in Hepatocellular Carcinoma: Classification, Survival Prediction, and Identification of Therapeutic Targets. Gastroenterology 127, S51–S55. doi: 10.1053/j.gastro.2004.09.015
Legendre, C., Reen, F. J., Mooij, M. J., McGlacken, G. P., Adams, C., O'Gara, F. (2012). Pseudomonas Aeruginosa Alkyl Quinolones Repress Hypoxia-Inducible Factor 1 (HIF-1) Signaling Through HIF-1alpha Degradation. Infect. Immun. 80, 3985–3992. doi: 10.1128/IAI.00554-12
Li, C., Wang, Y., Li, Y., Yu, Q., Jin, X., Wang, X., et al. (2018). HIF1alpha-Dependent Glycolysis Promotes Macrophage Functional Activities in Protecting Against Bacterial and Fungal Infection. Sci. Rep. 8, 3603. doi: 10.1038/s41598-018-22039-9
Lührmann, A., Newton, H. J., Bonazzi, M. (2017). Beginning to Understand the Role of the Type IV Secretion System Effector Proteins in Coxiella Burnetii Pathogenesis. Curr. topic. Microbiol. Immunol. 413, 243–268. doi: 10.1007/978-3-319-75241-9_10
Lührmann, A., Roy, C. R. (2007). Coxiella Burnetii Inhibits Activation of Host Cell Apoptosis Through a Mechanism That Involves Preventing Cytochrome C Release From Mitochondria. Infect. Immun. 75, 5282–5289. doi: 10.1128/IAI.00863-07
Lv, C., Wang, S., Lin, L., Wang, C., Zeng, K., Meng, Y., et al. (2021). USP14 Maintains HIF1-Alpha Stabilization via its Deubiquitination Activity in Hepatocellular Carcinoma. Cell Death Dis. 12, 803. doi: 10.1038/s41419-021-04089-6
Mahapatra, S., Gallaher, B., Smith, S. C., Graham, J. G., Voth, D. E., Shaw, E. I. (2016). Coxiella Burnetii Employs the Dot/Icm Type IV Secretion System to Modulate Host NF-Kappab/RelA Activation. Front. Cell Infect. Microbiol. 6, 188. doi: 10.3389/fcimb.2016.00188
Mahon, P. C., Hirota, K., Semenza, G. L. (2001). FIH-1: A Novel Protein That Interacts With HIF-1alpha and VHL to Mediate Repression of HIF-1 Transcriptional Activity. Genes Dev. 15, 2675–2686. doi: 10.1101/gad.924501
Majmundar, A. J., Wong, W. J., Simon, M. C. (2010). Hypoxia-Inducible Factors and the Response to Hypoxic Stress. Mol. Cell 40, 294–309. doi: 10.1016/j.molcel.2010.09.022
Malkov, M. I., Lee, C. T., Taylor, C. T. (2021). Regulation of the Hypoxia-Inducible Factor (HIF) by Pro-Inflammatory Cytokines. Cells 10, 2340. doi: 10.3390/cells10092340
Martinez, E., Cantet, F., Fava, L., Norville, I., Bonazzi, M. (2014). Identification of OmpA, a Coxiella Burnetii Protein Involved in Host Cell Invasion, by Multi-Phenotypic High-Content Screening. PLoS Pathog. 10, e1004013. doi: 10.1371/journal.ppat.1004013
Marxsen, J. H., Stengel, P., Doege, K., Heikkinen, P., Jokilehto, T., Wagner, T., et al. (2004). Hypoxia-Inducible Factor-1 (HIF-1) Promotes its Degradation by Induction of HIF-Alpha-Prolyl-4-Hydroxylases. Biochem. J. 381, 761–767. doi: 10.1042/BJ20040620
Maurin, M., Raoult, D. (1999). Q Fever. Clin. Microbiol. Rev. 12, 518–553. doi: 10.1128/CMR.12.4.518
Maxwell, P. H., Wiesener, M. S., Chang, G. W., Clifford, S. C., Vaux, E. C., Cockman, M. E., et al. (1999). The Tumour Suppressor Protein VHL Targets Hypoxia-Inducible Factors for Oxygen-Dependent Proteolysis. Nature 399, 271–275. doi: 10.1038/20459
Mills, E. L., Kelly, B., Logan, A., Costa, A. S. H., Varma, M., Bryant, C. E., et al. (2016). Succinate Dehydrogenase Supports Metabolic Repurposing of Mitochondria to Drive Inflammatory Macrophages. Cell 167, 457–470.e413. doi: 10.1016/j.cell.2016.08.064
Mishra, B. B., Rathinam, V. A., Martens, G. W., Martinot, A. J., Kornfeld, H., Fitzgerald, K. A., et al. (2013). Nitric Oxide Controls the Immunopathology of Tuberculosis by Inhibiting NLRP3 Inflammasome-Dependent Processing of IL-1beta. Nat. Immunol. 14, 52–60. doi: 10.1038/ni.2474
Nathan, C., Shiloh, M. U. (2000). Reactive Oxygen and Nitrogen Intermediates in the Relationship Between Mammalian Hosts and Microbial Pathogens. Proc. Natl. Acad. Sci. U. S. A. 97, 8841–8848. doi: 10.1073/pnas.97.16.8841
Obach, M., Navarro-Sabate, A., Caro, J., Kong, X., Duran, J., Gomez, M., et al. (2004). 6-Phosphofructo-2-Kinase (Pfkfb3) Gene Promoter Contains Hypoxia-Inducible Factor-1 Binding Sites Necessary for Transactivation in Response to Hypoxia. J. Biol. Chem. 279, 53562–53570. doi: 10.1074/jbc.M406096200
Ohh, M., Park, C. W., Ivan, M., Hoffman, M. A., Kim, T. Y., Huang, L. E., et al. (2000). Ubiquitination of Hypoxia-Inducible Factor Requires Direct Binding to the Beta-Domain of the Von Hippel-Lindau Protein. Nat. Cell Biol. 2, 423–427. doi: 10.1038/35017054
Pan, X., Lührmann, A., Satoh, A., Laskowski-Arce, M. A., Roy, C. R. (2008). Ankyrin Repeat Proteins Comprise a Diverse Family of Bacterial Type IV Effectors. Science 320, 1651–1654. doi: 10.1126/science.1158160
Pechstein, J., Schulze-Luehrmann, J., Lührmann, A. (2017). Coxiella Burnetii as a Useful Tool to Investigate Bacteria-Friendly Host Cell Compartments. Int. J. Med. Microbiol. 308, 77–83. doi: 10.1016/j.ijmm.2017.09.010
Peyssonnaux, C., Datta, V., Cramer, T., Doedens, A., Theodorakis, E. A., Gallo, R. L., et al. (2005). HIF-1alpha Expression Regulates the Bactericidal Capacity of Phagocytes. J. Clin. Invest. 115, 1806–1815. doi: 10.1172/JCI23865
Pugh, C. W. (2016). Modulation of the Hypoxic Response. Adv. Exp. Med. Biol. 903, 259–271. doi: 10.1007/978-1-4899-7678-9_18
Riess, T., Andersson, S. G., Lupas, A., Schaller, M., Schafer, A., Kyme, P., et al. (2004). Bartonella Adhesin a Mediates a Proangiogenic Host Cell Response. J. Exp. Med. 200, 1267–1278. doi: 10.1084/jem.20040500
Rius, J., Guma, M., Schachtrup, C., Akassoglou, K., Zinkernagel, A. S., Nizet, V., et al. (2008). NF-kappaB Links Innate Immunity to the Hypoxic Response Through Transcriptional Regulation of HIF-1alpha. Nature 453, 807–811. doi: 10.1038/nature06905
Rodriguez-Prados, J. C., Traves, P. G., Cuenca, J., Rico, D., Aragones, J., Martin-Sanz, P., et al. (2010). Substrate Fate in Activated Macrophages: A Comparison Between Innate, Classic, and Alternative Activation. J. Immunol. 185, 605–614. doi: 10.4049/jimmunol.0901698
Schäfer, W., Schmidt, T., Cordsmeier, A., Borges, V., Beare, P. A., Pechstein, J., et al. (2020). The Anti-Apoptotic Coxiella Burnetii Effector Protein AnkG is a Strain Specific Virulence Factor. Sci. Rep. 10, 15396. doi: 10.1038/s41598-020-72340-9
Semenza, G. L. (2014). Oxygen Sensing, Hypoxia-Inducible Factors, and Disease Pathophysiology. Annu. Rev. Pathol. 9, 47–71. doi: 10.1146/annurev-pathol-012513-104720
Sershen, C. L., Plimpton, S. J., May, E. E. (2016). Oxygen Modulates the Effectiveness of Granuloma Mediated Host Response to Mycobacterium Tuberculosis: A Multiscale Computational Biology Approach. Front. Cell Infect. Microbiol. 6, 6. doi: 10.3389/fcimb.2016.00006
Siegert, I., Schodel, J., Nairz, M., Schatz, V., Dettmer, K., Dick, C., et al. (2015). Ferritin-Mediated Iron Sequestration Stabilizes Hypoxia-Inducible Factor-1alpha Upon LPS Activation in the Presence of Ample Oxygen. Cell Rep. 13, 2048–2055. doi: 10.1016/j.celrep.2015.11.005
Strowitzki, M. J., Cummins, E. P., Taylor, C. T. (2019). Protein Hydroxylation by Hypoxia-Inducible Factor (HIF) Hydroxylases: Unique or Ubiquitous? Cells 8, 384. doi: 10.3390/cells8050384
Sukocheva, O. A., Manavis, J., Kok, T. W., Turra, M., Izzo, A., Blumbergs, P., et al. (2016). Coxiella Burnetii Dormancy in a Fatal Ten-Year Multisystem Dysfunctional Illness: Case Report. BMC Infect. Dis. 16, 165. doi: 10.1186/s12879-016-1497-z
Tannahill, G. M., Curtis, A. M., Adamik, J., Palsson-McDermott, E. M., McGettrick, A. F., Goel, G., et al. (2013). Succinate is an Inflammatory Signal That Induces IL-1beta Through HIF-1alpha. Nature 496, 238–242. doi: 10.1038/nature11986
Thomas, D. R., Newton, P., Lau, N., Newton, H. J. (2020). Interfering With Autophagy: The Opposing Strategies Deployed by Legionella Pneumophila and Coxiella Burnetii Effector Proteins. Front. Cell Infect. Microbiol. 10, 599762. doi: 10.3389/fcimb.2020.599762
Van den Brom, R., van Engelen, E., Roest, H. I., van der Hoek, W., Vellema, P. (2015). Coxiella Burnetii Infections in Sheep or Goats: An Opinionated Review. Vet. Microbiol. 181, 119–129. doi: 10.1016/j.vetmic.2015.07.011
Voth, D. E., Howe, D., Heinzen, R. A. (2007). Coxiella Burnetii Inhibits Apoptosis in Human THP-1 Cells and Monkey Primary Alveolar Macrophages. Infect. Immun. 75, 4263–4271. doi: 10.1128/IAI.00594-07
Wang, G. L., Jiang, B. H., Rue, E. A., Semenza, G. L. (1995). Hypoxia-Inducible Factor 1 is a Basic-Helix-Loop-Helix-PAS Heterodimer Regulated by Cellular O2 Tension. Proc. Natl. Acad. Sci. U. S. A. 92, 5510–5514. doi: 10.1073/pnas.92.12.5510
Werth, N., Beerlage, C., Rosenberger, C., Yazdi, A. S., Edelmann, M., Amr, A., et al. (2010). Activation of Hypoxia Inducible Factor 1 is a General Phenomenon in Infections With Human Pathogens. PLoS One 5, e11576. doi: 10.1371/journal.pone.0011576
Zamboni, D. S., Rabinovitch, M. (2003). Nitric Oxide Partially Controls Coxiella Burnetii Phase II Infection in Mouse Primary Macrophages. Infect. Immun. 71, 1225–1233. doi: 10.1128/IAI.71.3.1225-1233.2003
Zamboni, D. S., Campos, M. A., Torrecilhas, A. C., Kiss, K., Samuel, J. E., Golenbock, D. T., et al. (2004). Stimulation of Toll-Like Receptor 2 By Coxiella burnetii Is Required for Macrophage Production of Pro-Inflammatory Cytokines and Resistance to Infection. J. Biol. Chem. 279, 54405–54415. doi: 10.1074/jbc.M410340200
Keywords: Coxiella burnetii, HIF1α, T4SS, metabolism, apoptosis, inflammation
Citation: Hayek I, Szperlinski M and Lührmann A (2022) Coxiella burnetii Affects HIF1α Accumulation and HIF1α Target Gene Expression. Front. Cell. Infect. Microbiol. 12:867689. doi: 10.3389/fcimb.2022.867689
Received: 01 February 2022; Accepted: 21 April 2022;
Published: 09 June 2022.
Edited by:
Luís Jaime Mota, NOVA School of Science and Technology, PortugalReviewed by:
Anders Omsland, Washington State University, United StatesCopyright © 2022 Hayek, Szperlinski and Lührmann. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Anja Lührmann, YW5qYS5sdWVocm1hbm5AdWstZXJsYW5nZW4uZGU=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.