 Jéssica Alves de Cena
Jéssica Alves de Cena Jianying Zhang
Jianying Zhang Dongmei Deng
Dongmei Deng Nailê Damé-Teixeira
Nailê Damé-Teixeira Thuy Do
Thuy Do
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Cell. Infect. Microbiol., 31 May 2021
Sec. Microbiome in Health and Disease
Volume 11 - 2021 | https://doi.org/10.3389/fcimb.2021.689197
This article is part of the Research TopicApplications of Next Generation Sequencing (NGS) Technologies to Decipher the Oral Microbiome in Systemic Health and DiseaseView all 18 articles
Research on the human microbiome has mainly been restricted to the identification of most abundant microbiota associated with health or disease. Their abundance may reflect their capacity to exploit their niche, however, metabolic functions exerted by low-abundant microrganisms can impact the dysbiotic signature of local microbial habitats. This scoping review aims to map the literature regarding the management of low-abundant microorganisms in studies investigating human microbiome samples. A systematic literature search was performed in 5 electronic databases, as well as grey literature. We selected clinical microbiome studies targeting human participants of any age, from any body site. We also included studies with secondary data which originated from human biofilm samples. All of the papers used next-generation sequencing (NGS) techniques in their methodology. A total of 826 manuscripts were retrieved, of which 42 were included in this review and 22 reported low-abundant bacteria (LB) in samples taken from 7 body sites (breast, gut, oral cavity, skin, stomach, upper respiratory tract (URT), and vagina). Four studies reported microbes at abundance levels between 5 and 20%, 8 studies reported between 1 and 5%, and 18 studies reported below 1%. Fifteen papers mentioned fungi and/or archaea, and from those only 4 (fungi) and 2 (archaea) produced data regarding the abundance of these domains. While most studies were directed towards describing the taxonomy, diversity and abundance of the highly abundant species, low-abundant species have largely been overlooked. Indeed, most studies select a cut-off value at <1% for low-abundant organisms to be excluded in their analyses. This practice may compromise the true diversity and influence of all members of the human microbiota. Despite their low abundance and signature in biofilms, they may generate important markers contributing to dysbiosis, in a sort of ‘butterfly effect’. A detailed snapshot of the physiological, biological mechanisms at play, including virulence determinants in the context of a dysbiotic community, may help better understand the health-disease transition.
Advances in high-throughput sequencing approaches have revolutionised microbiology and enabled the characterization of the complex ecological contents of microbial communities, however, our understanding of the mechanisms impacting host-microbial homeostasis remains limited (Hajishengallis et al., 2012). Changes to the human gut microbial composition, for example, can influence host health and diseases, and may affect the microbiota at other body sites (Banerjee et al., 2018). A concept of pathogenicity influenced by both microorganisms and the host has been proposed in the damage-response framework (Casadevall and Pirofski, 2003).
Research on the human microbiome has mainly been restricted to comparisons of the most abundant organisms and the identification of a “core” microbiota associated with health or disease. Indeed, the core microbiome may reflect their capacity to exploit their niche, being favoured by nutrients, O2 concentrations, etc. to allow surface colonisation. However, opportunistic pathogens may contribute to the compositional and or functional shift towards dysbiosis and could be among the minority taxa. Key species could therefore easily be overlooked in next generation sequencing (NGS) analyses (Turnbaugh et al., 2007; Zerón, 2014).
Furthermore, studies using a 16S rRNA metagenomic approach are limited to the identification of bacteria and archaeae (arguably accurately to the genus level), leaving the view of the richness and diversity of the whole microbiome incomplete and underestimated (Brooks et al., 2015). This is certainly true for Methanobrevibacter smithii, a member of the Archaea domain in a relatively minor constituent of the gut microbiome that contributes to bacterial metabolism in ways that promote host dysbiosis (Hajishengallis et al., 2012). This species and its methanogenic relatives, though in low abundance, have been demonstrated to be capable of providing conditions for the growth of pathogenic bacteria in periodontal sites, driving to periodontitis (Lepp et al., 2004). The composition of the microbial communities can be misinterpreted regarding the presence of virus, archaea, and fungi, making it a challenge to gain a holistic view.
Subsequently, low-abundant microrganisms could be considered the “dark matter” of the human microbiome. Recent studies (Hajishengallis and Lamont, 2016; Wang et al., 2017; Banerjee et al., 2018; Stobernack, 2019; Berg et al., 2020; Xiao et al., 2020) are paying more attention to these organisms, and increasingly taking into account the “keystone species” concept, corresponding to organisms which effect on the community is disproportionately large compared to their relative abundance (Power et al., 1996). A similar concept in macroecology suggests species in low abundance have a major role in their respective community (Hajishengallis et al., 2012). Abundance is the factor differentiating keystone microorganisms from those that are dominant. A dominant species might affect the environment exclusively by its sheer abundance, while a keystone microorganism may influence metabolic functions of the microbiome, despite its low abundance. Examples of keystone pathogens are: Porphyromonas gingivalis associated with periodontitis (Holt and Ebersole, 2005; Perez-Chaparro et al., 2014; Burmistrz et al., 2015; Camelo-Castillo et al., 2015; Ai et al., 2017; Stobernack, 2019), Klebsiella pneumonia, Proteus mirabilis (Garrett et al., 2010), and Citrobacter rodentium (Bry et al., 2006) associated with intestinal inflammatory diseases; and Fusobacterium nucleatum (Kostic et al., 2013; Rubinstein et al., 2013) associated with colon cancer (Banerjee et al., 2018). Furthermore, studies investigating Bacteroides fragilis, a pro-oncogenic bacterium, have found it to be a minor constituent of the colon microbiota in terms of relative abundance. Its unique virulence characteristics, such as secretion of a zinc-dependent metalloprotease toxin, alter colonic epithelial cells and mucosal immune function to promote oncogenic mucosal events, in which in addition to the intraluminal environment, enhance the oncogenic process. This gave rise to the concept of “alpha-bugs”, due to its ability to be directly pro-oncogenic but also to be capable of remodeling the entire healthy microbiota (Sears and Pardoll, 2011; Hajishengallis et al., 2012). Thus, the identification of low-abundant organisms within a microbial population associated with disease could be crucial. Unless we have a more “complete” view of the microbiota, including an accurate detection of low-abundant species, our understanding of the microbiology remains limited, as well as our strategy to improve therapy designs/interventions in diseases with polymicrobial cause.
Studies of the minority microrganisms may reveal unique signatures, which could lead to diseases. Hence, a much deeper characterization of their presence in the microbiome in which they are involved is desirable. This scoping review aims to map the literature regarding the management of low-abundant organisms in studies investigating human samples. We aimed to determine: 1) How researchers classify organisms as low-abundant; 2) How they handled and processed NGS data of low-abundant organisms bioinformatically and 3) The distibution of low-abundant microorganisms among various body sites.
This is a scoping review to map the literature on low-abundant organisms in the human microbiome, conducted using the PRISMA Extension for Scoping Reviews (PRISMA-ScR) checklist (Tricco et al., 2018).
Systematic literature wide opened search was performed in electronic databases, also including the grey literature (Figure 1). General controlled vocabulary (MeSH Terms) and keywords were used and the searches had no language, year, or publication type restriction. The main terms included “microbiota”, “microbiome”, “human microbiota”, “low abundant”, “minority species”, “keystone”. The search strategy and the results retrieved in each electronic database are shown in Appendix 1. Duplicated references were removed by the reference manager EndNoteWeb (Clarivate Analytics, Mumbai) and then manually.
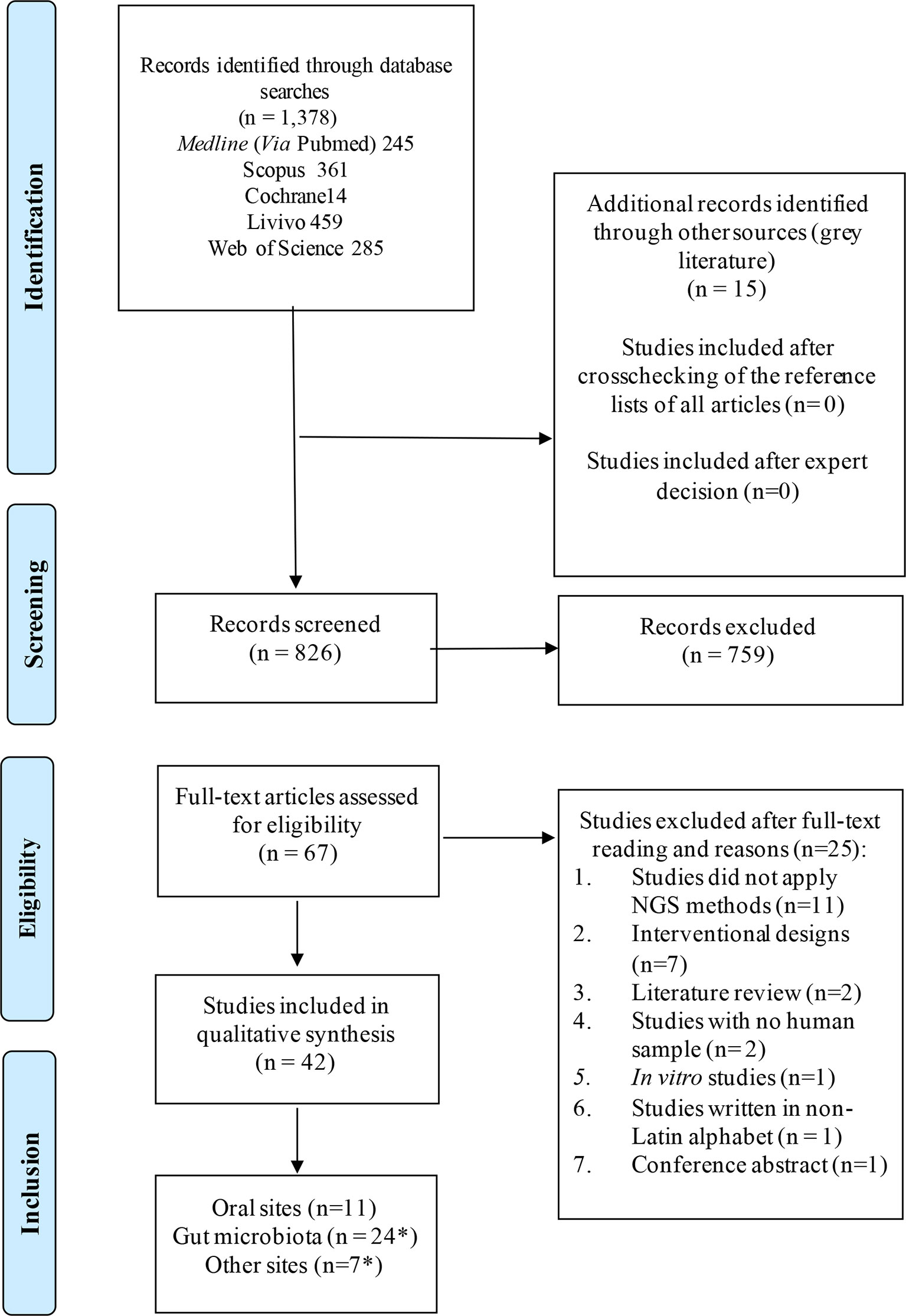
Figure 1 Flow diagram for study selection according to PRISMA guidelines. *Some studies sampled multiple sites in one study.
Studies were included if they satisfied all the following criteria: (1) clinical studies where the target population consisted of humans of any age who were donors of samples from any site; (2) the study design was either a observational study, case series, or any other type of clinical study or studies with secondary data originated from humans; and (3) studies with any term related to low-abundant organisms (e.g. keystones, minority species) in title or abstract.
Studies were excluded if: 1) Studies did not apply next-generation sequencing (NGS) methods to evaluate the microbiota; 2) They were designed as intervention studies; 3) They were literature review, conference abstracts, in vitro or animal studies, or any other kind of study carried out without human samples in a primary or secondary analysis; and 4) They were written in a non-Latin alphabet.
Two reviewers, JAC and JYZ, independently screened the eligibility of all identified titles and abstracts for inclusion in the full-text review at the Rayyan QCRI® (Qatar Computer Research Institute, Qatar). Any conflict that arose were resolved by a third reviewer. The same reviewers evaluated full-text articles for inclusion using the same inclusion and exclusion criteria. The list of selected articles was analysed to identify manuscripts that could have been lost during searches in the electronic database.
Data extraction was performed by the two reviewers independently, and included the following information: Author (year), country, design of the study, range of age of patients, sampling site, type of sample, the platform of sequencing; method of sequencing (16S rRNA or metagenomics or metranscriptomics), method of data analysis and bioinformatics; and abundance of species considered as low-abundant/minority microrganisms. All extracted data was checked by a third reviewer.
The systematic literature search resulted in 826 manuscripts of which 67 were considered for full-text review after removing duplicates and applying the eligibility criteria. Following full text reading, 42 studies remained (Figure 1; Table 1). Figure 2 shows the distribution of the papers by sampling site. Within them, the gastrointestinal tract and the oral cavity were the most studied ones. It may be due to the higher number of dysbiosis-related diseases or higher bacterial diversity in those sites, since only 10 out of the 42 articles exclusively analyzed samples from healthy individuals, and another 2 did not describe the status of health or disease, as they involved analysis of secondary data. The other sites included the vagina, respiratory system, skin, and blood. According to Hamady et al. (Hamady and Knight, 2009), the majority of microbiome studies describe the use of 16S rRNA gene sequencing for archaea and bacteria, and 18S rRNA gene sequencing for eukaryotes, which have limitations for the accurate identification to the species level.
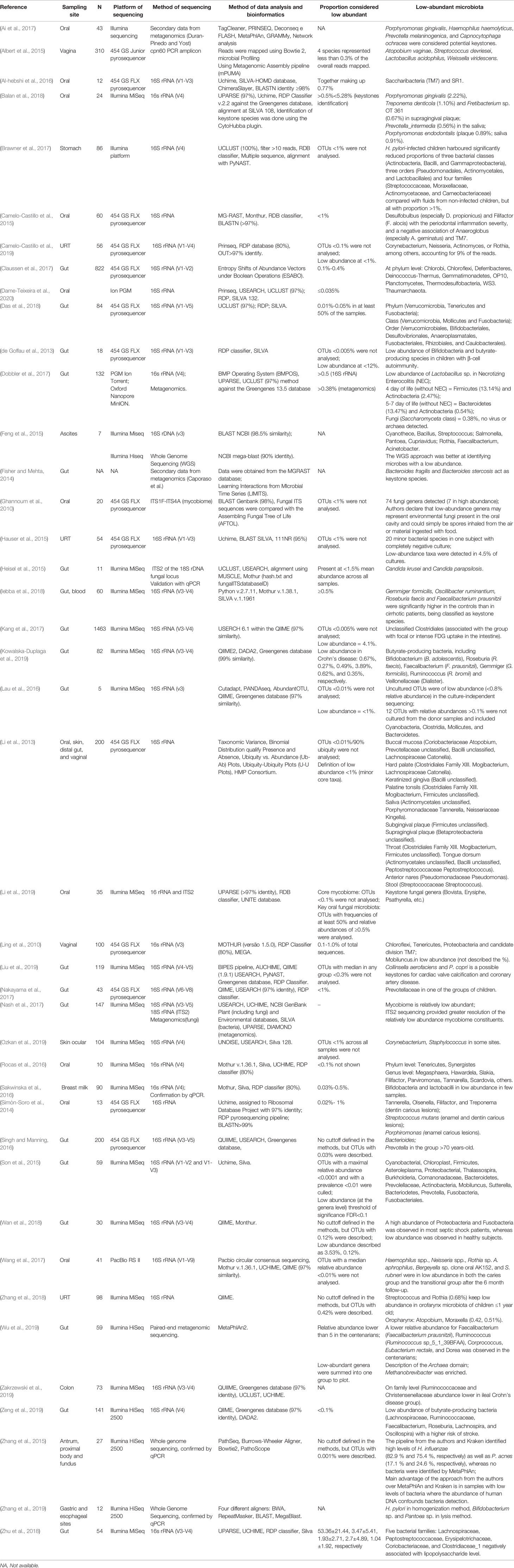
Table 1 Qualitative Data Synthesis of the Included Studies (n = 42).
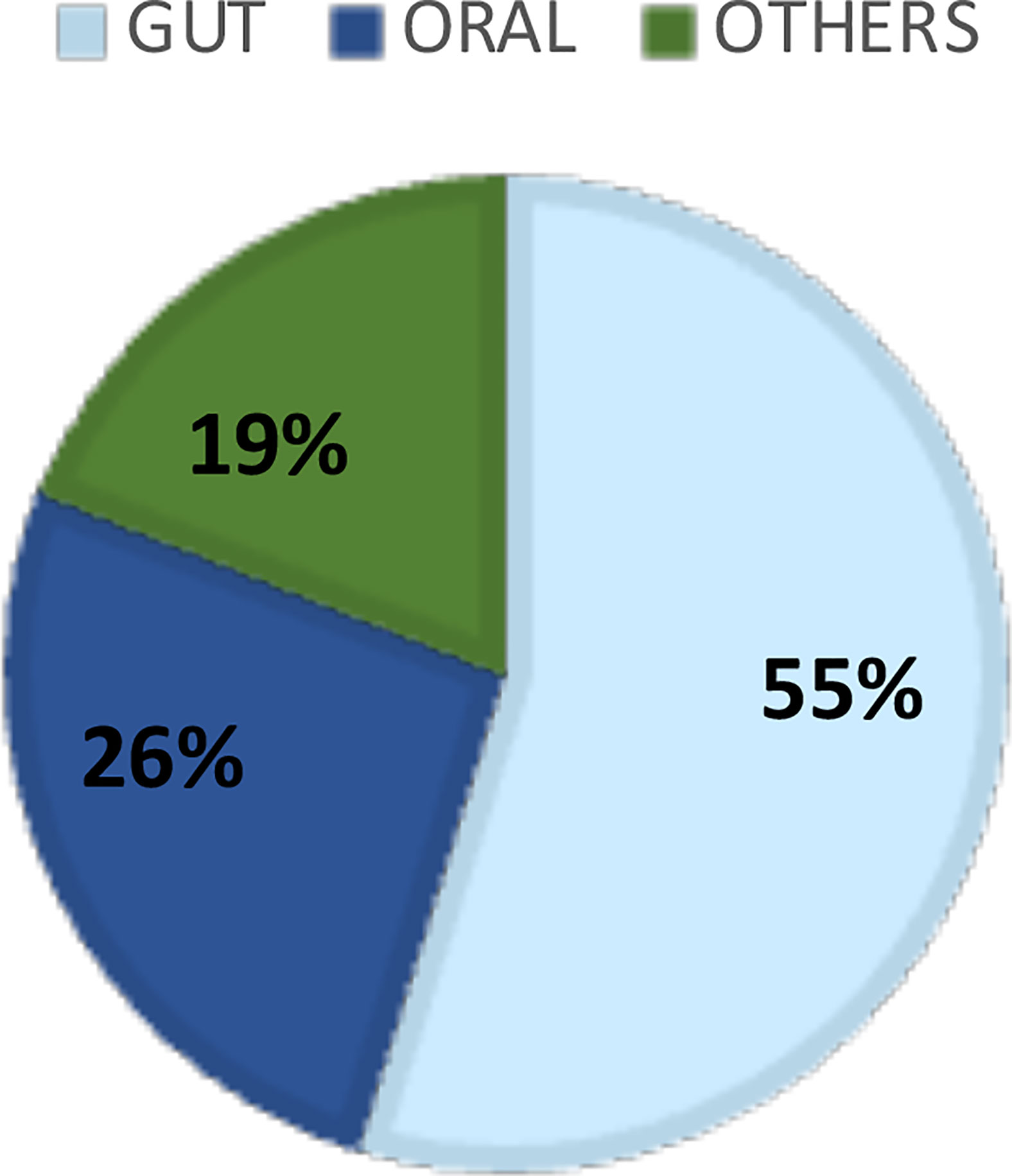
Figure 2 Distribution of the literature papers of low abundant organisms by sample’s sites.
Figure 3 shows the distribution of sequencing platforms used in the 42 selected articles. The most routinely used sequencing platforms were Illumina, followed by 454/Roche. Although these platforms are different in terms of biochemistry and in the way the matrix is generated, their workflows are conceptually similar (Shendure and Ji, 2008). A study of gut, mouth and skin samples from two subjects found that the composition of the gut and oral communities were not significantly dissimilar when either 454/Roche or Illumina (Figure 3) were used, albeit the communities of the skin were significantly different. This difference was attributed to bias associated with the primers (Caporaso et al., 2011).
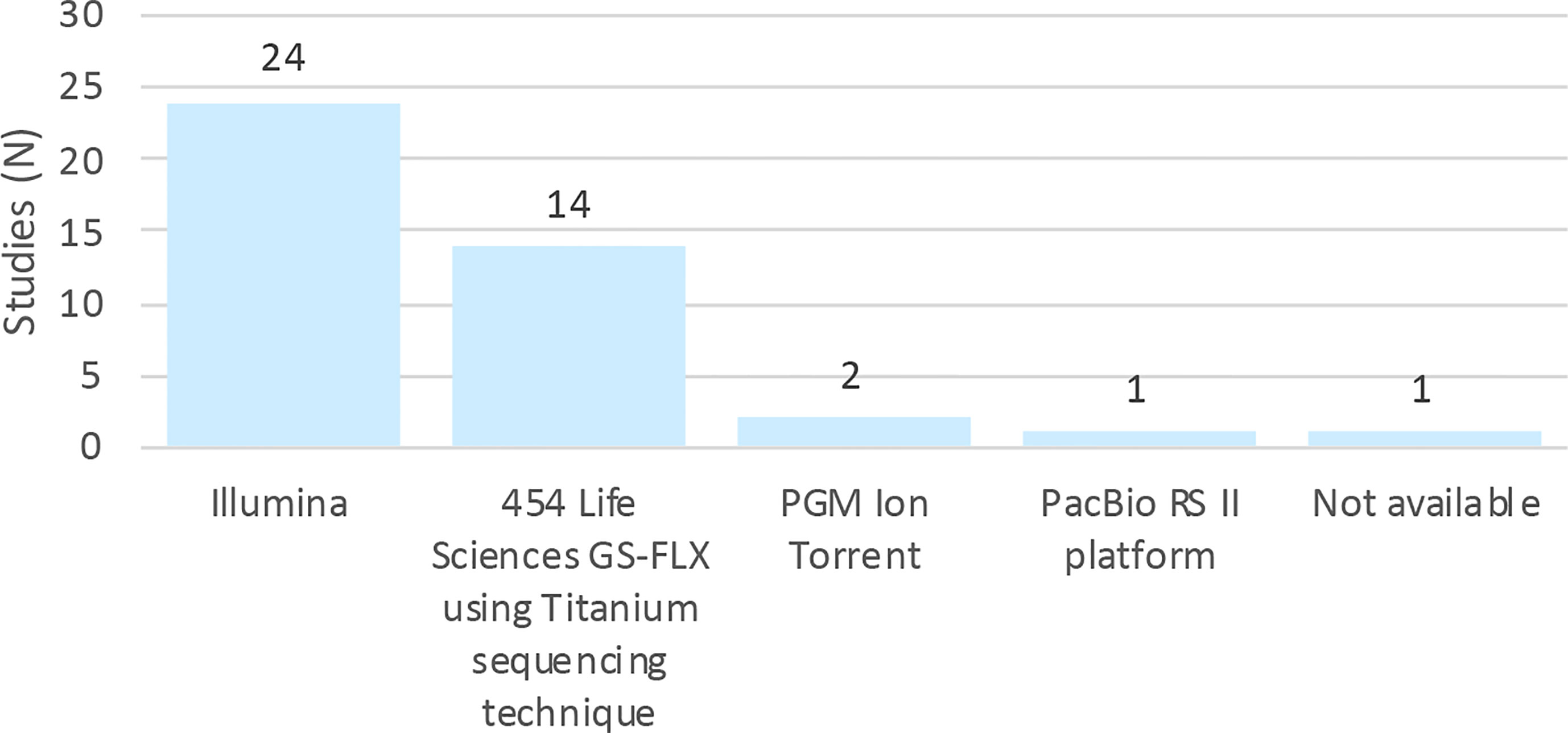
Figure 3 Distribution of studies by platforms of sequencing.
Out of 42 articles, 20 were excluded from the summary of sample site-related low abundant bacterial species, because the data on microbial abundance were unavailable or no information on low abundance rate was provided. In the remaining 22 studies, low-abundant bacteria (LB) have been reported in the biofilm samples taken from 7 body sites (breast, gut, oral cavity, skin, stomach, upper respiratory tract (URT), and vagina). LB were determined and displayed as the relative abundance of a given operational taxonomic unit (OTU), relative to the total sequencing reads. In total, 4 studies reported LB at abundance levels between 5 and 20%, 6 studies reported between 1 and 5%, and 16 studies reported below 1%. Here we summarized the information of those LB detected at abundance levels below 1%. The information on bacterial phyla can be extracted from all 22 studies, hence it is possible to summarize the major phyla of LB per sample site.
Table 2 summarizes how frequent a phylum was reported as LB (<1%) per site in the 22 studies. The frequency is indicated by the number of studies which have reported LB. In total, 6 different phyla have been reported as LB in more than 2 different studies or in more than 2 different body sites. Gut and oral cavity are the most examined body sites. Out of 6 different phyla, 5 phyla were reported in gut and 6 were reported in oral cavity. Actinobacteria and Firmicutes were the most frequently reported LB among various body sites. Actinobacteria has been found as LB in 6 different body sites. Firmicutes and Proteobacteria were found as LB in 5 different body sites. Compared to the gut, the oral cavity contains a site-specific LB phyla, Spirochaetes.
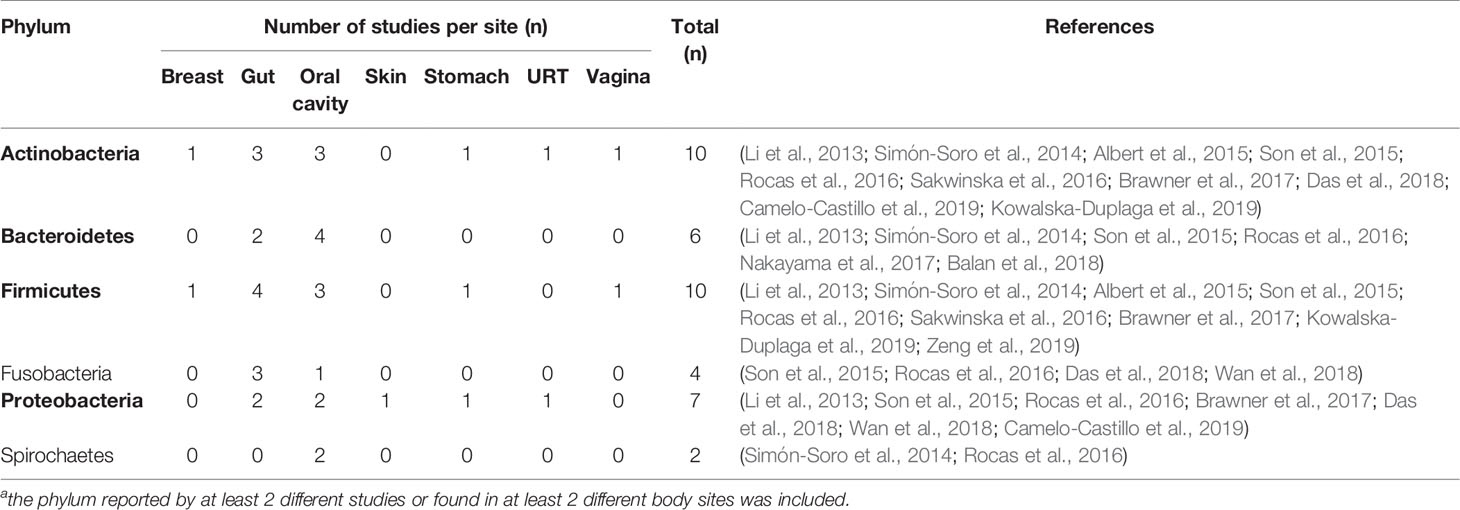
Table 2 Number of studiesa reported low abundant taxa (relative abundance <1%) at the level of phylum.
Table 3 shows the bacterial taxa at the genus level within the major LB phyla (Actinobacteria, Bacteroidetes, Firmicutes and Proteobacteria) (<1% abundance). The oral cavity and gut were the most studied body sites, where a low-abundant genus was detected in more than two studies. The reported LB at the genus level in gut was generally different from those of the oral cavity. Only 3 LB genera have been found in both gut and oral cavity, namely, Bifidobacterium, Prevotella and Streptococcus. No LB genus can be reliably identified either in the gut or the oral cavity, since the listed genera were only reported by 1 or 2 studies, which may infer on the diversity of the LB in the human body, or could be biased by sequencing/analysis methods employed.
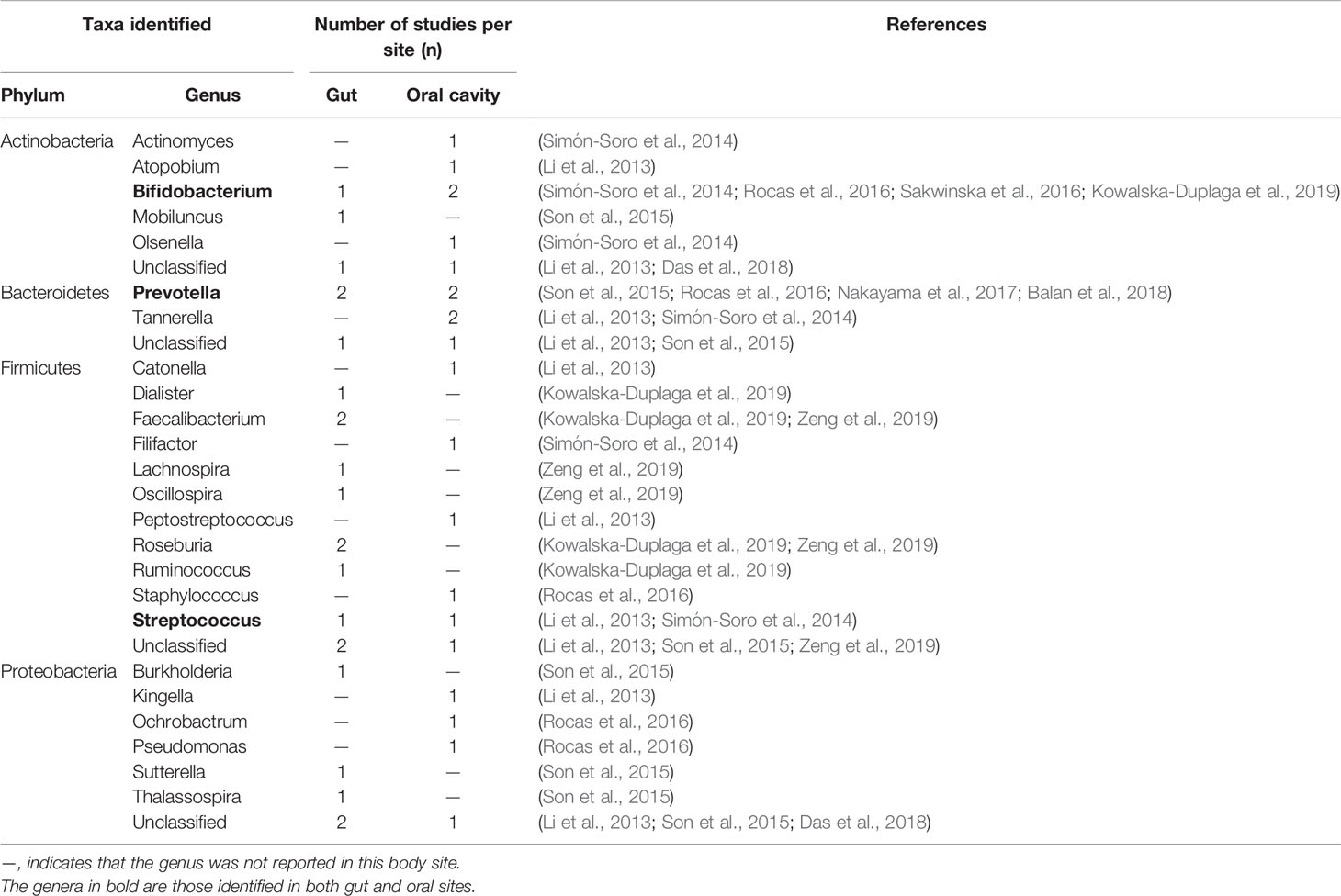
Table 3 Number of studies which reported low abundant taxa (relative abundance <1%) collected from gut and oral cavity.
Actinobacteria were most often reported as a low-abundant phylum among all body sites. In the gut, Actinobacteria are relatively scarce, but have a high degree of ecological connection and are positively correlated with the diversity of the intestinal microbiome, playing an important role in the biodegradation of complex starch. It may be involved in the prevention of dysbiosis in patients with inflammatory bowel disease (Trosvik and de Muinck, 2015). When very abundant, Actinobacteria are associated with obesity (White et al., 2009). In the oral cavity, members of this phylum are part of the healthy microbiota and their abundance varies at each oral sites, however in dental plaque, for example, their abundance is less than 1% (Peterson et al., 2013; Palmer, 2014).
Archaea and fungi (eukaryotes) are usually reported in low abundance, however, this detection should be viewed with caution and further studies are always encouraged to validate and confirm the data. From the 42 selected articles, only 15 mentioned fungi and/or archaea, and from those only 4 (fungi) and 2 (archaea) showed data regarding the abundance of these domains. Ghannoum et al. (2010) described that low-abundance genera may be transient, and represent environmental fungi present in the oral cavity and could simply be spores inhaled from the air or material ingested with food (Ghannoum et al., 2010). They have shown several species not described before in the oral cavity. Heisel et al. showed Candida krusei and Candida parapsilosis in >1.5% mean abundance in all analysed faecal samples (Heisel et al., 2015). Wu et al., 2019, using shotgun metagenomics, identified methanogenic archaea within the core microbiota, enriched in individuals aged >100 years old (Wu et al., 2019). This technique may therefore be preferrable to 16S rRNA to identify this domain of microrganisms.
The low abundance related to these domains in other studies may be linked to the sample collection method, detection probe, pair of primers used, sequencing technique, and low number of sequences registered in current databases (Ghannoum et al., 2010; Heisel et al., 2015; Dame-Teixeira et al., 2020). Furthermore, the study of the microbial community through the use of 16S rRNA sequencing and shotgun metagenomic methods allows analysis of the composition and genetic capabilities of the microbiota, but not the particularities of the role of low abundance in the microbial community, and of microbial community interactions (Centanni et al., 2018). Microbial communities are complex and constantly changing in response to their environment, influenced by various factors such as diet, use of antibiotics, exposure to transient microorganisms. In this case, other OMICS techniques can be used to understand how microbes react to the environment, including metatranscriptomics, proteomics and metabolomics. Those approaches give a holistic view of the sample content, and a clearer idea of inter-domain interactions within the human microbiome.
Since 1977, DNA-sequencing technology has evolved at a fast pace, and is reshaping our understanding of biology (Srivastava, 2011). Next generation sequencing (NGS) was introduced for the first time in 2005, extending the previous advantages achieved by Sanger sequencing, and facilitated the increase in generated data, while decreasing the cost of sequencing (Buermans and Den Dunnen, 2014). NGS is marked by the construction of libraries, enabling massively parallel sequencing, which has been increasingly simplified, and a higher throughput compared to Sanger sequencing (Ekblom and Galindo, 2011; Muzzey et al., 2015).
Nevertheless, NGS has some limitations including issues with alignment of short read sequences, detection of artifacts and microbial contaminants present in samples, in addition to the presence of human nucleic acids in clinical samples, thus limiting the analytical sensitivity of microbial detection (Davis et al., 2018). One solution to this limitation was presented as the use of targeted sequencing of the 16S rRNA gene. This gene is now considered as a reference in microbial ecology studies. However, the use of 16S rRNA-based molecular methods do not allow for a high resolution of microbiota identification, because there are biases introduced into molecular community analysis by many factors, such as sample handling, DNA extraction, PCR and partial sequence of the 16S rRNA gene (ranging between the V1 and V4 regions) (Case et al., 2007). To reduce contamination with sequence artifacts or low accuracy of read alignment, some studies remove sequence reads attributed to low-abundance operational taxonomic units (OTUs) obtained by amplicon sequencing of the 16S rRNA gene. However, it is necessary to perform the analyses with caution, because sequence data associated with these low-abundant taxa may be biologically significant. Therefore, it may not be recommended to exclude these data even if the distinction between expected and unexpected sequences is not always straightforward (Lazarevic et al., 2016).
While microbiome studies generally describe the taxonomy, diversity and abundance of the highly abundant microbes, low-abundant species have been overlooked. Most studies included in this scoping review select a cut-off value at <1% for an organism to be considered low abundant, although some studies have reported OTUs representing 0.003% of the relative abundance (Table 3). The choice of such cut-off value were attributed to low read count and or other considerations such as technical artefacts, contaminations, and the presence of transient species. However, by excluding these OTUs from the analysis, the full richness and diversity of the microbiota is underestimated. Camelo-Castillo et al. (2019) stated that only the OTUs representing over 0.1% of the total sequences of each sample were considered for their analysis, as low-frequency reads, including singletons, are more likely to represent sequencing errors, contaminants, or transient organisms without a biological role at the niche under study. Although artifacts and errors are expected, important signals from low-abundant members of microbial community, including keystone organisms, may be lost due to the current technical limitations provided by this strategy. As affirmed before, low-abundant species can be responsible for major functions on the microbial community such as processing certain secondary metabolites. An example comprises organisms from the Archaea domain, that can be detected with 16S rRNA deep sequencing but in very low abundance. Those microrganisms, particularly the methanogens, play a unique role by using hydrogen to produce methane, modulating the environment and were previously described as keystone pathogens associated with periodontal diseases (Camelo-Castillo et al., 2019).
To overcome this limitation, an interesting approach was applied by Li et al (2019), that defined a core microbiome based on high ubiquity taxa in conjunction with a characteristic of high abundance such that the significance of both measurements can be made with a sufficient degree of confidence across and within samples. Using this approach, they were able to classify OTUs with low abundance (<1%) that were highly prevalent across the samples. The authors proposed that larger sample size and sequencing depth are necessary, so that the detection of low abundant taxa may be considered non-spurious across the donors (Li et al., 2019). We believe that defining the ubiquity of the low-abundant microrganisms is a good strategy that should be better explored. A clearer cut-off point to confirm the presence and importance of such species should urgently be defined (minimum values of the sample size, as well as the ubiquity).
Another approach was recommended by Liu et al. (2013), and based on single-read-based, instead of assembly-based classification which has a higher resolution for the characterization of the composition and structure of microbiota, especially for species in low abundance. Their composition and phylogeny-based algorithm uses the strategy of composition comparison, and is capable of classifying millions of very short reads relatively quickly (Liu et al., 2013). Zhang et al (2019) also described two DNA extraction methods (using prolonged lysis and homogenizing methods) which presented marked differences specifically to the low abundance genera (Zhang et al., 2019), and might represent an important improvement in the field.
Metagenomic studies produce high-throughput sequence data that attempt to classify the taxonomy and function of all microbial communities and are greatly affected by the presence of sequencing errors that may influence the estimation of taxonomic diversity (Keegan et al., 2012). There are noise and errors in the sequencing data that can be influenced by the type of platform used. In the studies included in this review, the most commonly used platform was Illumina. With this platform, when errors occur, they are predominantly substitution-type and the error percentage for most Illumina sequence reads is approximately 0.5% (1 error in 200 bases) (Mardis, 2013). The Ion Torrent PGM and 454 GS Junior platforms produced a higher error rate associated with homopolymers around 1.5 and 0.38 errors per 100 bases, respectively (Loman et al., 2012). All platforms are considered suitable for metagenomic sequencing, but no instrument can generate completely accurate data sets, each technology has advantages and disadvantages (Luo et al., 2012). The length of reads generated, sequencing depth and error rates may be taken into account when choosing the most appropriate platform to use. For example, longer reads as those provided by MiSeq (Illumina), Ion Torrent, PacBio and Oxford Nanopore Technologies, are important to consider when carrying out 16S rRNA metagenomics, or genome sequencing (Winand et al., 2020).
There is currently no consensus in the literature on the classification of low-abundant organisms. Some studies have described such organisms being detected at less than 1% relative abundance, however, most studies use the same cutoff point (i.e. <1%) to exclude them, due to the risk of contamination or artifacts. This practice may compromise the identification of the true diversity of human microbiota. Domains other than Bacteria are neglected due to the cut-off, excluding OTUs with relative abundance lower than 0.1% or 1%. Representatives of Archaea, Fungi or Viruses are little explored. There is growing interest in developing new bioinformatics tools, such as single-read-based, instead of assembly-based, classification to obtain a higher resolution of the taxonomic analysis. Also, the ubiquity classification associated with the abundance could be a good strategy to identify the low-abundant microbiota. To achieve this, higher sequencing depths should be used in future microbiome investigations, as well as more holistic approaches including shotgun metagenomics should be employed to have a better view of the richness and diversity at play in health, disease and dysbiotic stages.
All authors listed have made a substantial, direct, and intellectual contribution to the work, and approved it for publication.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The authors are grateful to the Scientific Initiation Program from the Brazilian National Council for Scientific and Technological Development (CNPq), and to the UK's Academy of Medical Sciences, Newton International Fellowship (Grant no. NIF/R5/242) for their support.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fcimb.2021.689197/full#supplementary-material
Ai, D. M., Huang, R. C., Wen, J., Li, C., Zhu, J. P., Xia, L. C. (2017). Integrated Metagenomic Data Analysis Demonstrates That a Loss of Diversity in Oral Microbiota Is Associated With Periodontitis. BMC Genomics 18 (1), 1–15. doi: 10.1186/s12864-016-3254-5
Albert, A. Y., Chaban, B., Wagner, E. C., Schellenberg, J. J., Links, M. G., Van Schalkwyk, J., et al. (2015). A Study of the Vaginal Microbiome in Healthy Canadian Women Utilizing Cpn60-Based Molecular Profiling Reveals Distinct Gardnerella Subgroup Community State Types. PloS One 10 (8), e0135620. doi: 10.1371/journal.pone.0135620
Al-hebshi, N. N., Abdulhaq, A., Albarrag, A., Basode, V. K., Chen, T. (2016). Species-Level Core Oral Bacteriome Identified by 16S Rrna Pyrosequencing in a Healthy Young Arab Population. J. Oral. Microbiol. 8 (1), 31444. doi: 10.3402/jom.v8.31444
Balan, P., Chong, Y. S., Umashankar, S., Swarup, S., Loke, W. M., Lopez, V., et al. (2018). Keystone Species in Pregnancy Gingivitis: A Snapshot of Oral Microbiome During Pregnancy and Postpartum Period. Front. Microbiol. 9, 2360. doi: 10.3389/fmicb.2018.02360
Banerjee, S., Schlaeppi, K., van der Heijden, M. G. (2018). Keystone Taxa as Drivers of Microbiome Structure and Functioning. Nat. Rev. Microbiol. 16 (9), 567–576. doi: 10.1038/s41579-018-0024-1
Berg, G., Rybakova, D., Fischer, D., Cernava, T., Vergès, M.-C. C., Charles, T., et al. (2020). Microbiome Definition Re-Visited: Old Concepts and New Challenges. Microbiome 8 (1), 1–22. doi: 10.1186/s40168-020-00875-0
Brawner, K., Kumar, R., Serrano, C., Ptacek, T., Lefkowitz, E., Morrow, C., et al. (2017). Helicobacter Pylori Infection Is Associated With an Altered Gastric Microbiota in Children. Mucosal Immunol. 10 (5), 1169–1177. doi: 10.1038/mi.2016.131
Brooks, J. P., Edwards, D. J., Harwich, M. D., Rivera, M. C., Fettweis, J. M., Serrano, M. G., et al. (2015). The Truth About Metagenomics: Quantifying and Counteracting Bias in 16S rRNA Studies. BMC Microbiol. 15 (1), 66. doi: 10.1186/s12866-015-0351-6
Bry, L., Brigl, M., Brenner, M. B. (2006). Cd4+-T-Cell Effector Functions and Costimulatory Requirements Essential for Surviving Mucosal Infection With Citrobacter Rodentium. Infect. Immun. 74 (1), 673–681. doi: 10.1128/IAI.74.1.673-681.2006
Buermans, H., Den Dunnen, J. (2014). Next Generation Sequencing Technology: Advances and Applications. Biochim. Biophys. Acta (BBA)-Mol. Basis Dis. 1842 (10), 1932–1941. doi: 10.1016/j.bbadis.2014.06.015
Burmistrz, M., Dudek, B., Staniec, D., Rodriguez Martinez, J. I., Bochtler, M., Potempa, J., et al. (2015). Functional Analysis of Porphyromonas Gingivalis W83 Crispr-Cas Systems. J. Bacteriol. 197 (16), 2631–2641. doi: 10.1128/jb.00261-15
Burne, R., Zeng, L., Ahn, S., Palmer, S., Liu, Y., Lefebure, T., et al. (2012). Progress Dissecting the Oral Microbiome in Caries and Health. Adv. Dental Res. 24 (2), 77–80. doi: 10.1177/0022034512449462
Camelo-Castillo, A., Henares, D., Brotons, P., Galiana, A., Rodríguez, J. C., Mira, A., et al. (2019). Nasopharyngeal Microbiota in Children With Invasive Pneumococcal Disease: Identification of Bacteria With Potential Disease-Promoting and Protective Effects. Front. Microbiol. 10, 11. doi: 10.3389/fmicb.2019.00011
Camelo-Castillo, A., Novoa, L., Balsa-Castro, C., Blanco, J., Mira, A., Tomás, I. (2015). Relationship Between Periodontitis-Associated Subgingival Microbiota and Clinical Inflammation by 16S Pyrosequencing. J. Clin. Periodontol. 42 (12), 1074–1082. doi: 10.1111/jcpe.12470
Caporaso, J. G., Lauber, C. L., Costello, E. K., Berg-Lyons, D., Gonzalez, A., Stombaugh, J., et al. (2011). Moving Pictures of the Human Microbiome. Genome Biol. 12 (5), 1–8. doi: 10.1186/gb-2011-12-5-r50
Casadevall, A., Pirofski, X.-a. (2003). The Damage-Response Framework of Microbial Pathogenesis. Nat. Rev. Microbiol. 1 (1), 17–24. doi: 10.1038/nrmicro732
Case, R. J., Boucher, Y., Dahllöf, I., Holmström, C., Doolittle, W. F., Kjelleberg, S. (2007). Use of 16S rRNA and Rpob Genes as Molecular Markers for Microbial Ecology Studies. Appl. Environ. Microbiol. 73 (1), 278–288. doi: 10.1128/AEM.01177-06
Centanni, M., Lawley, B., Butts, C. A., Roy, N. C., Lee, J., Kelly, W. J., et al. (2018). Bifidobacterium Pseudolongum in the Ceca of Rats Fed Hi-Maize Starch Has Characteristics of a Keystone Species in Bifidobacterial Blooms. Appl. Environ. Microbiol. 84 (15), 11. doi: 10.1128/aem.00547-18
Claussen, J. C., Skiecevičienė, J., Wang, J., Rausch, P., Karlsen, T. H., Lieb, W., et al. (2017). Boolean Analysis Reveals Systematic Interactions Among Low-Abundance Species in the Human Gut Microbiome. PloS Comput. Biol. 13 (6), e1005361. doi: 10.1371/journal.pcbi.1005361
Dame-Teixeira, N., de Cena, J. A., Côrtes, D. A., Belmok, A., dos Anjos Borges, L. G., Marconatto, L., et al. (2020). Presence of Archaea in Dental Caries Biofilms. Arch. Oral. Biol. 110, 104606. doi: 10.1016/j.archoralbio.2019.104606
Das, B., Ghosh, T. S., Kedia, S., Rampal, R., Saxena, S., Bag, S., et al. (2018). Analysis of the Gut Microbiome of Rural and Urban Healthy Indians Living in Sea Level and High Altitude Areas. Sci. Rep. 8 (1), 1–15. doi: 10.1038/s41598-018-28550-3
Davis, N. M., Proctor, D. M., Holmes, S. P., Relman, D. A., Callahan, B. J. (2018). Simple Statistical Identification and Removal of Contaminant Sequences in Marker-Gene and Metagenomics Data. Microbiome 6 (1), 226. doi: 10.1186/s40168-018-0605-2
de Goffau, M. C., Luopajärvi, K., Knip, M., Ilonen, J., Ruohtula, T., Härkönen, T., et al. (2013). Fecal Microbiota Composition Differs Between Children With β-Cell Autoimmunity and Those Without. Diabetes 62 (4), 1238–1244. doi: 10.2337/db12-0526
Dobbler, P. T., Procianoy, R. S., Mai, V., Silveira, R. C., Corso, A. L., Rojas, B. S., et al. (2017). Low Microbial Diversity and Abnormal Microbial Succession Is Associated With Necrotizing Enterocolitis in Preterm Infants. Front. Microbiol. 8, 2243. doi: 10.3389/fmicb.2017.02243
Ekblom, R., Galindo, J. (2011). Applications of Next Generation Sequencing in Molecular Ecology of Non-Model Organisms. Heredity 107 (1), 1–15. doi: 10.1038/hdy.2010.152
Feng, Y., Chen, C.-L., Chen, T.-H., Liang, Y.-H., Chen, H.-L., Lin, C.-Y., et al. (2015). Application of Next-Generation Sequencing to Study Ascitic Microbiome in Cirrhotic Patients With or Without Spontaneous Bacterial Peritonitis. J. Microbiol. Immunol. Infect. 48 (5), 504–509. doi: 10.1016/j.jmii.2014.07.005
Fisher, C. K., Mehta, P. (2014). Identifying Keystone Species in the Human Gut Microbiome From Metagenomic Timeseries Using Sparse Linear Regression. PloS One 9 (7), 10. doi: 10.1371/journal.pone.0102451
Garrett, W. S., Gallini, C. A., Yatsunenko, T., Michaud, M., DuBois, A., Delaney, M. L., et al. (2010). Enterobacteriaceae Act in Concert With the Gut Microbiota to Induce Spontaneous and Maternally Transmitted Colitis. Cell Host Microbe 8 (3), 292–300. doi: 10.1016/j.chom.2010.08.004
Ghannoum, M. A., Jurevic, R. J., Mukherjee, P. K., Cui, F., Sikaroodi, M., Naqvi, A., et al. (2010). Characterization of the Oral Fungal Microbiome (Mycobiome) in Healthy Individuals. PloS Pathog. 6 (1), e1000713. doi: 10.1371/journal.ppat.1000713
Hajishengallis, G., Darveau, R. P., Curtis, M. A. (2012). The Keystone-Pathogen Hypothesis. Nat. Rev. Microbiol. 10 (10), 717–725. doi: 10.1038/nrmicro2873
Hajishengallis, G., Lamont, R. J. (2016). Dancing With the Stars: How Choreographed Bacterial Interactions Dictate Nososymbiocity and Give Rise to Keystone Pathogens, Accessory Pathogens, and Pathobionts. Trends Microbiol. 24 (6), 477–489. doi: 10.1016/j.tim.2016.02.010
Hamady, M., Knight, R. (2009). Microbial Community Profiling for Human Microbiome Projects: Tools, Techniques, and Challenges. Genome Res. 19 (7), 1141–1152. doi: 10.1101/gr.085464.108
Hauser, L. J., Feazel, L. M., Ir, D., Fang, R., Wagner, B. D., Robertson, C. E, et al. (2015). Sinus Culture Poorly Predicts Resident Microbiota. Int. Forum Allergy Rhinol. Wiley Online Library. 5, 3–9. doi: 10.1002/alr.21428
Heisel, T., Podgorski, H., Staley, C. M., Knights, D., Sadowsky, M. J., Gale, C. A. (2015). Complementary Amplicon-Based Genomic Approaches for the Study of Fungal Communities in Humans. PloS One 10 (2), e0116705. doi: 10.1371/journal.pone.0116705
Holt, S. C., Ebersole, J. L. (2005). Porphyromonas Gingivalis, Treponema Denticola, and Tannerella Forsythia: The ‘Red Complex’, a Prototype Polybacterial Pathogenic Consortium in Periodontitis. Periodontol. 2000 38 (1), 72–122. doi: 10.1111/j.1600-0757.2005.00113.x
Iebba, V., Guerrieri, F., Di Gregorio, V., Levrero, M., Gagliardi, A., Santangelo, F., et al. (2018). Combining Amplicon Sequencing and Metabolomics in Cirrhotic Patients Highlights Distinctive Microbiota Features Involved in Bacterial Translocation, Systemic Inflammation and Hepatic Encephalopathy. Sci. Rep. 8 (1), 1–14. doi: 10.1038/s41598-018-26509-y
Kang, J. Y., Kim, H.-N., Chang, Y., Yun, Y., Ryu, S., Shin, H., et al. (2017). Gut Microbiota and Physiologic Bowel 18 F-FDG Uptake. EJNMMI Res. 7 (1), 72. doi: 10.1186/s13550-017-0318-8
Keegan, K. P., Trimble, W. L., Wilkening, J., Wilke, A., Harrison, T., D’Souza, M., et al. (2012). A Platform-Independent Method for Detecting Errors in Metagenomic Sequencing Data: Drisee. PloS Comput. Biol. 8 (6), e1002541. doi: 10.1371/journal.pcbi.1002541
Kostic, A. D., Chun, E., Robertson, L., Glickman, J. N., Gallini, C. A., Michaud, M., et al. (2013). Fusobacterium Nucleatum Potentiates Intestinal Tumorigenesis and Modulates the Tumor-Immune Microenvironment. Cell Host Microbe 14 (2), 207–215. doi: 10.1016/j.chom.2013.07.007
Kowalska-Duplaga, K., Gosiewski, T., Kapusta, P., Sroka-Oleksiak, A., Wędrychowicz, A., Pieczarkowski, S., et al. (2019). Differences in the Intestinal Microbiome of Healthy Children and Patients With Newly Diagnosed Crohn’s Disease. Sci. Rep. 9 (1), 1–11. doi: 10.1038/s41598-019-55290-9
Lau, J. T., Whelan, F. J., Herath, I., Lee, C. H., Collins, S. M., Bercik, P., et al. (2016). Capturing the Diversity of the Human Gut Microbiota Through Culture-Enriched Molecular Profiling. Genome Med. 8 (1), 72. doi: 10.1186/s13073-016-0327-7
Lazarevic, V., Gaïa, N., Girard, M., Schrenzel, J. (2016). Decontamination of 16S Rrna Gene Amplicon Sequence Datasets Based on Bacterial Load Assessment by Qpcr. BMC Microbiol. 16 (1), 73. doi: 10.1186/s12866-016-0689-4
Lepp, P. W., Brinig, M. M., Ouverney, C. C., Palm, K., Armitage, G. C., Relman, D. A. (2004). Methanogenic Archaea and Human Periodontal Disease. Proc. Natl. Acad. Sci. U.S.A. 101 (16), 6176–6181. doi: 10.1073/pnas.0308766101
Li, K., Bihan, M., Methé, B. A. (2013). Analyses of the Stability and Core Taxonomic Memberships of the Human Microbiome. PloS One 8 (5), e63139. doi: 10.1371/journal.pone.0063139
Ling, Z., Kong, J., Liu, F., Zhu, H., Chen, X., Wang, Y., et al. (2010). Molecular Analysis of the Diversity of Vaginal Microbiota Associated With Bacterial Vaginosis. BMC Genomics 11 (1), 488. doi: 10.1186/1471-2164-11-488
Liu, Z., Li, J., Liu, H., Tang, Y., Zhan, Q., Lai, W., et al. (2019). The Intestinal Microbiota Associated With Cardiac Valve Calcification Differs From That of Coronary Artery Disease. Atherosclerosis 284, 121–128. doi: 10.1016/j.atherosclerosis.2018.11.038
Liu, J., Wang, H., Yang, H., Zhang, Y., Wang, J., Zhao, F., et al. (2013). Composition-Based Classification of Short Metagenomic Sequences Elucidates the Landscapes of Taxonomic and Functional Enrichment of Microorganisms. Nucleic Acids Res. 41 (1), e3–e3. doi: 10.1093/nar/gks828
Li, Y., Wang, K., Zhang, B., Tu, Q., Yao, Y., Cui, B., et al. (2019). Salivary Mycobiome Dysbiosis and its Potential Impact on Bacteriome Shifts and Host Immunity in Oral Lichen Planus. Int. J. Oral. Sci. 11 (2), 1–10. doi: 10.1038/s41368-019-0045-2
Loman, N. J., Misra, R. V., Dallman, T. J., Constantinidou, C., Gharbia, S. E., Wain, J., et al. (2012). Performance Comparison of Benchtop High-Throughput Sequencing Platforms. Nat. Biotechnol. 30 (5), 434–439. doi: 10.1038/nbt.2198
Luo, C., Tsementzi, D., Kyrpides, N., Read, T., Konstantinidis, K. T. (2012). Direct Comparisons of Illumina vs. Roche 454 Sequencing Technologies on the Same Microbial Community Dna Sample. PloS One 7 (2), e30087. doi: 10.1371/journal.pone.0030087
Mardis, E. R. (2013). Next-Generation Sequencing Platforms. Annu. Rev. Anal. Chem. 6, 287–303. doi: 10.1146/annurev-anchem-062012-092628
Muzzey, D., Evans, E. A., Lieber, C. (2015). Understanding the Basics of NGS: From Mechanism to Variant Calling. Curr. Genet. Med. Rep. 3 (4), 158–165. doi: 10.1007/s40142-015-0076-8
Nakayama, J., Yamamoto, A., Palermo-Conde, L. A., Higashi, K., Sonomoto, K., Tan, J., et al. (2017). Impact of Westernized Diet on Gut Microbiota in Children on Leyte Island. Front. Microbiol. 8, 197. doi: 10.3389/fmicb.2017.00197
Nash, A. K., Auchtung, T. A., Wong, M. C., Smith, D. P., Gesell, J. R., Ross, M. C., et al. (2017). The Gut Mycobiome of the Human Microbiome Project Healthy Cohort. Microbiome 5 (1), 153. doi: 10.1186/s40168-017-0373-4
Ozkan, J., Willcox, M., Wemheuer, B., Wilcsek, G., Coroneo, M., Thomas, T. (2019). Biogeography of the Human Ocular Microbiota. Ocul. Surf. 17 (1), 111–118. doi: 10.1016/j.jtos.2018.11.005
Palmer, R. J., Jr (2014). Composition and Development of Oral Bacterial Communities. Periodontol. 2000 64 (1), 20–39. doi: 10.1111/j.1600-0757.2012.00453.x
Perez-Chaparro, P. J., Goncalves, C., Figueiredo, L. C., Faveri, M., Lobao, E., Tamashiro, N., et al. (2014). Newly Identified Pathogens Associated With Periodontitis: A Systematic Review. J. Dent. Res. 93 (9), 846–858. doi: 10.1177/0022034514542468
Peterson, S. N., Snesrud, E., Liu, J., Ong, A. C., Kilian, M., Schork, N. J., et al. (2013). The Dental Plaque Microbiome in Health and Disease. PloS One 8 (3), e58487. doi: 10.1371/journal.pone.0058487
Power, M. E., Tilman, D., Estes, J. A., Menge, B. A., Bond, W. J., Mills, L. S., et al. (1996). Challenges in the Quest for Keystones: Identifying Keystone Species is Difficult—But Essential to Understanding How Loss of Species Will Affect Ecosystems. BioScience 46 (8), 609–620. doi: 10.2307/1312990
Rocas, I. N., Alves, F. R., Rachid, C. T., Lima, K. C., Assuncao, I. V., Gomes, P. N., et al. (2016). Microbiome of Deep Dentinal Caries Lesions in Teeth With Symptomatic Irreversible Pulpitis. PloS One 11 (5), e0154653. doi: 10.1371/journal.pone.0154653
Rubinstein, M. R., Wang, X., Liu, W., Hao, Y., Cai, G., Han, Y. W. (2013). Fusobacterium Nucleatum Promotes Colorectal Carcinogenesis by Modulating E-Cadherin/β-Catenin Signaling Via Its FadA Adhesin. Cell Host Microbe 14 (2), 195–206. doi: 10.1016/j.chom.2013.07.012
Sakwinska, O., Moine, D., Delley, M., Combremont, S., Rezzonico, E., Descombes, P., et al. (2016). Microbiota in Breast Milk of Chinese Lactating Mothers. PloS One 11 (8), e0160856. doi: 10.1371/journal.pone.0160856
Sears, C. L., Pardoll, D. M. (2011). Perspective: Alpha-Bugs, Their Microbial Partners, and the Link to Colon Cancer. J. Infect. Dis. 203 (3), 306–311. doi: 10.1093/jinfdis/jiq061
Shendure, J., Ji, H. (2008). Next-Generation DNA Sequencing. Nat. Biotechnol. 26 (10), 1135. doi: 10.1038/nbt1486
Simón-Soro, A., Guillen-Navarro, M., Mira, A. (2014). Metatranscriptomics Reveals Overall Active Bacterial Composition in Caries Lesions. J. Oral. Microbiol. 6 (1), 25443. doi: 10.3402/jom.v6.25443
Singh, P., Manning, S. D. (2016). Impact of Age and Sex on the Composition and Abundance of the Intestinal Microbiota in Individuals With and Without Enteric Infections. Ann. Epidemiol. 26 (5), 380–385. doi: 10.1016/j.annepidem.2016.03.007
Son, J. S., Zheng, L. J., Rowehl, L. M., Tian, X., Zhang, Y., Zhu, W., et al. (2015). Comparison of Fecal Microbiota in Children With Autism Spectrum Disorders and Neurotypical Siblings in the Simons Simplex Collection. PloS One 10 (10), e0137725. doi: 10.1371/journal.pone.0137725
Srivastava, A. (2011). Evolution & Detection of non-Coding RNA, and Transcriptome Analyses of Two non-Model Systems (University of Georgia).
Stobernack, T. (2019). Porphyromonas Gingivalis–an Oral Keystone Pathogen Challenging the Human Immune System (University of Groningen).
Tricco, A. C., Lillie, E., Zarin, W., O’Brien, K. K., Colquhoun, H., Levac, D., et al. (2018). Prisma Extension for Scoping Reviews (Prisma-Scr): Checklist and Explanation. Ann. Intern. Med. 169 (7), 467–473. doi: 10.7326/M18-0850
Trosvik, P., de Muinck, E. J. (2015). Ecology of Bacteria in the Human Gastrointestinal Tract-Identification of Keystone and Foundation Taxa. Microbiome 3, 1–12. doi: 10.1186/s40168-015-0107-4
Turnbaugh, P. J., Ley, R. E., Hamady, M., Fraser-Liggett, C. M., Knight, R., Gordon, J. I. (2007). The Human Microbiome Project. Nature 449 (7164), 804–810. doi: 10.1038/nature06244
Wang, B., Yao, M., Lv, L., Ling, Z., Li, L. (2017). The Human Microbiota in Health and Disease. Engineering 3 (1), 71–82. doi: 10.1016/J.ENG.2017.01.008
Wang, Y., Zhang, J., Chen, X., Jiang, W., Wang, S., Xu, L., et al. (2017). Profiling of Oral Microbiota in Early Childhood Caries Using Single-Molecule Real-Time Sequencing. Front. Microbiol. 8, 2244. doi: 10.3389/fmicb.2017.02244
Wan, Y.-D., Zhu, R.-X., Wu, Z.-Q., Lyu, S.-Y., Zhao, L.-X., Du, Z.-J., et al. (2018). Gut Microbiota Disruption in Septic Shock Patients: A Pilot Study. Med. Sci. Monit.: Int. Med. J. Exp. Clin. Res. 24, 8639. doi: 10.12659/MSM.911768
White, J. R., Nagarajan, N., Pop, M. (2009). Statistical Methods for Detecting Differentially Abundant Features in Clinical Metagenomic Samples. PloS Comput. Biol. 5 (4), e1000352. doi: 10.1371/journal.pcbi.1000352
Winand, R., Bogaerts, B., Hoffman, S., Lefevre, L., Delvoye, M., Van Braekel, J., et al. (2020). Targeting the 16s Rrna Gene for Bacterial Identification in Complex Mixed Samples: Comparative Evaluation of Second (Illumina) and Third (Oxford Nanopore Technologies) Generation Sequencing Technologies. Int. J. Mol. Sci. 21 (1), 298. doi: 10.3390/ijms21010298
Wu, L., Zeng, T., Zinellu, A., Rubino, S., Kelvin, D. J., Carru, C. (2019). A Cross-Sectional Study of Compositional and Functional Profiles of Gut Microbiota in Sardinian Centenarians. MSystems 4 (4), e00325–e00319. doi: 10.1128/mSystems.00325-19
Xiao, J., Fiscella, K. A., Gill, S. R. (2020). Oral Microbiome: Possible Harbinger for Children’s Health. Int. J. Oral. Sci. 12 (1), 1–13. doi: 10.1038/s41368-020-0082-x
Zakrzewski, M., Simms, L. A., Brown, A., Appleyard, M., Irwin, J., Waddell, N., et al. (2019). Il23r-Protective Coding Variant Promotes Beneficial Bacteria and Diversity in the Ileal Microbiome in Healthy Individuals Without Inflammatory Bowel Disease. J. Crohn’s Colitis 13 (4), 451–461. doi: 10.1093/ecco-jcc/jjy188
Zeng, X., Gao, X., Peng, Y., Wu, Q., Zhu, J., Tan, C., et al. (2019). Higher Risk of Stroke Is Correlated With Increased Opportunistic Pathogen Load and Reduced Levels of Butyrate-Producing Bacteria in the Gut. Front. Cell. Infect. Microbiol. 9, 4. doi: 10.3389/fcimb.2019.00004
Zerón, A. (2014). Genoma, Microbioma Y Epigenoma Humano. Una Visión Contemporánea De La Tríada Ecológica. Rev. ADM 71 (4), 161–172.
Zhang, C., Cleveland, K., Schnoll-Sussman, F., McClure, B., Bigg, M., Thakkar, P., et al. (2015). Identification of Low Abundance Microbiome in Clinical Samples Using Whole Genome Sequencing. Genome Biol. 16 (1), 265. doi: 10.1186/s13059-015-0821-z
Zhang, C., Thakkar, P. V., Powell, S. E., Sharma, P., Vennelaganti, S., Betel, D., et al. (2019). A Comparison of Homogenization vs. Enzymatic Lysis for Microbiome Profiling in Clinical Endoscopic Biopsy Tissue Samples. Front. Microbiol. 9, 3246. doi: 10.3389/fmicb.2018.03246
Zhang, Y., Wang, X., Li, H., Ni, C., Du, Z., Yan, F. (2018). Human Oral Microbiota and its Modulation for Oral Health. BioMed. Pharmacother. 99, 883–893. doi: 10.1016/j.biopha.2018.01.146
Keywords: next-generation sequencing, human microbiome, low-abundant microrganisms, scoping review, minority microbiota
Citation: Cena JA, Zhang J, Deng D, Damé-Teixeira N and Do T (2021) Low-Abundant Microorganisms: The Human Microbiome’s Dark Matter, a Scoping Review. Front. Cell. Infect. Microbiol. 11:689197. doi: 10.3389/fcimb.2021.689197
Received: 31 March 2021; Accepted: 13 May 2021;
Published: 31 May 2021.
Edited by:
Georgios N. Belibasakis, Karolinska Institutet (KI), SwedenReviewed by:
Maribasappa Karched, Kuwait University, KuwaitCopyright © 2021 Cena, Zhang, Deng, Damé-Teixeira and Do. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Nailê Damé-Teixeira, bmFpbGVkYW1lQGhvdG1haWwuY29t; bmFpbGVkYW1lQHVuYi5icg==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.