 Bart J. M. Grijmans
Bart J. M. Grijmans Sander B. van der Kooij
Sander B. van der Kooij Monica Varela
Monica Varela Annemarie H. Meijer
Annemarie H. Meijer- Institute of Biology Leiden, Leiden University, Leiden, Netherlands
Cells of the innate immune system continuously patrol the extracellular environment for potential microbial threats that are to be neutralized by phagocytosis and delivery to lysosomes. In addition, phagocytes employ autophagy as an innate immune mechanism against pathogens that succeed to escape the phagolysosomal pathway and invade the cytosol. In recent years, LC3-associated phagocytosis (LAP) has emerged as an intermediate between phagocytosis and autophagy. During LAP, phagocytes target extracellular microbes while using parts of the autophagic machinery to label the cargo-containing phagosomes for lysosomal degradation. LAP contributes greatly to host immunity against a multitude of bacterial pathogens. In the pursuit of survival, bacteria have developed elaborate strategies to disarm or circumvent the LAP process. In this review, we will outline the nature of the LAP mechanism and discuss recent insights into its interplay with bacterial pathogens.
Introduction
Throughout evolution, microbial pathogens and animal immune cells have developed elaborate mechanisms to face and withstand each other. Understanding these mechanisms lies at the heart of improving medical interventions against microbial infections. Phagocytes, specialized cells of the innate immune system, are characterized by their ability to engulf and intracellularly destroy foreign particles and dying cells. Engulfment and subsequent degradation of microbes is key to our innate, and ultimately adaptive defenses. Central to the phagocytic elimination of microbial invaders is the fusion of the phagosome with lysosomes, a process called phagosome maturation. Unless inhibited by virulence factors, an engulfed microbe will be exposed to an array of lysosomal enzymes — killing it within minutes (Fountain et al., 2021) .
Three different vesicle trafficking mechanisms are known to direct microbial pathogens to lysosomal degradation: phagocytosis, autophagy and LC3‐associated phagocytosis (LAP). All have overlapping characteristics but are initiated via distinct pathways, where cargo‐containing vesicles form and mature by different mechanisms. Phagocytosis, which was recognized as early as the 19th century, targets extracellular microbes via receptor‐mediated recognition (Rosales and Uribe‐Querol, 2017). Several pathogens have evolved strategies to subvert the phagocytic process, allowing them to establish a niche for their own proliferation (Flannagan et al., 2009). Some microbes, like Streptococcus pyogenes, can arrest ingestion by producing toxins or expressing antiphagocytic surface proteins (Brouwer et al., 2016), while others, like Mycobacterium tuberculosis, interfere with phagosome integrity to first establish an intravesicular replicative niche and subsequently escape the confines of the phagosomal vesicle (Simeone et al., 2021). Other microbes still, like Listeria monocytogenes, take advantage of the acidification of the phagosome, utilizing it to activate virulence-mediated disruption of the phagosomal membrane, leading to immediate invasion of the cytosol (Matereke and Okoh, 2020).
A second way for cells to effectively degrade microbial invaders is autophagy, strictly speaking macroautophagy. By definition, autophagy targets intracellular structures, such as protein aggregates or cytosolic bacteria, capturing them in a characteristic double‐membrane vesicle (Boya et al., 2013). Mediated by a group of conserved autophagy‐related proteins, a cup‐shaped double‐membrane complex is nucleated around a target structure. It extends to seal the target into a closed vesicle, the autophagosome. Similar to phagosomes, the transient autophagosomes mature by fusing with lysosomes. In recent decades, accumulating evidence has illustrated the extensive interactions between microbial pathogens and the host autophagic response, referred to as xenophagy (Deretic and Levine, 2009; Huang and Brumell, 2014). Intracellular pathogens have developed ingenious evasive mechanisms to avoid being killed in the autophagosome. Such strategies include the interference with autophagy‐initiating signaling, disruption of lysosome function and proteolytical inactivation of the autophagic machinery (Jiao and Sun, 2019). Furthermore, microbes have even evolved means to turn host autophagy to their own advantage, utilizing it to foster their own nutrient supply, replication, cellular egress and virulence (Kimmey and Stallings, 2016).
Since 2007, it has become clear that phagocytes have a third degradation mechanism to their disposal, which is now commonly referred to as LAP (Sanjuan et al., 2007). LAP has been described to exist at the crossroads of autophagy and phagocytosis, combining the strengths of both processes to ensure enhanced degradation of the engulfed cargo (Martinez, 2018; Heckmann and Green, 2019). Given its intricate role in anti-microbial immunity and preservation of homeostasis, LAP has sparked much interest in recent years. During LAP, which is initiated by receptor signaling, select parts of the autophagic machinery – particularly the ubiquitin-like protein LC3 (microtubule-associated proteins 1 A/1B light chain) – are specifically recruited to the single-membrane phagosome (Sanjuan et al., 2007). Early in the maturation process, an NADPH oxidase complex is assembled that generates reactive oxygen species (ROS) within the vesicle. Soon after LC3 is conjugated onto the phagosomal membrane, the phagosome (now termed LAPosome) fuses with lysosomes, leading to rapid clearance of the internalized material. LAP is often referred to as a form of non-canonical autophagy, but strictly speaking the term autophagy applies only in relation to the vesicular uptake of cytoplasmic cargo, while LAP targets vesicles with material coming directly from the extracellular environment.
Multiple lines of evidence have demonstrated that LAP mediates a variety of immunological functions that go beyond the elimination of pathogens. The process has been deemed important for the immunotolerant processing of dying cells, regulation of inflammatory responses, establishment of signaling compartments, and even attenuating autoimmunity (Martinez et al., 2015; Heckmann et al., 2017; Wong et al., 2021). With regard to human disease, LAP has drawn particular attention for its role in immunity to different classes of microbial pathogens (Chamilos et al., 2016; Besteiro, 2019; Jiao and Sun, 2019; Akoumianaki et al., 2021). In this review, we focus on the role of LAP in bacterial infectious diseases. We discuss the molecular mechanisms that orchestrate LAP, and provide an overview of its significance in fighting bacterial infections as well as its fragility in view of pathogenic evasion.,
Mechanisms of LAP Induction and Maturation
While classical autophagy and LAP have significant overlap in their utilization of the molecular machinery, induction of these processes is fundamentally distinct. LAP and related single membrane LC3 lipidation processes are triggered by the engagement of various surface receptors (Sanjuan et al., 2007; Martinez et al., 2015), including Toll-like receptors (TLRs), Dectin‐1, Dectin‐2, but also immunoglobulin receptors such as FcγR and scavenger receptors such as TIM4 (Sanjuan et al., 2007; Huang et al., 2009; Martinez et al., 2011; Ma et al., 2014; Lamprinaki et al., 2017). In addition, activation of the cytosolic innate immune sensor STING induces LC3 lipidation of single-membrane vesicles (Fischer et al., 2020). How these different cargo engagements and consequent signaling pathways activate the machinery required for LAP remains unclear. However, it has been well documented that LAP proceeds independently of the pre-initiation complex containing ULK1, ATG13, ATG101 and FIP200, which is crucial for autophagy induction (Martinez et al., 2011; Heckmann and Green, 2019). Indeed, LAP typically appears unresponsive to nutrient starvation and other autophagic signals associated with ULK1 activation (Sanjuan et al., 2007). Similar to phagocytosis but unlike classical autophagy, pathogens targeted by LAP are engulfed in a single‐membrane phagosome (Schille et al., 2018). This is one of the most significant ultrastructural differences that distinguishes LAPosomes from classical autophagosomes (Lai and Devenish, 2012).
After the pathogen is internalized, one of the first signaling complexes to associate with the budding phagosome is the class III phosphatidylinositol 3-kinase complex (PI3KC3), which ultimately delivers PI(3)P onto the phagosomal membrane (Matsunaga et al., 2009) (Figure 1A). The functional core of PI3KC3 is composed of VPS34 (the catalytic subunit), VPS15 and Beclin-1 (Backer, 2016). Following activation by VPS15 and Beclin-1, VPS34 generates PI(3)P from PI(3) via its kinase activity (Volinia et al., 1995; Petiot et al., 2000). The newly formed PI(3)P molecules disseminate throughout the phagosomal membrane, acting as a label for future LC3-conjugation (Martinez et al., 2011). Two critical proteins that specifically recruit PI3KC3 to the phagosome during LAP, are UVRAG and Rubicon (Martinez et al., 2015). While the PI3KC3 complex itself is non-specific for LAP, and is also involved in the activation of classical autophagy, Rubicon is essential for LAP maturation in contrast to its inhibitory role in autophagosome maturation (Martinez et al., 2015). In fact, Rubicon participates at multiple signaling steps relevant for LAP development through interaction with different binding partners (Matsunaga et al., 2009; Yang et al., 2012).
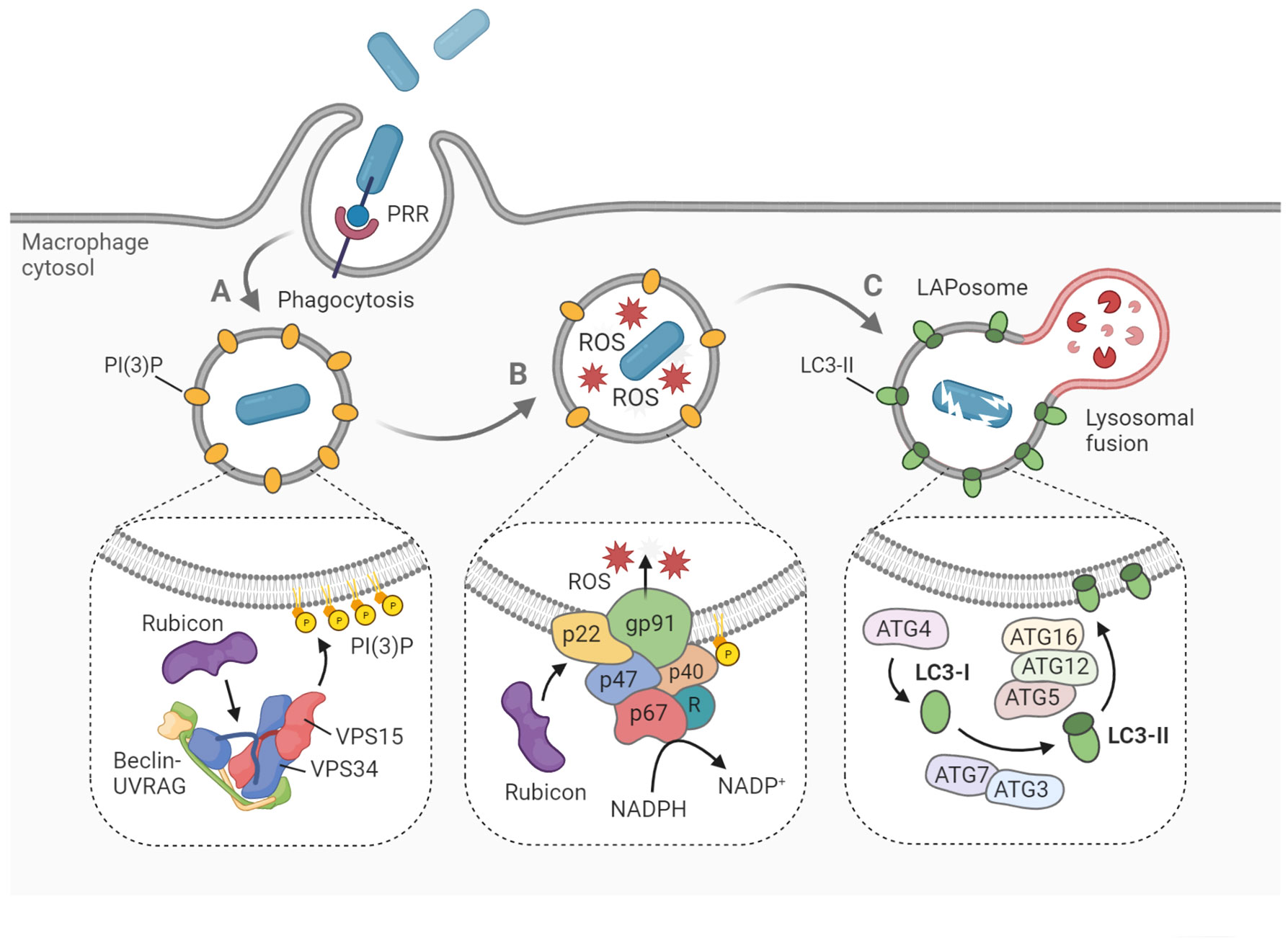
Figure 1 Hallmarks of LAP on the molecular level. LAP begins with pattern recognition receptor (PRR)-mediated phagocytosis of pathogens, dying cells and other particles. (A) The phagosome is marked with PI(3)P, a signaling lipid which is generated by the PI3KC3 complex, consisting of Beclin-1, UVRAG, VPS15, VPS34 and Rubicon. (B) Within the phagosome, ROS are produced by the NADPH oxidase complex. Rubicon stabilizes the complex via interaction with p22phox, while p40phox interacts with PI(3)P to recruit the remaining components. (C) Cytosolic LC3 is lipidated by the conjugation machinery to form LC3-II on the phagosomal membrane. Soon after, the LAPosome fuses with an available lysosome resulting in rapid degradation of the engulfed cargo. Figure created with BioRender.com.
Another hallmark of LAP, which also depends strictly on Rubicon activity, is the generation of reactive oxygen species (ROS) within the phagosome lumen (Martinez et al., 2015) (Figure 1B). ROS are produced by the NADPH oxidase 2 complex (NOX2), the only NADPH oxidase complex expressed in phagocytes (Bedard and Krause, 2007). The activity of NOX2 is dependent on the recruitment of four cytosolic subunits, namely p67phox, p47phox, p40phox and Rac1, to the two membrane-embedded subunits p22phox and gp91phox, which form the catalytic center. The p40 protein is capable of direct interaction with the PI(3)P present on the phagosome, functioning as a docking site for the other cytosolic subunits (Ellson et al., 2006). Rubicon is able to stabilize the NOX2 complex via direct interaction with p22, resulting in maximal ROS production (Yang et al., 2012). The ROS may serve several roles in the LAP process. Aside from their putative oxidative activity against the pathogen (Slauch, 2011), ROS are needed for recruitment of downstream LAP components, such as ATG7 and LC3 (Lam et al., 2013; Martinez et al., 2015). Furthermore, ROS generation by NOX2 has been shown to cause oxidative inactivation of ATG4B, thereby inhibiting the proteolytic release of LC3 and thus stabilizing the LAPosome (Ligeon et al., 2021). Further aspects of the mechanistic interplay between ROS signaling and LAP maturation are incompletely understood, though it is hypothesized that lipid peroxidation within the phagosome could serve a regulatory function (Holmström and Finkel, 2014).
After the phagosomal membrane is marked by PI(3)P and ROS have been produced, two conjugation systems are activated that will mediate the processing and incorporation of LC3 onto the phagosomal membrane (Figure 1C). Cytosolic pro-LC3 is converted into LC3-I by ATG4. Then, LC3-I is lipidated by ATG7-ATG3 and ATG12-ATG5-ATG16L1 via covalent attachment on phagosomal surface to form LC3-II (Martinez et al., 2015; Schille et al., 2018). Both LAP and classical autophagy are characterized by the association of LC3-II onto the target membrane. However, recruitment proceeds differently in both processes, as the target membrane is the phagophore in the case of autophagy, and the phagosome in the case of LAP (Herb et al., 2020). Furthermore, autophagy can maintain tissue homeostasis independent of LAP, which has been illustrated by the differential role of ATG16L1 in both processes. Specifically, autophagy requires the ATG5-binding and coiled coil domains of ATG16L1 but not the WD domain, whereas the WD domain is indispensable for LAP (Rai et al., 2019; Fischer et al., 2020; Wang et al., 2021). Following LC3-decoration, the LAPosome will rapidly fuse with lysosomes and acidify (Martinez et al., 2011). While it has been argued that LC3 family proteins play an important role in facilitating this lysosomal fusion, details about the vesicle fusion mechanism remain obscure (Martinez et al., 2015; McEwan et al., 2015; Nguyen and Yates, 2021).
Biological Functions of LAP
The primary function of LAP is to facilitate the fusion of phagosomes with lysosomes, assuring rapid degradation of the engulfed cargo and regulation of the appropriate immune response (Martinez, 2018). LAP and related single membrane LC3 lipidation processes exhibit a surprising antimicrobial versatility as it is required for successful processing of a wide variety of pathogens across different kingdoms, with the fungal pathogen Aspergillus fumigatus, the bacterial pathogen Listeria monocytogenes, the parasite Toxoplasma gondii, and Influenza A virus as notable examples (Martinez, 2018; Schille et al., 2018; Besteiro, 2019; Herb et al., 2020; Wang et al., 2021). Consequences of aberrant LAP for human disease is now an active field of research (Martinez, 2018; Upadhyay and Philips, 2019). In recent decades, interest in uncovering novel antimicrobial strategies has grown steadily, mainly due to the alarming prevalence of antibiotic resistance leading to incurable bacterial infections (Aslam et al., 2018).
In addition to its antimicrobial functions, LAP has been shown to be relevant for many other immunological processes, such as the clearance of dying cells and apoptotic remnants – a process known as efferocytosis. LAP enables professional phagocytes to process cellular debris in a remarkable immunosilent manner, by keeping levels of pro-inflammatory cytokines and associated signaling pathways at bay (Heckmann et al., 2017). Indeed, Rubicon-deficient mice show a defective clearance of apoptotic cells, resulting in an exaggerated inflammatory phenotype and ultimately the formation of auto-antibodies (Martinez et al., 2016). For humans, proper processing of cellular debris has been shown to be crucial for averting autoimmune disorders, such as systemic lupus erythematosus (SLE) (Muñoz et al., 2010). Intriguingly, genome-wide association studies among SLE patients have found a polymorphism in the ATG5 protein, suggesting that LAP or autophagy might play a critical role in the development of this disorder (Harley et al., 2008; Gateva et al., 2009). Furthermore, defects in LAP have been linked to numerous other inflammatory abnormalities, including atherosclerosis, visceral adiposity, and insulin resistance (Heckmann and Green, 2019).
Contrary to intuition, LAP may be a contributing factor in tumorigenesis, as it has been implicated in the establishment of a conducive microenvironment for cancerous cells. In mice, an increased LAP activity has been associated with tumor growth and aggressiveness (Asare et al., 2020). Indeed, high expression of Rubicon in cancer tissues predicts an adverse survival rate of patients with various cancer types. It is thought that the immunosuppressive signaling networks associated with LAP could be hijacked by developing cancer cells to bypass the immune response, thereby promoting their progression and metastatic potential. The implications of this have been reviewed elsewhere (Asare et al., 2020).
Interactions of Bacterial Pathogens With LAP
The diversity of evasive strategies adopted by different species of pathogens is testament to the complexity and effectiveness of the LAP process. In many cases, evasion mechanisms are only beginning to be discerned on the molecular level. Some bacterial pathogens circumvent LAP altogether by expressing effectors that impair their targeting, while others orchestrate their own internalization and survive inside phagosome (Figure 2). Below, we describe some notable examples of LAP-targeted bacterial pathogens and discuss how LAPosome formation and maturation may be modulated by virulence mechanisms of these pathogens (Table 1). We have included also cases that may represent different forms of single membrane LC3 lipidation closely resembling LAP.
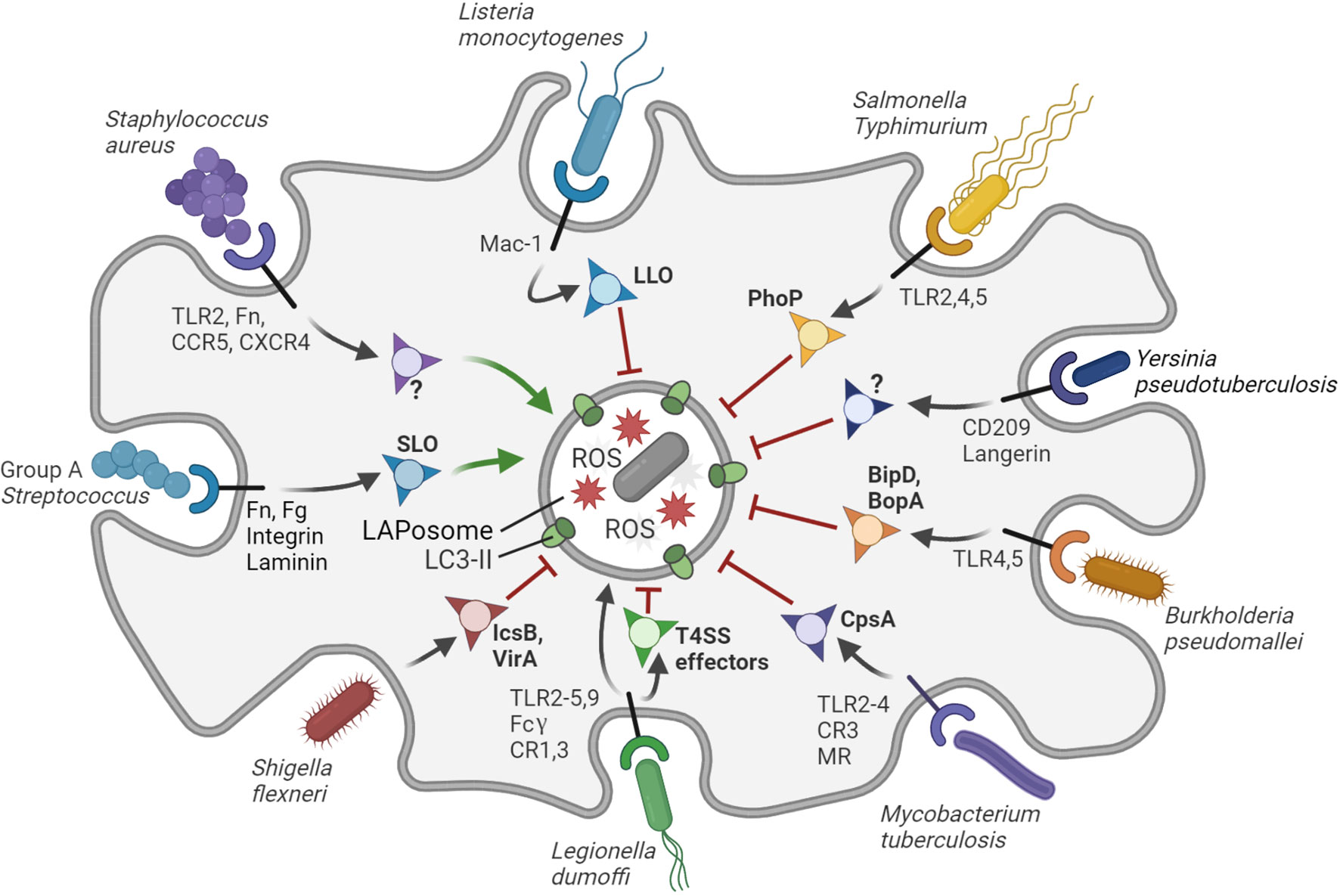
Figure 2 Interactions of bacterial pathogens with LAP. The LAPosome is a single membrane vesicle marked by LC3-II and producing ROS. LAP contributes to host defense, but bacterial pathogens have evolved diverse ways to inhibit (red arrows) or promote (green arrows) LAP to their own benefit. S. aureus promotes the formation of LAPosomes in neutrophils via an unknown virulence factor to establish a replicative niche. Group A Streptococcus promotes LAP via the virulence factor SLO to evade bactericidal xenophagy. L. dumoffi is effectively degraded in the LAPosome (black arrow), although it may inhibit LAP to some extent via T4SS effector proteins. All other bacterial pathogens shown in the figure can partially inhibit LAP in phagocytes or LAP-like processes in epithelial cells through the virulence factors indicated. Virulence factors that remain to be identified are indicated with question marks. The receptors that mediate entry and/or immune recognition by the host phagocytic cells are shown, except for S. flexneri, which attaches to surface proteins of M-cells in the gut epithelium. Figure created with BioRender.com.

Table 1 Overview of LAP-targeted bacterial pathogens and their evasion strategies.
Mycobacterium tuberculosis
Mycobacterium tuberculosis is the causative agent of acute or chronic manifestations of tuberculosis, the most lethal bacterial infectious disease today (WHO, 2020). M. tuberculosis is recognized and phagocytosed by macrophages via different surface receptors, including TLRs, mannose receptors and complement receptors (Schlesinger, 1993; Yu et al., 2014). Even though M. tuberculosis has been shown to be targeted by LAP, it is still not clear which fraction of phagosomes progresses to LAPosomes and whether this process enhances the ability of phagocytes to clear the pathogen or it is exploited by the pathogen for its intracellular survival (Köster et al., 2017; Köster et al., 2018).
LAP resistance of M. tuberculosis was found to be mediated by the virulence factor CpsA, which prevents recruitment of NOX2 to the pathogen-containing phagosome (Köster et al., 2017; Köster et al., 2018). While the inactivation of NOX2 by CpsA resulted in impaired lysosomal trafficking, reduced phagolysosome biogenesis and ultimately the survival and proliferation of intracellular M. tuberculosis, the deletion of CpsA in M. tuberculosis resulted in efficient degradation of the pathogen by LAP. M. tuberculosis is known to secrete several virulence factors that interfere with phagosome maturation and thus, it is likely to evade LAP in different ways. In this regard, the virulence factor NdkA has been shown to contribute to intracellular survival by interfering with phagosome maturation (Sun et al., 2010). Moreover, the presence of NdkA has been shown to decrease the recruitment of p67phox and Rac1 to the phagosome, interfering with ROS production by the NADPH oxidase complex and presumably undermining LAP (Sun et al., 2013).
In addition to inhibiting phagosome maturation, M. tuberculosis is able to conceal its presence by manipulating TLR recognition and therefore preventing its phagocytosis. It is known that TLR4 recognizes lipids, glycoproteins, secreted proteins and other surface ligands from M. tuberculosis, leading to fast phagocytosis of the pathogen (Stamm et al., 2015). Absence of phthiocerol dimycocerosate lipids (PDIM) in the M. tuberculosis cell wall induced an increase of TLR-dependent recruitment of microbicidal macrophages, indicating the inhibitory role of PDIM on pathogen recognition (Cambier et al., 2014). It has been suggested that this TLR recognition inhibition is due to masking of the mycobacterial pathogen associated molecular patterns (PAMPs) by PDIM (Cambier et al., 2014). The study of PDIM adds another dimension by which evasion of the LAP mechanism by M. tuberculosis is possible.
A screen in planarian flatworms identified a protein MORN2, of which the human ortholog was shown to play an important role in the LAP response of macrophages to M. tuberculosis, and also L. pneumophila and S. aureus (Abnave et al., 2014). MORN2 promotes LC3 recruitment to M. tuberculosis-containing phagosomes and their maturation into phagolysosomes. The role of LAP was confirmed by demonstrating the single-membrane nature of the bacteria-containing vesicles as well as the requirement of Atg5 and Beclin1 for LC3 recruitment, but not Ulk1 and Atg13 (Abnave et al., 2014). Further in line with the proposed role of MORN2 in LAP, its function was shown to depend on ROS (Morita et al., 2020). Using Escherichia coli and zymosan as alternative LAP substrates, SNARE proteins like SNAP-23 and syntaxin11 were implicated in vesicle fusions during MORN2-mediated LAP (Morita et al., 2020). Altogether, MORN2 emerges from this work as a positive regulator of LAP, which warrants further studies with M. tuberculosis and other pathogens.
In conclusion, while there is evidence that M. tuberculosis is actively targeted by LAP, its modes of evasion are still starting to be understood. Evasion of LAP by M. tuberculosis is likely to occur during the maturation of the phagosome while, among others virulence factors, CpsA and NdkA are secreted. Considering the diversity of virulence factors known to affect phagosome maturation, it is expected that M. tuberculosis mutant screens will soon reveal additional effectors critical for phagosome maturation and anti-LAP virulence. Additionally, evasion of LAP initiation via phagocytosis has been observed, a process mediated by effector molecules such as PDIM cell wall lipids. MORN2 seems a useful addition to Rubicon for further study as a host factor specifically promoting LAP.
Listeria monocytogenes
Listeria monocytogenes is an opportunistic bacterium that can cause severe food-borne diseases in immunocompromised individuals, pregnant women and newborns (Cossart and Lecuit, 1998; Lam et al., 2013). Clearance of L. monocytogenes is explicitly promoted by LAP and this host-pathogen interaction can be regarded as one of the most striking examples of the microbicidal power of the LAP pathway (Gluschko et al., 2018; Herb et al., 2018).
During infection, L. monocytogenes utilizes the virulence factors listeriolysin (LLO) and PlcA/B to escape from the phagosome and enter the cytosol, where it acquires actin-based motility (Cossart and Lecuit, 1998; Seveau, 2014). Within the cytosol, L. monocytogenes actively inhibits classical autophagy via IcsB, ActA and PlcA/B (Birmingham et al., 2008; Lam et al., 2013). LAP has become known as the key mechanism providing anti-Listeria immunity (Gluschko et al., 2018). Recognition of L. monocytogenes via the β2 integrin Mac-1 receptor activates LAP and the associated phagosomal ROS response. Interestingly, the same study also provided evidence that the execution of LAP is not influenced by virulence factors that inhibit classical autophagy and revealed a crucial role for acid sphingomyelinase, broadening our understanding of the LAP mechanism. The acid sphingmyelinase protein facilitates alterations in the lipid composition of the membrane, allowing the subsequent activation of the Nox2 complex, crucial for the ROS production and subsequent LC3 recruitment (Gluschko et al., 2018).
The possible evasion of LAP by L. monocytogenes is only beginning to be understood. Recent work showed that L. monocytogenes is able to suppress LAP by modulating mitochondrial calcium signaling (Li et al., 2021). After phagocytosis, L. monocytogenes induces mitochondrial calcium uptake by the mitochondrial Ca2+ uniporter (MCU) transporter. This increased calcium uptake promotes the production of acetyl-coenzyme A (acetyl-CoA) by pyruvate hydrogenase. Outside the mitochondrion, Rubicon is acetylated by acetyl-CoA resulting in decreased activity of Rubicon in the LAP pathway, thus acting in favour of bacterial survival. In agreement, a knockout of the MCU transporter abolishes calcium uptake, allowing LAP to overpower L. monocytogenes infection (Li et al., 2021). Together, these results show that L. monocytogenes is able to inhibit LAP through eliciting mitochondrial signaling, which adds to the growing connective network between mitochondrial metabolism and innate immune defense mechanisms (Li et al., 2021).
Interestingly, there are also cases known in which L. monocytogenes induces a prolonged infection which LAP fails to control. This might be achieved due to the formation of certain compartments termed spacious Listeria-containing phagosomes (SLAPs) via manipulation of the LAP mechanism. It is believed that these single membrane compartments provide a niche in which the bacteria are able to replicate and proliferate. The formation of this niche is possible due to failure of LAP to clear the infection and a lack of the expression of virulence factors that mediate escape from phagosomes into the cytosol (Birmingham et al., 2008; Lam et al., 2013). However, it should be noted that SLAPs were observed in immunodeficient or oncogenic transformed cells, and therefore their formation in healthy macrophages remains unclear.
Recent studies revealed that phagosome permeabilization by L. monocytogenes triggers another single membrane LC3 lipidation pathway, which has been named pore-forming toxin-induced non-canonical autophagy pathway (PINCA) (Mitchell et al., 2018; Gluschko et al., 2021). In bone marrow-derived macrophages, L. monocytogenes was shown to be targeted sequentially by multiple autophagic processes. The LLO-mediated perforation of phagosomes was shown to trigger LC3 recruitment in an ULK1-independent process. However, this PINCA response had no role in restricting bacteria growth, in contrast to subsequent xenophagy, which defends against L. monocytogenes bacteria upon invasion of the cytosol (Mitchell et al., 2018). PINCA is distinct from LAP, because it can occur in NOX2-deficient macrophages (Gluschko et al., 2021). Induction of LAP in PINCA-competent cells confirmed that LAP contributes to host defense, while no clear anti-Listeria function of PINCA could be identified (Gluschko et al., 2021).
To summarize, LAP provides anti-Listeria immunity in macrophages, while PINCA, the LC3 recruitment to permeabilized phagosomes, does not restrict bacterial growth (Gluschko et al., 2018; Herb et al., 2018; Mitchell et al., 2018; Gluschko et al., 2021). The host defense function of LAP is counteracted by bacterial LLO, the primary virulence factor that mediates invasion of the cytosol after phagocytosis, where L. monocytogenes has to defend itself against xenophagy (Seveau, 2014; Osborne and Brumell, 2017; Mitchell et al., 2018). In addition, it was recently reported that L. monocytogenes is able to suppress LAP by manipulating the MCU transporter and redirecting calcium signaling to inhibit the key LAP host factor, Rubicon (Li et al., 2021). Many cases of prolonged infections of L. monocytogenes are known, suggesting that bacterial virulence mechanism can modulate LAP to clear the way for SLAP biogenesis (Lam et al., 2013). The distinctive roles of LAP and PINCA and the mechanistic differences between these two processes require further dissection. It will be of great interest to investigate how these two mechanisms may also function side by side in infections with other pathogens that permeabilize phagosomes.
Salmonella typhimurium
Salmonella enterica serovar Typhimurium (S. typhimurium) is an intracellular pathogen that can invade both non-myeloid and phagocytic cells and is a major cause of gastroenteritis (Ibarra and Steele-Mortimer, 2009). Early studies on Salmonella infection in mouse macrophages and human epithelial cells already suggested that LAP could be a critical player in the immune response, because the triggering of TLR or Fc-gamma receptors induced LC3 recruitment on phagosomes in a manner dependent on ROS production (Huang et al., 2009). The requirement of phagocytic NADPH oxidase for LC3 recruitment to macrophage phagosomes was confirmed by knockdown of the Cyba component of NOX2 in a zebrafish embryo model of systemic S. typhimurium infection (Masud et al., 2019a). Furthermore, knockdown of Atg5 and Rubicon, but not the autophagy preinitiation factor Atg13, were shown to be required for LC3 recruitment and for the successful clearance of bacteria in the zebrafish model, providing in vivo evidence for the anti-Salmonella function of LAP (Masud et al., 2019a).
While LAP provides protection to S. typhimurium infection in zebrafish embryos, there is still a high mortality rate, indicating that the pathogen can resist LAP to a certain extent (Masud et al., 2019a). Several mutant S. typhimurium strains were screened to determine the possible role of virulence factors in LAP evasion (Masud et al., 2019b). None of the virulence factors tested, PhoP, PurA, FlhD, SipB and SsrB, appeared to be necessary for the host LAP response, as mutations in these factors did not abolish Rubicon-dependent GFP-LC3 recruitment (Masud et al., 2019b). However, quantitative differences in GFP-LC3 recruitment were observed between the wild type and mutant strains. The PhoP and PurA deficient strains, both attenuated in zebrafish and other animal models, respectively elicited higher and lower GFP-LC3 recruiment (Masud et al., 2019b; Garvis et al., 2001; Thompson et al., 2011; Dalebroux and Miller, 2014). The PhoP regulon has been reported to reduce TLR activation, serve a role in the inhibition of the phagolysosomal fusion, and mediate adaption to intra-macrophage stress (Garvis et al., 2001; Thompson et al., 2011; Dalebroux and Miller, 2014). Therefore, the higher levels of GFP-LC3 recruitment in infection with the ΔphoP mutant could suggest a role for PhoP in LAP evasion (Masud et al., 2019b). In contrast, in the case of ΔPurA mutant bacteria, a strongly reduced GFP-LC3 recruitment was observed, which might be explained by the virtually complete loss of virulence of this mutant, which could lead to rapid clearance of most of the bacterial population without inducing signals for LAP. (O’Callaghan et al., 1988; Masud et al., 2019b).
Mutation in the FlhD gene, which is crucial for flagella formation of S. typhimurium, strongly reduced GFP-LC3 recruitment in the zebrafish model. In line with results in mice, FlhD mutation also resulted in hypervirulence of the S. typhimurium pathogen in zebrafish (Fournier et al., 2009). An explanation for both the reduced GFP-LC3 recruitment and the hypervirulence could be that LAP induction is dependent on the recognition of flagellin by TLR5. However, to date no direct link between the signaling of TLR5 and LAP has been established, and therefore the role of the TLR ligand receptor interaction in LAP remains to be studied.
Finally, all the above-mentioned S. typhimurium strains displayed increased virulence in a Rubicon-deficient zebrafish host, with the notable exception of a ΔSsrB mutant (Masud et al., 2019b). SsrB is part of the bacterial regulatory system controlling expression of Salmonella Pathogenicity Island 2 (SPI2) effector molecules that are required for maintenance of the Salmonella-containing vacuole (Walthers et al., 2007). Knockdown of zebrafish Rubicon led to reduced GFP-LC3 recruitment towards ΔSsrB mutant bacteria, similar as observed with wild type bacteria or other virulence mutants. However, ΔSsrB survival was unaffected by Rubicon knockdown, suggesting that SPI2 effectors could be important for intracellular replication of S. typhimurium under conditions where LAP is impaired (Masud et al., 2019b).
To sum up, S. typhimurium is a pathogen which is targeted by the LAP pathway that is crucial for proper pathogen clearance, possibly triggered by TLR5-mediated recognition of flagella. Although successful engulfment and degradation is observed, it is possible that virulence factors like the PhoP/Q operon contribute to LAP evasion. Unlike other wild type or mutant Salmonella strains, ΔSsrB mutants, impaired in the expression of SPI2 effectors, were unable to display increased virulence in a LAP-deficient zebrafish host. The specific SPI2 effector(s) responsible for this phenotype remain to be established.
Legionella dumoffii
Legionella dumoffii is an intracellular pathogen which can reside in the vacuole after phagocytosis, and is closely related to the human lung disease pathogen, Legionella pneumophila (Horwitz, 1983). Phagocytosis of Legionella species is mediated by the CR1 and CR3 complement receptors and the Fcy receptor, and innate immune recognition of cell wall components, flagella and bacterial DNA is facilitated by among others the TLR2,3,4,5,9 receptors (Husmann and Johnson, 1992; Grigoryeva and Cianciotto, 2021). Upon phagocytosis, a subpopulation of L. dumoffii-containing single-membrane vesicles is decorated with LC3, which requires Rubicon and NOX2 activity, indicating that maturation of these vesicles occurs via the LAP pathway (Hubber et al., 2017). In addition, the initiation of the LAP response towards L. dumoffii requires pathogen recognition via TLR2 and diacylglycerol signaling. There was no interaction of L. dumoffii with ubiquitin receptors and LC3 decoration was independent of ULK1 kinase, thus arguing against a role for selective autophagy and supporting that a subpopulation of L. dumoffii resides in LAPosomes (Hubber et al., 2017).
Interestingly, the formation of L. dumoffii-containing LAPosomes is dependent on the presence of the bacterial type four secretion system (T4SS). However, independent of LAP, the majority of the bacteria-containing phagosomes are remodelled into a compartment that resembles the endoplasmatic reticulum, thereby inhibiting the fusion with lysosomes and allowing replication (Hubber et al., 2017). Similar to LAP this process is also mediated via the activity of T4SS, but it is not understood what determines if expression of T4SS leads to evasion of the immune system or directs bacteria to LAP-mediated degradation. To date L. dumoffii remains a relatively poorly studied pathogen compared to other pathogens. Further research involving L. dumoffii should be performed to create a more in depth understanding of the interaction between LAP and L. dumoffi.
Burkholderia pseudomallei
Burkholderia pseudomallei is a soil-dwelling pathogen that causes pneumonia, skin changes and sometimes severe inflammatory cascades and lethal sepsis, a condition known as melioidosis (Wiersinga et al., 2007). It is phagocytosed by macrophages, neutrophils and dendritic cells, and capable of invading epithelial cells (Horton et al., 2012). The pathogen is recognized by TLR2 and TLR4, but TLR2 has been shown to impact negatively on the host defense function, suggesting that this TLR is responsive for severe dysregulation of the immune system and/or facilitates the creation of a bacterial replication niche (Wiersinga et al., 2007). The type III secretion system (T3SS) of B. pseudomallei is required for its escape from phagosomes, permitting replication in the cytosol (Cullinane et al., 2008).
B. pseudomallei was found to co-localize with LC3 during infection of mouse RAW 264.7 macrophages and resides in single-membrane compartments, characterized as LAPosomes (Cullinane et al., 2008; Gong et al., 2011; Li et al., 2013). Starvation but not rapamycin treatment enhanced the residence B. pseudomallei in these LAPsomes, a process requiring Beclin 1 activity (Li et al., 2013). Treatment of RAW264.7 macrophages with lipopolysaccharide (LPS) from B. pseudomallei increased GFP-LC3 puncta formation, while removal of LPS decreased this response. Considering that the effect of B. pseudomallei LPS is mediated by TLR4 and unexpectedly also TLR2, it was proposed that LPS induces LAP in a TLR-dependent manner during B. pseudomallei infection (Wiersinga et al., 2007; Gong et al., 2011). By mediating the escape from phagosomes, the T3SS facilitates evasion of the LAP mechanism (Cullinane et al., 2008; Gong et al., 2011). Mutant bacteria for the bopA and bipD proteins, both crucial for the T3SS, show diminished escape from the phagosome, indicating the importance of a proper functioning T3SS for evasion of LAP (Cullinane et al., 2008; Gong et al., 2011).
The role of T3SS could in theory be exploited to increase the susceptibility of B. pseudomallei to LAP. T3SS-associated ATPases are known to be crucial for the proper function of the TTSS3 and therefore represent possible targets for modulating the interaction of the pathogen with LAP. Small-molecule inhibitors for the T3SS ATPase have been identified and are used to study the effect on B. pseudomallei infection and LAP. One of the ATPase inhibitors counteracted the escape of bacteria from the phagosome, leading to increased targeting by LAP and reduced bacterial survival. These promising results could be important for the development of therapies aimed against B. pseudomallei infections (Gong et al., 2015).
Yersinia pseudotuberculosis
Yersinia pseudotuberculosis is another food-born pathogen capable of causing an enteric illness. It can infect both epithelial cells and phagocytes by binding to integrins (Isberg and Leong, 1990; Pujol and Bliska, 2003). After invading a phagocyte, Y. pseudotuberculosis can survive inside the cell by manipulating the autophagy machinery and impairing the acidification of the autophagosome (Moreau et al., 2010). However, in epithelial cells Y. pseudotuberculosis was found to be captured in LC3-decorated, single-membrane and non-acidic vesicles. Despite that epithelial cells are non-phagocytic and lack the NOX2 complex required for LAP, the response of these cells to Y. pseudotuberculosis is reminiscent of LAP and could represent a related mechanism (Ligeon et al., 2014).
The study of Y. pseudotuberculosis in epithelial cells focused on the role of host derived, vesicle-associated membrane proteins (SNARE proteins) in the LAP-like response (Ligeon et al., 2014). At least two of the SNARE family members, VAMP3 and VAMP7, were found to be involved in the recruitment of LC3 to the pathogen-containing vesicles. Overexpression of VAMP3 resulted in an increase of Y. pseudotuberculosis bacteria localized in single-membrane vesicles. Conversely a knockdown of VAMP3 resulted in an increase of Y. pseudotuberculosis bacteria localized into double-membrane vesicles. These results suggest that a high concentration of VAMP3 increases LAP-like activity and a low concentration of VAMP3 increases the activity of the classical autophagy. In other words, VAMP3 appears to function as a molecular checkpoint for commitment to the single membrane pathway (LAP-like) or to the double membrane pathway (classical autophagy), dependent on its expression level. VAMP7 associates with the single membrane vesicles after the recruitment of VAMP3. Knockdown of VAMP7 led to a decrease in LC3 decoration of the single membrane compartments, suggesting that VAMP7 protein mediates LC3 recruitment during the LAP-like process. It should be noted that the VAMP7 protein also participates in the recruitment of LC3 during the classical form of autophagy, thereby suggesting a double role for VAMP7 of which the mechanism still remains unknown (Ligeon et al., 2014).
Evasion of the LAP-like response by Y. pseudotuberculosis is presumably mediated by blocking the acidification of the phagosome, something which is also seen in classical autophagy (Ligeon et al., 2014). Both the LAP-like process and autophagy are manipulated to establish a non-acidic niche, which raises the question how the manipulation of these two mechanisms is mediated and which processes contribute to their development. Multiple studies showed that SNARE proteins like VAMP3 and VAMP7 could be key to determine the maturation of different vesicular pathways (Fader et al., 2009; Itakura et al., 2012; Moreau et al., 2013).
Concluding, single membrane LC3 lipidation mechanism similar to LAP seems to target the Y. pseudotuberculosis pathogen in epithelial cells, but evasion of this mechanism by inhibition of LAPosome maturation is observed, leading to the formation of a replication niche (Ligeon et al., 2014). The VAMP3 protein seems to be a molecular switch for commitment to the single membrane or double membrane pathways. Additional evidence indicated a role for VAMP7 in LC3 recruitment during the LAP-like response, similar as in classical autophagy (Ligeon et al., 2014). It remains to be established whether or not this response also plays a prominent role in other cell types, including phagocytes.
Shigella flexneri
Shigella flexneri is a pathogen that invades epithelial cells and is targeted by a LAP-like mechanism early during infection, but is capable of effectively evading this host defense reponseby escaping into the cytosol and acquiring actin-based motility similar to L. monocytogenes (Baxt and Goldberg, 2014). It has been found that the presence of the T3SS is crucial to induce the uptake of S. flexneri, followed by the initiation of the LAP-like process (Campbell-Valois et al., 2015). IcsB and VirA are secreted effector proteins involved in the escape of the pathogen from the LC3-decorated vesicle into the cytosol, and therefore these virulence factors are also crucial for the evasion of the LAP-like pathway. (Baxt and Goldberg, 2014; Campbell-Valois et al., 2015).
Toca-1 is a host-derived protein required for the formation of actin tails that propel S. flexneri (Leung et al., 2008). The interaction of Toca-1 with IcsB was found to inhibit LC3 recruitment, presumably by inhibiting the ATG5 protein, which is crucial for the recruitment of LC3 to the phagosome (Baxt and Goldberg, 2014). Recent results also indicated that Toca-1, besides interacting with IcsB, also interacts with several other S. flexneri effectors, namely IpaB, OspC3 and IpgD. The function of these interactions and possible role in the evasion of the LAP-like response, autophagy and other aspects of S. flexneri pathogenesis remains to be further investigated (Miller et al., 2018).
Group A Streptococcus and Streptococcus pneumoniae
Group A Streptococci (GAS), mostly belonging to the species Streptococcus pyogenes, are commonly found among the bacteria colonizing the throat and skin, but they can also cause a range of mild to severe infections, including the deathly toxic shock syndrome (Henningham et al., 2012). Similarly, Streptococcus pneumoniae, which is not classified under GAS, generally colonizes the nasopharynx, but can become a cause of pneumonia, septicemia and meningitis (Bogaert et al., 2004). Streptococci adhere to various host cell surface receptors, among which fibronectin, fibrinogen, integrins and laminins (Brouwer et al., 2016). Recent studies have implicated LAP in the innate immune defense against both GAS and S. pneumoniae (Lu et al., 2017; Cheng et al., 2019; Inomata et al., 2020; Ogawa et al., 2020; Shizukuishi et al., 2020).
GAS is able to survive and replicate in endothelial cells. While these cells are autophagy competent under starvation, they were unable to sequester GAS in autophagosomes, which could be attributed to defective ubiquitin recruitment (Lu et al., 2017). The endothelial cells did capture GAS inside single membrane, LC3-associated vesicles. However, these GAS-containing vesicles failed to properly acidify after fusion with lysosomes and therefore bacterial clearance was impaired (Lu et al., 2017). NOX2 but not ULK1 was found to colocalize with the LC3-positive GAS-containing vesicles, indicating that they arise by LAP (Cheng et al., 2019). Inhibition of ROS production via NOX2, restored the vesicle acidification, redirected LAP to conventional anti-bacterial autophagy, and thereby reduced the intracellular growth of GAS. Furthermore, it was shown that streptolysin O (SLO) induces LAP and associated ROS production via β1 integrin. Thus, GAS evades the conventional, bacteriostatic autophagy route and induces a largely ineffective LAP response via its virulence factor SLO.
In the case of S. pneumoniae, LC3 association was investigated both in non-myeloid cells (fibroblasts) and in macrophages (Inomata et al., 2020; Ogawa et al., 2020; Shizukuishi et al., 2020). In non-myeloid cells it was observed that a LAP-like process and canonical autophagy are deployed sequentially, with the formation of LAPosome-like vesicles being indispensable for subsequent autophagosomes formation (Ogawa et al., 2020; Shizukuishi et al., 2020). In contrast to the LAP pathway, the S. pneumoniae-containing vesicles that resemble LAPosomes acquire LC3 independently of ROS. However, a feature shared with LAP is that their formation does not require FIP200, a component of the autophagy preinitiation complex. It was observed that that interactions between SQSTM1/p62 and ATG16L1 PcLV are required for the formation of the LAPosome-like vesicles and that LC3 and NDP52 (a member of the SQSTM1/p62 family) disappeared from these vesicles prior to the transition of the bacteria to autophagomes (Ogawa et al., 2020). What precisely distinguishes this LAP-like process from LAP, and whether the two processes can be operative simultaneously, requires further investigation.
In murine bone marrow-derived macrophages, a common LAP response to S. pneumoniae was observed where formation of LC3-positive, single membrane vesicles required Rubicon, NADPH oxidase, Atg5 and Atg7, but none of the autophagy preinitiation factors, Ulk1, FIP200, and Atg14 (Inomata et al., 2020). While highly efficient in macrophages from young mice, this LAP pathway was defective in macrophages from old mice, making them deficient in bacterial killing. Concomitant with the loss of LAP, macrophages from older mice also produced high levels of inflammatory cytokines. These interesting findings suggest that diminishing of LAP with age contributes to inflammation and infection susceptibility (Inomata et al., 2020).
Staphylococcus aureus
S. aureus can cause a wide range of diseases, from local skin infections to fatal bacteremia, often associated with antibiotic resistance (Lowy, 1998). While known for its extensive extracellular growth ability in infected tissues, intracellular growth stages in host phagocytes were recently found to be crucial for S. aureus pathogenicity (Prajsnar et al., 2012; McVicker et al., 2014). The internalization of S.aureus and its recognition is mediated by several surface proteins and receptors, including fibronectin, TLR2, and chemokine receptors like CCR5 and CXCR4 (Edwards et al., 2010; Bi et al., 2015; Tam et al., 2016).
Studies into the autophagy response to S. aureus led to different outcomes, pointing either to a host-beneficial effect or suggesting that the pathogen takes advantage of the host autophagy machinery (reviewed in Munoz-Sanchez et al., 2020). Similarly, the host LAP pathway has been found to be exploited to the pathogen’s benefit (Prajsnar et al., 2021). In a zebrafish systemic infection model, S. aureus was found to establish an intracellular niche in neutrophils. When internalized by these phagocytes, S. aureus was rapidly decorated by GFP-LC3, forming spacious GFP-LC3-positive vacuoles that did not acidify. Chemical and genetic disruption of NADPH oxidase prevented GFP-LC3 recruitment, indicating that the replication niche is formed by LAP, although the role of Rubicon was not addressed. Autophagy played an antagonistic role in this infection model, as GFP-Sqstm1 (p62) also decorated a subset of bacteria and Sqstm1 knockdown impaired host survival. Thus, despite a protective effect of selective autophagy, the prevailing LAP response in zebrafish neutrophils contributes to S. aureus pathogenesis and inhibition of this response improves host resistance (Prajsnar et al., 2021). The S. aureus virulence factors involved in generating the spacious LAPosomes and preventing acidification are yet to be uncovered.
Conclusions and Perspectives
When the LAP process was first described, its importance for microbial control was already underlined (Sanjuan et al., 2007). In the years that followed, it became clear that LAP constitutes a critical cornerstone for host defense against a variety of bacterial invaders. We now know that M. tuberculosis, L. monocytogenes, L. dumoffi, S. Typhimurium, B. pseudomallei, among other pathogens discussed in this review, are captured in a LC3-II-positive single-membrane phagosome and require Rubicon and NOX2-driven ROS production for their clearance. As the list of bacteria targeted by LAP continues to grow, efforts have been dedicated to determine how LAP affects the pathology of infectious disease. LAP has been best characterized in macrophages, yet LC3 lipidation of phagosomes has recently also been demonstrated in neutrophils, albeit as a mechanism of bacterial pathogenesis (Prajsnar et al., 2021). Most of the knowledge on LAP is based on genetic analyses of NOX2 and Rubicon, which also have LAP-independent roles in host defense and autophagy that complicate the interpretation of data. Furthermore, much remains to be discovered about the mechanisms downstream of NOX2 and Rubicon and about mechanisms independent of these two factors, especially because multiple pathways to single membrane LC3 lipidation seem to exist (Mitchell et al., 2018; Rai et al., 2019; Fischer et al., 2020, Gluschko et al., 2021 Wong et al., 2021).
Future research should lead to better understanding of the discrete mechanisms and functions of LAP and LAP-like processes, such as PINCA, which is triggered by phagosome permeabilization rather than by NOX2 activity (Mitchell et al., 2018; Gluschko et al., 2021). Another important area for future research is how LAP might work in concert with the closely related and recently discovered process, LC3-associated endocytosis (LANDO) (Heckmann and Green, 2019). LANDO has been shown to regulate the turnover of Aβ receptors in a murine model of Alzheimer’s disease. It will be of great interest to explore if LANDO and LAP also control levels of pattern recognition receptors and thereby contribute to the regulation of the innate immune response and pathogen clearance.
Although the different evasive strategies that bacteria use to circumvent or take advantage of LAP are progressively being unraveled, many questions about the molecular mechanisms that undergird these strategies remain unexplored. Strikingly, most bacterial pathogens targeted by LAP have evolved ways to specifically interfere with NOX2, signifying the central importance of NOX-derived ROS in LAP maturation. It is still difficult to say if this importance arises from the microbicidal or rather from the signaling functions of ROS (Holmström and Finkel, 2014), although this seems to differ between bacterial species (Herb and Schramm, 2021). Redox regulation of ATG proteins is indeed a prerequisite for the production of LC3-II during autophagy (Scherz-Shouval et al., 2007). Recently, it was discovered that NOX2 has a role in stabilizing the LAPosome itself by safeguarding LC3-II via redox regulation of ATG4B (Ligeon et al., 2021). Future studies should seek to answer how ROS contribute to pathogen clearance and engage parts of the LAP machinery, like the LC3 conjugation systems.
The ways in which LAP enhances phagosome-lysosome fusion are incompletely understood. Different bacterial effectors such as Mycobacterium CpsA and Legionella RavZ have been associated with impaired lysosomal trafficking during LAP (Choy et al., 2012; Köster et al., 2017). Such effectors may be critical for LAP evasion. However, as phagosome–lysosome fusion is a highly dynamic process that depends on membrane lipid composition and the coordinated action of Rab GTPases, tethering factors and SNAREs (Nguyen and Yates, 2021), details of the evasion strategies counteracting lysosomal fusion have yet to be substantiated.
At present, the machinery required for LAP can be specifically manipulated by various pharmacological or genetic means, such as the recently developed Rubicon inhibitor TIPTP (Kim et al., 2020), as well as Rubicon- and ATG16L1-deficient mouse lines (Martinez et al., 2016; Rai et al., 2019). Together, these techniques will be of great use to elucidate how bacterial species are targeted and killed by LAP, leaving aside the confounding effects of classical autophagy. Better knowledge about the antibacterial effects of LAP, and the comparison with antifungal and antiparasitic LAP mechanisms, could provide vital clues for developing novel intervention strategies in the ongoing battle against infectious diseases.
In most circumstances, cells that are proficient in LAP are generally well equipped to combat bacterial infection. However, some pathogens, with S. aureus as a notable example, are able to exploit LAP to generate a replication niche. In time, our understanding of LAP and its links with infectious disease will continue to increase in scope and diversity. It is, in the words of Shakespeare, a pathway lapp’d in proof – that is, clad in strong (proven) armor – when it comes to virulent bacteria that continue to undermine our vulnerable immune systems.
Author Contributions
BG and SK performed the literature research, wrote the manuscript, and designed the figures. MV made textual revisions. AM supervised the literature research and made textual revisions. All authors checked and approved the final version.
Funding
Our research related to this review is funded by the European Union’s Horizon2020 Marie Sklodowska-Curie projects H2020-MSCA-IF-2014-655424 and 721537-ImageInLife.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
We thank our colleagues at the Institute of Biology Leiden for helpful discussions.
References
Abnave, P., Mottola, G., Gimenez, G., Boucherit, N., Trouplin, V., Torre, C., et al. (2014). Screening in Planarians Identifies MORN2 as a Key Component in LC3-Associated Phagocytosis and Resistance to Bacterial Infection. Cell Host Microbe 16, 338–350. doi: 10.1016/j.chom.2014.08.002
Akoumianaki, T., Vaporidi, K., Diamantaki, E., Pène, F., Beau, R., Gresnigt, M. S., et al. (2021). Uncoupling of IL-6 Signaling and LC3-Associated Phagocytosis Drives Immunoparalysis During Sepsis. Cell Host Microbe 29, 1277–1293. doi: 10.1016/j.chom.2021.06.002
Asare, P. F., Roscioli, E., Hurtado, P. R., Tran, H. B., Mah, C. Y., Hodge, S. (2020). Lc3-Associated Phagocytosis (Lap): A Potentially Influential Mediator of Efferocytosis-Related Tumor Progression and Aggressiveness. Front. Oncol. 10. doi: 10.3389/fonc.2020.01298
Aslam, B., Wang, W., Arshad, M. I., Khurshid, M., Muzammil, S., Rasool, M. H., et al. (2018). Antibiotic Resistance: A Rundown of a Global Crisis. Infect. Drug Resist. 11, 1645. doi: 10.2147/IDR.S173867
Backer, J. M. (2016). The Intricate Regulation and Complex Functions of the Class Iii Phosphoinositide 3-Kinase Vps34. Biochem. J. 473, 2251–2271. doi: 10.1042/BCJ20160170
Baxt, L. A., Goldberg, M. B. (2014). Host and Bacterial Proteins That Repress Recruitment of Lc3 to Shigella Early During Infection. PloS One 9, e94653. doi: 10.1371/journal.pone.0094653
Bedard, K., Krause, K.-H. (2007). The Nox Family of Ros-Generating Nadph Oxidases: Physiology and Pathophysiology. Physiol. Rev. 87, 245–313. doi: 10.1152/physrev.00044.2005
Besteiro, S. (2019). The Role of Host Autophagy Machinery in Controlling Toxoplasma Infection. Virulence 10, 438–447. doi: 10.1080/21505594.2018.1518102
Bi, D., Qiao, L., Bergelson, I., Ek, C. J., Duan, L., Zhang, X., et al. (2015). Staphylococcus Epidermidis Bacteremia Induces Brain Injury in Neonatal Mice via Toll-Like Receptor 2-Dependent and -Independent Pathways. J. Infect. Dis. 212, 1480–1490. doi: 10.1093/infdis/jiv231
Birmingham, C. L., Higgins, D. E., Brumell, J. H. (2008). Avoiding Death by Autophagy: Interactions of Listeria Monocytogenes With the Macrophage Autophagy System. Autophagy 4, 368–371. doi: 10.4161/auto.5594
Bogaert, D., De Groot, R., Hermans, P. W. (2004). Streptococcus Pneumoniae Colonisation: The Key to Pneumococcal Disease. Lancet Infect. Dis. 4, 144–154. doi: 10.1016/S1473-3099(04)00938-7
Boya, P., Reggiori, F., Codogno, P. (2013). Emerging Regulation and Functions of Autophagy. Nat. Cell Biol. 15, 713–720. doi: 10.1038/ncb2788
Brouwer, S., Barnett, T. C., Rivera-Hernandez, T., Rohde, M., Walker, M. J. (2016). Streptococcus Pyogenes Adhesion and Colonization. FEBS Lett. 590, 3739–3757. doi: 10.1002/1873-3468.12254
Cambier, C., Takaki, K. K., Larson, R. P., Hernandez, R. E., Tobin, D. M., Urdahl, K. B., et al. (2014). Mycobacteria Manipulate Macrophage Recruitment Through Coordinated Use of Membrane Lipids. Nature 505, 218–222. doi: 10.1038/nature12799
Campbell-Valois, F.-X., Sachse, M., Sansonetti, P. J., Parsot, C. (2015). Escape of Actively Secreting Shigella Flexneri From Atg8/Lc3-Positive Vacuoles Formed During Cell-to-Cell Spread Is Facilitated by Icsb and Vira. MBio 6(3), e02567–14. doi: 10.1128/mBio.02567-14
Chamilos, G., Akoumianaki, T., Kyrmizi, I., Brakhage, A., Beauvais, A., Latge, J.-P. (2016). Melanin Targets Lc3-Associated Phagocytosis (Lap): A Novel Pathogenetic Mechanism in Fungal Disease. Autophagy 12, 888–889. doi: 10.1080/15548627.2016.1157242
Cheng, Y. L., Kuo, C. F., Lu, S. L., Omori, H., Wu, Y. N., Hsieh, C. L., et al. (2019). Group A Streptococcus Induces LAPosomes via SLO/β1 Integrin/NOX2/ROS Pathway in Endothelial Cells That Are Ineffective in Bacterial Killing and Suppress Xenophagy. mBio 10, e02148–e02119. doi: 10.1128/mBio.02148-19
Choy, A., Dancourt, J., Mugo, B., O’Connor, T. J., Isberg, R. R., Melia, T. J., et al. (2012). The Legionella Effector Ravz Inhibits Host Autophagy Through Irreversible Atg8 Deconjugation. Science 338, 1072–1076. doi: 10.1126/science.1227026
Cossart, P., Lecuit, M. (1998). Interactions of Listeria Monocytogenes With Mammalian Cells During Entry and Actin-Based Movement: Bacterial Factors, Cellular Ligands and Signaling. EMBO J. 17, 3797–3806. doi: 10.1093/emboj/17.14.3797
Cullinane, M., Gong, L., Li, X., Adler, N.-L., Tra, T., Wolvetang, E., et al. (2008). Stimulation of Autophagy Suppresses the Intracellular Survival of Burkholderia Pseudomallei in Mammalian Cell Lines. Autophagy 4, 744–753. doi: 10.4161/auto.6246
Dalebroux, Z. D., Miller, S. I. (2014). Salmonellae Phopq Regulation of the Outer Membrane to Resist Innate Immunity. Curr. Opin. Microbiol. 17, 106–113. doi: 10.1016/j.mib.2013.12.005
Deretic, V., Levine, B. (2009). Autophagy, Immunity, and Microbial Adaptations. Cell Host Microbe 5, 527–549. doi: 10.1016/j.chom.2009.05.016
Edwards, A. M., Potts, J., Josefsson, E., Massey, R. C. (2010). Staphylococcus Aureus Host Cell Invasion and Virulence in Sepsis Is Facilitated by the Multiple Repeats Within FnBPA. PloS Pathog. 6, e1000964. doi: 10.1371/journal.ppat.1000964
Ellson, C., Davidson, K., Anderson, K., Stephens, L. R., Hawkins, P. T. (2006). Ptdins3p Binding to the Px Domain of P40phox Is a Physiological Signal in Nadph Oxidase Activation. EMBO J. 25, 4468–4478. doi: 10.1038/sj.emboj.7601346
Fader, C. M., Sánchez, D. G., Mestre, M. B., Colombo, M. I. (2009). Ti-Vamp/Vamp7 and Vamp3/Cellubrevin: Two V-Snare Proteins Involved in Specific Steps of the Autophagy/Multivesicular Body Pathways. Biochim. Biophys. Acta (BBA)-Molecular Cell Res. 1793, 1901–1916. doi: 10.1016/j.bbamcr.2009.09.011
Fischer, T. D., Wang, C., Padman, B. S., Lazarou, M., Youle, R. J. (2020). STING Induces LC3B Lipidation Onto Single-Membrane Vesicles via the V-ATPase and ATG16L1-WD40 Domain. J. Cell Biol. 219, e202009128. doi: 10.1083/jcb.202009128
Flannagan, R. S., Cosío, G., Grinstein, S. (2009). Antimicrobial Mechanisms of Phagocytes and Bacterial Evasion Strategies. Nat. Rev. Microbiol. 7, 355–366. doi: 10.1038/nrmicro2128
Fountain, A., Inpanathan, S., Alves, P., Verdawala, M. B., Botelho, R. J. (2021). Phagosome Maturation in Macrophages: Eat, Digest, Adapt, and Repeat. Adv. Biol. Regul. 82, 100832. doi: 10.1016/j.jbior.2021.100832
Fournier, B., Williams, I. R., Gewirtz, A. T., Neish, A. S. (2009). Toll-Like Receptor 5-Dependent Regulation of Inflammation in Systemic Salmonella Enterica Serovar Typhimurium Infection. Infect Immun. 77, 4121–4129. doi: 10.1128/IAI.00656-09
Garvis, S. G., Beuzón, C. R., Holden, D. W. (2001). A Role for the Phop/Q Regulon in Inhibition of Fusion Between Lysosomes and Salmonella-Containing Vacuoles in Macrophages. Cell. Microbiol. 3, 731–744. doi: 10.1046/j.1462-5822.2001.00153.x
Gateva, V., Sandling, J. K., Hom, G., Taylor, K. E., Chung, S. A., Sun, X., et al. (2009). A Large-Scale Replication Study Identifies Tnip1, Prdm1, Jazf1, Uhrf1bp1 and Il10 as Risk Loci for Systemic Lupus Erythematosus. Nat. Genet. 41, 1228–1233. doi: 10.1038/ng.468
Gluschko, A., Farid, A., Herb, M., Grumme, D., Krönke, M., Schramm, M. (2021). Macrophages Target Listeria Monocytogenes by Two Discrete Non-Canonical Autophagy Pathways. Autophagy 2021, 1–18. doi: 10.1080/15548627.2021.1969765
Gluschko, A., Herb, M., Wiegmann, K., Krut, O., Neiss, W. F., Utermohlen, O., et al. (2018). The 2 Integrin Mac-1 Induces Protective Lc3-Associated Phagocytosis of Listeria Monocytogenes. Cell Host Microbe 23, 324–337. doi: 10.1016/j.chom.2018.01.018
Gong, L., Cullinane, M., Treerat, P., Ramm, G., Prescott, M., Adler, B., et al. (2011). The Burkholderia Pseudomallei Type Iii Secretion System and Bopa Are Required for Evasion of Lc3-Associated Phagocytosis. PloS One 6, e17852. doi: 10.1371/journal.pone.0017852
Gong, L., Lai, S.-C., Treerat, P., Prescott, M., Adler, B., Boyce, J. D., et al. (2015). Burkholderia Pseudomallei Type Iii Secretion System Cluster 3 Atpase Bsas, a Chemotherapeutic Target for Small-Molecule Atpase Inhibitors. Infect Immun. 83, 1276–1285. doi: 10.1128/IAI.03070-14
Grigoryeva, L. S., Cianciotto, N. P. (2021). Human Macrophages Utilize a Wide Range of Pathogen Recognition Receptors to Recognize Legionella Pneumophila, Including Toll-Like Receptor 4 Engaging Legionella Lipopolysaccharide and the Toll-Like Receptor 3 Nucleic-Acid Sensor. PloS Pathog. 17, e1009781. doi: 10.1371/journal.ppat.1009781
Harley, J. B., Alarcón-Riquelme, M. E., Criswell, L. A., Jacob, C. O., Kimberly, R. P., Moser, K. L., et al. (2008). Genome-Wide Association Scan in Women With Systemic Lupus Erythematosus Identifies Susceptibility Variants in Itgam, Pxk, Kiaa1542 and Other Loci. Nat. Genet. 40, 204–210. doi: 10.1038/ng.81
Heckmann, B. L., Boada-Romero, E., Cunha, L. D., Magne, J., Green, D. R. (2017). Lc3-Associated Phagocytosis and Inflammation. J. Mol. Biol. 429, 3561–3576. doi: 10.1016/j.jmb.2017.08.012
Heckmann, B. L., Green, D. R. (2019). Lc3-Associated Phagocytosis at a Glance. J. Cell Sci. 132(5), jcs222984. doi: 10.1242/jcs.222984
Henningham, A., Barnett, T. C., Maamary, P. G., Walker, M. J. (2012). Pathogenesis of Group A Streptococcal Infections. Discovery Med. 13, 329–342. doi: 10.1128/CMR.13.3.470
Herb, M., Gluschko, A., Schramm, M. (2018). Lc3-Associated Phagocytosis Initiated by Integrin Itgam-Itgb2/Mac-1 Enhances Immunity to Listeria Monocytogenes. Autophagy 14, 1462–1464. doi: 10.1080/15548627.2018.1475816
Herb, M., Gluschko, A., Schramm, M. (2020). Lc3-Associated Phagocytosis-the Highway to Hell for Phagocytosed Microbes. Cell Dev. Biol. 101, 68–76. doi: 10.1016/j.semcdb.2019.04.016
Herb, M., Schramm, M. (2021). Functions of ROS in Macrophages and Antimicrobial Immunity. Antioxidants 10, 313. doi: 10.3390/antiox10020313
Holmström, K. M., Finkel, T. (2014). Cellular Mechanisms and Physiological Consequences of Redox-Dependent Signalling. Nat. Rev. Mol. Cell Biol. 15, 411–421. doi: 10.1038/nrm3801
Horton, R. E., Morrison, N. A., Beacham, I. R., Peak, I. R. (2012). Interaction of Burkholderia Pseudomallei and Burkholderia Thailandensis With Human Monocyte-Derived Dendritic Cells. J. Med. Microbiol. 61, 607–614. doi: 10.1099/jmm.0.038588-0
Horwitz, M. A. (1983). Formation of a Novel Phagosome by the Legionnaires’ Disease Bacterium (Legionella Pneumophila) in Human Monocytes. J. Exp. Med. 158, 1319–1331. doi: 10.1084/jem.158.4.1319
Huang, J., Brumell, J. H. (2014). Bacteria–autophagy Interplay: A Battle for Survival. Nat. Rev. Microbiol. 12, 101–114. doi: 10.1038/nrmicro3160
Huang, J., Lam, G. Y., Steinberg, B. E., Dinauer, M. C., Magalhaes, M. A., Glogauer, M., et al. (2009). Activation of Antibacterial Autophagy by Nadph Oxidases. Proc. Natl. Acad. Sci. 106, 6226–6231. doi: 10.1073/pnas.0811045106
Hubber, A., Kubori, T., Coban, C., Matsuzawa, T., Ogawa, M., Kawabata, T., et al. (2017). Bacterial Secretion System Skews the Fate of Legionella-Containing Vacuoles Towards Lc3-Associated Phagocytosis. Sci. Rep. 7, 1–17. doi: 10.1038/srep44795
Husmann, L. K., Johnson, W. (1992). Adherence of Legionella Pneumophila to Guinea Pig Peritoneal Macrophages, J774 Mouse Macrophages, and Undifferentiated U937 Human Monocytes: Role of Fc and Complement Receptors. Infect Immun. 60, 5212–5218. doi: 10.1128/iai.60.12.5212-5218.1992
Ibarra, J. A., Steele-Mortimer, O. (2009). Salmonella–the Ultimate Insider. Salmonella Virulence Factors That Modulate Intracellular Survival. Cell. Microbiol. 11, 1579–1586. doi: 10.1111/j.1462-5822.2009.01368.x
Inomata, M., Xu, S., Chandra, P., Meydani, S. N., Takemura, G., Philips, J. A., et al. (2020). Macrophage LC3-Associated Phagocytosis Is an Immune Defense Against Streptococcus Pneumoniae That Diminishes With Host Aging. Proc. Natl. Acad. Sci. U.S.A. 117, 33561–33569. doi: 10.1073/pnas.2015368117
Isberg, R. R., Leong, J. M. (1990). Multiple 1 Chain Integrins Are Receptors for Invasin, a Protein That Promotes Bacterial Penetration Into Mammalian Cells. Cell 60, 861–871. doi: 10.1016/0092-8674(90)90099-Z
Itakura, E., Kishi-Itakura, C., Mizushima, N. (2012). The Hairpin-Type Tail-Anchored Snare Syntaxin 17 Targets to Autophagosomes for Fusion With Endosomes/Lysosomes. Cell 151, 1256–1269. doi: 10.1016/j.cell.2012.11.001
Jiao, Y., Sun, J. (2019). Bacterial Manipulation of Autophagic Responses in Infection and Inflammation. Front. Immunol. 10, 2821. doi: 10.3389/fimmu.2019.02821
Kim, Y.-R., Kim, J.-S., Gu, S.-J., Jo, S., Kim, S., Kim, S. Y., et al. (2020). Identification of Highly Potent and Selective Inhibitor, Tiptp, of the P22phox-Rubicon Axis as a Therapeutic Agent for Rheumatoid Arthritis. Sci. Rep. 10, 1–15. doi: 10.1038/s41598-020-61630-x
Kimmey, J. M., Stallings, C. L. (2016). Bacterial Pathogens Versus Autophagy: Implications for Therapeutic Interventions. Trends Mol. Med. 22, 1060–1076. doi: 10.1016/j.molmed.2016.10.008
Köster, S., Klevorn, T., Papavinasasundaram, K., Sassetti, C. M., Portal-Celhay, C., Philips, J. A. (2018). Consequence of Enhanced Lc3-Trafficking for a Live, Attenuated M. Tuberculosis Vaccine. Vaccine 36, 939–944. doi: 10.1016/j.vaccine.2018.01.012
Köster, S., Upadhyay, S., Chandra, P., Papavinasasundaram, K., Yang, G., Hassan, A., et al. (2017). Mycobacterium Tuberculosis Is Protected From Nadph Oxidase and Lc3-Associated Phagocytosis by the Lcp Protein Cpsa. Proc. Natl. Acad. Sci. 114, E8711–E8720. doi: 10.1073/pnas.1707792114
Lai, S.-C., Devenish, R. J. (2012). Lc3-Associated Phagocytosis (Lap): Connections With Host Autophagy. Cells 1, 396–408. doi: 10.3390/cells1030396
Lam, G. Y., Cemma, M., Muise, A. M., Higgins, D. E., Brumell, J. H. (2013). Host and Bacterial Factors That Regulate Lc3 Recruitment to Listeria Monocytogenes During the Early Stages of Macrophage Infection. Autophagy 9, 985–995. doi: 10.4161/auto.24406
Lamprinaki, D., Beasy, G., Zhekova, A., Wittmann, A., James, S., Dicks, J., et al. (2017). Lc3-Associated Phagocytosis Is Required for Dendritic Cell Inflammatory Cytokine Response to Gut Commensal Yeast Saccharomyces Cerevisiae. Front. Immunol. 8, 1397. doi: 10.3389/fimmu.2017.01397
Leung, Y., Ally, S., Goldberg, M. B. (2008). Bacterial Actin Assembly Requires Toca-1 to Relieve N-Wasp Autoinhibition. Cell Host Microbe 3, 39–47. doi: 10.1016/j.chom.2007.10.011
Ligeon, L.-A., Moreau, K., Barois, N., Bongiovanni, A., Lacorre, D.-A., Werkmeister, E., et al. (2014). Role of Vamp3 and Vamp7 in the Commitment of Yersinia Pseudotuberculosis to Lc3-Associated Pathways Involving Single-or Double-Membrane Vacuoles. Autophagy 10, 1588–1602. doi: 10.4161/auto.29411
Ligeon, L.-A., Pena-Francesch, M., Vanoaica, L. D., Núnez, N. G., Talwar, D., Dick, T. P., et al. (2021). Oxidation Inhibits Autophagy Protein Deconjugation From Phagosomes to Sustain Mhc Class Ii Restricted Antigen Presentation. Nat. Commun. 12, 1–13. doi: 10.1038/s41467-021-21829-6
Li, T., Kong, L., Li, X., Wu, S., Attri, K. S., Li, Y., et al. (2021). Listeria Monocytogenes Upregulates Mitochondrial Calcium Signalling to Inhibit Lc3-Associated Phagocytosis as a Survival Strategy. Nat. Microbiol. 6, 366–379. doi: 10.1038/s41564-020-00843-2
Li, X., Prescott, M., Adler, B., Boyce, J. D., Devenish, R. J. (2013). Beclin 1 Is Required for Starvation-Enhanced, But Not Rapamycin-Enhanced, LC3-Associated Phagocytosis of Burkholderia Pseudomallei in RAW 264.7 Cells. Infect Immun. 81, 271–277. doi: 10.1128/IAI.00834-12
Lowy, F. D. (1998). Staphylococcus Aureus Infections. New Engl. J. Med. 339, 520–532. doi: 10.1056/NEJM199808203390806
Lu, S. L., Kawabata, T., Cheng, Y. L., Omori, H., Hamasaki, M., Kusaba, T., et al. (2017). Endothelial Cells Are Intrinsically Defective in Xenophagy of Streptococcus Pyogenes. PloS Pathog. 13, e1006444. doi: 10.1371/journal.ppat.1006444
Ma, J., Becker, C., Reyes, C., Underhill, D. M. (2014). Cutting Edge: Fyco1 Recruitment to Dectin-1 Phagosomes Is Accelerated by Light Chain 3 Protein and Regulates Phagosome Maturation and Reactive Oxygen Production. J. Immunol. 192, 1356–1360. doi: 10.4049/jimmunol.1302835
Martinez, J. (2018). Lap it Up, Fuzz Ball: A Short History of Lc3-Associated Phagocytosis. Curr. Opin. Immunol. 55, 54–61. doi: 10.1016/j.coi.2018.09.011
Martinez, J., Almendinger, J., Oberst, A., Ness, R., Dillon, C. P., Fitzgerald, P., et al. (2011). Microtubule-Associated Protein 1 Light Chain 3 Alpha (Lc3)-Associated Phagocytosis Is Required for the Efficient Clearance of Dead Cells. Proc. Natl. Acad. Sci. 108, 17396–17401. doi: 10.1073/pnas.1113421108
Martinez, J., Cunha, L. D., Park, S., Yang, M., Lu, Q., Orchard, R., et al. (2016). Noncanonical Autophagy Inhibits the Autoinflammatory, Lupus-Like Response to Dying Cells. Nature 533, 115–119. doi: 10.1038/nature17950
Martinez, J., Malireddi, R. S., Lu, Q., Cunha, L. D., Pelletier, S., Gingras, S., et al. (2015). Molecular Characterization of Lc3-Associated Phagocytosis Reveals Distinct Roles for Rubicon, Nox2 and Autophagy Proteins. Nat. Cell Biol. 17, 893–906. doi: 10.1038/ncb3192
Masud, S., Prajsnar, T. K., Torraca, V., Lamers, G. E., Benning, M., van der Vaart, M., et al. (2019a). Macrophages Target Salmonella by Lc3-Associated Phagocytosis in a Systemic Infection Model. Autophagy 15, 796–812. doi: 10.1080/15548627.2019.1569297
Masud, S., van der Burg, L., Storm, L., Prajsnar, T. K., Meijer, A. H. (2019b). Rubicon-Dependent Lc3 Recruitment to Salmonella-Containing Phagosomes Is a Host Defense Mechanism Triggered Independently From Major Bacterial Virulence Factors. Front. Cell. Infect Microbiol. 9, 279. doi: 10.3389/fcimb.2019.00279
Matereke, L. T., Okoh, A. I. (2020). Listeria Monocytogenes Virulence, Antimicrobial Resistance and Environmental Persistence: A Review. Pathogens 9, 528. doi: 10.3390/pathogens9070528
Matsunaga, K., Saitoh, T., Tabata, K., Omori, H., Satoh, T., Kurotori, N., et al. (2009). Two Beclin 1-Binding Proteins, Atg14l and Rubicon, Reciprocally Regulate Autophagy at Different Stages. Nat. Cell Biol. 11, 385–396. doi: 10.1038/ncb1846
McEwan, D. G., Popovic, D., Gubas, A., Terawaki, S., Suzuki, H., Stadel, D., et al. (2015). Plekhm1 Regulates Autophagosome-Lysosome Fusion Through Hops Complex and Lc3/Gabarap Proteins. Mol. Cell 57, 39–54. doi: 10.1016/j.molcel.2014.11.006
McVicker, G., Prajsnar, T. K., Williams, A., Wagner, N. L., Boots, M., Renshaw, S. A., et al. (2014). Clonal Expansion During Staphylococcus Aureus Infection Dynamics Reveals the Effect of Antibiotic Intervention. PloS Pathog. 10, e1003959. doi: 10.1371/journal.ppat.1003959
Miller, K. A., Garza-Mayers, A. C., Leung, Y., Goldberg, M. B. (2018). Identification of Interactions Among Host and Bacterial Proteins and Evaluation of Their Role Early During Shigella Flexneri Infection. Microbiology 164, 540. doi: 10.1099/mic.0.000637
Mitchell, G., Cheng, M. I., Chen, C., Nguyen, B. N., Whiteley, A. T., Kianian, A., et al. (2018). Listeria Monocytogenes Triggers Noncanonical Autophagy Upon Phagocytosis, But Avoids Subsequent Growth-Restricting Xenophagy. Proc Natl. Acad. Sci. U.S.A. 115, E210–E217. doi: 10.1073/pnas.1716055115
Moreau, K., Lacas-Gervais, S., Fujita, N., Sebbane, F., Yoshimori, T., Simonet, M., et al. (2010). Autophagosomes Can Support Yersinia Pseudotuberculosis Replication in Macrophages. Cell. Microbiol. 12, 1108–1123. doi: 10.1111/j.1462-5822.2010.01456.x
Moreau, K., Renna, M., Rubinsztein, D. C. (2013). Connections Between Snares and Autophagy. Trends Biochem. Sci. 38, 57–63. doi: 10.1016/j.tibs.2012.11.004
Morita, M., Kajiye, M., Sakurai, C., Kubo, S., Takahashi, M., Kinoshita, D., et al. (2020). Characterization of MORN2 Stability and Regulatory Function in LC3-Associated Phagocytosis in Macrophages. Biol. Open 9, bio051029. doi: 10.1242/bio.051029
Muñoz, L. E., Lauber, K., Schiller, M., Manfredi, A. A., Herrmann, M. (2010). The Role of Defective Clearance of Apoptotic Cells in Systemic Autoimmunity. Nat. Rev. Rheumatol. 6, 280. doi: 10.1038/nrrheum.2010.46
Muñoz-Sánchez, S., van der Vaart, M., Meijer, A. H. (2020). Autophagy and Lc3-Associated Phagocytosis in Zebrafish Models of Bacterial Infections. Cells 9, 2372. doi: 10.3390/cells9112372
Nguyen, J. A., Yates, R. M. (2021). Better Together: Current Insights Into Phagosome-Lysosome Fusion. Front. Immunol. 12, 209. doi: 10.3389/fimmu.2021.636078
O’Callaghan, D., Maskell, D., Liew, F., Easmon, C., Dougan, G. (1988). Characterization of Aromatic-and Purine-Dependent Salmonella Typhimurium: Attention, Persistence, and Ability to Induce Protective Immunity in Balb/C Mice. Infect Immun. 56, 419–423. doi: 10.1128/iai.56.2.419-423.1988
Ogawa, M., Takada, N., Shizukuishi, S., Tomokiyo, M., Chang, B., Yoshida, M., et al. (2020). Streptococcus Pneumoniae Triggers Hierarchical Autophagy Through Reprogramming of LAPosome-Like Vesicles via NDP52-Delocalization. Commun. Biol. 3, 25. doi: 10.1038/s42003-020-0753-3
Osborne, S. E., Brumell, J. H. (2017). Listeriolysin O: From Bazooka to Swiss Army Knife. Philos. Trans. R. Soc. B: Biol. Sci. 372, 20160222. doi: 10.1098/rstb.2016.0222
Petiot, A., Ogier-Denis, E., Blommaart, E. F., Meijer, A. J., Codogno, P. (2000). Distinct Classes of Phosphatidylinositol 3’-Kinases Are Involved in Signaling Pathways That Control Macroautophagy in Ht-29 Cells. J. Biol. Chem. 275, 992–998. doi: 10.1074/jbc.275.2.992
Prajsnar, T. K., Hamilton, R., Garcia-Lara, J., McVicker, G., Williams, A., Boots, M., et al. (2012). A Privileged Intraphagocyte Niche Is Responsible for Disseminated Infection of Staphylococcus Aureus in a Zebrafish Model. Cell. Microbiol. 14, 1600–1619. doi: 10.1111/j.1462-5822.2012.01826.x
Prajsnar, T. K., Serba, J. J., Dekker, B. M., Gibson, J. F., Masud, S., Fleming, A., et al. (2021). The Autophagic Response to Staphylococcus Aureus Provides an Intracellular Niche in Neutrophils. Autophagy 17, 888–902. doi: 10.1080/15548627.2020.1739443
Pujol, C., Bliska, J. B. (2003). The Ability to Replicate in Macrophages Is Conserved Between Yersinia Pestis and Yersinia Pseudotuberculosis. Infect Immun. 71, 5892–5899. doi: 10.1128/IAI.71.10.5892-5899.2003
Rai, S., Arasteh, M., Jefferson, M., Pearson, T., Wang, Y., Zhang, W., et al. (2019). The Atg5-Binding and Coiled Coil Domains of Atg16l1 Maintain Autophagy and Tissue Homeostasis in Mice Independently of the Wd Domain Required for Lc3-Associated Phagocytosis. Autophagy 15, 599–612. doi: 10.1080/15548627.2018.1534507
Rosales, C., Uribe-Querol, E. (2017). Phagocytosis: A Fundamental Process in Immunity. BioMed. Res. Int. 2017. doi: 10.1155/2017/9042851
Sanjuan, M. A., Dillon, C. P., Tait, S. W., Moshiach, S., Dorsey, F., Connell, S., et al. (2007). Toll-Like Receptor Signalling in Macrophages Links the Autophagy Pathway to Phagocytosis. Nature 450, 1253–1257. doi: 10.1038/nature06421
Scherz-Shouval, R., Shvets, E., Fass, E., Shorer, H., Gil, L., Elazar, Z. (2007). Reactive Oxygen Species are Essential for Autophagy and Specifically Regulate the Activity of Atg4. EMBO J. 26, 1749–1760. doi: 10.1038/sj.emboj.7601623
Schille, S., Crauwels, P., Bohn, R., Bagola, K., Walther, P., van Zandbergen, G. (2018). Lc3-Associated Phagocytosis in Microbial Pathogenesis. Int. J. Med. Microbiol. 308, 228–236. doi: 10.1016/j.ijmm.2017.10.014
Schlesinger, L. (1993). Macrophage Phagocytosis of Virulent But Not Attenuated Strains of Mycobacterium Tuberculosis Is Mediated by Mannose Receptors in Addition to Complement Receptors. J. Immunol. 150, 2920–2930.
Seveau, S. (2014). Multifaceted Activity of Listeriolysin O, the Cholesterol-Dependent Cytolysin of Listeria Monocytogenes. MACPF/CDC Proteins-Agents of Defence, Attack and Invasion 161–195. doi: 10.1007/978-94-017-8881-6_9
Shizukuishi, S., Ogawa, M., Ryo, A., Ohnishi, M. (2020). The Multi-Step Mechanism and Biological Role of Noncanonical Autophagy Targeting Streptococcus Pneumoniae During the Early Stages of Infection. Autophagy 16, 1152–1153. doi: 10.1080/15548627.2020.1743937
Simeone, R., Sayes, F., Lawarée, E., Brosch, R. (2021). Breaching the Phagosome, the Case of the Tuberculosis Agent. Cell. Microbiol. 23, e13344. doi: 10.1111/cmi.13344
Slauch, J. M. (2011). How Does the Oxidative Burst of Macrophages Kill Bacteria? Still an Open Question. Mol. Microbiol. 80, 580–583. doi: 10.1111/j.1365-2958.2011.07612.x
Stamm, C. E., Collins, A. C., Shiloh, M. U. (2015). Sensing of M Ycobacterium Tuberculosis and Consequences to Both Host and Bacillus. Immunol. Rev. 264, 204–219. doi: 10.1111/imr.12263
Sun, J., Singh, V., Lau, A., Stokes, R. W., Obregón-Henao, A., Orme, I. M., et al. (2013). Mycobacterium Tuberculosis Nucleoside Diphosphate Kinase Inactivates Small Gtpases Leading to Evasion of Innate Immunity. PloS Pathog. 9, e1003499. doi: 10.1371/journal.ppat.1003499
Sun, J., Wang, X., Lau, A., Liao, T.-Y. A., Bucci, C., Hmama, Z. (2010). Mycobacterial Nucleoside Diphosphate Kinase Blocks Phagosome Maturation in Murine Raw 264.7 Macrophages. PloS One 5, e8769. doi: 10.1371/journal.pone.0008769
Tam, K., Schultz, M., Reyes-Robles, T., Vanwalscappel, B., Horton, J., Alonzo, F., et al. (2016). Staphylococcus Aureus Leukocidin LukED and HIV-1 Gp120 Target Different Sequence Determinants on CCR5. mBio 7, e02024–e02016. doi: 10.1128/mBio.02024-16
Thompson, J. A., Liu, M., Helaine, S., Holden, D. W. (2011). Contribution of the Phop/Q Regulon to Survival and Replication of Salmonella Enterica Serovar Typhimurium in Macrophages. Microbiology 157, 2084. doi: 10.1099/mic.0.048926-0
Upadhyay, S., Philips, J. A. (2019). Lc3-Associated Phagocytosis: Host Defense and Microbial Response. Curr. Opin. Immunol. 60, 81–90. doi: 10.1016/j.coi.2019.04.012
Volinia, S., Dhand, R., Vanhaesebroeck, B., MacDougall, L., Stein, R., Zvelebil, M., et al. (1995). A Human Phosphatidylinositol 3-Kinase Complex Related to the Yeast Vps34p-Vps15p Protein Sorting System. EMBO J. 14, 3339–3348. doi: 10.1002/j.1460-2075.1995.tb07340.x
Walthers, D., Carroll, R. K., Navarre, W. W., Libby, S. J., Fang, F. C., Kenney, L. J. (2007). The Response Regulator SsrB Activates Expression of Diverse Salmonella Pathogenicity Island 2 Promoters and Counters Silencing by the Nucleoid-Associated Protein H-Ns. Mol. Microbiol. 65, 477–493. doi: 10.1111/j.1365-2958.2007.05800.x
Wang, X., Sharma, P., Jefferson, M., Zhang, W., Bone, B., Kipar, A., et al. (2021). Non-Canonical Autophagy Functions of ATG16L1 in Epithelial Cells Limit Lethal Infection by Influenza A Virus. EMBO J. 40, e105543. doi: 10.15252/embj.2020105543
Wiersinga, W. J., Wieland, C. W., Dessing, M. C., Chantratita, N., Cheng, A. C., Limmathurotsakul, D., et al. (2007). Toll-Like Receptor 2 Impairs Host Defense in Gram-Negative Sepsis Caused by Burkholderia Pseudomallei (Melioidosis). PloS Med. 4, e248. doi: 10.1371/journal.pmed.0040248
Wong, S.-W., Upadhyay, S., Martinez, J. (2021). “Lc3-Associated Phagocytosis: Molecular Mechanisms and Pathological Consequences,” in Non-Canonical Autophagy (Durham, North Carolina, USA:Elsevier), 69–91.
Yang, C.-S., Lee, J.-S., Rodgers, M., Min, C.-K., Lee, J.-Y., Kim, H. J., et al. (2012). Autophagy Protein Rubicon Mediates Phagocytic Nadph Oxidase Activation in Response to Microbial Infection or Tlr Stimulation. Cell Host Microbe 11, 264–276. doi: 10.1016/j.chom.2012.01.018
Keywords: LC3-associated phagocytosis, macrophages, neutrophils, autophagy, innate immunity, intracellular pathogens, virulence mechanisms, immune evasion
Citation: Grijmans BJM, van der Kooij SB, Varela M and Meijer AH (2022) LAPped in Proof: LC3‐Associated Phagocytosis and the Arms Race Against Bacterial Pathogens. Front. Cell. Infect. Microbiol. 11:809121. doi: 10.3389/fcimb.2021.809121
Received: 04 November 2021; Accepted: 10 December 2021;
Published: 03 January 2022.
Edited by:
Hua Niu, Affiliated Hospital of Guilin Medical University, ChinaReviewed by:
Bradlee Heckmann, University of South Florida Health, United StatesGeorgios Chamilos, University of Crete, Greece
Alexander Gluschko, University Hospital Cologne, Germany
Copyright © 2022 Grijmans, van der Kooij, Varela and Meijer. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Annemarie H. Meijer, YS5oLm1laWplckBiaW9sb2d5LmxlaWRlbnVuaXYubmw=
†These authors have contributed equally to this work and share first authorship