 Fanghua Ye
Fanghua Ye Wen Zhang
Wen Zhang Jiajia Dong
Jiajia Dong Liangchun Yang
Liangchun Yang
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Cell. Infect. Microbiol., 21 October 2022
Sec. Clinical Microbiology
Volume 12 - 2022 | https://doi.org/10.3389/fcimb.2022.1002140
This article is part of the Research TopicThe Role of Host Genetics and Microbes in Paediatric InfectionsView all 4 articles
Mendelian susceptibility to mycobacterial diseases (MSMD) is a rare congenital immune deficiency characterized by susceptibility to weakly virulent mycobacteria. Loss-of-function (LOF) mutation of signal transducer and activator of transcription 1 (STAT1) is one of the common genetic causes of MSMD. In this study, we identified a patient who presented with multiple lymph node enlargements and multiple osteolytic disruptions. Mycobacterium gordonae infection was confirmed by metagenomic next-generation sequencing. Whole-exome sequencing identified a novel paternal heterozygous mutation in exon 22 of STAT1 (NM_007315.4, c.1892T>C, p.Val631Ala). This variant was confirmed pathogenic by multiple software predictions. Based on functional assays, STAT1 expression in STAT1V631A cells was not different from STAT1WT cells. But STAT1V631A mutation caused much lower activation of STAT1 when stimulated by interferon-γ (IFN-γ). Fluorescence localization analysis revealed that both STAT1V631A and STAT1WT proteins were located in the cytoplasm, and only a few STAT1V631A proteins were translocated to the nucleus in response to IFN-γ. These results suggest that STAT1V631A leads to LOF in IFN-γ-mediated mycobacterial immunity, resulting in MSMD. Treatment with antibiotics has achieved ideal disease control for this patient, and no adverse events occurred during follow-up. The STAT1 LOF deficiency is a genetic cause of MSMD, which should be considered in patients with mycobacterial disease, especially those with bone involvement.
Host genetics has been proved to play an important role in the susceptibility and severity of infectious diseases, which primarily influence the pathogen invasion and immune response (Ishii et al., 2008). Mendelian susceptibility to mycobacterial diseases (MSMD) known as a primary immunodeficiency disease (PIDs) characterized by susceptibility to weakly virulent mycobacteria such as non-tuberculous mycobacteria, Bacillus Calmette Guerin (BCG) and environmental mycobacteria, caused by a single gene mutation (Casanova and Abel, 2002; Bustamante et al., 2014). Presently, multiple genes have been identified, including X-linked (CYBB and NEMO) and autosomal genes (IFNGR1, IFNGR2, IL12B, IL12RB1, etc.), which are involved in the production or response to interferon-γ (IFN-γ) (Rosain et al., 2019; Ying et al., 2019; Bustamante, 2020; Mirzalieva et al., 2022).
Signal transducer and activator of transcription 1 (STAT1) belongs to the transcription factor STAT family, which is located on chromosome 2 and consists of 25 exons, contains the N-terminal domain, coiled-coil domain, DNA-binding domain, connection domain, SH2 domain, tail segment domain, and trans-activator domain (Horvath, 2000; Ihle, 2001). STAT1 is primarily activated by IFN in the response of the antimicrobial immune system. IFN-γ stimulation leads to STAT1 phosphorylation at the p.Tyr701 site, inducing its homodimerization to form γ-activating factor (GAF), which is translocated to the nucleus. GAF binds to γ-activating sequences to induce transcription of target genes to activate the antimicrobial immune response, followed by STAT1 dephosphorylation back to the cytoplasm (Vinkemeier, 2004). In contrast, IFN-α/β stimulation triggers STAT1 and STAT2 phosphorylation, leading to the formation of the interferon-stimulated genes factor-3 complex, which recognizes IFN-stimulated response element motifs in target genes and involves in the antiviral immune response (Dupuis et al., 2003).
Partial or complete autosomal recessive (AR) STAT1 deficiency disrupts the IFN-α/β and IFN-γ signaling pathways, which predisposes such patients to weakly virulent intracellular bacterial and viral infections (Immunodeficiency 31B, OMIM #613796). In contrast, autosomal dominant (AD) STAT1 deficiency has less of an impact on the anti-infective function of the IFN-α/β signaling pathways, and such patients may typically present with only mycobacterial infections (Immunodeficiency 31A, OMIM #614892). Intriguingly, the AD gain-of-function (GOF) STAT1 mutations may contribute to the auto-inflammatory response by enhancing the IFN-a/ß signaling pathway. This enhancement would inhibit interleukin (IL)-17-producing T-cell production and may develop chronic mucocutaneous candidiasis (Immunodeficiency 31C, OMIM # 614162) (Liu et al., 2011). Based on recent studies, loss-of-function (LOF) mutation of STAT1 is a genetic cause of MSMD that includes AD STAT1 deficiency and AR STAT1 deficiency, resulting in isolated MSMD or syndromic MSMD phenotype (Bustamante, 2020). Hence, genetic analysis and the pathogenic function of mutant genes are thus important to research.
In this study, we identified a novel paternal heterozygous STAT1 mutation (NM_007315.4, c.1892T>C, p.Val631Ala) in a patient with disseminated mycobacterial infection. Functional experiments verified that STAT1V631A provoked lessened STAT1 nuclear translocation and phosphorylation in response to IFN-γ, ultimately leading to MSMD. The relevant literature was also reviewed and analyzed, which expanded our knowledge of the role of STAT1 as a host genetic factor in mycobacterial infection, and provided an experience for clinicians and patients.
A female infant was diagnosed with disseminated mycobacterial infections in our hospital. After written informed consent was obtained from the parents, peripheral blood samples were collected from the proband and her healthy parents. Clinical case history, the laboratory, and image results were reviewed. She was treated and followed up regularly.
Genomic DNA was extracted from the peripheral blood of the patient and her family. WES, based on NovaSeq 6000 technology sequencing platform, using IDT xGen Exome Research Panel for a capture library building, double-end (Paired-End) sequencing strategy. Raw data>10G, Q30≥80%. Compared with the human genome information in the Single Nucleotide Polymorphism Database, the Human Gene Mutation Database (HGMD), and Exome Sequencing Project 6500 databases according to the American College of Medical Genetics and Genomics (ACMG) guidelines, and screened for suspicious variants. Sanger sequencing was performed on the ABI 3500 Dx platform to verify the WES results.
The tertiary structure of the STAT1 protein was predicted by AlphaFold1. The SH2 domain of STAT1 in this model has ideal confidence. All structure models were visualized using the molecular graphics program PyMol 2.1. The STAT1V631A structure was mutated by the PyMol mutagenesis tool. The electrostatic potential was calculated and gradient colored using the PyMol vacuum electrostatic program. The potential pathogenicity of the identified variants was predicted by multiple predictive tools such as PolyPhen2.22, SIFT3, and Mutation Taster4. Mutation Assessor5, REVEL6.
The human STAT1 coding sequence was cloned using reverse transcription PCR. The sequence of primer, forward: 5’-TTAAGCTTGGTACCGAGCTCGCCACCATGTCTCAGTGGTACG-3’; reverse: 5’-CCCTTGCTCACCATGGATCCGCTTCCCCCTCCTCCTACTGTGTTCATCATACTG
TCGAAT-3’. PCR products were ligated into the pcDNA3.1 vector between the kpnI and BamHI restriction sites using ClonExpress II One Step Cloning Kit (Vazyme, China). Mutations were introduced using Mut Express II Fast Mutagenesis Kit V2 (Vazyme, China) following the manufacturer’s instructions. Flag and GFP were tagged at the C-terminus of the expression plasmid. The constructed plasmids were verified by sequencing.
Human embryonic kidney 293T (HEK293T) cells were transfected using Lipofectamine 2000 (Thermo Fisher, USA) at a 1:1 ratio with either the WT or mutant plasmid. IFN-γ (105 IU/mL, Peprotech, USA) or IFN-α (105 IU/mL, MCE, USA) was used to stimulate the transfected cells for 30 or 60 minutes, or normal saline was used as a control. Western blot analysis was used to assess GAPDH, total STAT1 protein, and phospho-STAT1 (Y701). The primary antibodies used here included mouse anti-Flag (1:2000, Sigma, F1804), rabbit anti-STAT1 (phospho Y701) (1:1000, Abcam, ab109457), and rabbit anti-GAPDH (1:10000, Abcam, ab181602) antibody. The secondary antibodies were peroxidase-conjugated goat anti-rabbit IgG (1:10000, Jackson, #111-035-003) and HRP conjugated Goat Anti-Mouse IgG (1:5000, Servicebio, GB23301).
HEK293T cells were transfected with the WT or mutant plasmid. IFN-γ (105 IU/mL) or IFN-α (105 IU/mL) was used to stimulate the transfected cells for 60 minutes, or normal saline was used as a control. Then fixed with 4% paraformaldehyde and permeabilized with 0.3% Triton X-100 in PBS. After 5% Goat serum Albumin for 1 h, The nuclei were stained with 4,6-diamidino-2-phenylindole (Beyotime, China). Sublocalization of STAT1 was acquired by a confocal microscope (Zeiss, Germany).
Literature searches were conducted in PubMed and Web of Science library databases to find relevant articles published through May 1, 2022, to identify case studies reporting STAT1 LOF deficiency. Functional analysis confirmed the pathogenicity of mutation. Search themes include “STAT1” or “signaling sensor and transcriptional activator 1”, “loss-of-function” or “LOF”, “negative mutation” or “STAT1 deficiency”. All selected articles were reviewed for full text.
The proband was born as the first child uneventfully in a healthy non-consanguineous Chinese family. She was vaccinated with BCG after birth, without adverse reactions. There is no family history of recurrent infection, neoplastic disease, or any known genetic disorder. Her clinical case history is shown in Table 1. She presented with intermittent fever, multiple lymph node enlargements, and multiple osteolytic disruptions during her 1st year of life (Figure 1). High white blood cell and neutrophil counts were recorded in routine blood tests. The hematologic neoplastic disease was not confirmed because neither bone biopsy nor bone marrow biopsy uncovered tumor cells. Histological analysis of the left tibia revealed chronic inflammation. Nontuberculous mycobacteria DNA was detected by metagenomics next generation sequencing (mNGS) of biopsy tissue, and the strain was identified as Mycobacterium gordonae. Her condition continued to improve after treatment with linezolid, isoniazid, and rifampin.
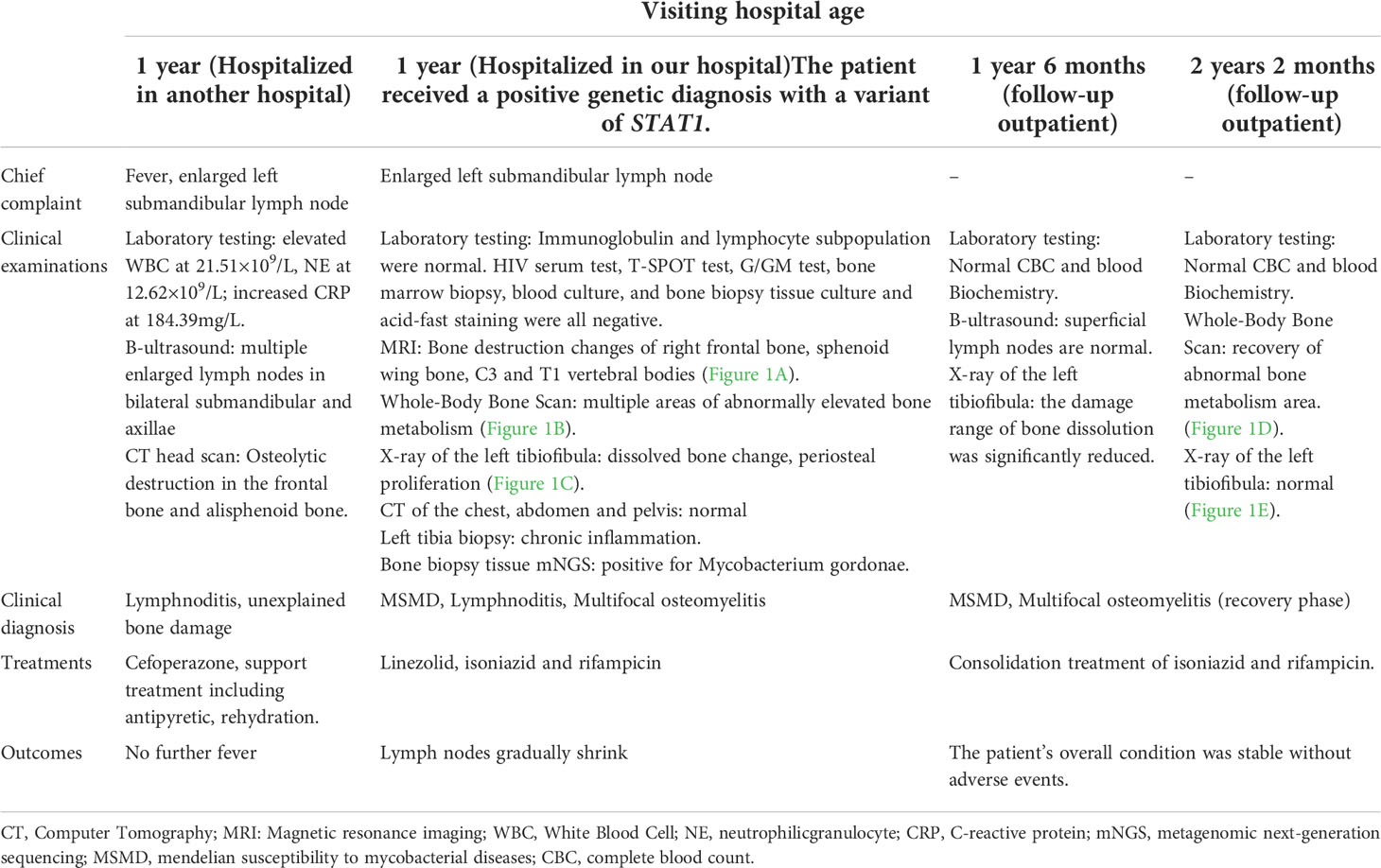
Table 1 Clinical case history and treatment process of our patient.
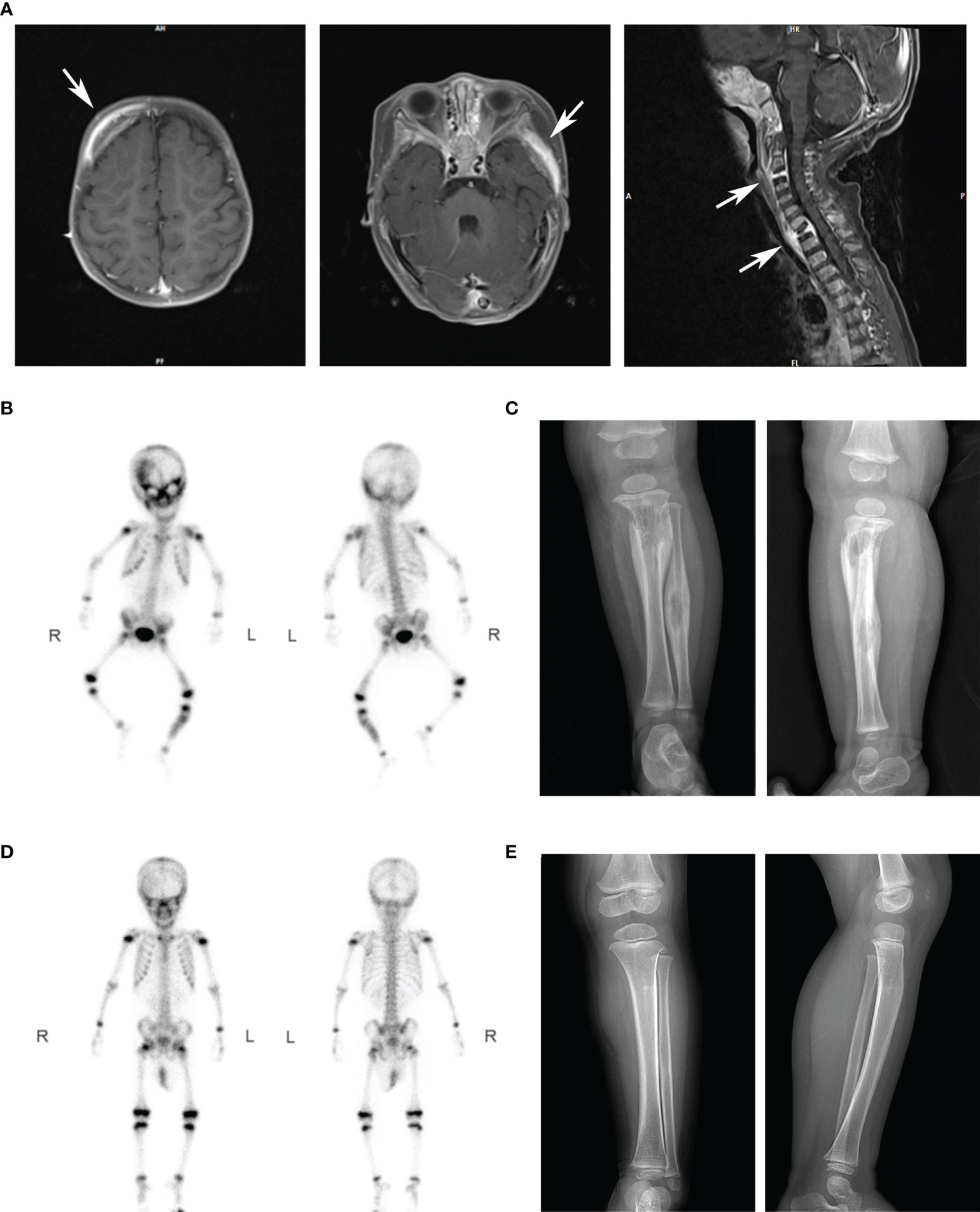
Figure 1 Imaging results of our patients in different periods. (A) The MRI scanning showed multiple osteolytic processes in the frontal temporal bone (left), pterygoid bone (middle), and C3 and T1 vertebrae (right). Arrows marked the diseased region. (B) After intravenous injection of 99mTc-MDP for 4 hours, anterior and posterior whole-body bone imaging was performed. Results showed abnormal concentration area of imaging agent distribution in multiple skulls (right frontal bone, bilateral orbital bone, sieve bone, pterygoid bone, maxillary bone, occipital bone, and temporal bone), right posterior ribs 10 and 11, left lower femur, and left middle and upper tibiofibular. (C) X-ray showed osteolytic changes and periosteal hyperplasia in the upper left tibia and middle left fibula (positive and lateral positions). (D) After antibiotics treatment, the abnormal bone metabolism area in the whole-body bone scan was significantly smaller than that before. (E) X-ray showed that the range of osteolytic damage was significantly reduced.
According to the age of onset and clinical phenotype, innate immune abnormalities were considered for the patient. Therefore, the whole-exome sequencing was performed for the patient and her parents. A novel paternal heterozygous variant in the STAT1 gene (NM_007315.4: c.1892T>C, p. Val631Ala) was identified, which was confirmed by Sanger sequencing (Figure 2A). Her healthy father was also found to be heterozygous for the p.V631A mutation, suggesting that this variant with low clinical penetrance, like the previously reported p.L706S and p.Q463H (Dupuis et al., 2001; Chapgier et al., 2006). The mutation was not found in the gnomAD, ClinVar, or HGMD. This mutation was located within the SH2 domain, resulting in a valine to alanine substitution at position 631. The alanine residue was not observed at this position in homologous sequences, suggesting high conservation among different species (Figure 2B).
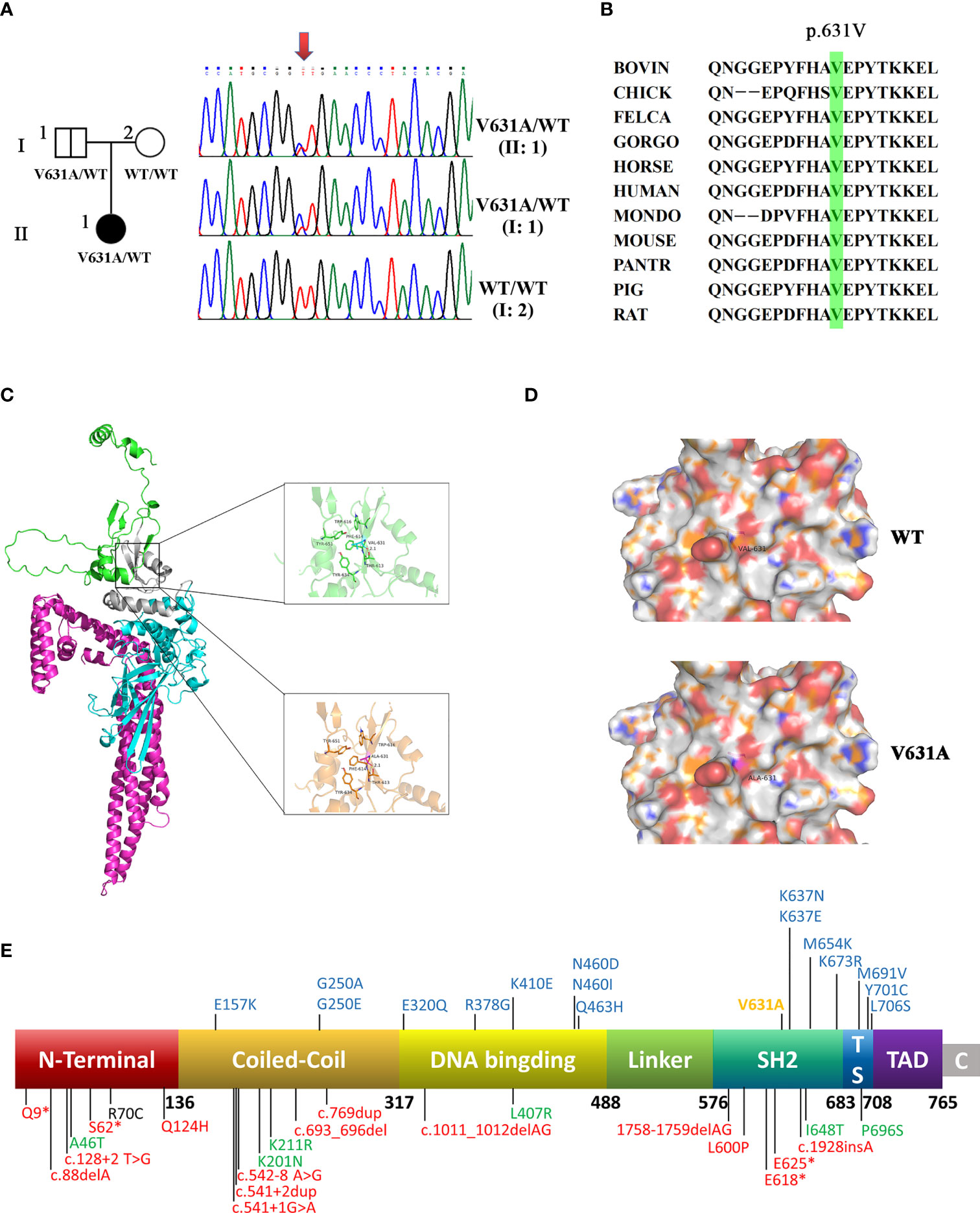
Figure 2 A novel heterozygous mutation was identified in STAT1. (A) The family pedigree. DNA sequence chromatograms of the patient and her family. Arrows marked the mutated site. c.1892T>C in exon 22 of STAT1 caused a conservative substitution of valine to alanine acid (p.V631A). (B) V631 amino acids in STAT1 protein are highly conserved during evolution. (C) The crystal structure of the STAT1 is shown in cartoon representation and colored for the coiled-coil domain (red), DNA-binding domain (cyan), linker domain (grey), and SH2 domain (green). Magnified pictures displayed the Val-631 (top), and Ala-631 (bottom). The R group of Ala-631 is methyl, which is less hydrophobic than the dimethyl group of Val-631, so the hydrophobic interaction with the surrounding hydrophobic amino acids will be weakened to a degree. (D) The calculated electrostatic potentials of STAT1WT and STAT1V631A are mapped on the surface and colored in a gradient from red (negative) to blue (positive). There is no significant change after mutation. (E) Domains of STAT1 protein: the coiled-coil domain (CCD), DNA-binding domain (DBD), linker domain (LD), SH2 domain (SH2D), tail segment domain (TSD), and trans-activator domain (TAD). AR forms of complete (red) and partial (green) STAT1 deficiency are shown below the protein. AD STAT1 LOF mutations are shown above the protein with blue (previous study) and yellow (current study).
Val-631 can form strong hydrophobic interactions with amino acid residues such as Trp-616, Tyr-651, Tyr-634, and Thr-613, which play a crucial role in stabilizing the protein. The mutant Ala-631 is less hydrophobic than Val-631, and the hydrophobic interaction with the surrounding hydrophobic amino acids will be weakened to a degree (Figure 2C). Both amino acids are hydrophobic, so there is no significant change on the electrostatic surface (Figure 2D).
STAT1V631A mutation was classified as a “variant of uncertain significance” (PM2_Supporting + PP2 + PP3) according to the 2015 ACMG guidelines (Richards et al., 2015). Prediction of protein function by bioinformatics software was performed. It was considered as “probably damaging” with PolyPhen2, “deleterious” with SIFT, “disease-causing automatic” with Mutation Taster, and “Medium” with Mutation Assessor. The REVEL score was 0.856. These results strongly indicated that the mutation might alter the STAT1 function.
We performed functional validation by transfecting either flag-tagged WT or mutant constructs into HEK293T cells. According to the intracellular fluorescence signals, the transfection efficiency of WT and mutant groups were comparable (Figures 3A, B). Western blot showed the total protein expression level of STAT1 was not different between STAT1WT and STAT1V631A cells (Figures 3B, C), which is consistent with the results of bioinformatics analysis of minor structural changes after mutation. The phosphorylated STAT1 of STAT1V631A cells was significantly lower than STAT1WT cells after IFN-γ and IFN-α stimulation (Figure 3C). The localization of STAT1 protein was detected by fluorescence microscopy. Before stimulation, the STAT1 proteins were located in the cytoplasm in both STAT1WT cells and STAT1V631A cells. After IFN-γ and IFN-α stimulation, most of the STAT1 was translocated to the nucleus in STAT1WT cells, but only a few STAT1 were translocated to the nucleus in STAT1V631A cells (Figure 3D). Collectively, these results suggested that the STAT1V631A represented a LOF mutation by weakening the phosphorylation and nuclear accumulation of STAT1.
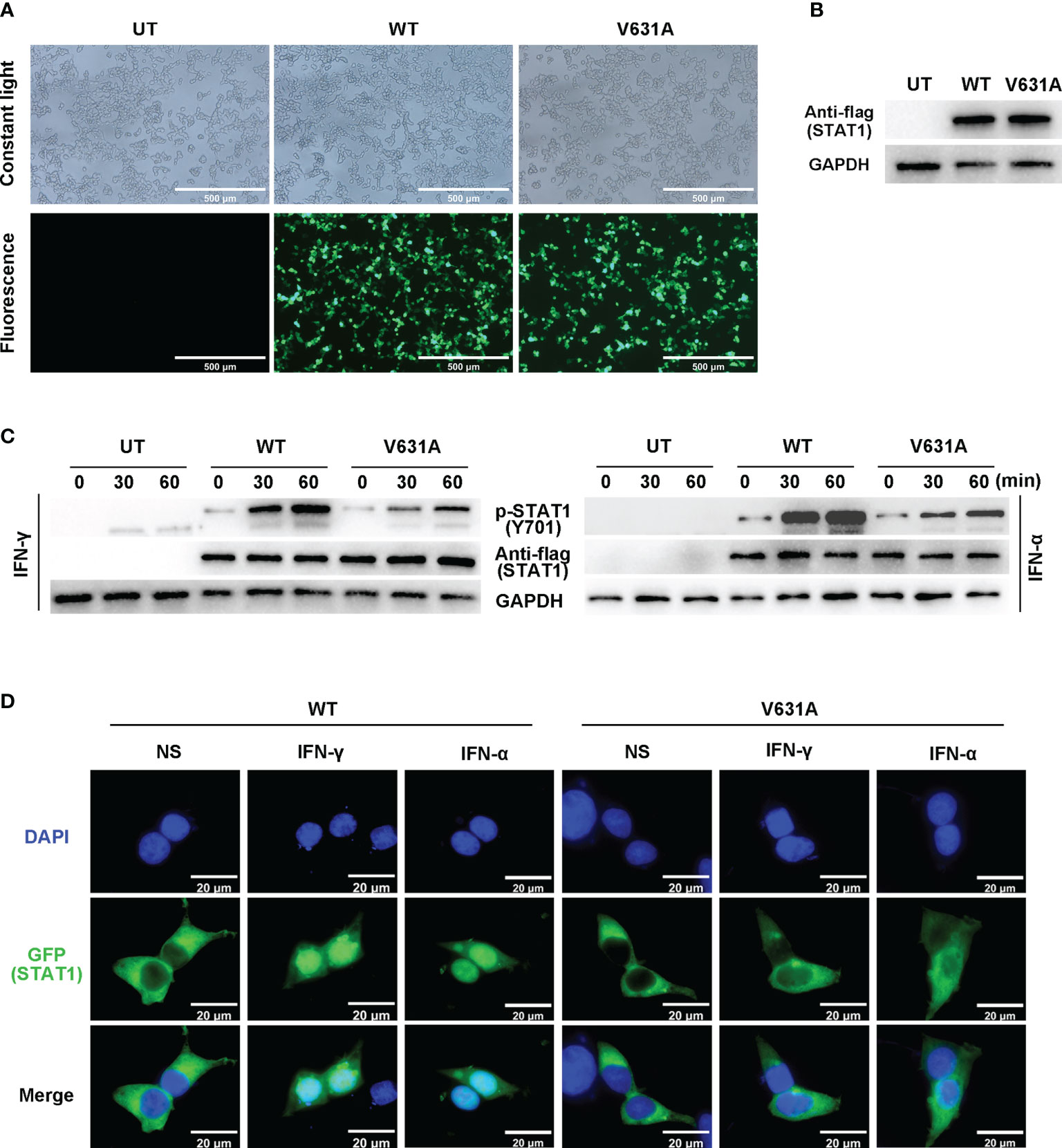
Figure 3 Functional assays of STAT1WT and STAT1V631A. (A) The cell growth diagram after transfection showed that there was no significant difference in GFP transfection efficiency among WT and V631A groups. UT, untreated. (B, C) Western blot shows the STAT1 expression (B) and phosphorylation (C) of STAT1 in the human embryonic kidney 293T (HEK293T) cells transfected with STAT1 constructs. The total STAT1 and GAPPDH were used as the loading controls. One experiment representative of three independent experiments is shown. (D) Localization of GFP-labeled STAT1 under non-stimulated (NS), IFN-γ stimulated (105 IU/mL, 60 mins), or IFN-α stimulated (105 IU/mL).
Here, we summarized the clinical features and genetic characteristics of symptomatic patients with STAT1 LOF deficiency in previous study (Table 2; Figure 2E, details in Supplementary 1). A total of 64 patients (31 AD STAT1 deficiency and 33 AR STAT1 deficiency) were selected for retrospective analysis (Dupuis et al., 2001; Chapgier et al., 2006; Sampaio et al., 2012; Tsumura et al., 2012; Hirata et al., 2013; Muriel-Vizcaino et al., 2016; Boudjemaa et al., 2017; Kagawa et al., 2017; Ueki et al., 2017; Ying et al., 2019; Bhattad et al., 2021; Le Voyer et al., 2021; Liu et al., 2022; Zhou et al., 2022). Clinical data for one patient were not available (Muriel-Vizcaino et al., 2016). Thirty-nine mutations have been reported and functionally validated, which were located throughout the STAT1 protein (Figure 2E), including 25 missense, four deletions, two insertion, four nonsense, and four splicing mutations.
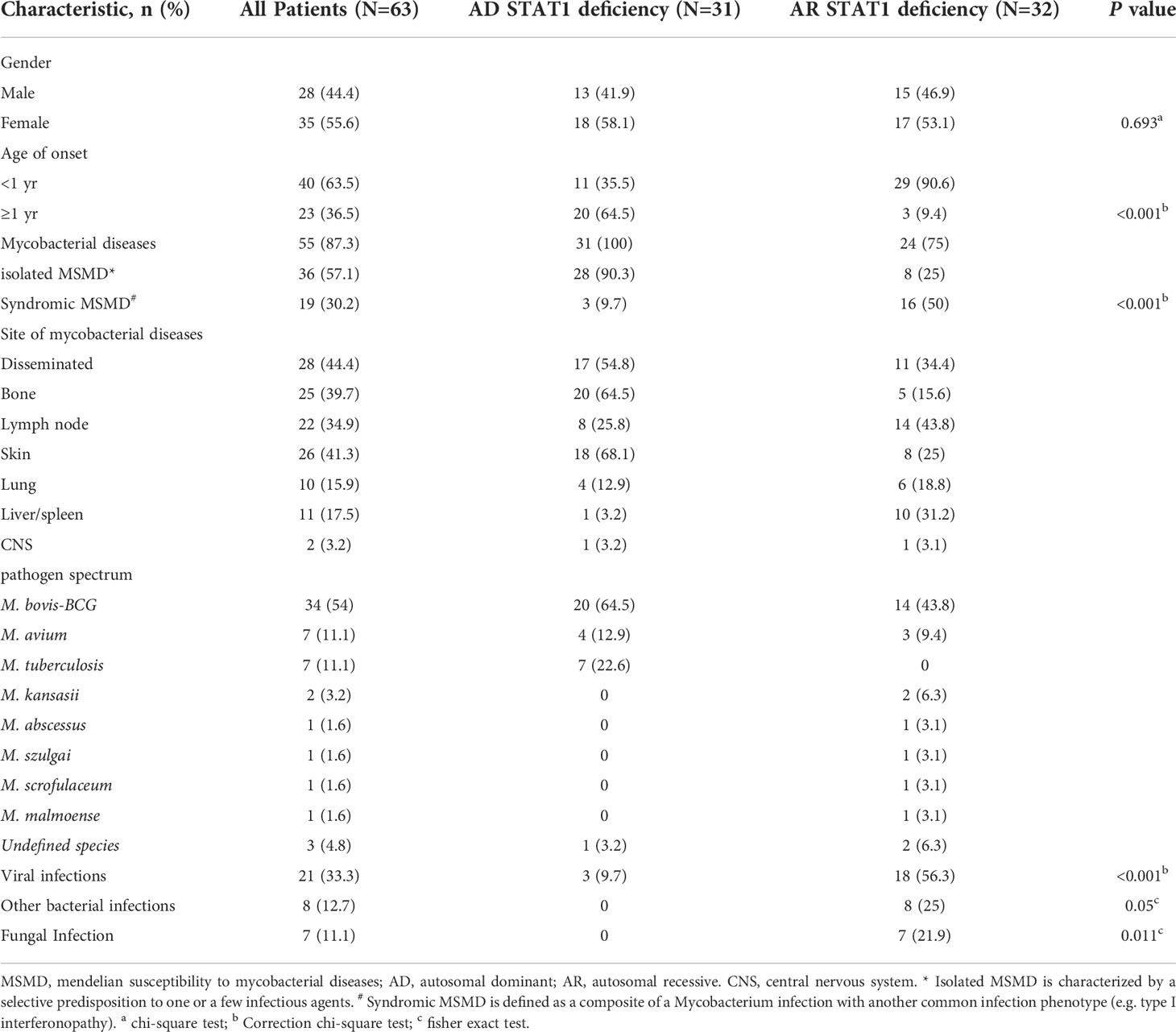
Table 2 Demographic and infectious phenotypes of the international cohort of patients with STAT1 LOF deficiency.
AD STAT1 deficiency manifests frequently as MSMD, virus infection in three cases, and bacterial or fungal infection in none. Some individuals with AD STAT1 deficiency are asymptomatic. In contrast, patients with AR STAT1 deficiency present with infections at a younger age and more often present with syndromic MSMD caused by other bacteria, viruses and fungi. MSMD in STAT1 LOF patients was most frequently caused by M. bovis-BCG (54%). The mycobacterial involved disseminated (44.4%), skin (41.3%), bone (39.7%), lymph node (34.9%), liver/spleen (17.5%), lung (15.9%), and central nervous system (3.2%) (Table 2).
Most patients with isolated MSMD were treated successfully with anti-mycobacterial therapy. Hepatitis, haemophagocytic lymphohistiocytosis (HLH), and disseminated intravascular coagulation are severe complications of AR STAT1 deficiency. Besides anti-infectives and antivirals, immunosuppressants and biologics are also used to treat AR STAT1 deficiency. Twelve patients with AR STAT1 deficiency received HSCT, four died, and none of the eight surviving patients developed viral or mycobacterial infections after transplantation (Le Voyer et al., 2021).
In this research, we identified a novel paternal heterozygous STAT1 mutation in the SH2 domain (p.V631A) associated with disseminated mycobacterial infection. Clinical features and functional studies confirm the loss of function of STAT1V631A in the IFN-γ pathway, leading to host susceptibility to mycobacterial disease. This is an enrichment of the clinical and molecular phenotypic studies of STAT1.
The SH2 domain (residues 577-683) is required for the recruitment of STAT1 to activated interferon receptors and plays an important role in Tyr701 phosphorylation. Phosphorylation-activated STAT1 forms a dimer that enters the nucleus and triggers high-affinity DNA binding and gene transcription (Boisson-Dupuis et al., 2012). Our study showed that V631A mutation reduced STAT1 phosphorylation and nuclear accumulation, consistent with other LOF heterozygous mutations located in the SH2 structural domain (e.g. K637E, K673R, and M654K) (Sampaio et al., 2012; Tsumura et al., 2012). Of four possible STAT1 dimers (WT/WT, WT/V631A, V631A/WT, V631A/V631A), only WT homodimers (WT/WT) formed normal, high-affinity phosphate dimers, which could enter the nucleus and participate in downstream signaling (Dupuis et al., 2001). In addition to the SH2 domain, STAT1 also includes DNA binding domain (residues 318-488) that mediate DNA binding and transcriptional activity, coil-coil domain (residues 137-317) that mediate the interaction between proteins and plays an important role in the formation and dephosphorylation of STAT1 dimer, tail segment domain (residues 684-708) that contain the key amino acid (Tyr701) phosphorylated by JAK kinase (Lim and Cao, 2006; Mao et al., 2005). The association of the variations with the phenotypes in these domains requires further investigation.
The genetic etiological diagnosis of MSMD relies primarily on NGS tests. Reduced cellular response to IFN-γ as a result of STAT1 deficiency is fundamental to susceptibility to mycobacterial disease. Widely, any STAT1 mutation may contribute to the mycobacterial disease phenotype (Bustamante et al., 2014). Contradictory and mysterious, several investigations have also noted the incidence of mycobacterial disease in patients with GOF of STAT1 (Toubiana et al., 2016). We reviewed the clinical data of patients with STAT1 LOF deficiency. A total of 36 STAT1 variants have been reported. With frameshift and nonsense mutations being highly associated with LOF. However, for a novel heterozygous missense mutation, it is difficult to identify whether the functional alteration is GOF or LOF merely from the mutation site and amino acid change. Most patients with autosomal dominant LOF mutations have a partial deletion of STAT1 phosphorylation levels in response to IFN-α or IFN-γ stimulation (Sampaio et al., 2012; Tsumura et al., 2012). However, a minority of patients have normal phosphorylation of STAT1 but exhibit impaired binding activity to DNA (Chapgier et al., 2006). Therefore, functional studies of heterozygous mutants are very important. Alanine scanning mutations established by Kagawa and colleagues are highly sensitive to estimating the function of STAT1 in the discoid and DNA-binding domains, which may be a valuable tool to assess an unknown mutation (Kagawa et al., 2017). However, for other domains or more types of variants, more prediction methods need to be developed.
The phenotypic categories of isolated/syndromic MSMD and genetic functional analysis contribute to our better understanding of the association between the genotype and phenotype (Bustamante, 2020). Our results showed that autosomal dominant LOF of STAT1 tends to cause isolated MSMD, whereas autosomal recessive STAT1 is mostly responsible for syndromic MSMD, the latter are commonly comprised of fatal complications. This will facilitate clinicians to manage such patients in a predictive manner. Literature review shows that MSMD occurs in patients with STAT1 LOF deficiency mainly in infancy, with recessive patients presenting earlier than dominant patients, mostly before 1 year of age. The incidence of multifocal osteomyelitis was high, and some patients even developed osteomyelitis without other organ involvement (Dupuis et al., 2001; Hirata et al., 2013; Boudjemaa et al., 2017; Kagawa et al., 2017). It was found that this may be due to impaired IFN-γ response and increased osteoclast differentiation in patients with STAT1 deficiency (Tsumura et al., 2022). Two cases of multifocal osteomyelitis had been misdiagnosed as neuroblastoma and Langerhans cell histiocytosis (Boudjemaa et al., 2017; Zhou et al., 2022). Despite histological analysis, another case of multifocal osteomyelitis was suspected sarcoidosis disease (Tsumura et al., 2022). The delay in diagnosis may be related to the low rate of positive cultures for mycobacteria. The mNGS method provides rapid, easy, and accurate identification of pathogenic bacteria and improves the detection of mycobacterial disease. Therefore, in clinical practice, STAT1 LOF deficiency needs to be considered in patients with mycobacterial osteomyelitis at a young age of onset. Molecular diagnostics in MSMD can provide families with genetic counseling and pave the way for a better understanding of treatment options for the pathogenesis of mycobacterial disease. For example, for patients with an incomplete lack of cellular response in STAT1 deficiency, antibiotics, antiviral agents or recombinant IFN-γ can achieve the desired efficacy (Sampaio et al., 2012; Hirata et al., 2013). In contrast, HSCT is curable in patients with complete STAT1 deficiency, who need to be alerted to fatal viral infections and serious complications (Naviglio et al., 2017; Le Voyer et al., 2021).
In conclusion, there are few reports about LOF of STAT1 variants to date. The molecular genetic mechanism of STAT1 related MSMD remains to be investigated. A study of the genetic background of the host can not only clarify the process of mycobacterial infection and immune response but also guide the prediction of the disease and the development of treatment strategies. Furthermore, with the rapid development of NGS, many new candidate genes and pathways critical for mycobacterial immunity may be identified.
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found below: CNGB, CNP0003320.
Written informed consent was obtained from the individual(s), and minor(s)’ legal guardian/next of kin, for the publication of any potentially identifiable images or data included in this article. This study was reviewed and approved by the Ethics Committee of Xiangya Hospital of Central South University.
FY and WZ performed the experiments and analyzed the data. FY wrote the manuscript. WD and HZ screened the relevant literature. JD, MP and CF collected and analyzed the clinical data. LY designed and guided the study and revised the manuscript critically. All authors contributed to the article and approved the submitted version.
This work was supported by the National Natural Science Foundation of China (NO.82000137, NO.82002118), and the Natural Science Foundation of Hunan Province (NO.2020JJ4918).
We thank the patient and her parents for their participation in this study. We thank the Medical Research Center of Xiangya Hospital Central South University for providing the platform and the personnel who work there for giving support and guidance.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fcimb.2022.1002140/full#supplementary-material
Bhattad, S., Unni, J., Varkey, S. (2021). MSMD in a 3-generation multiplex kindred due to autosomal dominant STAT1 deficiency. J. Clin. Immunol. 41 (1), 259–261. doi: 10.1007/s10875-020-00890-8
Boisson-Dupuis, S., Kong, X. F., Okada, S., Cypowyj, S., Puel, A., Abel, L., et al. (2012). Inborn errors of human STAT1: Allelic heterogeneity governs the diversity of immunological and infectious phenotypes. Curr. Opin. Immunol. 24 (4), 364–378. doi: 10.1016/j.coi.2012.04.011
Boudjemaa, S., Dainese, L., Heritier, S., Masserot, C., Hachemane, S., Casanova, J. L., et al. (2017). Disseminated bacillus calmette-guerin osteomyelitis in twin sisters related to STAT1 gene deficiency. Pediatr. Dev. Pathol. 20 (3), 255–261. doi: 10.1177/1093526616686255
Bustamante, J. (2020). Mendelian susceptibility to mycobacterial disease: Recent discoveries. Hum. Genet. 139 (6-7), 993–1000. doi: 10.1007/s00439-020-02120-y
Bustamante, J., Boisson-Dupuis, S., Abel, L., Casanova, J. L. (2014). Mendelian susceptibility to mycobacterial disease: Genetic, immunological, and clinical features of inborn errors of IFN-gamma immunity. Semin. Immunol. 26 (6), 454–470. doi: 10.1016/j.smim.2014.09.008
Casanova, J. L., Abel, L. (2002). Genetic dissection of immunity to mycobacteria: The human model. Annu. Rev. Immunol. 20, 581–620. doi: 10.1146/annurev.immunol.20.081501.125851
Chapgier, A., Boisson-Dupuis, S., Jouanguy, E., Vogt, G., Feinberg, J., Prochnicka-Chalufour, A., et al. (2006). Novel STAT1 alleles in otherwise healthy patients with mycobacterial disease. PLoS Genet. 2 (8), e131. doi: 10.1371/journal.pgen.0020131
Dupuis, S., Dargemont, C., Fieschi, C., Thomassin, N., Rosenzweig, S., Harris, J., et al. (2001). Impairment of mycobacterial but not viral immunity by a germline human STAT1 mutation. Science 293 (5528), 300–303. doi: 10.1126/science.1061154
Dupuis, S., Jouanguy, E., Al-Hajjar, S., Fieschi, C., Al-Mohsen, I. Z., Al-Jumaah, S., et al. (2003). Impaired response to interferon-alpha/beta and lethal viral disease in human STAT1 deficiency. Nat. Genet. 33 (3), 388–391. doi: 10.1038/ng1097
Hirata, O., Okada, S., Tsumura, M., Kagawa, R., Miki, M., Kawaguchi, H., et al. (2013). Heterozygosity for the Y701C STAT1 mutation in a multiplex kindred with multifocal osteomyelitis. Haematologica 98 (10), 1641–1649. doi: 10.3324/haematol.2013.083741
Horvath, C. M. (2000). STAT proteins and transcriptional responses to extracellular signals. Trends Biochem. Sci. 25 (10), 496–502. doi: 10.1016/s0968-0004(00)01624-8
Ihle, J. N. (2001). The stat family in cytokine signaling. Curr. Opin. Cell Biol. 13 (2), 211–217. doi: 10.1016/s0955-0674(00)00199-x
Ishii, K. J., Koyama, S., Nakagawa, A., Coban, C., Akira, S. (2008). Host innate immune receptors and beyond: Making sense of microbial infections. Cell Host Microbe 3 (6), 352–363. doi: 10.1016/j.chom.2008.05.003
Kagawa, R., Fujiki, R., Tsumura, M., Sakata, S., Nishimura, S., Itan, Y., et al. (2017). Alanine-scanning mutagenesis of human signal transducer and activator of transcription 1 to estimate loss- or gain-of-function variants. J. Allergy Clin. Immunol. 140 (1), 232–241. doi: 10.1016/j.jaci.2016.09.035
Le Voyer, T., Sakata, S., Tsumura, M., Khan, T., Esteve-Sole, A., Al-Saud, B. K., et al. (2021). Genetic, immunological, and clinical features of 32 patients with autosomal recessive STAT1 deficiency. J. Immunol. 207 (1), 133–152. doi: 10.4049/jimmunol.2001451
Lim, C. P., Cao, X. (2006). Structure, function, and regulation of STAT proteins. Mol. Biosyst. 2 (11), 536–550. doi: 10.1039/b606246f
Liu, L., Okada, S., Kong, X. F., Kreins, A. Y., Cypowyj, S., Abhyankar, A., et al. (2011). Gain-of-function human STAT1 mutations impair IL-17 immunity and underlie chronic mucocutaneous candidiasis. J. Exp. Med. 208 (8), 1635–1648. doi: 10.1084/jem.20110958
Liu, Z., Zhou, M., Yuan, C., Ni, Z., Liu, W., Tan, Y., et al. (2022). Two novel STAT1 mutations cause mendelian susceptibility to mycobacterial disease. Biochem. Biophys. Res. Commun. 591, 124–129. doi: 10.1016/j.bbrc.2021.11.036
Mao, X., Ren, Z., Parker, G. N., Sondermann, H., Pastorello, M. A., Wang, W., et al. (2005). Structural bases of unphosphorylated STAT1 association and receptor binding. Mol. Cell. 17 (6), 761–771. doi: 10.1016/j.molcel.2005.02.021
Mirzalieva, O., Juncker, M., Schwartzenburg, J., Desai, S. (2022). ISG15 and ISGylation in human diseases. Cells 11 (3), 538. doi: 10.3390/cells11030538
Muriel-Vizcaino, R., Yamazaki-Nakashimada, M., Lopez-Herrera, G., Santos-Argumedo, L., Ramirez-Alejo, N. (2016). Hemophagocytic lymphohistiocytosis as a complication in patients with MSMD. J. Clin. Immunol. 36 (5), 420–422. doi: 10.1007/s10875-016-0292-3
Naviglio, S., Soncini, E., Vairo, D., Lanfranchi, A., Badolato, R., Porta, F. (2017). Long-term survival after hematopoietic stem cell transplantation for complete STAT1 deficiency. J. Clin. Immunol. 37 (7), 701–706. doi: 10.1007/s10875-017-0430-6
Richards, S., Aziz, N., Bale, S., Bick, D., Das, S., Gastier-Foster, J., et al. (2015). Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American college of medical genetics and genomics and the association for molecular pathology. Genet. Med. 17 (5), 405–424. doi: 10.1038/gim.2015.30
Rosain, J., Kong, X. F., Martinez-Barricarte, R., Oleaga-Quintas, C., Ramirez-Alejo, N., Markle, J., et al. (2019). Mendelian susceptibility to mycobacterial disease: 2014-2018 update. Immunol. Cell Biol. 97 (4), 360–367. doi: 10.1111/imcb.12210
Sampaio, E. P., Bax, H. I., Hsu, A. P., Kristosturyan, E., Pechacek, J., Chandrasekaran, P., et al. (2012). A novel STAT1 mutation associated with disseminated mycobacterial disease. J. Clin. Immunol. 32 (4), 681–689. doi: 10.1007/s10875-012-9659-2
Toubiana, J., Okada, S., Hiller, J., Oleastro, M., Lagos, G. M., Aldave, B. J., et al. (2016). Heterozygous STAT1 gain-of-function mutations underlie an unexpectedly broad clinical phenotype. Blood 127 (25), 3154–3164. doi: 10.1182/blood-2015-11-679902
Tsumura, M., Miki, M., Mizoguchi, Y., Hirata, O., Nishimura, S., Tamaura, M., et al. (2022). Enhanced osteoclastogenesis in patients with MSMD due to impaired response to IFN-gamma. J. Allergy Clin. Immunol. 149 (1), 252–261. doi: 10.1016/j.jaci.2021.05.018
Tsumura, M., Okada, S., Sakai, H., Yasunaga, S., Ohtsubo, M., Murata, T., et al. (2012). Dominant-negative STAT1 SH2 domain mutations in unrelated patients with mendelian susceptibility to mycobacterial disease. Hum. Mutat. 33 (9), 1377–1387. doi: 10.1002/humu.22113
Ueki, M., Yamada, M., Ito, K., Tozawa, Y., Morino, S., Horikoshi, Y., et al. (2017). A heterozygous dominant-negative mutation in the coiled-coil domain of STAT1 is the cause of autosomal-dominant mendelian susceptibility to mycobacterial diseases. Clin. Immunol. 174, 24–31. doi: 10.1016/j.clim.2016.11.004
Vinkemeier, U. (2004). Getting the message across, STAT! design principles of a molecular signaling circuit. J. Cell Biol. 167 (2), 197–201. doi: 10.1083/jcb.200407163
Ying, W., Liu, D., Dong, X., Wang, W., Hui, X., Hou, J., et al. (2019). Current status of the management of mendelian susceptibility to mycobacterial disease in mainland china. J. Clin. Immunol. 39 (6), 600–610. doi: 10.1007/s10875-019-00672-x
Keywords: host genetics, STAT1, MSMD, loss-of-function, IFN-γ
Citation: Ye F, Zhang W, Dong J, Peng M, Fan C, Deng W, Zhang H and Yang L (2022) A novel STAT1 loss-of-function mutation associated with Mendelian susceptibility to mycobacterial disease. Front. Cell. Infect. Microbiol. 12:1002140. doi: 10.3389/fcimb.2022.1002140
Received: 24 July 2022; Accepted: 10 October 2022;
Published: 21 October 2022.
Edited by:
Wenhao Zhou, Fudan University, ChinaReviewed by:
Jacinta Bustamante, Université Paris Cité, FranceCopyright © 2022 Ye, Zhang, Dong, Peng, Fan, Deng, Zhang and Yang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Liangchun Yang, eWFuZ2xpYW5nY2h1bmdAMTYzLmNvbQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.