 R. Frèdi Langendonk
R. Frèdi Langendonk Daniel R. Neill
Daniel R. Neill Joanne L. Fothergill
Joanne L. Fothergill
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Cell. Infect. Microbiol., 16 April 2021
Sec. Molecular Bacterial Pathogenesis
Volume 11 - 2021 | https://doi.org/10.3389/fcimb.2021.665759
This article is part of the Research TopicPseudomonas aeruginosa Pathogenesis: Virulence, Antibiotic Tolerance and Resistance, Stress Responses and Host-pathogen Interactions.View all 7 articles
P. aeruginosa is classified as a priority one pathogen by the World Health Organisation, and new drugs are urgently needed, due to the emergence of multidrug-resistant (MDR) strains. Antimicrobial-resistant nosocomial pathogens such as P. aeruginosa pose unwavering and increasing threats. Antimicrobial stewardship has been a challenge during the COVID-19 pandemic, with a majority of those hospitalized with SARS-CoV2 infection given antibiotics as a safeguard against secondary bacterial infection. This increased usage, along with increased handling of sanitizers and disinfectants globally, may further accelerate the development and spread of cross-resistance to antibiotics. In addition, P. aeruginosa is the primary causative agent of morbidity and mortality in people with the life-shortening genetic disease cystic fibrosis (CF). Prolonged periods of selective pressure, associated with extended antibiotic treatment and the actions of host immune effectors, results in widespread adaptive and acquired resistance in P. aeruginosa found colonizing the lungs of people with CF. This review discusses the arsenal of resistance mechanisms utilized by P. aeruginosa, how these operate under high-stress environments such as the CF lung and how their interconnectedness can result in resistance to multiple antibiotic classes. Intrinsic, adaptive and acquired resistance mechanisms will be described, with a focus on how each layer of resistance can serve as a building block, contributing to multi-tiered resistance to antimicrobial activity. Recent progress in the development of anti-resistance adjuvant therapies, targeting one or more of these building blocks, should lead to novel strategies for combatting multidrug resistant P. aeruginosa. Anti-resistance adjuvant therapy holds great promise, not least because resistance against such therapeutics is predicted to be rare. The non-bactericidal nature of anti-resistance adjuvants reduce the selective pressures that drive resistance. Anti-resistance adjuvant therapy may also be advantageous in facilitating efficacious use of traditional antimicrobials, through enhanced penetration of the antibiotic into the bacterial cell. Promising anti-resistance adjuvant therapeutics and targets will be described, and key remaining challenges highlighted. As antimicrobial stewardship becomes more challenging in an era of emerging and re-emerging infectious diseases and global conflict, innovation in antibiotic adjuvant therapy can play an important role in extending the shelf-life of our existing antimicrobial therapeutic agents.
P. aeruginosa is a ubiquitous environmental bacterium capable of establishing opportunistic infections in both plants and animals, including humans (Mathee et al., 2008). It is the primary cause of Gram-negative nosocomial infections and of lung infections in people with cystic fibrosis (CF) (Pendleton et al., 2013) (WHO publishes list of bacteria for which new antibiotics are urgently needed), chronic obstructive pulmonary disease (COPD) and non-CF bronchiectasis (Streeter and Katouli, 2016). P. aeruginosa has a large genome of 5.5-7 million base pairs, with remarkable plasticity (Abril et al., 2019; Stover et al., 2000). Its ability to adapt to a range of environmental niches and its high nutritional versatility stems from this genome plasticity. In addition, a large variety of intrinsic and acquired resistance mechanisms exist within the P. aeruginosa population (El Zowalaty et al., 2015). Understanding these mechanisms of resistance and their interplay can help develop targeted therapies for combatting MDR P. aeruginosa infections.
Antibiotic resistance mechanisms can be broadly divided into three categories; intrinsic, acquired and adaptive. Intrinsic resistance mechanisms are those genetically encoded in the core genome of the organism, whereas adaptive resistance mechanisms are those induced by environmental stimuli, and acquired resistance arises from gain of resistance genes from other organisms or as a consequence of selection of advantageous mutations (Tenover, 2006).
Intrinsic resistance mechanisms of P. aeruginosa include its low outer membrane permeability (12- to 100- fold lower than that of Escherichia coli), the presence of antibiotic efflux pumps and β-lactamases, such as OXA-50 and AmpC (Bellido et al., 1992; Slama, 2008; Lister et al., 2009; Szczepanowski et al., 2009). Acquired resistance mechanisms from horizontal gene transfer include acquisition of transferable aminoglycoside modifying enzymes and β-lactamases, while acquired resistance as a result of de novo mutational events often takes the form of overexpression of efflux pumps and β-lactamases, along with decreased expression or modification of target sites and porins (Meletis and Bagkeri, 2013). Acquired resistance through adaptive mutations is common in CF isolates due to the prolonged periods of selective pressure during extended antibiotic treatment of chronic infection and the actions of host immune effectors (Oliver et al., 2000; Dettman et al., 2016). Adaptive resistance mechanisms are those that are induced through external stimuli, such as stress factors and the presence of certain antibiotics. This is different from acquired mutational resistance as adaptive resistance is transient and unstable. Adaptive resistance mechanisms are not permanent, unlike mutational events, and become inactive upon removal of the stress factor (Moradali et al., 2017). Adaptive resistance often involves regulatory pathways and leads to altered gene expression, changes in protein production or target alteration. For example, two-component signaling systems (TCSS), MexXY induction and biofilm formation (Hocquet et al., 2003; Fernández et al., 2010; Sun et al., 2018a; Coleman et al., 2020).
In this review, we will discuss the different resistance mechanisms P. aeruginosa employs to evade antibiotic action. Intrinsic resistance mechanisms, such as low-outer membrane permeability, efflux, lipopolysaccharide (LPS) modification and the bacterial enzyme AmpC, will be described, with a focus on how these contribute to high-level resistance via interplay with acquired mutations and adaptive mechanisms. In addition, we will outline the roles of bacterial enzymes and commonly acquired resistance mechanisms, as well as phenotypic contributions to resistance, such as motility and biofilm formation. Novel therapeutic efforts that directly target these resistance mechanisms will be reviewed and future priorities discussed.
Antibiotics must cross the bacterial membrane in order to act on intracellular targets (Figure 1) (Tipper, 1985; Tenson et al., 2003; Jacoby, 2005; Shakil et al., 2008). Accumulation of antibiotics in the cell is intrinsically diminished in P. aeruginosa due to its low outer membrane permeability (Lambert, 2002). The relative dearth of porins within the outer membrane (OM) of P. aeruginosa decreases the rate at which antibiotics can penetrate the cell (Trias et al., 1989). Porins not only function in transporting nutrients and other molecules across the membrane, but they also play a role in signaling, adhesion and stability of the membrane (Achouak et al., 2006). P. aeruginosa has 26 different types of porin, of which OprF is the most abundant in P. aeruginosa and has been implicated in a variety of functions (Chevalier et al., 2017).
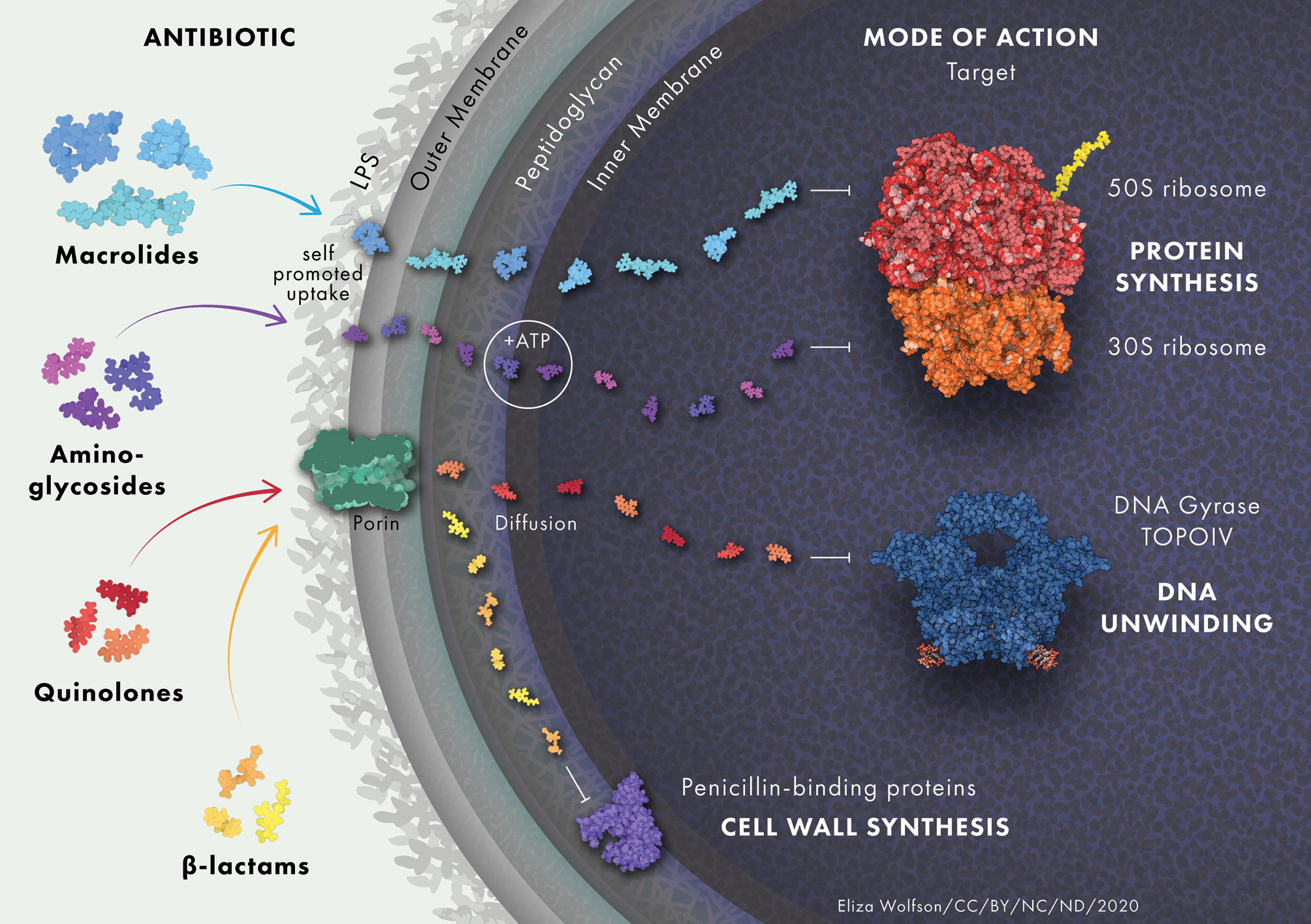
Figure 1 Antibiotic uptake in P. aeruginosa. Aminoglycosides diffuse through the outer membrane due to electrostatic interactions between the positively charged aminoglycoside and the negatively charged LPS. They undergo rapid energy-dependent accumulation into the cell with the use of electron transport and ATP hydrolysis. Once inside the cell, aminoglycosides bind to the 30S subunit of ribosomes to inhibit protein synthesis. β-lactam antibiotics target trans-peptidases on the cytoplasmic membrane that play a vital role in the assembly and cross-linking of cell wall peptidoglycan. Macrolides diffuse across the lipid bilayer due to their hydrophobic nature. They bind to the 50S ribosomal subunit and cause the dissociation of peptidyl-tRNA from the ribosome inside the cell. Quinolones inhibit DNA gyrase and DNA topoisomerase IV in Gram-negative and Gram-positive bacteria, respectively, leading to double stranded DNA breaks. Protein and drug structures were generated with Protein Imager (Tomasello et al., 2020).
OprF is present in two forms, the highly abundant two-domain closed channel and the single domain open channel. The open channel conformation occurs in <5% of the OprF protein population (Sugawara et al., 2012). The two-domain closed channel conformation is thought to interact with peptidoglycan and therefore to play a structural role by stabilizing the OM (Hancock et al., 1981; Woodruff and Hancock, 1989). The single domain open channel spontaneously transitions between different sub-conformations that are weakly conductive and exists for only a short time in its fully open state. The low occurrence of the open channel and its weak conductive state aid in the low permeability of P. aeruginosa (Nestorovich et al., 2006). Clinical OprF deficient mutants of P. aeruginosa with increased antibiotic resistance have been observed (Piddock et al., 1992). However, several studies have shown that complete loss of OprF alone does not seem to be a major route to antibiotic resistance amongst clinical isolates (Pumbwe et al., 1996; Brinkman et al., 1999; Pumbwe and Piddock, 2000). The direct role of OprF in antibiotic resistance is somewhat ambiguous, despite being implicated in quorum sensing and biofilm formation (Chevalier et al., 2017). Interestingly, OprF is detectable in CF patient sputum samples and is upregulated 40-fold in anaerobic conditions, in comparison to aerobic conditions. It has been found to be necessary for growth in the anaerobic environment of CF sputum (Yoon et al., 2002). Overexpression of OprF was also observed in a P. aeruginosa artificial sputum biofilm model of the CF lung and was shown to be necessary for the tight microcolony formation that occurs during the early stages of biofilm formation in the CF lung (Sriramulu et al., 2005). Therefore, although the direct role of OprF in antimicrobial resistance may be ambiguous, there is evidence of a wider role in the establishment of other P. aeruginosa resistance mechanisms during lung infections.
OprD is the second major porin protein in P. aeruginosa and one of the most well-characterized, due to its involvement in the entry of carbapenem antibiotics, mainly imipenem and meropenem (Triass and Nikaido, 1990; Yoneyama et al., 1995). Carbapenem resistance has been linked to mutations causing downregulation of oprD expression and to OprD amino acid substitution mutations found in hypermutator isolates (Richardot et al., 2015).
OprD has been found in high abundance in biofilm outer membrane vesicles (OMVs) (Park et al., 2015). OMVs bud from the outer-membrane and are filled with periplasmic components. They have several functions that serve to enhance bacterial survival, such as modulating host immunity, delivering virulence factors, acquiring nutrients and contributing to structural support in biofilms (Schwechheimer and Kuehn, 2015). OprD-rich OMVs are thought to absorb carbapenems through OprD, leading to accumulation in the OMV and simultaneously lowering the carbapenem concentration in the bacterial cell (Chevalier et al., 2017). Similarly, P. aeruginosa may package OprD into OMVs to decrease cellular levels of this porin and thereby reduce the passage of carbapenems across the membrane and into the cell (Park et al., 2015).
Chronic P. aeruginosa infection in CF can lead to reduced oxygen tension, due to nearby polymorphonuclear leukocyte activity, causing some bacteria to grow anaerobically (Hassett et al., 2009; Kolpen et al., 2017). Increased antibiotic tolerance is observed in P. aeruginosa that grow in well established, anoxic biofilms (Hill et al., 2005). Several studies have found strongly decreased expression of oprD in 48-hour and 96-hour anoxic biofilms, which may partly explain the enhanced resistance to carbapenems in biofilm (Dötsch et al., 2012; Tata et al., 2016). This is in contrast to oprF, which is upregulated under anoxic environments such as the CF lung (Bukhari and Aleanizy, 2020). Increased expression of oprF would not substantially increase membrane permeability, as the closed channel is the most common form of OprF. The increase in closed channel OprF porins along with the decrease in oprD expression may work in conjunction to further lower outer membrane permeability in P. aeruginosa and protect the cell from antibiotic permeation, although this has not been experimentally determined. Alternatively, the benefit derived from increased expression of oprF in the CF lung may relate to its role in QS signaling and/or as a host immune system sensor (Fito-Boncompte et al., 2011).
Khuata et al. found that sialylation occurs in multiple porin proteins in the presence of normal human serum. OprD was identified as a sialoglycoprotein and sialylation of OprD resulted in a decreased interaction with β-lactam antibiotics in comparison to non-sialylated OprD. It was also found that piperacillin and ceftazidime were able to penetrate non-sialylated OprD much more readily than the sialylated form. Accordingly, in the presence of normal human serum, increased survival in the presence of these antibiotics is observed, as a result of OprD sialylation (Khatua et al., 2014). These observations highlight the effect that mode of growth and host factors can have on the susceptibility of P. aeruginosa to antibiotics.
OprH is a 21.6 kDa porin protein and is the smallest porin found in P. aeruginosa (Kucharska et al., 2016). OprH interacts with LPS through electrostatic interactions and is implicated in polymyxin resistance through the low Mg2+ concentration sensing PhoPQ and PmrAB TCSS (Edrington et al., 2011; Kucharska et al., 2016). The genes for the PhoPQ TCSS form an operon with oprH. However, studies with oprH knockout P. aeruginosa have demonstrated that the porin is not essential for polymyxin resistance. Instead, OprH is thought to play a supplementary role in polymyxin resistance through stabilizing the OM during Mg2+ starvation (Macfarlene et al., 2000). OprH has also been linked with gentamicin resistance in small colony variants of P. aeruginosa, although it is not clear whether upregulation of the operon alone was responsible for the resistance phenotype, as several other genes were also upregulated (Wei et al., 2011).
Bacterial efflux pumps are categorized into five families, the two ancient super families that are made up of the adenosine triphosphate binding cassette (ABC) and the major facilitator super family (MFS), and three smaller families that consist of the multidrug and toxic compound extrusion family (MATE), the small MDR family (SMR) and the resistance nodulation-cell division family (RND) (Marquez, 2005). RND efflux pumps are the main multidrug efflux systems responsible for antibiotic extrusion and resistance in P. aeruginosa (Poole, 2001). P. aeruginosa possesses at least 12 RND efflux pumps, of which four are of clinical relevance and are the main contributors to antibiotic resistance through efflux (Stover et al., 2000; Alcalde-Rico et al., 2018), namely, MexAB-OprM, MexCD-OprJ, MexEF-OprN and MexXY-OprM (Dreier and Ruggerone, 2015) (Figure 2). All RND efflux pumps cross the cytoplasmic membrane, the periplasm and the OM (Nikaido, 2010). The RND efflux pump complex is comprised of the secondary active efflux pump protein embedded in the inner membrane, the outer membrane factor protein, and a membrane fusion protein that extends through the periplasm and connects the two (Daury et al., 2016; Hernando-Amado et al., 2016). Efflux pumps are powered by proton motor force and are able to export drugs from both the periplasm and the cytosol into the environment (Paulsen, 2003; Poole, 2008). Efflux and the low-permeability OM work in a cooperative manner, whereby those antibiotics that manage to permeate the OM are quickly extruded by efflux pumps, back into the environment (Nikaido, 2010).
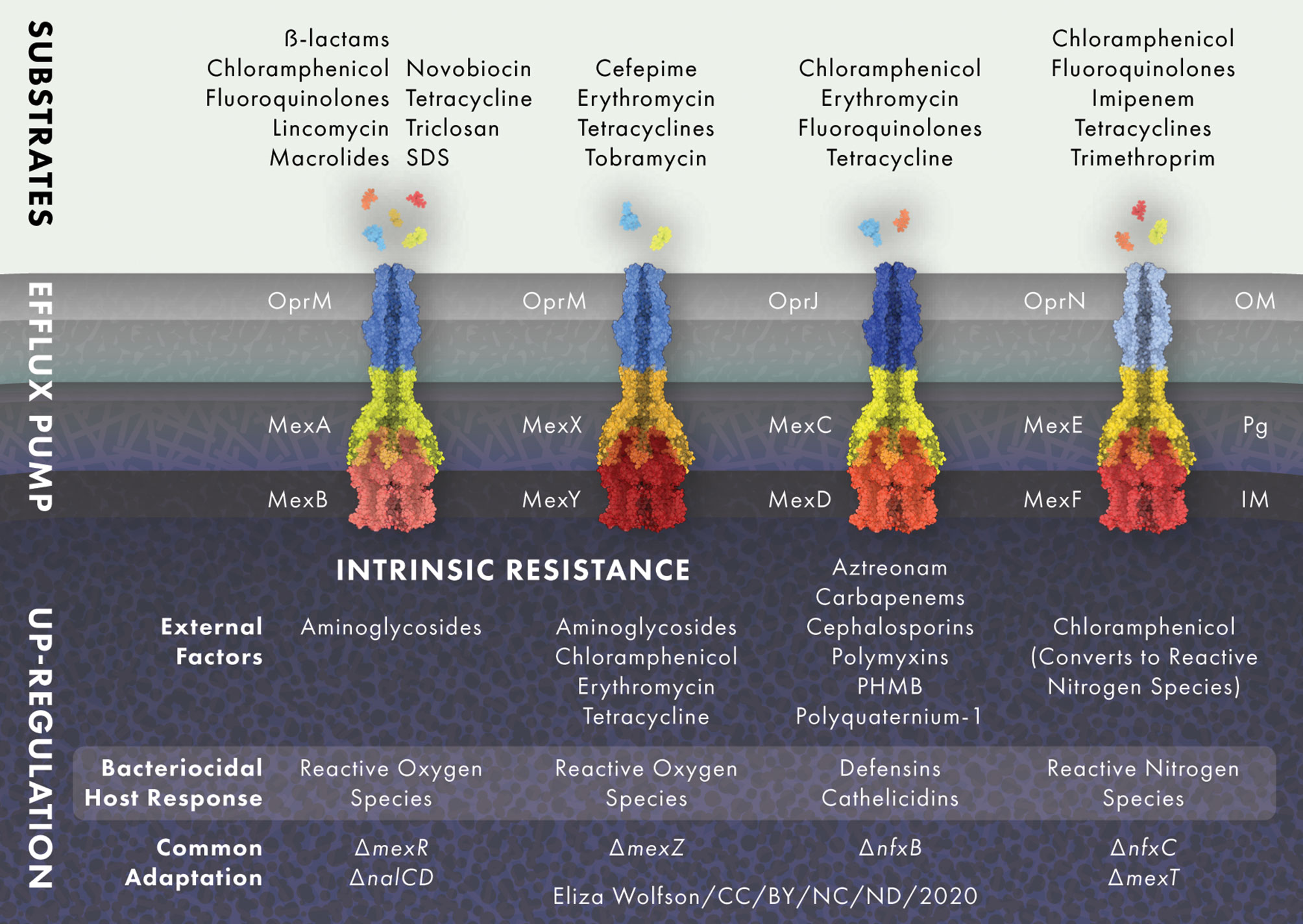
Figure 2 RND efflux pump expression is modulated in response to external stress. MexAB-OprM and MexXY-OprM are positively regulated by reactive oxygen species (ROS) and MexEF-OprN is induced by reactive nitrogen species (NOS), whilst MexCD-OprJ responds to membrane damaging agents and MexXY-OprM to ribosome blocking substances such as chloramphenicol, erythromycin, tetracycline and aminoglycoside antibiotics. Common adaptations leading to up-regulation of efflux pumps include; loss of active transcriptional regulators mexR, nalC and nalD for MexAB-OprM, loss of the transcriptional repressor mexZ for MexXY-OprM, loss of transcriptional regulator nfxB for the MexCD-OprJ efflux system and nfxC type mutations and/or loss of active transcriptional activator mexT for MexEF-OprN. Protein and drug structures were generated with Protein Imager (Tomasello et al., 2020).
Efflux pumps not only function in antimicrobial efflux, but are also recruited in response to cellular stress. Stress response signals including host factors, detergents and other endogenous inducers of the bacterial stress response may function as pressures for the selection of antimicrobial resistant efflux-expressing mutants, even in the absence of antimicrobials (Poole, 2014; Dreier and Ruggerone, 2015). For example, inflammation in the CF lung means P. aeruginosa is continuously exposed to reactive oxygen species (ROS), which is likely to drive the high prevalence of MexAB-OprM and MexXY-OprM over-expressing mutants (Poole, 2005) Additionally, in vitro studies have shown that ROS exposure selects for MexXY-OprM expressing, aminoglycoside resistant mutants (Fraud et al., 2008).
The MexAB-OprM efflux pump is constitutively active in P. aeruginosa wild-type strains and, along with other RND efflux systems, causes low level resistance but can also significantly contribute to the development of multi-drug resistance (Goli et al., 2016). MexAB-OprM extrudes carbapenems, chloramphenicol, fluoroquinolones, lincomycin, macrolides, novobiocin, penicillins, tetracyclines and all β-lactams (except imipenem and biapenem), as well as the antiseptic triclosan and the surfactant sodium dodecyl sulfate. MexAB-OprM null mutants are markedly susceptible to all of the aforementioned antimicrobials, whilst mutants that overproduce MexAB-OprM show high levels of resistance (Masuda and Ohya, 1992; Li et al., 1995; Srikumar et al., 1998; Köhler et al., 1999; Chen et al., 2016). MexAB-OprM consists of an inner membrane protein, MexA, and a fusion protein, MexB, that links MexA to the outer membrane protein OprM (Nehme et al., 2004). The efflux pump proteins are encoded in an operon, the expression of which is regulated by MexR (Poole et al., 1996a; Andrésen et al., 2010). MexR is encoded upstream of the mexAB-oprM operon and acts as a transcriptional repressor by binding to the mexA-mexR intergenic region near the promotors of mexR and a secondary promotor for mexA (Poole et al., 1996a; Evans et al., 2001). Loss of a functioning MexR causes hyperexpression of mexAB-oprM. Mutations in mexR found in clinical P. aeruginosa isolates can lead to the expression of MexR protein that is no longer able to dimerize or bind to DNA to repress mexAB-oprM expression. In addition, mutations leading to premature termination of the MexR peptide resulting in complete absence of the production of a functioning MexR have been identified in clinical isolates (Choudhury et al., 2016; Pan et al., 2016).
MexCD-OprJ is not normally expressed in wild-type strains and is functional in nfxB mutants only, it is therefore not classified as contributing to the natural resistance of P. aeruginosa (Poole et al., 1996b; Morita et al., 2001). Antibiotic substrates of the MexCD-OprJ efflux system are chloramphenicol, erythromycin, fluoroquinolones and tetracycline. The NfxB protein negatively regulates MexCD-OprJ and different mutations within the nfxB gene lead to varying levels of MexCD-OprJ expression, leading to clinical isolates with different levels of resistance to antimicrobials (Masuda et al., 1996).
The MexEF-OprN efflux system extrudes chloramphenicol, fluoroquinolones, tetracycline, trimethoprim and imipenem (Maseda et al., 2000; Sobel et al., 2005). The mexEF-oprN operon is regulated by the transcriptional activator MexT (Köhler et al., 1999). MexT positively regulates mexEF-oprN expression, in contrast to most other RND-efflux systems, which are negatively regulated by a repressor protein (Poole, 2005). However, often wild-type strains of P. aeruginosa carry an inert mexT gene encoding inactive MexT, causing the suppression of mexEF-oprN expression (Maseda et al., 2000). Besides acting as a positive regulator for the expression of the mexEF-oprN efflux system when MexT is active, it also acts as a repressor of oprD transcription. Active MexT leads to a reduction of the OprD porin in the bacterial membrane, causing increased resistance to carbapenem antibiotics (Ochs et al., 1999). The multidrug resistant nfxC-type mutant overexpresses the mexEF-oprN operon and is resistant to chloramphenicol, fluoroquinolones, tetracycline, trimethoprim and imipenem (Köhler et al., 1997).
The MexXY-OprM efflux system contributes to P. aeruginosa intrinsic resistance to aminoglycosides, tetracycline, erythromycin and cefepime (Masuda et al., 2000; Hocquet et al., 2006). MexXY-OprM is regulated by the MexZ repressor protein and mutations in or around mexZ lead to overproduction of MexXY-OprM (Llanes et al., 2004; Vogne et al., 2004). The mexZ gene is one of the most commonly mutated in isolates from people with CF, leading to loss of MexXY-OprM repression and thereby increasing its expression (Smith et al., 2006; Feliziani et al., 2010). Mutations in mexZ that arise during long-term infection in CF can result in resistance to tobramycin, a first-line antibiotic in CF clinics (Marvig et al., 2015; Prickett et al., 2017). One study looking at levels of mexY expression and mexZ mutation found that both were present at a significantly higher frequency in adults with chronic infection than in children with chronic or new infections and that mexZ mutation strongly correlated with aminoglycoside resistance (Prickett et al., 2017). This suggest that mexZ mutation undergoes positive selection during long term chronic infection.
LPS is a central component of the Gram negative bacterial membrane. It serves as the outer leaflet of the OM and is essential for host-pathogen interactions, virulence and antimicrobial resistance (Lam et al., 2011). P. aeruginosa LPS is composed of three domains: the core oligosaccharide, the lipid A portion and the O-antigen region (Gellatly and Hancock, 2013). Modification of LPS is a resistance mechanism that protects the bacterial cell against cationic antimicrobial peptides (CAPs) and polymyxin antibiotics (Baron et al., 2016). LPS modification in response to a low Mg2+ environment, or to exposure to CAPs and polymyxins, is achieved by the addition of a 4-amino-4-deoxy-L-arabinose (L-Ara4N) moiety to the phosphate unit on the lipid A group of the LPS. Modification of the lipid A arabinose subunit leads to a lower net negative charge of the bacterial membrane, which leads to a decrease in electrostatic interactions with positively charged antimicrobials. The decrease in electrostatic interaction between the antibiotic and the membrane prevents the drug from binding the LPS which would normally destabilize and disrupt the membrane (Falagas et al., 2010). LPS modification in P. aeruginosa is regulated by several two-component signaling systems (TCSS): PmrAB, PhoPQ, CprRS and ParRS (Fernández et al., 2010; Fernández et al., 2012). These TCSS regulate the transcription of the arnBCADTEF operon, which encodes the genes responsible for the addition of L-Ara4N to the lipid A group, in response to a variety of environmental signals (Skiada et al., 2011). PmrAB and PhoPQ regulate the activation of arnBCADTEF under Mg2+ deficient growth conditions (McPhee et al., 2003). ParRS and CprRS both play an active part in adaptive resistance against CAPs and can stimulate arnBCADTEF upregulation, independently of each other, in the presence of these peptides (Fernández et al., 2010; Fernández et al., 2012). Mutations in these regulatory TCSS that lead to the constitutive activity of the arn operon confer colistin resistance to P. aeruginosa (Sciuto and Imperi, 2018). One study found that mutations in the pmrB histidine kinase gene of the PmrAB TCSS commonly arise during the development of high level polymyxin resistance in experimental evolution experiments performed with CF isolates (Moskowitz et al., 2012; Jochumsen et al., 2016). Conversely, a missense mutation in pmrB in isolates taken from a murine model of P. aeruginosa chronic respiratory infection has been found to be associated with increased susceptibility to a wide range of antibiotic classes but also with resistance to the host antimicrobial enzyme lysozyme (Fothergill et al., 2014; Bricio-Moreno et al., 2018). High level resistance to polymyxin antibiotics is forged through epistatic interactions between multiple regulatory genes and can occur via several pathways (Jochumsen et al., 2016), but a functional arn operon and the aminoarabinosylation of the lipid A are requirements for polymyxin and colistin resistance in P. aeruginosa (Sciuto and Imperi, 2018).
β-lactam antibiotics interfere with cell wall synthesis and disrupt peptidoglycan recycling by inhibiting penicillin binding proteins (PBP), enzymes responsible for synthesis and maintenance of peptidoglycan (Sauvage et al., 2008; Johnson et al., 2013). β-lactamases are enzymes that interfere with this process by hydrolyzing the β-lactam ring that is present in all β-lactam antibiotics (Santajit and Indrawattana, 2016). β-lactamases are classified according to different systems, the Ambler classification groups enzymes according to amino acid sequence and the Bush-Jacoby-Medeiros system classifies enzymes according to function and phenotype (Bush and Jacoby, 2010).
The β-lactamase AmpC is an Ambler class C antibiotic inactivating enzyme. It contributes to the natural resistance of P. aeruginosa and is constitutively produced at basal levels (Juan et al., 2005). The chromosomal blaAmpCgene is drug inducible with aminopenicillins and cephalosporins, whereupon AmpC inactivates the β-lactam antibiotic through the process of hydrolysis (Masuda et al., 1999). AmpC induction occurs through two different mechanisms: perturbation of the peptidoglycan recycling pathway and through loss of PBP4 (Cavallari et al., 2013). Peptidoglycan is composed of cross-linked, alternating N-acetylglucosamine (GlcNAc) and N-acetylmuramic acid (MurNAc) residues (Vollmer et al., 2008). Peptidoglycan recycling requires the collaboration of PBPs, endopeptidases, carboxypeptidases, N-acetylmuramoyl-l-alanine amidases and lytic transglycosylases (LTs) (Johnson et al., 2013). LT’s cleave the β-1,4 glycosidic linkage between GlcNAc and MurNAc residues during growth and division, to generate new material to be inserted into the peptidoglycan layer. This cleavage causes the release of anhydromuropeptides (anhMPs) (Lee et al., 2013). The anhMPs are transported to the cytoplasm through an inner membrane permease AmpG, where they are processed for recycling back into the peptidoglycan biosynthesis pathway by the β-N-acetylglucosaminidase NagZ and the N-acetylmuramoyl-l-alanine amidase AmpD. Disruption of anhMP processing leads to their accumulation in the cytoplasm where they can bind to AmpR, the transcriptional regulator of AmpC. Two anhMPs, N-acetylglucosamine-1,6-anhydro-N-acetylmuramyl pentapeptide and 1,6-anhydro-N-acetylmuramyl pentapeptide, have been identified as signaling molecules that activate β-lactamase expression in P. aeruginosa (Lee et al., 2016).
Often β-lactamase resistance in clinical P. aeruginosa strains is caused by inactivation of the PBP4 encoding gene dacB, causing ampC overexpression and activation of the CreBC (BlrAB) TCSS (Moya et al., 2009). PBP4 functions to create an anhydromuropeptide (anhMP) that controls AmpR. Loss of PBP4 leads to the disappearance of this specific anhMP from the peptidoglycan recycling pathway, which changes AmpR regulation, thereby inducing ampC expression (Johnson et al., 2013). Upregulation of ampC is commonly observed amongst P. aeruginosa isolates from people with CF, it is selected for by clinical use of β-lactam antibiotics and the presence of hypermutable strains in the CF lung can hasten the emergence of this mutational overexpression (Mark et al., 2011; Llanes et al., 2013; López-Causapé et al., 2013; Aghazadeh et al., 2014).
Metallo β-lactamases (MBLs) belong to Ambler classification group B, they contain zinc in the active site, as opposed to a serine based hydrolytic system (Queenan and Bush, 2007). The most clinically relevant MBLs in P. aeruginosa are the Verona integron-encoded MBLs (VIM) and active-on-imipenem type MBLs (IMP). They are often located on integrons along with other resistance genes, which allows these genes to integrate into the chromosome or a plasmid and disseminate through bacterial populations (Palzkill, 2013). The presence of MBL causes high-level resistance against carbapenem antibiotics (Feng et al., 2017).
Extended spectrum β-lactamases (ESBL) hydrolyze oxyimino-aminothiazolyl cephalosporins (cefotaxime, cefuroxime, cefepime, ceftriaxone and ceftazidine) as well as penicillins and other cephalosporins, excluding cephamycins (Livermore and Brown, 2001). The most common ESBL in P. aeruginosa are the Ambler class A SHV-, VEB-, PER- and GES-type enzymes and the OXA-type enzymes from Ambler class D (Ortiz de la Rosa, et al., 2019). The presence of ESBLs confers a higher level of resistance to extended spectrum cephalosporins than AmpC, impermeability and efflux pump hyper-expression combined (Jiang et al., 2006). However, ESBL enzymes can be inhibited by clavulanic acid, sulbactam and tazobactam (Paterson and Bonomo, 2005). ESBL producing P. aeruginosa have been frequently isolated, although their detection is complicated by the activity of chromosomally encoded β-lactamases in P. aeruginosa (Livermore and Brown, 2001; Laudy et al., 2017).
Aminoglycoside modifying enzymes (AME) are capable of catalyzing the modification of the -OH or -NH2 groups of the 2-deoxystreptamine nucleus or the sugar moieties of aminoglycoside antibiotics (Ramirez and Tolmasky, 2010). There are three types of AMEs; acetyltransferases (AACs), nucleotidyltransferases (ANTs) and phosphorlyl transferases (APHs). All of these decrease the binding affinity of the antibiotic to its target by transferring a functional group onto the aminoglycoside (Azucena and Mobashery, 2001). All three types of AMEs have been observed in P. aeruginosa and are common determinants of aminoglycoside resistance in non-CF isolates (Poole, 2011). AMEs are not, however, the most common aminoglycoside resistance mechanism in people with CF. Instead, impermeability and efflux are the more commonly observed events (Islam et al., 2009). However, studies have observed a variable prevalence of these enzymes in between 5.6% and 52% of isolates (MacLeod et al., 2000; Aghazadeh et al., 2013).
16S rRNA methylases are another group of modifying enzymes carried by P. aeruginosa. These enzymes are carried on plasmids and are thought to have come from an aminoglycoside producing organism that utilizes these enzymes to protect its own ribosomes from aminoglycoside action (Doi and Arakawa, 2007). 16S rRNA methylases cause the methylation of a specific nucleotide of the 16s rRNA A-site, to protect the ribosome from aminoglycoside activity (Doi and Arakawa, 2007). Strains carrying 16s rRNA methylase genes show a high level of resistance to most clinically relevant aminoglycoside antibiotics. The prevalence of these enzymes is not common in CF and other respiratory associated infection isolates (Yokoyama et al., 2003; Tada et al., 2013; Francisco et al., 2015) However, rmtD carrying P. aeruginosa have been reported in Africa, Asia, Europe and South America and are, along with ESBLs, often associated with a pan-resistant genotype (Fontes et al., 2011; Urbanowicz et al., 2019).
DNA gyrase and topoisomerase IV are both involved in DNA replication and management of chromosome integrity (Drlica and Zhao, 1997). DNA gyrase functions to cleave double stranded DNA, introduce negative supercoiling and re-ligate the strand in an ATP-dependent reaction (Levine et al., 1998). Topoisomerase IV functions by decatenating and relaxing positively supercoiled DNA (Corbett et al., 2005). Fluoroquinolones like ciprofloxacin act on DNA gyrase and topoisomerase IV to prevent the re-ligation step, leaving the DNA cleaved and thereby acting as a bacteriostatic antibiotic, blocking DNA replication (Drlica, 1999).
Resistance against ciprofloxacin occurs most commonly through mutations in the DNA gyrase subunits GyrA and GyrB or the topoisomerase subunits ParC/ParD but also through the overexpression of efflux (Yang et al., 2015). Mutations in gyrA and gyrB are commonly identified in CF associated infections and are likely to arise during prolonged antibiotic treatment with ciprofloxacin (Fothergill et al., 2010; Marvig et al., 2015). The most frequently observed mutations in both CF and non-CF isolates, as well as in in vitro experiments, are the substitution of an amino acid at position 83 in GyrA from threonine to isoleucine and an amino acid substitution of serine to leucine at position 87 in ParC (Higgins et al., 2003; Lee et al., 2005; Bruchmann et al., 2013). The second most common mutation is GyrA position 87, where tyrosine is substituted by asparagine, glycine or tyrosine (Wydmuch et al., 2005). These positions of GyrA are important for the binding of ciprofloxacin to DNA gyrase and the amino acid substitutions greatly lower the affinity of this binding (Willmott and Maxwell, 1993; Aldred et al., 2014). Resistance to ciprofloxacin is higher when both mutations in gyrA and parC are present, in comparison to only gyrA mutation (Pasca et al., 2012). However, it is thought that gyrA mutation is necessary for high level resistance against ciprofloxacin, due to the strong affinity of the native form of the protein for this antimicrobial. When only gyrB, parC or parD mutations are present or when efflux is the main mechanism of ciprofloxacin resistance, only low level resistance is observed (Rehman et al., 2019). A combination of efflux, gyrAB and parCD mutation leads to a higher level of resistance and again highlights that resistance in P. aeruginosa is complex, often involving multiple mechanisms (Dunham et al., 2010; Bruchmann et al., 2013).
Mutations arising in the fusA1 gene lead to amino acid substitutions in a critical part of the translational machinery, elongation factor G (EF-G). Mutations within different domains of EF-G have been identified in clinical strains and are linked to decreased aminoglycoside susceptibility. Mutations in fusA1 have been reported in both CF and non-CF isolates globally (Del Barrio-Tofiño et al., 2017; Rees et al., 2019). The mutation is often paired with upregulation of efflux, mainly the MexXY-OprM efflux pump, leading to strong aminoglycoside resistance. However, on its own, fusA1 mutation can cause strains to be 4- to 8- fold more resistant to aminoglycosides. This was shown in vitro, where three fusA1 mutants in domains II, III and IV were created (Bolard et al., 2017). The molecular mechanism linking mutations within EF-G to increased aminoglycoside resistance remains unclear. Aminoglycosides do not bind to or target EF-G, thus resistance to aminoglycosides must be an indirect effect of EF-G alteration (Gutierrez et al., 2013).
Bacterial motility is a vital part of P. aeruginosa pathogenicity and plays a role in host colonization and establishment of infection (Yeung et al., 2012). P. aeruginosa employs several different forms of motility that are environmentally dependent and often require the use of type IV pili, flagella or rhamnolipid surfactants (Overhage et al., 2007; Sun et al., 2018a; Sun et al., 2018b). RNA-seq and transposon mutant studies have shown that both swarming and surfing phenotypes are associated with significant changes in gene expression, leading to an increase in antibiotic resistance. Swarming motility was linked to the altered regulation of 1,581 genes, including 104 regulatory genes, including transcription factors, TCSS and sigma factors (Coleman et al., 2020). Surfing motility was linked to altered regulation of up to 2,078 genes and out of these, 31 resistome genes were identified which were known to cause an increase in antibiotic susceptibility when mutated (Sun et al., 2018a).
Both swarming and surfing play a role in adaptive resistance. They are environmentally dependent and associated with conferring a high level of antibiotic resistance in comparison to alternative motility phenotypes (Wang et al., 2013; Sun et al., 2018a; Coleman et al., 2020). Swarming cells have been shown to be significantly more resistant to aminoglycosides, β-lactams, chloramphenicol, ciprofloxacin, tetracycline, ethoprim, erythromycin and azithromycin (Coleman et al., 2020). Surfing cells however, showed significantly higher resistance to polymyxins, aminoglycosides, fluoroquinolones, tetracycline, chloramphenicol, trimethoprim and several β-lactams, but not to macrolides (Sun et al., 2018a). On semi-solid surfaces with low nitrogen concentrations, P. aeruginosa is known to utilize swarming motility. The conditions under which P. aeruginosa exhibits swarming motility appear to mimic those of the lung, where semi-solid mucous overlays epithelial cells. Likewise, the CF and bronchiectasis lungs contain high levels of mucin, the component necessary for surfing motility (Sun et al., 2018a). Although P. aeruginosa motility is often lost in long term chronic illness such as in CF, a plethora of genetic heterogeneity exists in the bacterial populations residing in the CF lung. Furthermore, swarming and surfing motility may occur during early colonization and adaptation to the CF lung.
The biofilm mode of growth is a major impediment in the struggle to eradicate P. aeruginosa infection from the CF lung, due to the increased ability of bacteria in biofilms to withstand antibiotic treatment (Høiby et al., 2010). Biofilms are composed of bacteria surrounded by extracellular polymeric substances like exopolysaccharides, extracellular DNA and polypeptides (Rasamiravaka et al., 2015). This can lead to an increase in tolerance to antimicrobial agents of 100-1000 times, compared to planktonic cells (Ceri et al., 1999). Contributing features include quorum sensing, decreased ability to penetrate biofilm, presence of oxygen gradients, altered metabolism and slow bacterial growth rate (Olsen, 2015). Biofilms are common in chronic P. aeruginosa infection of the CF lung but have also been shown to be of significance in patients with COPD, bronchiectasis and chronic wounds (Costerton, 2001; Parameswaran and Sethi, 2012; Chalmers and Hill, 2013; Hassett et al., 2014; Rybtke et al., 2015). Airway mucins are found in abundance in people with CF, COPD and bronchiectasis. In vitro studies have shown that the presence of airway mucins are fundamental for the development of biofilm structures with enhanced tolerance to antimicrobials. Further, mucin may serve as a suitable attachment surface for P. aeruginosa biofilm formation (Landry et al., 2006; Müller et al., 2018).
Iglesias et al. investigated antibiotic pharmacodynamics within biofilm structures in the context of CF. Bacterial counts, metabolic activity and biomass of PAO1 biofilms grown in artificial sputum media (ASM) or trypticase soy-based medium were compared. They found that bacteria in ASM reached the same CFU and metabolic activity as biofilms formed in trypticase soy-based medium, although ASM biofilms grew slower and had a marginally higher biomass. When both biofilms were subjected to antibiotic treatment, ASM grown biofilms were substantially more resistant to tobramycin and colistin, but not ciprofloxacin and β-lactams (Diaz Iglesias and Van Bambeke, 2020). These studies indicate that environmental factors play an important role in determining biofilm structure and antimicrobial resistance. For comprehensive reviews on P. aeruginosa biofilms refer to: Olsen, 2015 and Maurice, Bedi and Sadikot, 2018 (Olsen, 2015; Maurice et al., 2018).
Multidrug resistance is often the result of the acquisition of external resistance genes and/or through mutational resistance (Diggle and Whiteley, 2020). Combining resistance genes encoding mechanisms previously described here, along with intrinsic resistance, can lead the development of MDR and XDR P. aeruginosa. MDR and XDR P. aeruginosa arise due to the compilation of resistance mechanisms, amassing building blocks into a barrier of resistance strategies that protect the bacteria from antimicrobial assault. Intrinsic resistance forms the base layer of this barrier, with additional acquired and adaptive resistance mechanisms forming further layers, collectively constructing a robust barrier against antimicrobials (Figure 3). Antibiotic resistance genes can quickly and easily be disseminated through P. aeruginosa populations en masse, via megaplasmids (>420kb). Megaplasmids contain dynamic accessory genomes where frequent recombination and duplication events take place, leading to diverse and adaptive multidrug resistance traits (Cazares et al., 2020). Accordingly, resistance to antimicrobial agents in P. aeruginosa clinical isolates is highly complex, with frequent interplay between intrinsic, adaptive and acquired resistance mechanisms. AmpC, low outer membrane permeability and efflux systems often work together in resistance to carbapenems, chloramphenicol, fluoroquinolones, macrolides, penicillins, tetracyclines and β-lactams and resistance may be enhanced through the accumulation of mutations leading to up- or down- regulation of each of these systems. The outer membrane porin OprH works in conjunction with the two-component signaling systems PhoPQ and PmrAB in modifying the bacterial LPS to regulate protection to polymyxin antibiotics. Protection against polymyxin antibiotics is additionally mediated via TCSS ParRS and/or CprRS in the presence of cationic antimicrobial peptides. Aminoglycoside resistance is achieved through mechanisms such as aminoglycoside modifying enzymes, fusA1 mutation, 16S rRNA methylation, along with MexXY-OprM upregulation. Likewise, gyrA and parC mutation leading to altered DNA gyrase and topoisomerase IV leads to fluoroquinolone resistance which can be heightened through the cooperation of MexAB-OprM and/or MexCD-OprJ, as well as MexEF-OprN. In addition, the presence of β-lactamases such as ESBLs and MBLs may further enhance carbapenem, cephalosporin and penicillin resistance. A perfect storm of highly resistant P. aeruginosa and a “dry pipeline” of traditional antimicrobials are driving innovation in novel therapeutic approaches that directly target resistance mechanisms. Counteracting these mechanisms could prolong the life of existing antimicrobials.
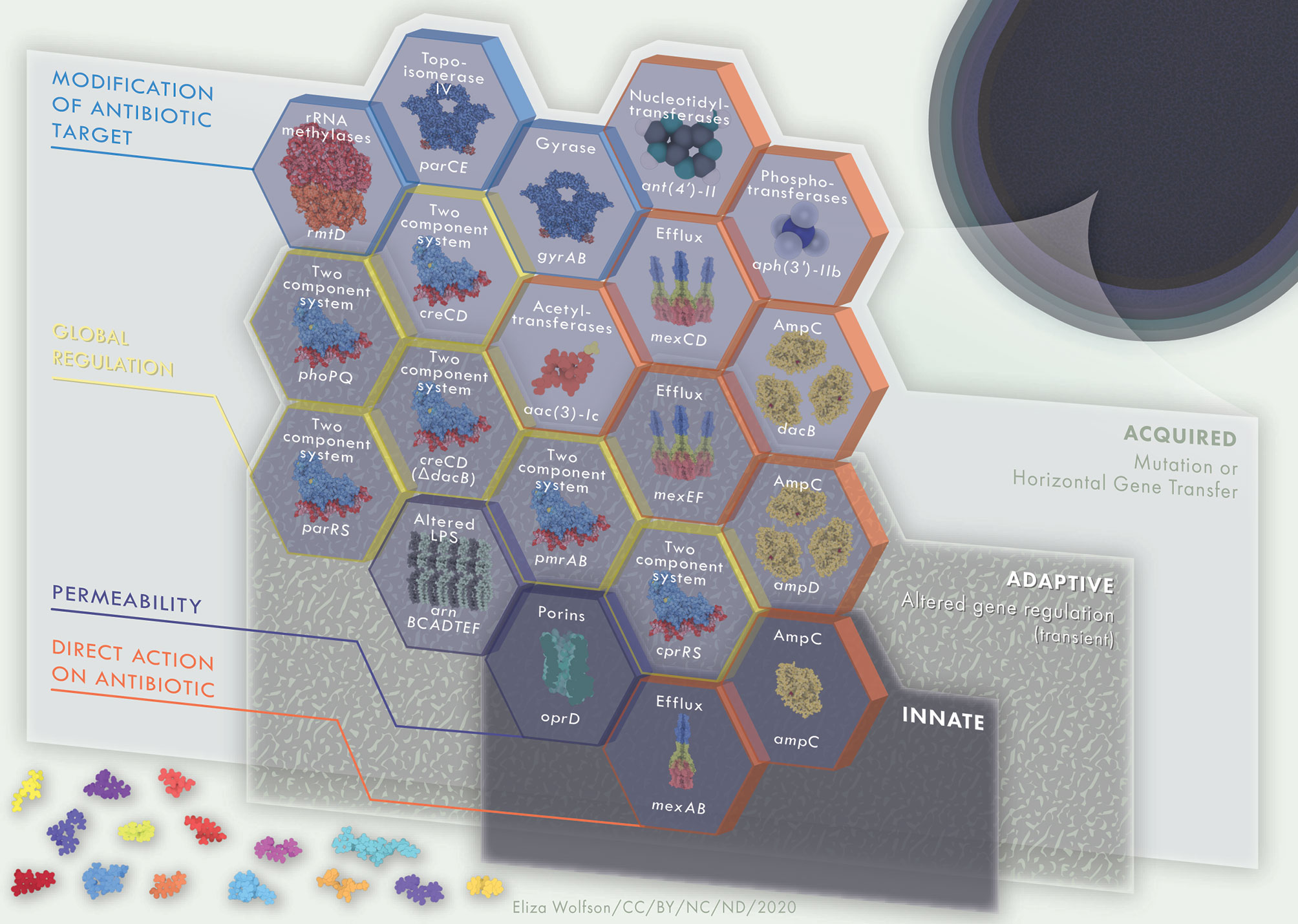
Figure 3 Multi-layered, interacting resistance mechanisms in P. aeruginosa. Innate (intrinsic) resistance mechanisms are encoded in the core genome of the organism, such as low outer membrane permeability, Mex-type efflux pumps and AmpC β-lactamase. Collectively, these comprise basal level resistance to antimicrobials, a foundation on which a variety of adaptive and acquired mechanisms of resistance may serve as building blocks to further enhance AMR in P. aeruginosa. Adaptive resistance mechanisms, including two-component regulatory systems, are environmentally dependent and will be expressed under certain conditions only. Mechanisms of resistance that are acquired, such as antibiotic modifying enzymes or mutations leading to antibiotic target modifications are strain dependent. The building blocks of innate, adaptive and acquired mechanisms of resistance contribute to a strong and multi-faceted protection against antimicrobial activity. Hexagon building blocks of resistance mechanisms are colored according to mechanism type; direct action on antibiotic, permeability, global regulation, modification of antibiotic target. Upper labels on hexagon building blocks describe resistance mechanism whilst lower labels define examples of each such systems. Protein and drug structures were generated with Protein Imager (Tomasello et al., 2020).
Increasing OM permeability to hydrophobic and amphiphilic compounds challenges the issue of intrinsic low-outer membrane permeability (Tümmler, 2019). For example, polymyxin B nonapeptide causes a 2- to 40-fold increase in susceptibility to ciprofloxacin, norfloxacin, and ofloxacin and 80- to 200-fold increase in susceptibility to nalidixic acid (Kubesch et al., 1987). Nalidixic acid is the precursor to ciprofloxacin, norfloxacin, and ofloxacin, and is not ordinarily more efficacious than the optimized antibiotics currently used in clinical practice. However, when the membrane becomes more permeable through polymyxin B nonapeptide treatment, nalidixic acid becomes a more powerful anti-pseudomonal (Tümmler, 2019). Polymyxin B nonapeptide is highly toxic and has therefore never been considered for clinical application (Tsubery et al., 2000). However, more than 30 years after the first report of OM sensitizers, three have been approved for clinical studies; the anti-protozoal pentamidine (Stokes et al., 2017) and the polymyxin B analogues SPR206 and SPR741 (Corbett et al., 2017; Vaara, 2019). None of these OM sensitizers, with the exception of SPR206, have been proven to have promising anti-pseudomonal activity. SPR206 is active against P. aeruginosa with a similar potency to polymyxin B and is currently undergoing phase-one clinical trial (Zhang et al., 2020). OM sensitizers appear to be a promising approach to resistance that can by-pass intrinsic, acquired and spontaneous resistance (Macnair and Brown, 2020). However, further investigation into additional non-toxic compounds that act on the P. aeruginosa OM is essential, especially for the treatment of polymyxin resistant isolates.
The most widely researched P. aeruginosa efflux-pump inhibiter (EPI) is Phe-Arg-β-naphthylamide (PAβN), a broad-spectrum peptidomimetic compound capable of interfering with all four clinically relevant P. aeruginosa RND efflux pumps. PAβN potentiates chloramphenicol, fluoroquinolones, macrolides, ketolides, oxazolidinones and rifampicin but not aminoglycosides or β-lactams (Lomovskaya and Bostian, 2006). The proposed mechanism of action for PAβN is that it functions as a substrate of the Mex-series efflux pumps and outcompetes the antibiotic for extrusion, preventing the antibiotic from leaving the cell (Mahmood et al., 2016). However, PAβN and derivatives of this compounds are not yet approved, as adverse toxicology and pharmacokinetic profiles were identified during phase 1 clinical trials (Renau et al., 2003). Instead, PAβN is solely utilized for research on EPI’s and AMR in vitro, rather than as a therapeutic, and may be used to validate the discovery of future EPI’s.
The pyridopyrimidine derivative D13-9001 is active against the MexAB-OprM efflux pump (Mahmood et al., 2016). D13-9001 has shown promising in vitro and in vivo activity, as well as high solubility and low-toxicity profiles (Yoshida et al., 2007). D13-9001 obstructs normal functioning of MexAB-OprM in two ways. Firstly, it prevents conformational changes by binding tightly to the hydrophobic trap. Secondly, it prevents substrate binding to MexB, through the interaction of the D13-9001 hydrophilic component and the substrate binding channel of MexB (Nakashima et al., 2013). EPIs will need to be broad-spectrum if they are to be used as an adjuvant to antibiotics that are substrates of several efflux-pumps. The specificity of D13-9001 would limit its usage to co-administration with antibiotics extruded exclusively by MexAB-OprM (Nakashima et al., 2013). However, a study by Ranjitkar et al. found that there are several mechanisms of resistance to D13-9001 potentiator activity in P. aeruginosa, when the agent is used together with carbenicillin, an antibiotic that is substrate specific to MexAB-OprM. Loss of potentiating activity of D13-9001 occurred rapidly due to a F628L substitution in mexB, which is known to play an important role in inhibitor binding (Ranjitkar et al., 2019).
Polyamines are aliphatic carbon chains containing several amino groups and are essential organic polycations present in every form of life. Polyamines are implicated in cell maintenance and viability and in the functioning of a wide array of organ systems, including, the nervous and immune systems (Sánchez-Jiménez et al., 2019).
Fleeman et al. identified a polyamine scaffold as a strong efflux pump inhibitor with no direct antimicrobial activity. Five lead agents were found to potentiate aztreonam, chloramphenicol and tetracycline by causing a 5- to 8-fold decrease in the MIC90 (Fleeman et al., 2018). In addition, the polyamine derivatives did not disrupt the bacterial membrane, unlike other polyamines, which can lead to the identification of false positives for EPIs (Fleeman et al., 2018; Reza et al., 2019). Moreover, polyamines did not display toxicity to mammalian cell lines and did not inhibit calcium channel activity in human kidney cells (Fleeman et al., 2018).
Phage therapy, the use of bacteriophages to infect and lyse bacterial cells, has been widely discussed (Chan et al., 2013; Gordillo Altamirano and Barr, 2019). Traditional phage therapy involves the administration of one, or a mixture, of phages that will invade the bacterial cell and clear infection (Waters et al., 2017). A different approach to phage therapy has been proposed, whereby phages would be used to steer antibiotic resistance evolution, selecting for phage resistance and antibiotic susceptibility. For example, the lytic Myoviridae bacteriophage, OMKO1, utilizes OprM of the multidrug efflux systems MexAB and MexXY as a receptor-binding site. Selection for resistance to OMKO1 bacteriophage attack creates an evolutionary trade-off in MDR P. aeruginosa, by changing the efflux pump mechanism, leading to an increased sensitivity to ciprofloxacin, tetracycline, ceftazidime and erythromycin, four drugs from different antibiotic classes (Chan et al., 2016). Phage steering can be achieved when the binding receptor for the bacteriophage is implicated in both antibiotic resistance and phage resistance. The advantage of this approach lies in the two distinct, and opposing, mechanisms leading to bacterial eradication (Gurney et al., 2020).
The prototypical example of successful anti-resistance therapeutics are the β-lactamase inhibitors. β-lactamase inhibitors such as clavulanic acid, sulbactam and tazobactam are widely used to combat resistance mediated by β-lactamases (Tooke et al., 2019). However, the majority of clinically used β-lactamase inhibitors have a limited spectrum and mainly target Ambler class A β-lactamases, excluding KPC-type β-lactamase. Progress has been made in the development of novel β-lactamase inhibitors with a wider spectrum of activity. Three novel β-lactamase inhibitors, avibactam, vaborbactam and relebactam, function against Ambler class A, C and D β-lactamases (Wong and Duin, 2017). However, only avibactam and relebactam are efficacious against P. aeruginosa infection (Aktaş et al., 2012; Barnes et al., 2018).
Lamut et al. designed 4,5,6,7-tetrahydrobenzo[d]thiazole-based DNA gyrase B inhibitors and incorporated these inhibitors with siderophore mimics. The siderophore mimic served as an inducer for increased uptake of the gyrase B inhibitors into the bacterial cytoplasm. Out of the ten gyrase B inhibitors tested against P. aeruginosa, four were able to inhibit ≥50% but only under iron-supplemented conditions, which is not reflective of the host environment during infection (Lamut et al., 2020). Several more attempts have been made at designing broad-spectrum anti-bacterial and anti-biofilm therapies targeting DNA gyrase or topoisomerase but none have shown good activity for P. aeruginosa (Dubey et al., 2012; Masih et al., 2020).
Murepavadin is a novel, non-lytic, species specific, outer-membrane protein targeting antibiotic for the treatment of P. aeruginosa infections, including those caused by MDR strains (Dale et al., 2018). Murepavadin is derived from the β-hairpin host defense molecule protegrin 1 (PG-1) and optimized to counteract unfavorable absorption, distribution, metabolism, excretion and toxicity (ADMET) properties normally associated with PG-1 (Obrecht et al., 2011). It is a macrocycle compound consisting of PG-1 loop sequences linked to a D-proline-L-proline sequence, the latter of which is important for its stability and subsequent strong antibacterial potential (Srinivas et al., 2010). Murepavadin functions through binding to the LPS transport protein D (LptD), an OMP necessary for LPS biogenesis in Gram-negative bacteria. The interaction between murepavadin and LptD causes inhibition of LPS transport, which leads to alterations of the LPS on the bacterial OM and eventually, cell death (Werneburg et al., 2012).
Murepavadin derivatives have been screened for activity against Gram-negative ESKAPE pathogens, including P. aeruginosa. These derivatives were initially shown to be effective, although the majority of leads were found to have high MIC values in the presence of 50% human serum and showed signs of lytic activity in human red blood cells. Therefore, compounds were generated consisting of β-hairpin macrocycles linked to the peptide macrocycle of polymyxin B. One of these compounds, compound 3, showed strong antimicrobial activity (MIC 0.5-2 μg/mL for P. aeruginosa isolates), low toxicity to mammalian cells, low plasma protein binding, good human plasma stability and no lytic activity towards human red blood cells. This compound was shown to perturb and permealise the bacterial membrane through interacting with the β-barrel domain of BamA in E. coli ATCC 25922 (Luther et al., 2019). BamA is part of the β-barrel assembly machinery BAM complex, which serves to fold and insert outer membrane proteins in the OM (Gu et al., 2016). The binding interaction between BamA and compound 3 locks BamA in its closed state through changing the conformational composite in the β-barrel lateral gate between open and closed states. It is not known what causes compound 3 to permeabilize the membrane. It may inhibit the folding activity of the BAM complex, leading to incorrectly folded proteins being misplaced in the inner membrane. Alternatively, BamA may only serve as an extra binding site for compound 3, thereby evading the LPS-modification resistance mechanism of Gram-negative pathogens (Luther et al., 2019).
Quorum sensing regulates a wide range of genes involved in virulence and bacterial adaptation (Kalia, 2013). For instance, QS is required for the surfing and swarming motility phenotypes associated with increased resistance to antimicrobials. The surfing phenotype is regulated via three QS systems in P. aeruginosa; Las, Rhl and Pqs (Sun et al., 2018b). In addition, QS has been found to influence tolerance to antibiotics in P. aeruginosa biofilms. QS provides structural rigidity through the regulation of Pel polysaccharides and eDNA release necessary for the extracellular polysaccharide matrix. In addition, the production of rhamnolipids, surfactants important for the establishment and maintenance of biofilms, is controlled under QS (de Kievit, 2009). Therefore, QS has been recognized as a significant potential target for developing anti-resistance therapies. Strategies to combat antimicrobial resistance by targeting adaptive resistance mechanisms have significant potential for reversing antibiotic resistance in P. aeruginosa. Adaptive resistance is often mediated through complex global regulatory systems, such as the QS system, and regulate an extensive set of genes involved in resistance. Targeting these regulatory systems may prevent the activation of expression of these resistance genes that would normally be expressed under the environmental conditions of infection.
Ajoene is a natural sulphur-containing compound extracted from garlic (Yoshida et al., 1998) that has been shown to modulate biofilm formation by inhibiting QS-induced production of virulence factors (Holm Jakobsen et al., 2012). It targets the Gac/Rsm component of QS, leading to a decreased expression of rsmZ and rsmY small regulatory RNAs. rsmZ and rsmY bind the global regulatory protein RsmA, and unbound RsmA represses the translation of genes by preventing ribosome binding to the Shine-Dalgarno site. Several genes involved in QS are under RsmA regulation, and low expression of rsmZ and rsmY in the presence of ajoene promotes RsmA mediated repression of these target genes (Jakobsen et al., 2017). However, the therapeutic applicability of ajoene is limited due to availability, instability, hydrophobicity and relatively high MIC values. Efforts are being undertaken to overcome these issues through modification, the use novel delivery systems and a targeted route of administration and through the development of synthetic ajoene analogues (Fong et al., 2017; Vadekeetil et al., 2019).
Another novel QS-inhibitor derived from a natural source is the plant flavonoid naringenin. Naringenin diminishes the production of QS-regulated virulence factors in P. aeruginosa by binding directly to LasR, thereby competing with the activator of LasR, N-(3-oxo-dodecanoyl)-l-homoserine lactone (HSL).
It is ineffective at outcompeting HSL when the activator is already bound to LasR. Thus, the QS-inhibitor will only sufficiently interfere with the QS response when administered during early exponential growth, when naringenin can compete with unbound HSL for LasR binding. Naringenin is only suitable for combatting P. aeruginosa populations at low cellular densities, which often does not represent the clinical infection scenario. The full potential of QS-inhibition will only be realized if an inhibitor is developed that is capable of targeting P. aeruginosa QS signaling regardless of bacterial density and/or QS status (Hernando-Amado et al., 2020).
Bacterial biofilms pose a physical barrier for drug penetration, which is one of the reasons that bacteria in a biofilm mode of growth are more resistant to antimicrobials. This phenomenon may be subverted with the use of nanocarriers that encapsulate antimicrobials and facilitate drug diffusion through the bacterial biofilm. In addition, nanocarriers can also protect drugs from degradation, ensure controlled drug release, and cause increased uptake by the drug target, leading to an overall higher efficiency of encapsulated drugs. Drug delivery methods can be diverse in chemical structure and nature (Table 1). Most published studies concur that encapsulated antibiotics are more effective at preventing or eradicating biofilm formation than their free drug counterpart (Alhariri et al., 2017; Shaaban et al., 2017; Zahra et al., 2017; Dai et al., 2018; Li et al., 2019), although several concluded that the activity of encapsulated and free antibiotic was equal (Severino et al., 2017; Wang et al., 2018; Hill et al., 2019).
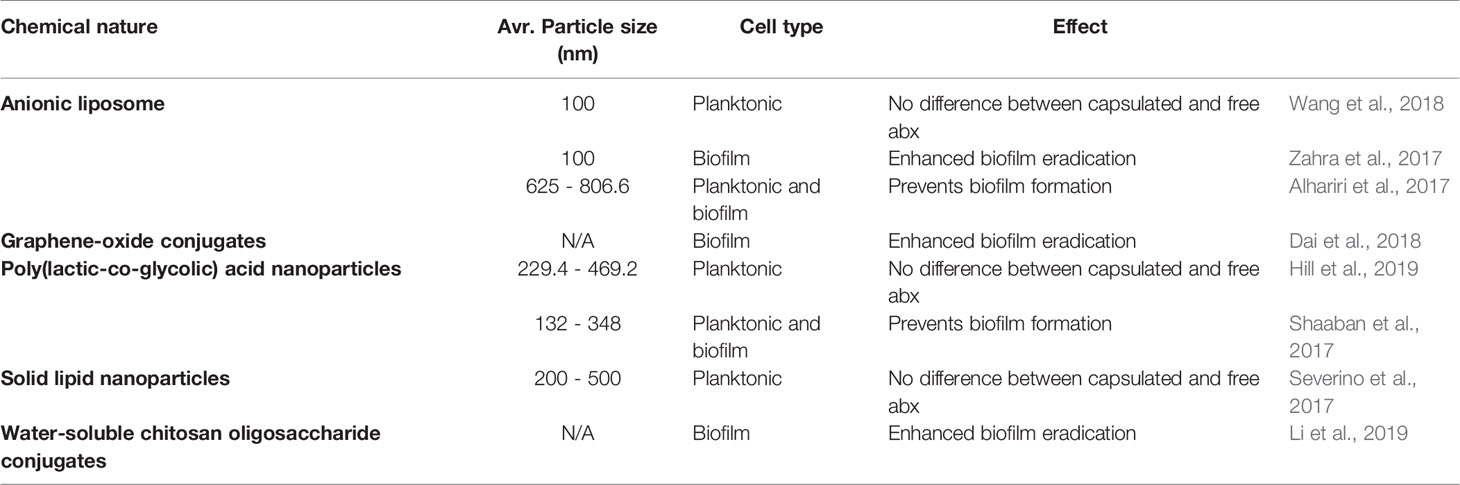
Table 1 Recent efforts in the design of drug delivery methods for anti-pseudomonal therapies.
Another promising antibiotic adjuvant targeting biofilms is the non-bactericidal, inhaled adjuvant, nitric oxide (NO). Exposure of P. aeruginosa biofilms to low-dose NO has been shown to cause dispersal of biofilms, rendering the infection susceptible to subsequent antibiotic treatment (Cai, 2020). NO functions by increasing bacterial phosphodiesterase activity which, in turn, leads to a reduction in the vital secondary signaling messenger, cyclic di-GMP. Cyclic di-GMP is vital for intracellular regulation of biofilm formation. Howlin et al. carried out in vitro biofilm studies using CF sputum clinical samples. Biofilms were treated with NO only, tobramycin only, tobramycin and ceftazidime, NO + tobramycin and NO + tobramycin and ceftazidime. Biofilms treated with NO showed a relative decrease in biofilm biomass and surface bound thickness in comparison to the untreated control. Further, treatment of biofilms with NO + tobramycin and NO + tobramycin and ceftazidime led to complete eradication of biofilm biomass and surface bound thickness. In comparison, biofilms treated with tobramycin or tobramycin and ceftazidime led to a 243% and 155% increase in biofilm biomass and a 199% and 174% increase in biofilm thickness, respectively, when compared to the untreated biofilm control. There is some evidence for the safety of NO administration in CF patients in vivo and NO is currently undergoing clinical trials to measure clinical efficacy (Howlin et al., 2017).
Poly-acetyl-arginyl-glucosamine (PAAG), also called SNSP113, is a novel inhaled adjuvant therapy currently undergoing phase one clinical trials. PAAG is a polycationic glycoprotein that functions by permeabilizing the bacterial membrane and is active against methicillin resistant Staphylococcus aureus (MRSA), Burkholderia spp., Mycobacterium spp. and E. coli. PAAG has been shown to effectively disperse Burkholderia cepacia complex biofilm structures extracted from the CF lung (Narayanaswamy et al., 2019). In P. aeruginosa, PAAG is has been shown to effectively eradicate persister cells, which is important for the prevention of recurrent P. aeruginosa infections and subsequent exacerbations in people with CF (Narayanaswamy et al., 2018). In addition to serving as an effective antibiotic adjuvant, PAAG also reduces inflammation and promotes viscoelasticity and mucociliary clearance, making it a suitable drug candidate to improve the quality of life for patients with a variety of mucus diseases (Fernandez-Petty et al., 2019).
The global overuse and misuse of antibiotics during the last 80 years has led to a profound increase in antimicrobial resistance. Between 2000 and 2010, global antibiotic consumption increased by nearly 70% and antibiotic resistant infections have accordingly become more pervasive, according to global epidemiological antimicrobial resistance surveillance networks (European Centre for Disease Prevention and Control, 2018) (Christaki et al., 2020). AMR is a complex, One Health issue, involving human, animal and environmental factors. The solution to AMR is therefore also likely to be a complex one, involving multiple strategies; maintaining AMR surveillance, containing AMR transmission, reducing selection pressure, developing novel antimicrobials or reverting antibiotic resistant microbes back to the susceptible phenotype with the use of antibiotic adjuvants (Hernando-Amado et al., 2019). Although progress in the development of naturally derived and peptide-based antimicrobials has been made (Mok et al., 2020; Upert et al., 2021). The conservation of existing antibiotics through careful stewardship is paramount to help mitigate the gap between the demand for new drugs and the diminishing supply pipeline. Antibiotic adjuvants will also play an important role in extending the shelf life of our existing antimicrobial therapeutic agents.
Adjuvant strategies targeting resistance mechanisms in P. aeruginosa could rejuvenate traditional antibiotic therapy by potentiating drug activity as well as slowing the development of antibiotic resistance. As described in this review, antimicrobial resistance in P. aeruginosa is regulated through a complex interplay of mechanisms. Resistance encoded in the core genome of P. aeruginosa, such as low outer membrane permeability, Mex-type efflux pumps and AmpC β-lactamase amount to the basal level of resistance against antimicrobials. This intrinsic resistance is present in the all P. aeruginosa strains and serves as a foundational level, which can be expanded upon. This expansion can be induced by environmental influences, such as host factors and signalling molecules, that switch on adaptive resistance mechanisms. Acquired resistance mechanisms, such as antibiotic target modifications generated via mutation, and antibiotic modifying enzymes or resistance plasmids, acquired by gene transfer, may serve as additional building blocks to expand the arsenal of resistance mechanisms a particular strain might carry. Several novel therapeutic strategies, targeting one or more of these mechanisms, have been described in this review. In light of recent findings, OM perturbants capable of sensitizing the Gram-negative bacterial membrane to previously non-active antibiotics seem an opportune strategy to combat resistance. OM perturbants can by-pass intrinsic as well as acquired and spontaneous resistance mechanisms, making them highly promising drug candidates, for which the development of resistance would be unlikely. However, efforts to finding perturbants suitable for targeting the P. aeruginosa membrane must be increased.
A second promising strategy is phage steering, which uses the natural predators of bacteria and the forces of evolutionary pressure to our advantage. Counterbalancing antibiotic resistance with phage susceptibility creates a double edged sword to circumvent key AMR mechanisms. In addition to these strategies, adjuvants targeting adaptive resistance mechanisms are worthy of consideration, due to the potential to disrupt multiple bacterial resistance and virulence processes with agents targeting a single regulator. Targeting global regulatory systems that would normally control the expression of resistance genes under infection conditions will prevent the activation of those genes with potential knock-on advantages in inhibition of virulence mechanisms. QS and two-component signaling systems are particularly attractive targets from this perspective, as are the regulators of biofilm formation.
There are several challenges in developing resistance-breaking therapy for P. aeruginosa infection. Firstly, due to its comparatively large genome and highly adaptive nature, a plethora of regulatory systems, as well as limited drug penetration and active efflux, many antibiotic adjuvants designed for Gram-negative pathogens do not show efficacy against P. aeruginosa. Secondly, toxicity has been proven to be the major hurdle for adjuvants designed against P. aeruginosa, leading to many being abandoned at early phases of development. Drug safety assessment is a long, expensive, but crucial process and toxicity is most likely where drug targets share structural similarity with human proteins. In this respect, bacterial signaling systems are good candidates, as prokaryotic and eukaryotic signaling systems are highly divergent, with eukaryotes lacking TCSS or phosphorelay systems.
As with all newly developed drugs designed to be used as combination therapy, care must be taken in determining the correct dosing and investigating clear synergy profiles. Drug levels necessary for synergy in vitro may not be achievable in vivo. Synergy in vivo may be affected by failure to obtain desired levels of drugs in the target tissue, drug metabolism or plasma protein binding. In addition, it is of paramount importance to evaluate drugs in relevant models that reflect the environmental conditions of infection. This will increase the predictive power of preclinical testing, reducing the costly progression of unpromising agents to clinical trials. The lack of well-validated in vivo models for testing CF anti-infective therapeutics limits the speed of development of new drugs. Murine models using cystic fibrosis transmembrane conductance regulator (CFTR) knockout animals, or transgenics in which the severe gut phenotype associated with loss of CFTR has been corrected are available, and have proved useful, but do not develop the characteristic features of acute and chronic P. aeruginosa infection seen in those with CF (Bragonzi, 2010; Semaniakou et al., 2019). Progress on the use of ferret and porcine infection models with mutated CFTR has been made, although these are limited by the availability of suitable immunobiology reagents (Keiser and Engelhardt, 2011). Intranasal administration of P. aeruginosa into the healthy murine lung often leads to either rapid clearance or sepsis. To create a model of persistent infection, it is usually necessary to immobilize P. aeruginosa. This can be achieved by encapsulating the bacteria into agar or alginate beads, where the bacteria are protected from clearance by immune effectors and where their mode of living more closely mimics bacterial biofilms present in chronic infection (Cigana et al., 2016). However, this bead model is technically demanding and requires surgical transtracheal instillation of the bead suspension, leading to additional complications and mortality not representative of bacterial infections in CF (Van Heeckeren and Schluchter, 2002). Alternatively, long term lung infection can be achieved using P. aeruginosa isolates from CF, some of which naturally establish chronic infection in mice, without the need for implantation into beads (Fothergill et al., 2014; Bricio-Moreno et al., 2018). This model has the advantages of using a natural infection route and having no requirement for surgical intervention, and offers opportunity to study lung infection over prolonged periods. However, the density of infection achieved in the lung is low, making some analyses challenging.
Questions remain regarding the commitment of governments and pharmaceutical manufacturers to ongoing investment in antibacterial drug development, particularly as financial and research priorities are reshuffled by the ongoing SARS-CoV2 crisis. Despite the understandable current emphasis on anti-viral agents and vaccines, it is important that we do not lose ground in the fight against AMR. Indeed, emerging evidence suggests that antibiotic use has increased dramatically in the COVID-19 era (Hsu, 2020). On top of this, increased usage of sanitizers and disinfectants globally may induce the development of cross-resistance to antibiotics. The ESKAPE pathogens, for which new medicines are urgently needed, continue to cause serious community-acquired and nosocomial infections, and if investment into research and drug development for these bacterial pathogens is diminished, it will exacerbate the global health and economic costs associated with the ongoing pandemic.
FL wrote the manuscript with supervision and input from all others. All authors contributed to the article and approved the submitted version.
FL is supported by a PhD studentship from the Rosetrees Trust (M750). DN is supported by a Sir Henry Dale Fellowship funded by the Wellcome Trust and the Royal Society (Grant number 204457/Z/16/Z).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Figures were designed and illustrated by Dr. Eliza Wolfson, https://lizawolfson.co.uk using molecular renderings by Protein Imager.
WHO publishes list of bacteria for which new antibiotics are urgently needed. Available at: https://www.who.int/news-room/detail/27-02-2017-who-publishes-list-of-bacteria-for-which-new-antibiotics-are-urgently-needed.
Abril D., Marquez-Ortiz R. A., Castro-Cardozo B., Moncayo-Ortiz J. I., Olarte Escobar N. M., Corredor Rozo Z. L., et al. (2019). Genome plasticity favours double chromosomal Tn4401b-bla KPC-2 transposon insertion in the Pseudomonas aeruginosa ST235 clone. BMC Microbiol. 19, 45. doi: 10.1186/s12866-019-1418-6
Achouak W., Heulin T., Pagès J.-M. (2006). Multiple facets of bacterial porins. FEMS Microbiol. Lett. 199, 1–7. doi: 10.1111/j.1574-6968.2001.tb10642.x
Aghazadeh M., Rezaee M. A., Nahaei M. R., Mahdian R., Pajand O., Saffari F., et al. (2013). Dissemination of Aminoglycoside-Modifying Enzymes and 16S rRNA Methylases Among Acinetobacter baumannii and Pseudomonas aeruginosa Isolates. Microb. Drug Resist. 19, 282–288. doi: 10.1089/mdr.2012.0223
Aghazadeh M., Hojabri Z., Mahdian R., Nahaei M. R., Rahmati M., Hojabri T., et al. (2014). Role of efflux pumps: MexAB-OprM and MexXY(-OprA), AmpC cephalosporinase and OprD porin in non-metallo-β-lactamase producing Pseudomonas aeruginosa isolated from cystic fibrosis and burn patients. Infect. Genet. Evol. 24, 187–192. doi: 10.1016/j.meegid.2014.03.018
Aktaş Z., Kayacan C., Oncul O. (2012). In vitro activity of avibactam (NXL104) in combination with β-lactams against Gram-negative bacteria, including OXA-48 β-lactamase-producing Klebsiella pneumoniae. Int. J. Antimicrob. Agents 39, 86–89. doi: 10.1016/j.ijantimicag.2011.09.012
Alcalde-Rico M., Olivares-Pacheco J., Alvarez-Ortega C., Cámara M., Martínez J. L. (2018). Role of the multidrug resistance efflux pump MexCD-OprJ in the Pseudomonas aeruginosa quorum sensing response. Front. Microbiol. 9. doi: 10.3389/fmicb.2018.02752
Aldred K. J., Kerns R. J., Osheroff N. (2014). Mechanism of Quinolone Action and Resistance. Biochemistry 53, 1565–1574. doi: 10.1021/bi5000564
Alhariri M., Majrashi M. A., Bahkali A. H., Almajed F. S., Azghani A. O., Khiyami M. A., et al. (2017). Efficacy of neutral and negatively charged liposome-loaded gentamicin on planktonic bacteria and biofilm communities. Int. J. Nanomed. 12, 6949–6961. doi: 10.2147/IJN.S141709
Andrésen C., Jalal S., Aili D., Wang Y., Islam S., Jarl A., et al. (2010). Critical biophysical properties in the Pseudomonas aeruginosa efflux gene regulator MexR are targeted by mutations conferring multidrug resistance. Protein Sci. 19, 680–692. doi: 10.1002/pro.343
Azucena E., Mobashery S. (2001). Aminoglycoside-modifying enzymes: Mechanisms of catalytic processes and inhibition. Drug Resist. Update 4, 106–117. doi: 10.1054/drup.2001.0197
Barnes M. D., Bethel C. R., Alsop J., Becka S. A., Rutter J. D., Papp-Wallace K. M., et al. (2018). Inactivation of the pseudomonas-derived cephalosporinase-3 (PDC-3) by Relebactam. Antimicrob. Agents Chemother. 62. doi: 10.1128/AAC.02406-17
Baron S., Hadjadj L., Rolain J. M., Olaitan A. O. (2016). Molecular mechanisms of polymyxin resistance: knowns and unknowns. Int. J. Antimicrob. Agents 48, 583–591. doi: 10.1016/j.ijantimicag.2016.06.023
Bellido F., Martin N. L., Siehnel R. J., Hancock R. E. W. (1992). Reevaluation, using intact cells, of the exclusion limit and role of porin OprF in Pseudomonas aeruginosa outer membrane permeability. J. Bacteriol. 174, 5196–5203. doi: 10.1128/JB.174.16.5196-5203.1992
Bolard A., Plésiat P., Jeannot K. (2017). Mutations in Gene fusA1 as a Novel Mechanism of Aminoglycoside Resistance in Clinical Strains of Pseudomonas aeruginosa. Antimicrob. Agents Chemother. 62. doi: 10.1128/AAC.01835-17
Bragonzi A. (2010). Murine models of acute and chronic lung infection with cystic fibrosis pathogens. Int. J. Med. Microbiol. 300, 584–593. doi: 10.1016/j.ijmm.2010.08.012
Bricio-Moreno L., Sheridan V. H., Goodhead I., Armstrong S., Wong J. K. L., Waters E. M., et al. (2018). Evolutionary trade-offs associated with loss of PmrB function in host-adapted Pseudomonas aeruginosa. Nat. Commun. 9, 1–12. doi: 10.1038/s41467-018-04996-x
Brinkman F. S., Schoofs G., Hancock R. E., De Mot R. (1999). Influence of a putative ECF sigma factor on expression of the major outer membrane protein, OprF, in Pseudomonas aeruginosa and Pseudomonas fluorescens. J. Bacteriol. 181, 4746–4754. doi: 10.1128/JB.181.16.4746-4754.1999
Bruchmann S., Dötsch A., Nouri B., Chaberny I. F., Häussler S. (2013). Quantitative contributions of target alteration and decreased drug accumulation to pseudomonas aeruginosa fluoroquinolone resistance. Antimicrob. Agents Chemother. 57, 1361–1368. doi: 10.1128/AAC.01581-12
Bukhari S. I., Aleanizy F. S. (2020). Association of OprF mutant and disturbance of biofilm and pyocyanin virulence in pseudomonas aeruginosa. Saudi Pharm. J. 28, 196–200. doi: 10.1016/j.jsps.2019.11.021
Bush K., Jacoby G. A. (2010). Updated functional classification of β-lactamases. Antimicrobial. Agents Chemother. 54, 969–976. doi: 10.1128/AAC.01009-09
Cai Y.-M., Webb J. S. (2020). Optimization of nitric oxide donors for investigating biofilm dispersal response in Pseudomonas aeruginosa clinical isolates. Appl. Microbiol. Biotechnol. 104, 8859–8869. doi: 10.1007/s00253-020-10859-7
Cavallari J. F., Lamers R. P., Scheurwater E. M., Matos A. L., Burrows L. L. (2013). Changes to its peptidoglycan-remodeling enzyme repertoire modulate β-lactam resistance in Pseudomonas aeruginosa. Antimicrob. Agents Chemother. 57, 3078–3084. doi: 10.1128/AAC.00268-13
Cazares A., Moore M. P., Hall J. P. J., Wright L. L., Grimes M., Emond-Rhéault J. G., et al. (2020). A megaplasmid family driving dissemination of multidrug resistance in Pseudomonas. Nat. Commun. 11, 1–13. doi: 10.1038/s41467-020-15081-7
Ceri H., Olson M. E., Stremick C., Read R. R., Morck D., Buret A., et al. (1999). The Calgary Biofilm Device: New technology for rapid determination of antibiotic susceptibilities of bacterial biofilms. J. Clin. Microbiol. 37, 1771–1776. doi: 10.1128/JCM.37.6.1771-1776.1999
Chalmers J. D., Hill A. T. (2013). Mechanisms of immune dysfunction and bacterial persistence in non-cystic fibrosis bronchiectasis. Mol. Immunol. 55, 27–34. doi: 10.1016/j.molimm.2012.09.011
Chan B. K., Abedon S. T., Loc-Carrillo C. (2013). Phage cocktails and the future of phage therapy. Future Microbiol. 8, 769–783. doi: 10.2217/fmb.13.47
Chan B. K., Sistrom M., Wertz J. E., Kortright K. E., Narayan D., Turner P. E., et al. (2016). Phage selection restores antibiotic sensitivity in MDR Pseudomonas aeruginosa. Sci. Rep. 6, 1–8. doi: 10.1038/srep26717
Chen W., Wang D., Zhou W., Sang H., Liu X., Ge Z., et al. (2016). Novobiocin binding to NalD induces the expression of the MexAB-OprM pump in Pseudomonas aeruginosa. Mol. Microbiol. 100, 749–758. doi: 10.1111/mmi.13346
Chevalier S., Bouffartigues E., Bodilis J., Maillot O., Lesouhaitier O., Feuilloley M. G. J., et al. (2017). Structure, function and regulation of Pseudomonas aeruginosa porins. FEMS Microbiol. Rev. 41, 698–722. doi: 10.1093/femsre/fux020
Choudhury D., Ghosh A., Dhar Chanda D., Das Talukdar A., Dutta Choudhury M., Paul D., et al. (2016). Premature Termination of MexR Leads to Overexpression of MexAB-OprM Efflux Pump in Pseudomonas aeruginosa in a Tertiary Referral Hospital in India. PloS One 11, e0149156. doi: 10.1371/journal.pone.0149156
Christaki E., Marcou M., Tofarides A. (2020). Antimicrobial Resistance in Bacteria: Mechanisms, Evolution, and Persistence. J. Mol. Evol. 88, 26–40. doi: 10.1007/s00239-019-09914-3
Cigana C., Lorè N. I., Riva C., De Fino I., Spagnuolo L., Sipione B., et al. (2016). Tracking the immunopathological response to Pseudomonas aeruginosa during respiratory infections. Sci. Rep. 6, 1–12. doi: 10.1038/srep21465
Coleman S. R., Blimkie T., Falsafi R., Hancock R. E. W. (2020). Multidrug adaptive resistance of Pseudomonas aeruginosa swarming cells. Antimicrob. Agents Chemother. 64. doi: 10.1128/AAC.01999-19
Corbett K. D., Schoeffler A. J., Thomsen N. D., Berger J. M. (2005). The structural basis for substrate specificity in DNA topoisomerase IV. J. Mol. Biol. 351, 545–561. doi: 10.1016/j.jmb.2005.06.029
Corbett D., Wise A., Langley T., Skinner K., Trimby E., Birchall S., et al. (2017). Potentiation of antibiotic activity by a novel cationic peptide: Potency and spectrum of activity of SPR741. Antimicrob. Agents Chemother. 61. doi: 10.1128/AAC.00200-17
Costerton J. W. (2001). Cystic fibrosis pathogenesis and the role of biofilms in persistent infection. Trends Microbiol. 9, 50–52. doi: 10.1016/S0966-842X(00)01918-1
Dai X., Zhao Y., Yu Y., Chen X., Wei X., Zhang X., et al. (2018). All-in-one NIR-activated nanoplatforms for enhanced bacterial biofilm eradication. Nanoscale 10, 18520–18530. doi: 10.1039/C8NR04748K
Dale G. E., Halabi A., Petersen-Sylla M., Wach A., Zwingelstein C. (2018). Pharmacokinetics, tolerability, and safety of murepavadin, a novel antipseudomonal antibiotic, in subjects with mild, moderate, or severe renal function impairment. Antimicrob. Agents Chemother. 62. doi: 10.1128/AAC.00490-18
Daury L., Orange F., Taveau J. C., Verchère A., Monlezun L., Gounou C., et al. (2016). Tripartite assembly of RND multidrug efflux pumps. Nat. Commun. 7. doi: 10.1038/ncomms10731
de Kievit T. R. (2009). Quorum sensing in Pseudomonas aeruginosa biofilms. Environ. Microbiol. 11, 279–288. doi: 10.1111/j.1462-2920.2008.01792.x
Del Barrio-Tofiño E., López-Causapé C., Cabot G., Rivera A., Benito N., Segura C., et al. (2017). Genomics and susceptibility profiles of extensively drug-resistant pseudomonas aeruginosa isolates from Spain. Antimicrob. Agents Chemother. 61. doi: 10.1128/AAC.01589-17
Dettman J. R., Sztepanacz J. L., Kassen R. (2016). The properties of spontaneous mutations in the opportunistic pathogen Pseudomonas aeruginosa. BMC Genomics 17, 27. doi: 10.1186/s12864-015-2244-3
Diaz Iglesias Y., Van Bambeke F. (2020). Activity of Antibiotics against Pseudomonas aeruginosa in an In Vitro Model of Biofilms in the Context of Cystic Fibrosis: Influence of the Culture Medium. Antimicrob. Agents Chemother. 64. doi: 10.1128/AAC.02204-19
Diggle S. P., Whiteley M. (2020). Microbe profile: Pseudomonas aeruginosa: Opportunistic pathogen and lab rat. Microbiol. (United Kingdom) 166, 30–33. doi: 10.1099/mic.0.000860
Doi Y., Arakawa Y. (2007). 16S Ribosomal RNA Methylation: Emerging Resistance Mechanism against Aminoglycosides. Clin. Infect. Dis. 45, 88–94. doi: 10.1086/518605
Dötsch A., Eckweiler D., Schniederjans M., Zimmermann A., Jensen V., Scharfe M., et al. (2012). The Pseudomonas aeruginosa Transcriptome in Planktonic Cultures and Static Biofilms Using RNA Sequencing. PloS One 7, e31092. doi: 10.1371/journal.pone.0031092
Dreier J., Ruggerone P. (2015). Interaction of antibacterial compounds with RND efflux pumps in Pseudomonas aeruginosa. Front. Microbiol. 6, 660. doi: 10.3389/fmicb.2015.00660
Drlica K., Zhao X. (1997). DNA Gyrase, Topoisomerase IV, and the 4-Quinolones. Microbiol. Mol. Biol. Rev. 61, 377–92. doi: 10.1128/.61.3.377-392.1997
Drlica K. (1999). Mechanism of fluoroquinolone action. Curr. Opin. Microbiol. 2, 504–508. doi: 10.1016/S1369-5274(99)00008-9
Dubey V., Pathak M., Bhat H. R., Singh U. P. (2012). Design, Facile Synthesis, and Antibacterial Activity of Hybrid 1,3,4-thiadiazole-1,3,5-triazine Derivatives Tethered via -S- Bridge. Chem. Biol. Drug Des. 80, 598–604. doi: 10.1111/j.1747-0285.2012.01433.x
Dunham S. A., McPherson C. J., Miller A. A. (2010). The relative contribution of efflux and target gene mutations to fluoroquinolone resistance in recent clinical isolates of Pseudomonas aeruginosa. Eur. J. Clin. Microbiol. Infect. Dis. 29, 279–288. doi: 10.1007/s10096-009-0852-z
Edrington T. C., Kintz E., Goldberg J. B., Tamm L. K. (2011). Structural basis for the interaction of lipopolysaccharide with outer membrane protein H (OprH) from Pseudomonas aeruginosa. J. Biol. Chem. 286, 39211–39223. doi: 10.1074/jbc.M111.280933
El Zowalaty M. E., Al Thani A. A., Webster T. J., El Zowalaty A. E., Schweizer H. P., Nasrallah G. K., et al. (2015). Pseudomonas aeruginosa: Arsenal of resistance mechanisms, decades of changing resistance profiles, and future antimicrobial therapies. Future Microbiol. 10, 1683–1706. doi: 10.2217/fmb.15.48
European Centre for Disease Prevention and Control (2018). Surveillance of antimicrobial resistance in Europe Annual report of the European Antimicrobial Resistance Surveillance Network (EARS-Net) 2017. ECDC: Surveillance Report. doi: 10.2900/230516
Evans K., Adewoye L., Poole K. (2001). MexR repressor of the mexAB-oprM multidrug efflux operon of Pseudomonas aeruginosa: Identification of MexR binding sites in the mexA-mexR intergenic region. J. Bacteriol. 183, 807–812. doi: 10.1128/JB.183.3.807-812.2001
Falagas M. E., Rafailidis P. I., Matthaiou D. K. (2010). Resistance to polymyxins: Mechanisms, frequency and treatment options. Drug Resist. Update 13, 132–138. doi: 10.1016/j.drup.2010.05.002
Feliziani S., Luján A. M., Moyano A. J., Sola C., Bocco J. L., Montanaro P., et al. (2010). Mucoidy, quorum sensing, mismatch repair and antibiotic resistance in pseudomonas aeruginosa from cystic fibrosis chronic airways infections. PloS One 5, 1–12. doi: 10.1371/journal.pone.0012669
Feng W., Sun F., Wang Q., Xiong W., Qiu X., Dai X., et al. (2017). Epidemiology and resistance characteristics of Pseudomonas aeruginosa isolates from the respiratory department of a hospital in China. J. Glob. Antimicrob. Resist. 8, 142–147. doi: 10.1016/j.jgar.2016.11.012
Fernández L., Gooderham W. J., Bains M., McPhee J. B., Wiegand I., Hancock R. E. W., et al. (2010). Adaptive resistance to the ‘last hope’ antibiotics polymyxin B and colistin in Pseudomonas aeruginosa is mediated by the novel two-component regulatory system ParR-ParS. Antimicrob. Agents Chemother. 54, 3372–3382. doi: 10.1128/AAC.00242-10
Fernández L., Jenssen H., Bains M., Wiegand I., Gooderham W. J., Hancock R. E. W., et al. (2012). The two-component system CprRS senses cationic peptides and triggers adaptive resistance in Pseudomonas aeruginosa independently of ParRS. Antimicrob. Agents Chemother. 56, 6212–6222. doi: 10.1128/AAC.01530-12
Fernandez-Petty C. M., Hughes G. W., Bowers H. L., Watson J. D., Rosen B. H., Townsend S. M., et al. (2019). A glycopolymer improves vascoelasticity and mucociliary transport of abnormal cystic fibrosis mucus. JCI Insight 4. doi: 10.1172/jci.insight.125954
Fito-Boncompte L., Chapalain A., Bouffartigues E., Chaker H., Lesouhaitier O., Gicquel G., et al. (2011). Full virulence of Pseudomonas aeruginosa requires OprF. Infect. Immun. 79, 1176–1186. doi: 10.1128/IAI.00850-10
Fleeman R. M., Debevec G., Antonen K., Adams J. L., Santos R. G., Welmaker G. S., et al. (2018). Identification of a Novel Polyamine Scaffold With Potent Efflux Pump Inhibition Activity Toward Multi-Drug Resistant Bacterial Pathogens. Front. Microbiol. 9, 1301. doi: 10.3389/fmicb.2018.01301
Fong J., Yuan M., Jakobsen T. H., Mortensen K. T., Delos Santos M. M. S., Chua S. L., et al. (2017). Disulfide Bond-Containing Ajoene Analogues As Novel Quorum Sensing Inhibitors of Pseudomonas aeruginosa. J. Med. Chem. 60, 215–227. doi: 10.1021/acs.jmedchem.6b01025
Fontes L. C., Neves P. R., Oliveira S., Silva K. C., Hachich E. M., Sato M. I. Z., et al. (2011). Isolation of Pseudomonas aeruginosa coproducing metallo-β-lactamase SPM-1 and 16S rRNA methylase RmtD1 in an urban river. Antimicrobial. Agents Chemother. 55, 3063–3064. doi: 10.1128/AAC.00138-11
Fothergill J. L., Mowat E., Ledson M. J., Walshaw M. J., Winstanley C. (2010). Fluctuations in phenotypes and genotypes within populations of Pseudomonas aeruginosa in the cystic fibrosis lung during pulmonary exacerbations. J. Med. Microbiol. 59, 472–481. doi: 10.1099/jmm.0.015875-0
Fothergill J. L., Neill D. R., Loman N., Winstanley C., Kadioglu A. (2014). Pseudomonas aeruginosa adaptation in the nasopharyngeal reservoir leads to migration and persistence in the lungs. Nat. Commun. 5, 4780. doi: 10.1038/ncomms5780
Francisco G. R., Nora S. T. R., Bueno M. F. C., Da Silva Filho L. V. R. F., De Oliveira Garcia D. (2015). Identification of aminoglycoside-resistant Pseudomonas aeruginosa producing RmtG 16S rRNA Methyltransferase in a cystic fibrosis patient. Antimicrob. Agents Chemother. 59, 2967–2968. doi: 10.1128/AAC.04607-14
Fraud S., Campigotto A. J., Chen Z., Poole K. (2008). MexCD-OprJ multidrug efflux system of Pseudomonas aeruginosa: Involvement in chlorhexidine resistance and induction by membrane-damaging agents dependent upon the AlgU stress response sigma factor. Antimicrob. Agents Chemother. 52, 4478–4482. doi: 10.1128/AAC.01072-08
Gellatly S. L., Hancock R. E. W. (2013). Pseudomonas aeruginosa: New insights into pathogenesis and host defenses. Pathog. Dis. 67, 159–173. doi: 10.1111/2049-632X.12033
Goli H. R., Nahaei M. R., Rezaee M. A., Hasani A., Samadi Kafil H., Aghazadeh M., et al. (2016). Contribution of mexAB-oprM and mexXY (-oprA) efflux operons in antibiotic resistance of clinical Pseudomonas aeruginosa isolates in Tabriz, Iran. Infect. Genet. Evol. 45, 75–82. doi: 10.1016/j.meegid.2016.08.022
Gordillo Altamirano F. L., and Barr J. J. (2019). Phage therapy in the postantibiotic era. Clin. Microbiol. Rev. 32. doi: 10.1128/CMR.00066-18
Gu Y., Li H., Dong H., Zeng Y., Zhang Z., Paterson N. G., et al. (2016). Structural basis of outer membrane protein insertion by the BAM complex. Nature 531, 64–69. doi: 10.1038/nature17199
Gurney J., Pradier L., Griffin J., Gougat-Barbera C., Chan B., Turner P., et al. (2020). Phage steering of antibiotic-resistance evolution in the bacterial pathogen Pseudomonas aeruginosa. Evol. Med. Public Heal. 2020, 148–157. doi: 10.1093/emph/eoaa026
Gutierrez B., Douthwaite S., Gonzalez-Zorn B. (2013). Indigenous and acquired modifications in the aminoglycoside binding sites of Pseudomonas aeruginosa rRNAs. RNA Biol. 10, 1324–1332. doi: 10.4161/rna.25984
Hancock R. E. W., Irvin R. T., Costerton J. W., Carey A. M. (1981). Pseudomonas aeruginosa outer membrane: Peptidoglycan-associated proteins. J. Bacteriol. 145, 628–631. doi: 10.1128/JB.145.1.628-631.1981
Hassett D. J., Sutton M. D., Schurr M. J., Herr A. B., Caldwell C. C., Matu J. O., et al. (2009). Pseudomonas aeruginosa hypoxic or anaerobic biofilm infections within cystic fibrosis airways. Trends Microbiol. 17, 130–138. doi: 10.1016/j.tim.2008.12.003
Hassett D. J., Borchers M. T., Panos R. J. (2014). Chronic Obstructive Pulmonary Disease (COPD): Evaluation From Clinical, Immunological and Bacterial Pathogenesis Perspectives. J. Microbiol. 52, 211–226. doi: 10.1007/s12275-014-4068-2
Hernando-Amado S., Blanco P., Alcalde-Rico M., Corona F., Reales-Calderón J. A., Sánchez M. B., et al. (2016). Multidrug efflux pumps as main players in intrinsic and acquired resistance to antimicrobials. Drug Resist. Updates 28, 13–27. doi: 10.1016/j.drup.2016.06.007
Hernando-Amado S., Coque T. M., Baquero F., Martínez J. L. (2019). Defining and combating antibiotic resistance from One Health and Global Health perspectives. Nat. Microbiol. 4, 1432–1442. doi: 10.1038/s41564-019-0503-9
Hernando-Amado S., Alcalde-Rico M., Gil-Gil T., Valverde J. R., Martínez J. L. (2020). Naringenin Inhibition of the Pseudomonas aeruginosa Quorum Sensing Response Is Based on Its Time-Dependent Competition With N-(3-Oxo-dodecanoyl)-L-homoserine Lactone for LasR Binding. Front. Mol. Biosci. 7. doi: 10.3389/fmolb.2020.00025
Higgins P. G., Fluit A. C., Milatovic D., Verhoef J., Schmitz F. J. (2003). Mutations in GyrA, ParC, MexR and NfxB in clinical isolates of Pseudomonas aeruginosa. Int. J. Antimicrob. Agents 21, 409–413. doi: 10.1016/S0924-8579(03)00009-8
Hill D., Rose B., Pajkos A., Robinson M., Bye P., Bell S., et al. (2005). Antibiotic susceptibilities of Pseudomonas aeruginosa isolates derived from patients with cystic fibrosis under aerobic, anaerobic, and biofilm conditions. J. Clin. Microbiol. 43, 5085–5090. doi: 10.1128/JCM.43.10.5085-5090.2005
Hill M., Cunningham R. N., Hathout R. M., Johnston C., Hardy J. G., Migaud M. E., et al. (2019). Formulation of antimicrobial tobramycin loaded PLGA nanoparticles via complexation with AOT. J. Funct. Biomater. 10. doi: 10.3390/jfb10020026
Hocquet D., Vogne C., El Garch F., Vejux A., Gotoh N., Lee A., et al. (2003). MexXy-OprM efflux pump is necessary for adaptive resistance of Pseudomonas aeruginosa to aminoglycosides. Antimicrob. Agents Chemother. 47, 1371–1375. doi: 10.1128/AAC.47.4.1371-1375.2003
Hocquet D., Nordmann P., El Garch F., Cabanne L., Plésiat P. (2006). Involvement of the MexXY-OprM efflux system in emergence of cefepime resistance in clinical strains of Pseudomonas aeruginosa. Antimicrob. Agents Chemother. 50, 1347–1351. doi: 10.1128/AAC.50.4.1347-1351.2006
Høiby N., Ciofu O., Bjarnsholt T. (2010). Pseudomonas aeruginosa biofilms in cystic fibrosis. Future Microbiol. 5, 1663–1674. doi: 10.2217/fmb.10.125
Holm Jakobsen T., van Gennip M., Phipps R.K., Shanmugham M.S., Christensen L.D., Alhede M., et al. (2012). Ajoene, a Sulfur-Rich Molecule from Garlic, Inhibits Genes Controlled by Quorum Sensing 56, 2314–2325. doi: 10.1128/AAC.05919-11
Howlin R. P., Cathie K., Hall-Stoodley L., Cornelius V., Duignan C., Allan R. N., et al. (2017). Low-Dose Nitric Oxide as Targeted Anti-biofilm Adjunctive Therapy to Treat Chronic Pseudomonas aeruginosa Infection in Cystic Fibrosis. Mol. Ther. 25, 2104–2116. doi: 10.1016/j.ymthe.2017.06.021
Hsu J. (2020). How covid-19 is accelerating the threat of antimicrobial resistance. BMJ 369. doi: 10.1136/bmj.m1983
Islam S., Oh H., Jalal S., Karpati F., Ciofu O., Høiby N., et al. (2009). Chromosomal mechanisms of aminoglycoside resistance in Pseudomonas aeruginosa isolates from cystic fibrosis patients. Clin. Microbiol. Infect. 15, 60–66. doi: 10.1111/j.1469-0691.2008.02097.x
Jacoby G. A. (2005). Mechanisms of Resistance to Quinolones. Clin. Infect. Dis. 41, S120–S126. doi: 10.1086/428052
Jakobsen T. H., Warming A. N., Vejborg R. M., Moscoso J. A., Stegger M., Lorenzen F., et al. (2017). A broad range quorum sensing inhibitor working through sRNA inhibition. Sci. Rep. 7, 1–12. doi: 10.1038/s41598-017-09886-8
Jiang X., Zhang Z., Li M., Zhou D., Ruan F., Lu Y., et al. (2006). Detection of extended-spectrum β-lactamases in clinical isolates of Pseudomonas aeruginosa. Antimicrob. Agents Chemother. 50, 2990–2995. doi: 10.1128/AAC.01511-05
Jochumsen N., Marvig R. L., Damkiær S., Jensen R. L., Paulander W., Molin S., et al. (2016). The evolution of antimicrobial peptide resistance in Pseudomonas aeruginosa is shaped by strong epistatic interactions. Nat. Commun. 7. doi: 10.1038/ncomms13002
Johnson J. W., Fisher J. F., Mobashery S. (2013). Bacterial cell-wall recycling. Ann. N. Y. Acad. Sci. 1277, 54–75. doi: 10.1111/j.1749-6632.2012.06813.x
Juan C., Maciá M. D., Gutiérrez O., Vidal C., Pérez J. L., Oliver A., et al. (2005). Molecular mechanisms of β-lactam resistance mediated by AmpC hyperproduction in Pseudomonas aeruginosa clinical strains. Antimicrob. Agents Chemother. 49, 4733–4738. doi: 10.1128/AAC.49.11.4733-4738.2005
Kalia V. C. (2013). Quorum sensing inhibitors: An overview. Biotechnol. Adv. 31, 224–245. doi: 10.1016/j.biotechadv.2012.10.004
Keiser N. W., Engelhardt J. F. (2011). New animal models of cystic fibrosis: What are they teaching us? Curr. Opin. Pulm. Med. 17, 478–483. doi: 10.1097/MCP.0b013e32834b14c9
Khatua B., Van Vleet J., Choudhury B. P., Chaudhry R., Mandal C. (2014). Sialylation of outer membrane porin protein D: A mechanistic basis of antibiotic uptake in Pseudomonas aeruginosa. Mol. Cell. Proteomics 13, 1412–1428. doi: 10.1074/mcp.M113.030999
Köhler T., Michéa-Hamzehpour M., Henze U., Gotoh N., Curty L. K., Pechère J. C., et al. (1997). Characterization of MexE-MexF-OprN, a positively regulated multidrug efflux system of Pseudomonas aeruginosa. Mol. Microbiol. 23, 345–354. doi: 10.1046/j.1365-2958.1997.2281594.x
Köhler T., Michea-Hamzehpour M., Epp S. F., Pechere J. C. (1999). Carbapenem activities against Pseudomonas aeruginosa: Respective contributions of OprD and efflux systems. Antimicrob. Agents Chemother. 43, 424–427. doi: 10.1128/AAC.43.2.424
Kolpen M., Lerche C. J., Kragh K. N., Sams T., Koren K., Jensen A. S., et al. (2017). Hyperbaric oxygen sensitizes anoxic Pseudomonas aeruginosa biofilm to ciprofloxacin. Antimicrob. Agents Chemother. 61. doi: 10.1128/AAC.01024-17
Kubesch P., Wehsling M., Tümmler B. (1987). Membrane permeability of Pseudomonas aeruginosa to 4-quinolones. Zentralbl. Bakteriol. Mikrobiol. Hyg. Abt. 1 Orig. A 265, 197–202. doi: 10.1016/S0176-6724(87)80166-9
Kucharska I., Liang B., Ursini N., Tamm L. K. (2016). Molecular Interactions of Lipopolysaccharide with an Outer Membrane Protein from Pseudomonas aeruginosa Probed by Solution NMR. Biochemistry 55, 5061–5072. doi: 10.1021/acs.biochem.6b00630
Lam J. S., Taylor V. L., Islam S. T., Hao Y., Kocíncová D. (2011). Genetic and Functional Diversity of Pseudomonas aeruginosa Lipopolysaccharide. Front. Microbiol. 2, 118. doi: 10.3389/fmicb.2011.00118
Lambert P. A. (2002). Mechanisms of antibiotic resistance in Pseudomonas aeruginosa. J. R. Soc. Med. Suppl. 95, 22–26.
Lamut A., Cruz C. D., Skok Ž., Barančoková M., Zidar N., Zega A., et al. (2020). Design, synthesis and biological evaluation of novel DNA gyrase inhibitors and their siderophore mimic conjugates. Bioorg. Chem. 95, 103550. doi: 10.1016/j.bioorg.2019.103550
Landry R. M., An D., Hupp J. T., Singh P. K., Parsek M. R. (2006). Mucin-Pseudomonas aeruginosa interactions promote biofilm formation and antibiotic resistance. Mol. Microbiol. 59, 142–151. doi: 10.1111/j.1365-2958.2005.04941.x
Laudy A. E., Róg P., Smolińska-Król K., Ćmiel M., Słoczyńska A., Patzer J., et al. (2017). Prevalence of ESBL-producing Pseudomonas aeruginosa isolates in Warsaw, Poland, detected by various phenotypic and genotypic methods. doi: 10.1371/journal.pone.0180121
Lee J. K., Lee Y. S., Park Y. K., Kim B. S. (2005). Alterations in the GyrA and GyrB subunits of topoisomerase II and the ParC and ParE subunits of topoisomerase IV in ciprofloxacin-resistant clinical isolates of Pseudomonas aeruginosa. Int. J. Antimicrob. Agents 25, 290–295. doi: 10.1016/j.ijantimicag.2004.11.012
Lee M., Wu J., Deng Y., Wang J., Wang C., Wang J., et al. (2013). Reactions of all escherichia coli lytic transglycosylases with bacterial cell wall. J. Am. Chem. Soc 135, 3311–3314. doi: 10.1021/ja309036q
Lee M., Dhar S., Debenedetti S., Hesek D., Boggess B., Blázquez B., et al. (2016). Muropeptides in Pseudomonas aeruginosa and their Role as Elicitors of β-Lactam-Antibiotic Resistance. Angew. Chem. Int. Ed. 55, 6882–6886. doi: 10.1002/anie.201601693
Levine C., Hiasa H., Marians K. J. (1998). DNA gyrase and topoisomerase IV: Biochemical activities, physiological roles during chromosome replication, and drug sensitivities. Biochim. Biophys. Acta Gene Struct. Expression 1400, 29–43. doi: 10.1016/S0167-4781(98)00126-2
Li X. Z., Nikaido H., Poole K. (1995). Role of MexA-MexB-OprM in antibiotic efflux in Pseudomonas aeruginosa. Antimicrob. Agents Chemother. 39, 1948–1953. doi: 10.1128/AAC.39.9.1948
Li R., Yuan X., Wei J., Zhang X., Cheng G., Wang Z. A., et al. (2019). Synthesis and Evaluation of a Chitosan Oligosaccharide-Streptomycin Conjugate against Pseudomonas aeruginosa Biofilms. Mar. Drugs 17. doi: 10.3390/md17010043
Lister P. D., Wolter D. J., Hanson N. D. (2009). Antibacterial-resistant Pseudomonas aeruginosa: clinical impact and complex regulation of chromosomally encoded resistance mechanisms. Clin. Microbiol. Rev. 22, 582–610. doi: 10.1128/CMR.00040-09
Livermore D. M., Brown D. F. J. (2001). Detection of β-lactamase-mediated resistance. J. Antimicrob. Chemother. 48, 59–64. doi: 10.1093/jac/48.suppl_1.59
Llanes C., Hocquet D., Vogne C., Benali-Baitich D., Neuwirth C., Plésiat P., et al. (2004). Clinical Strains of Pseudomonas aeruginosa Overproducing MexAB-OprM and MexXY Efflux Pumps Simultaneously. Antimicrob. Agents Chemother. 48, 1797–1802. doi: 10.1128/AAC.48.5.1797-1802.2004
Llanes C., Pourcel C., Richardot C., Plésiat P., Fichant G., Cavallo J.-D., et al. (2013). Diversity of β-lactam resistance mechanisms in cystic fibrosis isolates of Pseudomonas aeruginosa: a French multicentre study. J. Antimicrob. Chemother. 68, 1763–1771. doi: 10.1093/jac/dkt115
Lomovskaya O., Bostian K. A. (2006). Practical applications and feasibility of efflux pump inhibitors in the clinic - A vision for applied use. Biochem. Pharmacol. 71, 910–918. doi: 10.1016/j.bcp.2005.12.008
López-Causapé C., Rojo-Molinero E., Mulet X., Cabot G., Moyà B., Figuerola J., et al. (2013). Clonal Dissemination, Emergence of Mutator Lineages and Antibiotic Resistance Evolution in Pseudomonas aeruginosa Cystic Fibrosis Chronic Lung Infection. PloS One 8. doi: 10.1371/journal.pone.0071001
Luther A., Urfer M., Zahn M., Müller M., Wang S.-Y., Mondal M., et al. (2019). Chimeric peptidomimetic antibiotics against Gram-negative bacteria. Nature 576, 452–458. doi: 10.1038/s41586-019-1665-6
Macfarlene E. L. A., Kwasnicka A., Hancock R. E. W. (2000). Role of Pseudomonas aeruginosa Phop-PhoQ in resistance to antimicrobial cationic peptides and aminoglycosides. Microbiology 146, 2543–2554. doi: 10.1099/00221287-146-10-2543
MacLeod D. L., Nelson L. E., Shawar R. M., Lin B. B., Lockwood L. G., Dirk J. E., et al. (2000). Aminoglycoside-Resistance Mechanisms for Cystic Fibrosis Pseudomonas aeruginosa Isolates Are Unchanged by Long-Term, Intermittent, Inhaled Tobramycin Treatment. J. Infect. Dis. 181, 1180–1184. doi: 10.1086/315312
Macnair C. R., Brown E. D. (2020). Outer membrane disruption overcomes intrinsic, acquired, and spontaneous antibiotic resistance. MBio 11, 1–15. doi: 10.1128/mBio.01615-20
Mahmood H. Y., Jamshidi S., Mark Sutton J., Rahman K. M. (2016). Current Advances in Developing Inhibitors of Bacterial Multidrug Efflux Pumps. Curr. Med. Chem. 23, 1062–1081. doi: 10.2174/0929867323666160304150522
Mark B. L., Vocadlo D. J., Oliver A. (2011). Providing β-lactams a helping hand: targeting the AmpC β-lactamase induction pathway. Future Microbiol. 6, 1415–1427. doi: 10.2217/fmb.11.128
Marquez B. (2005). Bacterial efflux systems and efflux pumps inhibitors. Biochimie 87, 1137–1147. doi: 10.1016/j.biochi.2005.04.012
Marvig R. L., Sommer L. M., Molin S., Johansen H. K. (2015). Convergent evolution and adaptation of Pseudomonas aeruginosa within patients with cystic fibrosis. Nat. Genet. 47, 57–64. doi: 10.1038/ng.3148
Maseda H., Yoneyama H., Nakae T. (2000). Assignment of the substrate-selective subunits of the MexEF-OprN multidrug efflux pump of Pseudomonas aeruginosa. Antimicrob. Agents Chemother. 44, 658–664. doi: 10.1128/AAC.44.3.658-664.2000
Masih A., Shrivastava J. K., Bhat H. R., Singh U. P. (2020). Potent antibacterial activity of dihydydropyrimidine-1,3,5-triazines via inhibition of DNA gyrase and antifungal activity with favourable metabolic profile. Chem. Biol. Drug Des. cbdd.13695. 96 (2), 861–869. doi: 10.1111/cbdd.13695
Masuda N., Ohya S. (1992). Cross-resistance to meropenem, cephems, and quinolones in Pseudomonas aeruginosa. Antimicrob. Agents Chemother. 36, 1847–1851. doi: 10.1128/AAC.36.9.1847
Masuda N., Gotoh N., Ohya S., Nishino T. (1996). Quantitative correlation between susceptibility and OprJ production in NfxB mutants of Pseudomonas aeruginosa. Antimicrob. Agents Chemother. 40, 909–913. doi: 10.1128/AAC.40.4.909
Masuda N., Gotoh N., Ishii C., Sakagawa E., Ohya S., Nishino T., et al. (1999). Interplay between chromosomal β-lactamase and the MexAB-OprM efflux system in intrinsic resistance to β-lactams in Pseudomonas aeruginosa. Antimicrob. Agents Chemother. 43, 400–402. doi: 10.1128/AAC.43.2.400
Masuda N., Sakagawa E, Ohya S, Gotoh N, Tsujimoto H, Nishino T. (2000). Substrate specificities of MexAB-OprM, MexCD-OprJ, and MexXY-OprM efflux pumps in Pseudomonas aeruginosa. Antimicrob. Agents Chemother. 44, 3322–3327. doi: 10.1128/AAC.44.12.3322-3327.2000
Mathee K., Narasimhan G., Valdes C., Qiu X., Matewish J. M., Koehrsen M., et al. (2008). Dynamics of Pseudomonas aeruginosa genome evolution. Proc. Natl. Acad. Sci. 105, 3100–3105. doi: 10.1073/pnas.0711982105
Maurice N. M., Bedi B., Sadikot R. T. (2018). Pseudomonas aeruginosa biofilms: Host response and clinical implications in lung infections. Am. J. Respir. Cell Mol. Biol. 58, 428–439. doi: 10.1165/rcmb.2017-0321TR
McPhee J. B., Lewenza S., Hancock R. E. W. (2003). Cationic antimicrobial peptides activate a two-component regulatory system, PmrA-PmrB, that regulates resistance to polymyxin B and cationic antimicrobial peptides in Pseudomonas aeruginosa. Mol. Microbiol. 50, 205–217. doi: 10.1046/j.1365-2958.2003.03673.x
Meletis G., Bagkeri M. (2013). Pseudomonas aeruginosa: Multi-Drug-Resistance Development and Treatment Options. Infect. Control. doi: 10.5772/55616
Mok N., Chan S. Y., Liu S. Y., Chua S. L. (2020). Vanillin inhibits PqsR-mediated virulence in: Pseudomonas aeruginosa. Food Funct. 11, 6496–6508. doi: 10.1039/D0FO00046A
Moradali M. F., Ghods S., Rehm B. H. A. (2017). Pseudomonas aeruginosa lifestyle: A paradigm for adaptation, survival, and persistence. Front. Cell. Infect. Microbiol. 7, 39. doi: 10.3389/fcimb.2017.00039
Morita Y., Komori Y., Mima T., Kuroda T., Mizushima T., Tsuchiya T., et al. (2001). Construction of a series of mutants lacking all of the four major mex operons for multidrug efflux pumps or possessing each one of the operons from Pseudomonas aeruginosa PAO1: MexCD-OprJ is an inducible pump. FEMS Microbiol. Lett. 202, 139–143. doi: 10.1111/j.1574-6968.2001.tb10794.x
Moskowitz S. M., Brannon M. K., Dasgupta N., Pier M., Sgambati N., Miller A. K., et al. (2012). PmrB mutations promote polymyxin resistance of Pseudomonas aeruginosa isolated from colistin-treated cystic fibrosis patients. Antimicrob. Agents Chemother. 56, 1019–1030. doi: 10.1128/AAC.05829-11
Moya B., Dötsch A., Juan C., Blázquez J., Zamorano L., Haussler S., et al. (2009). β-lactam resistance response triggered by inactivation of a nonessential penicillin-binding protein. PloS Pathog. 5. doi: 10.1371/journal.ppat.1000353
Müller L., Murgia X., Siebenbürger L., Börger C., Schwarzkopf K., Sewald K., et al. (2018). Human airway mucus alters susceptibility of Pseudomonas aeruginosa biofilms to tobramycin, but not colistin. J. Antimicrob. Chemother. 73, 2762–2769. doi: 10.1093/jac/dky241
Nakashima R., Sakurai K., Yamasaki S., Hayashi K., Nagata C., Hoshino K., et al. (2013). Structural basis for the inhibition of bacterial multidrug exporters. Nature 500, 102–106. doi: 10.1038/nature12300
Narayanaswamy V. P., Duncan A. P., LiPuma J. J., Wiesmann W. P., Baker S. M., Townsend S. M., et al. (2019). In vitro activity of a novel glycopolymer against biofilms of burkholderia cepacia complex cystic fibrosis clinical isolates. Antimicrob. Agents Chemother. 63. doi: 10.1128/AAC.00498-19
Narayanaswamy V. P., Keagy L. L., Duris K., Wiesmann W., Loughran A. J., Townsend S. M., et al. (2018). Novel Glycopolymer Eradicates Antibiotic- and CCCP-Induced Persister Cells in Pseudomonas aeruginosa. Front. Microbiol. 9, 1724. doi: 10.3389/fmicb.2018.01724
Nehme D., Li X.-Z., Elliot R., Poole K. (2004). Assembly of the MexAB-OprM multidrug efflux system of Pseudomonas aeruginosa: identification and characterization of mutations in mexA compromising MexA multimerization and interaction with MexB. J. Bacteriol. 186, 2973–2983. doi: 10.1128/JB.186.10.2973-2983.2004
Nestorovich E. M., Sugawara E., Nikaido H., Bezrukov S. M. (2006). Pseudomonas aeruginosa porin OprF. Properties of the channel. J. Biol. Chem. 281, 16230–16237. doi: 10.1074/jbc.M600650200
Nikaido H. (2010). Structure and mechanism of RND-type multidrug efflux pumps. Adv. Enzymol. Relat. Areas Mol. Biol. 77 1, 1–60. doi: 10.1002/9780470920541.ch1
Obrecht D., Bernardini F., Dale G., Dembowsky K. (2011). Emerging New Therapeutics Against Key Gram-Negative Pathogens. Annu. Rep. Med. Chem. 46, 245–262. doi: 10.1016/B978-0-12-386009-5.00012-6
Ochs M. M., McCusker M. P., Bains M., Hancock R. E. W. (1999). Negative regulation of the Pseudomonas aeruginosa outer membrane porin OprD selective for imipenem and basic amino acids. Antimicrob. Agents Chemother. 43, 1085–1090. doi: 10.1128/AAC.43.5.1085
Oliver A., Cantón R., Campo P., Baquero F., Blázquez J. (2000). High frequency of hypermutable Pseudomonas aeruginosa in cystic fibrosis lung infection. Sci. (80-. ) 288, 1251–1253. doi: 10.1126/science.288.5469.1251
Olsen I. (2015). Biofilm-specific antibiotic tolerance and resistance. Eur. J. Clin. Microbiol. Infect. Dis. 34, 877–886. doi: 10.1007/s10096-015-2323-z
Ortiz de la Rosa J.-M., Nordmann P., Poirel L. (2019). ESBLs and resistance to ceftazidime/avibactam and ceftolozane/tazobactam combinations in Escherichia coli and Pseudomonas aeruginosa. J. Antimicrob. Chemother. 74, 1934–1939. doi: 10.1093/jac/dkz149
Overhage J., Lewenza S., Marr A. K., Hancock R. E. W. (2007). Identification of genes involved in swarming motility using a Pseudomonas aeruginosa PAO1 mini-Tn5-lux mutant library. J. Bacteriol. 189, 2164–2169. doi: 10.1128/JB.01623-06
Palzkill T. (2013). Metallo-β-lactamase structure and function. Ann. N. Y. Acad. Sci. 1277, 91–104. doi: 10.1111/j.1749-6632.2012.06796.x
Pan Y.-P., Xu Y.-H., Wang Z.-X., Fang Y.-P., Shen J.-L. (2016). Overexpression of MexAB-OprM efflux pump in carbapenem-resistant Pseudomonas aeruginosa. Arch. Microbiol. 198, 565–571. doi: 10.1007/s00203-016-1215-7
Parameswaran G. I., Sethi S. (2012). Pseudomonas infection in chronic obstructive pulmonary disease. Future Microbiol. 7, 1129–1132. doi: 10.2217/fmb.12.88
Park A. J., Murphy K., Surette M. D., Bandoro C., Krieger J. R., Taylor P., et al. (2015). Tracking the dynamic relationship between cellular systems and extracellular subproteomes in Pseudomonas aeruginosa biofilms. J. Proteome Res. 14, 4524–4537. doi: 10.1021/acs.jproteome.5b00262
Pasca M. R., Dalla Valle C., De Jesus Lopes Ribeiro A. L., Buroni S., Papaleo M. C., Bazzini S., et al. (2012). Evaluation of Fluoroquinolone Resistance Mechanisms in Pseudomonas aeruginosa Multidrug Resistance Clinical Isolates. Microb. Drug Resist. 18, 23–32. doi: 10.1089/mdr.2011.0019
Paterson D. L., Bonomo R. A. (2005). Extended-spectrum β-lactamases: A clinical update. Clin. Microbiol. Rev. 18, 657–686. doi: 10.1128/CMR.18.4.657-686.2005
Paulsen I. T. (2003). Multidrug efflux pumps and resistance: regulation and evolution. Curr. Opin. Microbiol. 6, 446–451. doi: 10.1016/j.mib.2003.08.005
Pendleton J. N., Gorman S. P., Gilmore B. F. (2013). Clinical relevance of the ESKAPE pathogens. Expert Rev. Anti-Infect. Ther. 11, 297–308. doi: 10.1586/eri.13.12
Piddock L. J. V., Hall M. C., Bellido F., Bains M., Hancock R. E. W. (1992). A pleiotropic, posttherapy, enoxacin-resistant mutant of Pseudomonas aeruginosa. Antimicrob. Agents Chemother. 36, 1057–1061. doi: 10.1128/AAC.36.5.1057
Poole K., Gotoh N., Tsujimoto H., Zhao Q., Wada A., Yamasaki T., et al. (1996a). Expression of the multidrug resistance operon mexA-mexB-oprM in Pseudomonas aeruginosa: mexR encodes a regulator of operon expression. Antimicrob. Agents Chemother. 40, 2021–2028. doi: 10.1128/AAC.40.9.2021
Poole K., Tetro K., Zhao Q., Neshat S., Heinrichs D. E., Bianco N., et al. (1996b). Overexpression of the mexC-mexD-oprJ efflux operon in nfxB -type multidrug-resistant strains of Pseudomonas aeruginosa. Mol. Microbiol. 21, 713–725. doi: 10.1046/j.1365-2958.1996.281397.x
Poole K. (2001). Multidrug Efflux Pumps and Antimicrobial Resistance in Pseudomonas aeruginosa and Related Organisms JMMB Symposium. J. Mol. Microbiol. Biotechnol. 613, 255–64.
Poole K. (2005). Aminoglycoside resistance in Pseudomonas aeruginosa. Antimicrobial. Agents Chemother. 49, 479–487. doi: 10.1128/AAC.49.2.479-487.2005
Poole K. (2008). Bacterial multidrug efflux pumps serve other functions. Microbe 3, 179–185. doi: 10.1128/microbe.3.179.1
Poole K. (2011). Pseudomonas aeruginosa: resistance to the max. Front. Microbiol. 2, 65. doi: 10.3389/fmicb.2011.00065
Poole K. (2014). Stress responses as determinants of antimicrobial resistance in Pseudomonas aeruginosa: multidrug efflux and more. Can. J. Microbiol. 60, 783–791. doi: 10.1139/cjm-2014-0666
Prickett M. H., Hauser A. R., McColley S. A., Cullina J., Potter E., Powers C., et al. (2017). Aminoglycoside resistance of Pseudomonas aeruginosa in cystic fibrosis results from convergent evolution in the mexZ gene. Thorax 72, 40–47. doi: 10.1136/thoraxjnl-2015-208027
Pumbwe L., Piddock L. J. V. (2000). Two efflux systems expressed simultaneously in multidrug-resistant Pseudomonas aeruginosa. Antimicrob. Agents Chemother. 44, 2861–2864. doi: 10.1128/AAC.44.10.2861-2864.2000
Pumbwe L., Everett M. J., Hancock R. E., Piddock L. J. (1996). Role of gyrA mutation and loss of OprF in the multiple antibiotic resistance phenotype of Pseudomonas aeruginosa G49. FEMS Microbiol. Lett. 143, 25–28. doi: 10.1111/j.1574-6968.1996.tb08456.x
Queenan A. M., Bush K. (2007). Carbapenemases: The versatile β-lactamases. Clin. Microbiol. Rev. 20, 440–458. doi: 10.1128/CMR.00001-07
Ramirez M. S., Tolmasky M. E. (2010). Aminoglycoside modifying enzymes. Drug Resist. Update 13, 151–171. doi: 10.1016/j.drup.2010.08.003
Ranjitkar S., Jones A. K., Mostafavi M., Zwirko Z., Iartchouk O., Barnes S. W., et al. (2019). Target (MexB)- and Efflux-Based Mechanisms Decreasing the Effectiveness of the Efflux Pump Inhibitor D13-9001 in Pseudomonas aeruginosa PAO1: Uncovering a New Role for MexMN-OprM in Efflux of β-Lactams and a Novel Regulatory Circuit (MmnRS) Controlling MexMN Expression. Antimicrob. Agents Chemother. 63. doi: 10.1128/AAC.01718-18
Rasamiravaka T., Labtani Q., Duez P., El Jaziri M. (2015). The formation of biofilms by pseudomonas aeruginosa: A review of the natural and synthetic compounds interfering with control mechanisms. BioMed. Res. Int. 2015. doi: 10.1155/2015/759348
Rees V. E., Deveson Lucas D. S., López-Causapé C., Huang Y., Kotsimbos T., Bulitta J. B., et al. (2019). Characterization of Hypermutator Pseudomonas aeruginosa Isolates from Patients with Cystic Fibrosis in Australia. Antimicrob. Agents Chemother. 63. doi: 10.1128/AAC.02538-18
Rehman A., Patrick W. M., Lamont I. L. (2019). Mechanisms of ciprofloxacin resistance in pseudomonas aeruginosa: New approaches to an old problem. J. Med. Microbiol. 68, 1–10. doi: 10.1099/jmm.0.000873
Renau T. E., Léger R., Filonova L., Flamme E. M., Wang M., Yen R., et al. (2003). Conformationally-restricted analogues of efflux pump inhibitors that potentiate the activity of levofloxacin in Pseudomonas aeruginosa. Bioorg. Med. Chem. Lett. 13, 2755–2758. doi: 10.1016/S0960-894X(03)00556-0
Reza A., Mark Sutton J., Rahman K. M. (2019). Effectiveness of efflux pump inhibitors as biofilm disruptors and resistance breakers in gram- negative (ESKAPEE) bacteria. Antibiotics 8, 229. doi: 10.3390/antibiotics8040229
Richardot C., Plésiat P., Fournier D., Monlezun L., Broutin I., Llanes C., et al. (2015). Carbapenem resistance in cystic fibrosis strains of Pseudomonas aeruginosa as a result of amino acid substitutions in porin OprD. Int. J. Antimicrob. Agents 45, 529–532. doi: 10.1016/j.ijantimicag.2014.12.029
Rybtke M., Hultqvist L. D., Givskov M., Tolker-Nielsen T. (2015). Pseudomonas aeruginosa Biofilm Infections: Community Structure, Antimicrobial Tolerance and Immune Response. J. Mol. Biol. 427, 3628–3645. doi: 10.1016/j.jmb.2015.08.016
Sánchez-Jiménez F., Medina M. Á., Villalobos-Rueda L., Urdiales J. L. (2019). Polyamines in mammalian pathophysiology. Cell. Mol. Life Sci. 76, 3987–4008. doi: 10.1007/s00018-019-03196-0
Santajit S., Indrawattana N. (2016). Mechanisms of Antimicrobial Resistance in ESKAPE Pathogens. BioMed. Res. Int. 2016. doi: 10.1155/2016/2475067
Sauvage E., Kerff F., Terrak M., Ayala J. A., Charlier P. (2008). The penicillin-binding proteins: Structure and role in peptidoglycan biosynthesis. FEMS Microbiol. Rev. 32, 234–258. doi: 10.1111/j.1574-6976.2008.00105.x
Schwechheimer C., Kuehn M. J. (2015). Outer-membrane vesicles from Gram-negative bacteria: Biogenesis and functions. Nat. Rev. Microbiol. 13, 605–619. doi: 10.1038/nrmicro3525
Sciuto A., Imperi F. (2018). Aminoarabinosylation of lipid a is critical for the development of colistin resistance in pseudomonas aeruginosa. Antimicrob. Agents Chemother. 62. doi: 10.1128/AAC.01820-17
Semaniakou A., Croll R. P., Chappe V. (2019). Animal models in the pathophysiology of cystic fibrosis. Front. Pharmacol. 9, 1475. doi: 10.3389/fphar.2018.01475
Severino P., Silveira E. F., Loureiro K., Chaud M. V., Antonini D., Lancellotti M., et al. (2017). Antimicrobial activity of polymyxin-loaded solid lipid nanoparticles (PLX-SLN): Characterization of physicochemical properties and in vitro efficacy. Eur. J. Pharm. Sci. 106, 177–184. doi: 10.1016/j.ejps.2017.05.063
Shaaban M. I., Shaker M. A., Mady F. M. (2017). Imipenem/cilastatin encapsulated polymeric nanoparticles for destroying carbapenem-resistant bacterial isolates. J. Nanobiotechnol. 15, 1–12. doi: 10.1186/s12951-017-0262-9
Shakil S., Khan R., Zarrilli R., Khan A. U. (2008). Aminoglycosides versus bacteria - A description of the action, resistance mechanism, and nosocomial battleground. J. Biomed. Sci. 15, 5–14. doi: 10.1007/s11373-007-9194-y
Skiada A., Markogiannakis A., Plachouras D., Daikos G. L. (2011). Adaptive resistance to cationic compounds in Pseudomonas aeruginosa. Int. J. Antimicrobial. Agents 37, 187–193. doi: 10.1016/j.ijantimicag.2010.11.019
Slama T. G. (2008). Gram-negative antibiotic resistance: there is a price to pay. Crit. Care 12 Suppl 4, S4. doi: 10.1186/cc6820
Smith E. E., Buckley D. G., Wu Z., Saenphimmachak C., Hoffman L. R., D’Argenio D. A., et al. (2006). Genetic adaptation by Pseudomonas aeruginosa to the airways of cystic fibrosis patients. Proc. Natl. Acad. Sci. 103, 8487–8492. doi: 10.1073/pnas.0602138103
Sobel M. L., Neshat S., Poole K. (2005). Mutations in PA2491 (mexS) promote MexT-dependent mexEF-oprN expression and multidrug resistance in a clinical strain of Pseudomonas aeruginosa. J. Bacteriol. 187, 1246–1253. doi: 10.1128/JB.187.4.1246-1253.2005
Srikumar R., Kon T., Gotoh N., Poole K. (1998). Expression of Pseudomonas aeruginosa multidrug efflux pumps mexA-mexB- OprM and MexC-MexD-OprJ in a multidrug-sensitive Escherichia coli strain. Antimicrob. Agents Chemother. 42, 65–71. doi: 10.1128/AAC.42.1.65
Srinivas N., Jetter P., Ueberbacher B. J., Werneburg M., Zerbe K., Steinmann J., et al. (2010). Peptidomimetic antibiotics target outer-membrane biogenesis in pseudomonas aeruginosa. Science 327, 1010–1013. doi: 10.1126/science.1182749
Sriramulu D. D., Lünsdorf H., Lam J. S., Römling U. (2005). Microcolony formation: A novel biofilm model of Pseudomonas aeruginosa for the cystic fibrosis lung. J. Med. Microbiol. 54, 667–676. doi: 10.1099/jmm.0.45969-0
Stokes J. M., Macnair C. R., Ilyas B., French S., Côté J. P., Bouwman C., et al. (2017). Pentamidine sensitizes Gram-negative pathogens to antibiotics and overcomes acquired colistin resistance. Nat. Microbiol. 2. doi: 10.1038/nmicrobiol.2017.28
Stover C. K., Pham X. Q., Erwin A. L., Mizoguchi S. D., Warrener P., Hickey M. J., et al. (2000). Complete genome sequence of Pseudomonas aeruginosa PAO1, an opportunistic pathogen. Nature 406, 959. doi: 10.1038/35023079
Streeter K., Katouli M. (2016). Pseudomonas aeruginosa: A review of their Pathogenesis and Prevalence in Clinical Settings and the Environment. Infect. Epidemiol. Med. 2, 25–32. doi: 10.18869/modares.iem.2.1.25
Sugawara E., Nagano K., Nikaido H. (2012). Alternative folding pathways of the major porin OprF of Pseudomonas aeruginosa. FEBS J. 279, 910–918. doi: 10.1111/j.1742-4658.2012.08481.x
Sun E., Gill E. E., Falsafi R., Yeung A., Liu S., Hancock R. E. W., et al. (2018a). Broad-spectrum adaptive antibiotic resistance associated with pseudomonas aeruginosa mucin-dependent surfing motility. Antimicrob. Agents Chemother. 62. doi: 10.1128/AAC.00848-18
Sun E., Liu S., Hancock R. E. W. (2018b). Surfing Motility: a Conserved yet Diverse Adaptation among Motile Bacteria. Journal of Bacteriology 200 (23). doi: 10.1128/JB.00394-18
Szczepanowski R., Linke B., Krahn I., Gartemann K. H., Gützkow T., Eichler W., et al. (2009). Detection of 140 clinically relevant antibiotic-resistance genes in the plasmid metagenome of wastewater treatment plant bacteria showing reduced susceptibility to selected antibiotics. Microbiology 155, 2306–2319. doi: 10.1099/mic.0.028233-0
Tada T., Miyoshi-Akiyama T., Kato Y., Ohmagari N., Takeshita N., Hung N. V., et al. (2013). Emergence of 16S rRNA methylase-producing Acinetobacter baumannii and Pseudomonas aeruginosa isolates in hospitals in Vietnam. BMC Infect. Dis. 13, 251. doi: 10.1186/1471-2334-13-251
Tata M., Wolfinger M. T., Amman F., Roschanski N., Dötsch A., Sonnleitner E., et al. (2016). RNASeq Based Transcriptional Profiling of Pseudomonas aeruginosa PA14 after Short- and Long-Term Anoxic Cultivation in Synthetic Cystic Fibrosis Sputum Medium. PloS One 11, e0147811. doi: 10.1371/journal.pone.0147811
Tenover F. C. (2006). Mechanisms of Antimicrobial Resistance in Bacteria. Am. J. Med. 119, S3–10. doi: 10.1016/j.amjmed.2006.03.011
Tenson T., Lovmar M., Ehrenberg M. (2003). The mechanism of action of macrolides, lincosamides and streptogramin B reveals the nascent peptide exit path in the ribosome. J. Mol. Biol. 330, 1005–1014. doi: 10.1016/S0022-2836(03)00662-4
Tipper D. J. (1985). Mode of action of β-lactam antibiotics. Pharmacol. Ther. 27, 1–35. doi: 10.1016/0163-7258(85)90062-2
Tomasello G., Armenia I., Molla G. (2020). The Protein Imager: a full-featured online molecular viewer interface with server-side HQ-rendering capabilities. Bioinformatics 36, 2909–2911. doi: 10.1093/bioinformatics/btaa009
Tooke C. L., Hinchliffe P., Bragginton E. C., Colenso C. K., Hirvonen V. H. A., Takebayashi Y., et al. (2019). β-Lactamases and β-Lactamase Inhibitors in the 21st Century. J. Mol. Biol. 431, 3472–3500. doi: 10.1016/j.jmb.2019.04.002
Trias J., Dufresne J., Levesque R. C., Nikaido H. (1989). Decreased outer membrane permeability in imipenem-resistant mutants of Pseudomonas aeruginosa. Antimicrob. Agents Chemother. 33, 1201–1206. doi: 10.1128/AAC.33.8.1202
Triass J., Nikaido H. (1990). Protein D2 Channel of the Pseudomonas aeruginosa Outer Membrane Has a Binding Site for Basic Amino Acids and Peptides. J. Biol. Chem. 265, 15680–15684. doi: 10.1016/S0021-9258(18)55452-1
Tsubery H., Ofek I., Cohen S., Fridkin M. (2000). The functional association of polymyxin B with bacterial lipopolysaccharide is stereospecific: Studies on polymyxin B nonapeptide. Biochemistry 39, 11837–11844. doi: 10.1021/bi000386q
Tümmler B. (2019). Emerging therapies against infections with pseudomonas aeruginosa [version 1; peer review: 2 approved]. F1000Research 8. doi: 10.12688/f1000research.19509.1
Upert G., Luther A., Obrecht D., Ermert P. (2021). Emerging peptide antibiotics with therapeutic potential. Med. Drug Discovery 9, 100078. doi: 10.1016/j.medidd.2020.100078
Urbanowicz P., Izdebski R., Baraniak A., Żabicka D., Ziółkowski G., Hryniewicz W., et al. (2019). Pseudomonas aeruginosa with NDM-1, DIM-1 and PME-1 β-lactamases, and RmtD3 16S rRNA methylase, encoded by new genomic islands. J. Antimicrob. Chemother. 74, 3117–3119. doi: 10.1093/jac/dkz262
Vaara M. (2019). Polymyxin derivatives that sensitize Gram-negative bacteria to other antibiotics. Molecules 24, 249. doi: 10.3390/molecules24020249
Vadekeetil A., Chhibber S., Harjai K. (2019). Efficacy of intravesical targeting of novel quorum sensing inhibitor nanoparticles against Pseudomonas aeruginosa biofilm-associated murine pyelonephritis. J. Drug Targeting 27, 995–1003. doi: 10.1080/1061186X.2019.1574802
Van Heeckeren A. M., Schluchter M. D. (2002). Murine models of chronic Pseudomonas aeruginosa lung infection. Lab. Anim. 36, 291–312. doi: 10.1258/002367702320162405
Vogne C., Aires J. R., Bailly C., Hocquet D., Plésiat P. (2004). Role of the Multidrug Efflux System MexXY in the Emergence of Moderate Resistance to Aminoglycosides among Pseudomonas aeruginosa Isolates from Patients with Cystic Fibrosis. Antimicrob. Agents Chemother. 48, 1676–1680. doi: 10.1128/AAC.48.5.1676-1680.2004
Vollmer W., Blanot D., De Pedro M. A. (2008). Peptidoglycan structure and architecture. FEMS Microbiol. Rev. 32, 149–167. doi: 10.1111/j.1574-6976.2007.00094.x
Wang L., Zhang C., Gong F., Li H., Xie X., Xia C., et al. (2013). Influence of pseudomonas aeruginosa pvdQ gene on altering antibiotic susceptibility under swarming conditions. Curr. Microbiol. 66, 152–161. doi: 10.1007/s00284-012-0217-1
Wang S., Yu S., Lin Y., Zou P., Chai G., Yu H. H., et al. (2018). Co-Delivery of Ciprofloxacin and Colistin in Liposomal Formulations with Enhanced In Vitro Antimicrobial Activities against Multidrug Resistant Pseudomonas aeruginosa. Pharm. Res. 35, 187. doi: 10.1007/s11095-018-2464-8
Waters E. M., Neill D. R., Kaman B., Sahota J. S., Clokie M. R. J., Winstanley C., et al. (2017). Phage therapy is highly effective against chronic lung infections with Pseudomonas aeruginosa. Thorax 72, 666–667. doi: 10.1136/thoraxjnl-2016-209265
Wei Q., Tarighi S., Dötsch A., Häussler S., Müsken M., Wright V. J., et al. (2011). Phenotypic and genome-wide analysis of an antibiotic-resistant small colony variant (SCV) of pseudomonas aeruginosa. PloS One 6. doi: 10.1371/journal.pone.0029276
Werneburg M., Zerbe K., Juhas M., Bigler L., Stalder U., Kaech A., et al. (2012). Inhibition of Lipopolysaccharide Transport to the Outer Membrane in Pseudomonas aeruginosa by Peptidomimetic Antibiotics. ChemBioChem 13, 1767–1775. doi: 10.1002/cbic.201200276
Willmott C. J. R., Maxwell A. (1993). A single point mutation in the DNA gyrase A protein greatly reduces binding of fluoroquinolones to the gyrase-DNA complex. Antimicrobial. Agents Chemother. 37, 126–127. doi: 10.1128/AAC.37.1.126
Wong D., Duin D.V. (2017). Novel Beta-Lactamase Inhibitors: Unlocking Their Potential in Therapy. Drugs 77, 615–628. doi: 10.1007/s40265-017-0725-1
Woodruff W. A., Hancock R. E. W. (1989). Pseudomonas aeruginosa outer membrane protein F: Structural role and relationship to the Escherichia coli omP protein. J. Bacteriol. 171, 3304–3309. doi: 10.1128/JB.171.6.3304-3309.1989
Wydmuch Z., Skowronek-Ciołek O., Cholewa K., Mazurek U., Pacha J., Kepa M., et al. (2005). GyrA mutations in ciprofloxacin-resistant clinical isolates of Pseudomonas aeruginosa in a Silesian Hospital in Poland. Polish J. Microbiol. 54, 201—206.
Yang X., Xing B., Liang C., Ye Z., Zhang Y. (2015). Prevalence and fluoroquinolone resistance of pseudomonas aeruginosa in a hospital of South China. Int. J. Clin. Exp. Med. 8, 1386–1390.
Yeung A. T. Y., Parayno A., Hancock R. E. W. (2012). Mucin promotes rapid surface motility in pseudomonas aeruginosa. MBio 3. doi: 10.1128/mBio.00073-12
Yokoyama K., Doi Y., Yamane K., Kurokawa H., Shibata N., Shibayama K., et al. (2003). Acquisition of 16S rRNA methylase gene in Pseudomonas aeruginosa. Lancet 362, 1888–1893. doi: 10.1016/S0140-6736(03)14959-8
Yoneyama H., Yamano Y., Nakae T. (1995). Role of Porins in the Antibiotic Susceptibility of Pseudomonas aeruginosa: Construction of Mutants with Deletions in the Multiple Porin Genes. Biochem. Biophys. Res. Commun. 213, 88–95. doi: 10.1006/bbrc.1995.2102
Yoon S. S., Hennigan R. F., Hilliard G. M., Ochsner U. A., Parvatiyar K., Kamani M. C., et al. (2002). Pseudomonas aeruginosa anaerobic respiration in biofilms: Relationships to cystic fibrosis pathogenesis. Dev. Cell 3, 593–603. doi: 10.1016/S1534-5807(02)00295-2
Yoshida H., Iwata N., Katsuzaki H., Naganawa R., Ishikawa K., Fukuda H., et al. (1998). Antimicrobial Activity of a Compound Isolated from an Oil-Macerated Garlic Extract. Biosci. Biotechnol. Biochem. 62. doi: 10.1271/bbb.62.1014
Yoshida K., Nakayama K., Ohtsuka M., Kuru N., Yokomizo Y., Sakamoto A., et al. (2007). MexAB-OprM specific efflux pump inhibitors in Pseudomonas aeruginosa. Part 7: Highly soluble and in vivo active quaternary ammonium analogue D13-9001, a potential preclinical candidate. Bioorg. Med. Chem. 15, 7087–7097. doi: 10.1016/j.bmc.2007.07.039
Zahra M.-J., Hamed H., Mohammad R.-Y., Nosratollah Z., Akbarzadeh A., Morteza M., et al. (2017). Evaluation and study of antimicrobial activity of nanoliposomal meropenem against Pseudomonas aeruginosa isolates. Artif. Cells Nanomed. Biotechnol. 45, 975–980. doi: 10.1080/21691401.2016.1198362
Keywords: multidrug-resistance (MDR), resistance mechanism interplay, antimicrobial resistance (AMR), resistance-breaking therapy, adjuvant therapies, Pseudomonas aeruginosa
Citation: Langendonk RF, Neill DR and Fothergill JL (2021) The Building Blocks of Antimicrobial Resistance in Pseudomonas aeruginosa: Implications for Current Resistance-Breaking Therapies. Front. Cell. Infect. Microbiol. 11:665759. doi: 10.3389/fcimb.2021.665759
Received: 08 February 2021; Accepted: 29 March 2021;
Published: 16 April 2021.
Edited by:
Melanie Ghoul, University of Oxford, United KingdomReviewed by:
Oana Ciofu, University of Copenhagen, DenmarkCopyright © 2021 Langendonk, Neill and Fothergill. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: R. Frèdi Langendonk, Zi5lLmxhbmdlbmRvbmtAbGl2ZXJwb29sLmFjLnVr
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.