 Qian Li1,2,3†
Qian Li1,2,3† Taif Shah1,2†
Taif Shah1,2† Binghui Wang1,2Linyu Qu1,2Rui Wang1,2Yutong Hou1,2
Binghui Wang1,2Linyu Qu1,2Rui Wang1,2Yutong Hou1,2 Zulqarnain Baloch1,2Xueshan Xia1,2*
Zulqarnain Baloch1,2Xueshan Xia1,2*- 1Faculty of Life Science and Technology, Kunming University of Science and Technology, Kunming, Yunnan, China
- 2Affiliated Anning First People’s Hospital, Kunming University of Science and Technology, Kunming, China
- 3The First Affiliated Hospital & Clinical Medical College, Dali University, Dali, Yunnan, China
Coronaviruses (CoVs) continuously evolve, crossing species barriers and spreading across host ranges. Over the last two decades, several CoVs (HCoV-229E, HCoV-NL63, HCoV-HKU1, HCoV-OC43, SARS-CoV, MERS-CoV, and SARS-CoV-2) have emerged in animals and mammals, causing significant economic and human life losses. Due to CoV cross-species transmission and the evolution of novel viruses, it is critical to identify their natural reservoiurs and the circumstances under which their transmission occurs. In this review, we use genetic and ecological data to disentangle the evolution of various CoVs in wildlife, humans, and domestic mammals. We thoroughly investigate several host species and outline the epidemiology of CoVs toward specific hosts. We also discuss the cross-species transmission of CoVs at the interface of wildlife, animals, and humans. Clarifying the epidemiology and diversity of species reservoirs will significantly impact our ability to respond to the future emergence of CoVs in humans and domestic animals.
Brief introduction
Coronaviruses (CoVs) are positive-sense, linear, non-segmented, single-stranded RNA viruses (Shah et al., 2022). These viruses are grouped into the order Nidovirales, family Coronaviridae, and subfamily Coronavirinae, with a genome size of about 27~32 kb. The subfamily has been further classified into genera: alpha Human CoVs (HCoV-229E and HCoV-NL63); beta HCoVs (severe acute respiratory syndrome (SARS)-CoV, Middle East respiratory syndrome (MERS)-CoV, HCoV-HKU1, and HCoV-OC43); gamma-CoVs and delta-CoVs (Zhang et al., 2018). These viruses cause respiratory, digestive, and even nervous system disorders in humans, animals (rats, pigs, turtles, whales, etc.), and poultry (Dijkman et al., 2013; Sipulwa et al., 2016; Mulabbi et al., 2021). The average incubation period of this disease is about 5.2 days (Li et al., 2020a), exhibiting manifestations in humans, including respiratory tract disorders, sneezing, dry cough, sore throat, etc. In addition, COVID-19 patients experienced diarrhea, while SARS or MERS cases have a low probability of developing such disease symptoms (Lee et al., 2003; Assiri et al., 2013).
SARS-CoV-2 has given a 5% mortality rate to the current global pneumonia compared to SARS-CoV (10% mortality) and MERS-CoV (34% mortality) (Gabutti et al., 2020; Wang et al., 2020b). Until the SARS emergence in China in 2002–2003, CoVs had been of greater concern to agriculture than public health. During the SARS-CoV outbreak, numerous scientific efforts have focused on identifying and characterizing additional CoVs in wildlife and mammals. Consequently, several novel CoVs have been reported in bats, and poultry over the past two decades (Walsh et al., 2013). The current SARS-CoV-2 outbreak has again put CoVs in the global spotlight and is marked as the most highly pathogenic human virus after SARS-CoV and MERS-CoV. Indeed, many unidentified CoVs may circulate in bats (natural reservoirs) before transmission to humans, animals, or poultry via intermediate hosts (Zhu et al., 2022). Therefore, long-term research efforts will instigate the exploration of pathogenic HCoVs that may provide an essential theoretical basis for preventing new outbreaks in the future.
The current review discusses CoVs’ taxonomic, phylogenetic, and epidemiological characteristics. We also highlighted the evolution of CoVs that co-exist with their hosts for long periods to cause persistent infection. Moreover, we describe CoV’s host cellular receptors, their entry mechanisms into the host cell. Finally, we discuss CoVs-vulnerable hosts and cross-species transmission.
Taxonomic and diversity of CoVs
The International Committee on Taxonomy of Viruses (ICTV) first proposed the order Nidovirales (in Latin, nido means nest) in 1996 (Pringle, 1996), which primarily comprises the Coronaviridae and Arteriviridae viral families (Pringle, 1996). Later, improved detection techniques and metagenomics included more novel viruses in the Nidovirales order (Ge et al., 2012; Shi et al., 2018), which profoundly changed the Virus Classification System. Currently, eight suborders (Arnidovirineae, Abnidovirineae, Cornidovirineae, Nanidovirineae, Mesnidovirineae, Monidovirineae, Ronidovirineae, and Tornidovirineae) are included in the Nidovirales order (Walker et al., 2019). These suborders comprise fourteen families, twenty-five subfamilies, thirty-nine genera, sixty-five subgenera, and one hundred and nine species. Based on genotypic and serological characteristics, CoVs were further classified into alpha, beta, gamma, and delta genera. A study reported seven different CoVs, i.e., alpha CoVs (HCoV-229E and HCoV-NL63) and beta CoVs (HCoV-HKU1, HCoV-OC43, SARS-CoV, MERS-CoV, and SARS-CoV-2), infecting humans (Kesheh et al., 2022; Li et al., 2022a). Genera alpha and beta CoVs included subgroups A, B, C, and D that infect humans and animals (cats, cows, dogs, horses, mice, pigs, etc.), whereas gamma and delta infect birds along with other mammals (Kesheh et al., 2022; Li et al., 2022a). Due to their high pathogenicity, alpha- and beta HCoVs have attracted more attention (Schoeman et al., 2021). Gamma and delta avian CoVs infect chicken, duck, goose, pigeon, sparrow, and turkey with a 5% infection rate (Wille and Holmes, 2020). Due to their wide distribution and strong migratory characteristics, bats may increase the risk of disease transmission to animals or humans. Recently, with the occurrence of SARS-CoV2, relevant departments have strengthened safe bat migration and detection mechanisms.
Epidemiology of CoVs
SARS, the first emerging epidemic of the twenty-first century (lasting approximately six months), first appeared in China and quickly spread to other countries, including Europe and America. All the SARS cases experienced high-grade fever (>100.3°F), headache, aches, diarrhea, feeling uncomfortable, cough, respiratory disorders, and pneumonia (in severe cases). SARS-CoV is most commonly transmitted via inhalation of respiratory droplets from an infected person to healthier ones or by touching virus-contaminated surfaces. CDC’s collaboration with WHO and other global partners addressed SARS’s global crisis by providing emergency health services, deploying medical specialists to assist with on-site investigations, and providing extensive clinical testing for SARS patients (CDC, 2022). Apart from the SARS-CoV outbreak, MERS-CoV reportedly infect human with similar clinical symptoms to SARS-CoV (de Wit et al., 2016).
The epidemic of animal and poultry CoVs causes significant economic challenges, and their transmission may threaten the human population due to their close association. In 1984, McNulty and his team detected Bovine CoV (BCoV) in calf lungs suffering from pneumonia, diarrhea, and respiratory disorders in America, Europe, Asia, and Africa (Zhu et al., 2022), revealing CoV transmission among poultry and birds. The transmission of viruses from poultry to birds represents a new paradigm at the livestock-wildlife interface. Researchers discovered three avian CoV species in 297 wild birds in Egypt between early 2014 and late 2015. The genetic characterization revealed a close relationship with the country’s most commonly used live-attenuated vaccines, highlighting extensive vaccine strain spillover in poultry (Rohaim et al., 2017). In 2015, researchers in Italy investigated the genetic diversity of CoVs in quail, pheasant, and partridge. Avian CoV (gamma CoVs) were found in quail that had not been immunized against the Massachusetts serotype of the Infectious Bronchitis Virus (IBV) and in pheasants that had been vaccinated with IBV. Avian CoVs found in quail and pheasants were linked to IBV-793B and IBV types based on gene sequence homologies encoding spike proteins. RdRp nucleotide sequence analyses revealed quail susceptibility to Delta CoVs, implying that avian CoVs from quail and pheasants share spike gene homology with chicken IBV (Torres et al., 2017). A study reported a novel avian IBV in commercial chicken in Brazil (Fraga et al., 2013) and domestic peafowl in Guangdong Province, China (Liu et al., 2005). In Poland, the overall CoV prevalence in wild birds was 4.15%, with orders Anseriformes (3.51%) and Charadriiformes (5.59%) being the primary reservoirs. Gamma CoVs were more frequently detected (3.5%) in six bird orders (Anseriformes, Charadriiformes, Columbiformes, Galliformes, Gruiformes, and Passeriformes) than delta CoVs (0.7%), which requires a taxonomic update. CoV detection in Anseriformes (3.51%) and Charadriiformes (3.1%) belonged to the subgenera Igacovirus and Brangacovirus, respectively. Most of these were igacoviruses belonging to the duck-CoV-2714 and two avian CoV-9203 phylogenetic groups. In addition, a porcine delta CoV (PDCoV) HKU15, was identified in Hong Kong in 2012 (Woo et al., 2012), infecting pig intestinal epithelia and causing severe diarrhea and vomiting (Ma et al., 2015). Several PDCoV strains were later detected in the United States (Alhamo et al., 2022), Canada (Ajayi et al., 2018), South Korea (Chung et al., 2017), Thailand (Janetanakit et al., 2016), China (Huang et al., 2020a), Japan (Suzuki et al., 2018), Vietnam (Le et al., 2018), etc., posing a severe threat to humans population associated with the swine industry. So far, all members of the delta CoV genus have been found in birds (Saeng-Chuto et al., 2017), implying that birds serve as the natural reservoir for these viruses (Jung et al., 2015; Ma et al., 2015). The close association of the PDCoV genome with the sparrow CoV-HKU17 genome might be attributed to the recombination between Bulbul-CoV HKU11 (Lau et al., 2018). Therefore, identifying novel pathogenic PDCoV may provide opportunities to define the underlying mechanism that allows viruses to cross the host-range barrier. The close interaction of swine and humans endangers public health, as evidenced by the swine-originated influenza virus outbreak (Smith et al., 2009). The prevention and control strategies for PDCoV cross-species transmission among wildlife and livestock demand further investigation to unveil the future dispersal history of CoVs, evolution, and their cross-species transmission.
All highly pathogenic alpha- and beta HCoVs and animal CoVs primarily originate in bats before transmission to humans or animals. For example, SARS-CoV originated in bats (Graham and Baric, 2010), while MERS-CoV originated in camels before being transmitted to humans (Dudas et al., 2018). The current pathogenic novel SARS-CoV-2, first reported in Wuhan, China, is responsible for a significant public health crisis and global economic and social challenges (Gabutti et al., 2020; Zhu et al., 2020). Many of the first COVID-19 patients claimed to have visited an animal market in Wuhan, implying that the virus may have been transmitted to them by animals at the market (Zhou et al., 2020). Although the exact route of SARS-CoV-2 transmission from bat reservoirs to humans is unknown (Lu et al., 2020; Zhou et al., 2020), recent evidence points to raccoons as the intermediate mammalian host between bats and humans (Worobey et al., 2022). Because of its high contagiousness, SARS-CoV-2 spreads rapidly across countries, claiming substantial life losses (WHO, 2020). The emergence of SARS-CoV in 2002, MERS-CoV in 2012, and the current SARS-CoV-2 outbreak in 2019 provide evidence for future CoV outbreaks.
Evolution of CoVs
Adaptive evolution is the only way for CoVs to co-exist with their hosts over a long period for successful infection. CoVs have a high error rate of replication (much higher than DNA viruses), resulting in a high mutation rate because they are single-stranded RNA compared with double-stranded DNA structures. Almost all CoVs originating from bats cause various diseases in poultry, animals, and humans (Su et al., 2016; Alluwaimi et al., 2020). Before SARS-CoV emergence, the two HCoV prototypes (HCoV-OC43 and HCoV-229E) were primarily associated with the common cold (Weiss and Navas-Martin, 2005). In contrast, the HCoV variants HCoV-NL63 and HCoV-HKU1 (Zhou et al., 2020) were associated with mild respiratory and enteric diseases in humans (Liu et al., 2021). Bats originated SARS-CoV, which developed in civet cats (as an intermediate host) (Banerjee et al., 2020), and MERS-CoV is the most infectious HCoV that causes severe infection (Yin and Wunderink, 2018). Given the prevalence of the SARS-like virus in bats and their vast genetic diversity, coexistence, and frequent CoV recombination, new mutations are expected in the future. CoVs, cross-host species, and barriers spread over a wide area over time. In rare cases, viruses transmit across host species for infection. In recent years, the chance of cross-species transmission of mammalian viruses has increased due to increased human-animal contact. The viruses crossing the animal-human species barrier are the avian influenza virus, Hantavirus, hemorrhagic fever virus, insect-borne virus, monkeypox virus, Nipa and Hendra viruses, and SARS-CoVs (Louz et al., 2005; Olival et al., 2017).
CoVs have low mutation rates and high degrees of genetic diversity (Liu et al., 2017) compared to other single-stranded RNA viruses (Dudas and Rambaut, 2016; Su et al., 2016). The first reports of CoV recombination in the 1980s focused on infections with different murine mouse hepatitis viruses (Keck et al., 1988). CoV genome analyses from natural infections have yielded evidence that recombination, particularly between divergent CoVs within individual subgenera, significantly contributes to CoV evolution (Lee and Jackwood, 2000). For example, a complex recombinant has been observed between the Alpha CoV-1 (canine CoV), which primarily infects dogs (Lednicky et al., 2022), the feline CoV, which infects cats (Herrewegh et al., 1998), and the transmissible gastroenteritis virus from swine, which infects pigs (Decaro et al., 2009). Copy-choice is the most common CoVs recombination mechanism in which a viral RNA-dependent RNA polymerase (RdRp) is interrupted during replication, drops off the copying RNA template, and re-engages with a different RNA template at a homologous position before replication (Cheng and Nagy, 2003). During replication, such template switches (recombination breakpoints) transform recombinant daughter genomes with different sequences derived from parental genomes, most likely giving viruses more evolutionary options than mutation alone (Simon-Loriere et al., 2009). Many newly arising mutations within the genomes of genetically compact CoV are expected to have negative fitness consequences, as are mutations that occur between genetically divergent viruses (Drummond et al., 2005). Recombination frequently disrupts favorable co-evolved interactions (referred to as epistatic interactions) within the genome by transferring nucleotide sequences into genomic backgrounds with which they did not co-evolve. For example, recombination (pairing nucleotides to form biologically functional genomic secondary structures) could disrupt epistatic interactions (Martin et al., 2005; de Klerk et al., 2022). However, because recombination generally occurs between fully functional genomes, the range of potential negative fitness consequences should be less extreme than that of newly arising mutations (Drummond et al., 2005). Recombination frequency among CoVs is so high that it occurs without external stress and becomes the dominant virus population (Lai, 1992; Simon-Loriere and Holmes, 2011).
A total of 93 different CoV RdRp protein sequences (that exhibited ~90% similarity) were retrieved from the NCBI database to construct a phylogenetic tree. All the RdRp protein sequences were compared for similarities based on their amino acid residues. CLUSTALW-aligned RdRp sequences were used to construct a tree for understanding RdRp homology from different CoVs. Bats and pigs are the most important natural reservoirs for alpha, beta, gamma, and delta CoVs. The phylogenetic tree shows that alpha and beta CoV genera had high sequence similarity, followed by gamma and delta (Figure 1).
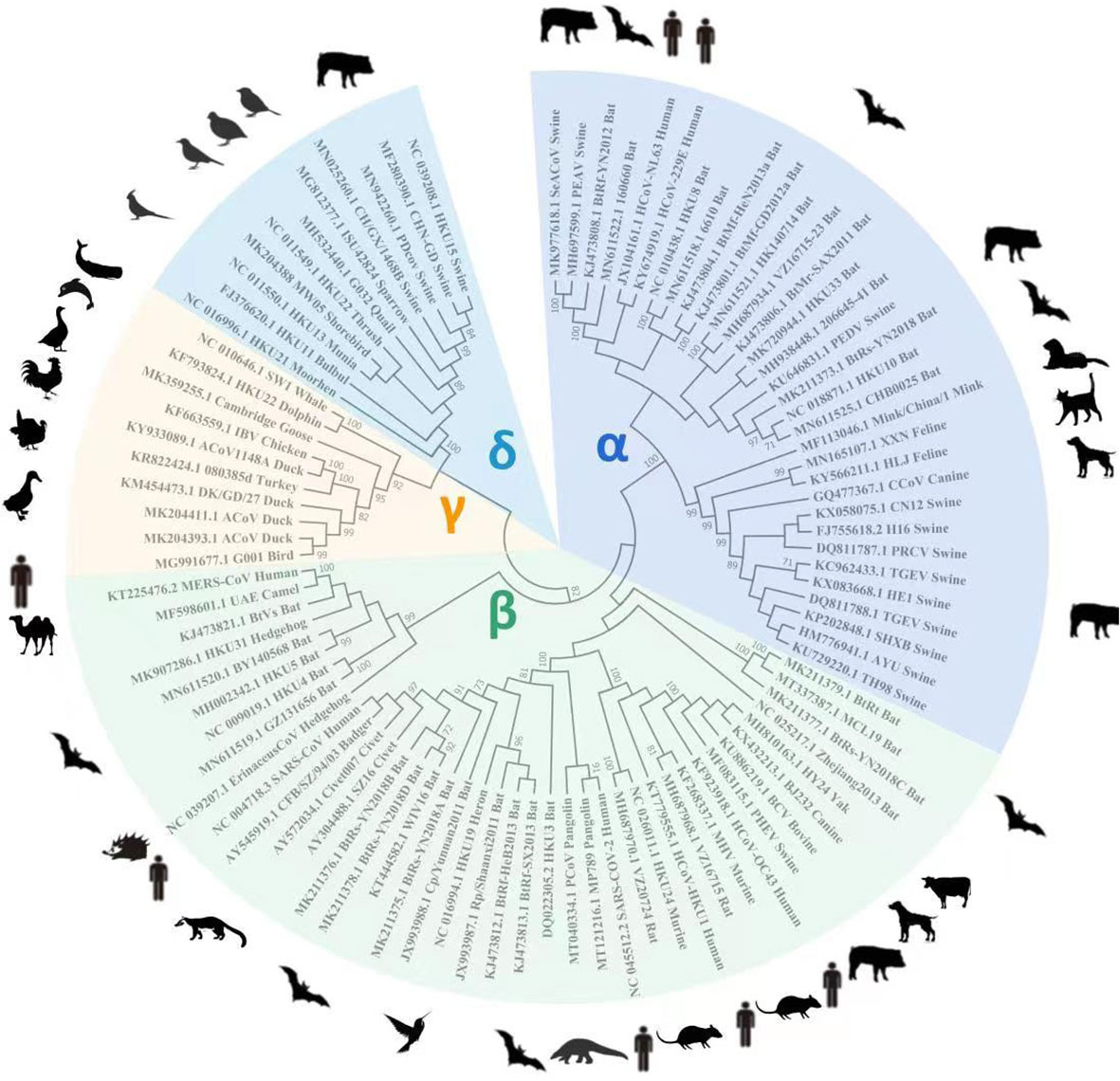
Figure 1 For RdRp gene homology analysis, 93 known RdRp gene amino acid sequences were retrieved from the NCBI database to construct a phylogenetic tree using Mega X software. The tree shows alpha and beta CoVs genera had the highest RdRp amino acid sequence homology, followed by gamma and delta viruses.
CoVs are positive-sense single-stranded RNA viruses with giant RNA genomes (ranging from 27 to 32 kb in length) capable of trans-species movement, as evidenced by the SARS-CoV outbreak (Drosten et al., 2003; Ksiazek et al., 2003). In addition, the emergence of MERS-CoV in 2012 shows that CoVs continue to cause severe human disease (Zaki et al., 2012). The discovery of 3′–5′ exoribonuclease activity within the non-structural nsp14 exoribonuclease, critical for CoV high-fidelity replication, calls into question the long-held belief that RNA viruses cannot proofread and raises the possibility of an entirely new model for how these viruses regulate replication fidelity. RNA viruses rely primarily on low RdRp gene fidelity to facilitate their adaptation to host environments (Domingo et al., 2012). The average mutation rate of RNA viruses is estimated to be around one mutation per genome per round of replication (Drake and Holland, 1999; Malpica et al., 2002). Thus, allowing for enormous population diversity, the low fidelity of RdRp-mediated replication imposes constraints on the maintenance of genomic integrity, theoretically limiting the size of RNA virus genomes to around 15 kb in length (Domingo et al., 2012). Large population sizes, rapid replication cycles, resistance to mutations, recombination, and compact genomes are some of the mechanisms that RNA viruses have evolved to partially circumvent these constraints (Graci et al., 2012; Lauring et al., 2013). These re-assorting mutations generate novel CoV phenotypes with expanded characteristics (Forni et al., 2017). For example, CoV genomic RNA plasticity provides numerous benefits, including the virus’s rapid adaptability to unusual host species with increased virulence. According to scientific evidence, almost all CoVs that infect humans are primarily derived from bats; thus, bats play an essential role in their evolution toward new host species. The jumping of alpha- and beta CoVs from bats to other mammals resulted in introducing these viruses into new hosts (Woo et al., 2012). This “jumping” phenomenon is common among CoVs (Tong et al., 2009; Anthony et al., 2017; Mulabbi et al., 2021), but the underlying molecular mechanism for novel phenotypes transferring to new hosts requires further investigation.
To track the outbreak history and evolution, the CoV infection rate among 1016 diarrheal cattle in Heilongjiang Province, China, was 15.45% in 2020, significantly correlated with age, breeding type, etc. (Lv et al., 2021). A new IBV isolate was isolated from vaccinated chicken in Guangxi, China, in 2016, indicating constant virus evolution and emphasizing the importance of monitoring new IBVs (Lv et al., 2021). In 2019, 3160 poultry samples from 14 Chinese provinces were tested for CoVs. Of the total analyzed samples, 593 were positive for avian CoVs, including 485 avian IBVs, 72 duck CoVs, and 36 pigeon CoVs, demonstrating distinct CoVs in the country. The current study demands more research into these viruses to better understand their cross-species transmission and clinical significance. (Li et al., 2021). According to a study, the avian IBV outbreak in southern China from 1985 to 2017 experienced significant changes in genetic diversity and genotypes, indicating the importance of a long-term virus monitoring system and the urgency of developing new vaccines to combat emerging IBV strains (Fan et al., 2019). More CoV monitoring and research are required to understand virus diversity and evolution better.
CoVs receptor and host cellular entry mechanism
The spike (S) protein, composed of approximately 1273 amino acids and 150~200 kDa, shows varying degrees of conservation among the CoVs (Yadav et al., 2021). Any conformational changes in S domains may alter viral pathogenesis and host cellular entry mechanisms (Aoe, 2020; Huang et al., 2020b). Following synthesis, the S protein forms a homotrimeric assemblage that contains several domains, including a receptor binding site, etc., that interact with the angiotensin-converting enzyme-2 (ACE2) host receptor (Casalino et al., 2020; Moreira et al., 2020). ACE2 is a type I transmembrane protein found in the kidney, heart, intestines, and lungs. It aids in regulating the renin-angiotensin system by balancing angiotensin and protecting against lung injury and pulmonary hypertension. SARS-CoV, HCoV-NL63, and SARS-CoV-2 use the host cellular ACE2 receptor to enter the host cell cytoplasm (Hofmann et al., 2005; Wan et al., 2020). The virus’s binding to the host surface reduces the ACE2 expression level, resulting in pulmonary injury and even death in severe cases (Ge et al., 2013). In addition, dipeptidyl peptidase-4 (DPP4), a conserved glycoprotein expressed in the kidney, respiratory tract, immune cells, etc. (Danta, 2020), helps in MERS-CoV surface receptor function and mediates viral entry into the host cell cytoplasm (Ghosh et al., 2021).
TMPRSS2 is another host cellular protease generally expressed in the epithelium of the respiratory, digestive, and reproductive tracts (David et al., 2020), priming SARS-CoV-2 S attachment with the host cell. To begin the proteolytically active fusion process, TMPRSS2 cleaves the proximal S2 subdomain, exposing the fusion peptide (Dong et al., 2020). In addition, the virus uses the S1 to bind to the host, triggering the effects of TMPRSS2 on the S cleavage and priming membrane fusion for viral entry (Djomkam et al., 2020; Ju et al., 2020; Walls et al., 2020). In addition to TMPRSS2, members of the C-type lectin mediate viral entry into the host cell to initiate pathogenesis (Amraei et al., 2021). Moreover, the metabotropic glutamate receptor (mGluR2) is a GPCR-class glycoprotein expressed on presynaptic and postsynaptic cells in the CNS (Singh et al., 2021). To mediate SARS-CoV-2 internalization, MGluR2 interacted directly with the virus S protein. In contrast, SARS-CoV-2 penetration was reduced by knocking out mGluR2, and the virus binding capability remains stable (Wang et al., 2021a). Unlike mGluR2, which directly interacts with the SARS-CoV-2 S receptor, AXL specifically interacts with the SARS-CoV-2 N-terminal domain (Wang et al., 2021b) for penetration into the host cell. This virus surface protein also shields the pathogen from antibody neutralization (Gu et al., 2022; Hoffmann and Pöhlmann, 2022). CEACAM1 is another transmembrane protein belonging to the carcinoembryonic antigen family of the immunoglobulins that acts as a receptor for CoVs. The binding of CoV S to the CEACAM1a domain causes a conformational change in the S1 domain, facilitating virus entry into the host cell (Haan et al., 2006). In a study, molecular modeling and docking simulation addressed the interaction between CD13-related domains and the SARS-CoV S receptor. The binding of the SARS-CoV S protein’s D757-R761 motif to the P585-A653 domain of CD13 was simulated, implying that CD13 could be a SARS-CoV S receptor associated with SARS infection (Yu et al., 2003). Some of the CoV species and different host cellular receptors are shown in Table 1.
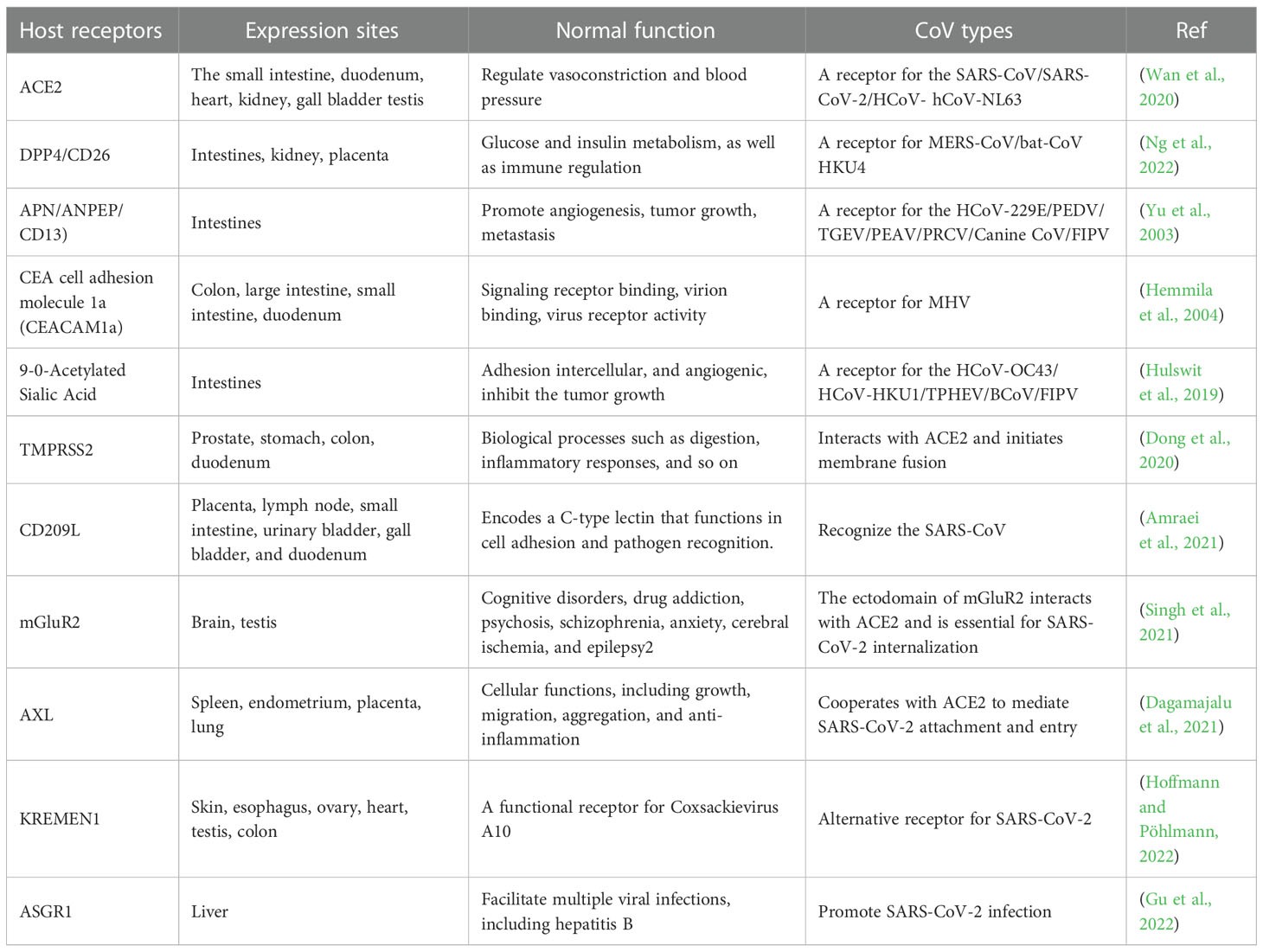
Table 1 Characteristics of host cellular receptors for various CoVs.
CoVs-susceptible hosts
Several CoVs have been confirmed to be primarily hosted by bats, rodents, porcine, mice, birds, etc. (de Wilde et al., 2018). Alpha- and beta CoVs are primarily found in mammals, whereas gamma and delta CoVs are mostly found in birds. Identifying virus-carrying hosts will be critical to disease prevention and control strategies. Humans are vulnerable to at least seven types of human CoVs (HCoV-229E, HCoV-NL63, HCoV-HKU1, HCoV-OC43, SARS, MERS, and SARS-CoV-2), primarily transmitting through respiratory droplets, direct contact with infected people, animals, body fluids, or touching contaminated surfaces (Milewska et al., 2014; Ye et al., 2020). CoV-29E or HCoV-OC43 causes approximately 30% of common colds in humans, particularly children (Dijkman et al., 2008; Walsh et al., 2013). The other three viruses (SARS-CoV, MERS-CoV, and SARS-CoV-2) cause severe respiratory disorders in humans and even death in severe cases (van der Hoek, 2007; de Groot et al., 2013). Bats typically feed on flying insects, prefer to live in caves, abandoned mines, tunnels, hollow trees, etc., and are widespread on the planet. There are over 1,300 bat species found worldwide that harbor several pathogenic viruses (including SARS-CoV and, MERS-CoV, SARS-CoV-2) that cause human diseases. Their gregarious living habits provide possible survival conditions and cell types for spreading zoonotic viral infections among birds, animals, and humans (Plowright et al., 2015). The ability of bats to migrate over long distances facilitates the exchange of viruses or their genetic material between bats and other animals, indicating that different living environments lead to the evolution of diverse CoVs. Over the last 20 years, several CoVs (SARS-CoV, MERS-CoV, SARS-CoV-2, etc.) have jumped between bats, animals, or humans, resulting in diseases with high fatality (Wong et al., 2021). Rodentia is one of the most diverse mammals, with over 2000 species carrying a wide range of microorganisms that are the primary source of infectious diseases. Many rodents live close to humans and pose significant zoonotic risks. Only one rodent species has been identified as carrying CoV, indicating that rodents are the reservoir for RNA viruses. Murine CoV (mCoV) is a well-studied CoV archetype (Cheever et al., 1949). Following the isolation of the first MHV from mice in 1949, a mutant sialodacryoadenitis-CoV (SDAV) was discovered in 1970 (members of the beta CoV) (Wang et al., 2015). Notably, rodent-associated CoVs account for most of the known HCoV-HKU1 and HCoV-OC43 genetic diversity, which causes enteric and respiratory diseases in humans and domestic animals (Grabherr et al., 2021). As a result, further research into the rodent’s potential role in the evolution of CoVs is required to unravel the mysteries of future viral outbreaks (Han et al., 2015). Notably, murine thrive in urban areas and are well-adapted to coexistence with wildlife and humans (Langer and Giessen, 2007; Bartak et al., 2021), and they may spread unidentified CoVs in the community. Such viruses include swine enteric CoVs (SeCoVs), Transmissible gastroenteritis virus (TGEV), porcine epidemic diarrhea virus (PEDV), etc. (Pan et al., 2017; Cheng et al., 2020; Li et al., 2022b). The porcine respiratory CoV (PRCV) and TGEV are closely related to the feline and canine CoVs (Liu and Wang, 2021). Porcine hemagglutinating encephalomyelitis virus (PHEV) infections are common in pigs, causing encephalomyelitis and severe vomiting in piglets (Liu and Wang, 2021). Porcine enteric alpha CoV (PEAV) was first discovered in southeast China in 2017 and shares 90% aa homology with bat CoV-HKU2 (Gong et al., 2017). Later, a PEAV was identified and isolated in China (Yong-Le et al.). PEDV and PDCoV are the most pathogenic porcine viruses, threatening the swine industry and causing watery diarrhea in piglets (Surapong et al., 2019). The coinfection of PDCoV and PEDV may result in higher piglet mortality. Moreover, CoVs cause respiratory, intestinal, and systemic diseases in many other hosts. Most clinical manifestations are mild, but a few can have serious public health consequences. In cattle and wild ruminants, Genera beta CoV causes respiratory and intestinal disorders (Vlasova and Saif, 2021). Avian infectious bronchitis is a contagious upper respiratory tract disease in chickens caused by gamma CoV (infectious bronchitis virus—IBV) that significantly impacts the poultry industry globally (Legnardi et al., 2020). Feline CoV (FeCoV) is well-known for causing minor intestinal infections in domestic cats (Oguzoglu et al., 2021). Canine respiratory CoV (CRCoV) causes respiratory diseases in dogs (Priestnall, 2020), whereas ferret entero-CoV (FrECoV), alpaca CoV, and equine CoV (ECoV) are pathogenic to humans and their companion animals (Csiszar et al., 2020; Gilmutdinov et al., 2020; Islam et al., 2021).
CoVs cross-species transmission
The interaction between the virus and the host is the primary driving force behind virus evolution. A virus with a faster rate of evolution may be better suited for host and cross-species transmission. CoVs are sufficient to provide more base space for gene mutations. Mutations at the gene level can be used to adapt to the host body’s environment and jump to new hosts, promoting cross-species transmission and forming a wide host distribution (Olival et al., 2017). As stated earlier, bat species are the primary reservoirs for many pathogenic viruses, including CoVs. Hosts and viruses have either co-evolved or been paired through spillover, and ample studies show how reservoir hosts like bats have likely co-evolved with CoVs (Wessner, 2010). It has been established that initial CoV transmission occurs from bats, but the underlying transmission mechanism from these mammals to humans requires further investigation. Literature has shown how intermediate hosts played a crucial role in the transmission and evolution of CoVs and other related viruses (Graham and Baric, 2010; Cotten et al., 2013; Enserink, 2013).
The close identity of alpaca CoVs and HCoV-229E with African bat CoVs (Corman et al., 2015) implies that humans may have contact with these bats in their natural habitats rather than alpacas, which do not share habitats. Further investigation reveals that camelid CoVs shared genomic similarities with HCoV-229E, implying the evolution of a new viral genotype toward humans. A mutation (deletion/insertion) in the S gene sequence facilitates the transfer of bat-associated HCoV-229E to humans, establishing camelids as the primary zoonotic source for humans (Corman et al., 2018). Artiodactyls, carnivores, lagomorphs, perissodactyls, primates, and rodents have all been found to carry HCoV-OC43 (Mulabbi et al., 2021). The best representative of these CoVs are bovine CoVs, beta CoVs, and HCoV-OC43 reported in livestock, which might transmit to humans. The S gene mutation (deletion or insertion), like other CoVs, reflects HCoV-OC43 adaptation to the human environment. Like beta CoVs, HCoV-OC43 has an ancestral link to rodents (but not bats) due to their close similarity to MHV, indicating their origin in rodents (Corman et al., 2018). However, the transmission mechanism of HCoV-OC43 from rodents to bovines is unknown, whereas HCoV-NL63 is related to some bat-associated CoVs of the Vespertillionidae and Hipposideridae families (Pfefferle et al., 2009). So far, no zoonotic reservoir has been identified as the underlying transmission mechanism of HCoV-NL43 to humans. Moreover, there are no sequence similarities between rodent-associated HCoV-HKU1 and other animal CoVs (Woo et al., 2005; Wang et al., 2015). Like HCoV-NL63, the HCoV-HKU1 transmission mechanism from animals to humans is unknown. Further, rodent-associated HCoV-HKU1 shares no sequence similarities with other animal species (Woo et al., 2005; Wang et al., 2015). In light of the recent SARS-CoV, MERS-CoV, and SARS-CoV-2 outbreaks, new insights into CoV transmission patterns will require extensive research in future viral outbreaks (Guan et al., 2003).
Bats have been identified as a natural reservoir for an increasing number of emerging zoonotic viruses. A study identified the horseshoe bat (genus Rhinolophus) as the reservoir host for many viruses with a close genetic relationship to SARS-like COVs (SLCoV). Additional novel bat SLCoV detection has shed new light on the origin and transmission of various SARS among wild mammals (Wang et al., 2006). These animals were thought to harbor SARS-CoV in their markets, likely where the virus originated before causing human disease (Weiss and Navas-Martin, 2005; Zhou et al., 2020). Although SARS-CoV has been discovered to infect monkeys and domestic cats, there is no evidence of virus transmission from these animals to humans (Chan et al., 2013; Sabir et al., 2016), suggesting the virus originated in a wild, natural reservoir. Due to the sequence identity of MERS-CoV, with bat-CoV-HKU4 and KHU5, the first attempt focused on bats (Chan et al., 2013). According to the molecular and serological investigation, MERS was first reported in camels in Saudi Arabia, Oman, and Qatar (Chan et al., 2013; Mulabbi et al., 2021). In Saudi Arabia, dromedary camels harbor several CoV lineages, including the one responsible for human outbreaks. The infectious MERS-CoV strain isolated from these camels demonstrates the outbreak’s severity (Sabir et al., 2016). It was speculated that the close association of dromedary camels with humans might result in a novel HCoVs outbreak (as the current SARS-CoV-2 pandemic)—most likely due to human interactions with bats or other intermediate hosts (Wang et al., 2005; Baseler et al., 2016).
The SARS-CoV-2 outbreak in humans has been linked to China’s Wuhan Seafood Market, where several birds, rodents, and rabbits were sold (Mackenzie and Smith, 2020; Zhou et al., 2020). SARS-CoV was closely related to bats that originated SLCoV-ZC45 and SLCoV-ZXC21, which had previously been identified in China (Hu et al., 2018), indicating that bats are natural reservoirs for such viruses. SARS-CoV-2, like other HCoVs, is likely to transfer to an intermediate host in the same market before spreading to humans (Li et al., 2020b). This is because there is no evidence of human contact with the virus-carrying bats, which are known to hibernate in December (Li et al., 2020b; Shah et al., 2022). Next-generation sequencing revealed SARS-CoV’s resemblance with previously identified SLCoV-ZC45 and SLCoV-ZXC21 (Hu et al., 2018), revealing bats as their natural reservoir.
CoVs human-to-human transmission
Once zoonotic CoVs cross the species barrier for human infections, their survival in humans depends on their ability to sustain transmission from person to person through aerosolized particles or physical contact with infected people or objects. The CoVs responsible for human outbreaks are HCoV-229E, HCoV-HKU1, HCoV-NL63, HCoV-OC43, SARS-CoV, MERS-CoV, and SARS-CoV-2, which spread via air droplets, direct contact with an infected person, or contaminated surfaces. The viruses primarily replicate in the respiratory tract and then spread to other body organs (Li et al., 2020bb; Mulabbi et al., 2021). Furthermore, virus transmission from infected individuals to healthy individuals depends on the virus’s stability and adaptation to a new host or environment. It has been reported that HCoV-NL63 can survive in an aqueous solution and the respiratory tract for at least seven days (Müller et al., 2008) at 20°C and 50% humidity (Chowell et al., 2015; Hunter et al., 2016). MERS-CoV can survive for 48 hours at 20°C (40% humidity), while SARS-CoV can survive for five days at 22°C (50% humidity). Both viruses lose viability when temperatures and humidity rise (van Doremalen et al., 2013). Because of this stability, viruses can be transmitted via aerosols; for example, SARS is more stable and has a longer transmission via aerosol than MERS. In addition, these viruses are stable on inanimate surfaces and infect humans after touching the contaminated surfaces (Lee and Wong, 2015), explaining human infections that were not close to patients during their respective outbreaks. SARS-CoV can be excreted in feces and remains infectious in sewerage for up to 48 hours, implying that viral shading in stool and clogged sewerage canals, as well as the possibility of fecal-oral transmission, may be a potential transmission route when people come into contact with contaminated sewerage (Wang et al., 2005; Young et al., 2020). The disposal of virus-containing waste in landfills can contaminate drinking water, necessitating the centralized disposal of environmentally sound units. SARS-CoV-2 can be transmitted from an infected person to a healthy person, as evidenced by infected families and medical staff who did not visit the market where the virus was primarily reported (Carlos et al., 2020; Wang et al., 2020a). The ability of the SARS-CoV-2 infection to remain alive on surfaces for days allows virus transmission and fomites (van Doremalen et al., 2020). In contrast to SARS and MERS, which cause intestinal infections at later stages; whereas, SARS-CoV-2-infected individuals may harbor the virus in their intestines at any stage of infection. The detection of SARS in the oral cavity, anus and body fluids demonstrates an alternative transmission route (Zhang et al., 2020), which suggests additional research to understand virus dynamics and transmission better. The cross-species transmission of some known CoVs among various animals is shown in Figure 2.
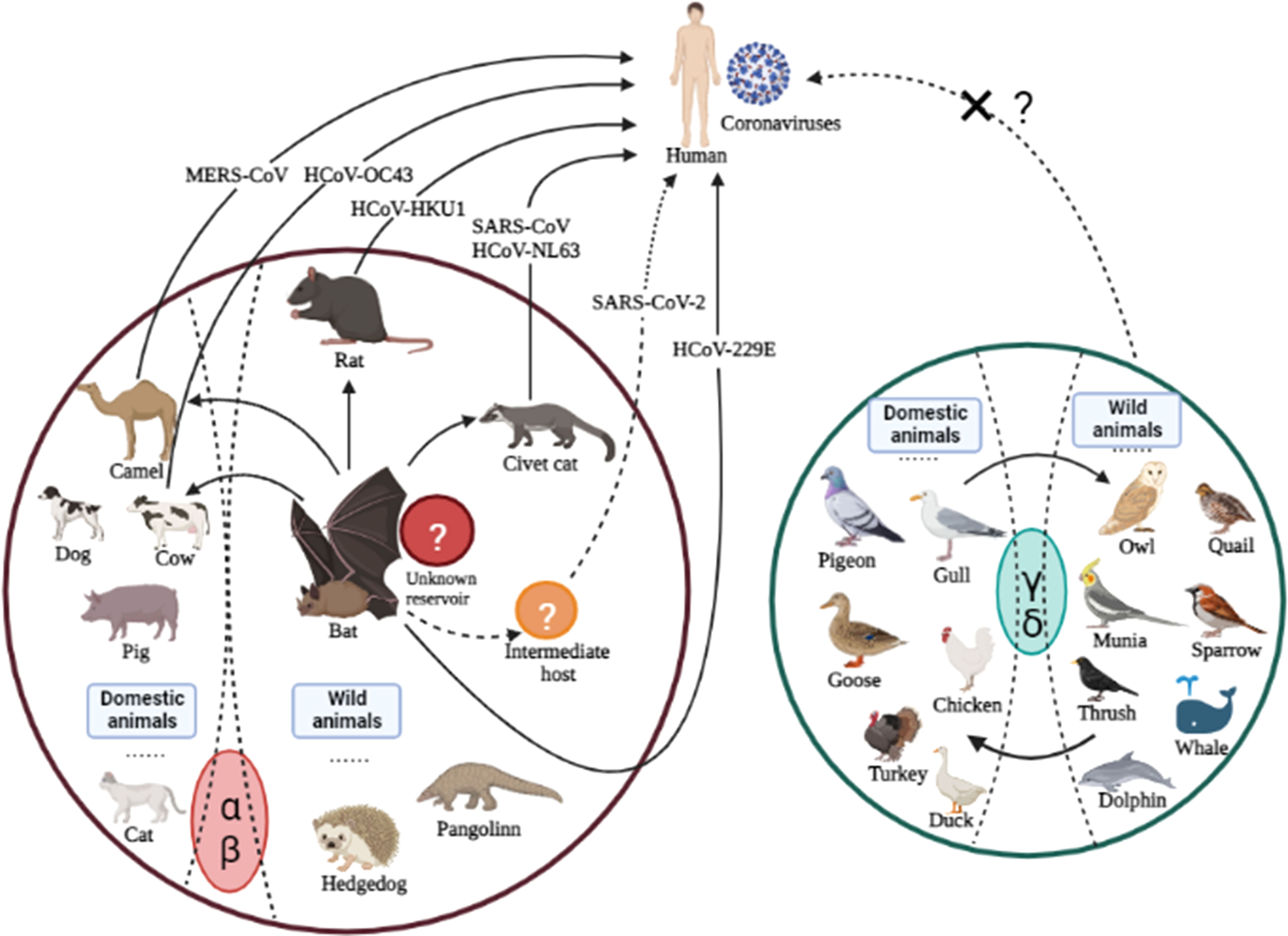
Figure 2 Cross-species transmission of CoVs is common among various animals. The arrows indicate the direction of virus transmission. The transmission sources of seven CoVs (all belonging to genera α and β) infecting humans are depicted are intragenus mutual transmission. There have been no reports of human infections with genera γ or δ, which calls for additional research in the future.
Conclusion and future perspective
COV infection in humans causes mild to severe acute respiratory illnesses with high fatality rates. CoVs, like other emerging pathogens, pose a great public health challenge due to the lack of information before they jump from primary sources, i.e., wildlife to birds, humans, or other mammals.
Humans are encroaching on new habitats, and increasing their interactions with wildlife (and the diseases they carry) has serious public health implications. Diseases that spread from animals to humans are rising; for example, SARS-CoV spread from bats and civet cats, Spanish influenza affected birds and other mammals before transferring to humans, and SARS-CoV-2 spread from a wildlife market to humans. Thus, zoonotic spillover of viruses is always possible; thus, harmonious development can effectively prevent viral zoonosis by maintaining a barrier between human society and the virus’s natural host (Albery et al., 2021). The most significant challenge is developing preventive therapies and vaccines in time for new viral outbreaks. Since bats naturally harbor a variety of CoVs, ongoing efforts should be made to identify and characterize bat-associated viruses and the risk of transmission to domestic animals and humans. Identifying the viruses’ underlying cross-host species transmission mechanisms that link bats to humans is critical. Apart from the indirect transmission, there is always the possibility of virus transmission from bats to humans, which needs to be thoroughly investigated. In addition to identifying intermediate hosts, viral surveillance studies should be expanded to wildlife, birds, animals, and mammals before they spread to humans. Mutations (sequence deletion/insertion) and recombination in ssRNA viruses contribute to the emergence of novel pathogenic viruses capable of infecting humans. Consequently, ongoing monitoring of virus mutations is required to ensure that their emergence does not catch humans off guard, and preventive measures to prevent zoonotic viral transmission are recommended. Stringent regulations to monitor wildlife trading, barriers around wild mammals and animals, and a general shift in community cultural practices should all be monitored for this purpose. These can be supplemented with other commonly used methods, such as infected person isolation and restricting public mobility in CoV-endemic areas. The One Health concept is recommended for approaching and eradicating future public health risks from emerging novel viral outbreaks.
Author contributions
QL and TS performed the initial draft preparation and revision. XX, TS, LQ, BW, RW, ZB, and YH made suggestions for the writing of the manuscript and revision. All authors actively contributed to the article and approved the submitted version.
Funding
This study was supported by grants from the National Natural Science Foundation of China (No. 81860592), Yunnan Key R&D Program (No. 202103AQ100001), Yunnan Major Scientific and Technological Projects (No. 202202AG050013) and Yunnan Province Basic Research Program Projects (No. 202101AS070028).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Ajayi, T., Dara, R., Misener, M., Pasma, T., Moser, L., Poljak, Z. (2018). Herd-level prevalence and incidence of porcine epidemic diarrhoea virus (PEDV) and porcine deltacoronavirus (PDCoV) in swine herds in Ontario, Canada. Transbound Emerg. Dis. 65 (5), 1197–1207. doi: 10.1111/tbed.12858
Albery, G. F., Becker, D. J., Brierley, L., Brook, C. E., Christofferson, R. C., Cohen, L. E., et al. (2021). The science of the host-virus network. Nat. Microbiol. 6 (12), 1483–1492. doi: 10.1038/s41564-021-00999-5
Alhamo, M. A., Boley, P. A., Liu, M., Niu, X., Yadav, K. K., Lee, C., et al. (2022). Characterization of the cross-species transmission potential for porcine deltacoronaviruses expressing sparrow coronavirus spike protein in commercial poultry. Viruses 14 (6). doi: 10.3390/v14061225
Alluwaimi, A. M., Alshubaith, I. H., Al-Ali, A. M., Abohelaika, S. (2020). The coronaviruses of animals and birds: Their zoonosis, vaccines, and models for SARS-CoV and SARS-CoV2. Front. Vet. Sci. 7. doi: 10.3389/fvets.2020.582287
Amraei, R., Yin, W., Napoleon, M. A., Suder, E. L., Berrigan, J., Zhao, Q., et al. (2021). CD209L/L-SIGN and CD209/DC-SIGN act as receptors for SARS-CoV-2. ACS Cent. Sci. 7 (7), 1156–1165. doi: 10.1021/acscentsci.0c01537
Anthony, S. J., Gilardi, K., Menachery, V. D., Goldstein, T., Ssebide, B., Mbabazi, R., et al. (2017). Further evidence for bats as the evolutionary source of middle East respiratory syndrome coronavirus. mBio 8 (2). doi: 10.1128/mBio.00373-17
Aoe, T. (2020). Pathological aspects of COVID-19 as a conformational disease and the use of pharmacological chaperones as a potential therapeutic strategy. Front. Pharmacol. 11. doi: 10.3389/fphar.2020.01095
Assiri, A., Al-Tawfiq, J. A., Al-Rabeeah, A. A., Al-Rabiah, F. A., Al-Hajjar, S., Al-Barrak, A., et al. (2013). Epidemiological, demographic, and clinical characteristics of 47 cases of middle East respiratory syndrome coronavirus disease from Saudi Arabia: a descriptive study. Lancet Infect. Dis. 13 (9), 752–761. doi: 10.1016/S1473-3099(13)70204-4
Banerjee, A., Subudhi, S., Rapin, N., Lew, J., Jain, R., Falzarano, D., et al. (2020). Selection of viral variants during persistent infection of insectivorous bat cells with middle East respiratory syndrome coronavirus. Sci. Rep. 10 (1), 7257. doi: 10.1038/s41598-020-64264-1
Bartak, M., Słońska, A., Bańbura, M. W., Cymerys, J. (2021). SDAV, the rat coronavirus–how much do we know about it in the light of potential zoonoses. Viruses 13 (10). doi: 10.3390/v13101995
Baseler, L. J., Falzarano, D., Scott, D. P., Rosenke, R., Thomas, T., Munster, V. J., et al. (2016). An acute immune response to middle East respiratory syndrome coronavirus replication contributes to viral pathogenicity. Am. J. Pathol. 186 (3), 630–638. doi: 10.1016/j.ajpath.2015.10.025
Carlos, W. G., Dela Cruz, C. S., Cao, B., Pasnick, S., Jamil, S. (2020). Novel wuhan, (2019-nCoV) coronavirus. Am. J. Respir. Crit. Care Med. 201 (4), P7–p8. doi: 10.1164/rccm.2014P7
Casalino, L., Gaieb, Z., Goldsmith, J. A., Hjorth, C. K., Dommer, A. C., Harbison, A. M., et al. (2020). Beyond shielding: The roles of glycans in the SARS-CoV-2 spike protein. ACS Cent Sci. 6 (10), 1722–1734. doi: 10.1021/acscentsci.0c01056
CDC (2022) Severe acute respiratory syndrome (SARS): SARS basics fact sheet. Available at: https://www.cdc.gov/sars/about/fs-sars.html (Accessed 2022-09-04).
Chan, J. F., Lau, S. K., Woo, P. C. (2013). The emerging novel middle East respiratory syndrome coronavirus: the "knowns" and "unknowns". J. Formos Med. Assoc. 112 (7), 372–381. doi: 10.1016/j.jfma.2013.05.010
Cheever, F. S., Daniels, J. B., Pappenheimer, A. M., Bailey, O. T. (1949). A murine virus (JHM) causing disseminated encephalomyelitis with extensive destruction of myelin. J Exp Med. 90 (3), 181–194. doi: 10.1084/jem.90.3.181
Cheng, C. P., Nagy, P. D. (2003). Mechanism of RNA recombination in carmo- and tombusviruses: evidence for template switching by the RNA-dependent RNA polymerase in vitro. J. Virol. 77 (22), 12033–12047. doi: 10.1128/jvi.77.22.12033-12047.2003
Cheng, S., Wu, H., Chen, Z. (2020). Evolution of transmissible gastroenteritis virus (TGEV): A codon usage perspective. Int. J. Mol. Sci. 21 (21). doi: 10.3390/ijms21217898
Chowell, G., Abdirizak, F., Lee, S., Lee, J., Jung, E., Nishiura, H., et al. (2015). Transmission characteristics of MERS and SARS in the healthcare setting: a comparative study. BMC Med. 13, 210. doi: 10.1186/s12916-015-0450-0
Chung, H. C., Nguyen, V. G., Oh, W. T., My Le, H. T., Moon, H. J., Lee, J. H., et al. (2017). Complete genome sequences of porcine deltacoronavirus strains DH1/2016 and DH2/2016 isolated in south Korea. Genome Announc 5 (18). doi: 10.1128/genomeA.01706-16
Corman, V. M., Baldwin, H. J., Tateno, A. F., Zerbinati, R. M., Annan, A., Owusu, M., et al. (2015). Evidence for an ancestral association of human coronavirus 229E with bats. J. Virol. 89 (23), 11858–11870. doi: 10.1128/jvi.01755-15
Corman, V. M., Muth, D., Niemeyer, D., Drosten, C. (2018). Hosts and sources of endemic human coronaviruses. Adv. Virus Res. 100, 163–188. doi: 10.1016/bs.aivir.2018.01.001
Cotten, M., Watson, S. J., Kellam, P., Al-Rabeeah, A. A., Makhdoom, H. Q., Assiri, A., et al. (2013). Transmission and evolution of the middle East respiratory syndrome coronavirus in Saudi Arabia: a descriptive genomic study. Lancet 382 (9909), 1993–2002. doi: 10.1016/s0140-6736(13)61887-5
Csiszar, A., Jakab, F., Valencak, T. G., Lanszki, Z., Tóth, G. E., Kemenesi, G., et al. (2020). Companion animals likely do not spread COVID-19 but may get infected themselves. GeroScience 42 (5), 1229–1236. doi: 10.1007/s11357-020-00248-3
Dagamajalu, S., Rex, D. A. B., Palollathil, A., Shetty, R., Bhat, G., Cheung, L. W. T., et al. (2021). A pathway map of AXL receptor-mediated signaling network. J. Cell Commun. Signal 15 (1), 143–148. doi: 10.1007/s12079-020-00580-5
Danta, C. C. (2020). Dipeptidyl peptidase-4: A potential therapeutic target in diabetic kidney disease with SARS-CoV-2 infection. ACS Pharmacol. Transl. Sci. 3 (5), 1020–1022. doi: 10.1021/acsptsci.0c00097
David, A., Khanna, T., Beykou, M., Hanna, G., Sternberg, M. J. E. (2020). Structure, function and variants analysis of the androgen-regulated TMPRSS2, a drug target candidate for COVID-19 infection. BioRxiv. doi: 10.1101/2020.05.26.116608
Decaro, N., Mari, V., Campolo, M., Lorusso, A., Camero, M., Elia, G., et al. (2009). Recombinant canine coronaviruses related to transmissible gastroenteritis virus of swine are circulating in dogs. J. Virol. 83 (3), 1532–1537. doi: 10.1128/jvi.01937-08
de Groot, R. J., Baker, S. C., Baric, R. S., Brown, C. S., Drosten, C., Enjuanes, L., et al. (2013). Middle East respiratory syndrome coronavirus (MERS-CoV): Announcement of the coronavirus study group. J. OF Virol. 87 (14), 7790–7792. doi: 10.1128/JVI.01244-13
de Klerk, A., Swanepoel, P., Lourens, R., Zondo, M., Abodunran, I., Lytras, S., et al. (2022). Conserved recombination patterns across coronavirus subgenera. Virus Evol. 8 (2), veac054. doi: 10.1093/ve/veac054
de Wilde, A. H., Snijder, E. J., Kikkert, M., van Hemert, M. J. (2018). Host factors in coronavirus replication. Curr. Top. Microbiol. Immunol. 419, 1–42. doi: 10.1007/82_2017_25
de Wit, E., van Doremalen, N., Falzarano, D., Munster, V. J. (2016). SARS and MERS: recent insights into emerging coronaviruses. Nat. Rev. Microbiol. 14 (8), 523–534. doi: 10.1038/nrmicro.2016.81
Dijkman, R., Jebbink, M. F., Idrissi, N., Pyrc, K., Muller, M. A., Kuijpers, T. W., et al. (2008). Human coronavirus NL63 and 229E seroconversion in children. J. Clin. Microbiol. 46 (7), 2368–2373. doi: 10.1128/JCM.00533-08
Dijkman, R., Jebbink, M. F., Koekkoek, S. M., Deijs, M., Jónsdóttir, H. R., Molenkamp, R., et al. (2013). Isolation and characterization of current human coronavirus strains in primary human epithelial cell cultures reveal differences in target cell tropism. J. Virol. 87 (11), 6081–6090. doi: 10.1128/jvi.03368-12
Djomkam, A. L. Z., Olwal, C. O., Sala, T. B., Paemka, L. (2020). Commentary: SARS-CoV-2 cell entry depends on ACE2 and TMPRSS2 and is blocked by a clinically proven protease inhibitor. Front. Oncol. 10, 1448. doi: 10.3389/fonc.2020.01448
Domingo, E., Sheldon, J., Perales, C. (2012). Viral quasispecies evolution. Microbiol. Mol. Biol. Rev. 76 (2), 159–216. doi: 10.1128/mmbr.05023-11
Dong, M., Zhang, J., Ma, X., Tan, J., Chen, L., Liu, S., et al. (2020). ACE2, TMPRSS2 distribution and extrapulmonary organ injury in patients with COVID-19. BioMed. Pharmacother. 131, 110678. doi: 10.1016/j.biopha.2020.110678
Drake, J. W., Holland, J. J. (1999). Mutation rates among RNA viruses. Proc. Natl. Acad. Sci. U.S.A. 96 (24), 13910–13913. doi: 10.1073/pnas.96.24.13910
Drosten, C., Günther, S., Preiser, W., van der Werf, S., Brodt, H.-R., Becker, S., et al. (2003). Identification of a novel coronavirus in patients with severe acute respiratory syndrome. New Engl. J. Med. 348 (20), 1967–1976. doi: 10.1056/NEJMoa030747
Drummond, D. A., Silberg, J. J., Meyer, M. M., Wilke, C. O., Arnold, F. H. (2005). On the conservative nature of intragenic recombination. Proc. Natl. Acad. Sci. U.S.A. 102 (15), 5380–5385. doi: 10.1073/pnas.0500729102
Dudas, G., Carvalho, L. M., Rambaut, A., Bedford, T. (2018). MERS-CoV spillover at the camel-human interface. Elife 7. doi: 10.7554/eLife.31257
Dudas, G., Rambaut, A. (2016). MERS-CoV recombination: implications about the reservoir and potential for adaptation. Virus Evol. 2 (1), vev023. doi: 10.1093/ve/vev023
Enserink, M. (2013). Infectious diseases. amid heightened concerns, new name for novel coronavirus emerges. Science 340 (6133), 673. doi: 10.1126/science.340.6133.673
Fan, W., Tang, N., Dong, Z., Chen, J., Zhang, W., Zhao, C., et al. (2019). Genetic analysis of avian coronavirus infectious bronchitis virus in yellow chickens in southern China over the past decade: Revealing the changes of genetic diversity, dominant genotypes, and selection pressure. Viruses 11 (10). doi: 10.3390/v11100898
Forni, D., Cagliani, R., Clerici, M., Sironi, M. (2017). Molecular evolution of human coronavirus genomes. Trends Microbiol. 25 (1), 35–48. doi: 10.1016/j.tim.2016.09.001
Fraga, A. P., Balestrin, E., Ikuta, N., Fonseca, A. S., Spilki, F. R., Canal, C. W., et al. (2013). Emergence of a new genotype of avian infectious bronchitis virus in Brazil. Avian Dis. 57 (2), 225–232. doi: 10.1637/10346-090412-Reg.1
Gabutti, G., d'Anchera, E., Sandri, F., Savio, M., Stefanati, A. (2020). Coronavirus: Update related to the current outbreak of COVID-19. Infect. Dis. Ther. 9 (2), 241–253. doi: 10.1007/s40121-020-00295-5
Ge, X. Y., Li, J. L., Yang, X. L., Chmura, A. A., Zhu, G., Epstein, J. H., et al. (2013). Isolation and characterization of a bat SARS-like coronavirus that uses the ACE2 receptor. Nature 503 (7477), 535–538. doi: 10.1038/nature12711
Ge, X., Li, Y., Yang, X., Zhang, H., Zhou, P., Zhang, Y., et al. (2012). Metagenomic analysis of viruses from bat fecal samples reveals many novel viruses in insectivorous bats in China. J. Virol. 86 (8), 4620–4630. doi: 10.1128/jvi.06671-11
Ghosh, P., Jayaram, S., Patwardhan, D., Marimuthu, S., Lenehan, P., Venkatakrishnan, A., et al. (2021). Diversity of coronavirus receptors. 2021080071. doi: 10.20944/preprints202108.0071.v1
Gilmutdinov, R., Shalamova, G., Domolazov, S. (2020). Coronaviruses of wild animals in Russia. E3S Web Conferences 203 (1), 01013. doi: 10.1051/e3sconf/202020301013
Gong, L., Li, J., Zhou, Q., Xu, Z., Chen, L., Zhang, Y., et al. (2017). A new Bat-HKU2–like coronavirus in swine, chin. Emerging Infect. Dis. J. 23 (9), 1607. doi: 10.3201/eid2309.170915
Grabherr, S., Ludewig, B., Pikor, N. B. (2021). Insights into coronavirus immunity taught by the murine coronavirus. Eur. J. Immunol. 51 (5), 1062–1070. doi: 10.1002/eji.202048984
Graci, J. D., Gnädig, N. F., Galarraga, J. E., Castro, C., Vignuzzi, M., Cameron, C. E. (2012). Mutational robustness of an RNA virus influences sensitivity to lethal mutagenesis. J. Virol. 86 (5), 2869–2873. doi: 10.1128/jvi.05712-11
Graham, R. L., Baric, R. S. (2010). Recombination, reservoirs, and the modular spike: mechanisms of coronavirus cross-species transmission. J. Virol. 84 (7), 3134–3146. doi: 10.1128/jvi.01394-09
Guan, Y., Zheng, B. J., He, Y. Q., Liu, X. L., Zhuang, Z. X., Cheung, C. L., et al. (2003). Isolation and characterization of viruses related to the SARS coronavirus from animals in southern China. Science 302 (5643), 276–278. doi: 10.1126/science.1087139
Gu, Y., Cao, J., Zhang, X., Gao, H., Wang, Y., Wang, J., et al. (2022). Receptome profiling identifies KREMEN1 and ASGR1 as alternative functional receptors of SARS-CoV-2. Cell Res. 32 (1), 24–37. doi: 10.1038/s41422-021-00595-6
Haan, C. D., Lintelo, E. T., Li, Z., Raaben, M., Wurdinger, T., Bosch, B. J., et al. (2006). Cooperative involvement of the S1 and S2 subunits of the murine coronavirus spike protein in receptor binding and extended host range. J. Virol. 80 (22), 10909–10918. doi: 10.1128/JVI.00950-06
Han, B. A., Schmidt, J. P., Bowden, S. E., Drake, J. M. (2015). Rodent reservoirs of future zoonotic diseases. Proc Natl Acad Sci U. S. A. 112 (22), 7039–7044.
Hemmila, E., Turbide, C., Olson, M., Jothy, S., Holmes, K. V., Beauchemin, N. (2004). Ceacam1a-/- mice are completely resistant to infection by murine coronavirus mouse hepatitis virus A59. J. Virol. 78 (18), 10156–10165. doi: 10.1128/jvi.78.18.10156-10165.2004
Herrewegh, A. A., Smeenk, I., Horzinek, M. C., Rottier, P. J., de Groot, R. J. (1998). Feline coronavirus type II strains 79-1683 and 79-1146 originate from a double recombination between feline coronavirus type I and canine coronavirus. J. Virol. 72 (5), 4508–4514. doi: 10.1128/jvi.72.5.4508-4514.1998
Hoffmann, M., Pöhlmann, S. (2022). Novel SARS-CoV-2 receptors: ASGR1 and KREMEN1. Cell Res. 32 (1), 1–2. doi: 10.1038/s41422-021-00603-9
Hofmann, H., Pyrc, K., Van, D., Geier, M., Berkhout, B., Pohlmann, S. (2005). Human coronavirus NL63 employs the severe acute respiratory syndrome coronavirus receptor for cellular entry. Proc. Natl. Acad. Sci. United States America 102 (22), 7988–7993. doi: 10.1073/pnas.0409465102
Huang, Y., Yang, C., Xu, X. F., Xu, W., Liu, S. W. (2020b). Structural and functional properties of SARS-CoV-2 spike protein: potential antivirus drug development for COVID-19. Acta Pharmacol. Sin. 41 (9), 1141–1149. doi: 10.1038/s41401-020-0485-4
Huang, H., Yin, Y., Wang, W., Cao, L., Sun, W., Shi, K., et al. (2020a). Emergence of Thailand-like strains of porcine deltacoronavirus in guangxi province, China. Vet. Med. Sci. 6 (4), 854–859. doi: 10.1002/vms3.283
Hulswit, R. J. G., Lang, Y., Bakkers, M. J. G., Li, W., Li, Z., Schouten, A., et al. (2019). Human coronaviruses OC43 and HKU1 bind to 9-o-acetylated sialic acids via a conserved receptor-binding site in spike protein domain a. Proc. Natl. Acad. Sci. U.S.A. 116 (7), 2681–2690. doi: 10.1073/pnas.1809667116
Hunter, J. C., Nguyen, D., Aden, B., Al Bandar, Z., Al Dhaheri, W., Abu Elkheir, K., et al. (2016). Transmission of middle East respiratory syndrome coronavirus infections in healthcare settings, Abu Dhabi. Emerg. Infect. Dis. 22 (4), 647–656. doi: 10.3201/eid2204.151615
Hu, D., Zhu, C., Ai, L., He, T., Wang, Y., Ye, F., et al. (2018). Genomic characterization and infectivity of a novel SARS-like coronavirus in Chinese bats. Emerg. Microbes Infect. 7 (1), 154. doi: 10.1038/s41426-018-0155-5
Islam, A., Ferdous, J., Islam, S., Abu Sayeed, M., Choudhury, S. D., Saha, O., et al. (2021). Evolutionary dynamics and epidemiology of endemic and emerging coronaviruses in humans, domestic animals, and wildlife. Viruses 13 (10). doi: 10.3390/v13101908
Janetanakit, T., Lumyai, M., Bunpapong, N., Boonyapisitsopa, S., Chaiyawong, S., Nonthabenjawan, N., et al. (2016). Porcine deltacoronavirus, thailan. Emerg. Infect. Dis. 22 (4), 757–759. doi: 10.3201/eid2204.151852
Jung, K., Hu, H., Eyerly, B., Lu, Z., Chepngeno, J., Saif, L. J. (2015). Pathogenicity of 2 porcine deltacoronavirus strains in gnotobiotic pigs. Emerg. Infect. Dis. 21 (4), 650–654. doi: 10.3201/eid2104.141859
Ju, B., Zhang, Q., Ge, J., Wang, R., Zhang, L. (2020). Human neutralizing antibodies elicited by SARS-CoV-2 infection. Nature 115-119. doi: 10.1038/s41586-020-2380-z
Keck, J. G., Soe, L. H., Makino, S., Stohlman, S. A., Lai, M. M. (1988). RNA Recombination of murine coronaviruses: recombination between fusion-positive mouse hepatitis virus A59 and fusion-negative mouse hepatitis virus 2. J. Virol. 62 (6), 1989–1998. doi: 10.1128/jvi.62.6.1989-1998.1988
Kesheh, M. M., Hosseini, P., Soltani, S., Zandi, M. (2022). An overview on the seven pathogenic human coronaviruses. Rev. Med. Virol. 32 (2), e2282. doi: 10.1002/rmv.2282
Ksiazek, T. G., Erdman, D., Goldsmith, C. S., Zaki, S. R., Peret, T., Emery, S., et al. (2003). A novel coronavirus associated with severe acute respiratory syndrome. N Engl. J. Med. 348 (20), 1953–1966. doi: 10.1056/NEJMoa030781
Lai, M. M. (1992). RNA Recombination in animal and plant viruses. Microbiol. Rev. 56 (1), 61–79. doi: 10.1128/mr.56.1.61-79.1992
Langer, P., Giessen (2007). Mammal species of the world: A taxonomic and geographic reference. Mamm. Biol. 72 (3), 191. doi: 10.1016/j.mambio.2006.02.003
Lauring, A. S., Frydman, J., Andino, R. (2013). The role of mutational robustness in RNA virus evolution. Nat. Rev. Microbiol. 11 (5), 327–336. doi: 10.1038/nrmicro3003
Lau, S. K. P., Wong, E. Y. M., Tsang, C. C., Ahmed, S. S., Au-Yeung, R. K. H., Yuen, K. Y., et al. (2018). Discovery and sequence analysis of four deltacoronaviruses from birds in the middle East reveal interspecies jumping with recombination as a potential mechanism for avian-to-Avian and avian-to-Mammalian transmission. J. Virol. 92 (15). doi: 10.1128/jvi.00265-18
Lednicky, J. A., Tagliamonte, M. S., White, S. K., Blohm, G. M., Alam, M. M., Iovine, N. M., et al. (2022). Isolation of a novel recombinant canine coronavirus from a visitor to Haiti: Further evidence of transmission of coronaviruses of zoonotic origin to humans. Clin. Infect. Dis. 75 (1), e1184–e1187. doi: 10.1093/cid/ciab924
Lee, N., Hui, D., Wu, A., Chan, P., Cameron, P., Joynt, G. M., et al. (2003). A major outbreak of severe acute respiratory syndrome in Hong Kong. New Engl. J. OF Med. 348 (20), 1986–1994. doi: 10.1056/NEJMoa030685
Lee, C. W., Jackwood, M. W. (2000). Evidence of genetic diversity generated by recombination among avian coronavirus IBV. Arch. Virol. 145 (10), 2135–2148. doi: 10.1007/s007050070044
Lee, S. S., Wong, N. S. (2015). Probable transmission chains of middle East respiratory syndrome coronavirus and the multiple generations of secondary infection in south Korea. Int. J. Infect. Dis. 38, 65–67. doi: 10.1016/j.ijid.2015.07.014
Legnardi, M., Tucciarone, C. M., Franzo, G., Cecchinato, M. (2020). Infectious bronchitis virus evolution, diagnosis and control. Veterinary Sci. 7 (2), 79. doi: 10.3390/vetsci7020079
Le, V. P., Song, S., An, B. H., Park, G. N., Pham, N. T., Le, D. Q., et al. (2018). A novel strain of porcine deltacoronavirus in Vietnam. Arch. Virol. 163 (1), 203–207. doi: 10.1007/s00705-017-3594-8
Li, Q., Guan, X., Wu, P., Wang, X., Zhou, L., Tong, Y., et al. (2020a). Early transmission dynamics in wuhan, China, of novel coronavirus-infected pneumonia. New Engl. J. Med. 382 (13), 1199–1207. doi: 10.1056/NEJMoa2001316
Li, C. X., Noreen, S., Zhang, L. X., Saeed, M., Wu, P. F., Ijaz, M., et al. (2022a). A critical analysis of SARS-CoV-2 (COVID-19) complexities, emerging variants, and therapeutic interventions and vaccination strategies. BioMed. Pharmacother. 146, 112550. doi: 10.1016/j.biopha.2021.112550
Liu, S., Chen, J., Chen, J., Kong, X., Shao, Y., Han, Z., et al. (2005). Isolation of avian infectious bronchitis coronavirus from domestic peafowl (Pavo cristatus) and teal (Anas). J. Gen. Virol. 86 (3), 719–725. doi: 10.1099/vir.0.80546-0
Liu, D. X., Liang, J. Q., Fung, T. S. (2021). Human coronavirus-229E, -OC43, -NL63, and -HKU1 (Coronaviridae). Encyclopedia Virol. 428-440, 75–86. doi: 10.1016/B978-0-12-809633-8.21501-X
Liu, P., Shi, L., Zhang, W., He, J., Liu, C., Zhao, C., et al. (2017). Prevalence and genetic diversity analysis of human coronaviruses among cross-border children. Virol. J. 14 (1), 230. doi: 10.1186/s12985-017-0896-0
Liu, Q., Wang, H. Y. (2021). Porcine enteric coronaviruses: an updated overview of the pathogenesis, prevalence, and diagnosis. VETERINARY Res. Commun. 45 (2-3), 75–86. doi: 10.1007/s11259-021-09808-0
Li, X., Zai, J., Zhao, Q., Nie, Q., Li, Y., Foley, B. T., et al. (2020b). Evolutionary history, potential intermediate animal host, and cross-species analyses of SARS-CoV-2. J. Med. Virol. 92 (6), 602–611. doi: 10.1002/jmv.25731
Li, G., Zhai, S. L., Zhou, X., Chen, T. B., Niu, J. W., Xie, Y. S., et al. (2022b). Phylogeography and evolutionary dynamics analysis of porcine delta-coronavirus with host expansion to humans. Transbound Emerg. Dis. 69 (5), 1670–1681. doi: 10.1111/tbed.14503
Li, Y., Zhuang, Q., Jiang, L., Jiang, W., Peng, C., Jiang, N., et al. (2021). Traceable surveillance and genetic diversity analysis of coronaviruses in poultry from China in 2019. Virus Res. 306, 198566. doi: 10.1016/j.virusres.2021.198566
Louz, D., Bergmans, H. E., Loos, B. P., Hoeben, R. C. (2005). Cross-species transfer of viruses: implications for the use of viral vectors in biomedical research, gene therapy and as live-virus vaccines. J. Gene Med. 7 (10), 1263–1274. doi: 10.1002/jgm.794
Lu, R., Zhao, X., Li, J., Niu, P., Yang, B., Wu, H., et al. (2020). Genomic characterisation and epidemiology of 2019 novel coronavirus: implications for virus origins and receptor binding. Lancet 395 (10224), 565–574. doi: 10.1016/s0140-6736(20)30251-8
Lv, D., Dong, Z. H., Fan, W. S., Tang, N., Wang, L., Wei, L. P., et al. (2021). Identification of a novel avian coronavirus infectious bronchitis virus variant with three-nucleotide-deletion in nucleocapsid gene in China. J. OF VETERINARY Med. Sci. 83 (10), 1608–1619. doi: 10.1292/jvms.21-0351
Mackenzie, J. S., Smith, D. W. (2020). COVID-19: a novel zoonotic disease caused by a coronavirus from China: what we know and what we don't. Microbiol. Aust. MA20013. doi: 10.1071/ma20013
Malpica, J. M., Fraile, A., Moreno, I., Obies, C. I., Drake, J. W., García-Arenal, F. (2002). The rate and character of spontaneous mutation in an RNA virus. Genetics 162 (4), 1505–1511. doi: 10.1093/genetics/162.4.1505
Martin, D. P., van der Walt, E., Posada, D., Rybicki, E. P. (2005). The evolutionary value of recombination is constrained by genome modularity. PLoS Genet. 1 (4), e51. doi: 10.1371/journal.pgen.0010051
Ma, Y., Zhang, Y., Liang, X., Lou, F., Oglesbee, M., Krakowka, S., et al. (2015). Origin, evolution, and virulence of porcine deltacoronaviruses in the united states. mBio 6 (2), e00064. doi: 10.1128/mBio.00064-15
Milewska, A., Zarebski, M., Nowak, P., Stozek, K., Potempa, J., Pyrc, K. (2014). Human coronavirus NL63 utilizes heparan sulfate proteoglycans for attachment to target cells. J. OF Virol. 88 (22), 13221–13230. doi: 10.1128/JVI.02078-14
Moreira, R. A., Guzman, H. V., Boopathi, S., Baker, J. L., Poma, A. B. (2020). Characterization of structural and energetic differences between conformations of the SARS-CoV-2 spike protein. Materials (Basel) 13 (23). doi: 10.3390/ma13235362
Mulabbi, E. N., Tweyongyere, R., Byarugaba, D. K. (2021). The history of the emergence and transmission of human coronaviruses. Onderstepoort J. Vet. Res. 88 (1), e1–e8. doi: 10.4102/ojvr.v88i1.1872
Müller, A., Tillmann, R. L., Müller, A., Simon, A., Schildgen, O. (2008). Stability of human metapneumovirus and human coronavirus NL63 on medical instruments and in the patient environment. J. Hosp Infect. 69 (4), 406–408. doi: 10.1016/j.jhin.2008.04.017
Ng, L., Wong, S. K., Huang, Z., Lam, C. S., Chow, A. K., Foo, D. C., et al. (2022). CD26 induces colorectal cancer angiogenesis and metastasis through CAV1/MMP1 signaling. Int. J. Mol. Sci. 23 (3). doi: 10.3390/ijms23031181
Oguzoglu, T. C., Koc, B. T., Akkutay-Yoldar, A. Z. (2021). Triple viral infections in the same cats: Feline coronavirus, feline parvovirus, feline foamy virus. Rev. MVZ CORDOBA 26 (3). doi: 10.21897/rmvz.2182
Olival, K. J., Hosseini, P. R., Zambrana-Torrelio, C., Ross, N., Bogich, T. L., Daszak, P. (2017). Host and viral traits predict zoonotic spillover from mammals. NATURE 546 (7660), 646–64+. doi: 10.1038/nature22975
Pan, Y., Tian, X., Qin, P., Wang, B., Zhao, P., Yang, Y. L., et al. (2017). Discovery of a novel swine enteric alphacoronavirus (SeACoV) in southern China. Veterinary Microbiol. 15, 15–21. doi: 10.1016/j.vetmic.2017.09.020
Pfefferle, S., Oppong, S., Drexler, J. F., Gloza-Rausch, F., Ipsen, A., Seebens, A., et al. (2009). Distant relatives of severe acute respiratory syndrome coronavirus and close relatives of human coronavirus 229E in bats, Ghana. Emerg. Infect. Dis. 15 (9), 1377–1384. doi: 10.3201/eid1509.090224
Plowright, R. K., Eby, P., Hudson, P. J., Smith, I. L., Westcott, D., Bryden, W. L., et al. (2015). Ecological dynamics of emerging bat virus spillover. Proc. Biol. 282 (282). doi: 10.1098/rspb.2014.2124
Priestnall, S. L. (2020). Canine respiratory coronavirus: A naturally occurring model of COVID-19? Veterinary Pathol. 57 (4), 467–471. doi: 10.1177/0300985820926485
Pringle, C. R. (1996). Virus taxonomy 1996 - a bulletin from the xth international congress of virology in Jerusalem. Arch. Virol. 141 (11), 2251–2256. doi: 10.1007/bf01718231
Rohaim, M. A., El Naggar, R. F., Helal, A. M., Hussein, H. A., Munir, M. (2017). Reverse spillover of avian viral vaccine strains from domesticated poultry to wild birds. Vaccine 35 (28), 3523–3527. doi: 10.1016/j.vaccine.2017.05.033
Sabir, J. S., Lam, T. T., Ahmed, M. M., Li, L., Shen, Y., Abo-Aba, S. E., et al. (2016). Co-Circulation of three camel coronavirus species and recombination of MERS-CoVs in Saudi Arabia. Science 351 (6268), 81–84. doi: 10.1126/science.aac8608
Saeng-Chuto, K., Lorsirigool, A., Temeeyasen, G., Vui, D. T., Stott, C. J., Madapong, A., et al. (2017). Different lineage of porcine deltacoronavirus in Thailand, Vietnam and lao PDR in 2015. Transbound Emerg. Dis. 64 (1), 3–10. doi: 10.1111/tbed.12585
Schoeman, D., Gordon, B., Fielding, B. C. (2021). Pathogenic human coronaviruses. Reference Module Biomed. Sci. 2021, 241–258. doi: 10.1016/B978-0-12-818731-9.00052-5
Shah, T., Xia, K.-Y., Shah, Z., Baloch, Z. (2022). Therapeutic mechanisms and impact of traditional Chinese medicine on COVID-19 and other influenza diseases. Pharmacol. Res. - Modern Chin. Med. 2, 100029. doi: 10.1016/j.prmcm.2021.100029
Shi, M., Lin, X. D., Chen, X., Tian, J. H., Chen, L. J., Li, K., et al. (2018). The evolutionary history of vertebrate RNA viruses. Nature 556 (7700), 197–202. doi: 10.1038/s41586-018-0012-7
Simon-Loriere, E., Galetto, R., Hamoudi, M., Archer, J., Lefeuvre, P., Martin, D. P., et al. (2009). Molecular mechanisms of recombination restriction in the envelope gene of the human immunodeficiency virus. PLoS Pathog. 5 (5), e1000418. doi: 10.1371/journal.ppat.1000418
Simon-Loriere, E., Holmes, E. C. (2011). Why do RNA viruses recombine? Nat. Rev. Microbiol. 9 (8), 617–626. doi: 10.1038/nrmicro2614
Singh, D. R., Pandey, K., Mishra, A. K., Pandey, P., Vivcharuk, V. (2021). Glutamate binding triggers monomerization of unliganded mGluR2 dimers. Arch. Biochem. Biophys. 697, 108632. doi: 10.1016/j.abb.2020.108632
Sipulwa, L. A., Ongus, J. R., Coldren, R. L., Bulimo, W. D. (2016). Molecular characterization of human coronaviruses and their circulation dynamics in kenya 2009-2012. Virol. J. 13, 18. doi: 10.1186/s12985-016-0474-x
Smith, G. J., Vijaykrishna, D., Bahl, J., Lycett, S. J., Worobey, M., Pybus, O. G., et al. (2009). Origins and evolutionary genomics of the 2009 swine-origin H1N1 influenza a epidemic. Nature 459 (7250), 1122–1125. doi: 10.1038/nature08182
Su, S., Wong, G., Shi, W., Liu, J., Lai, A. C. K., Zhou, J., et al. (2016). Epidemiology, genetic recombination, and pathogenesis of coronaviruses. Trends Microbiol. 24 (6), 490–502. doi: 10.1016/j.tim.2016.03.003
Suzuki, T., Shibahara, T., Imai, N., Yamamoto, T., Ohashi, S. (2018). Genetic characterization and pathogenicity of Japanese porcine deltacoronavirus. Infect. Genet. Evol. 61, 176–182. doi: 10.1016/j.meegid.2018.03.030
Tong, S., Conrardy, C., Ruone, S., Kuzmin, I. V., Guo, X., Tao, Y., et al. (2009). Detection of novel SARS-like and other coronaviruses in bats from Kenya. Emerg. Infect. Dis. 15 (3), 482–485. doi: 10.3201/eid1503.081013
Torres, C. A., Listorti, V., Lupini, C., Franzo, G., Drigo, M., Catelli, E., et al. (2017). Gamma and deltacoronaviruses in quail and pheasants from northern Italy1. Poultry Sci. 96 (3), 717–722. doi: 10.3382/ps/pew332
van Doremalen, N., Bushmaker, T., Morris, D. H., Holbrook, M. G., Gamble, A., Williamson, B. N., et al. (2020). Aerosol and surface stability of SARS-CoV-2 as compared with SARS-CoV-1. N Engl. J. Med. 382 (16). doi: 10.1056/NEJMc2004973
van Doremalen, N., Bushmaker, T., Munster, V. J. (2013). Stability of middle East respiratory syndrome coronavirus (MERS-CoV) under different environmental conditions. Euro Surveill 18 (38), 1564–1567. doi: 10.2807/1560-7917.es2013.18.38.20590
Vlasova, A. N., Saif, L. J. (2021). Bovine coronavirus and the associated diseases. Front. Veterinary Sci. 8, 643220. doi: 10.3389/fvets.2021.643220
Walker, P. J., Siddell, S. G., Lefkowitz, E. J., Mushegian, A. R., Dempsey, D. M., Dutilh, B. E., et al. (2019). Changes to virus taxonomy and the international code of virus classification and nomenclature ratified by the international committee on taxonomy of viruses, (2019). Arch. Virol. 164 (9), 2417–2429. doi: 10.1007/s00705-019-04306-w
Walls, A. C., Park, Y. J., Tortorici, M. A., Wall, A., Mcguire, A. T., Veesler, D. (2020). Structure, function and antigenicity of the SARS-CoV-2 spike glycoprotein. Cell 181 (2), 281–292. doi: 10.1016/j.cell.2020.02.058
Walsh, E. E., Shin, J. H., Falsey, A. R. (2013). Clinical impact of human coronaviruses 229E and OC43 infection in diverse adult populations. J. Infect. Dis. 208 (10), 1634–1642. doi: 10.1093/infdis/jit393
Wang, W., Lin, X. D., Guo, W. P., Zhou, R. H., Wang, M. R., Wang, C. Q., et al. (2015). Discovery, diversity and evolution of novel coronaviruses sampled from rodents in China. Virology 474, 19–27. doi: 10.1016/j.virol.2014.10.017
Wang, B., Potter, S. J., Lin, Y., Cunningham, A. L., Dwyer, D. E., Su, Y., et al. (2005). Rapid and sensitive detection of severe acute respiratory syndrome coronavirus by rolling circle amplification. J. Clin. Microbiol. 43 (5), 2339–2344. doi: 10.1128/jcm.43.5.2339-2344.2005
Wang, S., Qiu, Z., Hou, Y., Deng, X., Xu, W., Zheng, T., et al. (2021b). AXL is a candidate receptor for SARS-CoV-2 that promotes infection of pulmonary and bronchial epithelial cells. Cell Res. 31 (2), 126–140. doi: 10.1038/s41422-020-00460-y
Wang, N., Shang, J., Jiang, S., Du, L. (2020b). Subunit vaccines against emerging pathogenic human coronaviruses. Front. Microbiol. 11. doi: 10.3389/fmicb.2020.00298
Wang, L. F., Shi, Z., Zhang, S., Field, H., Daszak, P., Eaton, B. T. (2006). Review of bats and SARS. Emerg. Infect. Dis. 12 (12), 1834–1840. doi: 10.3201/eid1212.060401
Wang, J., Yang, G., Wang, X., Wen, Z., Shuai, L., Luo, J., et al. (2021a). SARS-CoV-2 uses metabotropic glutamate receptor subtype 2 as an internalization factor to infect cells. Cell Discovery 7 (1), 119. doi: 10.1038/s41421-021-00357-z
Wan, Y., Shang, J., Graham, R., Baric, R. S., Li, F. (2020). Receptor recognition by the novel coronavirus from wuhan: an analysis based on decade-long structural studies of SARS coronavirus. J. Virol. 94 (7). doi: 10.1128/JVI.00127-20
Weiss, S. R., Navas-Martin, S. (2005). Coronavirus pathogenesis and the emerging pathogen severe acute respiratory syndrome coronavirus. Microbiol. Mol. Biol. Rev. 69 (4), 635–664. doi: 10.1128/mmbr.69.4.635-664.2005
WHO (2020) WHO director-general's opening remarks at the media briefing on COVID-19-20 November 2020. Available at: https://reliefweb.int/report/world/who-director-generals-opening-remarks-media-briefing-covid-19-20-november-2020?gclid=EAIaIQobChMIh7m_58n8-QIVjwByCh14RwKZEAAYASAAEgLePPD_BwE (Accessed 2022-09-05).
Wille, M., Holmes, E. C. (2020). Wild birds as reservoirs for diverse and abundant gamma- and deltacoronaviruses. FEMS Microbiol. Rev. 44 (5), 631–644. doi: 10.1093/femsre/fuaa026
Wong, A. C. P., Lau, S. K. P., Woo, P. C. Y. (2021). Interspecies jumping of bat coronaviruses. Viruses 13 (11). doi: 10.3390/v13112188
Woo, P. C., Lau, S. K., Chu, C. M., Chan, K. H., Tsoi, H. W., Huang, Y., et al. (2005). Characterization and complete genome sequence of a novel coronavirus, coronavirus HKU1, from patients with pneumonia. J. Virol. 79 (2), 884–895. doi: 10.1128/jvi.79.2.884-895.2005
Woo, P. C., Lau, S. K., Lam, C. S., Lau, C. C., Tsang, A. K., Lau, J. H., et al. (2012). Discovery of seven novel mammalian and avian coronaviruses in the genus deltacoronavirus supports bat coronaviruses as the gene source of alphacoronavirus and betacoronavirus and avian coronaviruses as the gene source of gammacoronavirus and deltacoronavirus. J. Virol. 86 (7), 3995–4008. doi: 10.1128/jvi.06540-11
Worobey, M., Levy, J. I., Malpica Serrano, L., Crits-Christoph, A., Pekar, J. E., Goldstein, S. A., et al. (2022). The huanan seafood wholesale market in wuhan was the early epicenter of the COVID-19 pandemic. Science 377 (6609), 951–959. doi: 10.1126/science.abp8715
Yadav, R., Chaudhary, J. K., Jain, N., Chaudhary, P. K., Khanra, S., Dhamija, P., et al. (2021). Role of structural and non-structural proteins and therapeutic targets of SARS-CoV-2 for COVID-19. Cells 10 (4). doi: 10.3390/cells10040821
Ye, Z. W., Yuan, S., Yuen, K. S., Fung, S. Y., Jin, D. Y. (2020). Zoonotic origins of human coronaviruses. Int. J. Biol. Sci. 16 (10), 1686–1697. doi: 10.7150/ijbs.45472
Yin, Y., Wunderink, R. G. (2018). MERS, SARS and other coronaviruses as causes of pneumonia. Respirology 23 (2), 130–137. doi: 10.1111/resp.13196
Young, B. E., Ong, S. W. X., Kalimuddin, S., Low, J. G., Tan, S. Y., Loh, J., et al. (2020). Epidemiologic features and clinical course of patients infected with SARS-CoV-2 in Singapore. JAMA 323 (15), 1488–1494. doi: 10.1001/jama.2020.3204
Yu, X. J., Luo, C., Lin, J. C., Hao, P., He, Y. Y., Guo, Z. M., et al. (2003). Putative hAPN receptor binding sites in SARS_CoV spike protein. Acta Pharmacol. Sin. 24 (6), 481–488.
Zaki, A. M., van Boheemen, S., Bestebroer, T. M., Osterhaus, A. D., Fouchier, R. A. (2012). Isolation of a novel coronavirus from a man with pneumonia in Saudi Arabia. N Engl. J. Med. 367 (19), 1814–1820. doi: 10.1056/NEJMoa1211721
Zhang, W., Du, R. H., Li, B., Zheng, X. S., Yang, X. L., Hu, B., et al. (2020). Molecular and serological investigation of 2019-nCoV infected patients: implication of multiple shedding routes. Emerg. Microbes Infect. 9 (1), 386–389. doi: 10.1080/22221751.2020.1729071
Zhang, S. F., Tuo, J. L., Huang, X. B., Zhu, X., Zhang, D. M., Zhou, K., et al. (2018). Epidemiology characteristics of human coronaviruses in patients with respiratory infection symptoms and phylogenetic analysis of HCoV-OC43 during 2010-2015 in guangzhou. PLoS One 13 (1), e0191789. doi: 10.1371/journal.pone.0191789
Zhou, P., Yang, X. L., Wang, X. G., Hu, B., Zhang, L., Zhang, W., et al. (2020). A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature 579 (7798), 270–273. doi: 10.1038/s41586-020-2012-7
Zhu, Q., Li, B., Sun, D. (2022). Advances in bovine coronavirus epidemiology. Viruses 14 (5). doi: 10.3390/v14051109
Keywords: coronaviruses, epidemiology, evolution, cross-species transmission, zoonosis
Citation: Li Q, Shah T, Wang B, Qu L, Wang R, Hou Y, Baloch Z and Xia X (2023) Cross-species transmission, evolution and zoonotic potential of coronaviruses. Front. Cell. Infect. Microbiol. 12:1081370. doi: 10.3389/fcimb.2022.1081370
Received: 01 November 2022; Accepted: 19 December 2022;
Published: 06 January 2023.
Edited by:
Yongfen Xu, Institut Pasteur of Shanghai, Chinese Academy of Sciences (CAS), ChinaReviewed by:
Sonia Zuñiga, National Center for Biotechnology, Spanish National Research Council (CSIC), SpainShengyan Gao, University of Texas Southwestern Medical Center, United States
Copyright © 2023 Li, Shah, Wang, Qu, Wang, Hou, Baloch and Xia. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Xueshan Xia, b2xpdmVyeGlhMjAwMEBhbGl5dW4uY29t
†These authors have contributed equally to this work