 Pan Lu
Pan Lu Jialiang Chen
Jialiang Chen Zhenpeng Li
Zhenpeng Li Jingyun Zhang
Jingyun Zhang Biao Kan
Biao Kan Bo Pang
Bo Pang
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Cell. Infect. Microbiol. , 31 March 2022
Sec. Clinical Microbiology
Volume 12 - 2022 | https://doi.org/10.3389/fcimb.2022.863435
This article is part of the Research Topic Point-of-Care Testing for Infectious and Foodborne Pathogens View all 16 articles
There is a growing demand for rapid, sensitive, field-deployable nucleic acid tests for cholera, which usually occurs in rural areas. In this study, we developed a Cas12a-assisted rapid isothermal detection (CARID) system for the detection of toxigenic V. cholerae serogroups O1 and O139 by combining recombinase-aided amplification and CRISPR-Cas (clustered regularly interspaced short palindromic repeats and CRISPR-associated proteins). The results can be determined by fluorescence signal and visualized by lateral flow dipstick. We identified 154 V. cholerae strains and 129 strains of other intestinal diarrheagenic bacteria with a 100% coincidence rate. The limit of detection of CARID was 20 copies/reaction of V. cholerae genomic DNA, which is comparable to that of polymerase chain reaction (PCR) and qPCR. Multiple-CARID was also established for efficiency and economic considerations with an acceptable decrease in sensitivity. Simulated sample tests showed that CARID is suitable for complex samples. In conclusion, CARID is a rapid, sensitive, economically efficient, and portable method for the detection of V. cholerae, which makes it suitable for field responses to cholera.
Cholera remains a threat to public health in many countries with poor sanitation or that are short of safe water (Deen et al., 2020). It has been estimated that there are roughly 1.3 to 4.0 million cases of cholera annually, with 21,000 to 143,000 deaths worldwide (Ali et al., 2015). Vibrio cholerae, the causative agent of cholera, can be transmitted through contaminated water and/or food (Sack et al., 2004). Cholera toxin (CT), an important pathogenic factor of V. cholerae, is encoded by ctxA and ctxB, and V. cholerae that carry ctxA and ctxB are referred to as toxigenic strains (Bharati and Ganguly, 2011). To date, only the toxigenic serogroups O1 and O139 are known to have caused epidemics and pandemics of cholera, even though there are more than 200 serogroups of V. cholerae (Harris et al., 2012; Chowdhury et al., 2017).
Early detection and confirmation of cholera cases are critical for rapid implementation of interventions. However, traditional culture methods for isolation and identification of V. cholerae can take three days or more and require extensive laboratory infrastructure and experienced staff (Ramamurthy et al., 2020). While molecular methods such as polymerase chain reaction (PCR) and quantitative real-time PCR (qPCR) have accelerated the diagnosis process, they rely heavily on expensive instruments and require elaborate experimental conditions, which are often difficult to obtain in rural areas. Therefore, rapid and point-of-care testing methods are essential for timely cholera detection and control.
CRISPR-Cas (clustered regularly interspaced short palindromic repeats and CRISPR-associated proteins) systems are adaptive immune systems consisting of Cas effector proteins and CRISPR RNAs (crRNAs) that are widely distributed in archaea and bacteria (Li and Du, 2011; Terns and Terns, 2011). CRISPR-Cas relies on crRNAs for sequence-specific detection and silencing of foreign nucleic acids, thereby protecting organisms from viruses and phages (Barrangou et al., 2007; Al-Attar et al., 2011; Bhaya et al., 2011; Wiedenheft et al., 2012). CRISPR-Cas12a systems can cleave single-stranded DNA (ssDNA) indiscriminately when bound to target sequences under the guidance of crRNA in vitro (Chen et al., 2018); therefore, this principle has been used in the specific detection of pathogens (Chen et al., 2018; Gootenberg et al., 2018; Li et al., 2018). For double-stranded target DNA activators, the prerequisite for Cas12a to effectively cleave non-target DNA is identification of a short T-rich (5 ‘-TTTN-3’) protospacer-adjacent motif (PAM) in the target strand (Zetsche et al., 2015; Chen et al., 2018), which provides theoretical guidance for the design of crRNA.
Because the detection ability of CRISPR-Cas systems relies on the template provided, it is usually combined with several amplification methods, such as recombinase polymerase amplification (RPA) (Chen et al., 2018), PCR (Li et al., 2018) and recombinase-aided amplification (RAA) (Xiao et al., 2021). RAA is a sensitive, rapid, low-cost isothermal amplification technique that can be completed within 15–30 min at 37 to 42°C without using large, expensive instruments (Shen et al., 2019; Wang et al., 2020).
Based on the detection principles of CRISPR-Cas12a and RAA, we developed CARID (Cas12a-assisted rapid isothermal detection) for the rapid visual detection of toxigenic V. cholerae serogroups O1 and O139 (Figure 1). Using simulated samples, we demonstrated that CARID could be completed within an hour, and that the results could be visualized through fluorescent signals or lateral flow dipstick (LFD). The method established in this study can detect the ctxA gene and identify the O1 and O139 serogroups with a specificity of 100%. The minimum detectable genomic concentration is 20 copies/reaction, and the sensitivity is comparable to that of PCR and qPCR.
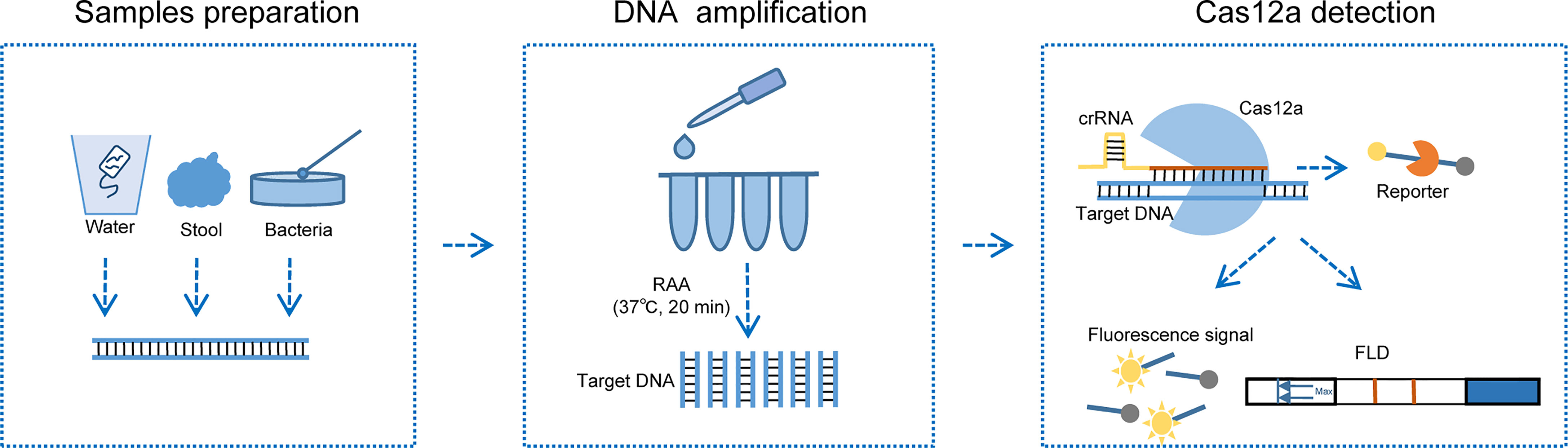
Figure 1 Schematic diagram of CARID detecting V. cholerae. Nucleic acid can be extracted from V. cholerae contaminated river water samples, stool, and clones cultured on LB agar plates. RAA amplifies the template at 37°C for 20 min, after which it is added to the Cas12a reaction mixture and incubated at 37°C for 30 min. The fluorescent signal and lateral flow dipstick can be used to determine the results.
A total of 154 strains of V. cholerae were included in this study, namely, 112 strains of V. cholerae carrying cholera toxin (22 strains of serogroup O1 and 90 strains of serogroup O139) and 42 strains of V. cholerae without CT (22 strains of serogroup O1 and 20 strains of serogroup O139). These strains were isolated from cholera patients and environmental samples. Oral consent was obtained from each eligible patient (or the legal guardian of the patient, if the patient is <18 years of age). Other intestinal diarrheagenic bacteria, namely, 20 strains of Vibrio parahaemolyticus, 14 strains of Vibrio alginolyticus, 13 strains of Vibrio mimicus, 14 strains of Vibrio furnis, 10 strains of Vibrio fluvialis, 14 strains of Escherichia coli, 15 strains of Salmonella enteritidis, 14 strains of Shigella, and 15 strains of Yersinia enterocolitica were also evaluated in this study.
To prepare recombinant LbCas12a proteins (Zetsche et al., 2015), protein sequences were codon-optimized for E. coli expression. Synthetic oligonucleotide fragments were then cloned into bacterial protein expression vector pET-28a and transfected into E. coli BL21. E. coli cells were first grown with 50 µg/ml Kanamycin at 37°C until OD600 reached 0.5–0.6, followed by incubation with 0.5 mM IPTG at 21°C for 16 h to induce LbCas12a expression. The cultures were then lysed by sonication, filtered through 0.22-µm filters, and applied to a nickel column (Ni-NTA agarose, Qiagen, Germany) for protein purification. The eluted protein was stored in a storage solution (500 mM NaCl, 20 mM Sodium Acetate, 0.1 mM EDTA, 0.1 mM DTT, 50% Glycerol). Purified LbCas12a proteins were examined by SDS-PAGE and protein concentrations were determined using a Pierce™ BCA Protein Assay Kit (Thermo Scientific, Shanghai, China).
DNA was extracted from bacterial colonies cultured overnight using the boiling method, after which the extracted nucleic acid was dissolved in 50 µl ddH2O. Target DNA was amplified using an RAA isothermal amplification kit (Qitian, Jiangsu, China) followed by Cas12a detection. Briefly, 50 µl RAA reactions contained 2 µl template, 0.4 µM forward and reverse primer, 1× reaction buffer, nuclease-free water, lyophilized powder reaction unit, and 2.5 µl of 280 mM magnesium acetate. The RAA mix was incubated at 37°C for 20 min. For the fluorescence assay, the amplification product was added to CRISPR reaction mix consisting of 1× CRISPR reaction buffer (10 mM Tris–HCl, 10 mM MgCl2, pH = 8.0), 100 nM crRNA, 100 nM Cas12a, and 200 nM ssDNA reporter (5’-6FAM-TTATT-BHQI-3’). This final reaction was incubated at 37°C and monitored for fluorescence for 30 min using a Gentier 96E qPCR machine (TIANLONG, Xian, China). Samples that produced fluorescence at levels 2.0-fold or greater above the NC were considered positive. For the LFD detection reactions, the ssDNA reporter was replaced with 5’-6FAM-TTATT-Biotin-3’. The final reaction system was incubated at 37°C for 30 min, after which the reaction products were diluted with ddH2O and then inserted into the LFD (Tiosbio, Beijing, China) to read the results. A band on the T test line can be judged as positive.
To evaluate the sensitivity of CARID, V. cholerae genomic DNA was extracted using a Wizard Genomic DNA Purification Kit (Promega, Madison, WI, USA) and diluted with ddH2O to concentrations of 107 to 1 copies/µl (DNA copies number were determined using following formula: (6.02 × 1023) × (ng/µl × 10−9)/(DNA length × 660) = copies/µl). Diluted samples were then used as templates for CARID detection, PCR, and qPCR. PCR was conducted using TaKaRa Premix Taq™ (TaKaRa, Dalian, China) and a 50-µL reaction system that contained 1×Premix Taq, 0.4 µM forward primer, 0.4 µM reverse primer, and 2 µl of template. The reaction conditions were 95°C for 5 min, followed by 40 cycles of 95°C for 10 s, 50°C for 30 s, and 72°C for 30 s, and then 72°C for 10 min. A qPCR assay was performed in a 50-µl volume using Premix Ex TaqTM (TaKaRa, Dalian, China). The reaction system contained 1× Premix Ex Taq (Probe qPCR), 0.4 µM forward primer, 0.4 µM reverse primer, 0.4 µM probe, and 2 µl of template. The reaction conditions for qPCR were 95°C for 30 s, followed by 40 cycles of 95°C for 10 s and 60°C for 30 s. The amplification process was conducted using a Gentier 96E qPCR machine (Tianlong, Xian, China).
One toxigenic serogroup O1 and one toxigenic serogroup O139 of V. cholerae was cultured on nutrient agar overnight. Individual colonies were then selected and incubated in Luria–Bertani (LB) broth (Oxoid, Basingstoke, UK) at 37°C with shaking (200 rpm) to a concentration of OD600 = 1.0 (approximately 1 × 109 colony forming units (CFU)/ml), after which the bacteria were gradient-diluted into a bacterial suspension in PBS with a concentration of 109–10 CFU/ml. Simulated water samples with bacterial concentrations of 107–10−1 CFU/ml were prepared by adding 50 µl of bacterial suspension to 5 ml of pre-mixed and sterilized river water (4.5 ml of river water and 0.5 ml of 10× 1% sodium chloride alkaline peptone water). The simulated water samples were then shaken at 200 rpm and 37°C for 4 h, and 1 ml of bacterial suspension was taken every hour for nucleic acid extraction and CARID detection as described above. Fluorescent signal detection and LFD were then used to visualize the results.
The bacterial deposits of 109–10 CFU were mixed with 250-mg stool samples of healthy adults, after which nucleic acid was extracted using a QIAamp® Power Fecal® Pro DNA Kit (Qiagen, Germany) according to the manufacturer instructions. The extracted nucleic acid was then dissolved in 50 µl ddH2O, after which 2-µl aliquots were used as templates for detection of the ctxA and O-PS specific genes. Fluorescent signal detection and LFD were used to visualize the results.
The multiple RAA system included 1× reaction buffer, 0.4 µM forward and reverse primers for V. cholerae ctxA, serogroups O1 and O139 O-PS specific genes, and the RAA reagent. After preparing the system, 2 µl of DNA template and 2.5 µl of 280 mM magnesium acetate solution were added and samples were then incubated at 37°C for 30 min. CrRNA was then used to detect ctxA, O1, and O139 O-PS specific genes in the product. Fluorescent signal detection and LFD were used to visualize the results.
The GraphPad Prism 9 software was used for statistical analyses and preparation of figures. Analyses were based on three replicate values. Unpaired t-tests were used to identify differences between two groups.
Primers and crRNA were designed according to the conserved regions of ctxA, rfb-O1, and rfb-O139 genes of V. cholerae. Conserved fragments containing the PAM sequence (5’-TTTN-3’) were selected as the target to design primers and guide sequences. CrRNA contains a conserved stem-loop structure that is necessary for forming the Cas12a–crRNA complex, and the adaptive crRNA stem-loop sequence of Cas12a is different for each Cas12a type (Zetsche et al., 2015). After the guide sequence was designed, a stem-loop sequence homologous to LbCas12a was added to its 5’ end to ensure that the protein had strong cleavage ability. The primers and RNA sequences are not published herein because they are currently being patented.
The LbCas12a protein is composed of 1,228 amino acids with a molecular weight of 143,035 g/mol. The results of SDS-PAGE analysis of purified LbCas12a protein were consistent with the expected size of the target protein (Figure 2). To verify the protein activity of LbCas12a, the target DNA (ctxA: 691bp, O1: 1033bp, O139: 965bp) was amplified by PCR and then added into the CRISPR reaction system to detect its fluorescent signal. The CRISPR reaction could detect fluorescent signals in the absence of buffer, but the fluorescence value was much lower than when the buffer is included (Figure 3A). This indicates that buffer significantly enhanced the accessory cleavage ability of the protein. The LbCas12a protein, crRNA, reporter, and target DNA were obligate for successful detection by the fluorescence method or the LFD method (Figures 3A, B). The CRISPR reaction products were detected by electrophoresis. The LbCas12a protein cleaved the target DNA successfully under the guidance of crRNA. Taken together, these findings indicated that ctxA-crRNA, O1-crRNA, and O139-crRNA were suitable for detection of the target DNA (Figure 3C).
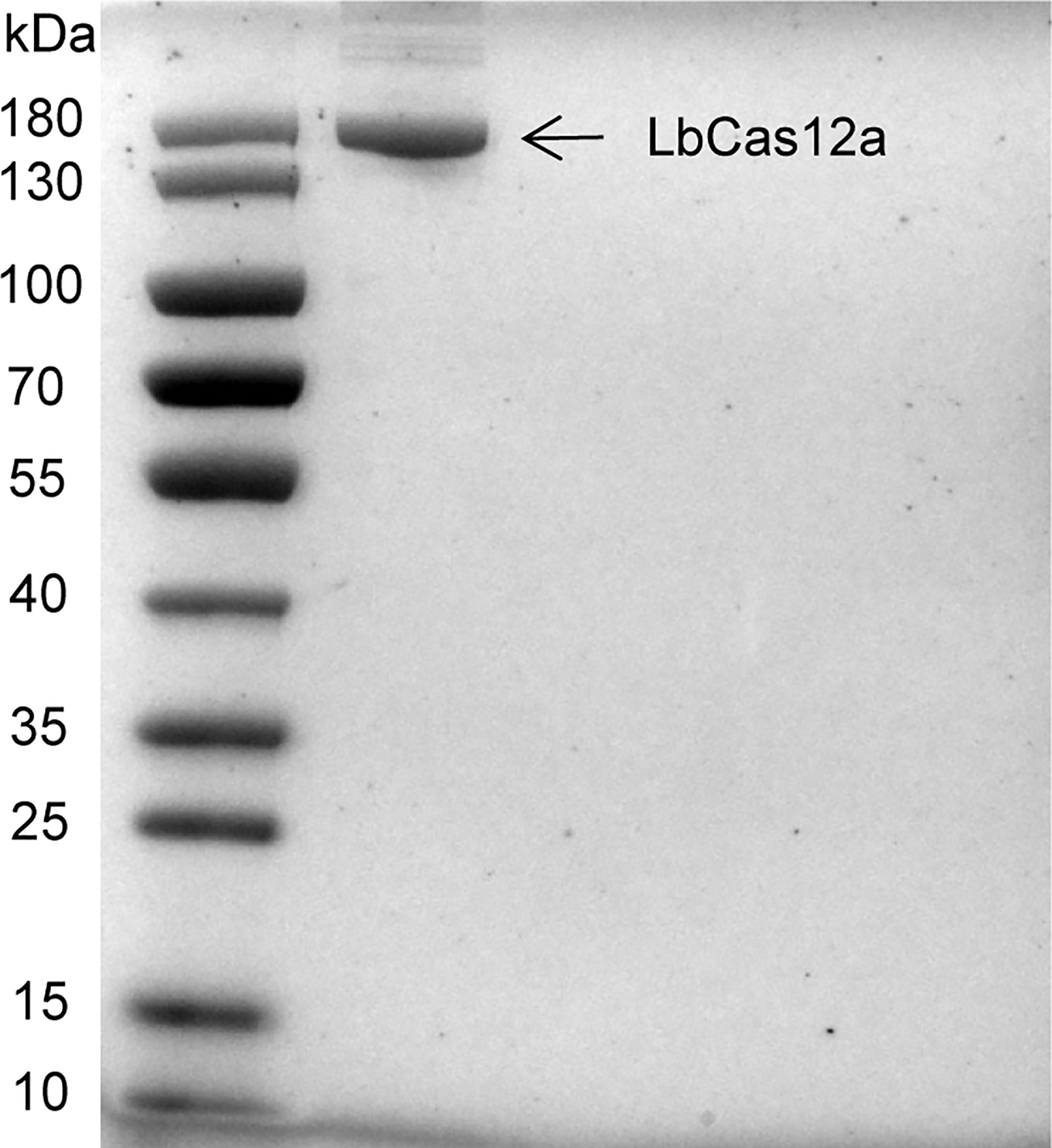
Figure 2 Purification of Cas12 proteins. SDS-PAGE gel of LbCas12 proteins was used in this study.
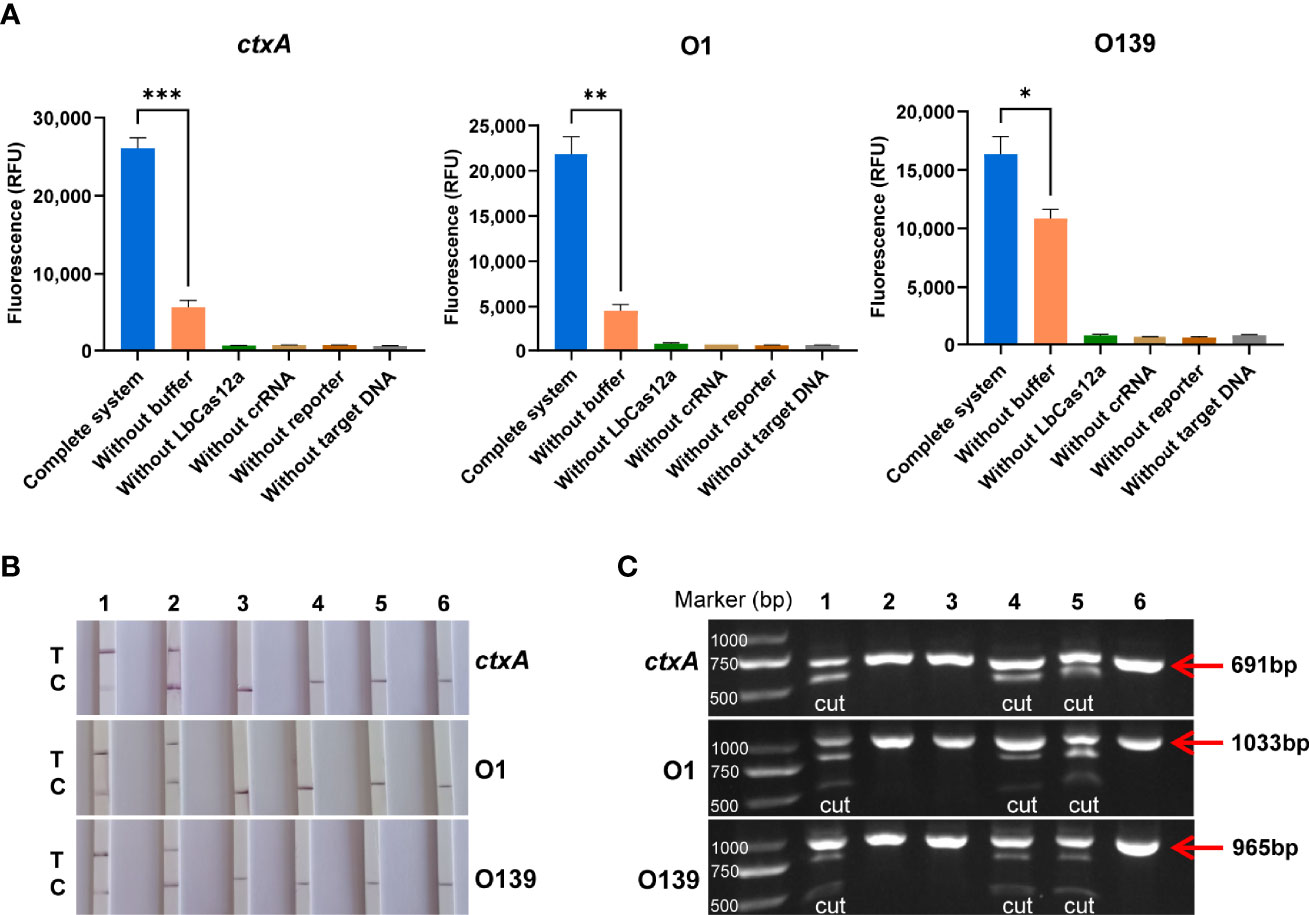
Figure 3 Validation of LbCas12a protein activity and crRNA adaptation. (A) Fluorescence value of the complete Cas12a system, and the absence of buffer, Cas12a, crRNA, reporter or target DNA (n = 3 technical replicates; two-tailed Student’s t-test; *p < 0.05; **p < 0.01; ***p < 0.001; bars represent the mean ± SEM). (B) LFD results of the complete Cas12a system and the absence of buffer, Cas12a, crRNA, reporter, or target DNA (T is the test band and C is the control band). 1 is the complete system, 2, is the system without buffer, and 3–6 are systems without LbCas12a protein, crRNA, reporter, and target DNA, respectively. (C) Cas12a cutting target DNA was detected by agarose gel electrophoresis. 1 is the complete system, 2–5 are systems without Cas12a, crRNA, buffer, and reporter, respectively, and 6 is the target DNA alone.
A total of 154 strains of V. cholerae were used to evaluate the specificity of CARID, and 112 toxigenic V. cholerae and 42 nontoxigenic strains were successfully identified. Forty-four serogroup O1 strains and 110 serogroup O139 strains were also successfully identified. The identification rate was 100%, which was consistent with whole-genome sequencing data. The RAA primers and crRNA of ctxA, O1, and O139 were used to detect other diarrheagenic bacteria, namely, E. coli, Salmonella enteritidis, Shigella, Y. enterocolitica and other Vibrios to confirm the specificity of the method. There were no positive results for these bacteria, and the coincidence rate was 100% (Table 1). The results of the fluorescence assay were consistent with those of the LFD.
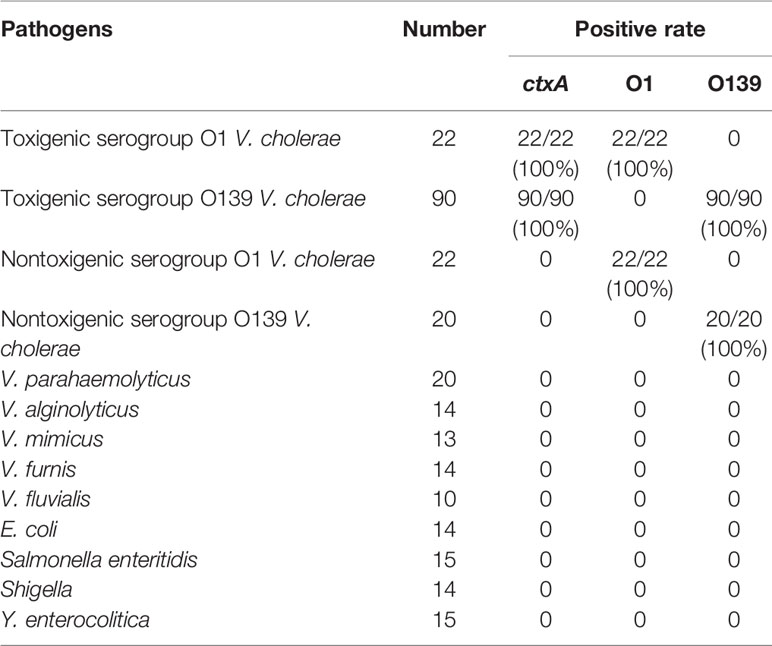
Table 1 Specificity of CARID for detection of V. cholerae and other intestinal diarrheagenic bacteria.
A series of samples containing V. cholerae genomic DNA that had been diluted from 107 to 1 copies/µl were analyzed by CARID, PCR, and qPCR. Furthermore, the primers of RAA were used in the PCR experiment. The results showed that CARID could detect 20 copies/reaction (Figure 4A). However, only weakly positive or negative results were observed when samples containing 20 copies/reaction of genome DNA were tested by LFD (Figure 4B). When the genome concentration was 20 copies/reaction, the CT values of ctxA, O1, and O139 O-PS specific genes were 34.15 ± 0.92, 32.63 ± 0.40, and 34.19 ± 1.08 (mean ± SD), respectively, for qPCR (Figure 4C). Moreover, only weak bands were amplified by PCR at a genomic concentration of 200 copies/reaction (Figure 4D).
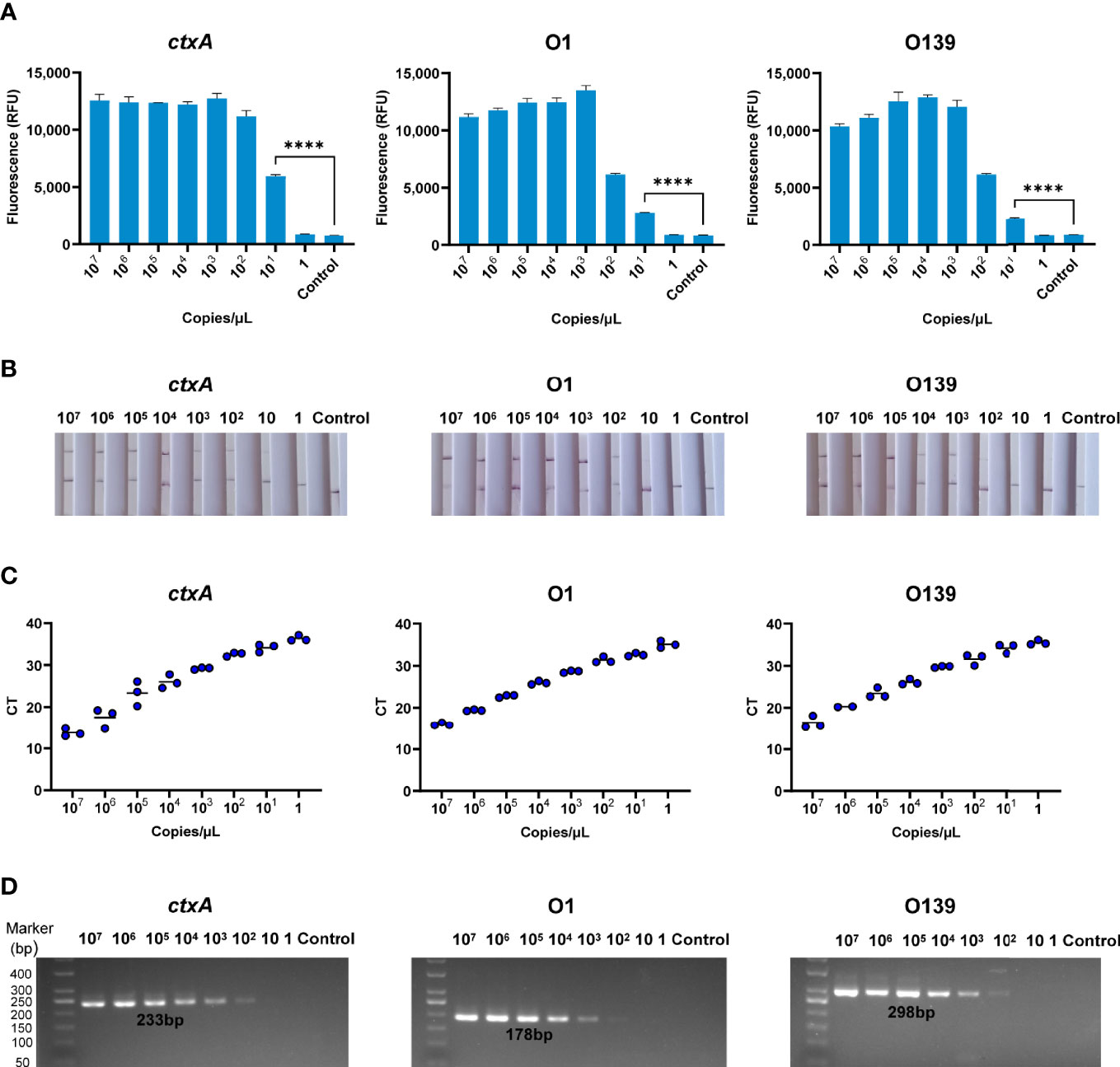
Figure 4 Sensitivity of CARID for detection of V. cholerae. (A) The 107–1 copies/μl V. cholerae templates were detected by fluorescence method of CARID (n = 3 technical replicates; two-tailed Student’s t-test; ****p <0.0001; bars represent the mean ± SEM). (B) The 107–1 copies/μl V. cholerae templates were detected by LFD. (C) QPCR detected ctxA, O1, and O139 genes from 107–1 copies/μl V. cholerae templates. (D) PCR detected ctxA, O1, and O139 genes from 107–1 copies/μl V. cholerae templates.
A series of simulated river water samples containing 107–10−1 CFU/ml toxigenic V. cholerae was used to evaluate the limit of detection (LOD) of CARID. For the toxigenic serogroup O1, positive results for ctxA and O1 O-PS specific genes were obtained in samples containing 104 CFU/ml of V. cholerae. Samples containing 103 CFU/ml could be detected after 1 h of culture, while 10 CFU/ml could be detected after 2 h of culture, and 1 CFU/ml could be detected after 4 h. In contrast to ctxA and O1, O139 O-PS specific genes could be detected in river water containing 105 CFU/ml before culture and 102 CFU/ml after 2 h of culture (Figure 5). Overall, river water samples containing 1 CFU/ml of V. cholerae could be successfully detected by CARID after culture for 4 h with alkaline peptone water.
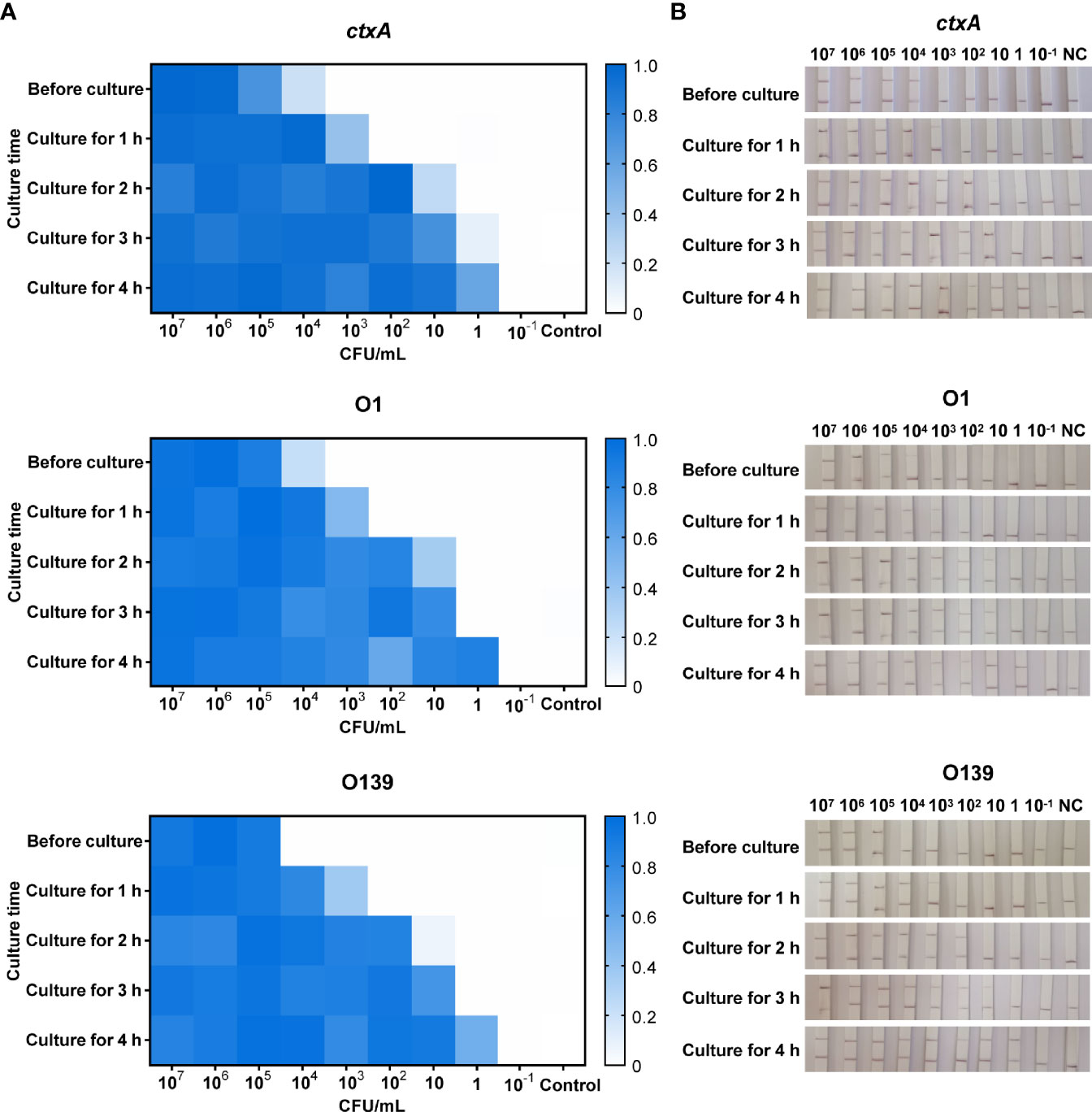
Figure 5 CARID detection of simulated river water samples. (A) Simulated water samples were tested by fluorescent CARID before culture and after culture for 1–4 h. The fluorescent signal of each sample was normalized against the negative control of the same batch. The heatmap represents normalized mean fluorescence values (n = 3 technical replicates). (B) Simulated water samples were tested by the LFD method of CARID before culture and after culture for 1–4 h.
Application of CARID to stool samples containing 109–10 CFU/250 mg bacteria revealed the ctxA gene could be detected at 103 CFU/250 mg bacteria (equivalent to 4 × 103 CFU/g). In addition, the O1 and O139 genes could be detected from simulated stool samples at 4 × 104 CFU/g bacteria (Figure 6).
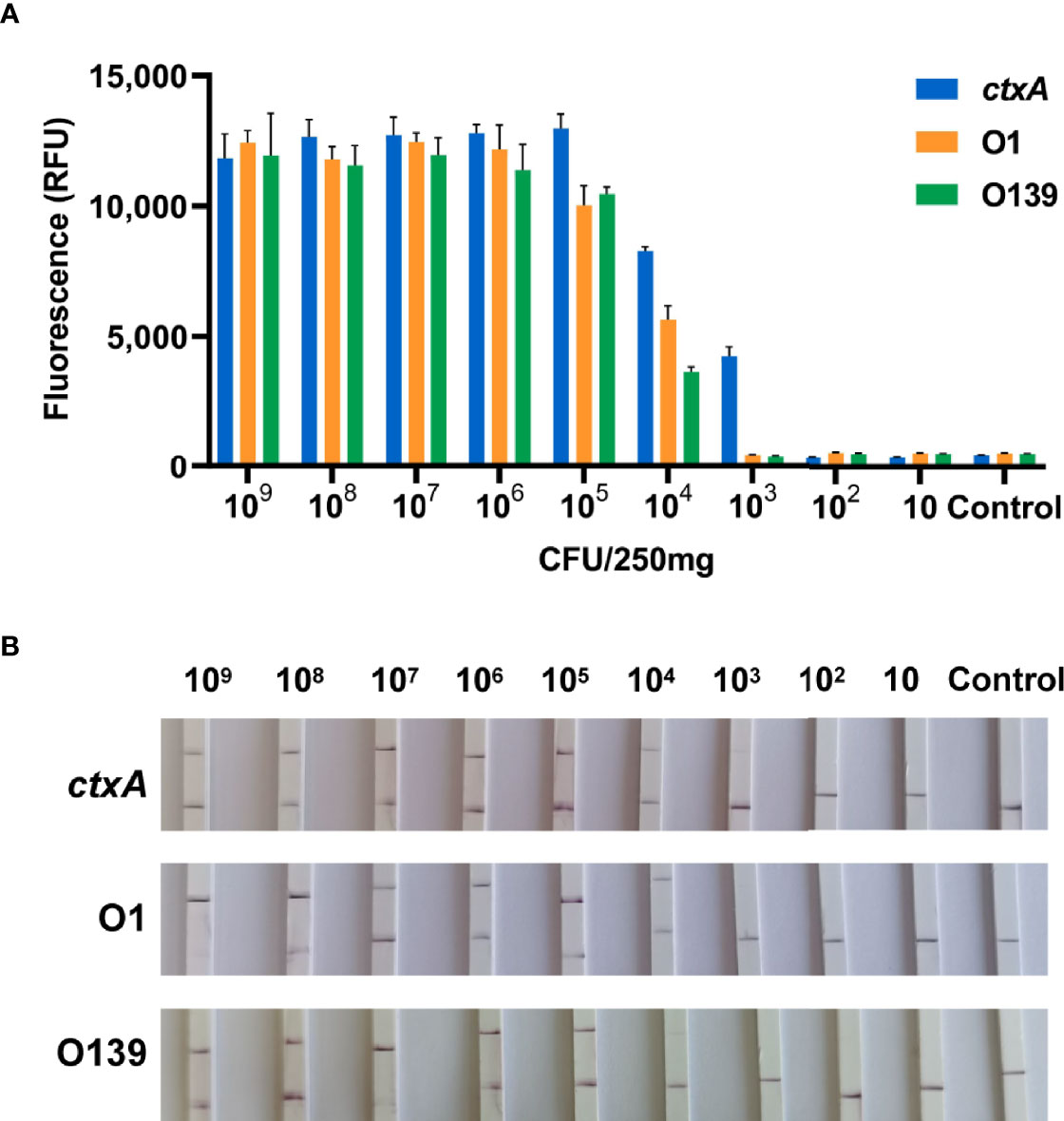
Figure 6 CARID detection of simulated stool samples. (A) Templates extracted from stool containing 109–10 CFU/250 mg V. cholerae were analyzed by fluorescent CARID (n = 3 technical replicates; bars represent the mean ± SEM). (B) Templates extracted from stool containing 109–10 CFU/250 mg V. cholerae were analyzed by the LFD method.
Owing to efficiency and economic considerations, we set up multiple-CARID for simultaneous detection of ctxA and O-PS specific genes. Using the protocol described in the Materials and Methods section, a nucleic acid mixture of toxigenic V. cholerae serogroups O1 and O139 was analyzed, and positive results were obtained using all three CRISPR reactions (Figures 7A, B). To evaluate the LOD of multiple-CARID, 103–10 copies/µl of V. cholerae genomic DNA was examined, and 10 repeats were performed for each dose. The lowest dose point with a 100% detection rate was taken as the LOD. For the fluorescence assay, ctxA and O1 O-PS specific genes showed higher sensitivity than O139, with 200 copies/reaction of V. cholerae DNA consistently detected in 10 out of 10 tests, and 20 copies/reaction detected in 7 out of 10 tests. However, an O139 O-PS specific gene was detected in 9 out of 10 tests of samples containing 200 copies/reaction of V. cholerae DNA (Figure 7C). When LFD analysis was conducted, ctxA and O1 O-PS specific genes were detected from samples containing 200 copies/reaction of V. cholerae DNA with clear bands in all 10 replicates. In comparison, O139 O-PS specific genes were detected in samples containing 2,000 copies/reaction of V. cholerae DNA with 100% detection rates Figure 7D.
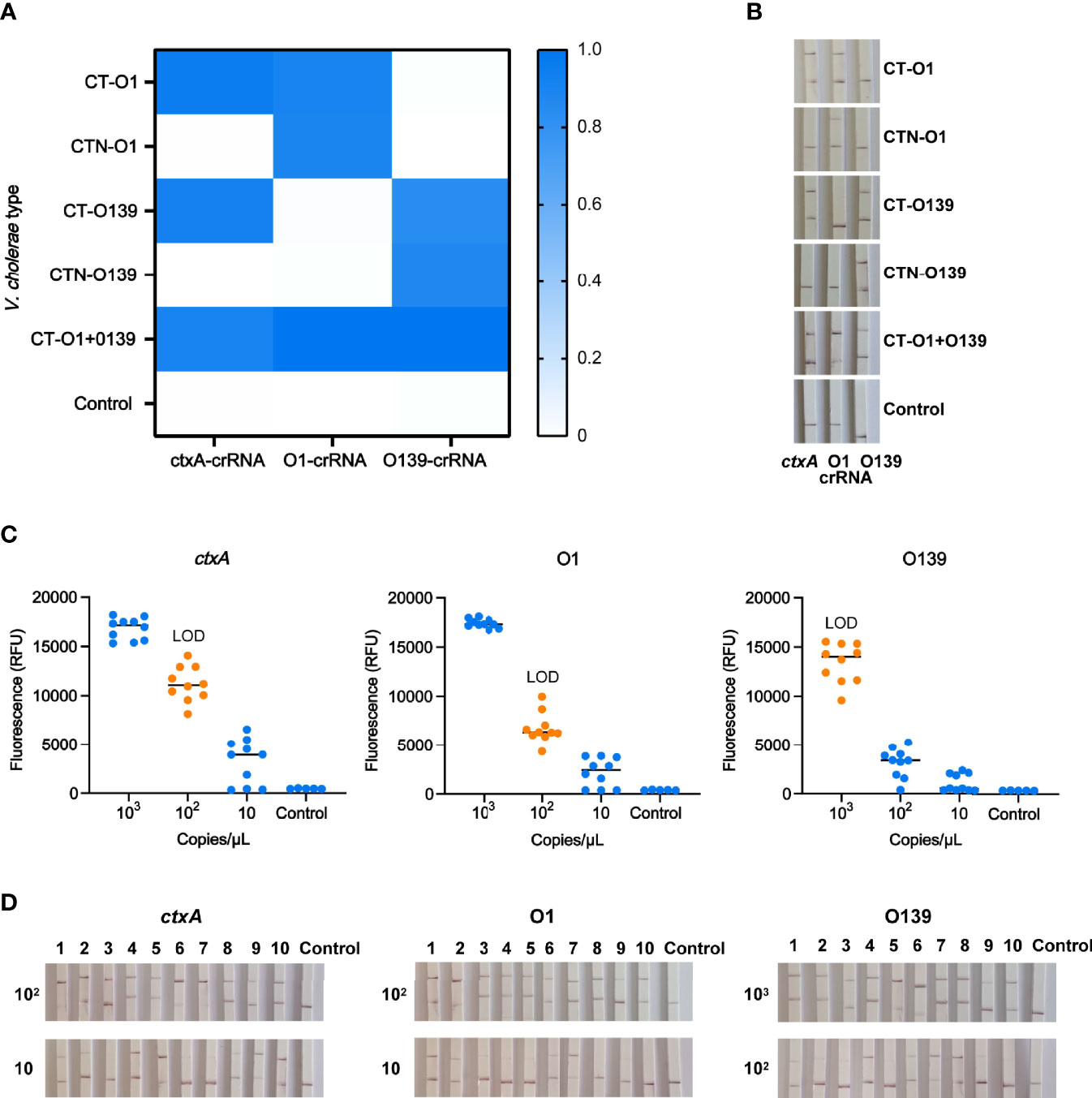
Figure 7 Identification and typing of V. cholerae with multiple-CARID. (A) Fluorescent CARID for detection of ctxA and identification of serogroups O1 and O139. The heatmap represents normalized mean fluorescence values (n = 3 technical replicates). CT-O1 refers to V. cholerae serogroup O1 containing cholera toxin; CTN-O1 refers to V. cholerae serogroup O1 without cholera toxin. CT-O139 refers to O139 containing cholera toxin; CTN-O139 refers to O139 without cholera toxin. CT-O1 + O139 refers to mixed V. cholerae serogroups O1 and O139 CT. (B) LFD method of CARID for detection of ctxA and identification of serogroups O1 and O139. (C) LOD of the fluorescent method of multiple-CARID. Ten repetitions were performed. (D) LOD of the LFD method of multiple-CARID.
Cholera often occurs in areas with poor sanitation, limited resources, and low testing capabilities, and V. cholerae serogroups O1 and O139 are the predominant agents of cholera epidemics (Deen et al., 2020). Accordingly, rapid field detection and pathogen identification are critical to controlling cholera. In the present study, we developed a visual detection method for toxigenic V. cholerae under isothermal conditions based on the principle of CRISPR-Cas12a. The developed method successfully detected toxigenic V. cholerae and identified serogroups O1 and O139 in less than 1 h, which is ideal for rapid responses in cholera control. The buffer can significantly improve the fluorescence value, which supported that the addition of a buffer with Mg2+ significantly improved the reaction efficiency of Cas12a (Wu et al., 2020). To confirm that the system works effectively, we set the detection time of the Cas12a reaction to 30 min; however, 10 min was found to be sufficient to produce robust results. Nevertheless, our findings indicate that the efficiency of the test could be improved in the future.
The specificity of CARID for detection of V. cholerae and other intestinal diarrheagenic bacteria was comparable to those of other methods. The detection limits were 20 copies/reaction and 200 copies/reaction, respectively, for fluorescent signal and LFD detection, which are comparable to the detection limits of PCR and qPCR. In areas of cholera outbreaks, polluted water is the main infectious medium (Rafique et al., 2016). In the present study, CARID could detect the target pathogens from simulated river water samples with the concentration of 104–105 CFU/ml V. cholerae, while the infection dose of Vibrio cholerae in humans is 108–1011 cells (Nelson et al., 2009). Our results showed that, after 4 h of enrichment, V. cholerae at an initial concentration of 1 CFU/ml could reach the detection limit of CARID. Therefore, for water samples, we recommend an enrichment step, which can improve the detection rate. Patients with symptoms of cholera usually shed 107–109 cells of Vibrio cholerae per gram of stool (Nelson et al., 2009), and the detection limit of CARID is 4 × 103 CFU/g of stool. Therefore, these findings indicate that CARID is sensitive enough for application in cholera control and for analysis of surveillance samples. The LOD of simultaneous detection of ctxA, O1, and O139 serogroup-specific genes in a single amplification was 200–2,000 copies/reaction, which was comparable to single amplifications. Hence, the multiplex protocol is both time-saving and economically efficient.
A major concern of the CRISPR-Cas-mediated detection protocol is that cross-contamination transfer of amplicons can lead to aerosol contamination; therefore, we recommend that the configuration system, addition of templates, and transfer of amplification products be conducted in different experimental areas. For example, in SARS-COV-2 detection with CRISPR-Cas12a, Chen et al. (2020) effectively avoided aerosol contamination of the amplicon by covering the isothermal amplification reaction solution with mineral oil and pre-adding CRISPR reagents to the tube lid, which avoided opening of the tube cap during the entire process Chen et al. (2020). In addition, lab-on-a-chip and microfluidics can also be used to develop portable, sensitive, and cost-effective biosensing systems for Cas12a-mediated detection (Khizar et al., 2020; Ramachandran et al., 2020). In such systems, the RAA and CRISPR reagents can be integrated onto a chip in advance, which enables the application of CRISPR-Cas-mediated steps in various settings, namely, clinics, mobile testing stations, and rural areas.
The original contributions presented in the study are included in the article/supplementary material. Further inquiries can be directed to the corresponding author.
BP conceived and designed the study. PL and JC contributed to the experiment. ZhenL contributed to the analysis of bioinformatics. PL and BP contributed to writing the manuscript. JZ, ZheL, and BK contributed to the design of the experiment. All authors listed have made a substantial, direct, and intellectual contribution to the work and approved it for publication.
This work was supported by the National Science and Technology Major Project (2018ZX10714-002).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
We thank Professor Weili Liang and Meiying Yan for their helpful discussion.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fcimb.2022.863435/full#supplementary-material
Al-Attar, S., Westra, E. R., van der Oost, J., Brouns, S. J. J. (2011). Clustered Regularly Interspaced Short Palindromic Repeats (CRISPRs): The Hallmark of an Ingenious Antiviral Defense Mechanism in Prokaryotes. Biol. Chem. 392 (4), 277–289. doi: 10.1515/bc.2011.042
Ali, M., Nelson, A. R., Lopez, A. L., Sack, D. A. (2015). Updated Global Burden of Cholera in Endemic Countries. PloS Negl. Trop. Dis. 9, e0003832. doi: 10.1371/journal.pntd.0003832
Barrangou, R., Fremaux, C., Deveau, H., Richards, M., Boyaval, P., Moineau, S., et al. (2007). CRISPR Provides Acquired Resistance Against Viruses in Prokaryotes. Science 315, 1709–1712. doi: 10.1126/science.1138140
Bharati, K., Ganguly, N. K. (2011). Cholera Toxin: A Paradigm of a Multifunctional Protein. Indian J. Med. Res. 133, 179–187.
Bhaya, D., Davison, M., Barrangou, R. (2011). CRISPR-Cas Systems in Bacteria and Archaea: Versatile Small RNAs for Adaptive Defense and Regulation. Annu. Rev. Genet. 45, 273–297. doi: 10.1146/annurev-genet-110410-132430
Chen, J. S., Ma, E., Harrington, L. B., Da Costa, M., Tian, X., Palefsky, J. M., et al. (2018). CRISPR-Cas12a Target Binding Unleashes Indiscriminate Single-Stranded DNase Activity. Science 360, 436–439. doi: 10.1126/science.aar6245
Chen, Y, Shi, Y, Chen, Y, Yang, Z, Wu, H, Zhou, Z, et al (2020). Contamination-Free Visual Detection of SARS-CoV-2 With CRISPR/Cas12a: A Promising Method in the Point-of-Care Detection. Biosens. Bioelectron. 169, 112642 doi: 10.1016/j.bios.2020.112642
Chowdhury, F. R., Nur, Z., Hassan, N., von Seidlein, L., Dunachie, S. (2017). Pandemics, Pathogenicity and Changing Molecular Epidemiology of Cholera in the Era of Global Warming. Ann. Clin. Microbiol. Antimicrob. 16, 10. doi: 10.1186/s12941-017-0185-1
Deen, J., Mengel, M. A., Clemens, J. D. (2020). Epidemiology of Cholera. Vaccine 38, A31–A40. doi: 10.1016/j.vaccine.2019.07.078
Gootenberg, J. S., Abudayyeh, O. O., Kellner, M. J., Joung, J., Collins, J. J., Zhang, F. (2018). Multiplexed and Portable Nucleic Acid Detection Platform With Cas13, Cas12a, and Csm6. Science 360, 439–444. doi: 10.1126/science.aaq0179
Harris, J. B., LaRocque, R. C., Qadri, F., Ryan, E. T., Calderwood, S. B. (2012). Cholera. Lancet 379, 2466–2476. doi: 10.1016/S0140-6736(12)60436-X
Khizar, S., Ben Halima, H., Ahmad, N. M., Zine, N., Errachid, A., Elaissari, A. (2020). Magnetic Nanoparticles in Microfluidic and Sensing: From Transport to Detection. Electrophoresis 41, 1206–1224. doi: 10.1002/elps.201900377
Li, S.-Y., Cheng, Q.-X., Wang, J.-M., Li, X.-Y., Zhang, Z.-L., Gao, S., et al. (2018). CRISPR-Cas12a-Assisted Nucleic Acid Detection. Cell Discov. 4, 20. doi: 10.1038/s41421-018-0028-z
Li, T.-M., Du, B. (2011). CRISPR-Cas System and Coevolution of Bacteria and Phages: CRISPR-Cas System and Coevolution of Bacteria and Phages. Hereditas (Beijing) 33, 213–218. doi: 10.3724/SP.J.1005.2011.00213
Nelson, E. J., Harris, J. B., Glenn Morris, J., Calderwood, S. B., Camilli, A. (2009). Cholera Transmission: The Host, Pathogen and Bacteriophage Dynamic. Nat. Rev. Microbiol. 7, 693–702. doi: 10.1038/nrmicro2204
Rafique, R., Rashid, M., Monira, S., Rahman, Z., Mahmud, T., Mustafiz, M., et al. (2016). Transmission of Infectious Vibrio Cholerae Through Drinking Water Among the Household Contacts of Cholera Patients (CHoBI7 Trial). Front. Microbiol. 7. doi: 10.3389/fmicb.2016.01635
Ramachandran, A., Huyke, D. A., Sharma, E., Sahoo, M. K., Huang, C., Banaei, N., et al. (2020). Electric Field-Driven Microfluidics for Rapid CRISPR-Based Diagnostics and its Application to Detection of SARS-CoV-2. Proc. Natl. Acad. Sci. U. S. A. 117, 29518–29525. doi: 10.1073/pnas.2010254117
Ramamurthy, T., Das, B., Chakraborty, S., Mukhopadhyay, A. K., Sack, D. A. (2020). Diagnostic Techniques for Rapid Detection of Vibrio Cholerae O1/O139. Vaccine 38, A73–A82. doi: 10.1016/j.vaccine.2019.07.099
Sack, D. A., Sack, R. B., Nair, G. B., Siddique, A. (2004). Cholera. Lancet 363, 223–233. doi: 10.1016/S0140-6736(03)15328-7
Shen, X., Qiu, F., Shen, L.-P., Yan, T., Zhao, M., Qi, J.-J., et al. (2019). A Rapid and Sensitive Recombinase Aided Amplification Assay to Detect Hepatitis B Virus Without DNA Extraction. BMC Infect. Dis. 19, 229. doi: 10.1186/s12879-019-3814-9
Terns, M. P., Terns, R. M. (2011). CRISPR-Based Adaptive Immune Systems. Curr. Opin. Microbiol. 14, 321–327. doi: 10.1016/j.mib.2011.03.005
Wang, J., Cai, K., He, X., Shen, X., Wang, J., Liu, J., et al. (2020). Multiple-Centre Clinical Evaluation of an Ultrafast Single-Tube Assay for SARS-CoV-2 RNA. Clin. Microbiol. Infect. 26, 1076–1081. doi: 10.1016/j.cmi.2020.05.007
Wiedenheft, B., Sternberg, S. H., Doudna, J. A. (2012). RNA-Guided Genetic Silencing Systems in Bacteria and Archaea. Nature 482, 331–338. doi: 10.1038/nature10886
Wu, H., He, J., Zhang, F., Ping, J., Wu, J. (2020). Contamination-Free Visual Detection of CaMV35S Promoter Amplicon Using CRISPR/Cas12a Coupled With a Designed Reaction Vessel: Rapid, Specific and Sensitive. Anal. Chim Acta 1096, 130–137. doi: 10.1016/j.aca.2019.10.042
Xiao, X., Lin, Z., Huang, X., Lu, J., Zhou, Y., Zheng, L., et al. (2021). Rapid and Sensitive Detection of Vibrio Vulnificus Using CRISPR/Cas12a Combined With a Recombinase-Aided Amplification Assay. Front. Microbiol. 12. doi: 10.3389/fmicb.2021.767315
Keywords: CARID, CRISPR-Cas, RAA, detection, Vibrio cholerae, cholera
Citation: Lu P, Chen J, Li Z, Li Z, Zhang J, Kan B and Pang B (2022) Visual Identification and Serotyping of Toxigenic Vibrio cholerae Serogroups O1 and O139 With CARID. Front. Cell. Infect. Microbiol. 12:863435. doi: 10.3389/fcimb.2022.863435
Received: 27 January 2022; Accepted: 07 March 2022;
Published: 31 March 2022.
Edited by:
Ciprian Iliescu, National Institute for Research and Development in Microtechnologies, RomaniaReviewed by:
Md Nur Hossain, Bangladesh Council of Scientific and Industrial Research, BangladeshCopyright © 2022 Lu, Chen, Li, Li, Zhang, Kan and Pang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Bo Pang, cGFuZ2JvQGljZGMuY24=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.