 Bálint Gergely Szabó1,2†
Bálint Gergely Szabó1,2† Rebeka Kiss1†Nóra Makra3Kinga Pénzes3Eszter Vad1Katalin Kamotsay1
Rebeka Kiss1†Nóra Makra3Kinga Pénzes3Eszter Vad1Katalin Kamotsay1 Dóra Szabó3
Dóra Szabó3 Eszter Ostorházi3*
Eszter Ostorházi3*- 1South Pest Central Hospital, National Institute of Hematology and Infectious Diseases, Budapest, Hungary
- 2Departmental Group of Infectious Diseases, Department of Haematology and Internal Medicine, Semmelweis University, Budapest, Hungary
- 3Institute of Medical Microbiology, Semmelweis University, Budapest, Hungary
Background: Characteristics of the blood microbiota among adult patients with community-acquired sepsis are poorly understood. Our aim was to analyze the composition of blood microbiota in adult patients with community-acquired sepsis, and correlate changes with non-septic control patients.
Methods: A prospective observational study was carried out by including adult patients hospitalized for community-acquired sepsis at our center between January and November 2019, by random selection from a pool of eligible patients. Study inclusion was done on the day of sepsis diagnosis. Community acquisition was ascertained by a priori exclusion criteria; sepsis was defined according to the SEPSIS-3 definitions. Each included patient was matched with non-septic control patients by age and gender in a 1:1 fashion enrolled from the general population. Conventional culturing with BacT/ALERT system and 16S rRNA microbiota analysis were performed from blood samples taken in a same time from a patient. Abundance data was analyzed by the CosmosID HUB Microbiome software.
Results: Altogether, 13 hospitalized patients were included, 6/13 (46.2%) with sepsis and 7/13 (53.8%) with septic shock at diagnosis. The most prevalent etiopathogen isolated from blood cultures was Escherichia coli, patients mostly had intraabdominal septic source. At day 28, all-cause mortality was 15.4% (2/13). Compared to non-septic control patients, a relative scarcity of Faecalibacterium, Blautia, Coprococcus and Roseburia genera, with an abundance of Enhydrobacter, Pseudomonas and Micrococcus genera was observed among septic patients. Relative differences between septic vs. non-septic patients were more obvious at the phylum level, mainly driven by Firmicutes (25.7% vs. 63.1%; p<0.01) and Proteobacteria (36.9% vs. 16.6%; p<0.01). The alpha diversity, quantified by the Chao1 index showed statistically significant difference between septic vs. non-septic patients (126 ± 51 vs. 66 ± 26; p<0.01). The Bray-Curtis beta diversity, reported by principal coordinate analysis of total hit frequencies, revealed 2 potentially separate clusters among septic vs. non-septic patients.
Conclusion: In adult patients with community-acquired sepsis, specific changes in the composition and abundance of blood microbiota could be detected by 16S rRNA metagenome sequencing, compared to non-septic control patients. Traditional blood culture results only partially correlate with microbiota test results.
1. Introduction
Sepsis is a dysregulated host immune response arising from an infection, which may lead to multi-systemic inflammation, organ failure and ultimately, patient death (Singer et al., 2016). According to recent data from the Centers for Disease Control and Prevention, as high as 87% of sepsis present before the patient is admitted to a healthcare facility, pointing to a significant community-level acquision in a relevant proportion of all cases (CDC, 2022). By most literature sources, community-acquired sepsis is defined as a case of sepsis with any severity (given by the either ACCP/SCCM SIRS based or the SEPSIS-3 consensus criteria), and a clinical onset between 48 to 72 hours after hospital admission, in a patient without any recent connection to healthcare facilities or nosocomial activities (Szabo et al., 2019).
According to the literature, the spectrum of etiopathogens most frequently isolated from clinical cases of community-acquired sepsis in immunocompetent adults is similar across different studies: the main isolated organism is Escherichia coli, followed by Staphylococcus aureus, Streptococcus pneumoniae and Klebsiella pneumoniae, lastly other Streptococci and Gram negative bacteria. However, by utilizing commercially available classical culturing or non-culture based microbiological methods (eg. antigen detection, serology or polymerase chain reaction [PCR] assays), an etiology could only be documented in 60–75% of all cases, even with concomitant bacteremia rates of community-acquired sepsis falling between 15 and 35% (Goncalves-Pereira et al., 2013; Sogaard et al., 2015; Henriksen et al., 2017). These limitations might affect clinical risk stratification and antimicrobial stewardship efforts of targeted de-escalation.
In recent years, the existence of an “eubiotic” blood microbiota has been proposed in healthy human individuals by the literature. One review assessed that its three chief components, detected mainly by 16s rRNA gene sequencing at the phyla level, were Proteobacteria, Actinobacteria and Firmicutes, as they contributed to >70% of the blood microbiota, while Bacteroidetes, Fusobacteria, Cyanobacteria, Verrucomicrobia and Acidobacteria only constitute the minority. In contrast, Firmicutes and Bacteroidetes are the typical phyla of the human gut microbiome (Suparan et al., 2022). Despite encouraging results, another review found that the main limitation to properly interpret the phylogenetic diversity of the healthy human blood microbiota lies in significant inter-research variability (Castillo et al., 2019). In septic patients, a “dysbiotic” shift of blood microbiota has been observed by some authors, with a higher ratio of the phyla Proteobacteria and Bacteroidetes, along with a reduction in Actinobacteria and a rise in Agrococcus (Paisse et al., 2016; Gosiewski et al., 2017). However, characteristic changes of the blood microbiota abundance among adults with community-acquired sepsis are still poorly understood, despite the vast rates of this clinical scenario among all documented septic cases, and needs further evaluation. Therefore, our aim was to assess the composition and changes of blood microbiota in adult patients with community-acquired sepsis, and correlate these changes to non-septic control patients.
2. Materials and methods
2.1. Study design, patient enrollment and patient–control matching
A prospective observational study was carried out by including patients from a cohort of eligible patients by computerized random selection on the day of sepsis diagnosis ascertainment. All consecutive adult (≥18 years of age at diagnosis) patients hospitalized for community-acquired sepsis at the study site between January and November 2019 were eligible for inclusion. The study site is a national-level referral center with >200 ward and intensive care unit beds for infectious and malignant hematological diseases. The study was in accordance with the Helsinki Declaration and national ethical standards. The study protocol was approved by the Institutional Review Board of Semmelweis University (Permission Number: SE RKEB: 87/2019) and South Pest Central Hospital, National Institute of Hematology and Infectious Diseases (Permission Number: ESZSZK EB 37/2016). Written informed consent was obtained from each included patient.
Sepsis was defined according to the SEPSIS-3 consensus definitions (Singer et al., 2016). Community acquisition was ascertained if sepsis onset or diagnosis was within ≤72 h from hospital admission or transportation occurred to the study site from the community or another hospital within the time frame, plus by applying a priori exclusion criteria, detailed in extenso in an earlier publication of our group (Szabo et al., 2019). Briefly, patients with nosocomial infections of any kind, recent hospitalizations, regular outpatient visits to clinics or long-term care facility residence were excluded. Each included septic patient was matched by age and gender (or best available option) with a non-septic control patient in a 1:1 ratio, enrolled from the general non-hospitalized population.
2.2. Data collection, follow-up and outcomes
Patient-level data were anonymously transferred to a standardized electronic case report form by manual extraction from healthcare charts. Data collected were: 1) age and gender, 2) comorbidities, 3) clinical presentation (symptom duration before hospitalization, severity and source of sepsis, length of hospitalization), 4) laboratory results (peripheral white blood cell count, absolute neutrophil granulocyte and lymphocyte counts, serum C-reactive protein [CRP] and procalcitonin [PCT] levels), 5) microbiological results (blood culture and 16S rRNA microbiota analyzes), 6) types and durations of empirical and targeted antimicrobial therapies, 7) clinical outcomes.
At the study site, diagnostic and therapeutic approach of septic patients was facilitated by an annually updated in-house written protocol since 2016. Patients were followed up for a total of 28 days, a post-discharge follow-up was done by telephone, e-mails and cross-checking in the National eHealth Infrastructure (social security database of Hungary). Clinical outcomes were all-cause mortality and need for intensive care unit (ICU) admission. Baseline variables were collected on the day of sepsis diagnosis, microbiological data and clinical outcomes were ascertained at 28 days post-admission.
2.3. Traditional blood culture method
From each participant, up to 10 mL per bottle of venous whole blood was inoculated into BacT/ALERT FA (bioMérieux, Marcy-l’Étoile, France) culture bottles for the cultivation of aerobic bacteria and fungi (2 bottles per patient), and another 10 mL of whole blood was inoculated into FN bottles for the cultivation of anaerobic bacteria promptly after sepsis diagnosis (2 bottles per patient). The skin site was mechanically cleaned with soap, and disinfected with 10% povidone-iodine and 2% chlorhexidine digluconate + 95% ethyl alcohol solutions three times before venipuncture. Until transportation to the laboratory, inoculated bottles were stored at +24°C. At the laboratory, these were then placed in an automated microbial detection system at +37°C (BACT/ALERT 3D, bioMérieux, Marcy-l’Étoile, France). After a positive signal of potential growth was detected, a Gram stained smear was prepared and aerobic, anaerobic and fungal cultivation began on the appropriate solid media. Negative blood culture results were interpreted on the basis of negative microscopic and culture procedures, after incubation of the bottles for 3 weeks. Identification of an organisms isolated from a positive blood culture was accomplished by matrix-assisted laser desorption/ionization time-of-flight mass spectrometry (MALDI/TOF–MS). Antibiotic susceptibility testing of isolates was carried out according to the recommendations given by the European Committee on Antimicrobial Susceptibility Testing (EUCAST). Apart from blood cultures, other relevant clinical samples were also collected from patients for non-culture based methods (e.g. in vitro urinary antigen detection for S. pneumoniae and L. pneumophila, in vitro toxin detection for C. difficile etc.), as determined by the clinical context at the discretion of the attending physician. A pathogen was accepted as a causative organism of sepsis if it was microbiologically identified from a clinically relevant sample during a compatible case presentation. The traditional blood culture and non-culture based methods were carried out at the Core Microbiology Laboratory of South Pest Central Hospital, National Institute of Hematology and Infectious Diseases (Budapest, Hungary).
2.4. 16S rRNA microbiota analysis
From each patient, at least 3-3 mLs of whole blood were collected into citrate filled VACUETTE collection tubes (Greiner Bio-One, Stonehouse, UK), and after tube rotation, immediately frozen to –80°C. Skin site preparation was performed as presented earlier. DNA isolation was performed by NucleoSpin Blood, Mini kit (Macherey-Nagel, Allentown, PA, USA) according to the instructions of the manufacturer. Concentration of genomic DNA was measured using a Qubit2.0 Fluorometer with Qubit dsDNA HS Assay Kit (Thermo Fisher Scientific, Waltham, MA, USA). Bacterial DNA was amplified with tagged primers covering the V3-V4 region of the bacterial 16S rRNA gene. PCR and DNA purifications were performed according to Illumina’s protocol. PCR product libraries were assessed using DNA 1000 Kit with Agilent 2100 Bioanalyzer (Agilent Technologies, Waldbronn, Germany). Equimolar concentrations of libraries were pooled and sequenced on Illumina MiSeq platform (Illumina, San Diego, CA, USA) using MiSeq Reagent Kit v3 (600 cycles PE). To ensure reproducibility, each blood sample was independently extracted. To avoid false results (e.g. contamination) and to increase the reliability of the study, all analysis procedures were done in duplicate from 2 different isolated DNA samples of each donor. In order to evaluate the contribution of extraneous DNA from reagents, extraction negative controls and PCR negative controls were included in every run. Raw sequencing data were retrieved from the Illumina BaseSpace and the data were analyzed by the CosmosID bioinformatics platform (Yan et al., 2019), (CosmosId, 2022). The 16S rRNA metagenome sequencing was carried out at the Institute of Medical Microbiology, Semmelweis University (Budapest, Hungary).
2.5. Statistical analysis
Continuous variables are expressed as median ± interquartile regions (IQR) with minimum–maximum ranges, categorical variables are expressed as absolute numbers and percentages relative to their respective groups (n, %). The Wilcoxon Rank Sum test for Chao1 Alpha diversity, and PERMANOVA analysis for Bray-Curtis PCoA Beta diversity was used for statistical testing between cohorts of blood samples, by applying the statistical analysis support of CosmosID bioinformatics platform. Statistical significance was decided upon at a two-tailed p value of ≤0.05.
3. Results
3.1. Baseline and clinical characteristics of septic patients
Altogether, 13 septic patients were enrolled in the study. Baseline characteristics are reported in Table 1. The median age of the cohort was 70 ± 6 (59–83) years, 53.8% (7/13) of patients were females. Prevalent comorbidities were chronic heart disease (5/13, 38.5%) and atherosclerosis (4/13, 30.8%). At inclusion, sepsis and septic shock was diagnosed in 6/13 (46.2%) and 7/13 (53.8%) patients, respectively. All patients had an identified source, most frequently with an intraabdominal septic focus (7/13, 53.8%), and surgical or radio-interventional source control was sought in each case when clinically feasible. For comparison, baseline characteristics of the 13 non-septic control patients are reported in Supplementary Table 1.
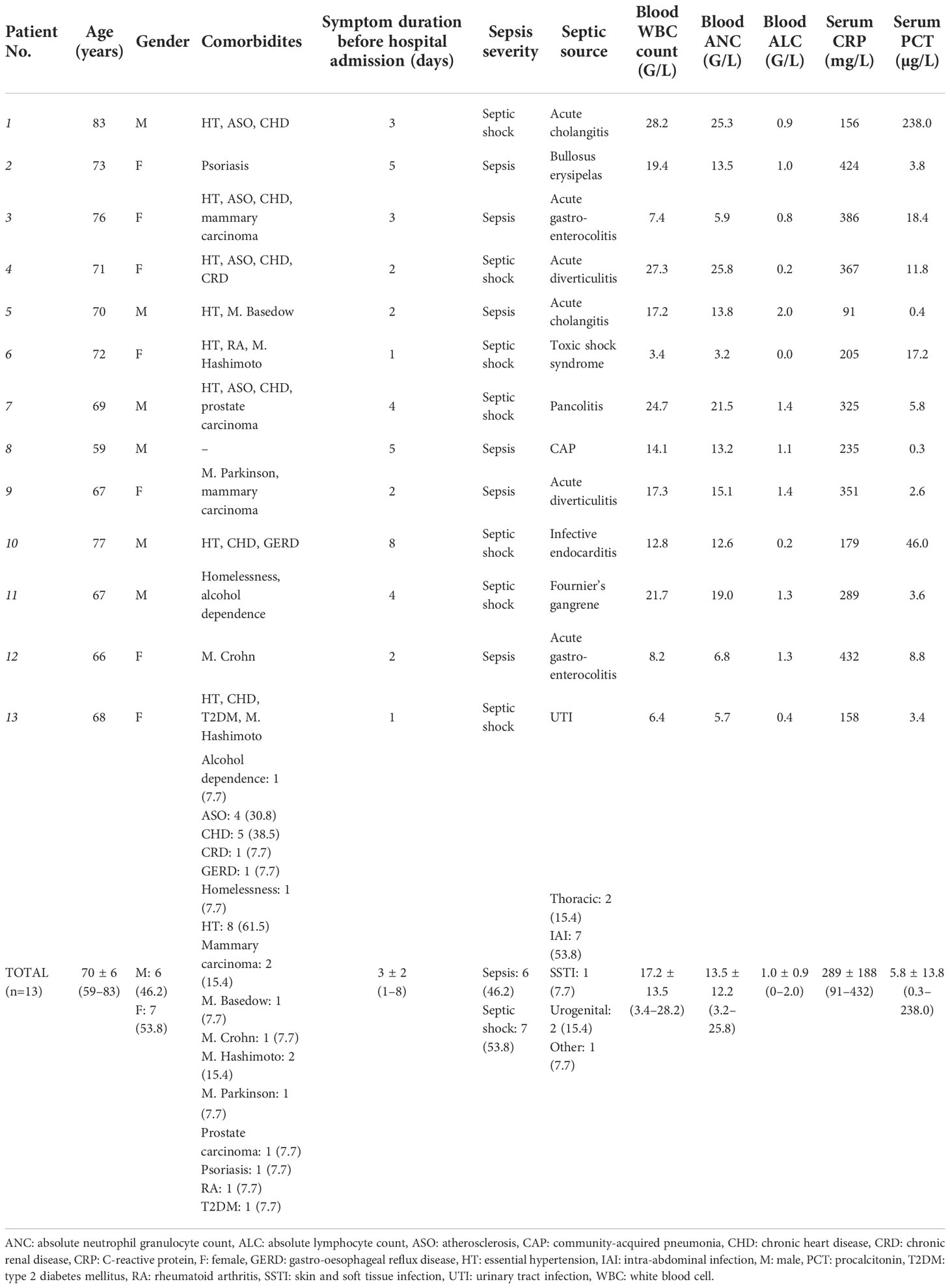
Table 1 Baseline characteristics of septic patients included in the study. Septic sources were regrouped according to their gross anatomical regions under the total count.
Microbiological characteristics, antimicrobial therapies and outcomes of septic patients are reported in Table 2. Blood cultures proved to be negative in 53.8% (7/13), while E. coli was isolated in 30.8% (4/13) of cases. Fungaemia was not detected. Non-culture based methods documented further etiologies (Clostridioides diffcile and Legionella pneumophila) in 2/7 (28.6%) of blood culture negative cases. Seven antimicrobials were initiated empirically, with ceftriaxone (9/13, 69.2%) and metronidazole (4/13, 30.8%) is most cases. At 28 days post-admission, all-cause mortality was 15.4% (2/13), while the need for ICU admission was 30.8% (4/13).
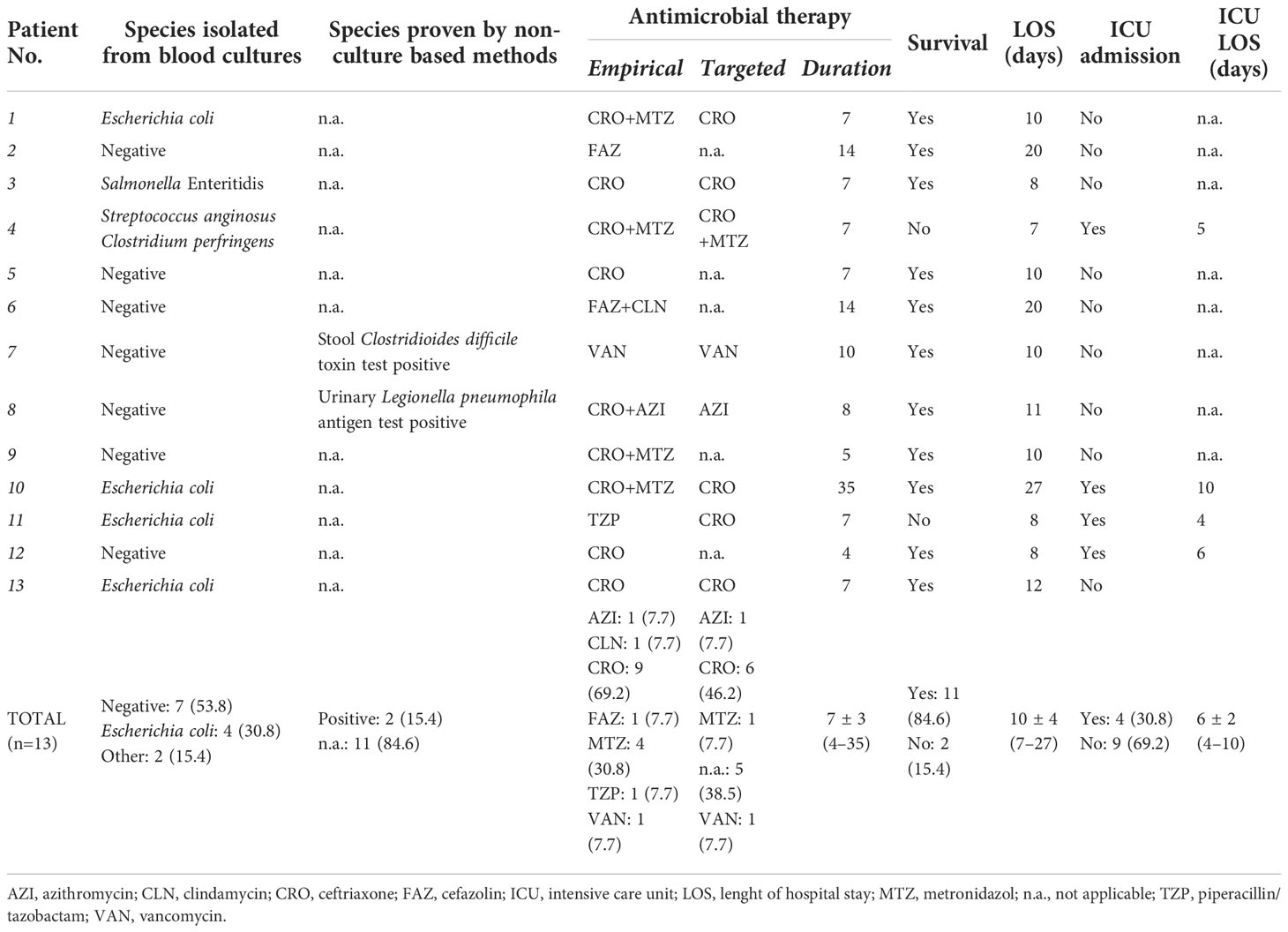
Table 2 Microbiological characteristics, antimicrobial therapies and outcomes of septic patients included in the study.
3.2. Microbiota analysis of septic and non-septic control blood samples
3.2.1. Results of DNA recovery from samples
The median of isolated DNA from blood samples was 24.72 ± 18.31 (13.11–37.24) ng/µL containing human and microbial DNA. After 16S rRNA PCR, a median DNA amount of 3.61 ± 0.65 (3.22–4.87) ng/µL, and after indexing PCR, a median DNA amount of 4.87 ± 1.78 (3.76–6.23) ng/µL was amplified. The average length of index PCR products was 664 ± 53 base pairs. A total of 15.2 million valid sequences were obtained, resulting in 10.7 million high-quality reads. The median number of reads within one sample was 169411 ± 29511 (132487–195364) in the control patients’ group, 206754 ± 21332 (187999–245872) in the septic patients’ subgroup and 335678 ± 23521 (319442–351263) in the septic shock patients’ subgroup.
3.2.2. Diversity indices among septic patients and non-septic controls
The Chao1 alfa diversity was higher in the septic group, compared to the control group in a statistically significant manner (Figures 1A, B). By further subgrouping within the septic group, the alpha diversity of both the non-shock and septic shock subgroups is significantly higher than the alpha diversity of the control group. However, there was no statistically significant difference detected in the alpha diversity of the two septic subgroups.
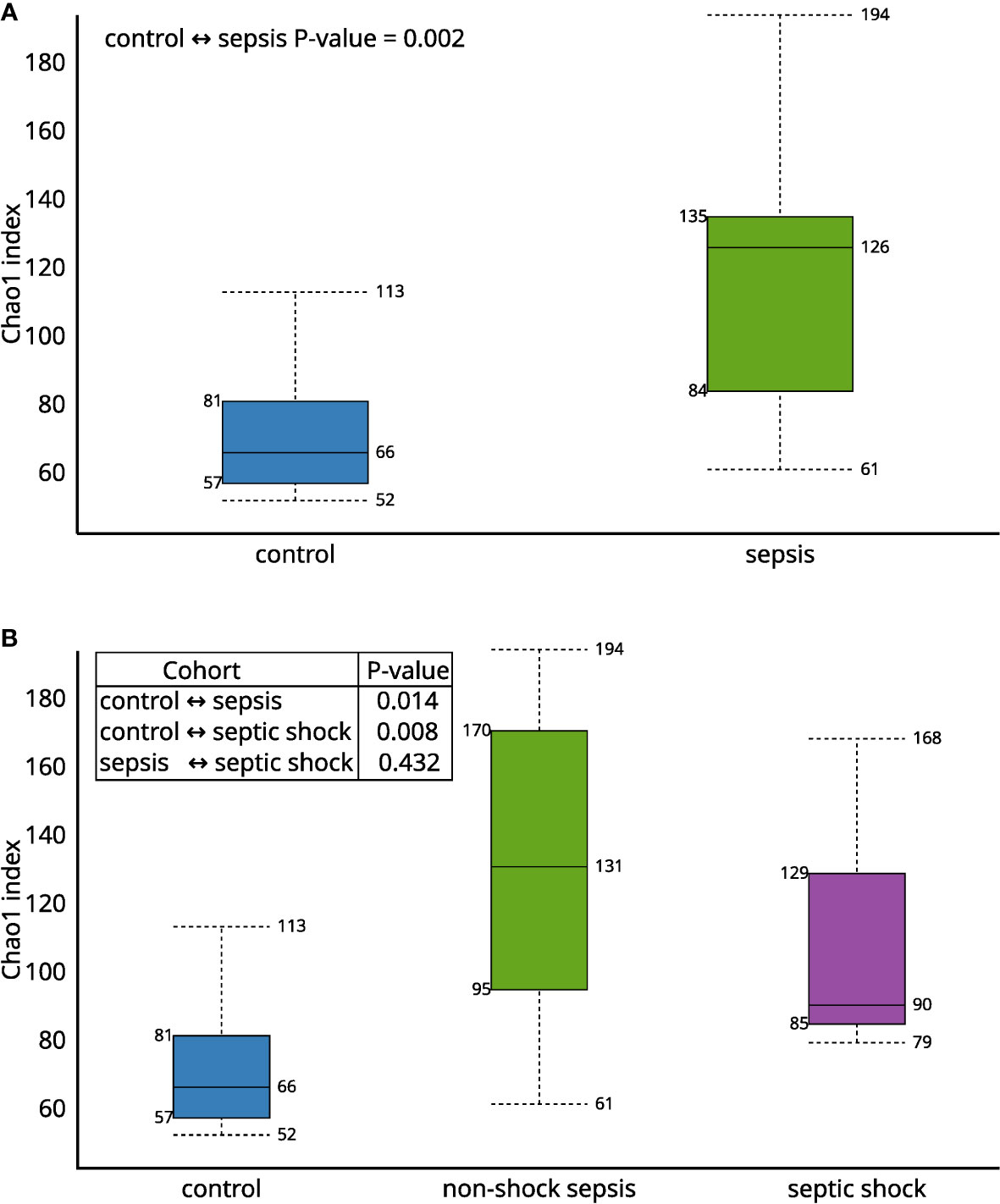
Figure 1 The Chao1 alfa diversity between (A) septic and control patients; (B) patients with non-shock sepsis, septic shock and control patients: The difference of alpha diversities between control/septic, control/non-shock septic and control/septic shock patients are significant. There is no significant difference between the two septic subgroups.
The Bray-Curtis beta diversity Principal Component Analysis (PCoA) shows significantly distinct clusters among septic vs. non-septic patients (Figure 2A), however, although there is a trend in the shift of septic subgroups compared to the control group, the two subgroups do not form new clusters that are significantly different from each other (Figure 2B).
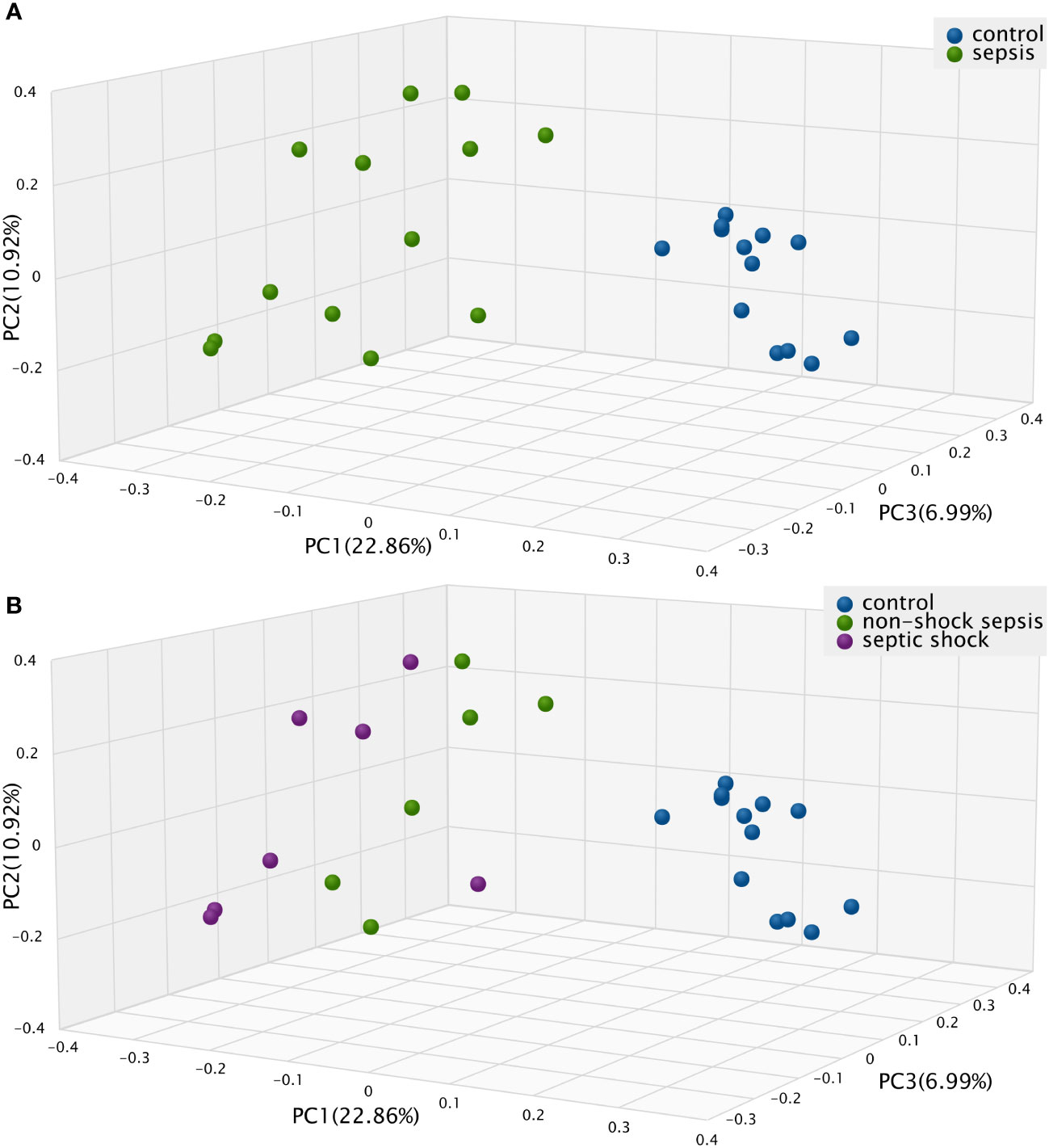
Figure 2 The Bray-Curtis Principal Component Analysis (PCoA) Beta diversity between (A) septic and control patients; (B) patients with non-shock sepsis, septic shock and control patients: The difference of beta diversities between control/septic, control/non-shock septic and control/septic shock patients are significant. There is no significant difference between the two septic subgroups.
3.2.3. Differential abundance among septic patients and non-septic controls
The comparison of the values of the septic and non-septic control groups resulted in a significant difference in the abundance of phyla Firmicutes (25.7% vs. 63.1%; p<0.01), Proteobacteria (36.9% vs. 16.6%; p<0.01) and Actinobacteria (22.1% vs. 9.9%; p<0.01) (Figure 3). Furthermore, phylum abundance differences showed a gradual decrease among Firmicutes, and an increasing trend among Proteobacteria in the control, non-shock sepsis and septic shock groups, respectively
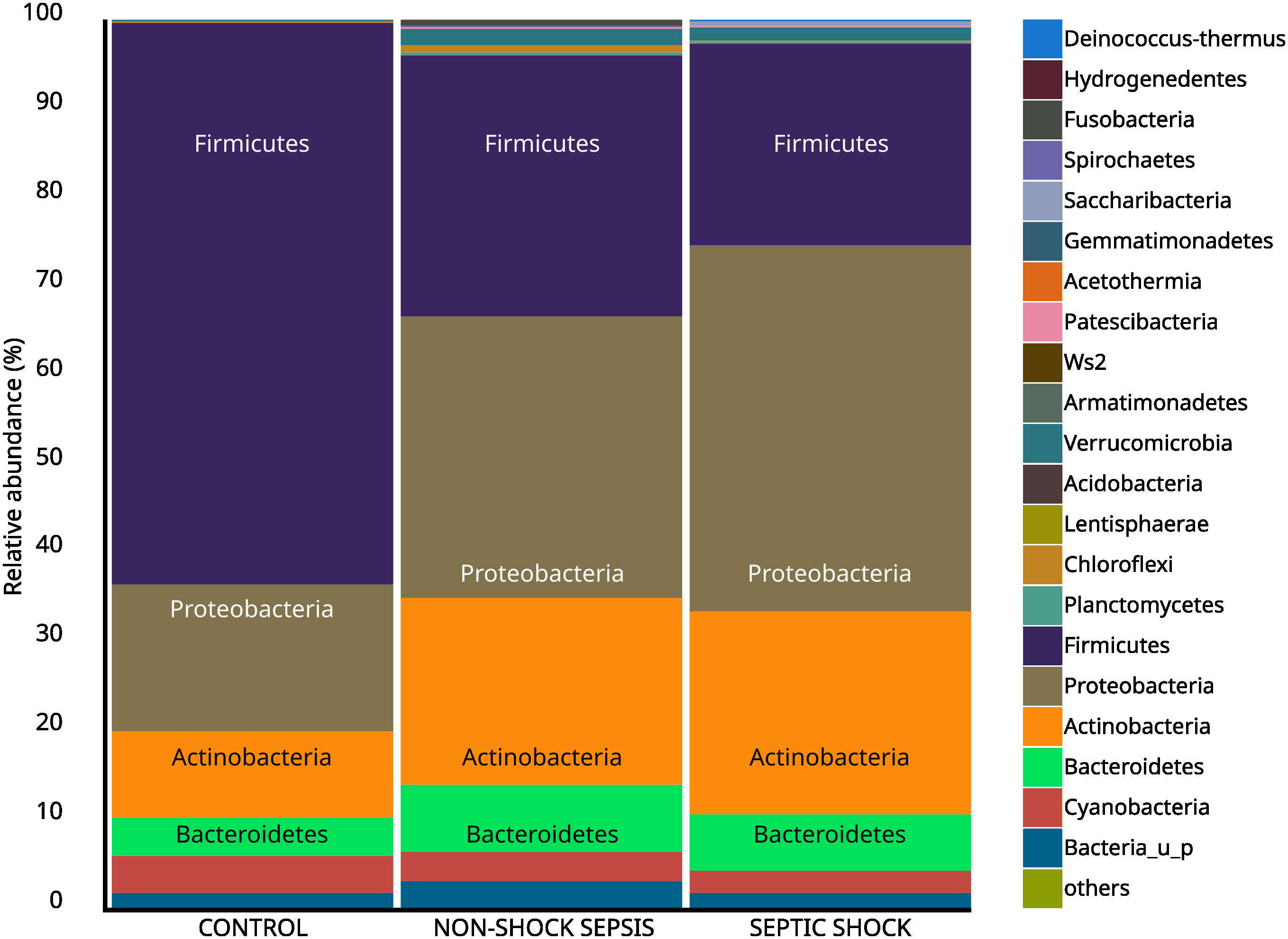
Figure 3 Abundance of taxa at the phylum level among non-septic control, non-shock sepsis and septic shock patients.
Only six of thirteen traditional blood culturing resulted in positive isolates which partially correlated with microbiota abundance results. Table 3. showed Escherichia coli as the most prevalent isolated etiopathogen (4/7, 57.1%) of blood culture samples, however, the highest abundance of Escherichia coli was only obtained during 2/4 (50.0%) microbiota analyzes from related blood samples. Furthermore, in septic patients, the 3 most abundant genera belonged mainly to the Proteobacteria phylum in microbiota analysis. Bacteria isolated from blood cultures also appeared in the microbiota analysis for each patient, but their abundance varied on a wide scale from 0.11% to 17.43%. Salmonella Enteritidis and Clostridium perfringens, which were interpreted as etiopathogens based on blood culture results, appeared only in the microbiota analysis of septic patients. In contrast, Streptococcus anginosus and Escherichia coli taxa were present with varying abundance in the control and septic groups, but these differences were not statistically significant. Surprisingly, contrary to positive non-culture based results of urinary and stool tests, Clostridioides difficile and Legionella pneumophila taxa were not detected during blood microbiota analysis of the affected patients. Comparing the results of Table 3 and Figure 4A, bacteria interpreted as etiopathogens based on cultured or non-cultured routine laboratory results were confirmed to be the most frequently present only in Sepsis 1 and Sepsis 10 patients by microbiome analysis.
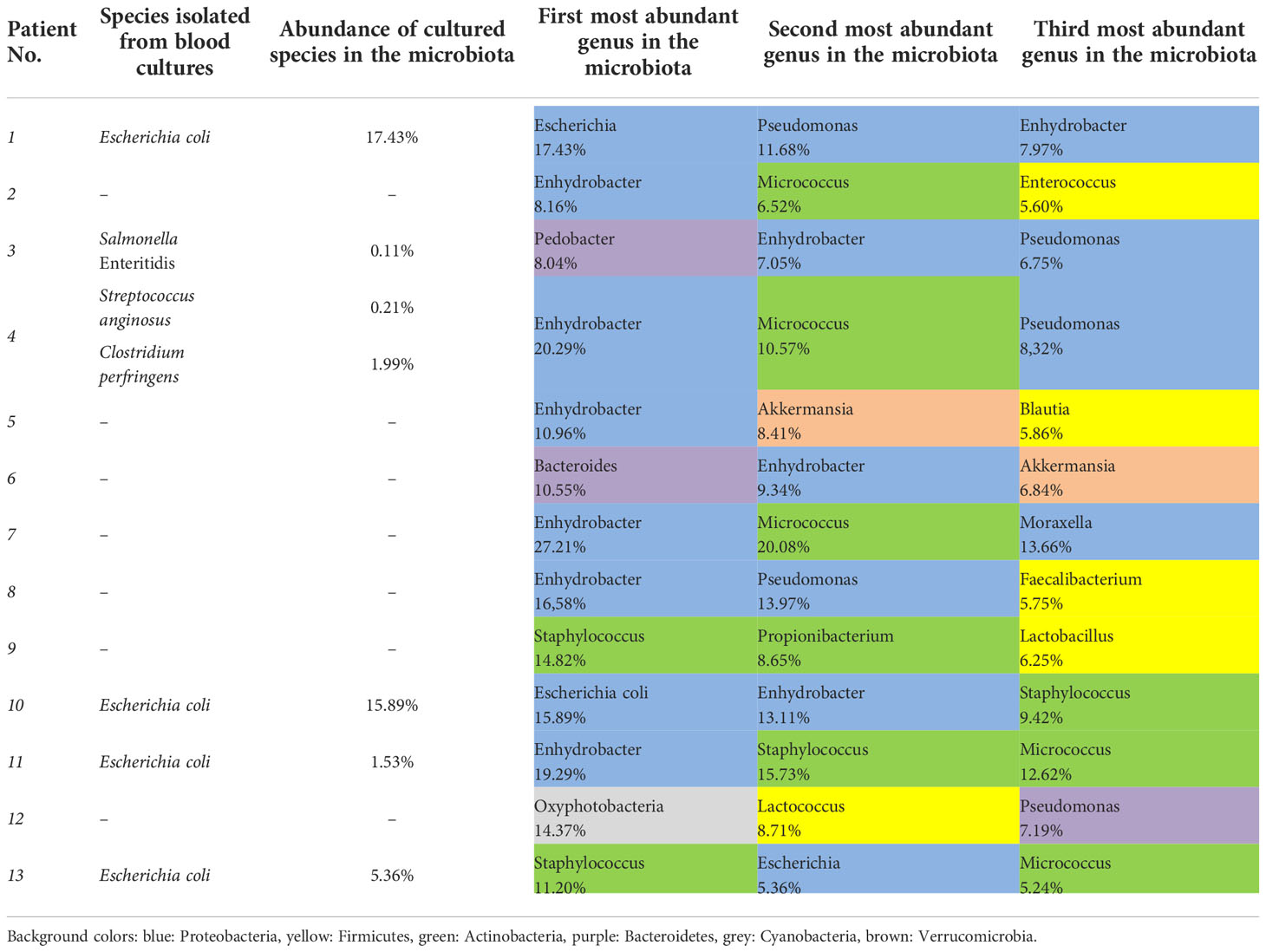
Table 3 Results of species isolated from blood cultures and abundance during microbiota analysis in septic patients.
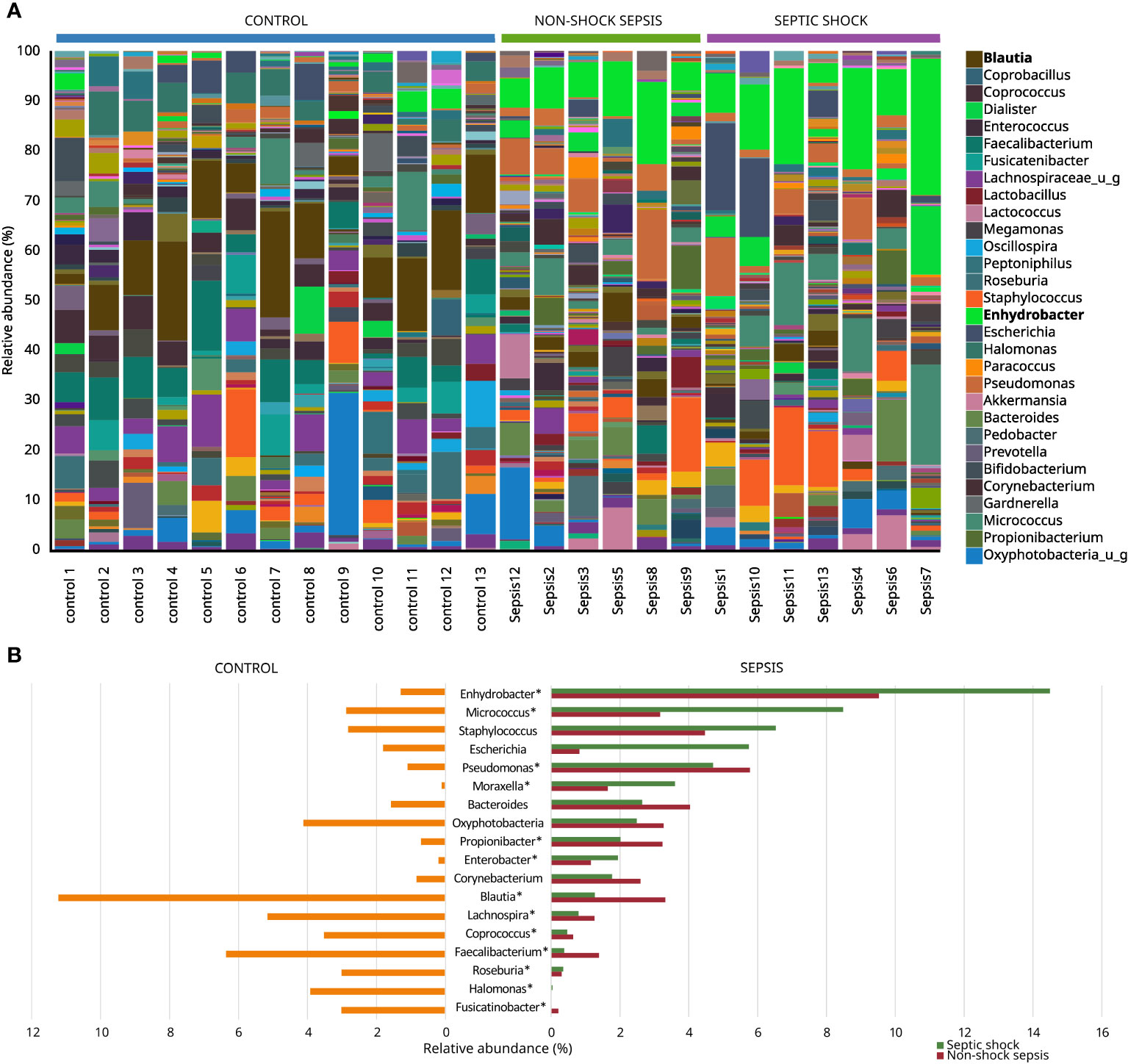
Figure 4 Most abundant genera among non-septic control, non-shock sepsis and septic shock patients (A), and genera with the highest overall relative abundance among non-septic control and septic patients (B). * means significant difference.
The microbiota of non-septic control patients contained the Enhydrobacter genus in 8/13 (61.5%) cases with an abundance of 1.18−4.07%, and this taxon was also detected in 11/13 (84.6%) septic cases with a statistically significantly higher abundance of 3.92−27.21% (p<0.001). The abundances of Micrococcus (0.37%−20.08% vs. 0.00%−11.64%; p=0.03) and Pseudomonas (0.59%−13.97% vs. 0.17%−2.55%; p=0.04) genera were also statistically significantly higher in the sepsis group, compared to non-septic controls. Although with low abundance values, the genera Moraxella, Propionibacterium and Enterobacter also occurred in significantly higher quantities in the sepsis group, while the abundance of the genera Corynebacterium, Bacteroides and Staphylococcus did not show any statistically significant difference. The genera with high abundance in the control group, such as Blautia, Faecalibacterium, Coprococcus, Lachnospira, Roseburia, Halomonas and Fusicatinobacter were all present in significantly higher amounts in this group, compared to septic patients (Figures 4A, B).
4. Discussion
4.1. Main findings of the present study
In our prospective observational study, whole blood samples originated from 13 hospitalized patients with community-acquired sepsis were examined with traditional blood culturing, in parallel to 16S rRNA microbiota analysis. To our knowledge, this is the first study evaluating the composition and changes of blood microbiota in adult patients with community-acquired sepsis, with correlation to non-septic control patients.
We found that Chao1 alpha diversity index showed a significant difference and the Bray-Curtis beta diversity revealed 2 significantly separate clusters between septic and control patients. We also found that relative microbiota differences between patients were more pronounced at the phylum level, mainly by Firmicutes (25.7% vs. 63.1%; p<0.01) and Proteobacteria (36.9% vs. 16.6%; p<0.01). Interestingly, the most prevalent etiopathogen isolated from blood cultures was Escherichia coli, from mainly intraabdominal septic sources, but only six of thirteen traditional blood culturing resulted in positive isolates correlated with microbiota abundance results.
Moreover, a relative scarcity of Faecalibacterium, Blautia, Coprococcus and Roseburia genera, with an abundance of Enhydrobacter, Pseudomonas and Micrococcus genera was observed among septic patients. Faecalibacterium genus has a sole known species, Faecalibacterium prausnitzii, a Gram positive anaerobic rod with a high abundance in the human colon. By fermenting dietary fibers to butyrate and other short-chain fatty acids, the “eubiotic” presence of F. prausnitzii in the gut homeostasis might translate to anti-inflammatory properties, such as improvement of the intestinal colonization resistance and mucosal permeability. In contrast, decreased abundance of F. prausnitzii has been associated with inflammatory bowel diseases and colorectal cancer. Similar to Faecalibacterium genus, members of the Blautia (21 identified species), Roseburia (10 identified species) and Coprococcus (5 identified species) genera are also butyrate-producing Gram positive anaerobic bacteria, most abundant in the human feces (Machiels et al., 2014; Martin et al., 2018). The present study did not analyze the change in the gut microbiome of sepsis, but it could be noted that the abundance decrement in the 4 taxa in the blood of our patients might partially be related to a similar microbiome change in the gut, as this notion was already proven by others (Du et al., 2021; Yu et al., 2022). We might hypothesize that the decreased amount of these “eubiotic” genera could be biomarkers of intestinal dysfunction during sepsis, and their relative scarcity may indicate an ongoing facilitation of systemic inflammation and invasion of other microbes to deeper tissues and the bloodstream, but this idea has yet to be proven. Enhydrobacter genus with its one identified species, Enhydrobacter aerosaccus, a Gram negative facultative anaerobic rod, was found to be abundant in blood samples of most patients with sepsis. As Enhydrobacter aerosaccus was first isolated from an eutrophic lake in 1987, it may be surprising to identify this bacterium in several cases. However, there are increasing amount of data detailing Enhydrobacter aerosaccus as a possible member of the human skin microbiota. Profiled by age, the composition of the skin microbiome confirmed an increased presence of Enhydrobacter at older ages (Kim et al., 2021). A positive correlation was demonstrated between the vulnerability of the skin barrier and the abundance of Enhydrobacter genus (Zhou et al., 2022). Information exists about this taxon not only as a component of the skin microbiota, but the increasing level of Enhydrobacter genus in blood microbiota was proved to be associated with cardiovascular disease mortality (Lawrence et al., 2022). Knowing these correlations, it is not surprising that the blood microbiota of our septic and septic shock patients belonging to the older age group contained an increasing amount of the Enhydrobacter genus compared to the blood samples of healthy controls.
4.2. Previous studies from the literature
The notion that the human blood is physiologically not sterile goes back to the 1960s, when the presence of viable bacteria was first documented in blood specimens of healthy individuals. Since then, numerous studies have aimed to prove this hypothesis by examining different clinical preparations of blood samples with several culture and non-culture based microbiological methods, including radioactive nucleoside and amino acid uptake (outdated), classical culturing of whole and filtrated blood, fluorescence in situ hybridization (FISH) probing, and most recently, PCR based amplification and sequencing of different bacterial DNA and RNA targets, including 16S rRNA genes and cell-free DNA (Castillo et al., 2019)
Interpretation of blood microbiota data is somewhat conflicting. It should be noted that the type of specimen may influence the diversity and abundance profiles found in the blood, creating a microbiologically relevant difference between results of each study. The types of blood specimens could be grouped as whole blood, cellular components such as leukocytes, erythrocytes, the buffy coat, mononuclear cells and neutrophil granulocytes, and non-cellular components, such as serum, plasma, and extracellular vesicles. However, it is generally thought that analysis of whole blood might be the most representative specimen for blood microbiota, since it consists of all blood elements (Suparan et al., 2022). According to published data, the most abundant phyla of whole blood “eubiotic” microbiota was Proteobacteria, followed by Firmicutes and Actinobacteria, but results are highly discordant on the genera level, identifying relevant microbiological variability, probably originating from oral and skin communities (Paisse et al., 2016; Whittle et al., 2018) Lastly, comparative data showed that factors affecting the profiles of blood microbiota might be age, gender and geographic location (D’Aquila et al., 2021; Panaiotov et al., 2021).
Most previous literature data about the blood microbiota during sepsis are derived from studies enrolling patients from the healthcare setting, including potential sources of nosocomial acquisition. In one of the first observational studies reporting on the blood microbiota changes during sepsis, Gosiewski et al. collected whole blood from 23 healthy volunteers and 62 septic patients recently undergoing cardiothoracis surgical procedures, and analyzed samples by 16S rRNA metagenomic next-generation sequencing. They reported that all blood samples contained bacterial DNA at detectable levels, and a higher diversity and bacterial predominance of the order Bifidobacteriales was proven among healthy individuals. Moreover, the abundance of the phyla Proteobacteria decreased in blood samples from healthy subjects, while the phyla Actinobacteria decreased in septic patients (Gosiewski et al., 2017).
Other researchers sought to assess the possible origins of blood microbiota in septic patients among the microbiological changes which characterize this group. Wang et al. recruited 204 cases with recent surgical intervention, and collected blood samples for high‐throughput DNA sequencing to detect whole blood and neutrophil-specific microbiota. The authors found that both whole blood and neutrophil-derived microbiota of surgical patients reflected diverse composition, with an abundance of the phyla Proteobacteria, Firmicutes and Bacteroidetes, and the origin of approximately 80% of blood bacteriome could be traced back to the gut microbiome Furthermore, the presence of Agrococcus, Polynucleobacter and Acidovorax in the blood positively correlated with sepsis-related organ dysfunction and serum lactate levels. The authors therefore concluded that the dysbiotic state of blood microbiota among surgical patients with sepsis might partially originate from the gut microbiome, and Agrococcus may play a role in the clinical progression of sepsis (Wang et al., 2021).
These findings were also mirrored in recent reviews, pointing out that the frequent loss of “eubiotic” Bacteroidetes and Firmicutes, and novel abundance of Proteobacteria could be documented after the onset of sepsis. Moreover, the disproportionate shift from the dominance of Faecalibacterium and other commensals may facilitate the colonization of the intestinal microbiome by healthcare-associated pathogens during sepsis (Adelman et al., 2020; Miller et al., 2021).
Also, it has been proposed that the intestinal microbiome and its derived metabolic products are a relevant modulator of the host immune response, thereby promoting homeostasis and interfering with systemic infection. The lack of an overwhelming host immune response generated against members of the circulating blood microbiota of non-septic, healthy patients might partially be explainable by central and peripheral immune tolerance directed against these “eubiotic” components with low virulence, probably induced by a cross-talk of the gut microbiome, but this phenomenon needs further research clarification (Niu and Chen, 2021).
4.3. Limitations of the study
Our study has limitations. Firstly, the relatively low number of enrolled septic patients calls for an overall cautious interpretation of our results. Second, a near-perfect matching of an adequate control group might be challenging, as the blood microbiota may become altered by chronic comorbidities to show an already “dysbiotic” state. Third, the time between potential exposure to infectious agents and diagnosis of sepsis was not unambiguously explorable for some patients. Lastly, undetected blood sampling or handling errors might have biased our final microbiological results.
5. Conclusion
In conclusion, adult patients hospitalized with community-acquired sepsis may harbor specific “dysbiotic” changes in the taxonomic composition and abundance of blood microbiota, compared to non-septic control patients. Furthermore, traditional blood culturing results might only partially correlate with results from 16S rRNA metagenome sequencing. More studies are possibly warranted to shed light on the relevance of these phenomena.
Data availability statement
The microbiological datasets generated during the current study are available in the Short Read Archive (SRA) of National Center for Biotechnology Information (NCBI) under accession number: PRJNA886752 (free available from 06.12.2022.). Anonymized clinical data of patients are available from the corresponding author on reasonable request.
Ethics statement
The studies involving human participants were reviewed and approved by Institutional Review Board of Semmelweis University (Permission Number: SE RKEB: 87/2019) South Pest Central Hospital, National Institute of Hematology and Infectious Diseases (Permission Number: ESZSZK EB 37/2016). The patients/participants provided their written informed consent to participate in this study.
Author contributions
Conceptualization, BS, RK, DS, and EO. Data curation, BS, RK, and EO. Funding acquisition, BS and DS. Investigation, NM, KP, EV, KK, and EO. Methodology, BS, RK, EO, NM, and KP. Project administration, BS, RK, and EO. Resources, BS, RK, EV, KK, EO, and DS. Software, EO and DS. Supervision, BS and EO. Validation, BS, RK, EV, KK, EO, and DS. Visualization, EO. Writing – original draft, BS, RK, and EO. Writing – review and editing, BS, RK, DS, and EO. All authors read and approved the final manuscript. All authors agree to be accountable for the content of the work.
Acknowledgments
We express our gratitude to Gábor Fekete for his help in editing the figures.
Funding
This work was supported by Semmelweis University –Eötvös Lóránd Research Network, Human Microbiota Study Group No. ,,0272” and "BGSz received the PhD Doctorate Grant from Semmelweis University (EFOP-3.6.3-VEKOP-16-2017-00009) and was supported by the New National Excellence Program of the Ministry of Innovation and Technology of Hungary (ÚNKP-19-3-I-SE-74), and the National Grant for Youth Excellence of the Ministry of Human Capacities, Human Resource Support Operator of Hungary (NTP-NFTÖ-21-B-0338).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fcimb.2022.1067476/full#supplementary-material
Footnotes
- ^ CRP, C-reactive protein; EUCAST, European Committee on Antimicrobial Susceptibility Testing; DNA, deoxyribonucleic acid; FISH, fluorescence in situ hybridization; ICU, intensive care unit; IQR, interquartile region; MALDI/TOF–MS, matrix-assisted laser desorption/ionization time-of-flight mass spectrometry; PCoA, Principal Component Analysis; PCR, polymerase chain reaction; PCT, procalcitonin; RNA, ribonucleic acid; rRNA, ribosomal ribonucleic acid.
References
Adelman, M. W., Woodworth, M. H., Langelier, C., Busch, L. M., Kempker, J. A., Kraft, C. S., et al. (2020). The gut microbiome's role in the development, maintenance, and outcomes of sepsis. Crit. Care 24, 278. doi: 10.1186/s13054-020-02989-1
Castillo, D. J., Rifkin, R. F., Cowan, D. A., Potgieter, M. (2019). The healthy human blood microbiome: Fact or fiction? Front. Cell. Infect. Microbiol. 9. doi: 10.3389/fcimb.2019.00148
Centers for Disease Control and Prevention. What is sepsis? [Online, accessed Dec 1, 2022.] Webpage: https://www.cdc.gov/sepsis/what-is-sepsis.html
D'Aquila, P., Giacconi, R., Malavolta, M., Piacenza, F., Burkle, A., Villanueva, M. M., et al. (2021). Microbiome in blood samples from the general population recruited in the MARK-AGE project: A pilot study. Front. Microbiol. 12, 707515. doi: 10.3389/fmicb.2021.707515
Du, B., Shen, N., Tao, Y., Sun, S., Zhang, F., Ren, H., et al. (2021). Analysis of gut microbiota alteration and application as an auxiliary prognostic marker for sepsis in children: a pilot study. Transl. Pediatr. 10, 1647–1657. doi: 10.21037/tp-21-51
Goncalves-Pereira, J., Povoa, P. R., Lobo, C., Carneiro, A. H. (2013). Bloodstream infections as a marker of community-acquired sepsis severity. results from the Portuguese community-acquired sepsis study (SACiUCI study). Clin. Microbiol. Infect. 19, 242–248. doi: 10.1111/j.1469-0691.2012.03776.x
Gosiewski, T., Ludwig-Galezowska, A. H., Huminska, K., Sroka-Oleksiak, A., Radkowski, P., Salamon, D., et al. (2017). Comprehensive detection and identification of bacterial DNA in the blood of patients with sepsis and healthy volunteers using next-generation sequencing method - the observation of DNAemia. Eur. J. Clin. Microbiol. Infect. Dis. 36, 329–336. doi: 10.1007/s10096-016-2805-7
Henriksen, D. P., Pottegard, A., Laursen, C. B., Jensen, T. G., Hallas, J., Pedersen, C., et al. (2017). Intermediate-term and long-term mortality among acute medical patients hospitalized with community-acquired sepsis: a population-based study. Eur. J. Emergency Med. 24, 404–410. doi: 10.1097/MEJ.0000000000000379
Kim, J. H., Son, S. M., Park, H., Kim, B. K., Choi, I. S., Kim, H., et al. (2021). Taxonomic profiling of skin microbiome and correlation with clinical skin parameters in healthy koreans. Sci. Rep. 11, 16269. doi: 10.1038/s41598-021-95734-9
Lawrence, G., Midtervoll, I., Samuelsen, S. O., Kristoffersen, A. K., Enersen, M., Haheim, L. L. (2022). The blood microbiome and its association to cardiovascular disease mortality: case-cohort study. BMC Cardiovasc. Disord. 22, 344. doi: 10.1186/s12872-022-02791-7
Machiels, K., Joossens, M., Sabino, J., De Preter, V., Arijs, I., Eeckhaut, V., et al. (2014). A decrease of the butyrate-producing species roseburia hominis and faecalibacterium prausnitzii defines dysbiosis in patients with ulcerative colitis. Gut 63, 1275–1283. doi: 10.1136/gutjnl-2013-304833
Martin, R., Bermudez-Humaran, L. G., Langella, P. (2018). Searching for the bacterial effector: The example of the multi-skilled commensal bacterium faecalibacterium prausnitzii. Front. Microbiol. 9, 346. doi: 10.3389/fmicb.2018.00346
Miller, W. D., Keskey, R., Alverdy, J. C. (2021). Sepsis and the microbiome: A vicious cycle. J. Infect. Dis. 223, S264–S269. doi: 10.1093/infdis/jiaa682
Niu, M., Chen, P. (2021). Crosstalk between gut microbiota and sepsis. Burns Trauma 9, tkab036. doi: 10.1093/burnst/tkab036
Paisse, S., Valle, C., Servant, F., Courtney, M., Burcelin, R., Amar, J., et al. (2016). Comprehensive description of blood microbiome from healthy donors assessed by 16S targeted metagenomic sequencing. Transfusion 56, 1138–1147. doi: 10.1111/trf.13477
Panaiotov, S., Hodzhev, Y., Tsafarova, B., Tolchkov, V., Kalfin, R. (2021). Culturable and non-culturable blood microbiota of healthy individuals. Microorganisms 9, 1–12. doi: 10.3390/microorganisms9071464
Singer, M., Deutschman, C. S., Seymour, C. W., Shankar-Hari, M., Annane, D., Bauer, M., et al. (2016). The third international consensus definitions for sepsis and septic shock (Sepsis-3). JAMA 315, 801–810. doi: 10.1001/jama.2016.0287
Sogaard, M., Thomsen, R. W., Bang, R. B., Schonheyder, H. C., Norgaard, M. (2015). Trends in length of stay, mortality and readmission among patients with community-acquired bacteraemia. Clin. Microbiol. Infect. 21, 789.e1–789.e7. doi: 10.1016/j.cmi.2015.05.018
Suparan, K., Sriwichaiin, S., Chattipakorn, N., Chattipakorn, S. C. (2022). Human blood bacteriome: Eubiotic and dysbiotic. States Health Dis. Cells 11, 1–29. doi: 10.3390/cells11132015
Szabo, B. G., Kiss, R., Lenart, K. S., Marosi, B., Vad, E., Lakatos, B., et al. (2019). Clinical and microbiological characteristics and outcomes of community-acquired sepsis among adults: a single center, 1-year retrospective observational cohort study from Hungary. BMC Infect. Dis. 19, 584. doi: 10.1186/s12879-019-4219-5
Wang, C., Li, Q., Tang, C., Zhao, X., He, Q., Tang, X., et al. (2021). Characterization of the blood and neutrophil-specific microbiomes and exploration of potential bacterial biomarkers for sepsis in surgical patients. Immun. Inflammation Dis. 9, 1343–1357. doi: 10.1002/iid3.483
Whittle, E., Leonard, M. O., Harrison, R., Gant, T. W., Tonge, D. P. (2018). Multi-method characterization of the human circulating microbiome. Front. Microbiol. 9, 3266. doi: 10.3389/fmicb.2018.03266
Yan, Q., Wi, Y. M., Thoendel, M. J., Raval, Y. S., Greenwood-Quaintance, K. E., Abdel, M. P., et al. (2019). Evaluation of the CosmosID bioinformatics platform for prosthetic joint-associated sonicate fluid shotgun metagenomic data analysis. J. Clin. Microbiol. 57, 1–13. doi: 10.1128/JCM.01182-18
Yu, J., Li, H., Zhao, J., Huang, Y., Liu, C., Yang, P., et al. (2022). Alterations of the gut microbiome in Chinese zhuang ethnic patients with sepsis. Mediators Inflammation 2022, 2808249. doi: 10.1155/2022/2808249
Keywords: sepsis, blood microbiota, blood culture, 16S rRNA, community-acquired sepsis
Citation: Szabó BG, Kiss R, Makra N, Pénzes K, Vad E, Kamotsay K, Szabó D and Ostorházi E (2022) Composition and changes of blood microbiota in adult patients with community-acquired sepsis: A pilot study from bench to bedside. Front. Cell. Infect. Microbiol. 12:1067476. doi: 10.3389/fcimb.2022.1067476
Received: 11 October 2022; Accepted: 29 November 2022;
Published: 13 December 2022.
Edited by:
Seyed Davar Siadat, Pasteur Institute of Iran (PII), IranReviewed by:
Davide Carcione, ASST Valle Olona, ItalyPartha Chattopadhyay, Council of Scientific and Industrial Research (CSIR), India
Samira Tarashi, Pasteur Institute of Iran (PII), Iran
Copyright © 2022 Szabó, Kiss, Makra, Pénzes, Vad, Kamotsay, Szabó and Ostorházi. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Eszter Ostorházi, b3N0b3JoYXppLmVzenRlckBtZWQuc2VtbWVsd2Vpcy11bml2Lmh1
†These authors have contributed equally to this work and share first authorship