 Natalie K. Boyd1,2
Natalie K. Boyd1,2 Christopher R. Frei
Christopher R. Frei
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Cell. Infect. Microbiol. , 17 May 2021
Sec. Clinical Microbiology
Volume 11 - 2021 | https://doi.org/10.3389/fcimb.2021.684515
This article is part of the Research Topic Drug Re-Purposing for the Treatment of Bacterial Infections View all 5 articles
Drug repurposing, or identifying new uses for existing drugs, has emerged as an alternative to traditional drug discovery processes involving de novo synthesis. Drugs that are currently approved or under development for non-antibiotic indications may possess antibiotic properties, and therefore may have repurposing potential, either alone or in combination with an antibiotic. They might also serve as “antibiotic adjuvants” to enhance the activity of certain antibiotics.
Traditional drug discovery strategies aim to identify the next new chemical or molecular entity that possesses a novel mechanism of action. For any promising new compound, the path from initial discovery to market launch is sluggish, costly, and fraught with a multitude of barriers. Moving a new drug from pre-clinical phases to market generally requires a minimum timeframe of 10-12 years and over $2 billion in resources (DiMasi, 2014; DiMasi et al., 2016). Additionally, the probability of success is low, with only 1-2 drugs from an initial 10,000 compounds reaching Federal Drug Administration (FDA) approval. Following target identification and validation, high-throughput screening (HTS) assays are developed and run against compound libraries to generate ‘hits’, which are the compounds that demonstrate the desired activity or interaction with the target of interest. Each hit series undergoes additional screening and/or chemical modifications to become more ‘druggable’ lead compounds before in vitro and in vivo pharmacokinetic testing is performed in animal models (preclinical). Generally, only a handful of drug candidates from the initial 10,000 compounds enter clinical trials. Success rates for drugs entering Phase I clinical trials have approximately 10% chance of gaining FDA approval for the desired indication (Mullard, 2016).
Milestones of antibiotic discovery and development can offer insights into future solutions. The pre-antibiotic era bears striking resemblance to circumstances of today, regarding a need for: 1) novel, effective antibiotics, 2) large scale collaboration, and 3) efficient processes/timelines for antibiotic approvals.
The discovery of penicillin in 1928 is regarded as one of the most significant medical and scientific breakthroughs in history (Ligon, 2004a; Ligon, 2004b; Kardos and Demain, 2011; Lobanovska and Pilla, 2017). It represents one of history’s earliest examples of translating a scientific discovery into medicine. The story of how penicillin was developed is as important as the discovery of the drug itself. Overcoming the major barriers during that time helped establish methods that led to next-generation penicillins and development of other antibiotic classes (Kardos and Demain, 2011; Lobanovska and Pilla, 2017).
When a fungal contaminant (Penicillium notatum) on a petri dish was found to produce a potent substance that inhibited growth of Staphylococcus aureus, Alexander Fleming had unknowingly discovered a life-saving antibiotic. He published his findings the following year (Fleming, 1929), but surprisingly, the world did not take notice right away. Furthermore, there were problems with the drug itself. Penicillin was chemically unstable and difficult to isolate from the mold, raising serious doubts about its potential as a therapeutic agent.
More than ten years later, in 1939, a group of scientists at Oxford University (Howard Florey, Ernest Chain, Norman Heatley, and Edward Abraham) took on this challenge, and successfully developed a procedure for isolating and purifying penicillin (Chain et al., 2005), thereby enabling extraction of sufficient material to conduct in vivo efficacy studies (Chain et al., 1993). Clinical trials began in 1941, demonstrating drug stability and efficacy against Streptococcus pyogenes and Staphylococcus aureus, with no signs of toxicity (Ligon, 2004a; Ligon, 2004b; Lobanovska and Pilla, 2017).
With World War II underway, there was an established need for penicillin use in wounded soldiers and civilians (Fraser, 1984; Ligon, 2004a; Ligon, 2004b); thus, creating an intense sense of urgency for large-scale production. Funding, however, was limited in England for a task of this size, leading the Oxford team to relocate to the United States for assistance. Collaborative efforts soon began at an unprecedented level between the United States and United Kingdom that included academic and government entities teaming up with multiple pharmaceutical companies (including Merck, Squibb, Lilly, and Pfizer to start). Through their combined resources and expertise, they developed new procedures for the purification and mass production of penicillin - and just in time for D-Day (1944) (Swann, 1983; Fraser, 1984). These were ground-breaking techniques (i.e. submerged fermentation, microbial strain production, mutational strain improvement) representing a combination of scientific disciplines including microbiology, biochemistry, and chemical engineering, that quickly became integral for the development of subsequent antibiotics (Richards, 1964).
Shortly after the war in 1946, penicillin became widely available by prescription, which revolutionized medicine. Providers were able to, for the first time; effectively treat previously incurable diseases such as rheumatic fever, scarlet fever, syphilis, severe wounds, and infections with aggressive pathogens such as Staphylococcus or Streptococcus spp (Dowling and Lepper, 1951; Armstrong et al., 1999; Kardos and Demain, 2011; Aminov, 2017). Alexander Fleming’s serendipitous discovery of penicillin was the breakthrough of the century; however, it took an international collaboration composed of government, academia, and industry scientists to translate this discovery into one of the most important medical treatments in history.
The drug discovery landscape was forever changed after the arrival of penicillin. Not only did it save thousands of lives, it also ushered in an era of natural products discovery (Wright, 2014; Moloney, 2016). Building on the work of Fleming, microbiologist Selman Waksman sought to find more sources of antibiotic-producing microbes from soil. His approach involved the screening of soil-derived bacteria (mostly Actinomycetes spp.) against susceptible test organisms and evaluating zones of inhibited growth on an overlay plate (Schatz et al., 2005). This method is similar to Fleming’s discovery of penicillin; however, Waksman applied a more systematic, deliberate screening approach, while Fleming’s discovery of an antibiotic-producing mold was accidental. This new screening approach, otherwise known as the ‘Waksman platform’ led to the discovery of an important antibiotic streptomycin, which exhibited in vitro activity against Gram-positive and Gram-negative bacteria (Jones et al., 1944). Though penicillin was highly effective and in frequent use at the time, its antibacterial activity was primarily limited to Gram-positive bacteria. Streptomycin, the first of the aminoglycoside antibiotic class, was also the first drug with activity against Mycobacterium tuberculosis.
After the successful launch of streptomycin, the Waksman platform quickly became the quintessential tool for antibiotic discovery at the time, and ultimately the most successful and widely adopted antibiotic discovery platform to date. Discovery of other antibiotics occurred shortly thereafter, and continued over the next 20 years, famously referred to as the ‘golden age’ of antibiotics (Lewis, 2013; Lyddiard et al., 2016). In fact, the bulk of antibiotics in use today are from natural products or their semisynthetic derivatives that were discovered by this method of mining through soil-derived compounds (Moloney, 2016; Mohr, 2016; Katz and Baltz, 2016). Vancomycin, clindamycin, rifampin, tetracycline, and daptomycin are among a few important natural product antibiotics discovered during this era that remain in use today (Table 1).
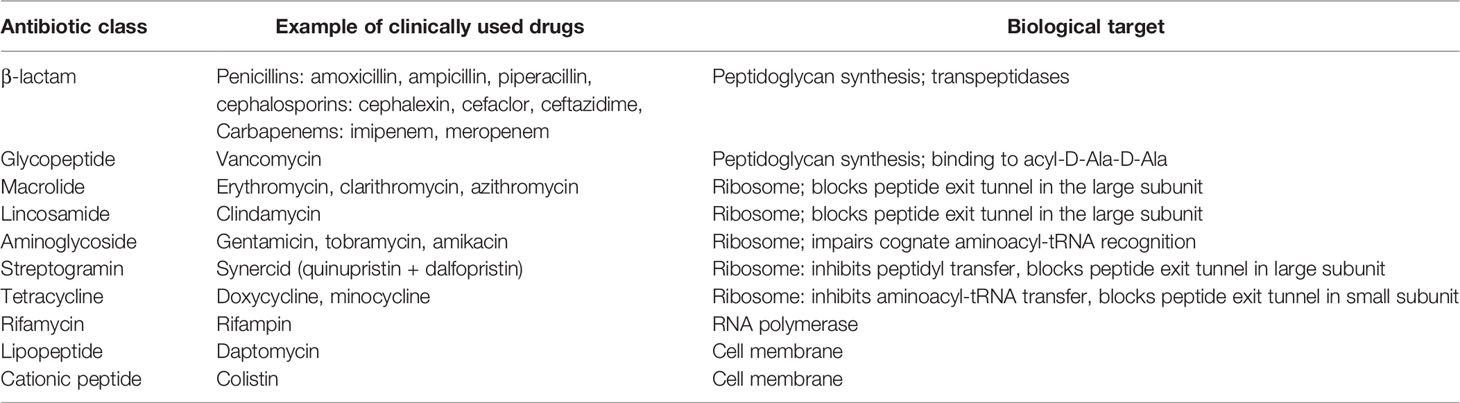
Table 1 Antibiotics derived from natural products (Lewis, 2013; Wright, 2014).
During the golden age of antibiotics, from 1940 through 1960s, the antibiotic development pipeline flourished (Walsh and Wencewicz, 2014). In fact, the rapidity of new antibiotics discovered at the time appeared to be outpacing the spread of antibiotic resistance. However, the majority of antibiotics developed during this period were through natural product discovery, a few synthetic antibiotic classes or “scaffolds” were also developed with success and remain in used today: fluoroquinolones (ciprofloxacin, levofloxacin), sulfonamides (sulfamethoxazole), oxazolidinones (linezolid), and nitroimidazole (metronidazole) (Wright, 2014; Lewis, 2017). Rapid advances in biotechnology gave rise to HTS in the early 1990s (Kubinyi, 1995). Plus, with the advancements in medicinal chemistry, molecular biology, and arrival of genomic tools, the pharmaceutical industry was seemingly more equipped than ever to discover the next wave of novel antibiotic compounds. Expectations were high for productivity as the industry moved away from laboriously mining soil for naturally-occurring compounds, opting instead for target-based HTS of synthetic compounds (Silver, 2011; Lewis, 2013). However, these high-tech platforms yielded only disappointing returns. What occurred instead was nearly a 40-year innovation gap. After the introduction of nalidixic acid in 1962, no new structural classes of antibiotics were developed again until linezolid in 2000 (Walsh, 2003; Fischbach and Walsh, 2009).
A combination of several important factors is likely to blame for the antibiotic innovation gap or discover void lasting several decades:
This drug discovery platform was a success for approximately 20 years. Unfortunately, mining through soil microbes eventually led to frequent re-isolation or rediscovery of known compounds (Katz and Baltz, 2016). After yielding diminished returns, the platform was abandoned. Still, many experts now advocate for a revival of this platform, as synthetic approaches have been unable to replace the success of natural product drug discovery. Furthermore, soil and marine environments may still be promising untapped sources for antibiotic compounds. Metagenomic analyses have shown that 99% of bacteria from soil and marine samples are “uncultured,” meaning they do not grow under normal laboratory conditions (Rappe and Giovannoni, 2003; Schloss and Handelsman, 2004). Recently, investigators unveiled the discovery of a new antibiotic, teixobactin (Ling et al., 2015; Fiers et al., 2017), using a method similar to the Waksman platform, but with a modified technique for isolating and growing uncultured bacteria (Nichols et al., 2010).
Much of the focus in the pharmaceutical industry during the 1960s and 1970s shifted from novel discovery of compounds to the chemical tailoring of existing antibiotics to create successive generations of antibiotics (2nd, 3rd, 4th, etc.). This has been important for improving the efficacy and/or pharmacological properties of antibiotics but has not lead to any new molecular entities or novel antibiotic scaffolds (Walsh, 2003; Aminov, 2017; Lewis, 2017).
Poor absorption was a major source of attrition during drug discovery in the early 1990s (Kola and Landis, 2004). Renowned medicinal chemist, Christopher Lipinski, along with his team, sought to determine the physiochemical properties of compounds that best predict absorption and advancement to clinical stages of development. Lipinski analyzed properties of compounds that had emerged from Phase I and entered Phase II clinical studies, accessed through the World Drug Index (WDI) and United States Adopted Name (USAN) databases (Lipinski et al., 2001). More than 90% of compounds that reached Phase II status had the following physiochemical parameters: i) molecular weight < 500, ii) number of hydrogen-bond donors < 5, iii) number of hydrogen-bond acceptors < 10, and iv) calculated octanol-water partition coefficient < 5. These characteristics, which became known as Lipinski’s rule of five are associated with solubility and permeability, and therefore increased absorption (Lipinski, 2000). This rule-based approach for synthesizing or screening new compounds was widely adopted by the pharmaceutical industry. Antibiotics, however, are unique molecules, and have always been an exception to the Lipinski rules. Unlike drugs developed for other therapeutic areas, antibiotic drug candidates must be able to penetrate bacterial cells, and not just human cells. Lipinski’s rules do not account for this critical physiochemical property (O’Shea and Moser, 2008; Lewis, 2013). In fact, the widespread use of Lipinski’s ‘rule of five’ may have inadvertently selected against discovery of new antibiotic compounds.
Antibiotics during the Golden Age were discovered empirically using in vitro growth inhibition assays, in which phenotypic endpoints were recorded as bacterial ‘growth’ or ‘no growth’ (Waksman et al., 1946; Ligon, 2004a; Ligon, 2004b; Moloney, 2016; Katz and Baltz, 2016). Mechanisms of action were usually determined later, often many years after approval - a significant downside to using traditional whole-cell phenotypic assays. Following the arrival of genomics, bioinformatics, and high throughput screening, drug screening strategies shifted from phenotypic to molecular target-based platforms, thereby enabling target identification and validation of important disease-related targets (Flordellis et al., 2006; Lewis, 2013). A target-based method involves the in vitro interaction between a drug candidate and a defined/validated target (e.g. enzyme or receptor) in a cell-free system. Other distinguishing characteristics between phenotypic and target-based screening is described in Table 2.

Table 2 Comparing target-based and phenotypic-based screens (Swinney and Anthony, 2011; Zheng et al., 2013; Bell et al., 2015; Wagner, 2016; Moffat et al., 2017).
Perceived at the time as the more sophisticated and more promising screening platform for anti-infective research and development, target-based screens soon became widely favored in industry. However, this highly anticipated method was met with disappointing results - no new antibiotics emerged from these platforms. Payne et al. published a highly influential article, in which the authors provided candid insight on experiences with target-based screening platforms at GlaxoSmithKline (GSK) (Payne et al., 2007). Between 1995 and 2001, a total of 67 HTS campaigns using genomic-derived antibiotic targets were run against a large compound collection, consisting of 260,000 to 530,000 compounds. This was an unprecedented amount of effort and resources for screening only one single therapeutic area. Only 16 ‘hits’ were identified, five of which became ‘lead’ compounds; but ultimately none of them reached clinical trial phases. Payne et al. concluded that whole-cell phenotypic assays, rather than target-based, are more likely to produce successful leads (Payne et al., 2007). The authors discussed several possibilities for the poor performing results, one of which is an inability to translate in vitro activity observed from target-based assays to activity that occurs with live bacterial cells. Target-based screening can produce many ‘hits.’ However, if these compounds cannot overcome the permeability barriers and tendencies for efflux pump activity in bacteria, then none of them, not one single hit, will progress to a lead compound (Livermore and British Society for Antimicrobial Chemotherapy Working Party on The Urgent Need: Regenerating Antibacterial Drug D, Development, 2011; Aminov, 2017; Lewis, 2017; Moffat et al., 2017; Singh et al., 2017). According to Dr. Kim Lewis, Ph.D., Distinguished Professor of Biology and Director of Antimicrobial Discovery Center at Northeastern University, “…simply doing more high-throughput screening or adding yet another target to the long list of potential ones will not do” (Lewis, 2017).
Although the cell permeability hurdle was specific to bacterial cells, the lower productivity from target-based screens does not appear to be limited to the development of antibiotics. An analysis of FDA drug approvals between 1999 and 2008 revealed a higher number of first-in-class compounds (i.e. new molecular entities) discovered through phenotypic screening compared to molecular target-based approach (Swinney and Anthony, 2011). From a total of 50 new in-class drugs, 28 (56%) were discovered using a phenotypic approach, while 17 (34%) were from target-based methods. One area in which target screening appears to be more successful, however, is in the field of cancer. Between 1999 and 2013, 31 of the 48 first in-class oncology drugs were discovered through target-based screens, 21 of which were kinase inhibitors (Moffat et al., 2014).
Despite having fallen out of favor more than two decades ago, and replaced with molecular target-based platforms, phenotypic screening has been undergoing a resurgence (Zheng et al., 2013; Wagner, 2016; Moffat et al., 2017). The ideal screening strategy to improve productivity in antibiotic drug discovery, however, is one that combines advantages of both phenotypic and target screening, while circumventing their limitations (Zheng et al., 2013; Farha and Brown, 2015; Matano et al., 2016). This can be accomplished through a number of ways. One approach is through the use of a parent wildtype bacterial strain paired with a mutant or modified strain, in which a specific target or mechanism of interest has been altered (Farha and Brown, 2015). Comparing the in vitro response of a wildtype/mutant pair to drug candidates can preferentially reveal compounds that inhibit a target or pathway. This method allows for a more hypothesis-driven phenotypic approach, for which the screen hits are biologically active, and the mechanism of action can also be deduced. There are increasing reports of using this integrated strategy, some referring to it as target-, pathway-, or mechanism-based whole cell screens (Testa and Johnson, 2012; Gengenbacher and Dick, 2015; Matano et al., 2016; Bonnett et al., 2016).
Antimicrobial research and development (R&D) programs were becoming unattractive investments. After the ‘high-tech’ target-based platforms failed to produce new antibiotics, the anti-infective divisions within the pharmaceutical industry began shutting down (Payne et al., 2007; Tommasi et al., 2015; Lewis, 2017). Turning a profit may take years, possibly not until final years of the patent life, depending on the circumstances (Fisher and Mobashery, 2016). Furthermore, antibiotics in general tend to have short durations of therapy (≤ 14 days). In contrast, chronic conditions, such as diabetes or hypertension may require daily treatment for many years, if not for life.
Pharmaceutical companies estimate risk/benefit and profitability of a developing product using a metric known as net present value (NPV). This is a summation of Research and Development (R&D) costs and expected value of future revenue (Sciarretta et al., 2016). A threshold target NPV of $200 million is recommended for an antibiotic to be an attractive investment and comparable to other therapeutic classes (Sharma and Towse, 2011). However, the predicted NPV of an antibiotic is estimated at negative $50 million, meaning the developmental costs would exceed projected earnings (Sharma and Towse, 2011). Among the more recently launched antibiotics, Avycaz (ceftazidime/avibactam) and Teflaro (ceftaroline fosamil) had the highest sales at ~ $80 and ~$50 million, respectively, two years post-launch. Meanwhile, other popular non-antibiotic drugs had sales ranging from $500 million to over $1 billion (Fernandes and Martens, 2017). Thus, even for antibiotics that bring in revenue, the return of investment is low compared to other popular ‘blockbuster’ treatments for other conditions.
Antibiotic resistance is an extremely complicated problem. The urgency to develop new antibiotics is almost entirely driven by escalating resistance rates (Theuretzbacher, 2011). The first sign of penicillin resistance was observed in 1940, several years before penicillin was available for widespread use (1945), when a penicillin-inactivating enzyme (penicillinase) was discovered in an E. coli strain (Abraham and Chain, 1988). In 1942, penicillin resistance was noted in four clinical strains of S. aureus, also by a penicillinase (Rammelkamp and Stolzer, 1961). Unfortunately, this was only the beginning. As each new antibiotic was launched into market, reports of resistance followed shortly thereafter. Over time, this pattern began occurring in a variety of bacterial pathogens, spanning several decades. Today, there is no shortage of antibiotic resistant bacteria, but there is a shortage of effective treatment options.
Until better control measures are in place and more novel antibiotics are available, the threat of resistance will loom, putting an expiration date on each and every antibiotic in use.
The emergence and spread of resistant bacteria, coupled with the paucity of new antibiotics, has evolved into a global health crisis (French, 2010; Lushniak, 2014; Rossolini et al., 2014; Brown and Wright, 2016; Martens and Demain, 2017). The Centers for Disease Control and Prevention (CDC) estimated that two million patients per year in the U.S. have infections associated with drug-resistant bacteria and 23,000 die annually as a result (Centers for Disease Control and Prevention, 2013). If antimicrobial resistance continues its current trajectory, an estimated 10 million deaths worldwide are predicted by 2050 (surpassing cancer) (Review on Antimicrobial Resistance, 2015). Industry sponsors, regulatory agencies, and organizations at the national and international level are taking action to overcome hurdles that led to a dry antibiotic pipeline. Just as the Oxford group discovered during their wartime efforts of mass-producing penicillin, public-private collaborations are critical to successfully revive the antibiotic pipeline and bring new antibiotics to patients in need (Luepke and Mohr, 2017; Luepke et al., 2017).
In an effort to improve regulatory processes for drug approvals, the FDA developed four expedited drug review pathways (Table 3) for the treatment of life-threatening/serious or rare conditions: i) accelerated approval, ii) priority review, iii) fast track, and iv) breakthrough therapy (Guidance for industry: expedited programs for serious conditions – drugs and biologics, 2014; Hwang et al., 2017). Bacterial infections were not specifically addressed in these pathways. That changed, however, in 2012 when the Generating Antibiotic Incentives Now (GAIN) act was signed into law. Under the GAIN act, industry sponsors can petition the FDA for a Qualified Infectious Disease Product (QIDP) designation, defined as an antibacterial or antifungal drug for human use intended to treat serious or life-threatening infections. Antibiotics with QIDP designation can receive both fast track and priority review status (Brown, 2013). The FDA is expected to have frequent meetings and written communications with the sponsor and provide guidance along the way on developing pathogen-focused antibiotics. QIDP antibiotics are also eligible for five additional years of exclusivity, which allows the sponsor a longer timeframe to recoup development costs.

Table 3 FDA expedited regulatory pathways (Darrow et al., 2014; Kesselheim and Darrow, 2015; Sinha and Kesselheim, 2016).
In addition to expedited approval programs, another pathway, the Limited Population Antimicrobial Drug (LPAD) pathway, was signed into law in 2016 as a provision to the 21st Century Cures Act (Stone, 2015; Sinha and Kesselheim, 2016). Established with the intention to streamline antibiotic development, LPAD allows faster access to antibiotics for patients with serious or life-threatening bacterial infections in which no appropriate treatment options exist. The drug’s safety and effectiveness can be studied in significantly smaller, more rapid, and less expensive clinical trials using this mechanism, which is similar to the orphan drug approval process (Simoens et al., 2012; Kwok and Koenigbauer, 2015). Data considered acceptable for drug approval using this pathway can include a combination of non-clinical, in vitro susceptibility, pharmacokinetic-pharmacodynamic, and phase II data (Rex et al., 2013). An antibiotic approved by this pathway must have “Limited Population” in the labeling of the drug.
The GAIN act and LPAD pathway are important milestones for revitalizing the antibiotic pipeline. However, some remain skeptical about the benefits of these expedited programs and have concern that speed is being favored over safety. Critics argue that expedited approval has been granted for drugs that did not meet qualifying criteria (i.e. life-threatening/serious, urgent unmet medical need, breakthrough) and have also pointed out a lack of oversight (or enforcement) of post-marketing surveillance studies for approvals based on surrogate markers (Herper; Frank et al., 2014; Kesselheim and Darrow, 2015; Kesselheim et al., 2015; Kim and Prasad, 2016; Chary and Pandian, 2017; Mostaghim et al., 2017). Others have questioned how safety and efficacy can adequately be assessed from such limited data (Light and Lexchin, 2015).
Streamlining drug approval processes is an important strategy to help bring new antibiotics to market in a shorter timeline, but this is only one of several measures needed to combat antibiotic resistance. Several national and international initiatives aimed at incentivizing antibiotic R&D are currently underway, including tax credits, market exclusivity extension, public-private partnerships, and reimbursements (Brogan and Mossialos, 2016; Sciarretta et al., 2016; Luepke and Mohr, 2017; Luepke et al., 2017). Still, despite an expanding antibiotic pipeline, experts remain concerned that these measures will simply not be enough and that we will be outmatched by worsening antibiotic resistance rates (Antibacterial agents in clinical development: an analysis of the antibacterial clinical development pipeline, including tuberculosis 2017; Breaking throught the wall: A call for concerted action on antibiotics research and development, 2017; Simpkin et al., 2017).
As of September 2017, 48 new antibiotics were in Phase I to Phase III development (Antibiotics currently in global clinical development, 2018). While this news was initially encouraging, further investigation revealed a more sobering outlook. First, only approximately 20-30% will translate to a marketable product, given the success rates of an antibiotic moving through development (Payne et al., 2007; Thomas et al., 2017). Second, most of these antibiotics do not have a novel mechanism of action but are instead modifications of existing antibiotic classes (Antibiotics currently in global clinical development, 2018). Third, only 38% of the antibiotics in development are expected to be active against ESKAPE pathogens (Enterococcus faecium, Staphylococcus aureus, Klebsiella pneumoniae, Acinetobacter baumanii, Pseudomonas aeruginosa, and Enterobacter), which have been regarded as high priority for more than a decade (Boucher et al., 2009; Thomas et al., 2017). These factors, combined with the efficacy and safety uncertainties of expedited/LPAD pathway approvals leave little optimism for the future of antibiotics.
Perhaps an alternative solution to the scientific, regulatory, and safety barriers may be to seek new therapeutic uses from existing drugs, also known as drug repurposing or drug repositioning (Beachy et al., 2014). This is becoming an increasingly attractive translational strategy to expedite therapies into the clinic, circumventing much of the early phases of drug development. Drug repurposing is, in itself, an expedited process.
Drug repurposing provides a way for pharmaceutical companies to reduce costs, increase efficiency, and minimize investment and safety risks (Tobinick, 2009; Strittmatter, 2014; Pryor and Cabreiro, 2015). Developing a repurposed drug, as opposed to a newly discovered compound, has the potential to save more than $1 billion and reduce the time to FDA approval by 50% (Scannell et al., 2012; Beachy et al., 2014). Using this strategy can also bring a failed drug back to life, often in the form of a different indication, thereby adding value back to a lost investment.
There is no ‘standardized’ definition for drug repurposing. However, the term does have many other names, which has caused some confusion: “repositioning”, “reprofiling”, “redirecting”, “rediscovery”, and “redeployment”, to name a few (Langedijk et al., 2015). These terms are generally considered interchangeable, with the exception of one – “drug rescue.” This is specifically applied to drugs that failed their intended indication and have successfully been developed for an unrelated indication (Cavalla and Singal, 2012; Medina-Franco et al., 2013). There is also some disagreement as to whether the terms “drug repurposing” and “drug repositioning” are synonymous. The source of some of this confusion is that the wording does not indicate whether a drug failed, was withdrawn, or was abandoned (i.e., early versus late development stages). Even the wording “existing drugs” is not very clear, since this could be interpreted as drug candidates in development or FDA approved drugs. Clearly, the terminology is a work in progress. Dr. Hermann Mucke, PhD, editor of Drug Repurposing, Rescue, and Repositioning, suggested that the term “drug repurposing” be used as a ‘catch-all’ phrase to describe the general concept of “developing an active pharmaceutical ingredient, at any stage of the life cycle and regardless of the success or misfortune it has encountered so far; to serve a therapeutic purpose that is significantly different from the originally intended one.” (Mucke, 2017) For the sake of simplicity, we will follow this suggestion and use the wording “drug repurposing” as an all-encompassing term.
Drug repurposing is not a new concept. In fact, it accounts for approximately 30% of all FDA approved drugs in recent years (Jin and Wong, 2014). Repurposing is a rapidly emerging field, galvanizing interest from both industry sponsors and government agencies. In 2012, the National Center for Advancing Translational Sciences (NCATS) launched a drug repurposing program, entitled “Discovering New Therapeutic Uses for Existing Molecules.” (Allison, 2012) Recognizing the value of drug repurposing, as well as the value of public-private collaborations, NCATS established a three-way partnership between academia, industry, and government with the goal to identify new therapeutic uses of propriety assets (drugs/biologics) across a range of human diseases in areas of unmet clinical need. Drug repurposing partnerships are established through the use of template agreements. AstraZeneca, Janssen Research, Pfizer, and Sanofi are a few of the companies making their assets available to researchers. Essentially, NCATS acts as the ‘matchmaker’ between the ideas from academia and experimental assets from pharmaceutical companies.
Successful drug repurposing may lead to three possible outcomes: (1) new indications for shelved candidates, (2) line extension for existing drugs, and (3) new targets and new indications for existing drugs (Tobinick, 2009; Beachy et al., 2014). Shelved drugs may have failed due to efficacy or safety reasons or were discontinued by the sponsor for strategic reasons. ‘Existing drugs’ refers to those that are FDA approved and currently available. Drugs for which there is pre-existing knowledge of safety and toxicity data (i.e. cleared Phase I trials) are generally the most ideal candidates for repurposing, as this significantly de-risks the clinical development phases. Furthermore, sponsors can leverage pre-existing safety and/or efficacy data to streamline the regulatory approval process of their drug through another expedited pathway, known as the 505(b)(2) pathway. Established in 1984, this pathway is used when changes have been made to previously approved drugs. Ceftazidime-avibactam, which is a combination of an approved cephalosporin (ceftazidime) and a novel β-lactamase inhibitor (avibactam) was approved in 2015 using the 505(b)(2) pathway (Hwang and Kesselheim, 2016). Ceftazidime-avibactam was also a QIDP designated drug, therefore receiving priority review, fast-track designation, and an additional five years of exclusivity.
Repurposing has led to a number of successful drug launches (Table 4) (Padhy and Gupta, 2011; Sardana et al., 2011; Barratt and Frail, 2012; Carlson-Banning et al., 2013; DiMasi, 2013; Beachy et al., 2014; Heslop et al., 2015; Rumore, 2016). Many of these success stories were the result of serendipitous re-discoveries, having once been abandoned or shelved after failing during early development for the intended indication. Zidovudine, for example, was originally developed as an anticancer agent, but development came to a halt after testing in animal models was unsuccessful. Years later, zidovudine was found to have potent in vitro activity against HIV, and after a rigorous clinical trial, was fast-tracked to FDA approval in 1987 (Yarchoan and Broder, 1987). This marked the beginning of the antiretroviral era, paving the way for discovery of additional life-saving antiretroviral drugs (Broder, 2010). Probably the most notable example of reviving a discontinued drug is the case of thalidomide. Originally marketed in 1957 to pregnant women for treatment of morning sickness, thalidomide was found to have devastating teratogenic effects, causing more than 10,000 birth defects (McBride, 1976; Vargesson, 2015), and was withdrawn in 1961. Thalidomide resurfaced in the late 1990s due to growing interest in the drug for treatment of erythema nodosum leprosum, an indication for which it received FDA approval in 1998 (pregnant women were excluded from study population). This launched an aggressive effort by the sponsor (Celgene) to pursue additional indications and to develop analogues that lacked the teratogenic side effects. In 2003, thalidomide was also approved for treatment of multiple myeloma and quickly became Celgene’s blockbuster drug (Novac, 2013). The thalidomide case, a redeployment of a once considered disastrous drug, demonstrates that repurposing possibilities can arise even from the unlikeliest of sources. This fuels hope that similar repurposing successes will be possible with antimicrobials.
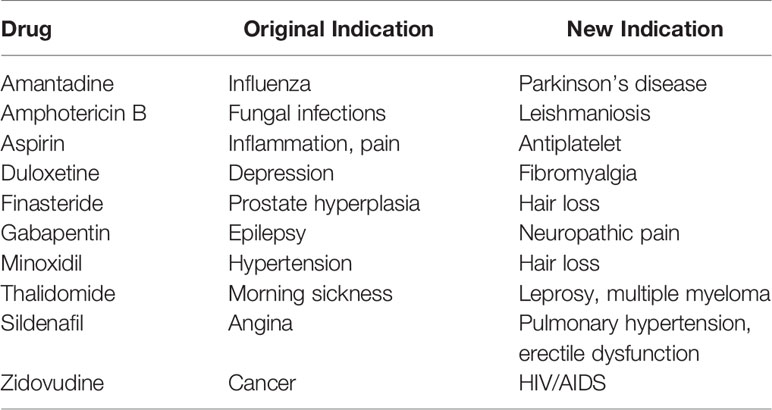
Table 4 Examples of drugs successfully repurposed or repositioned.
No drugs to date have been repurposed as antibiotics. However, a number of existing drugs have demonstrated in vitro activity against bacterial pathogens and are therefore known as “non-antibiotics.” They exist among a range of drug classes (Mazumdar et al., 2010; Carlson-Banning et al., 2013; Enserink, 2014; Perlmutter et al., 2014; Thangamani et al., 2015; Schneider et al., 2017; Stewart, 2015; Ruiz et al., 2017), and their microbiological activity is highly variable depending on the specific drug. In many cases, non-antibiotic concentrations used during in vitro testing exceed human plasma levels (Sun, 2015). In those instances, it is unlikely a non-antibiotic would be pursued (alone) as a therapeutic agent, given the lack of potent in vitro activity. That being said, plasma concentration is not the only metric for drug exposure. Drugs with a large volume of distribution have more drug distributed into the tissues than in plasma and could be useful for soft-tissue infections rather than bloodstream infections.
Whether or not a non-antibiotic demonstrates antibacterial activity, directly, as monotherapy, is only one part of the story. Antibacterial properties of a drug can also be apparent when in combination with an antibiotic. When presence of a non-antibiotic drug enhances the in vitro activity of an antibiotic, that drug is referred to as an “antibiotic adjuvant” or “antibiotic potentiator.” (Ejim et al., 2011) Some investigators refer to them as “antibiotic helper compounds,” “resistance modulators,” or “resistance breakers.” (Gibbons and Udo, 2000; Mazumdar et al., 2010; Bernal et al., 2013; Brown, 2015; Stenger et al., 2015; de Araujo et al., 2016) Adjuvants not only potentiate antibiotic activity, but they can also minimize or even prevent antibiotic resistance (Kalan and Wright, 2011; Gill et al., 2015).
Use of combination regimens for the prevention or reversal of antibiotic resistance is already a common strategy, but is typically performed with the combination of two antibiotics, rather than an antibiotic plus antibiotic adjuvant. The goal with co-administration of two antibiotics is to achieve synergistic activity, or when the sum of in vitro activity of two drugs is greater than with either agent alone (Pillai et al., 2005). The ideal scenario is to administer reduced doses of both antibiotics to decrease the risk of toxicity. However, given the increasing presence of multi-drug resistant pathogens (e.g., Pseudomonas aeruginosa), this is not a realistic treatment approach. Though it is possible that one of the combined antibiotics could be administered at a reduced dose, this applies only to certain types of infections and/or specific bacterial pathogens, such as the use of ampicillin with low-dose gentamicin for enterococcal endocarditis (Le and Bayer, 2003).
An antibiotic plus antibiotic adjuvant combination differs from antibiotic-antibiotic combination in that the adjuvant itself may have little to no in vitro activity against bacteria, especially at clinically relevant doses (Kalan and Wright, 2011; Gill et al., 2015). The primary action of the adjuvant is to enhance antibiotic activity. Gill et al. has characterized adjuvant mechanisms by dividing them into anti-resistance and anti-virulence (Gill et al., 2015). Quorum sensing inhibitors have demonstrated some success in vitro and in murine models for treatment and prevention of biofilm formation (Chow et al., 2014; Sully et al., 2014). Meanwhile several anti-toxin antibodies are undergoing clinical trials. Efflux pumps have become a source of considerable research, particularly the NorA efflux pump that confers a multidrug resistant phenotype of S. aureus (Gibbons et al., 2003; Couto et al., 2008; Felicetti et al., 2017; Zimmermann et al., 2017).
Unfortunately, drug development is notoriously riddled with barriers and has a high failure rate (DiMasi, 2014; DiMasi et al., 2016). One of the major benefits of drug repurposing is that it can significantly reduce this lengthy timeline, thereby making treatments more readily available for clinical unmet needs (Ashburn and Thor, 2004; Barratt and Frail, 2012; Austin and Gadhia, 2017). Drug repurposing is simply a more expedited form of traditional drug development.
The conventional understanding of translational science is that it is a “bench-to-bedside” forward movement that begins in a laboratory and ends with the treatment of patients (Fernandez-Moure, 2016; Fort et al., 2017). Not only is drug repurposing a translational process, but it is also the embodiment of a concept known as “reverse translation.” This is defined loosely as the use of scientific research from an earlier phase to answer questions arising from clinical data (Shakhnovich, 2018). Reverse translation typically begins with patients, in which a question is identified during patient care experiences or clinical trials, and works backwards to test the new hypothesis. Reverse translation is therefore often referred to as a “bedside-to-bench” approach, as it occasionally requires revisiting basic sciences to answer a clinically relevant question (Becker and Funk, 2018). The same principle applies with drug repurposing, in which existing drugs are reevaluated for new indications. Drugs that tend to have the highest probability of success and shortest turnaround time to a new indication are those that are reevaluated within the clinical development phases (i.e., T1 - T3) that have at least cleared phase I clinical trials (i.e., safety and tolerability). Even drugs that failed to meet clinical efficacy endpoints for their desired indications can be excellent candidates for repurposing.
Discovery of an unanticipated drug effect (e.g., a side effect or therapeutic effect unrelated to the primary indication) occurs most commonly during patient care. Investigation of this unexpected effect is a frequent driver for working backwards to reevaluate an old drug. Though it is more ideal to stay within the T1-T3 phases during reevaluation, many of these clinically relevant questions require investigations at the basic science level (Becker and Funk, 2018; McWilliam et al., 2018).
The traditional process of drug discovery and development of a novel compound has a notoriously low probability of success. Drug repurposing, a reverse translational process, is a strategy that can dramatically increase this probability. Using pre-existing scientific knowledge, usually in the form of human clinical data, drug repurposing has the potential to turn even a failed drug into a success.
Study concept and design: NB and CF. Interpretation of data: all authors. Drafting of the manuscript: NB. Critical revision of the manuscript for important intellectual content: all authors. Study supervision: NB and CF.
CF was supported, in part, by an NIH Clinical and Translational Science Award (National Center for Advancing Translational Sciences, UL1 TR002645) while the study was being conducted.
The views expressed in this article are those of the authors and do not necessarily represent the views of the Department of Veterans Affairs, the National Institutes of Health, or the authors’ affiliated institutions.
CF’s institutions have received grant money, for CF to perform research, from AstraZeneca Pharmaceuticals, in the last 3 years.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Abraham, E. P., Chain, E. (1988). An Enzyme From Bacteria Able to Destroy Penicillin. 1940 Rev. Infect. Dis. 10, 677–678. doi: 10.1038/146837a0
Allison, M. (2012). NCATS Launches Drug Repurposing Program. Nat. Biotechnol. 30, 571–572. doi: 10.1038/nbt0712-571a
Aminov, R. (2017). History of Antimicrobial Drug Discovery: Major Classes and Health Impact. Biochem. Pharmacol. 133, 4–19. doi: 10.1016/j.bcp.2016.10.001
Armstrong, G. L., Conn, L. A., Pinner, R. W. (1999). Trends in Infectious Disease Mortality in the United States During the 20th Century. JAMA 281, 61–66. doi: 10.1001/jama.281.1.61
Ashburn, T. T., Thor, K. B. (2004). Drug Repositioning: Identifying and Developing New Uses for Existing Drugs. Nat. Rev. Drug Discov. 3, 673–683. doi: 10.1038/nrd1468
Austin, B. A., Gadhia, A. D. (2017). “New Therapeutic Uses for Existing Drugs,” in Rare Diseases Epidemiology: Update and Overview, vol. 1031 . Eds. Posada de la Paz, M., Taruscio, D., Croft, S. (Cham, Switzerland: Springer), 233–247. doi: 10.1007/978-3-319-67144-4_14
(2014) Guidance for Industry: Expedited Programs for Serious Conditions – Drugs and Biologics (U.S. Food and Drug Adminstration: Center for Drug Evaluation and Research, Center for Biologics Evaluation and Research). Available at: http://www.fda.gov/downloads/drugs/guidancecomplianceregulatoryinformation/guidances/ucm358301.pdf (Accessed March 15, 2021).
(2017) Antibacterial Agents in Clinical Development: An Analysis of the Antibacterial Clinical Development Pipeline, Including Tuberculosis (World Health Organization). Available at: http://www.who.int/medicines/news/2017/IAU_AntibacterialAgentsClinicalDevelopment_webfinal_2017_09_19.pdf (Accessed March 15, 2021).
(2017) Breaking Throught the Wall: A Call for Concerted Action on Antibiotics Research and Development (The Boston Consulting Group). Available at: https://www.bundesgesundheitsministerium.de/fileadmin/Dateien/5_Publikationen/Gesundheit/Berichte/GUARD_Follow_Up_Report_Full_Report_final.pdf (Accessed March 15, 2021).
(2018) Antibiotics Currently in Global Clinical Development (The Pew Charitable Trusts). Available at: http://www.pewtrusts.org/en/research-and-analysis/data-visualizations/2014/antibiotics-currently-in-clinical-development (Accessed March 15, 2021).
Barratt, M. J., Frail, D. (2012). Drug Repositioning: Bringing New Life to Shelved Assets and Existing Drugs (Hoboken, N.J: John Wiley & Sons). doi: 10.1002/9781118274408
Beachy, S. H., Johnson, S. G., Olson, S., Berger, A. C. (2014). Drug Repurposing and Repositioning: Workshop Summary (Washington, D.C: The National Academies Press).
Becker, M. L., Funk, R. S. (2018). Reverse Translation in Advancing Pharmacotherapy in Pediatric Rheumatology: A Logical Approach in Rare Diseases With Limited Resources. Clin. Transl. Sci. 11, 106–108. doi: 10.1111/cts.12513
Bell, A., Fecke, W., Williams, C. (2015). “Influence of Phenotypic and Target-Based Screening Strategies on Compound Attrition and Project Choice,” in Attrition in the Pharmaceutical Industry. Eds. Alex, A., CJ, H., DA, S. (Hoboken, New Jersey, USA: John Wiley & Sons), 229–263. doi: 10.1002/9781118819586.ch9
Bernal, P., Molina-Santiago, C., Daddaoua, A., Llamas, M. A. (2013). Antibiotic Adjuvants: Identification and Clinical Use. Microb. Biotechnol. 6, 445–449. doi: 10.1111/1751-7915.12044
Bonnett, S. A., Ollinger, J., Chandrasekera, S., Florio, S., O’Malley, T., Files, M., et al. (2016). A Target-Based Whole Cell Screen Approach to Identify Potential Inhibitors of Mycobacterium Tuberculosis Signal Peptidase. ACS Infect. Dis. 2, 893–902. doi: 10.1021/acsinfecdis.6b00075
Boucher, H. W., Talbot, G. H., Bradley, J. S., Edwards, J. E., Gilbert, D., Rice, L. B., et al. (2009). Bad Bugs, No Drugs: No ESKAPE! An Update From the Infectious Diseases Society of America. Clin. Infect. Dis. 48, 1–12. doi: 10.1086/595011
Broder, S. (2010). The Development of Antiretroviral Therapy and its Impact on the HIV-1/AIDS Pandemic. Antiviral Res. 85, 1–18. doi: 10.1016/j.antiviral.2009.10.002
Brogan, D. M., Mossialos, E. (2016). A Critical Analysis of the Review on Antimicrobial Resistance Report and the Infectious Disease Financing Facility. Global Health 12, 8. doi: 10.1186/s12992-016-0147-y
Brown, E. D. (2013). Is the GAIN Act a Turning Point in New Antibiotic Discovery? Can. J. Microbiol. 59, 153–156. doi: 10.1139/cjm-2013-0089
Brown, D. (2015). Antibiotic Resistance Breakers: can Repurposed Drugs Fill the Antibiotic Discovery Void? Nat. Rev. Drug Discov. 14, 821–832. doi: 10.1038/nrd4675
Brown, E. D., Wright, G. D. (2016). Antibacterial Drug Discovery in the Resistance Era. Nature 529, 336–343. doi: 10.1038/nature17042
Carlson-Banning, K. M., Chou, A., Liu, Z., Hamill, R. J., Song, Y., Zechiedrich, L. (2013). Toward Repurposing Ciclopirox as an Antibiotic Against Drug-Resistant Acinetobacter Baumannii, Escherichia Coli, and Klebsiella Pneumoniae. PLoS One 8, e69646. doi: 10.1371/journal.pone.0069646
Cavalla, D., Singal, C. (2012). Retrospective Clinical Analysis for Drug Rescue: for New Indications or Stratified Patient Groups. Drug Discov. Today 17, 104–109. doi: 10.1016/j.drudis.2011.09.019
Centers for Disease Control and Prevention (2013) Antibiotic Resistance Threats in the United States. Available at: https://www.cdc.gov/drugresistance/threat-report-2013/ (Accessed March 15, 2021).
Chain, E., Florey, H. W., Adelaide, M. B., Gardner, A. D., Heatley, N. G., Jennings, M. A., et al. (1993). Penicillin as a Chemotherapeutic Agent. 1940. Clin. Orthop. Relat. Res., 3–7. doi: 10.1097/00003086-199310000-00002
Chain, E., Florey, H. W., Gardner, A. D., Heatley, N. G., Jennings, M. A., Orr-Ewing, J., et al. (2005). The CLASSIC: Penicillin as a Chemotherapeutic Agent. 1940. Clin. Orthop. Relat. Res. 439, 23–26. doi: 10.1097/01.blo.0000183429.83168.07
Chary, K. V., Pandian, K. (2017). Accelerated Approval of Drugs: Ethics Versus Efficacy. Indian J. Med. Ethics 2, 244–247. doi: 10.20529/IJME.2017.062
Chow, J. Y., Yang, Y., Tay, S. B., Chua, K. L., Yew, W. S. (2014). Disruption of Biofilm Formation by the Human Pathogen Acinetobacter Baumannii Using Engineered Quorum-Quenching Lactonases. Antimicrob. Agents Chemother. 58, 1802–1805. doi: 10.1128/AAC.02410-13
Couto, I., Costa, S. S., Viveiros, M., Martins, M., Amaral, L. (2008). Efflux-Mediated Response of Staphylococcus Aureus Exposed to Ethidium Bromide. J. Antimicrob. Chemother. 62, 504–513. doi: 10.1093/jac/dkn217
Darrow, J. J., Avorn, J., Kesselheim, A. S. (2014). New FDA Breakthrough-Drug Category–Implications for Patients. N. Engl. J. Med. 370, 1252–1258. doi: 10.1056/NEJMhle1311493
de Araujo, R. S., Barbosa-Filho, J. M., Scotti, M. T., Scotti, L., da Cruz, R. M., Falcao-Silva Vdos, S., et al. (2016). Modulation of Drug Resistance in Staphylococcus Aureus With Coumarin Derivatives. Sci. (Cairo) 2016, 6894758. doi: 10.1155/2016/6894758
DiMasi, J. A. (2013). Innovating by Developing New Uses of Already-Approved Drugs: Trends in the Marketing Approval of Supplemental Indications. Clin. Ther. 35, 808–818. doi: 10.1016/j.clinthera.2013.04.004
DiMasi, J. A. (2014). Pharmaceutical R&D Performance by Firm Size: Approval Success Rates and Economic Returns. Am. J. Ther. 21, 26–34. doi: 10.1097/MJT.0b013e318269198f
DiMasi, J. A., Grabowski, H. G., Hansen, R. W. (2016). Innovation in the Pharmaceutical Industry: New Estimates of R&D Costs. J. Health Econ. 47, 20–33. doi: 10.1016/j.jhealeco.2016.01.012
Dowling, H. F., Lepper, M. H. (1951). The Effect of Antibiotics (Penicillin, Aureomycin, and Terramycin) on the Fatality Rate and Incidence of Complications in Pneumococcic Pneumonia; a Comparison With Other Methods of Therapy. Am. J. Med. Sci. 222, 396–403. doi: 10.1097/00000441-195110000-00005
Ejim, L., Farha, M. A., Falconer, S. B., Wildenhain, J., Coombes, B. K., Tyers, M., et al. (2011). Combinations of Antibiotics and Nonantibiotic Drugs Enhance Antimicrobial Efficacy. Nat. Chem. Biol. 7, 348–350. doi: 10.1038/nchembio.559
Enserink, M. (2014). Infectious Diseases. Debate Erupts on ‘Repurposed’ Drugs for Ebola. Science 345, 718–719. doi: 10.1126/science.345.6198.718
Farha, M. A., Brown, E. D. (2015). Unconventional Screening Approaches for Antibiotic Discovery. Ann. N. Y. Acad. Sci. 1354, 54–66. doi: 10.1111/nyas.12803
Felicetti, T., Cannalire, R., Burali, M. S., Massari, S., Manfroni, G., Barreca, M. L., et al. (2017). Searching for Novel Inhibitors of the S. Aureus NorA Efflux Pump: Synthesis and Biological Evaluation of the 3-phenyl-1,4-benzothiazine Analogues. ChemMedChem 12, 1293–1302. doi: 10.1002/cmdc.201700286
Fernandes, P., Martens, E. (2017). Antibiotics in Late Clinical Development. Biochem. Pharmacol. 133, 152–163. doi: 10.1016/j.bcp.2016.09.025
Fernandez-Moure, J. S. (2016). Lost in Translation: The Gap in Scientific Advancements and Clinical Application. Front. Bioeng. Biotechnol. 4, 43. doi: 10.3389/fbioe.2016.00043
Fiers, W. D., Craighead, M., Singh, I. (2017). Teixobactin and its Analogues: A New Hope in Antibiotic Discovery. ACS Infect. Dis. 3, 688–690. doi: 10.1021/acsinfecdis.7b00108
Fischbach, M. A., Walsh, C. T. (2009). Antibiotics for Emerging Pathogens. Science 325, 1089–1093. doi: 10.1126/science.1176667
Fisher, J. F., Mobashery, S. (2016). Endless Resistance. Endless Antibiot. Med. Chem. Comm. 7, 37–49. doi: 10.1039/C5MD00394F
Fleming, A. (1980). Classics in Infectious Diseases: On the Antibacterial Action of Cultures of a Penicillium, with Special Reference to Their Use in the Isolation of B. influenzae by Alexander Fleming, Reprinted from the British Journal of Experimental Pathology 10, 226-236, 1929. Rev. Infect. Dis. 2, 129–139. doi: 10.1093/clinids/2.1.129
Flordellis, C. S., Manolis, A. S., Paris, H., Karabinis, A. (2006). Rethinking Target Discovery in Polygenic Diseases. Curr. Top. Med. Chem. 6, 1791–1798. doi: 10.2174/156802606778194226
Fort, D. G., Herr, T. M., Shaw, P. L., Gutzman, K. E., Starren, J. B. (2017). Mapping the Evolving Definitions of Translational Research. J. Clin. Transl. Sci. 1, 60–66. doi: 10.1017/cts.2016.10
Frank, C., Himmelstein, D. U., Woolhandler, S., Bor, D. H., Wolfe, S. M., Heymann, O., et al. (2014). Era of Faster FDA Drug Approval has Also Seen Increased Black-Box Warnings and Market Withdrawals. Health Aff. (Millwood) 33, 1453–1459. doi: 10.1377/hlthaff.2014.0122
Fraser, I. (1984). Penicillin: Early Trials in War Casualties. Br. Med. J. (Clin. Res. Ed) 289, 1723–1725. doi: 10.1136/bmj.289.6460.1723
French, G. L. (2010). The Continuing Crisis in Antibiotic Resistance. Int. J. Antimicrob. Agents 36 Suppl 3, S3–S7. doi: 10.1016/S0924-8579(10)70003-0
Gengenbacher, M., Dick, T. (2015). Antibacterial Drug Discovery: Doing it Right. Chem. Biol. 22, 5–6. doi: 10.1016/j.chembiol.2014.12.005
Gibbons, S., Oluwatuyi, M., Kaatz, G. W. (2003). A Novel Inhibitor of Multidrug Efflux Pumps in Staphylococcus Aureus. J. Antimicrob. Chemother. 51, 13–17. doi: 10.1093/jac/dkg044
Gibbons, S., Udo, E. E. (2000). The Effect of Reserpine, a Modulator of Multidrug Efflux Pumps, on the In Vitro Activity of Tetracycline Against Clinical Isolates of Methicillin Resistant Staphylococcus Aureus (MRSA) Possessing the Tet(K) Determinant. Phytother. Res. 14, 139–140. doi: 10.1002/(SICI)1099-1573(200003)14:2<139::AID-PTR608>3.0.CO;2-8
Gill, E. E., Franco, O. L., Hancock, R. E. (2015). Antibiotic Adjuvants: Diverse Strategies for Controlling Drug-Resistant Pathogens. Chem. Biol. Drug Des. 85, 56–78. doi: 10.1111/cbdd.12478
Herper, M. The FDA is Basically Approving Everything. Here’s the Data to Prove it (Forbes). Available at: http://www.forbes.com/sites/matthewherper/2015/08/20/the-fda-is-basically-approving-everything-heres-the-data-to-prove-it/415ec59d4938 (Accessed March 15, 2021).
Heslop, E., Csimma, C., Straub, V., McCall, J., Nagaraju, K., Wagner, K. R., et al. (2015). Tact. The Treat-NMD Advisory Committee for Therapeutics (TACT): An Innovative De-Risking Model to Foster Orphan Drug Development. Orphanet J. Rare Dis. 10, 49. doi: 10.1186/s13023-015-0258-1
Hwang, T. J., Darrow, J. J., Kesselheim, A. S. (2017). The FDA’s Expedited Programs and Clinical Development Times for Novel Therapeutics, 2012-2016. JAMA 318, 2137–2138. doi: 10.1001/jama.2017.14896
Hwang, T. J., Kesselheim, A. S. (2016). Leveraging Novel and Existing Pathways to Approve New Therapeutics to Treat Serious Drug-Resistant Infections. Am. J. Law Med. 42, 429–450. doi: 10.1177/0098858816658275
Jin, G., Wong, S. T. (2014). Toward Better Drug Repositioning: Prioritizing and Integrating Existing Methods Into Efficient Pipelines. Drug Discov.Today 19, 637–644. doi: 10.1016/j.drudis.2013.11.005
Jones, D., Metzger, H. J., Schatz, A., Waksman, S. A. (1944). Control of Gram-negative Bacteria in Experimental Animals by Streptomycin. Science 100, 103–105. doi: 10.1126/science.100.2588.103
Kalan, L., Wright, G. D. (2011). Antibiotic Adjuvants: Multicomponent Anti-Infective Strategies. Expert Rev. Mol. Med. 13, e5. doi: 10.1017/S1462399410001766
Kardos, N., Demain, A. L. (2011). Penicillin: The Medicine With the Greatest Impact on Therapeutic Outcomes. Appl. Microbiol. Biotechnol. 92, 677–687. doi: 10.1007/s00253-011-3587-6
Katz, L., Baltz, R. H. (2016). Natural Product Discovery: Past, Present, and Future. J. Ind. Microbiol. Biotechnol. 43, 155–176. doi: 10.1007/s10295-015-1723-5
Kesselheim, A. S., Darrow, J. J. (2015). FDA Designations for Therapeutics and Their Impact on Drug Development and Regulatory Review Outcomes. Clin. Pharmacol. Ther. 97, 29–36. doi: 10.1002/cpt.1
Kesselheim, A. S., Wang, B., Franklin, J. M., Darrow, J. J. (2015). Trends in Utilization of FDA Expedited Drug Development and Approval Programs, 1987-2014: Cohort Study. BMJ 351, h4633. doi: 10.1136/bmj.h4633
Kim, C., Prasad, V. (2016). Strength of Validation for Surrogate End Points Used in the US Food and Drug Administration’s Approval of Oncology Drugs. Mayo Clin. Proc. 91 (6), 713–25. doi: 10.1016/j.mayocp.2016.02.012
Kola, I., Landis, J. (2004). Can the Pharmaceutical Industry Reduce Attrition Rates? Nat. Rev. Drug Discov. 3, 711–715. doi: 10.1038/nrd1470
Kwok, A. K., Koenigbauer, F. M. (2015). Incentives to Repurpose Existing Drugs for Orphan Indications. ACS Med. Chem. Lett. 6, 828–830. doi: 10.1021/acsmedchemlett.5b00276
Langedijk, J., Mantel-Teeuwisse, A. K., Slijkerman, D. S., Schutjens, M. H. (2015). Drug Repositioning and Repurposing: Terminology and Definitions in Literature. Drug Discov. Today 20, 1027–1034. doi: 10.1016/j.drudis.2015.05.001
Le, T., Bayer, A. S. (2003). Combination Antibiotic Therapy for Infective Endocarditis. Clin. Infect. Dis. 36, 615–621. doi: 10.1086/367661
Lewis, K. (2013). Platforms for Antibiotic Discovery. Nat. Rev. Drug Discov. 12, 371–387. doi: 10.1038/nrd3975
Lewis, K. (2017). New Approaches to Antimicrobial Discovery. Biochem. Pharmacol. 134, 87–98. doi: 10.1016/j.bcp.2016.11.002
Ligon, B. L. (2004a). Penicillin: its Discovery and Early Development. Semin. Pediatr. Infect. Dis. 15, 52–57. doi: 10.1053/j.spid.2004.02.001
Ligon, B. L. (2004b). Sir Alexander Fleming: Scottish Researcher Who Discovered Penicillin. Semin. Pediatr. Infect. Dis. 15, 58–64. doi: 10.1053/j.spid.2004.02.002
Ling, L. L., Schneider, T., Peoples, A. J., Spoering, A. L., Engels, I., Conlon, B. P., et al. (2015). A New Antibiotic Kills Pathogens Without Detectable Resistance. Nature 517, 455–459. doi: 10.1038/nature14098
Lipinski, C. A. (2000). Drug-Like Properties and the Causes of Poor Solubility and Poor Permeability. J. Pharmacol. Toxicol. Methods 44, 235–249. doi: 10.1016/S1056-8719(00)00107-6
Lipinski, C. A., Lombardo, F., Dominy, B. W., Feeney, P. J. (2001). Experimental and Computational Approaches to Estimate Solubility and Permeability in Drug Discovery and Development Settings. Adv. Drug Deliv. Rev. 46, 3–26. doi: 10.1016/S0169-409X(00)00129-0
Livermore, D. M., British Society for Antimicrobial Chemotherapy Working Party on The Urgent Need: Regenerating Antibacterial Drug D, Development (2011). Discovery Research: The Scientific Challenge of Finding New Antibiotics. J. Antimicrob. Chemother. 66, 1941–1944. doi: 10.1093/jac/dkr262
Lobanovska, M., Pilla, G. (2017). Penicillin’s Discovery and Antibiotic Resistance: Lessons for the Future? Yale J. Biol. Med. 90, 135–145.
Luepke, K. H., Mohr, J. F. ,. 3. (2017). The Antibiotic Pipeline: Reviving Research and Development and Speeding Drugs to Market. Expert Rev. Anti Infect. Ther. 15, 425–433. doi: 10.1080/14787210.2017.1308251
Luepke, K. H., Suda, K. J., Boucher, H., Russo, R. L., Bonney, M. W., Hunt, T. D., et al. (2017). Past, Present, and Future of Antibacterial Economics: Increasing Bacterial Resistance, Limited Antibiotic Pipeline, and Societal Implications. Pharmacotherapy 37, 71–84. doi: 10.1002/phar.1868
Lushniak, B. D. (2014). Antibiotic Resistance: A Public Health Crisis. Public Health Rep. 129, 314–316. doi: 10.1177/003335491412900402
Lyddiard, D., Jones, G. L., Greatrex, B. W. (2016). Keeping it Simple: Lessons From the Golden Era of Antibiotic Discovery. FEMS Microbiol. Lett. 363, 1–3. doi: 10.1093/femsle/fnw084
Martens, E., Demain, A. L. (2017). The Antibiotic Resistance Crisis, With a Focus on the United States. J. Antibiot (Tokyo) 70, 520–526. doi: 10.1038/ja.2017.30
Matano, L. M., Morris, H. G., Wood, B. M., Meredith, T. C., Walker, S. (2016). Accelerating the Discovery of Antibacterial Compounds Using Pathway-Directed Whole Cell Screening. Bioorg. Med. Chem. 24, 6307–6314. doi: 10.1016/j.bmc.2016.08.003
Mazumdar, K., Asok Kumar, K., Dutta, N. K. (2010). Potential Role of the Cardiovascular non-Antibiotic (Helper Compound) Amlodipine in the Treatment of Microbial Infections: Scope and Hope for the Future. Int. J. Antimicrob. Agents 36, 295–302. doi: 10.1016/j.ijantimicag.2010.05.003
McBride, W. G. (1976). Studies of the Etiology of Thalidomide Dysmorphogenesis. Teratology 14, 71–87. doi: 10.1002/tera.1420140110
McWilliam, S. J., Antoine, D. J., Pirmohamed, M. (2018). Repurposing Statins for Renal Protection: Is it a Class Effect? Clin. Transl. Sci. 11, 100–102. doi: 10.1111/cts.12521
Medina-Franco, J. L., Giulianotti, M. A., Welmaker, G. S., Houghten, R. A. (2013). Shifting From the Single to the Multitarget Paradigm in Drug Discovery. Drug Discov. Today 18, 495–501. doi: 10.1016/j.drudis.2013.01.008
Moffat, J. G., Rudolph, J., Bailey, D. (2014). Phenotypic Screening in Cancer Drug Discovery - Past, Present and Future. Nat. Rev. Drug Discov. 13, 588–602. doi: 10.1038/nrd4366
Moffat, J. G., Vincent, F., Lee, J. A., Eder, J., Prunotto, M. (2017). Opportunities and Challenges in Phenotypic Drug Discovery: An Industry Perspective. Nat. Rev. Drug Discov. 16, 531–543. doi: 10.1038/nrd.2017.111
Mohr, K. I. (2016). History of Antibiotics Research. Curr. Top. Microbiol. Immunol. 398, 237–272. doi: 10.1007/82_2016_499
Moloney, M. G. (2016). Natural Products as a Source for Novel Antibiotics. Trends Pharmacol. Sci. 37, 689–701. doi: 10.1016/j.tips.2016.05.001
Mostaghim, S. R., Gagne, J. J., Kesselheim, A. S. (2017). Safety Related Label Changes for New Drugs After Approval in the US Through Expedited Regulatory Pathways: Retrospective Cohort Study. BMJ 358, j3837. doi: 10.1136/bmj.j3837
Mucke, H. (2017). Old Compounds, New Uses, New Ways: Many Ways. Assay Drug Dev. Technol. 15, 353. doi: 10.1089/adt.2017.29067.hmu
Mullard, A. (2016). Parsing Clinical Success Rates. Nat. Rev. Drug Discovery 15, 447. doi: 10.1038/nrd.2016.136
Nichols, D., Cahoon, N., Trakhtenberg, E. M., Pham, L., Mehta, A., Belanger, A., et al. (2010). Use of Ichip for High-Throughput in Situ Cultivation of “Uncultivable” Microbial Species. Appl. Environ. Microbiol. 76, 2445–2450. doi: 10.1128/AEM.01754-09
Novac, N. (2013). Challenges and Opportunities of Drug Repositioning. Trends Pharmacol. Sci. 34, 267–272. doi: 10.1016/j.tips.2013.03.004
O’Shea, R., Moser, H. E. (2008). Physicochemical Properties of Antibacterial Compounds: Implications for Drug Discovery. J. Med. Chem. 51, 2871–2878. doi: 10.1021/jm700967e
Padhy, B. M., Gupta, Y. K. (2011). Drug Repositioning: Re-Investigating Existing Drugs for New Therapeutic Indications. J. Postgrad. Med. 57, 153–160. doi: 10.4103/0022-3859.81870
Payne, D. J., Gwynn, M. N., Holmes, D. J., Pompliano, D. L. (2007). Drugs for Bad Bugs: Confronting the Challenges of Antibacterial Discovery. Nat. Rev. Drug Discov. 6, 29–40. doi: 10.1038/nrd2201
Perlmutter, J. I., Forbes, L. T., Krysan, D. J., Ebsworth-Mojica, K., Colquhoun, J. M., Wang, J. L., et al. (2014). Repurposing the Antihistamine Terfenadine for Antimicrobial Activity Against Staphylococcus Aureus. J. Med. Chem. 57, 8540–8562. doi: 10.1021/jm5010682
Pillai, S. K., Moellering, R. C., Eliopoulos, G. M. (2005). “Antimicrobial Combinations,” in Antibiotics in Laboratory Medicine, vol. 5th . Ed. Lorian, V. (Philadelphia, PA: Lippincott Williams & Wilkins), 365 – 440.
Pryor, R., Cabreiro, F. (2015). Repurposing Metformin: An Old Drug With New Tricks in its Binding Pockets. Biochem. J. 471, 307–322. doi: 10.1042/BJ20150497
Rammelkamp, C. H., Jr., Stolzer, B. L. (1961). The Latent Period Before the Onset of Acute Rheumatic Fever. Yale J. Biol. Med. 34, 386–398.
Rappe, M. S., Giovannoni, S. J. (2003). The Uncultured Microbial Majority. Annu. Rev. Microbiol. 57, 369–394. doi: 10.1146/annurev.micro.57.030502.090759
Review on Antimicrobial Resistance (2015) Securing New Drugs for Future Generations: The Pipeline of Antibiotics. Available at: https://amr-review.org/sites/default/files/SECURING%20NEW%20DRUGS%20FOR%20FUTURE%20GENERATIONS%20FINAL%20WEB_0.pdf (Accessed March 15, 2021).
Rex, J. H., Eisenstein, B. I., Alder, J., Goldberger, M., Meyer, R., Dane, A., et al. (2013). A Comprehensive Regulatory Framework to Address the Unmet Need for New Antibacterial Treatments. Lancet Infect. Dis. 13, 269–275. doi: 10.1016/S1473-3099(12)70293-1
Richards, A. N. (1964). Production of Penicillin in the United States (1941-1946). Nature 201, 441–445. doi: 10.1038/201441a0
Rossolini, G. M., Arena, F., Pecile, P., Pollini, S. (2014). Update on the Antibiotic Resistance Crisis. Curr. Opin. Pharmacol. 18, 56–60. doi: 10.1016/j.coph.2014.09.006
Ruiz, J., Castro, I., Calabuig, E., Salavert, M. (2017). Non-Antibiotic Treatment for Infectious Diseases. Rev. Esp. Quimioter 30 Suppl 1, 66–71.
Rumore, M. M. (2016). Medication Repurposing in Pediatric Patients: Teaching Old Drugs New Tricks. J. Pediatr. Pharmacol. Ther. 21, 36–53. doi: 10.5863/1551-6776-21.1.36
Sardana, D., Zhu, C., Zhang, M., Gudivada, R. C., Yang, L., Jegga, A. G. (2011). Drug Repositioning for Orphan Diseases. Brief Bioinform. 12, 346–356. doi: 10.1093/bib/bbr021
Scannell, J. W., Blanckley, A., Boldon, H., Warrington, B. (2012). Diagnosing the Decline in Pharmaceutical R&D Efficiency. Nat. Rev. Drug Discov. 11, 191–200. doi: 10.1038/nrd3681
Schatz, A., Bugie, E., Waksman, S. A. (2005). Streptomycin, a Substance Exhibiting Antibiotic Activity Against Gram-positive and Gram-negative Bacteria. Clin. Orthop. Relat. Res. 437, 3–6. doi: 10.1097/01.blo.0000175887.98112.fe
Schloss, P. D., Handelsman, J. (2004). Status of the Microbial Census. Microbiol. Mol. Biol. Rev. 68, 686–691. doi: 10.1128/MMBR.68.4.686-691.2004
Schneider, E. K., Reyes-Ortega, F., Velkov, T., Li, J. (2017). Antibiotic-non-Antibiotic Combinations for Combating Extremely Drug-Resistant Gram-negative ‘Superbugs’. Essays Biochem. 61, 115–125. doi: 10.1042/EBC20160058
Sciarretta, K., Rottingen, J. A., Opalska, A., Van Hengel, A. J., Larsen, J. (2016). Economic Incentives for Antibacterial Drug Development: Literature Review and Considerations From the Transatlantic Task Force on Antimicrobial Resistance. Clin. Infect. Dis. 63, 1470–1474. doi: 10.1093/cid/ciw593
Shakhnovich, V. (2018). It’s Time to Reverse Our Thinking: The Reverse Translation Research Paradigm. Clin. Transl. Sci. 11, 98–99. doi: 10.1111/cts.12538
Sharma, P., Towse, A. (2011) New Drugs to Tackle Antimicrobial Resistance: Analysis of EU Policy Options. Available at: https://www.ohe.org/publications/new-drugs-tackle-antimicrobial-resistance-analysis-eu-policyoptions (Accessed March 15, 2021). doi: 10.2139/ssrn.2640028
Silver, L. L. (2011). Challenges of Antibacterial Discovery. Clin. Microbiol. Rev. 24, 71–109. doi: 10.1128/CMR.00030-10
Simoens, S., Cassiman, D., Dooms, M., Picavet, E. (2012). Orphan Drugs for Rare Diseases: Is it Time to Revisit Their Special Market Access Status? Drugs 72, 1437–1443. doi: 10.2165/11635320-000000000-00000
Simpkin, V. L., Renwick, M. J., Kelly, R., Mossialos, E. (2017). Incentivising Innovation in Antibiotic Drug Discovery and Development: Progress, Challenges and Next Steps. J. Antibiot (Tokyo) 70, 1087–1096. doi: 10.1038/ja.2017.124
Singh, S. B., Young, K., Silver, L. L. (2017). What is an “Ideal” Antibiotic? Discovery Challenges and Path Forward. Biochem. Pharmacol. 133, 63–73. doi: 10.1016/j.bcp.2017.01.003
Sinha, M. S., Kesselheim, A. S. (2016). Regulatory Incentives for Antibiotic Drug Development: A Review of Recent Proposals. Bioorg. Med. Chem. 24, 6446–6451. doi: 10.1016/j.bmc.2016.08.033
Stenger, M., Hendel, K., Bollen, P., Licht, P. B., Kolmos, H. J., Klitgaard, J. K. (2015). Assessments of Thioridazine as a Helper Compound to Dicloxacillin Against Methicillin-Resistant Staphylococcus Aureus: In Vivo Trials in a Mouse Peritonitis Model. PLoS One 10, e0135571. doi: 10.1371/journal.pone.0135571
Stewart, P. S. (2015). Prospects for Anti-Biofilm Pharmaceuticals. Pharmaceut. (Basel) 8, 504–511. doi: 10.3390/ph8030504
Strittmatter, S. M. (2014). Overcoming Drug Development Bottlenecks With Repurposing: Old Drugs Learn New Tricks. Nat. Med. 20, 590–591. doi: 10.1038/nm.3595
Sully, E. K., Malachowa, N., Elmore, B. O., Alexander, S. M., Femling, J. K., Gray, B. M., et al. (2014). Selective Chemical Inhibition of Agr Quorum Sensing in Staphylococcus Aureus Promotes Host Defense With Minimal Impact on Resistance. PLoS Pathog. 10, e1004174. doi: 10.1371/journal.ppat.1004174
Sun, L. H. (2015) Scientists Find Way to Disarm Deadly Bacteria Without Destroying the Good Ones in Your Gut (The Washington Post). Available at: https://www.washingtonpost.com/news/to-your-health/wp/2015/09/23/scientists-find-way-to-disarm-deadly-bacteria-without-destroying-the-good-ones-in-your-gut/?utm_term=.0328b1024dce (Accessed March 15, 2021).
Swann, J. P. (1983). The Search for Synthetic Penicillin During World War Ii. Br. J. Hist Sci. 16, 154–190. doi: 10.1017/S0007087400026789
Swinney, D. C., Anthony, J. (2011). How Were New Medicines Discovered? Nat. Rev. Drug Discov. 10, 507–519. doi: 10.1038/nrd3480
Testa, C. A., Johnson, L. J. (2012). A Whole-Cell Phenotypic Screening Platform for Identifying Methylerythritol Phosphate Pathway-Selective Inhibitors as Novel Antibacterial Agents. Antimicrob. Agents Chemother. 56, 4906–4913. doi: 10.1128/AAC.00987-12
Thangamani, S., Mohammad, H., Younis, W., Seleem, M. N. (2015). Drug Repurposing for the Treatment of Staphylococcal Infections. Curr. Pharm. Des. 21, 2089–2100. doi: 10.2174/1381612821666150310104416
Theuretzbacher, U. (2011). Resistance Drives Antibacterial Drug Development. Curr. Opin. Pharmacol. 11, 433–438. doi: 10.1016/j.coph.2011.07.008
Thomas, D. W., Burns, J., Audette, J., Carroll, A., Dow-Hygelund, C., Hay, M. (2017) Clinical Development Success Rates 2006–2015 (BIO Industry Analysis). Available at: https://www.bio.org/sites/default/files/Clinical%20Development%20Success%20Rates%202006-2015%20-%20BIO,%20Biomedtracker,%20Amplion%202016.pdf (Accessed March 15, 2021).
Tobinick, E. L. (2009). The Value of Drug Repositioning in the Current Pharmaceutical Market. Drug News Perspect. 22, 119–125. doi: 10.1358/dnp.2009.22.2.1303818
Tommasi, R., Brown, D. G., Walkup, G. K., Manchester, J. I., Miller, A. A. (2015). Eskapeing the Labyrinth of Antibacterial Discovery. Nat. Rev. Drug Discov. 14, 529–542. doi: 10.1038/nrd4572
Vargesson, N. (2015). Thalidomide-Induced Teratogenesis: History and Mechanisms. Birth Defects Res. C. Embryo Today 105, 140–156. doi: 10.1002/bdrc.21096
Wagner, B. K. (2016). The Resurgence of Phenotypic Screening in Drug Discovery and Development. Expert Opin. Drug Discov. 11, 121–125. doi: 10.1517/17460441.2016.1122589
Waksman, S. A., Reilly, H. C., Johnstone, D. B. (1946). Isolation of Streptomycin-Producing Strains of Streptomyces Griseus. J. Bacteriol. 52, 393–397. doi: 10.1128/JB.52.3.393-397.1946
Walsh, C. (2003). Where Will New Antibiotics Come From? Nat. Rev. Microbiol. 1, 65–70. doi: 10.1038/nrmicro727
Walsh, C. T., Wencewicz, T. A. (2014). Prospects for New Antibiotics: A Molecule-Centered Perspective. J. Antibiot. (Tokyo) 67, 7–22. doi: 10.1038/ja.2013.49
Wright, G. D. (2014). Something Old, Something New: Revisiting Natural Products in Antibiotic Drug Discovery. Can. J. Microbiol. 60, 147–154. doi: 10.1139/cjm-2014-0063
Yarchoan, R., Broder, S. (1987). Development of Antiretroviral Therapy for the Acquired Immunodeficiency Syndrome and Related Disorders. A. Prog. Rep. N. Engl. J. Med. 316, 557–564. doi: 10.1056/NEJM198702263160925
Zheng, W., Thorne, N., McKew, J. C. (2013). Phenotypic Screens as a Renewed Approach for Drug Discovery. Drug Discov. Today 18, 1067–1073. doi: 10.1016/j.drudis.2013.07.001
Keywords: drug repurposing, antibiotics, drug development, translational science, reverse translation
Citation: Boyd NK, Teng C and Frei CR (2021) Brief Overview of Approaches and Challenges in New Antibiotic Development: A Focus On Drug Repurposing. Front. Cell. Infect. Microbiol. 11:684515. doi: 10.3389/fcimb.2021.684515
Received: 23 March 2021; Accepted: 04 May 2021;
Published: 17 May 2021.
Edited by:
Costas C. Papagiannitsis, University of Thessaly, GreeceReviewed by:
Jaroslav Hrabak, Charles University, CzechiaCopyright © 2021 Boyd, Teng and Frei. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Christopher R. Frei, ZnJlaWNAdXRoc2NzYS5lZHU=; orcid.org/0000-0002-0692-4787
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.