 Xi He1†‡
Xi He1†‡ Daibin Zhong2‡
Daibin Zhong2‡ Chunyan Zou3Liang Pi1
Chunyan Zou3Liang Pi1 Luyi Zhao1Yucheng Qin4Maohua Pan4
Luyi Zhao1Yucheng Qin4Maohua Pan4 Siqi Wang1
Siqi Wang1 Weiling Zeng1Zheng Xiang1Xi Chen1Yanrui Wu5Yu Si1
Weiling Zeng1Zheng Xiang1Xi Chen1Yanrui Wu5Yu Si1 Liwang Cui6Yaming Huang7*
Liwang Cui6Yaming Huang7* Guiyun Yan2*Zhaoqing Yang1*
Guiyun Yan2*Zhaoqing Yang1*- 1Department of Pathogen Biology and Immunology, Kunming Medical University, Kunming, China
- 2Program in Public Health, College of Health Sciences, University of California at Irvine, Irvine, CA, United States
- 3Department of Electrocardiogram, Guangxi Zhuang Autonomous Region People’s Hospital, Nanning, China
- 4Department of Infectious Diseases, Shanglin County People’s Hospital, Shanglin, China
- 5Department of Cell Biology & Genetics, Kunming Medical University, Kunming, China
- 6Department of Internal Medicine, Morsani College of Medicine, University of South Florida, Tampa, FL, United States
- 7Department of Protozoa, Guangxi Zhuang Autonomous Region Center for Disease Prevention and Control, Nanning, China
Imported malaria and recurrent infections are becoming an emerging issue in many malaria non-endemic countries. This study aimed to determine the molecular patterns of the imported malaria infections and recurrence. Blood samples were collected from patients with imported malaria infections during 2016–2018 in Guangxi Zhuang Autonomous Region, China. Next-generation amplicon deep-sequencing approaches were used to assess parasite genetic diversity, multiplexity of infection, relapse, recrudescence, and antimalarial drug resistance. A total of 44 imported malaria cases were examined during the study, of which 35 (79.5%) had recurrent malaria infections within 1 year. The majority (91.4%) had one recurrent malaria episode, whereas two patients had two recurrences and one patient had three recurrences. A total of 19 recurrence patterns (the species responsible for primary and successive clinical episodes) were found in patients returning from malaria epidemic countries. Four parasite species were detected with a higher than usual proportion (46.2%) of non-falciparum infections or mixed-species infections. An increasing trend of recurrence infections and reduced drug treatment efficacy were observed among the cases of imported malaria. The high recurrence rate and complex patterns of imported malaria from Africa to non-endemic countries have the potential to initiate local transmission, thereby undermining efforts to eliminate locally acquired malaria. Our findings highlight the power of amplicon deep-sequencing applications in molecular epidemiological studies of the imported malaria recurrences.
Introduction
Malaria remains a major public health problem in sub-Saharan Africa. In 2019, an estimated 229 million cases and 409,000 deaths of malaria occurred globally with the vast majority of cases (94%) in the African region, followed by South-East Asia (3%) (WHO, 2020). In addition, imported malaria infection is emerging in many malaria-free countries including China, which have recently become malaria-free. Historically, the Guangxi Zhuang Autonomous Region was one of the four highest malaria-endemic areas in southern China (Yang et al., 2018). Guangxi had no indigenous malaria cases reported since 2013. Imported malaria is increasing due to increasing travel for tourism and trade in high-risk areas; Guangxi remains the area of China at highest risk for imported malaria (Lin et al., 2017; Jianhai et al., 2020). During 2011 and 2018 in Guangxi, a total of 3,943 imported malaria cases were reported and most of them originated from Africa (e.g., Ghana, Congo, and Cameroon), where Plasmodium falciparum is the predominant parasite species (Lin Kang-Ming et al., 2019). The increase in imported malaria cases poses a major challenge for the malaria elimination program in China (Lai et al., 2019).
Accurate identification and assessment of imported malaria parasite multiplicity of infection (MOI), genetic diversity in relapse and recrudescence, and antimalarial drug resistance are essential for malaria treatment, prognosis, and prevention of malaria transmission. To improve control, we require information about the parasite’s diversity, transmission dynamics, relapse frequency, and mechanisms of adaptation to environmental and interventional pressures (Apinjoh et al., 2019). Traditional polymerase chain reaction (PCR)-based methods relying on DNA sequence length polymorphisms, such as antigenic polymorphic markers merozoite surface proteins (msp1 and msp2), apical membrane antigen (ama1), circumsporozoite protein (csp), and microsatellites, have poor sensitivity for detection of less abundant parasite infections (Imwong et al., 2005; Kadekoppala and Holder, 2010; Lin et al., 2012; Lin et al., 2015), while next-generation sequencing of the PCR amplicon using Illumina HiSeq platforms provides a more sensitive molecular approach to determining the genetic diversity of Plasmodium infections and allows for more accurate identification and assessment. Amplicon deep sequencing (ADS) is more sensitive and reliable for detection of minority clone infections and determination of MOI (Flaherty et al., 2012; Early et al., 2019; Gruenberg et al., 2019), recrudescence and relapse (Lin et al., 2015; Zhong et al., 2018; Gruenberg et al., 2019), quantification of drug-resistance alleles (Bhatnagar et al., 2019; Brazeau et al., 2019; Gaye et al., 2020), and investigations of parasite microgeographic epidemiology (Koepfli and Mueller, 2017; Hemming-Schroeder et al., 2020).
This study was designed to answer the following questions: 1) What is the species composition of malaria parasites that cause imported malaria cases? 2) What proportion of these infections represent recrudescence or relapse? It is needed to establish whether the recurrent/relapsing infections were due to a new or existing infection. 3) Which mutations were associated with recrudescent infection in imported and recurrent episodes? To answer the first question, we conducted PCR and ADS using the mitochondrial mtDNA cytochrome b (CYTB) gene and small-subunit ribosomal (18S rRNA), referred to here as SSU gene markers for species identifications. To answer the second question, we performed ADS using msp1 and ama1 markers for understanding the genetic diversity between new and relapsing infections. Finally, we examined the gene mutations of the P. falciparum Kelch-domain protein (pfk13), chloroquine resistance transporter (pfcrt), and multidrug resistance protein-1 (pfmdr1) by Sanger sequencing. Since the effectiveness of primaquine (PQ) for the radical cure of vivax malaria is influenced by CYP2D6 activity (He et al., 2019), we wanted to determine whether the failure of PQ might be linked to CYP2D6 genotypes associated with poor metabolism of PQ. The cytochrome P450 isoenzyme 2D6 gene in P. vivax infected patients was also detected.
Materials and Methods
Study Design
During 2016 and 2018, imported malaria infections in Shanglin County, Guangxi, were diagnosed by microscopic examination of Giemsa-stained thick and thin blood films. The questionnaires including gender, age, occupation, travel history, and date of diagnosis were recorded. Malaria patients were admitted for inpatient treatment following the guidelines of the Chinese Center for Disease Control and Prevention (CDC) and WHO recommendations on the diagnosis and treatment of uncomplicated and severe malaria (WHO, 2015) (Supplementary Methods). All patients who had two or more times malaria attack episodes were included in the study (Supplementary Table 1). The human subject protocol for this study was approved by the Shanglin Hospital Institutional Review Board. All participants supplied written informed consent.
Sample Collection and Nested-PCR Amplification
Two hundred microliters of venous blood was collected from patients at the time of diagnosis at initial and repeated attacks. Malaria parasite DNA was extracted from whole blood using the High Pure PCR Template Preparation Kit (Roche, Mannheim, Germany) following the manufacturer’s instruction and eluted in 100 μl of water. The nested PCR method was used to identify the Plasmodium species with species-specific primers for P. falciparum, P. vivax, P. malariae, P. ovale, and P. knowlesi (Miguel-Oteo et al., 2017). The amplification reactions were conducted in 25 μl, containing 3 µl DNA template, 0.5 μM of each forward and reverse primer, 2 μl dNTP, 5 μl buffer, and 0.5 μl PrimeSTAR GAX DNA polymerase (Takara Bio Inc., Japan). The PCR amplification was performed at 94°C for 3 min, 35 cycles of 94°C for 30 s, 60°C for 30 s, and 72°C for 1 min, and a final extension at 72°C for 6 min. The amplified products were visualized using agarose gel electrophoresis.
PCR Amplification and Deep Sequencing
Four target genes, including mitochondrial cytochrome b (CYTB) gene, small subunit ribosomal RNA (SSU) gene, the merozoite surface protein 1 (msp1) gene, and apical membrane antigen 1 (ama1) gene, were used to design seven sets of PCR primers based on the highly variable regions of SSU, CYTB, pvmsp1, pvama1, pomsp1, pfmsp1, and pfama1 genes (Supplementary Table 2). PCR products (<400 bp) were prepared for next-generation sequencing by a two-step PCR approach following the previous reports (Lalremruata et al., 2017; Zhong et al., 2018). Briefly, the first-round PCR primers appended the overhang adapter sequence to the 5′ end of forward and reverse target gene-specific primers, while the second-round PCR used a universal primer with barcode primers that appended the 5′ end. Each sample was amplified in duplicate using a unique barcode (MIDs). The PCR primers and length of amplicon deep sequencing are listed in Supplementary Table 2. PCR amplification was performed in a total reaction volume of 25 μl reaction in duplicate following the protocols previously described (Zhong et al., 2018). Plasmodium falciparum laboratory strain 3D7 (MRA-102G) was used as control. Amplicons were cleaned and normalized to 1 ng/µl concentration using the SequalPrep Normalization Plate Kit (Thermo Fisher Scientific, Inc., Waltham, MA, USA). Amplicon deep sequencing was performed on an Illumina MiSeq platform in paired-end mode using a MiSeq Reagent Kit v3 PE300 (UCI Genomics High-Throughput Facility, Irvine, CA) with PhiX control (Illumina, PhiX Control v3) and the minimum target read depths of 10,000×.
CYP2D6 Genotyping and Sequencing of Antimalarial Drug-Resistance Genes
The CYP2D6 gene in P. vivax-infected patients was analyzed by PCR amplification of the full-length CYP2D6-coding region using previously described primers and sequenced the PCR products (He et al., 2019). Patients’ CYP2D6 allele variants and the overall genotype activity score were evaluated by the CYP2D6 allele-naming database (www.Pharmvar.org/gene/CYP2D6). The full-length pfk13 gene was amplified and sequenced using a previously published protocol (Zhang et al., 2016). The P. falciparum chloroquine resistance transporter gene (pfcrt) and the P. falciparum multidrug resistance protein-1 (pfmdr1) gene were amplified and sequenced using previously described primers (Li et al., 2015).
Data Analysis
Deep-sequence data extraction, processing, and analyses were performed using the SeekDeep-targeted amplicon bioinformatics pipeline (Hathaway et al., 2017). Sequencing reads were separated by sample-specific barcodes and clustered according to markers, samples, and replicates. A cluster cutoff threshold of >2.0% in frequency, and a minimum read count of 10 for all markers were used to estimate haplotypes and frequency based on the accuracy of classification in positive controls and sample replicates. Classification of recurrences was conducted based on WHO classification of responses to treatment (WHO, 2009) and shared haplotypes between the initial and recurrent infections (Kyabayinze et al., 2003; Nyachieo et al., 2005). Analysis of haplotype and nucleotide diversity was performed using DnaSP v5. MEGA v7 was used to create a UPGMA phylogenetic tree. BioEdit v7 was used to align DNA sequences. All the case information was collected and input into SAS JMP 14.0 software (SAS Inc., Cary, NC), and statistical analyses were performed. The differences were compared by chi-square or Fisher’s exact tests at a two-sided p value <0.05 for statistical significance.
Results
Molecular Determination of the Malaria Parasite Species
A total of 93 samples from 44 patients were confirmed for malaria infections by nested PCR. All the patients were male between the ages of 22 and 60 years and had travel history from abroad with most of the destination countries (96.8%) in Africa. ADS of the CYTB and SSU markers was performed in 81 and 85 samples, respectively. An average sequencing coverage depth for the major haplotypes was 20,069 and 11,109 for CYTB and SSU markers, respectively (Supplementary Table 3). For CYTB, no polymorphism was detected within any species except for P. ovale, which includes two subspecies P. ovale curtisi and P. ovale wallikeri, whereas high polymorphism was found in the SSU marker with the number of haplotypes ranging from 1 to 5 in the five parasite species and similarity >99% compared to sequences in GenBank. The phylogenetic tree of CYTB comprises five clusters for the five Plasmodium species (Figure 1A), whereas a mixed pattern of SSU tree was observed in the five species (Figure 1B). Among the 93 samples, ADS identified 50 (53.8%) P. falciparum, 14 (15.1%) P. vivax, 9 (9.7%) P. ovale curtisi, 4 (4.3%) P. ovale wallikeri, 1 (1.1%) P. malariae, and 15 (16.1%) mixed-species infections (Supplementary Table 4). The proportion of P. ovale curtisi to P. ovale wallikeri is 2.25 (9/4) in single-species infections and 4.5 (9/2) in mixed-species infections. Six out of the 15 mixed species were identified by the SSU marker, which showed various frequencies of different haplotypes, and most of them had predominant P. ovale curtisi mixed with less abundant P. falciparum (Supplementary Figure 1A, B). The other mixed-species infections were determined by combined markers. The haplotype sequences of CYTB and SSU markers were deposited at NCBI GenBank with accession numbers MT942975–MT942996.
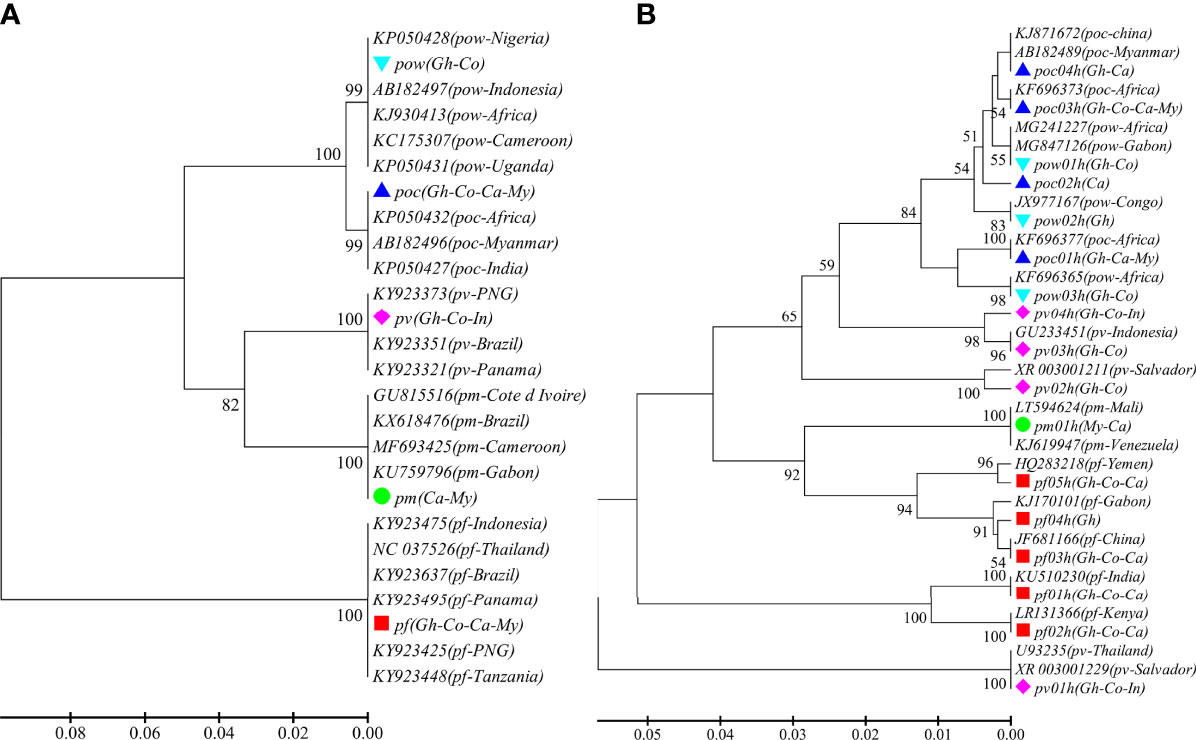
Figure 1 The phylogenetic UPGMA tree of haplotypes determined by amplicon deep sequencing. (A) CYTB phylogenetic tree for the 5 species and 21 sequences retrieved from GenBank (KY923637, KY923475, KY923495, KY923425, KY923448, NC_037526, KX618476, MF693425, GU815516, KU759796, KP050432, KP050427, AB182496, FJ409565, KJ930413, AB182497, KP050428, KP050431, KY923351, KY923321, KY923373). (B) SSU phylogenetic tree for the haplotypes of the 5 species and 19 sequences retrieved from GenBank (GU233451, JF681166, KJ170101, KJ619947, KU510230, MG241227, MG847126, U93235, XR_003001229, XR_003001211, LR131366, LT594624, KJ871672, AB182489, KF696373, KF696377, JX977167, KF696365, HQ283218). pf, Plasmodium falciparum; pv, P. vivax; pm, P. malariae; poc, P. ovale curtisi; and pow, P. ovale wallikeri. Different color markers represent different species identified in this study. Gh, Ghana; Co, Congo; Ca, Cameroon; My, Myanmar; and In, Indonesia.
Recurrent Species Patterns of Imported Malaria Cases in China
Among the 44 patients investigated, three patients (6.8%) had a second malaria attack within the first week of returning to China. Thirty-five patients (79.5%) had recurrent malaria infections later, but within 1 year of their return. The majority (91.4%) had one recurrent malaria episode, whereas two patients had two recurrences and one patient had three recurrences (Table 1). A total of 19 recurrence patterns were found in patients returning from malaria epidemic countries, including Ghana, Congo, and Cameroon. The average interval of malaria recurrence was 68.6 ± 11.6, ranging from 9 to 250 days. Overall, there were 26 (59.0%) not completely cured in all malaria species, 16 (36.4%) relapse in non-falciparum malaria, and 3 (6.8%) due to P. falciparum parasite dormancy or present at low-level parasitemia (Muehlenbachs et al., 2007; Malvy et al., 2018) (Supplementary Table 5). Among these, 27.2% (12/44) were recurrent infections. In addition, there were three patients who had tested positive malaria from 3 to 6 days, including two patients who showed to be coinfected with other species and six patients who had traveled abroad and repeated infections after previous malaria attack (Supplementary Table 6).
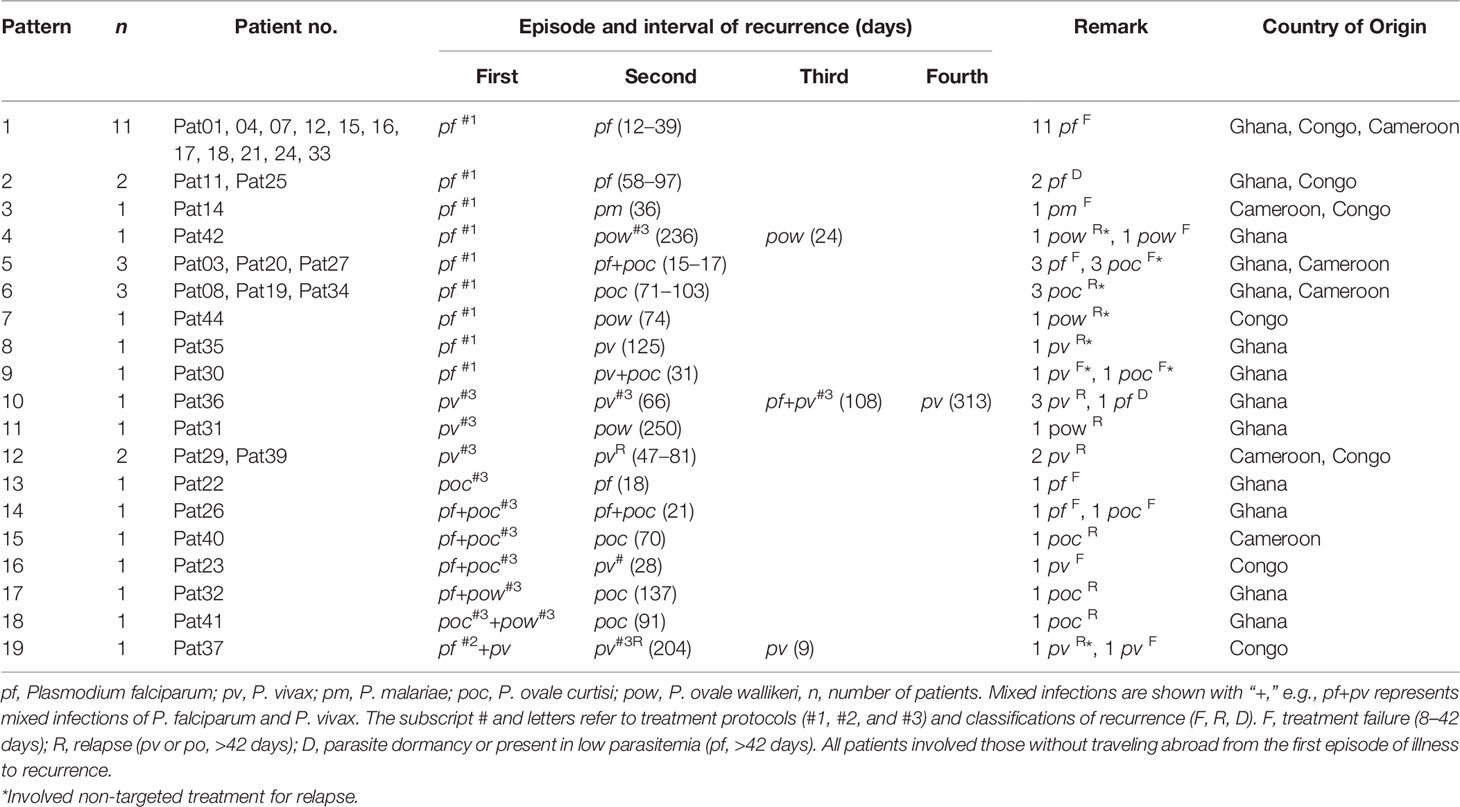
Table 1 Recurrence patterns of the imported malaria cases.
Genetic Diversity and MOI of P. falciparum Determined by pfmsp1 and pfama1 Markers
A total of 49 and 33 P. falciparum samples were performed by deep sequencing of pfmsp1 and pfama1 markers, respectively. An average sequencing coverage depth for the major haplotypes was 11,136 and 8,656 for pfmsp1 and pfama1 markers, respectively (Supplementary Table 3). pfmsp1 showed a total of 10 length polymorphic fragments (266, 284, 293, 302, 311, 320, 329, 347, 356, and 392 bp) in the 27 predominant haplotypes within samples (Supplementary Figure 2), whereas pfama1 had an identical length of 390 bp in the 20 predominant haplotypes within samples (Supplementary Table 7). Seventy percent (42/60) of pfmsp1 haplotypes were new haplotypes, whereas only 11.1% (3/27) of pfama1 haplotypes were new haplotypes compared to the sequences available in GenBank. Among the 20 predominant haplotypes of pfama1, a total of 31 segregating sites were found with a nucleotide diversity Pi = 0.025 and a haplotype diversity Hd = 0.960. Overall, the number of clones ranged from 1 to 6 with a mean MOI of 2.2 ± 0.2, 95% CI [1.7, 2.6] determined for the 50 P. falciparum samples by combining the two markers. Of these, 44% (22/50) samples had multiclonal infections (Supplementary Figure 3). The pfmsp1 and pfama1 sequences of predominant haplotypes were deposited at GenBank with accession numbers MT947241–MT947300 and MT947313–MT947339.
Genetic Diversity and MOI of P. vivax and P. ovale Determined by msp1 and ama1 Markers
A total of 14 and 15 P. vivax samples were performed for deep sequencing of pvmsp1 and pvama1 markers, respectively. An average sequencing coverage depth for the major haplotypes was 9,259 and 9,250 for pvmsp1 and pvama1 markers, respectively (Supplementary Table 3). No length polymorphism was found within samples in both pvmsp1 amplicon (309 bp) and pvama1 amplicon (288 bp). Seven haplotypes were identified for pvmsp1 and 5 for pvama1. Two of the seven pvmsp1 haplotypes were new, whereas none of the five pvama1 haplotypes were new compared to those sequences in the GenBank (Supplementary Table 7). Molecular phylogenetic analysis indicated that most haplotypes were shared among Africa and Asia (Figure 2). Overall, MOI = 1 (single infection) was found for all the 15 P. vivax samples by combining the two markers. For P. ovale, a total of 14 samples were subjected to deep sequencing of the pomsp1 marker. An average sequencing coverage depth for the major haplotypes was 10,759 joined reads (Supplementary Table 3). pomsp1 showed a total of four length polymorphic fragments (289, 316, 331, and 352 bp) and five haplotypes (four for poc and one for pow). Four of the five pomsp1 haplotypes were novel haplotypes compared to those sequences in GenBank. Phylogenetic tree analysis indicated that Ghana had all five haplotypes, whereas Congo, Ghana, and Myanmar had one haplotype for each country (Figure 3). The pvmsp1, pvama1, and pomsp1 sequences of predominant haplotypes were deposited at NCBI GenBank with accession numbers MT947301–MT947312 and MT947340–MT947344.
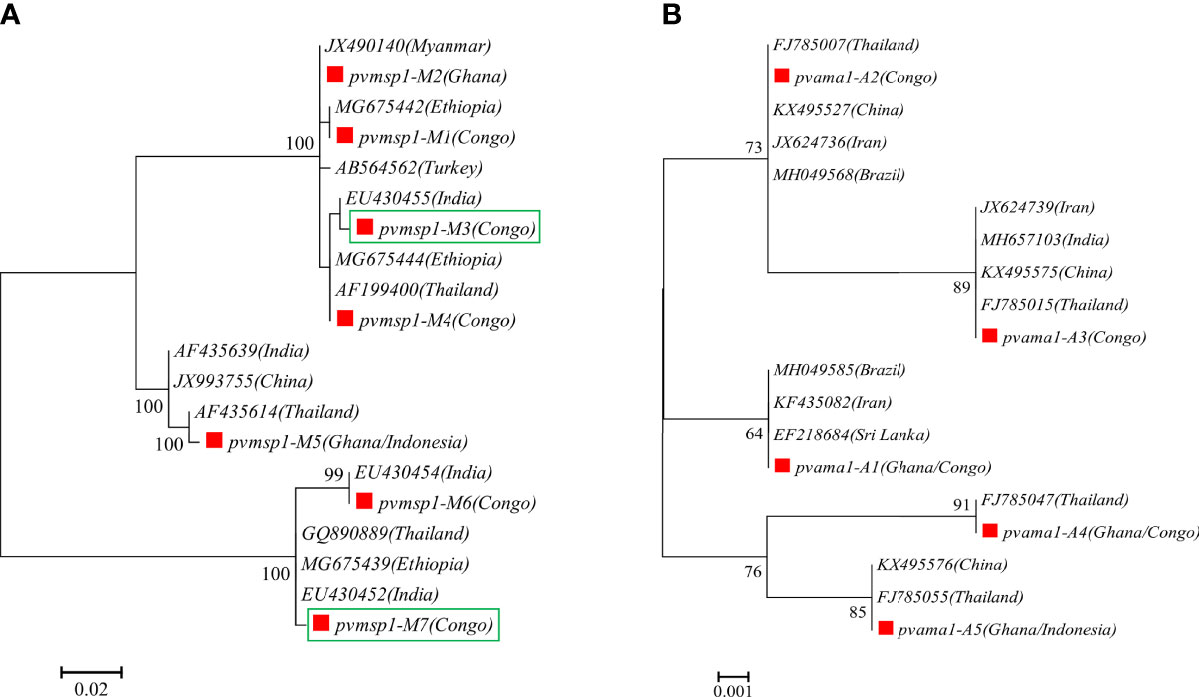
Figure 2 Molecular phylogenetic analysis by the maximum likelihood method based on haplotypes determined by amplicon deep sequencing in P. vivax. (A) pvmsp1 phylogenetic tree for the seven haplotypes (M1–M7) and 13 sequences retrieved from GenBank (AB564562, AF199400, AF435614, AF435639, EU430452, EU430454, EU430455, GQ890889, JX490140, JX993755, MG675439, MG675442, MG675444). (B) pvama1 phylogenetic tree for the five haplotypes (A1–A5) and 14 sequences retrieved from GenBank (MH049585, FJ785007, FJ785015, FJ785055, MH049568, FJ785047, EF218684, JX624736, JX624739, MH657103, KX495527, KX495575, KX495576, KF435082). pf, P. falciparum; pv, P. vivax; pm, P. malariae; poc, P. ovale curtisi; and pow, P. ovale wallikeri. Red square color mark represents haplotypes identified in this study. The new haplotypes are showing in green boxes.
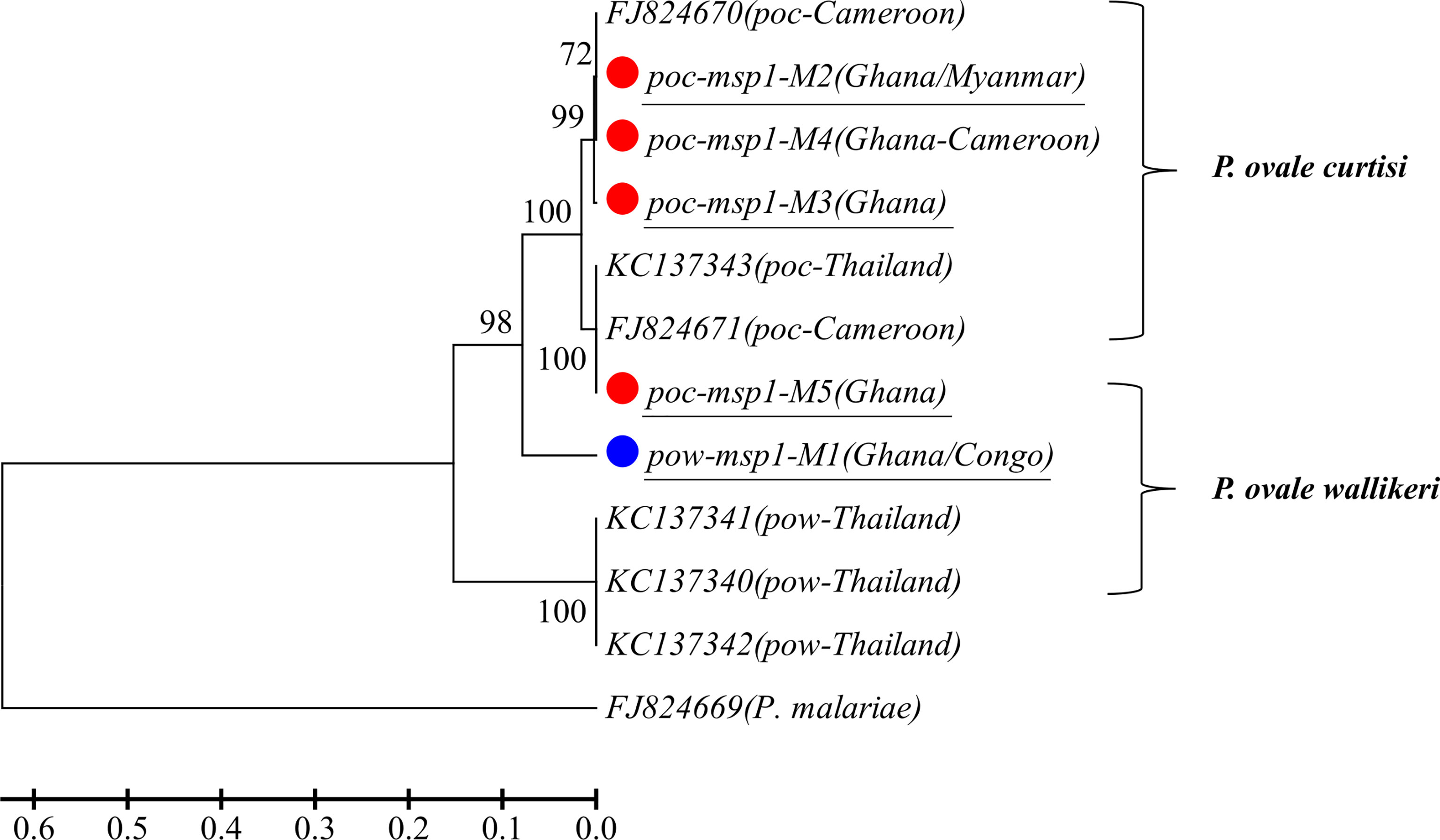
Figure 3 Phylogenetic tree analysis of msp1 haplotypes (M1–M5) determined by amplicon deep sequencing in P. ovale. poc, P. ovale curtisi; pow, P. ovale wallikeri. P. malariae is used as outgroup. Color mark represents haplotypes identified in this study. The novel haplotypes show the underlined label.
Comparison of Haplotypes Between First and Recurrent Malaria Episodes by msp1 and ama1 Markers
The deep sequencing of pfmsp1 and pfama1 on the first and second episodes was successfully performed for a total of 12 patients infected with P. falciparum (Table 2). Combining pfmsp1 and pfama1 haplotypes, three patients had identical genotypes and three patients had a reduced number of haplotypes from the first to the second episode, whereas two patients had partial changes of haplotype number at recurrence. One patient had an increased number of haplotypes, and three patients had changed haplotypes at recurrence. Overall, one patient failed treatment within 14 days. Nine patients had a second infection 15 to 39 days posttreatment and shared one or more haplotypes with the first episode. The remaining two patients showed different haplotypes between the first and second episodes and were considered as indeterminate. The pie chart showed some examples of detailed recurring patterns by the changes of pfmsp1 haplotype frequencies (Figure 4).

Table 2 Comparisons of pfmsp1 and pfama1 combined haplotypes between first malaria episode and recurrent malaria.
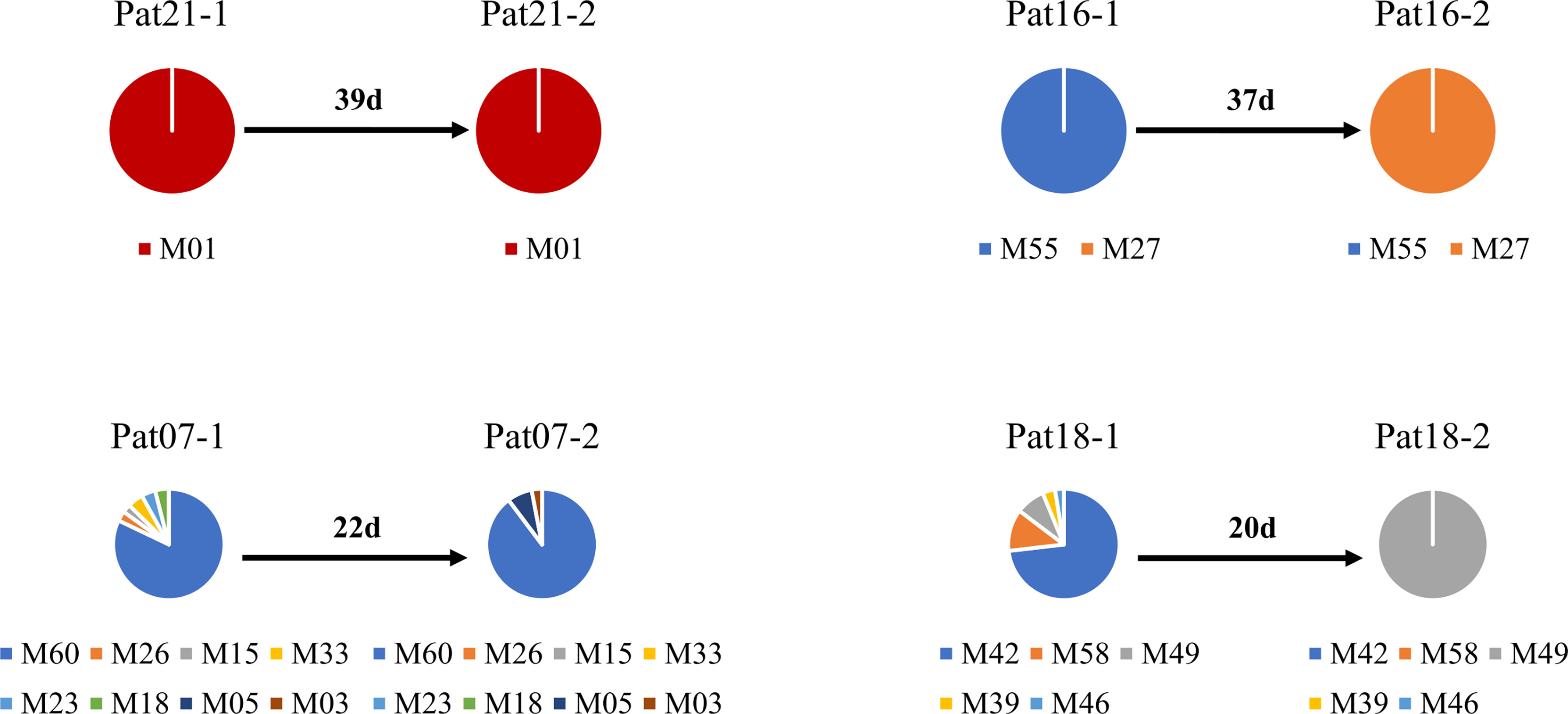
Figure 4 Pie chart shows the recurring patterns of P. falciparum by pfmsp1 haplotypes (“M” with two-digit numbers). Malaria patient episodes are indicated above each pie chart; the number above the arrow indicates malaria recurrence intervals (days). Additional information is shown in Supplementary Table 1.
The deep sequencing of pvmsp1 and pvama1 on the first and recurrent episodes was successfully performed for a total of five patients infected with P. vivax. Of these, three patients underwent two rounds of treatments, whereas one patient had three rounds of treatments and one had four rounds of treatments. All-round of treatments were the same prescriptions (Supplementary_Methods). Except Pat37 who had an early treatment failure after the second episode, all patients had a recurrent infection from 47 to 503 days in which four had identical combined pvmsp1 and pvama1 haplotypes, while four had different haplotypes between two episodes (Figure 5). Although Pat38 had additional travel history to Ghana, the haplotype (M5+A5) of the recurrent episode was identical to the first episode and different from Ghana as identified in this study. Overall, all the five P. vivax recurrences except the one as treatment failure could probably be considered relapses since these patients did not travel after the first episode, and there is no local malaria transmission. For P. ovale, there was one patient (Pat42) identified by pomsp1 with identical genotypes between second episode and third episode with a time length interval of 24 days, whereas the other patients had only one episode successfully genotyped.
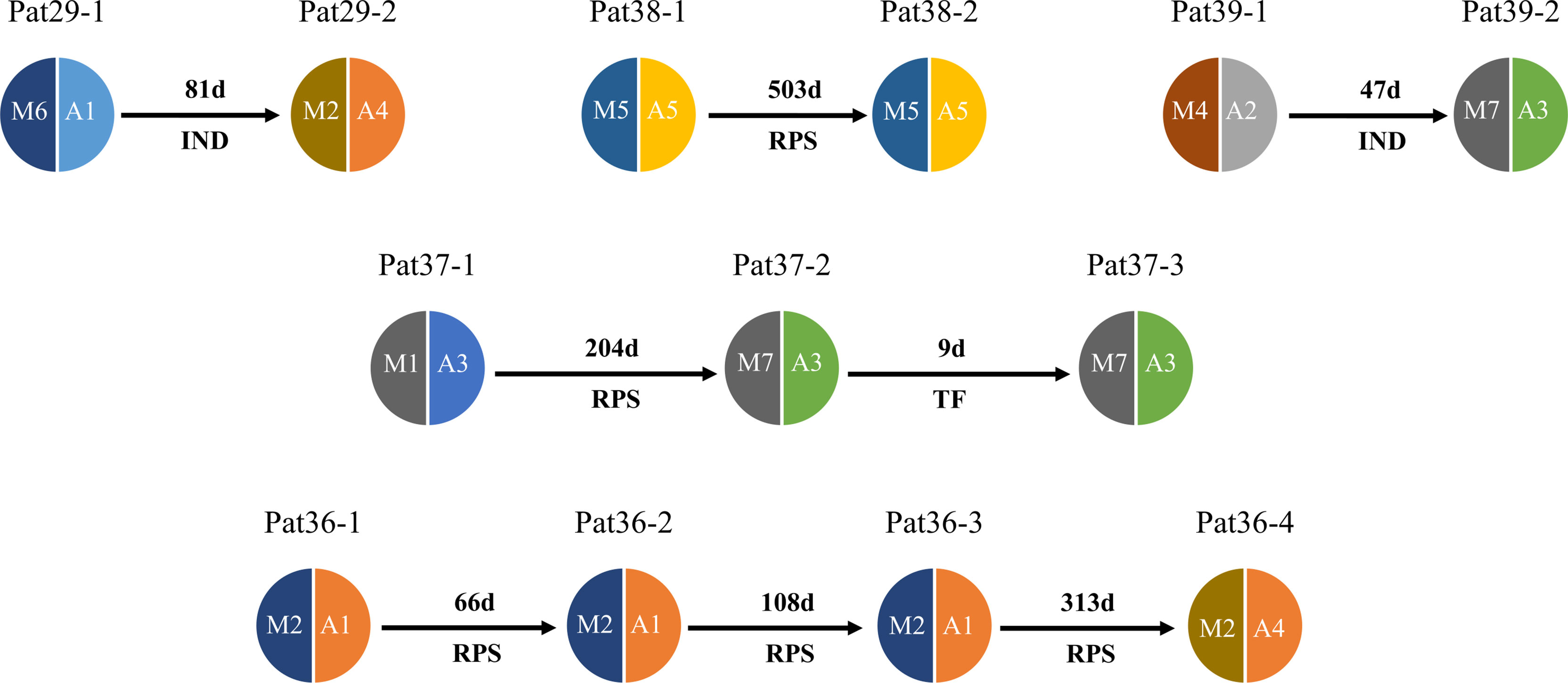
Figure 5 Pie chart shows haplotype changes between first episode and recurrence of P. vivax. Malaria patient episodes are indicated above each pie chart; arrows indicate recurrence interval (days); M1–M7 represent pvmsp1 haplotypes, A1–A5 represent pvmasp1 haplotypes. RPS = relapse (defined by shared one or more haplotypes after 42 days post treatment), TF = treatment failure (within 42 days), IND, indeterminate (no shared haplotype). Additional information is shown in Supplementary Table 1.
Mutations of Antimalarial Drug Resistance Genes in P. falciparum Infections
A total of eight patients were successfully genotyped for mutations in the five antimalaria drug resistance genes (Supplementary Table 8). All the patients had recurrence and travel history before the first malaria attack from Ghana, except one (Pat07) from Congo. However, Pat18 had malaria recrudesces and showed reduced genotypes 20 days after treatment, suggesting the role of parasite-resistant genotypes. There was no statistically significant difference between the first episode and recurrent infections in the mutation frequencies of all the antimalaria drug resistance genes (Fisher’s exact test, p > 0.05). The sample size for patients with recurrence may not be enough to exclude its association with mutations.
Profiles of CYP2D6 Gene in P. vivax Patients
A total of four patients were successfully genotyped for mutations of the CYP2D6 gene in P. vivax patients. The results indicated that those patients’ CYP2D6 genotypes correspond to *4N/*2A, *2A/*4N, *2A/*36, and *4N/*15 allele variants, respectively (Supplementary Table 1). The overall genotype activity scores were 1, 1, 1, and 0, respectively. The CYP2D6 genotype activity scores ≤ 1.0 were considered poor primaquine metabolizers in P. vivax infection (Baird et al., 2018).
Discussion
Imported malaria cases to areas free of malaria transmission provide a unique opportunity to investigate patterns of malaria reappearance. Next-generation targeted amplicon deep-sequencing approaches empower a high-throughput, quantitative method for identification of Plasmodium species and mixed-species infections as well as detection of multiclonal infections, including minor alleles present within a sample (Koepfli and Mueller, 2017; Lalremruata et al., 2017; Lerch et al., 2017; Zhong et al., 2018). In the study, mixed-species infections were detected in 15 out of the 93 malaria episodes (~16%). Most of these were P. ovale curtisi coinfected with P. falciparum. This result is consistent with the previous study in parasite samples from Thailand and West Africa (Saralamba et al., 2019). A high proportion of mixed-species infections was also reported in other studies using ADS, in which up to quadruple mixed-species infections were observed (Lalremruata et al., 2017). Compared to CYTB, small-subunit ribosomal RNA (18S rRNA) had much higher polymorphism and multiple different haplotypes or multiple copies (up to 5) within the sample as seen in this study. Although both markers can be used to discriminate P. ovale subspecies, no mixed-species infection was detected by the CYTB marker when up to 15 mixed-species infections were found by using the SSU marker. Likewise, this SSU marker should be a good candidate and well-suited molecular target for the detection and discrimination of all Plasmodium species. Compared to ama1, msp1 had a higher polymorphism similar to our previous study in Kenya samples (Zhong et al., 2018), which is also a good candidate marker for amplicon deep sequencing to detect multiclonal infections.
Although P. falciparum infections were responsible for the majority of imported malaria cases in China, there was a large proportion (25.9% for P. ovale and 19.4% for P. vivax including mixed species) of non-falciparum infections cases in this study. Recently, P. ovale infections have become more common in Africa, suggesting that P. ovale is becoming an increasingly important malaria parasite, highlighting the need for attention toward non-falciparum malaria (Yman et al., 2019; Zhou et al., 2019; Lin et al., 2020). A large proportion of P. ovale infections and mixed-species infections were also evidenced in the imported malaria cases from Africa to China as seen in the current study. There is very limited genomic information of P. ovale available in the NCBI sequence database. In this study, we found a total of seven haplotypes (four for P. ovale curtisi and three for P. ovale wallikeri) by the SSU marker, five haplotypes (four for P. ovale curtisi and one for P. ovale wallikeri) from the pomsp1 marker. Among these, four out of the five pomsp1 haplotypes were identified as novel haplotypes (all from P. ovale curtisi); its potential role in antimalaria resistance needs to be studied further. The high proportion of P. vivax infections and relapse was observed in the study, possibly due to positive Duffy antigens elevating susceptibility to vivax malaria in Asian patients (Miller et al., 1976; de Carvalho and de Carvalho, 2011; Howes et al., 2011).
We found a very complex pattern of malaria reappearance in these imported malaria cases by examining msp1 and ama1 markers. For P. falciparum, the same genotypes were detected from 15 to 39 days after ACT, while a reduced genotype was observed from 15 to 21 days after ACT. Starting from 17 days, an increased number of genotypes were observed in Patient 20, whereas totally different genotypes were found from 13 to 37 days after ACT. These complex patterns of reappearance may be an indication of early treatment failures (Borrmann et al., 2003; Jackson et al., 2006; Huang et al., 2012; Madamet et al., 2017), antimalarial drug resistance (Lu et al., 2017), late treatment failures (Borrmann et al., 2003; Sondén et al., 2016), and ring-stage parasites associated with artemisinin resistance (Zhang et al., 2017). However, there was no resistance-associated pfk13 mutation detected except for N-terminal insertion (NN) and K189T/N; their roles in resistance are not yet determined (Group, 2019; Zhao et al., 2019). The changed allele observed in reappearance might be explained by early detection failure due to the presence of minor alleles in multiclonal infections, or parasites remaining dormant and relapsing during pregnancy (Theunissen et al., 2009; Malvy et al., 2018). Some of the tested samples had P. falciparum drug-resistance alleles to chloroquine with the changed multiclonal infections detected, further supporting the presence of resistant parasites below the limit of detection. For P. vivax, a long-latency period of 503 days between first episode and relapse was observed with the same combined haplotypes from Indonesia (Pat38), although this patient had an additional trip to Ghana. The short or long latency relapses after primaquine treatment for P. vivax infections could be explained by parasite drug resistance (Baird, 2004) or patients with a poor metabolizer CYP2D6 variant for three-fourths of vivax cases, as detected in the current study. There is little information about drug resistance in P. ovale; further study is needed to investigate the association between genetic and drug resistance in this species.
Conclusion
This study elucidated patterns of malaria reappearance, especially for the less studied non-falciparum malaria parasite species. A high proportion of P. vivax and P. ovale parasites carriers, as well as a high proportion of mixed-species infections, may have important implications for non-falciparum malaria control and treatment strategies. The high recurrence rate and unexpectedly complex patterns of imported malaria from Africa to non-endemic countries pose a risk for increased local transmission and might undermine malaria control and elimination. Appropriate interventions and monitoring and accurate diagnostics using molecular methods are essential for imported malaria management. Finally, the study highlighted the power of ADS applications in molecular epidemiological studies of imported malaria.
Data Availability Statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/Supplementary Material.
Ethics Statement
The studies involving human participants were reviewed and approved by the Ethical Review Committee of Shanglin People’s Hospital, Guangxi, China. The patients/participants provided their written informed consent to participate in this study.
Author Contributions
GY, ZY, and DZ conceived and designed the study. XH wrote the first draft of the paper. DZ and LC helped in performing data analysis and drafted the manuscript. XH, CZ, LP, LZ, YQ, MP, SW, WZ, ZX, XC, YW, YS, and YH conducted the sample collection and experiments. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by grants (31860604 and U1802286) from the National Natural Science Foundation of China and by a grant (2018ZF0081) from Major Science and Technology Projects of Yunnan and Technology Cooperation-Yunnan International Science and Technology Cooperation Base (202003AE140004). LC was supported by a grant U19AI089672 from the National Institutes of Health, USA. GY was supported by the National Institutes of Health USA (U19AI129326). CZ was funded by a grant from the Youth Fund Project of People’s Hospital of Guangxi Zhuang Autonomous Region, China (QN2017-10). MP was funded by a grant (ZC20153012) from the Science and Technology Bureau Programs of Nanning, Guangxi, China. YQ was funded by the Scientific Research Project of Health Committee of Guangxi Zhuang Autonomous Region (20191525). WZ was supported by the Education Department Fund of Yunnan Province (2019J1184). XC and ZX were sponsored by the Yunnan Applied Basic Research Projects-Union Foundation (2018FE001-190 and 2019FE001-015, respectively). YW was supported by the Hundred-Talent Program of Kunming Medical University (60117190439), and the Foundation of the Education Department of Yunnan Province (2018JS151) and the Creative Experimental Project of Kunming Medical University and Yunnan Province.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
We want to thank the staff at the Shanglin Hospital for collecting data on hematology and blood chemistry for the vivax patients, and local CDC personnel for the directly observed therapy. We also thank the Associate Editor Shruthi Sridhar Vembar and two reviewers for their constructive comments that improved this manuscript.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fcimb.2021.725859/full#supplementary-material
References
Apinjoh, T. O., Ouattara, A., Titanji, V. P. K., Djimde, A., Amambua-Ngwa, A. (2019). Genetic Diversity and Drug Resistance Surveillance of Plasmodium falciparum for Malaria Elimination: Is There an Ideal Tool for Resource-Limited Sub-Saharan Africa? Malar. J. 18 (1), 217. doi: 10.1186/s12936-019-2844-5
Baird, J. K. (2004). Chloroquine Resistance in Plasmodium vivax. Antimicrob. Agents Chemother. 48 (11), 4075–4083. doi: 10.1128/AAC.48.11.4075-4083.2004
Baird, J. K., Battle, K. E., Howes, R. E. (2018). Primaquine Ineligibility in Anti-Relapse Therapy of Plasmodium vivax Malaria: The Problem of G6PD Deficiency and Cytochrome P-450 2D6 Polymorphisms. Malar. J. 17 (1), 42. doi: 10.1186/s12936-018-2190-z
Bhatnagar, B., Orwick, S., Heerema, N. A., Walker, A. R., Mims, A. S., Blachly, J. S., et al. (2019). NPM1 Mutations Using Deep Amplicon Sequencing and Broad Next Generation Sequencing at the Time of Complete Remission Is Informative to Predicting Risk of Relapse Following Intensive Chemotherapy. Blood 134 (Supplement_1), 1329–1329. doi: 10.1182/blood-2019-130856
Borrmann, S., Adegnika, A. A., Missinou, M. A., Binder, R. K., Issifou, S., Schindler, A., et al. (2003). Short-Course Artesunate Treatment of Uncomplicated Plasmodium falciparum Malaria in Gabon. Antimicrob. Agents Chemother. 47 (3), 901–904. doi: 10.1128/aac.47.3.901-904.2003
Brazeau, N. F., Assefa, A., Mohammed, H., Seme, H., Tsadik, A. G., Parr, J. B., et al. (2019). Pooled Deep Sequencing of Drug Resistance Loci From Plasmodium falciparum Parasites Across Ethiopia. Am. J. Trop. Med. Hyg. 101 (5), 1139–1143. doi: 10.4269/ajtmh.19-0142
de Carvalho, G. B., de Carvalho, G. B. (2011). Duffy Blood Group System and the Malaria Adaptation Process in Humans. Rev. Bras. Hematol. Hemoter. 33 (1), 55–64. doi: 10.5581/1516-8484.20110016
Early, A. M., Daniels, R. F., Farrell, T. M., Grimsby, J., Volkman, S. K., Wirth, D. F., et al. (2019). Detection of Low-Density Plasmodium falciparum Infections Using Amplicon Deep Sequencing. Malar. J. 18 (1), 219. doi: 10.1186/s12936-019-2856-1
Flaherty, P., Natsoulis, G., Muralidharan, O., Winters, M., Buenrostro, J., Bell, J., et al. (2012). Ultrasensitive Detection of Rare Mutations Using Next-Generation Targeted Resequencing. Nucleic Acids Res. 40 (1), e2. doi: 10.1093/nar/gkr861
Gaye, A., Sy, M., Ndiaye, T., Siddle, K. J., Park, D. J., Deme, A. B., et al. (2020). Amplicon Deep Sequencing of Kelch13 in Plasmodium falciparum Isolates From Senegal. Malar. J. 19 (1), 134. doi: 10.1186/s12936-020-03193-w
Group, W.K.G.-P.S (2019). Association of Mutations in the Plasmodium falciparum Kelch13 Gene (Pf3D7_1343700) With Parasite Clearance Rates After Artemisinin-Based Treatments-A WWARN Individual Patient Data Meta-Analysis. BMC Med. 17 (1), 1–1. doi: 10.1186/s12916-018-1207-3
Gruenberg, M., Lerch, A., Beck, H.-P., Felger, I. (2019). Amplicon Deep Sequencing Improves Plasmodium falciparum Genotyping in Clinical Trials of Antimalarial Drugs. Sci. Rep. 9 (1), 17790. doi: 10.1038/s41598-019-54203-0
Hathaway, N. J., Parobek, C. M., Juliano, J. J., Bailey, J. A. (2017). SeekDeep: Single-Base Resolution De Novo Clustering for Amplicon Deep Sequencing. Nucleic Acids Res. 46 (4), e21–e21. doi: 10.1093/nar/gkx1201
Hemming-Schroeder, E., Zhong, D., Kibret, S., Chie, A., Lee, M.-C., Zhou, G., et al. (2020). Microgeographic Epidemiology of Malaria Parasites in an Irrigated Area of Western Kenya by Deep Amplicon Sequencing. J. Infect. Dis. 223 (8), 1456–1465. doi: 10.1093/infdis/jiaa520
He, X., Pan, M., Zeng, W., Zou, C., Pi, L., Qin, Y., et al. (2019). Multiple Relapses of Plasmodium vivax Malaria Acquired From West Africa and Association With Poor Metabolizer CYP2D6 Variant: A Case Report. BMC Infect. Dis. 19 (1), 704. doi: 10.1186/s12879-019-4357-9
Howes, R. E., Patil, A. P., Piel, F. B., Nyangiri, O. A., Kabaria, C. W., Gething, P. W., et al. (2011). The Global Distribution of the Duffy Blood Group. Nat. Commun. 2, 266–266. doi: 10.1038/ncomms1265
Huang, F., Tang, L., Yang, H., Zhou, S., Sun, X., Liu, H. (2012). Therapeutic Efficacy of Artesunate in the Treatment of Uncomplicated Plasmodium falciparum Malaria and Anti-Malarial, Drug-Resistance Marker Polymorphisms in Populations Near the China-Myanmar Border. Malar. J. 11, 278–278. doi: 10.1186/1475-2875-11-278
Imwong, M., Pukrittayakamee, S., Grüner, A. C., Rénia, L., Letourneur, F., Looareesuwan, S., et al. (2005). Practical PCR Genotyping Protocols for Plasmodium vivax Using Pvcs and Pvmsp1. Malar. J. 4 (1), 20. doi: 10.1186/1475-2875-4-20
Jackson, Y., Chappuis, F., Loutan, L., Taylor, W. (2006). Malaria Treatment Failures After Artemisinin-Based Therapy in Three Expatriates: Could Improved Manufacturer Information Help to Decrease the Risk of Treatment Failure? Malar. J. 5 (1), 81. doi: 10.1186/1475-2875-5-81
Jianhai, Y., Li, Z., Jun, F., Shuisen, Z., Zhigui, X. (2020). Malaria Diagnosis and Verification — China, 2017-2018. Chin. Center. Dis. Control. Prev. CCDC. Weekly. 2 (17), 285–288. doi: 10.46234/ccdcw2020.073
Kadekoppala, M., Holder, A. A. (2010). Merozoite Surface Proteins of the Malaria Parasite: The MSP1 Complex and the MSP7 Family. Int. J. Parasitol. 40 (10), 1155–1161. doi: 10.1016/j.ijpara.2010.04.008
Koepfli, C., Mueller, I. (2017). Malaria Epidemiology at the Clone Level. Trends Parasitol. 33 (12), 974–985. doi: 10.1016/j.pt.2017.08.013
Kyabayinze, D., Cattamanchi, A., Kamya, M. R., Rosenthal, P. J., Dorsey, G. (2003). Validation of a Simplified Method for Using Molecular Markers to Predict Sulfadoxine-Pyrimethamine Treatment Failure in African Children With Falciparum Malaria. Am. J. Trop. Med. Hyg. 69 (3), 247–252. doi: 10.4269/ajtmh.2003.69.247
Lai, S., Sun, J., Ruktanonchai, N. W., Zhou, S., Yu, J., Routledge, I., et al. (2019). Changing Epidemiology and Challenges of Malaria in China Towards Elimination. Malar. J. 18 (1), 107–107. doi: 10.1186/s12936-019-2736-8
Lalremruata, A., Jeyaraj, S., Engleitner, T., Joanny, F., Lang, A., Bélard, S., et al. (2017). Species and Genotype Diversity of Plasmodium in Malaria Patients From Gabon Analysed by Next Generation Sequencing. Malar. J. 16 (1), 398. doi: 10.1186/s12936-017-2044-0
Lerch, A., Koepfli, C., Hofmann, N. E., Messerli, C., Wilcox, S., Kattenberg, J. H., et al. (2017). Development of Amplicon Deep Sequencing Markers and Data Analysis Pipeline for Genotyping Multi-Clonal Malaria Infections. BMC Genomics 18 (1), 864. doi: 10.1186/s12864-017-4260-y
Li, J., Chen, J., Xie, D., Eyi, U. M., Matesa, R. A., Obono, M. M. O., et al. (2015). Molecular Mutation Profile of Pfcrt and Pfmdr1 in Plasmodium falciparum Isolates From Bioko Island, Equatorial Guinea. Infect. Genet. Evol. 36, 552–556. doi: 10.1016/j.meegid.2015.08.039
Lin, J. T., Hathaway, N. J., Saunders, D. L., Lon, C., Balasubramanian, S., Kharabora, O., et al. (2015). Using Amplicon Deep Sequencing to Detect Genetic Signatures of Plasmodium vivax Relapse. J. Infect. Dis. 212 (6), 999–1008. doi: 10.1093/infdis/jiv142
Lin, J. T., Juliano, J. J., Kharabora, O., Sem, R., Lin, F.-C., Muth, S., et al. (2012). Individual Plasmodium vivax msp1 Variants Within Polyclonal P. Vivax Infections Display Different Propensities for Relapse. J. Clin. Microbiol. 50 (4), 1449–1451. doi: 10.1128/JCM.06212-11
Lin Kang-Ming, ,. L. J., Wei, S.-J., Zhang, W.-W., Feng, X.-Y., Yan, H., Wei, H.-Y., et al. (2019). Epidemic Situation and Control Strategy of Malaria in Guangxi Zhuang Autonomous Region From 2011 to 2018. Chin. J. Schisto. Cont. 31 (3), 315–318. doi: 10.16250/j.32.1374.2019047
Lin, J. T., Parr, J. B., Ngasala, B. (2020). Non-Falciparum Malaria in Africa and Learning From Plasmodium vivax in Asia. Clin. Infect. Dis. 70 (9), 2018–2019. doi: 10.1093/cid/ciz780
Lin, K., Wei, H., Jiang, W., Li, J., Zhang, W., Wei, S., et al. (2017). Malaria in the Guangxi Zhuang Autonomous Region in China: A Twelve-Year Surveillance Data Study. Am. J. Trop. Med. Hyg. 97 (4), 1163–1169. doi: 10.4269/ajtmh.16-0260
Lu, F., Culleton, R., Zhang, M., Ramaprasad, A., von Seidlein, L., Zhou, H., et al. (2017). Emergence of Indigenous Artemisinin-Resistant Plasmodium falciparum in Africa. N. Engl. J. Med. 376 (10), 991–993. doi: 10.1056/NEJMc1612765
Madamet, M., Kounta, M. B., Wade, K. A., Lo, G., Diawara, S., Fall, M., et al. (2017). Absence of Association Between Polymorphisms in the K13 Gene and the Presence of Plasmodium falciparum Parasites at Day 3 After Treatment With Artemisinin Derivatives in Senegal. Int. J. Antimicrob. Agents 49 (6), 754–756. doi: 10.1016/j.ijantimicag.2017.01.032
Malvy, D., Torrentino-Madamet, M., Ollivier, C., Receveur, M.-C., Jeddi, F., Delhaes, L., et al. (2018). Plasmodium falciparum Recrudescence Two Years After Treatment of an Uncomplicated Infection Without Return to an Area Where Malaria Is Endemic. Antimicrob. Agents Chemother. 62 (2), e01892–e01817. doi: 10.1128/AAC.01892-17
Miguel-Oteo, M., Jiram, A. I., Ta-Tang, T. H., Lanza, M., Hisam, S., Rubio, J. M. (2017). Nested Multiplex PCR for Identification and Detection of Human Plasmodium Species Including Plasmodium knowlesi. Asian Pac. J. Trop. Med. 10 (3), 299–304. doi: 10.1016/j.apjtm.2017.03.014
Miller, L. H., Mason, S. J., Clyde, D. F., McGinniss, M. H. (1976). The Resistance Factor to Plasmodium vivax in Blacks. The Duffy-Blood-Group Genotype, FyFy. N. Engl. J. Med. 295 (6), 302–304. doi: 10.1056/nejm197608052950602
Muehlenbachs, A., Mutabingwa, T. K., Fried, M., Duffy, P. E. (2007). An Unusual Presentation of Placental Malaria: A Single Persisting Nidus of Sequestered Parasites. Hum. Pathol. 38 (3), 520–523. doi: 10.1016/j.humpath.2006.09.016
Nyachieo, A., Van Overmeir, C., Laurent, T., Dujardin, J. C., D’Alessandro, U. (2005). Plasmodium falciparum Genotyping by Microsatellites as a Method to Distinguish Between Recrudescent and New Infections. Am. J. Trop. Med. Hyg. 73 (1), 210–213. doi: 10.4269/ajtmh.2005.73.210
Saralamba, N., Nosten, F., Sutherland, C. J., Arez, A. P., Snounou, G., White, N. J., et al. (2019). Genetic Dissociation of Three Antigenic Genes in Plasmodium ovale curtisi and Plasmodium ovale wallikeri. PloS One 14 (6), e0217795. doi: 10.1371/journal.pone.0217795
Sondén, K., Wyss, K., Jovel, I., Vieira da Silva, A., Pohanka, A., Asghar, M., et al. (2016). High Rate of Treatment Failures in Nonimmune Travelers Treated With Artemether-Lumefantrine for Uncomplicated Plasmodium falciparum Malaria in Sweden: Retrospective Comparative Analysis of Effectiveness and Case Series. Clin. Infect. Dis. 64 (2), 199–206. doi: 10.1093/cid/ciw710
Theunissen, C., Janssens, P., Demulder, A., Nouboussié, D., Van Esbroeck, M., Van Gompel, A., et al. (2009). Falciparum Malaria in Patient 9 Years After Leaving Malaria-Endemic Area. Emerging. Infect. Dis. J. 15 (1), 115. doi: 10.3201/eid1501.080909
WHO (2009). Methods for Surveillance of Antimalarial Drug Efficacy (Geneva, Switzerland: World Health Organization). Available at: https://www.who.int/malaria/publications/atoz/9789241597531/en/. Global Malaria Programme.
WHO (2015). Guidelines for the Treatment of Malaria. 3rd ed. (Geneva, Switzerland: World Health Organization). Available at: https://www.who.int/malaria/publications/atoz/9789241549127/en/. Global Malaria Programme.
WHO (2020). World Malaria Report 2020 (Geneva, Switzerland: World Health Organization). Available at: https://www.who.int/publications/i/item/9789240015791. Global Malaria Programme.
Yang, Y., Liu, Y., Xie, Z., Wu, S., Yang, L., Li, W., et al. (2018). Epidemiology of Malaria in Yulin, South China 1999–2016: Imported Malaria Threatens Zero Local Case Status. Vector. Borne. Zoonotic. Dis. 18 (10), 533–538. doi: 10.1089/vbz.2017.2236
Yman, V., Wandell, G., Mutemi, D. D., Miglar, A., Asghar, M., Hammar, U., et al. (2019). Persistent Transmission of Plasmodium malariae and Plasmodium ovale Species in an Area of Declining Plasmodium falciparum Transmission in Eastern Tanzania. PloS Neglect. Trop. Dis. 13 (5), e0007414. doi: 10.1371/journal.pntd.0007414
Zhang, J., Feng, G.-H., Zou, C.-Y., Su, P.-C., Liu, H.-E., Yang, Z.-Q. (2017). Overview of the Improvement of the Ring-Stage Survival Assay-A Novel Phenotypic Assay for the Detection of Artemisinin-Resistant. Plasmodium. falciparum. Zool. Res. 38 (6), 317–320. doi: 10.24272/j.issn.2095-8137.2017.075
Zhang, Y.-M., Wu, Y.-R., Hu, Y., Wang, L.-Q., Ruan, Y.-H., Ma, N., et al. (2016). In Vitro Susceptibility of Plasmodium falciparum to Artemisinin Drugs and K13-Propeller Polymorphisms at the China-Myanmar Border. Chin. J. Zoonoses. 32 (3), 219–223. doi: 10.3969/j.issn.1002-2694.2016.03.002
Zhao, Y., Liu, Z., Soe, M. T., Wang, L., Soe, T. N., Wei, H., et al. (2019). Genetic Variations Associated With Drug Resistance Markers in Asymptomatic Plasmodium falciparum Infections in Myanmar. Genes (Basel) 10 (9), 692. doi: 10.3390/genes10090692
Zhong, D., Lo, E., Wang, X., Yewhalaw, D., Zhou, G., Atieli, H. E., et al. (2018). Multiplicity and Molecular Epidemiology of Plasmodium vivax and Plasmodium falciparum Infections in East Africa. Malar. J. 17 (1), 185–185. doi: 10.1186/s12936-018-2337-y
Keywords: imported malaria, mixed-species infection, relapse, recrudescence, multiplexity of infection, drug resistance
Citation: He X, Zhong D, Zou C, Pi L, Zhao L, Qin Y, Pan M, Wang S, Zeng W, Xiang Z, Chen X, Wu Y, Si Y, Cui L, Huang Y, Yan G and Yang Z (2021) Unraveling the Complexity of Imported Malaria Infections by Amplicon Deep Sequencing. Front. Cell. Infect. Microbiol. 11:725859. doi: 10.3389/fcimb.2021.725859
Received: 15 June 2021; Accepted: 16 August 2021;
Published: 14 September 2021.
Edited by:
Shruthi Sridhar Vembar, Institut Pasteur, FranceReviewed by:
Jaishree Raman, National Institute of Communicable Diseases (NICD), South AfricaJianxia Tang, Jiangsu Institute of Parasitic Diseases (JIPD), China
Copyright © 2021 He, Zhong, Zou, Pi, Zhao, Qin, Pan, Wang, Zeng, Xiang, Chen, Wu, Si, Cui, Huang, Yan and Yang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yaming Huang, MTcyNDUwMTk2NEBxcS5jb20=; Guiyun Yan, Z3VpeXVueUB1Y2kuZWR1; Zhaoqing Yang, emhhb3Fpbmd5OTJAaG90bWFpbC5jb20=
†Present address: Xi He, Department of Laboratory Medicine, The People’s Hospital of Dujiangyan, Sichuan, China
‡These authors have contributed equally to this work and share first authorship