 Xianxiu Wan1
Xianxiu Wan1 Nisha Jain Garg1,2*
Nisha Jain Garg1,2*- 1Department of Microbiology and Immunology, University of Texas Medical Branch, Galveston, TX, United States
- 2Institute for Human Infections and Immunity, University of Texas Medical Branch, Galveston, TX, United States
Trypanosoma cruzi is a digenetic parasite that requires triatomines and mammalian host to complete its life cycle. T. cruzi replication in mammalian host induces immune-mediated cytotoxic proinflammatory reactions and cellular injuries, which are the common source of reactive oxygen species (ROS) and reactive nitrogen species (RNS) during the acute parasitemic phase. Mitochondrial dysfunction of electron transport chain has been proposed as a major source of superoxide release in the chronic phase of infection, which renders myocardium exposed to sustained oxidative stress and contributes to Chagas disease pathology. Sirtuin 1 (SIRT1) is a class III histone deacetylase that acts as a sensor of redox changes and shapes the mitochondrial metabolism and inflammatory response in the host. In this review, we discuss the molecular mechanisms by which SIRT1 can potentially improve mitochondrial function and control oxidative and inflammatory stress in Chagas disease.
Introduction
Trypanosoma cruzi (T. cruzi) infection leads to the development of Chagas disease (CD) that is one of the most frequent causes of heart failure and sudden death in the Americas. There are three stages in Chagas disease: the acute phase, the early chronic phase that is also referred as indeterminate phase, and the late chronic disease phase. Shortly after exposure to the parasite, infected individuals develop acute parasitemia when trypomastigotes and amastigotes can be easily detected by microscopic examination of the blood and/or cerebrospinal fluid. In most of the infected individuals, immune response is sufficiently active to control acute parasitemia within 2 to 4 months post-exposure ([[NoAuthor]]). Infected individuals then appear healthy with almost no clinical symptoms of cardiac involvement and may remain in this phase for decades. However, approximately 1/3 of the infected persons progress to clinically symptomatic, chronic disease phase presented with cardiomyopathy and heart failure (Dias et al., 2016; Rassi et al., 2017). Chronically infected individuals may also develop digestive or neurological disorders.
Sirtuins, initially named as silent information regulator 2 (Sir2) proteins, are defined as class III histone deacetylases that utilize nicotinamide adenine dinucleotide (NAD+) as substrate. In the deacetylase reaction, sirtuins hydrolyze one NAD+ molecule and produce deacetylated substrate, nicotinamide (NAM), and O-acetyl-ADP ribose (Landry et al., 2000). The sirtuin family of proteins in humans consists of SIRT1–SIRT7 that are highly conserved, both structurally and functionally. Sirtuins are ubiquitously expressed in all organs including the blood, brain, heart, kidney, lung, liver, ovary, skeletal muscle, spleen, and testis, though the level of expression varies in different organs. The catalytic core deacetylase domain is highly conserved in sirtuins; however, they differ in sequence, subcellular location, enzyme activity, substrate specificity and physiological functions (Frye, 1999; Michishita et al., 2005) (Figure 1). For example, SIRT1, SIRT6, and SIRT7 are generally found in a nuclear compartment and known to modulate gene expression through transcription factors, co-factors, or histones (Liszt et al., 2005; Ford et al., 2006; Mostoslavsky et al., 2006; Michishita et al., 2008; Zhong et al., 2010). SIRT2 is primarily localized in the cytoplasm, and it was shown to regulate oligodendrocyte differentiation and cell cycle (Dryden et al., 2003; Li et al., 2007). The remaining known sirtuins (SIRT3, SIRT4, and SIRT5) are predominantly found in mitochondrial compartment and shown to regulate metabolic enzyme activities and oxidative stress pathways (Ahuja et al., 2007; Nakamura et al., 2008).
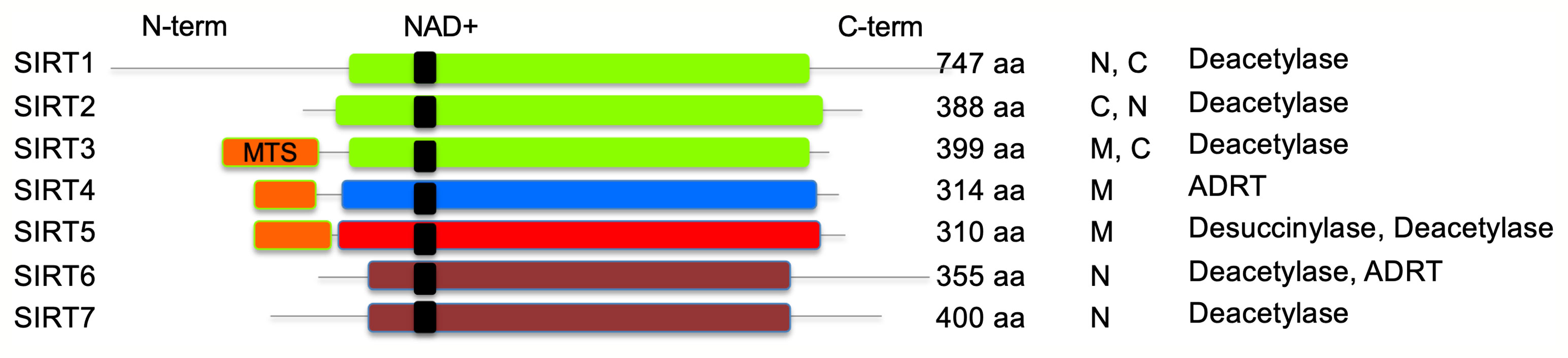
Figure 1 Schematic representation of human sirtuins. SIRT1–3 (class I), SIRT4 (class II), SIRT5 (class III), and SIRT6–7 (class IV) are shown. NAD+ binding region is presented in black. MTS, mitochondria-targeting sequence is shown in orange; N, nuclear; C, cytoplasmic; ADRT, ADP-ribosyltransferase. Catalytic histidine and zinc-coordinating cysteines are not shown.
SIRT1 is the closest homolog of yeast Sir2, and it is the most studied sirtuin (Frye, 2000). At the cellular level, SIRT1 shuttles between the nucleus and cytoplasm depending on environmental conditions (Hou et al., 2010). SIRT1 has two nuclear localization signals and a coiled-coil domain in addition to the core deacetylase domain. Many of the endogenous substrates of SIRT1, including p53, NBS1, p65, c-Jun, and c-Myc play a role in transcriptional regulation of gene expression (Yeung et al., 2004; Solomon et al., 2006; Yuan et al., 2007; Gao and Ye, 2008; Yuan et al., 2009). While SIRT1 core deacetylase domain determines its enzymatic activity, other domains may influence the binding to NAD+ substrate and target proteins. SIRT1-mediated deacetylation activity regulates several proteins and their translation, and it is involved in critical physiological processes including cell proliferation, genomic stability, metabolism, and antioxidant/oxidant response. SIRT1 has been reported to associate with chronic inflammatory diseases, metabolic dysfunctions, neurodegenerative diseases, and cardiovascular dysfunction (Boutant and Canto, 2014; Chang and Guarente, 2014; Herskovits and Guarente, 2014; Hubbard and Sinclair, 2014).
T. cruzi is a eukaryotic parasite that consists of genes encoding for two sirtuins, named TcSir2rp1 and TcSir2rp3 that are identified to be localized in cytosol and mitochondria, respectively (Moretti et al., 2015). Inhibition and overexpression studies suggest that TcSir2rp1 and TcSir2rp3 seemingly have opposite roles in parasite growth and differentiation (Moretti et al., 2015). A panel of sirtuin inhibitors has been screened for their activity against TcSir2rp1 and TcSir2rp3 (Matutino Bastos et al., 2020) and broad-range SIRT inhibitors (salermide or nictoinamide) were shown to inhibit T. cruzi growth and differentiation in culture and in vivo in mice (Moretti et al., 2015). Thus, SIRTs are required for parasite growth and survival in the mammalian host, and SIRT inhibitors are developed as anti-parasite therapies.
In this review, we discuss various roles of SIRT1 with a focus on its beneficial effects in regulating Chagas disease pathogenesis.
Mitochondria Dysfunction in Experimental Models of CD and Infected Humans
Heart function is supported by a high rate of ATP production through mitochondrial oxidative phosphorylation pathway. Cardiomyocytes have a high copy number of mitochondrial DNA (mtDNA) that encodes essential components of respiratory complexes to support energy demands of the heart (Gustafsson and Gottlieb, 2008). Mitochondrial biogenesis encompasses processes and events resulting in replication, maintenance, and function of mitochondria. Peroxisome proliferator-activated receptor gamma coactivator 1 (PGC-1α) is a member of the PGC family of transcription coactivators, and it is designated as a master regulator of mitochondrial biogenesis and oxidative metabolism (Finck and Kelly, 2006). PGC-1α is preferentially expressed in the heart, kidneys, and skeletal muscle tissues that have high oxidative capacity and abundant mitochondria. PGC-1α functions as an adapter or scaffold protein and docks onto the transcription factor targets or protein complexes to drive the expression of nuclear DNA and mtDNA encoded proteins involved in mitochondrial biogenesis and oxidative phosphorylation (Ventura-Clapier et al., 2008). The estrogen-related nuclear orphan receptors (ERR-α and ERR-γ) are also activated by PGC-1α, which enhances the expression of proteins involved in fatty acid uptake, fatty acid oxidation, and ATP production and transportation (Shao et al., 2010). PGC-1 also binds to and co-activates nuclear respiratory factors 1 and 2 (NRF-1 and NRF-2) that maintain redox homeostasis and upregulate the mitochondrial transcription factor A (TFAM)-mediated mtDNA replication and transcription (Gureev et al., 2019)
The impairment of mitochondrial membrane phospholipids, DNA, or proteins can all affect the mitochondrial oxidative phosphorylation capacity. We have documented significant decline in the activities of the respiratory complex I and complex III in cultured human cardiomyocytes and myocardium of mice infected with T. cruzi (Vyatkina et al., 2004). Quantitative assays and light and electron microscopic analysis of the human plasma and serum samples (Wen et al., 2006) the myocardial biopsies of human CD patients (Wan et al., 2012) and T. cruzi-infected experimental animals showed that mitochondrial degenerative changes occurred in early T. cruzi infection and were exacerbated with progression of disease (Wen et al., 2006; Wen et al., 2008; Wan et al., 2012). No changes in PGC-1α levels were noted; however, its activity in signaling NRF1/2 and TFAM-mediated mitochondrial biogenesis was clearly compromised as was evidenced by the findings of a decline in cardiac mtDNA content, mtDNA synthesis, and oxidative phosphorylation mediated ATP synthesis in Chagas hearts (Wan et al., 2012) (Figure 2).
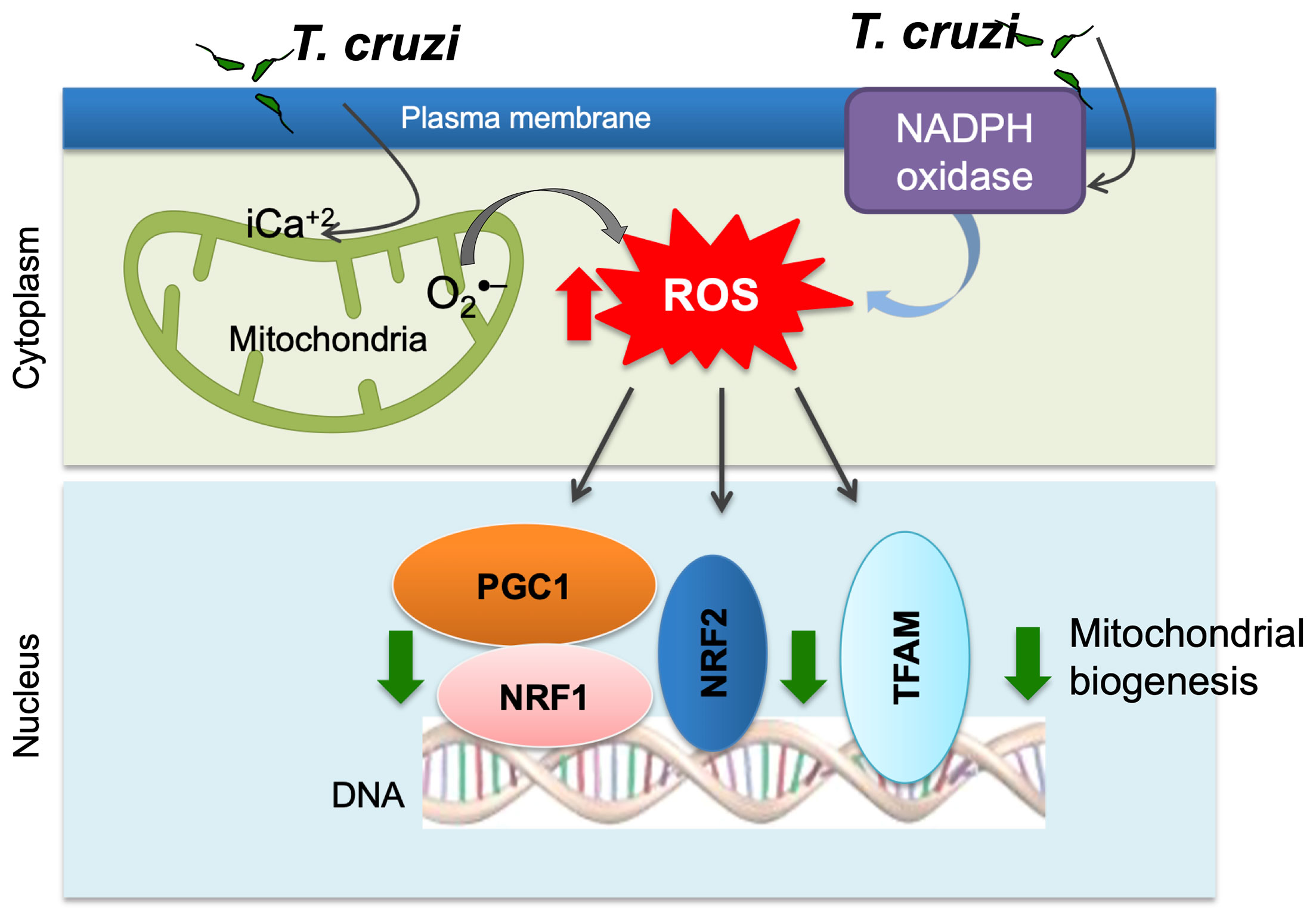
Figure 2 Potential mechanism of mitochondrial dysfunction in Chagas disease. T. cruzi uptake by monocytes/macrophages activates NADPH oxidase-mediated superoxide and ROS production. T. cruzi induces intracellular Ca2+ flux that causes mitochondrial membrane permeability transition, respiratory complex inefficiency, and increased leakage of electron from electron transport chain to oxygen, resulting in superoxide production. ROS suppress mitochondrial biogenesis through inhibition of PGC-1α-mediated transcriptional activation of NRF1/2 and TFAM that maintain redox homeostasis and mtDNA replication and transcription.
Oxidative Stress in CD
High oxidative stress results from increase in the ROS and RNS production above the physiologically relevant threshold levels. ROS describe a variety of free radicals, including superoxide (O2•−), hydroxyl radical (•OH), and hydrogen peroxide (H2O2) derived from molecular oxygen. ROS are formed in various organs and tissues through the action of specific oxidases [e.g., NADPH oxidase (NOX2)], peroxidases (e.g., myeloperoxidase), and as a by-products of mitochondrial electron transport chain (Altenhofer et al., 2012). RNS are a family of molecules derived from nitric oxide (•NO) produced by neuronal, endothelial, and inducible isoforms of nitric oxide synthase (NOS). Phagosomal activation of NOX2 and iNOS serves as primary source of O2•− and •NO that react together to form highly stable and cytotoxic peroxynitrite (ONOO−) required for direct killing of T. cruzi in macrophages (Alvarez et al., 2011). While ROS/RNS play an important role in signaling host defense and regulation of pH and ion concentration in the phagosome and in cell differentiation (Bedard and Krause, 2007), excessive oxidative/nitrosative stress can cause a wide range of pathological processes and cell and tissue injury in the host. In T. cruzi infection, NOX2 components were detected at the plasma membrane of peritoneal macrophages (de Carvalho and de Souza, 1987). However, splenocytes of infected mice and macrophages in vitro infected with T. cruzi exhibited low levels of ROS and •NO production that allowed parasite survival (Koo et al., 2016). Myeloperoxidase and nitrite levels were increased in circulation of T. cruzi-infected mice (Dhiman et al., 2008) and humans (Dhiman et al., 2009), though their relevance in parasite control is not clear.
Mitochondrial respiratory chain is the site for leakage of electrons to oxygen and superoxide production. In context of CD, we have documented that infection by T. cruzi results in intracellular Ca2+ flux, changes in mitochondrial membrane potential, and a decline in the activities of the respiratory complexes. Consequently increase in electrons’ leakage to O2 and increased O2•− formation were noted in murine cardiomyocytes (Gupta et al., 2009; Wen and Garg, 2010) and almost all muscle tissues of acutely infected mice (Gupta et al., 2009). Mitochondrial defects of complex III and resultant increase in ROS release persisted in the heart tissue of chronically infected mice and Chagas patients (Wen et al., 2008; Wen and Garg, 2010; Wan et al., 2012; Dhiman et al., 2013), thus suggesting that mitochondria are a major source of oxidative stress after acute infection phase and during chronic phase of disease progression. ROS-induced oxidative adducts were conducive to proinflammatory macrophage activation (Choudhuri and Garg, 2020).
The overall cellular oxidative stress is an outcome of the relative rate of ROS production and the rate by which ROS are reduced by antioxidants. The critical antioxidants in cardiomyocytes including Mn2+ superoxide dismutase (MnSOD), glutathione peroxidase, catalase, and glutathione were enhanced in various muscle tissues of acutely infected mice (Wen et al., 2004). However, progression of chronic phase in infected mice and humans was associated with sustained or increase in mitochondrial ROS release and oxidative stress markers (e.g., glutathione disulfide, lipid peroxide, protein carbonyl, 4-hydroxynonenal) as well as non-responsive or decreased antioxidants (especially MnSOD) activity (Perez-Fuentes et al., 2003; Budni et al., 2013; Dhiman et al., 2013). Moreover, treatment with an antioxidant controlled the oxidative insult in infected patients (Ribeiro et al., 2010) and preserved the mitochondrial and cardiac function in infected mice (Wen et al., 2006; Wen et al., 2010). Phenyl-N-tert-butylnitrone (PBN) is a spin-trapping antioxidant. Treatment of T. cruzi-infected mice and rats with PBN was beneficial in arresting the myocardial oxidative lesions and preserving the rate of oxidative phosphorylation and ATP synthesis (Wen et al., 2006; Wen et al., 2010). Likewise, MnSOD transgenic mice were better equipped in controlling the inflammatory infiltrate and oxidative stress and cardiac hypertrophy that are hallmarks of chronic CD (Dhiman et al., 2013). All these observations suggest that inefficient antioxidant response, along with mitochondrial dysfunction, contributes to sustained oxidative and inflammatory stress in Chagas cardiomyopathy (Figure 3).
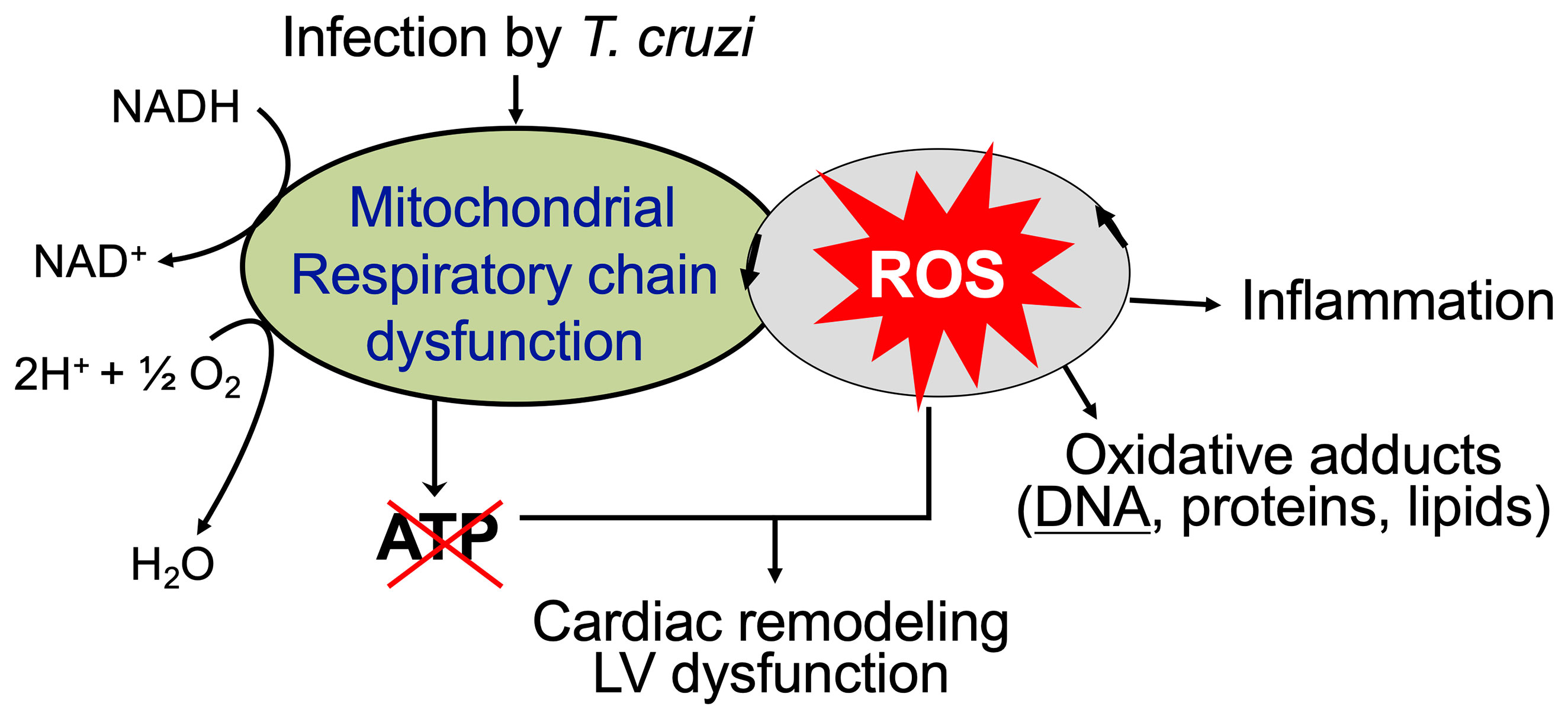
Figure 3 Mitochondrial ROS contribute to cardiac damage, inflammation, and remodeling in chronic Chagas cardiomyopathy. Mitochondrial dysfunction of electron transport chain is sustained in chronically infected mice and humans that results in persistence of ROS production and a decline in oxidative phosphorylation and ATP production. ROS-induced oxidative adducts signal inflammatory responses. Antioxidants capable of scavenging ROS have a potential to control cardiac damage and remodeling in chronic CD.
SIRT1: A Potential Adjuvant Therapy in CD
SIRT1 Agonists
Resveratrol and SRT1720 are the most studied SIRT1 agonists in scientific literature. Resveratrol (3,5,4′-trihydroxy-trans-stilbene) is a natural phytoallexin found in grapes’ skin, berries, peanuts, and roots of rhubarb, and other plants. Resveratrol protects the plants from fungal infection, and it was first described in 1940s. In 2003, a drug screen for small molecule activators of SIRT1 identified fifteen SIRT1 activators, and resveratrol was the most potent one. The anti-aging properties of resveratrol were evidenced by an increase in the life span of yeast, nematodes, fish, and flies etc. (Valenzano et al., 2006; Agarwal and Baur, 2011). Others documented that resveratrol is broadly beneficial in inhibiting the growth of cancer cells in culture (Saunier et al., 2017), and in preventing or slowing down the neurodegenerative disorders, cardiac involvement, and cancer in rodents (Hung et al., 2000; Ignatowicz and Baer-Dubowska, 2001; Shigematsu et al., 2003). Moreover, resveratrol treatment of mice improved the running time and increased oxygen consumption in the skeletal muscle, which was associated with activation of genes for oxidative phosphorylation and mitochondrial biogenesis (Guo et al., 2014). The authors attributed these benefits to activation of SIRT1 and PGC-1α and found that resveratrol treatment prevented the diet-induced-obesity and insulin resistance in mice. Cumulatively, many studies have shown that resveratrol offers a multitude of benefits including increase in mitochondrial content and antioxidant capacity, reduced inflammation, improved metabolic and vascular function (Thirunavukkarasu et al., 2007; Barger et al., 2008; Bereswill et al., 2010; Guo et al., 2014).
SRT1720 was first identified in a high-throughput in vitro fluorescence polarization assay which was used for screening SIRT1 agonists (Milne et al., 2007). SRT1720 is ~1,000-fold more potent than resveratrol in activating SIRT1 activity. SRT1720 exerts its effects by binding to SIRT1–substrate complex and potentiate deacetylation of SIRT1 target proteins (Milne et al., 2007; Feige et al., 2008). C2C12 cells treated with SRT1720 exhibited increased expression of citrate synthase activity and ATP levels indicating the improvement of mitochondrial biogenesis (Smith et al., 2009). In vivo studies in obese mice and rats showed that SRT1720 treatment improved the insulin sensitivity, regulated plasma glucose levels, and enhanced the mitochondrial oxidative metabolism in various tissues, including skeletal muscle, liver, and brown adipose tissue (Minor et al., 2011; Durkacz et al., 1980; D’Amours et al., 1999). We demonstrated that SIRT1 activity was decreased in mice infected with T. cruzi, and treatment with SRT1720 during the indeterminate phase, i.e., after the immune control of circulating, blood parasitemia and before the onset of chronic disease phase, preserved the cardiac structure and function in CD mice (Wan et al., 2016).
Some studies have noted that resveratrol and SRT1720 exhibit off-target activities against many enzymes, ion channels, receptors, and transporters and do not directly influence SIRT1-mediated deacetylation on target peptides (Pacholec et al., 2010). Nevertheless, because of its small molecular size and potent activity, SRT1720 is actively being studied for the treatment of metabolic and chronic diseases.
SIRT1 Improves Mitochondrial Biogenesis
One of the primary targets of SIRT1 deacetylation is PGC-1α. PGC-1α undergoes post-translational modifications including acetylation and phosphorylation, and it serves as master regulator of the mitochondrial biogenesis and oxidative metabolism. Studies have shown that deacetylation of PGC-1α is dependent on SIRT1 activity (Sims et al., 1981; Gerhart-Hines et al., 2007; Amat et al., 2009; Gurd, 2011; Guo et al., 2014). Mutation of the acetylation sites, which sealed PGC-1α in deacetylated state, markedly enhanced its basal activity (Sims et al., 1981). It was suggested that SIRT1 may physically and functionally interact with PGC-1α through ADP-ribosyl transferase domain of SIRT1, and mutations in this domain prevent SIRT1’s interactions with PGC-1α (Gurd, 2011). SIRT1-activated PGC-1α rapidly translocates to the nucleus where it co-activates the NRF1/NRF2 and TFAM to regulate the expression of genes encoding key components of respiration, mtDNA transcription, and mtDNA replication machineries. SIRT1 also acts as a sensor of changes in nutrient and energy metabolism. SIRT1 was activated by fasting, and SIRT1 and PGC-1α interaction enhanced the expression of hepatic gluconeogenic genes (Sims et al., 1981). SIRT1 deacetylation of PGC-1α in skeletal muscle was required for signaling the expression of enzymes involved in mitochondrial fatty acid oxidation (Gerhart-Hines et al., 2007).
Poly(ADP-ribose) polymerase (PARP) family members are recognized for their function in maintaining DNA and RNA metabolism. Among the 17 known members of the PARP family, PARP1 is most active enzymatically. PARP1 is activated upon sensing the oxidized DNA (Beck et al., 2014), and it utilizes NAD as a substrate to catalyze the formation of poly(ADP-ribose) (PAR) chains on itself and other proteins (D’Amours et al., 1999; Hassa et al., 2006; Messner et al., 2010). SIRT1 and PARP1 compete for NAD+ availability; crosstalk between these proteins is inevitable (Luna et al., 2013). PARP1 knockout mice exhibited an increase in mitochondrial content and energy expenditure and were protected against metabolic disease (Hassa et al., 2006) confirming that PARP1 influences SIRT1 activity in maintaining mitochondrial homeostasis. In mice chronically infected with T. cruzi, myocardial PARP1 expression was increased, and it compromised the polymerase gamma activity resulting in a decline in mtDNA content, mtDNA-encoded gene expression, and oxidative phosphorylation capacity (Wan et al., 2016; Wen et al., 2018). Consequently, an increase in mitochondrial ROS and oxidative stress was noted in Chagas myocardium. Genetic deletion of PARP1 or treatment with PARP1 inhibitor or SIRT1 agonist improved the mitochondrial biogenesis, oxidative phosphorylation, and left ventricular function in chronic CD (Wan et al., 2016; Wen et al., 2018). These studies indicate the SIRT1–PARP1 imbalance is a major contributor to myocardial mitochondrial stress in CD.
SIRT1 Mitigates Oxidative Stress
Oxidative stress is characterized by increased intracellular levels of ROS and decreased antioxidant capacity (Johnson and Giulivi, 2005). Oxidative stress plays a key role in the pathophysiology of many diseases, including atherosclerosis, diabetic mellitus, and myocardial dysfunction of infectious and non-infectious etiologies (Deng et al., 2008).
SIRT1 has been demonstrated to control ROS levels by its regulatory effects on mitochondrial electron transport chain (discussed above). SIRT1 also inhibited NF-κB transcriptional activation and reduced the expression of gp91phox and p22phox encoding for NADPH oxidase subunits, and thus prevented the production of reactive oxygen and nitrogen radicals by phagocytes (Xie et al., 1994; Sakitani et al., 1998; Anrather et al., 2006; Manea et al., 2007; Manea et al., 2010). Quercetin enhanced the SIRT1 expression, increased AMP-activated protein kinase (AMPK) activity, and decreased NADPH production, thus providing protective effects against the hyperglycemia-induced oxidant damage in HUVEC cells (Hung et al., 2015). Conversely, SIRT1 inhibition led to increase in NOX2 activity and ROS production and contributed to endothelial dysfunction (Wosniak et al., 2009; Zarzuelo et al., 2013).
At a molecular level, SIRT1 influences antioxidant status through its effects on transcription factors. FoXO (Forkhead box) family of transcription factors consists of FoxO1, FoxO3, FoxO4, and FoxO6 (Kaestner et al., 2000). FoXO proteins regulate many cellular processes, including stress resistance, energy metabolism, cell cycle, and cell death. In a number of studies, SIRT1 is shown to deacetylate FoxO1, FoxO3a, and FoxO4, and activate FoXO-regulated antioxidants in mitochondria (e.g., MnSOD, peroxiredoxins 3 and 5), peroxisomes (catalase), and plasma (e.g., selenoprotein P, ceruloplasmin) (Zhang and Tarleton, 1999; Brunet et al., 2004; van der Horst et al., 2004; Sengupta et al., 2011; Xiong et al., 2011; Yamamoto and Sadoshima, 2011). SIRT1-mediated deacetylation of FoXO1 and FoXO4 enhanced their DNA-binding ability to induce antioxidants’ gene expression (Daitoku et al., 2004; van der Horst et al., 2004). FoxO1 depletion by siRNA inhibited SIRT1 expression in vascular smooth muscle cells and HEK293 cells suggesting that FoxO1 provides positive feedback signal for SIRT1 activity (Xiong et al., 2011).
Nuclear Factor, Erythroid 2 Like 2 (NFE2L2, also referred as NRF2 in literature) bind to the antioxidant response element (ARE) sequences in promoters of the antioxidant genes and plays an important role in activation of cellular antioxidant defense. It has been reported that melatonin signals SIRT1-dependent transcriptional activation of NRF2 to exert anti-oxidative effects in the developing rat brain and BV2 cells, and SIRT1 inhibitor significantly decreased the SIRT1 and NRF2 expression in BV2 cells (Shah et al., 2017). SIRT1 was also demonstrated to enhance the activation of the NFE2L2 (NRF2)/ARE antioxidant pathway and inhibit the apoptosis of type II alveolar epithelial cells (Ding et al., 2016). We noted that T. cruzi induced ROS had a negative effect on the expression, nuclear translocation as well as ARE binding and transcriptional activity of NFE2L2 in cardiomyocytes (Wen et al., 2017). Subsequently, expression of several of the NFE2L2-regulated antioxidants, including gamma-glutamyl cysteine synthase, heme oxygenase-1, glutamate-cysteine ligase, thioredoxin, glutathione S transferase, and NADPH dehydrogenase quinone 1 was decreased in cardiomyocytes and myocardium of mice infected with T. cruzi (Wen et al., 2017). Though we did not examine the effects of SIRT1 agonist on NFE2L2 in this study; however, preserving the NFE2L2 activity by other means established the antioxidant/oxidant balance and preserved the mitochondrial and cardiac health in CD mice (Wen et al., 2017).
SIRT1 Controls Inflammation
The NF-κB transcription factor family consists of five proteins including p65 (RelA), RelB, c-Rel, p105/p50 (NF-κB1), and p100/52 (NF-κB2). NF-κB transcription factors regulate the expression of a large number of genes involved in inflammation, cell proliferation, cell differentiation, and cell survival. In the absence of stimuli, NF-κB remains in an inactive form in the cytoplasm through association with inhibitory IκB proteins. IκB kinase (IKK)-mediated IκB phosphorylation leads to its degradation and released NF-κB translocates to nucleus to carry out transcription of target genes (Nakajima and Kitamura, 2013). SIRT1 deacetylates the p65/RelA at Lys310 residue and inhibits NF-κB activity (Yeung et al., 2004). SIRT1 can also inhibit NF-κB by activating AMPK and peroxisome proliferator-activated receptor α, which inactivate the NF-κB pathway and prevent ROS generation. Resveratrol treatment enhanced the SIRT1 activity, and repressed NF-κB and ROS generation in HUVEC cells treated with TNF-α (Pan et al., 2016). NF-κB can also suppress SIRT1 activity (Kauppinen et al., 2013), thus suggesting that a feedback regulation of SIRT1 and NF-κB determines the inflammatory and oxidative status in a cell.
Acute inflammation occurs over seconds to days to eliminate the cause of tissue injury (e.g., microbes) and reinstates the immune homeostasis. Innate immune cells including monocytes/macrophages, granulocytes, and dendritic cells express toll-like receptors (TLRs), nod like receptors (NLRs), rig-I-like receptors (RLRs), and other receptors that sense pathogen-associated molecular patterns (PAMPs) and signal transcription activation of cytokines and chemokines (Dalpke and Heeg, 2002). Further, TLR sensing of PAMPs rapidly stimulates the expression and transcriptional activation of Hypoxia Inducible Factor 1 Subunit Alpha (HIF-1α), which switches mitochondrial glucose oxidation to glycolysis and increase in glucose level for supporting the expression of proinflammatory genes (Koo and Garg, 2019). Transcription regulation of the switching from the proinflammatory to anti-inflammatory and immuno-regulatory state requires SIRT1-dependent deacetylation of NF-κB and histone proteins (e.g., H1K26, H4K16, H3K9, and H3K14) and recruitment of new methyltransferases (Liu and McCall, 2013). SIRT1 also regulates acute inflammatory process by mediating a metabolic switch (Krawczyk et al., 2010; Michalek et al., 2011; Liu et al., 2012) through activating PGC-1α that increases the flux of fatty acids and transfer of fatty acids into mitochondria and supports fatty acid oxidation as an energy source for mitochondrial function (Liu and McCall, 2013).
Chronic inflammation is caused by persistent low-level of pathogenic or non-pathogenic stimuli. A decline in SIRT1 activity is noted in many chronic inflammatory diseases. Examples include fat deposits in obesity (Schug and Li, 2010), lungs in chronic obstructive pulmonary disease (COPD) (Rajendrasozhan et al., 2008), brain in Alzheimer’s disease (Qin et al., 2006), arteries in atherosclerosis (Gorenne et al., 2013), and skin in aging (Massudi et al., 2012). SIRT1 inhibition likely contributes to chronic inflammation due to hosts’ inability to shut down NF-κB-dependent gene expression of proinflammatory cytokines (Serrano-Marco et al., 2012). Yet, the mechanisms of SIRT1 decline during chronic inflammation are not very clear. Enhancing NAD+ levels (Imai and Guarente, 2014) or activating SIRT1 by resveratrol suppressed chronic inflammation and helped establish metabolic homeostasis (Haigis and Sinclair, 2010). It is suggested that cellular bioenergetics and SIRT1 coordinate to provide a regulation axis (positive and negative) that controls host’s inflammatory response to pathogens as well as maintain cellular metabolic homeostasis (Liu et al., 2011).
We showed that yolk-sac-derived CD11b+ F4/80+ monocytes/macrophages were increased in spleen and heart tissue of CD mice, and these cells displayed surface markers of inflammatory phenotype (CD80+/CD64+ > CD200+/CD206+) as well as inflammatory functional response, evidenced by increase in the cytokines’ expression (IL-6 + TNF-α >> Arg-1 + IL-10) (Wan et al., 2019). When treated with SRT1720, splenic expansion and myocardial infiltration of proinflammatory monocytes/macrophages were controlled in CD mice. SRT1720 did not alter the inherent capability of macrophages to respond to T. cruzi infection. Instead, SRT1720 treatment diminished the T. cruzi-induced expression and/or phosphorylation of focal adhesion kinase (FAK) non-receptor kinase and the downstream transcription factors (Pu.1, c-Myb, and Runx1) that are known to participate in macrophage proliferation and migration and Notch1 that is involved in macrophage’s functional activation. Studies in cultured macrophages showed that SIRT1 agonist or FAK inhibitor abrogated the NF-κB transcriptional activity and inflammatory cytokine gene expression in macrophages infected with T. cruzi, and thus provided further evidence of agonistic effects of SIRT1 on FAK signaling of transcription factors involved in macrophage functional activation (Wan et al., 2019).
Conclusive Remarks
Inflammatory and oxidative stress and mitochondrial dysfunction are recognized of pathological importance in chronic Chagas cardiomyopathy. We have shown that SIRT1 activity is compromised in the myocardium during CD progression, and treatment of infected mice with SRT1720 for 3-weeks in indeterminate phase ameliorated the left ventricular dysfunction. The benefits of SRT1720 were associated with control of chronic oxidative stress and proinflammatory differentiation of macrophages in spleen of chronically infected mice (Figure 4). Similar benefits in improving the cardiac outcomes in infected mice were obtained by inhibiting PARP1 that competes with SIRT1 for the NAD+ substrate. Whether PARP1–SIRT1 imbalance occurs in human Chagas disease and whether PARP1 inhibitors or SIRT1 agonists will be useful in improving the cardiac outcomes in human Chagas disease remain to be investigated in future studies.
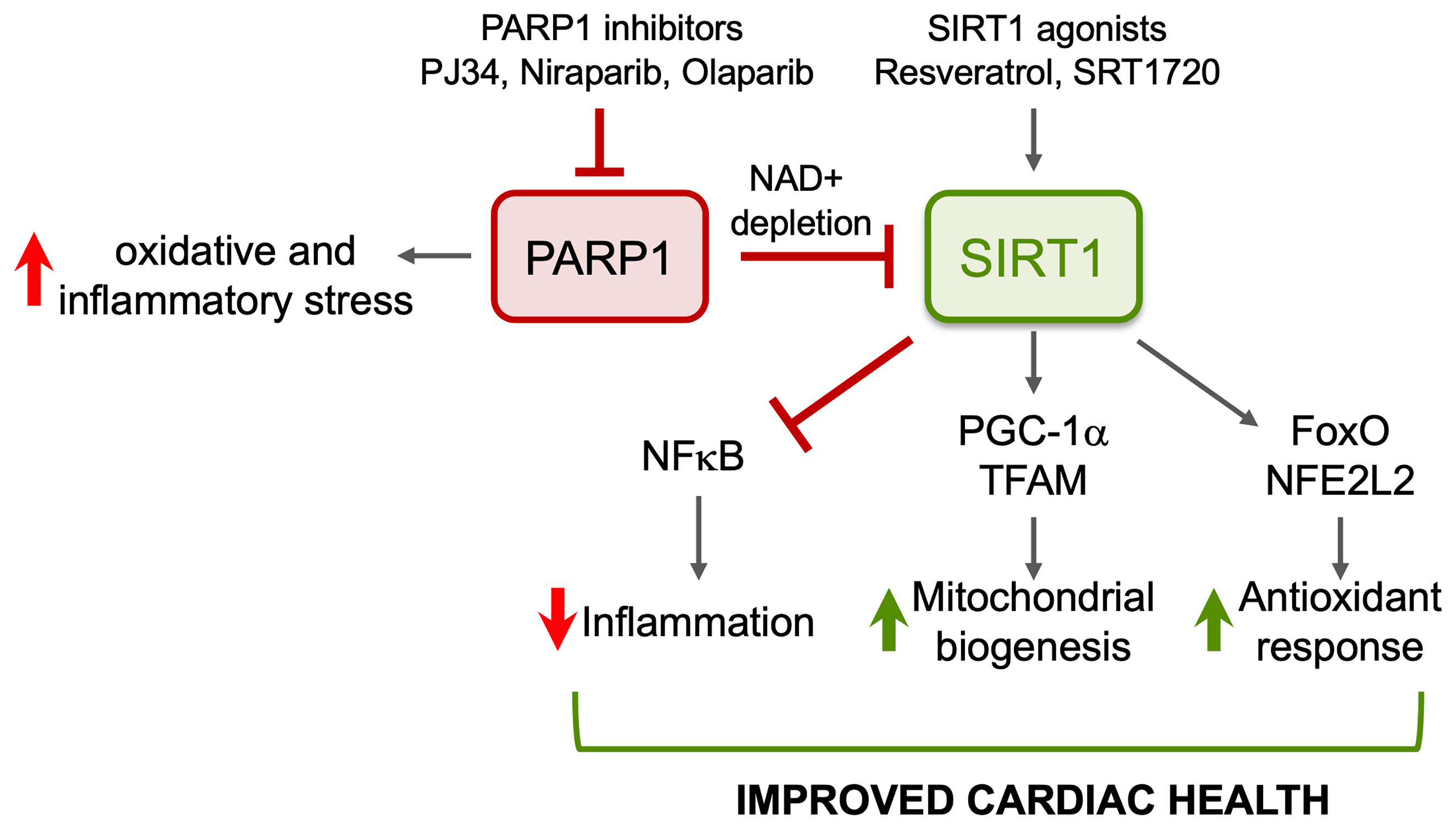
Figure 4 Potential benefits of SIRT1 agonists in CD. ROS induced oxidative adducts cause PARP1 hyperactivation that suppresses mitochondrial DNA replication and transcription and signals inflammation. PARP1 suppresses SIRT1 activity in Chagasic heart through depletion of NAD+ pool. Pharmacological inhibitors of PARP1 or activators of SIRT1 will enhance SIRT1-mediated deacetylation and will have cytoprotective effects in Chagas disease through inhibition of oxidative stress and inflammation and induction of antioxidant response and mitochondrial biogenesis.
The mechanism(s) connecting SIRT1 to tissue inflammation/oxidative stress have been enigmatic. The bone marrow and splenic myeloid progenitor cells give rise to proinflammatory macrophages in Chagas heart. However, how SIRT1 deficiency and SIRT1 agonists regulate the proliferation and differentiation of monocytes of different lineages into macrophages with diverse phenotypic and functional activation remains to be studied. Likewise, how the crosstalk or substrate allocation between SIRT1 and PARP1 is regulated is not known, and further studies will be needed to understand their precise role in human health and disease, especially as related to Chagas disease.
Author Contributions
XW wrote the first draft. XW and NG edited and formatted the later drafts. All authors contributed to the article and approved the submitted version.
Funding
NJG has been supported by grants from the National Institute of Allergy and Infectious Diseases (R01AI136031; R01AI054578) and National Heart Lung and Blood Institute (R01HL094802) of the National Institutes of Health. XW was supported by American Heart Association pre-doctoral fellowship and Kempner post-doctoral fellowship. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Abbreviations
CD, Chagas disease; mtDNA, mitochondrial DNA; PGC-1α, peroxisome proliferator–activated receptor gamma coactivator 1; RNS, reactive nitrogen species; ROS, reactive oxygen species; SIRT1, sirtuin 1.
References
Agarwal, B., Baur, J. A. (2011). Resveratrol and Life Extension. Ann. N. Y. Acad. Sci. 1215, 138–143. doi: 10.1111/j.1749-6632.2010.05850.x
Ahuja, N., Schwer, B., Carobbio, S., Waltregny, D., North, B. J., Castronovo, V., et al. (2007). Regulation of Insulin Secretion by SIRT4, A Mitochondrial ADP-Ribosyltransferase. J. Biol. Chem. 282, 33583–33592. doi: 10.1074/jbc.M705488200
Altenhofer, S., Kleikers, P. W., Radermacher, K. A., Scheurer, P., Rob Hermans, J. J., Schiffers, P., et al. (2012). The NOX Toolbox: Validating the Role of NADPH Oxidases in Physiology and Disease. Cell. Mol. Life Sci. C. M. L. S. 69, 2327–2343. doi: 10.1007/s00018-012-1010-9
Alvarez, M. N., Peluffo, G., Piacenza, L., Radi, R. (2011). Intraphagosomal Peroxynitrite as a Macrophage-Derived Cytotoxin Against Internalized Trypanosoma Cruzi: Consequences for Oxidative Killing and Role of Microbial Peroxiredoxins in Infectivity. J. Biol. Chem. 286, 6627–6640. doi: 10.1074/jbc.M110.167247
Amat, R., Planavila, A., Chen, S. L., Iglesias, R., Giralt, M., Villarroya, F. (2009). SIRT1 Controls the Transcription of the Peroxisome Proliferator-Activated Receptor-Gamma Co-activator-1alpha (PGE-1alpha) Gene in Skeletal Muscle Through the PGC-1alpha Autoregulatory Loop and Interaction With Myod. J. Biol. Chem. 284, 21872–21880. doi: 10.1074/jbc.M109.022749
Anrather, J., Racchumi, G., Iadecola, C. (2006). NF-Kappab Regulates Phagocytic NADPH Oxidase by Inducing the Expression of Gp91phox. J. Biol. Chem. 281, 5657–5667. doi: 10.1074/jbc.M506172200
Barger, J. L., Kayo, T., Pugh, T. D., Prolla, T. A., Weindruch, R. (2008). Short-Term Consumption of a Resveratrol-Containing Nutraceutical Mixture Mimics Gene Expression of Long-Term Caloric Restriction in Mouse Heart. Exp. Gerontol 43, 859–866. doi: 10.1016/j.exger.2008.06.013
Beck, C., Robert, I., Reina-San-Martin, B., Schreiber, V., Dantzer, F. (2014). Poly(ADP-Ribose) Polymerases in Double-Strand Break Repair: Focus on PARP1, PARP2 and PARP3. Exp. Cell Res. 329, 18–25. doi: 10.1016/j.yexcr.2014.07.003
Bedard, K., Krause, K. H. (2007). The NOX Family of ROS-generating NADPH Oxidases: Physiology and Pathophysiology. Physiol. Rev. 87, 245–313. doi: 10.1152/physrev.00044.2005
Bereswill, S., Munoz, M., Fischer, A., Plickert, R., Haag, L. M., Otto, B., et al. (2010). Anti-Inflammatory Effects of Resveratrol, Curcumin and Simvastatin in Acute Small Intestinal Inflammation. PloS One 5, e15099. doi: 10.1371/journal.pone.0015099
Boutant, M., Canto, C. (2014). SIRT1 Metabolic Actions: Integrating Recent Advances From Mouse Models. Mol. Metab. 3, 5–18. doi: 10.1016/j.molmet.2013.10.006
Brunet, A., Sweeney, L. B., Sturgill, J. F., Chua, K. F., Greer, P. L., Lin, Y., et al. (2004). Stress-Dependent Regulation of FOXO Transcription Factors by the SIRT1 Deacetylase. Science 303, 2011–2015. doi: 10.1126/science.1094637
Budni, P., Pedrosa, R. C., Dalmarco, E. M., Dalmarco, J. B., Frode, T. S., Wilhelm Filho, D. (2013). Carvedilol Enhances the Antioxidant Effect of Vitamins E and C in Chronic Chagas Heart Disease. Arq. Bras. Cardiol. 101, 304–310. doi: 10.5935/abc.20130184
Chang, H. C., Guarente, L. (2014). SIRT1 and Other Sirtuins in Metabolism. Trends Endocrinol. Metabol: TEM 25, 138–145. doi: 10.1016/j.tem.2013.12.001
Choudhuri, S., Garg, N. J. (2020). Parp1-cGAS-NF-kappaB Pathway of Proinflammatory Macrophage Activation by Extracellular Vesicles Released During Trypanosoma Cruzi Infection and Chagas Disease. PloS Pathog. 16, e1008474. doi: 10.1371/journal.ppat.1008474
Daitoku, H., Hatta, M., Matsuzaki, H., Aratani, S., Ohshima, T., Miyagishi, M., et al. (2004). Silent Information Regulator 2 Potentiates Foxo1-Mediated Transcription Through Its Deacetylase Activity. Proc. Natl. Acad. Sci. U. S. A. 101, 10042–10047. doi: 10.1073/pnas.0400593101
Dalpke, A., Heeg, K. (2002). Signal Integration Following Toll-Like Receptor Triggering. Crit. Rev. Immunol. 22, 217–250. doi: 10.1615/CritRevImmunol.v22.i3.40
D’Amours, D., Desnoyers, S., D’Silva, I., Poirier, G. G. (1999). Poly(ADP-Ribosyl)Ation Reactions in the Regulation of Nuclear Functions. Biochem. J. 342 ( Pt 2), 249–268. doi: 10.1073/pnas.0400593101
de Carvalho, T. U., de Souza, W. (1987). Cytochemical Localization of NADH and NADPH Oxidases During Interaction of Trypanosoma Cruzi With Activated Macrophages. Parasitol. Res. 73, 213–217. doi: 10.1007/BF00578506
Deng, J., Wang, G., Huang, Q., Yan, Y., Li, K., Tan, W., et al. (2008). Oxidative Stress-Induced Leaky Sarcoplasmic Reticulum Underlying Acute Heart Failure in Severe Burn Trauma. Free Radical Biol. Med. 44, 375–385. doi: 10.1016/j.freeradbiomed.2007.09.023
Dhiman, M., Coronado, Y. A., Vallejo, C. K., Petersen, J. R., Ejilemele, A., Nunez, S., et al. (2013). Innate Immune Responses and Antioxidant/Oxidant Imbalance are Major Determinants of Human Chagas Disease. PloS NTD 7, e2364. doi: 10.1371/journal.pntd.0002364
Dhiman, M., Estrada-Franco, J. G., Pando, J., Ramirez-Aguilar, F., Spratt, H., Vasquez-Corzo, S., et al. (2009). Increased Myeloperoxidase Activity and Protein Nitration Are Indicators of Inflammation in Chagasic Patients. Clin. Vaccine Immunol. 16, 660–666. doi: 10.1128/CVI.00019-09
Dhiman, M., Nakayasu, E. S., Madaiah, Y. H., Reynolds, B. K., Wen, J. J., Almeida, I. C., et al. (2008). Enhanced Nitrosative Stress During Trypanosoma Cruzi Infection Causes Nitrotyrosine Modification of Host Proteins: Implications in Chagas’ Disease. Am. J. Pathol. 173, 728–740. doi: 10.2353/ajpath.2008.080047
Dhiman, M., Wan X-X, P. L. V., Vargas, G., Garg, N. J. (2013). MnSODtg Mice Control Myocardial Inflammatory and Oxidative Stress and Remodeling Responses Elicited in Chronic Chagas Disease. J. Am. Heart Assoc. 2, e000302. doi: 10.1161/JAHA.113.000302
Dias, J. C., Ramos, A. N., Jr., Gontijo, E. D., Luquetti, A., Shikanai-Yasuda, M. A., Coura, J. R., et al. (2016). 2 Nd Brazilian Consensus on Chagas Disease, 2015. Rev. Soc. Bras. Med. Trop. 49 (Suppl 1), 3–60. doi: 10.1590/0037-8682-0505-2016
Ding, Y. W., Zhao, G. J., Li, X. L., Hong, G. L., Li, M. F., Qiu, Q. M., et al. (2016). SIRT1 Exerts Protective Effects Against Paraquat-Induced Injury in Mouse Type II Alveolar Epithelial Cells by Deacetylating NRF2 In Vitro. Int. J. Mol. Med. 37, 1049–1058. doi: 10.3892/ijmm.2016.2503
Dryden, S. C., Nahhas, F. A., Nowak, J. E., Goustin, A. S., Tainsky, M. A. (2003). Role for Human SIRT2 NAD-Dependent Deacetylase Activity in Control of Mitotic Exit in the Cell Cycle. Mol. Cell. Biol. 23, 3173–3185. doi: 10.1128/MCB.23.9.3173-3185.2003
Durkacz, B. W., Omidiji, O., Gray, D. A., Shall, S. (1980). (ADP-Ribose)N Participates in DNA Excision Repair. Nature 283, 593–596. doi: 10.1038/283593a0
Feige, J. N., Lagouge, M., Canto, C., Strehle, A., Houten, S. M., Milne, J. C., et al. (2008). Specific SIRT1 Activation Mimics Low Energy Levels and Protects Against Diet-Induced Metabolic Disorders by Enhancing Fat Oxidation. Cell Metab. 8, 347–358. doi: 10.1016/j.cmet.2008.08.017
Finck, B. N., Kelly, D. P. (2006). PGC-1 Coactivators: Inducible Regulators of Energy Metabolism in Health and Disease. J. Clin. Invest. 116, 615–622. doi: 10.1172/JCI27794
Ford, E., Voit, R., Liszt, G., Magin, C., Grummt, I., Guarente, L. (2006). Mammalian Sir2 Homolog SIRT7 Is an Activator of RNA Polymerase I Transcription. Genes Dev. 20, 1075–1080. doi: 10.1101/gad.1399706
Frye, R. A. (1999). Characterization of Five Human cDNAs With Homology to the Yeast SIR2 Gene: Sir2-like Proteins (Sirtuins) Metabolize NAD and May Have Protein ADP-ribosyltransferase Activity. Biochem. Biophys. Res. Commun. 260, 273–279. doi: 10.1006/bbrc.1999.0897
Frye, R. A. (2000). Phylogenetic Classification of Prokaryotic and Eukaryotic Sir2-like Proteins. Biochem. Biophys. Res. Commun. 273, 793–798. doi: 10.1006/bbrc.2000.3000
Gao, Z., Ye, J. (2008). Inhibition of Transcriptional Activity of c-JUN by SIRT1. Biochem. Biophys. Res. Commun. 376, 793–796. doi: 10.1016/j.bbrc.2008.09.079
Gerhart-Hines, Z., Rodgers, J. T., Bare, O., Lerin, C., Kim, S. H., Mostoslavsky, R., et al. (2007). Metabolic Control of Muscle Mitochondrial Function and Fatty Acid Oxidation Through SIRT1/PGC-1alpha. EMBO J. 26, 1913–1923. doi: 10.1038/sj.emboj.7601633
Gorenne, I., Kumar, S., Gray, K., Figg, N., Yu, H., Mercer, J., et al. (2013). Vascular Smooth Muscle Cell Sirtuin 1 Protects Against DNA Damage and Inhibits Atherosclerosis. Circulation 127, 386–396. doi: 10.1161/CIRCULATIONAHA.112.124404
Guo, P., Pi, H., Xu, S., Zhang, L., Li, Y., Li, M., et al. (2014). Melatonin Improves Mitochondrial Function by Promoting MT1/SIRT1/PGC-1 Alpha-Dependent Mitochondrial Biogenesis in Cadmium-Induced Hepatotoxicity In Vitro. Toxicol. Sci. Off. J. Soc. Toxicol. 142, 182–195. doi: 10.1093/toxsci/kfu164
Gupta, S., Bhatia, V., Wen, J.-J., Wu, Y., Huang, M.-H., Garg, N. J. (2009). Trypanosoma Cruzi Infection Disturbs Mitochondrial Membrane Potential and ROS Production Rate in Cardiomyocytes. Free Radic. Biol. Med. 47, 1414–1421. doi: 10.1016/j.freeradbiomed.2009.08.008
Gurd, B. J. (2011). Deacetylation of PGC-1 Alpha by SIRT1: Importance for Skeletal Muscle Function and Exercise-Induced Mitochondrial Biogenesis. Appl. Physiol. Nutr. Me 36, 589–597. doi: 10.1139/h11-070
Gureev, A. P., Shaforostova, E. A., Popov, V. N. (2019). Regulation of Mitochondrial Biogenesis as a Way for Active Longevity: Interaction Between the Nrf2 and PGC-1alpha Signaling Pathways. Front. Genet. 10, 435. doi: 10.3389/fgene.2019.00435
Gustafsson, A. B., Gottlieb, R. A. (2008). Heart Mitochondria: Gates of Life and Death. Cardiovasc. Res. 77, 334–343. doi: 10.1093/cvr/cvm005
Haigis, M. C., Sinclair, D. A. (2010). Mammalian Sirtuins: Biological Insights and Disease Relevance. Annu. Rev. Pathol. 5, 253–295. doi: 10.1146/annurev.pathol.4.110807.092250
Hassa, P. O., Haenni, S. S., Elser, M., Hottiger, M. O. (2006). Nuclear ADP-Ribosylation Reactions in Mammalian Cells: Where Are We Today and Where Are We Going? Microbiol. Mol. Biol. Rev. MMBR 70, 789–829. doi: 10.1128/MMBR.00040-05
Herskovits, A. Z., Guarente, L. (2014). SIRT1 in Neurodevelopment and Brain Senescence. Neuron 81, 471–483. doi: 10.1016/j.neuron.2014.01.028
Hou, J., Chong, Z. Z., Shang, Y. C., Maiese, K. (2010). Early Apoptotic Vascular Signaling Is Determined by Sirt1 Through Nuclear Shuttling, Forkhead Trafficking, Bad, and Mitochondrial Caspase Activation. Curr. Neurovascular Res. 7, 95–112. doi: 10.2174/156720210791184899
Hubbard, B. P., Sinclair, D. A. (2014). Small Molecule SIRT1 Activators for the Treatment of Aging and Age-Related Diseases. Trends Pharmacol. Sci. 35, 146–154. doi: 10.1016/j.tips.2013.12.004
Hung, C. H., Chan, S. H., Chu, P. M., Tsai, K. L. (2015). Quercetin is a Potent Anti-Atherosclerotic Compound by Activation of SIRT1 Signaling Under oxLDL Stimulation. Mol. Nutr. Food Res. 59, 1905–1917. doi: 10.1002/mnfr.201500144
Hung, L. M., Chen, J. K., Huang, S. S., Lee, R. S., Su, M. J. (2000). Cardioprotective Effect of Resveratrol, A Natural Antioxidant Derived From Grapes. Cardiovasc. Res. 47, 549–555. doi: 10.1016/S0008-6363(00)00102-4
Ignatowicz, E., Baer-Dubowska, W. (2001). Resveratrol, A Natural Chemopreventive Agent Against Degenerative Diseases. Pol. J. Pharmacol. 53, 557–569.
Imai, S., Guarente, L. (2014). NAD+ and Sirtuins in Aging and Disease. Trends Cell Biol. 24, 464–471. doi: 10.1016/j.tcb.2014.04.002
Johnson, F., Giulivi, C. (2005). Superoxide Dismutases and Their Impact Upon Human Health. Mol. Aspects Med. 26, 340–352. doi: 10.1016/j.mam.2005.07.006
Kaestner, K. H., Knochel, W., Martinez, D. E. (2000). Unified Nomenclature for the Winged Helix/Forkhead Transcription Factors. Genes Dev. 14, 142–146. doi: 10.1101/gad.14.2.142
Kauppinen, A., Suuronen, T., Ojala, J., Kaarniranta, K., Salminen, A. (2013). Antagonistic Crosstalk Between NF-kappaB and SIRT1 in the Regulation of Inflammation and Metabolic Disorders. Cell. Signalling 25, 1939–1948. doi: 10.1016/j.cellsig.2013.06.007
Koo, S. J., Chowdhury, I. H., Szczesny, B., Wan, X., Garg, N. J. (2016). Macrophages Promote Oxidative Metabolism to Drive Nitric Oxide Generation in Response to Trypanosoma Cruzi. Infect. Immun. 84, 3527–3541. doi: 10.1128/IAI.00809-16
Koo, S. J., Garg, N. J. (2019). Metabolic Programming of Macrophage Functions and Pathogens Control. Redox. Biol. 24, 101198. doi: 10.1016/j.redox.2019.101198
Krawczyk, C. M., Holowka, T., Sun, J., Blagih, J., Amiel, E., DeBerardinis, R. J., et al. (2010). Toll-Like Receptor-Induced Changes in Glycolytic Metabolism Regulate Dendritic Cell Activation. Blood 115, 4742–4749. doi: 10.1182/blood-2009-10-249540
Landry, J., Sutton, A., Tafrov, S. T., Heller, R. C., Stebbins, J., Pillus, L., et al. (2000). The Silencing Protein SIR2 and its Homologs Are NAD-Dependent Protein Deacetylases. Proc. Natl. Acad. Sci. U. S. A. 97, 5807–5811. doi: 10.1073/pnas.110148297
Liszt, G., Ford, E., Kurtev, M., Guarente, L. (2005). Mouse Sir2 Homolog SIRT6 is a Nuclear ADP-Ribosyltransferase. J. Biol. Chem. 280, 21313–21320. doi: 10.1074/jbc.M413296200
Liu, T. F., McCall, C. E. (2013). Deacetylation by SIRT1 Reprograms Inflammation and Cancer. Genes Cancer 4, 135–147. doi: 10.1177/1947601913476948
Liu, T. F., Vachharajani, V. T., Yoza, B. K., McCall, C. E. (2012). NAD+ -Dependent Sirtuin 1 and 6 Proteins Coordinate a Switch From Glucose to Fatty Acid Oxidation During the Acute Inflammatory Response. J. Biol. Chem. 287, 25758–25769. doi: 10.1074/jbc.M112.362343
Liu, T. F., Yoza, B. K., El Gazzar, M., Vachharajani, V. T., McCall, C. E. (2011). NAD+-Dependent SIRT1 Deacetylase Participates in Epigenetic Reprogramming During Endotoxin Tolerance. J. Biol. Chem. 286, 9856–9864. doi: 10.1074/jbc.M110.196790
Li, W., Zhang, B., Tang, J., Cao, Q., Wu, Y., Wu, C., et al. (2007). Sirtuin 2, a Mammalian Homolog of Yeast Silent Information Regulator-2 Longevity Regulator, Is an Oligodendroglial Protein That Decelerates Cell Differentiation Through Deacetylating Alpha-Tubulin. J. Neurosci. Off. J. Soc. Neurosci. 27, 2606–2616. doi: 10.1523/JNEUROSCI.4181-06.2007
Luna, A., Aladjem, M. I., Kohn, K. W. (2013). SIRT1/PARP1 Crosstalk: Connecting DNA Damage and Metabolism. Genome Integr. 4, 6. doi: 10.1186/2041-9414-4-6
Manea, A., Manea, S. A., Gafencu, A. V., Raicu, M. (2007). Regulation of NADPH Oxidase Subunit p22(phox) by NF-kB in Human Aortic Smooth Muscle Cells. Arch. Physiol. Biochem. 113, 163–172. doi: 10.1080/13813450701531235
Manea, A., Tanase, L. I., Raicu, M., Simionescu, M. (2010). Transcriptional Regulation of NADPH Oxidase Isoforms, Nox1 and Nox4, by Nuclear Factor-Kappab in Human Aortic Smooth Muscle Cells. Biochem. Biophys. Res. Commun. 396, 901–907. doi: 10.1016/j.bbrc.2010.05.019
Massudi, H., Grant, R., Braidy, N., Guest, J., Farnsworth, B., Guillemin, G. J. (2012). Age-Associated Changes in Oxidative Stress and NAD+ Metabolism in Human Tissue. PloS One 7, e42357. doi: 10.1371/journal.pone.0042357
Matutino Bastos, T., Botelho Pereira Soares, M., Haddad Franco, C., Alcantara, L., Antonini, L., Sabatino, M., et al. (2020). Identification of Inhibitors to Trypanosoma Cruzi Sirtuins Based on Compounds Developed to Human Enzymes. Int. J. Mol. Sci. 21, 3659–3675. doi: 10.3390/ijms21103659
Messner, S., Altmeyer, M., Zhao, H., Pozivil, A., Roschitzki, B., Gehrig, P., et al. (2010). Parp1 ADP-ribosylates Lysine Residues of the Core Histone Tails. Nucleic Acids Res. 38, 6350–6362. doi: 10.1093/nar/gkq463
Michalek, R. D., Gerriets, V. A., Jacobs, S. R., Macintyre, A. N., MacIver, N. J., Mason, E. F., et al. (2011). Cutting Edge: Distinct Glycolytic and Lipid Oxidative Metabolic Programs Are Essential for Effector and Regulatory CD4+ T Cell Subsets. J. Immunol. 186, 3299–3303. doi: 10.4049/jimmunol.1003613
Michishita, E., McCord, R. A., Berber, E., Kioi, M., Padilla-Nash, H., Damian, M., et al. (2008). SIRT6 Is a Histone H3 Lysine 9 Deacetylase That Modulates Telomeric Chromatin. Nature 452, 492–496. doi: 10.1038/nature06736
Michishita, E., Park, J. Y., Burneskis, J. M., Barrett, J. C., Horikawa, I. (2005). Evolutionarily Conserved and Nonconserved Cellular Localizations and Functions of Human SIRT Proteins. Mol. Biol. Cell 16, 4623–4635. doi: 10.1091/mbc.e05-01-0033
Milne, J. C., Lambert, P. D., Schenk, S., Carney, D. P., Smith, J. J., Gagne, D. J., et al. (2007). Small Molecule Activators of SIRT1 as Therapeutics for the Treatment of Type 2 Diabetes. Nature 450, 712–716. doi: 10.1038/nature06261
Minor, R. K., Baur, J. A., Gomes, A. P., Ward, T. M., Csiszar, A., Mercken, E. M., et al. (2011). SRT1720 Improves Survival and Healthspan of Obese Mice. Sci. Rep. 1, 70. doi: 10.1038/srep00070
Moretti, N. S., da Silva Augusto, L., Clemente, T. M., Antunes, R. P., Yoshida, N., Torrecilhas, A. C., et al. (2015). Characterization of Trypanosoma Cruzi Sirtuins as Possible Drug Targets for Chagas Disease. Antimicrob. Agents Chemother. 59, 4669–4679. doi: 10.1128/AAC.04694-14
Mostoslavsky, R., Chua, K. F., Lombard, D. B., Pang, W. W., Fischer, M. R., Gellon, L., et al. (2006). Genomic Instability and Aging-Like Phenotype in the Absence of Mammalian SIRT6. Cell 124, 315–329. doi: 10.1016/j.cell.2005.11.044
Nakajima, S., Kitamura, M. (2013). Bidirectional Regulation of NF-kappaB by Reactive Oxygen Species: A Role of Unfolded Protein Response. Free Radical Biol. Med. 65, 162–174. doi: 10.1016/j.freeradbiomed.2013.06.020
Nakamura, Y., Ogura, M., Tanaka, D., Inagaki, N. (2008). Localization of Mouse Mitochondrial SIRT Proteins: Shift of SIRT3 to Nucleus by Co-Expression With SIRT5. Biochem. Biophys. Res. Commun. 366, 174–179. doi: 10.1016/j.bbrc.2007.11.122
Pacholec, M., Bleasdale, J. E., Chrunyk, B., Cunningham, D., Flynn, D., Garofalo, R. S., et al. (2010). Srt1720, SRT2183, SRT1460, and Resveratrol are Not Direct Activators of SIRT1. J. Biol. Chem. 285, 8340–8351. doi: 10.1074/jbc.M109.088682
Pan, W., Yu, H., Huang, S., Zhu, P. (2016). Resveratrol Protects Against TNF-alpha-Induced Injury in Human Umbilical Endothelial Cells Through Promoting Sirtuin-1-Induced Repression of NF-KB and p38 Mapk. PloS One 11, e0147034. doi: 10.1371/journal.pone.0147034
Perez-Fuentes, R., Guegan, J. F., Barnabe, C., Lopez-Colombo, A., Salgado-Rosas, H., Torres-Rasgado, E., et al. (2003). Severity of Chronic Chagas Disease is Associated With Cytokine/Antioxidant Imbalance in Chronically Infected Individuals. Int. J. Parasitol 33, 293–299. doi: 10.1016/S0020-7519(02)00283-7
Qin, W., Yang, T., Ho, L., Zhao, Z., Wang, J., Chen, L., et al. (2006). Neuronal SIRT1 Activation as a Novel Mechanism Underlying the Prevention of Alzheimer Disease Amyloid Neuropathology by Calorie Restriction. J. Biol. Chem. 281, 21745–21754. doi: 10.1074/jbc.M602909200
Rajendrasozhan, S., Yang, S. R., Kinnula, V. L., Rahman, I. (2008). SIRT1, an Antiinflammatory and Antiaging Protein, Is Decreased in Lungs of Patients With Chronic Obstructive Pulmonary Disease. Am. J. Respir. Crit. Care Med. 177, 861–870. doi: 10.1164/rccm.200708-1269OC
Rassi, A., Jr., Marin, J. A. N., Rassi, A. (2017). Chronic Chagas Cardiomyopathy: A Review of the Main Pathogenic Mechanisms and the Efficacy of Aetiological Treatment Following the BENznidazole Evaluation for Interrupting Trypanosomiasis (BENEFIT) Trial. Mem. Inst. Oswaldo Cruz 112, 224–235. doi: 10.1590/0074-02760160334
Ribeiro, C. M., Budni, P., Pedrosa, R. C., Farias, M. S., Parisotto, E. B., Dalmarco, E. M., et al. (2010). Antioxidant Therapy Attenuates Oxidative Insult Caused by Benzonidazole in Chronic Chagas’ Heart Disease. Int. J. Cardiol. 145, 27–33. doi: 10.1016/j.ijcard.2009.06.033
Sakitani, K., Nishizawa, M., Inoue, K., Masu, Y., Okumura, T., Ito, S. (1998). Synergistic Regulation of Inducible Nitric Oxide Synthase Gene by CCAAT/enhancer-binding Protein Beta and Nuclear Factor-Kappab in Hepatocytes. Genes Cells Devoted. Mol. Cell. Mech. 3, 321–330. doi: 10.1046/j.1365-2443.1998.00193.x
Saunier, E., Antonio, S., Regazzetti, A., Auzeil, N., Laprevote, O., Shay, J. W., et al. (2017). Resveratrol Reverses the Warburg Effect by Targeting the Pyruvate Dehydrogenase Complex in Colon Cancer Cells. Sci. Rep. 7, 6945. doi: 10.1038/s41598-017-07006-0
Schug, T. T., Li, X. (2010). Surprising Sirtuin Crosstalk in the Heart. Aging 2, 129–132. doi: 10.18632/aging.100128
Sengupta, A., Molkentin, J. D., Paik, J. H., DePinho, R. A., Yutzey, K. E. (2011). Foxo Transcription Factors Promote Cardiomyocyte Survival Upon Induction of Oxidative Stress. J. Biol. Chem. 286, 7468–7478. doi: 10.1074/jbc.M110.179242
Serrano-Marco, L., Chacon, M. R., Maymo-Masip, E., Barroso, E., Salvado, L., Wabitsch, M., et al. (2012). TNF-Alpha Inhibits PPARbeta/delta Activity and SIRT1 Expression Through NF-kappaB in Human Adipocytes. Biochim. Biophys. Acta 1821, 1177–1185. doi: 10.1016/j.bbalip.2012.05.006
Shah, S. A., Khan, M., Jo, M. H., Jo, M. G., Amin, F. U., Kim, M. O. (2017). Melatonin Stimulates the SIRT1/Nrf2 Signaling Pathway Counteracting Lipopolysaccharide (LPS)-Induced Oxidative Stress to Rescue Postnatal Rat Brain. CNS Neurosci. Ther. 23, 33–44. doi: 10.1111/cns.12588
Shao, D., Liu, Y., Liu, X., Zhu, L., Cui, Y., Cui, A., et al. (2010). PGC-1 Beta-Regulated Mitochondrial Biogenesis and Function in Myotubes Is Mediated by NRF-1 and ERR Alpha. Mitochondrion 10, 516–527. doi: 10.1016/j.mito.2010.05.012
Shigematsu, S., Ishida, S., Hara, M., Takahashi, N., Yoshimatsu, H., Sakata, T., et al. (2003). Resveratrol, A Red Wine Constituent Polyphenol, Prevents Superoxide-Dependent Inflammatory Responses Induced by Ischemia/Reperfusion, Platelet-Activating Factor, or Oxidants. Free Radic. Biol. Med. 34, 810–817. doi: 10.1016/S0891-5849(02)01430-2
Sims, J. L., Berger, S. J., Berger, N. A. (1981). Effects of Nicotinamide on NAD and Poly(ADP-Ribose) Metabolism in DNA-Damaged Human Lymphocytes. J. Supramol. Structure Cell. Biochem. 16, 281–288. doi: 10.1002/jsscb.1981.380160308
Smith, J. J., Kenney, R. D., Gagne, D. J., Frushour, B. P., Ladd, W., Galonek, H. L., et al. (2009). Small Molecule Activators of SIRT1 Replicate Signaling Pathways Triggered by Calorie Restriction In Vivo. BMC Syst. Biol. 3, 31. doi: 10.1186/1752-0509-3-31
Solomon, J. M., Pasupuleti, R., Xu, L., McDonagh, T., Curtis, R., DiStefano, P. S., et al. (2006). Inhibition of SIRT1 Catalytic Activity Increases p53 Acetylation But Does Not Alter Cell Survival Following DNA Damage. Mol. Cell. Biol. 26, 28–38. doi: 10.1128/MCB.26.1.28-38.2006
Thirunavukkarasu, M., Penumathsa, S. V., Koneru, S., Juhasz, B., Zhan, L., Otani, H., et al. (2007). Resveratrol Alleviates Cardiac Dysfunction in Streptozotocin-Induced Diabetes: Role of Nitric Oxide, Thioredoxin, and Heme Oxygenase. Free Radical Biol. Med. 43, 720–729. doi: 10.1016/j.freeradbiomed.2007.05.004
Valenzano, D. R., Terzibasi, E., Genade, T., Cattaneo, A., Domenici, L., Cellerino, A. (2006). Resveratrol Prolongs Lifespan and Retards the Onset of Age-Related Markers in a Short-Lived Vertebrate. Curr. Biol. 16, 296–300. doi: 10.1016/j.cub.2005.12.038
van der Horst, A., Tertoolen, L. G., de Vries-Smits, L. M., Frye, R. A., Medema, R. H., Burgering, B. M. (2004). FOXO4 Is Acetylated Upon Peroxide Stress and Deacetylated by the Longevity Protein Hsir2(SIRT1). J. Biol. Chem. 279, 28873–28879. doi: 10.1074/jbc.M401138200
Ventura-Clapier, R., Garnier, A., Veksler, V. (2008). Transcriptional Control of Mitochondrial Biogenesis: The Central Role of PGC-1alpha. Cardiovasc. Res. 79, 208–217. doi: 10.1093/cvr/cvn098
Vyatkina, G., Bhatia, V., Gerstner, A., Papaconstantinou, J., Garg, N. J. (2004). Impaired Mitochondrial Respiratory Chain and Bioenergetics During Chagasic Cardiomyopathy Development. Biochim. Biophys. Acta 1689, 162–173. doi: 10.1016/j.bbadis.2004.03.005
Wan, X., Chowdhury, I. H., Jie, Z., Choudhuri, S., Garg, N. J. (2019). Origin of Monocytes/Macrophages Contributing to Chronic Inflammation in Chagas Disease: SIRT1 Inhibition of FAK-Nfκb-Dependent Proliferation and Proinflammatory Activation of Macrophages. Cells 9, 80. doi: 10.3390/cells9010080
Wan, X.-X., Gupta, S., Zago, M. P., Davidson, M. M., Dousset, P., Amoroso, A., et al. (2012). Defects of mtDNA Replication Impaired the Mitochondrial Biogenesis During Trypanosoma Cruzi Infection in Human Cardiomyocytes and Chagasic Patients: The Role of Nrf1/2 and Antioxidant Response. J. Am. Heart Assoc. 1, e003855. doi: 10.1161/JAHA.112.003855
Wan, X., Wen, J. J., Koo, S. J., Liang, L. Y., Garg, N. J. (2016). SIRT1-PGC1alpha-Nfkappab Pathway of Oxidative and Inflammatory Stress During Trypanosoma Cruzi Infection: Benefits of SIRT1-Targeted Therapy in Improving Heart Function in Chagas Disease. PloS Pathog. 12, e1005954. doi: 10.1371/journal.ppat.1005954
Wen, J.-J., Bhatia, V., Popov, V. L., Garg, N. J. (2006). Phenyl-Alpha-Tert-Butyl Nitrone Reverses Mitochondrial Decay in Acute Chagas Disease. Am. J. Pathol. 169, 1953–1964. doi: 10.2353/ajpath.2006.060475
Wen, J. J., Dhiman, M., Whorton, E. B., Garg, N. J. (2008). Tissue-Specific Oxidative Imbalance and Mitochondrial Dysfunction During Trypanosoma Cruzi Infection in Mice. Microbes Infect. 10, 1201–1209. doi: 10.1016/j.micinf.2008.06.013
Wen, J.-J., Garg, N. J. (2010). Mitochondrial Complex III Defects Contribute to Inefficient Respiration and ATP Synthesis in the Myocardium of Trypanosoma Cruzi-Infected Mice. Antioxid. Redox. Signal. 12, 27–37. doi: 10.1089/ars.2008.2418
Wen, J.-J., Gupta, S., Guan, Z., Dhiman, M., Condon, D., Lui, C. Y., et al. (2010). Phenyl-Alpha-Tert-Butyl-Nitrone and Benzonidazole Treatment Controlled the Mitochondrial Oxidative Stress and Evolution of Cardiomyopathy in Chronic Chagasic Rats. J. Am. Coll. Cardiol. 55, 2499–2508. doi: 10.1016/j.jacc.2010.02.030
Wen, J. J., Porter, C., Garg, N. J. (2017). Inhibition of NFE2L2-Antioxidant Response Element Pathway by Mitochondrial Reactive Oxygen Species Contributes to Development of Cardiomyopathy and Left Ventricular Dysfunction in Chagas Disease. Antioxid. Redox. Signal. 27, 550–566. doi: 10.1089/ars.2016.6831
Wen, J.-J., Vyatkina, G., Garg, N. J. (2004). Oxidative Damage During Chagasic Cardiomyopathy Development: Role of Mitochondrial Oxidant Release and Inefficient Antioxidant Defense. Free Radic. Biol. Med. 37, 1821–1833. doi: 10.1016/j.freeradbiomed.2004.08.018
Wen, J.-J., Yachelini, P. C., Sembaj, A., Manzur, R. E., Garg, N. J. (2006). Increased Oxidative Stress is Correlated With Mitochondrial Dysfunction in Chagasic Patients. Free Rad. Biol. Med. 41, 270–276. doi: 10.1016/j.freeradbiomed.2006.04.009
Wen, J. J., Yin, Y. W., Garg, N. J. (2018). PARP1 Depletion Improves Mitochondrial and Heart Function in Chagas Disease: Effects on POLG Dependent mtDNA Maintenance. PloS Pathog. 14, e1007065. doi: 10.1371/journal.ppat.1007065
World Health Organization. (2015). Chagas Disease in Latin America: An Epidemiological Update Based on 2010 Estimates. Wkly Epidemiol. Rec 90, 33–44.
Wosniak, J., Jr., Santos, C. X., Kowaltowski, A. J., Laurindo, F. R. (2009). Cross-Talk Between Mitochondria and NADPH Oxidase: Effects of Mild Mitochondrial Dysfunction on Angiotensin II-mediated Increase in Nox Isoform Expression and Activity in Vascular Smooth Muscle Cells. Antioxid. Redox Signaling 11, 1265–1278. doi: 10.1089/ars.2009.2392
Xie, Q. W., Kashiwabara, Y., Nathan, C. (1994). Role of Transcription Factor NF-kappa B/Rel in Induction of Nitric Oxide Synthase. J. Biol. Chem. 269, 4705–4708. doi: 10.1016/S0021-9258(17)37600-7
Xiong, S., Salazar, G., Patrushev, N., Alexander, R. W. (2011). FoxO1 Mediates an Autofeedback Loop Regulating SIRT1 Expression. J. Biol. Chem. 286, 5289–5299. doi: 10.1074/jbc.M110.163667
Yamamoto, T., Sadoshima, J. (2011). Protection of the Heart Against Ischemia/Reperfusion by Silent Information Regulator 1. Trends Cardiovasc. Med. 21, 27–32. doi: 10.1016/j.tcm.2012.01.005
Yeung, F., Hoberg, J. E., Ramsey, C. S., Keller, M. D., Jones, D. R., Frye, R. A., et al. (2004). Modulation of NF-kappaB-dependent Transcription and Cell Survival by the SIRT1 Deacetylase. EMBO J. 23, 2369–2380. doi: 10.1038/sj.emboj.7600244
Yuan, J., Minter-Dykhouse, K., Lou, Z. (2009). A c-Myc-SIRT1 Feedback Loop Regulates Cell Growth and Transformation. J. Cell Biol. 185, 203–211. doi: 10.1083/jcb.200809167
Yuan, Z., Zhang, X., Sengupta, N., Lane, W. S., Seto, E. (2007). SIRT1 Regulates the Function of the Nijmegen Breakage Syndrome Protein. Mol. Cell 27, 149–162. doi: 10.1016/j.molcel.2007.05.029
Zarzuelo, M. J., Lopez-Sepulveda, R., Sanchez, M., Romero, M., Gomez-Guzman, M., Ungvary, Z., et al. (2013). SIRT1 Inhibits NADPH Oxidase Activation and Protects Endothelial Function in the Rat Aorta: Implications for Vascular Aging. Biochem. Pharmacol. 85, 1288–1296. doi: 10.1016/j.bcp.2013.02.015
Zhang, L., Tarleton, R. L. (1999). Parasite Persistence Correlates With Disease Severity and Localization in Chronic Chagas’ Disease. J. Infect. Dis. 180, 480–486. doi: 10.1086/314889
Keywords: peroxisome proliferator-activated receptor gamma coactivator 1, reactive oxygen species, sirtuin, Chagas disease, mitochondrial dysfunction
Citation: Wan X and Garg NJ (2021) Sirtuin Control of Mitochondrial Dysfunction, Oxidative Stress, and Inflammation in Chagas Disease Models. Front. Cell. Infect. Microbiol. 11:693051. doi: 10.3389/fcimb.2021.693051
Received: 09 April 2021; Accepted: 21 May 2021;
Published: 09 June 2021.
Edited by:
Mario Alberto Rodriguez, Instituto Politécnico Nacional de México (CINVESTAV), MexicoReviewed by:
Juan Diego Maya, University of Chile, ChileAna Rosa Pérez, Consejo Nacional de Investigaciones Científicas y Técnicas (CONICET) Argentina
Andres Sanchez Alberti, Consejo Nacional de Investigaciones Científicas y Técnicas (CONICET), Argentina
Copyright © 2021 Wan and Garg. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Nisha Jain Garg, bmlnYXJnQHV0bWIuZWR1