 Haifa Bichiou1,2
Haifa Bichiou1,2 Cyrine Bouabid
Cyrine Bouabid Imen Rabhi
Imen Rabhi Lamia Guizani-Tabbane
Lamia Guizani-Tabbane- 1Laboratory of Medical Parasitology, Biotechnology and Biomolecules (PMBB), Institut Pasteur de Tunis, Tunis, Tunisia
- 2Faculty of Sciences of Tunis, Université de Tunis El Manar, Tunis, Tunisia
- 3Biotechnology Department, Higher Institute of Biotechnology at Sidi-Thabet (ISBST), Biotechpole Sidi-Thabet- University of Manouba, Tunis, Tunisia
Leishmaniasis is a group of heterogenous diseases considered as an important public health problem in several countries. This neglected disease is caused by over 20 parasite species of the protozoa belonging to the Leishmania genus and is spread by the bite of a female phlebotomine sandfly. Depending on the parasite specie and the immune status of the patient, leishmaniasis can present a wide spectrum of clinical manifestations. As an obligate intracellular parasite, Leishmania colonize phagocytic cells, mainly the macrophages that orchestrate the host immune response and determine the fate of the infection. Once inside macrophages, Leishmania triggers different signaling pathways that regulate the immune and metabolic response of the host cells. Various transcription factors regulate such immune-metabolic responses and the associated leishmanicidal and inflammatory reaction against the invading parasite. In this review, we will highlight the most important transcription factors involved in these responses, their interactions and their impact on the establishment and the progression of the immune response along with their effect on the physiopathology of the disease.
Introduction
Leishmania spp. are protozoan parasites that affects millions of people around the world. They are the causative agent of leishmaniasis, a group of heterogeneous diseases endemic in various countries. Depending on the parasite specie, and the immune status of the patient, leishmaniasis can present a spectrum of clinical manifestations. In the mammalian host, this parasite targets the immune cells that represent the first line of defense against infections and uses macrophage as a final host making Leishmania-macrophages interactions central to the outcome of the disease.
Different Leishmania species trigger distinct immune responses and macrophage polarization, upon their infection by the parasite, highly impact the infection outcome.
In the site of infection, recruited monocytes differentiate into macrophages that show extensive plasticity, depending on converging signals delivered by the numerous factors (microbial products, cytokines (IFNγ, TNFα) and growth factors, present in the local tissue environment. Macrophages would then adopt a spectrum of distinct polarization states, among which the M1 inflammatory and the M2 anti-inflammatory cells are the extremes (Xue et al., 2014).
The « classically activated » or M1 macrophages polarization are important for the control of infections by intracellular pathogens. They are characterized by the production of proinflammatory cytokines such as TNFα, IL-1β, IL-6, IL-12, IL-18, IL-23, and Type 1 IFN, but also by the generation of reactive oxygen and nitrogen species that ensure efficient microbial killing. The « alternatively activated” M2 macrophages are elicited by type 2 cytokines (IL-4, IL-10, IL-13) and are inflammation-resolving macrophages with anti-parasitic and tissue repair functions (Murray, 2017).
These macrophage polarization phenotypes are also characterized by two distinct metabolic signatures. Indeed, in resting macrophages, glucose is metabolized to pyruvate which is transported into the mitochondria where it is oxidized into acetyl coenzyme A (acetyl-CoA) to fuel the tricarboxylic acid (TCA) cycle. The latter generates NADH, which provides electrons to the mitochondrial electron transport chain so that oxidative phosphorylation (OXPHOS) can progress and generate ATP. Upon infection, the metabolic profile switches from oxidative phosphorylation to aerobic glycolysis and the macrophages rely on glycolysis for the production of ATP (Kelly and O’Neill, 2015). Glycolysis provides the glucose-6-phosphate, an enhancer of the Pentose Phosphate Pathway (PPP) and a booster of NADPH production. This latter is then used by the NADPH oxidase for ROS and nitric oxide generation, two pathogens-killing agents. Metabolism reprogramming embraces also different other metabolism including lipid, and M1 macrophages have been shown to drive fatty acid synthesis (Zhu et al., 2015). By contrast, M2 macrophages rely on fatty acid oxidation in the mitochondria to generate acetyl-CoA, NADH, and FADH2 used to produce ATP through OXPHOS, assuring the long-term need in energy of these macrophages.
Along with this role in energy generation, cellular metabolism is emerging as an important determinant of the functional phenotype acquired by macrophages and a key regulator of macrophage immune response and inflammation (Kelly and O’Neill, 2015; Zhu et al., 2015). For instance, the role of L-arginine metabolism in regulating the microbicidal activity of macrophages through the production of nitric oxide or the generation of ornithine and polyamines is well documented and was one of the first characteristics used to define macrophage subsets (Muxel et al., 2017).
Progress has been made in defining the molecular pathways that underlie M2 versus M1 polarization and the different signaling molecules and transcription factors involved in regulating macrophage metabolic profiles, polarization and function.
Functional control of macrophages largely occurs at the transcriptional level and different transcription factors have been implicated in the polarization and activation of macrophages in response to infection. Signal transducers and activators of transcription (STATs) (STAT1), interferon regulatory factors (IRF) and nuclear factor kappa B (NF-κB) together with hypoxia-Inducible Factor (HIFα), are major players in inflammation and in the M1 polarization of macrophage during infection whereas IRF4, STAT6 and Peroxisome proliferator-activated receptor (PPAR) activated by IL-4, control M2 gene expression (Murray, 2017).
In this review we will summarize key mechanisms of Leishmania-macrophage interactions, focusing on the contribution of transcription factors (Table 1) to macrophage responses and to the physiopathology of the disease.

Table 1 Key transcription factors that modulate macrophages response to Leishmania infection.
Transcriptomic Signature of Leishmania Infected Macrophages
When infecting macrophages, microbes induce marked changes in gene expression programme and shape the phenotype and function of the infected cell. These changes have been investigated for different intracellular pathogens and the macrophagic transcriptomic signature identified for various bacteria, viruses and parasites. Different genes expression profiles have been compared and pathogens-mediated signaling have been shown to share a common host-transcriptional-response (Jenner and Young, 2005), which includes the group of genes that activate immune response, genes encoding inflammatory cytokines and chemokines and those stimulated by Interferon. This common host transcriptional-response is also shared by the transcriptomic profile of Leishmania infected macrophages (Buates and Matlashewski, 2001; Chaussabel et al., 2003; Rodriguez et al., 2004; Osorio y Fortea et al., 2009) except for the IFNγ activated genes involved in the development of the microbicidal activities and promoted by the Janus kinase signal transducer and activator of transcription (JAK/STAT) pathway.
Interferon gamma receptor ligation induces the activation of JAK1 and JAK2 tyrosine kinases, constitutively associated to the receptor. These kinases phosphorylate, on the receptor, specific tyrosine residues that serve as a docking site for STATI transcription factor which is also a target of JAKs. Once phosphorylated by JAKs, STATI form dimers and translocate to the nucleus to bind the gamma interferon activation site (GAS) elements, present on different promoters of several genes including those encoding for NOS2, the MHC class II transactivator (CIITA) and IL-12 cytokine.
Leishmania-infected macrophages fail to respond to IFNγ a key cytokine that empower macrophages to clear the parasite and this failure is the result of the IFNγ signaling pathway inhibition. Indeed, in both differentiated U-937 cells and human monocytes infected by Leishmania donovani, the tyrosine phosphorylation, Jakl and Jak2 activation and Statl phosphorylation normally induced by IFNγ, were selectively impaired (Nandan and Reiner, 1995). Similar response was observed when macrophages were infected by other Leishmania species. Several mechanisms have been proposed to explain the inhibition of JAK-STAT pathway during Leishmania infection. This IFNγ signaling pathway inhibition was suggested to be the result of a decreased expression of IFN-gamma R-alpha protein after infection (Ray et al., 2000). The co-immunoprecipitation of JAK2 with the Leishmania induced-tyrosine phosphatase (P’TP) SHP-1, suggests that this phosphatase may be at the origin of the IFN-gamma-inducible JAK2 unresponsiveness (Blanchette et al., 1999). However, additional mechanisms must participate to the inhibition of IFNγ signaling as Leishmania parasite retains its capability to inhibit STATI activation in SHP-1-deficient macrophages suggesting a SHP-1 independent mechanism (Forget et al., 2006). Leishmania induced STATI inactivation could be also the consequence of proteasome-mediated degradation of this transcription factor since proteasome inhibitors rescued STAT1 nuclear translocation and restored STAT1 protein level (Forget et al., 2005). It could also be the result of a defective nuclear translocation of STATI due to a compromised IFN-induced STAT1 association with the nuclear transport adaptor importin-5 as described for L. donovani amastigote (Matte and Descoteaux, 2010).
The suppressor of cytokine signaling (SOCS) proteins by binding to JAK or cytokine receptors and suppressing STAT phosphorylation are one of the crucial negative regulators of JAK/STAT signaling pathways. SOCS have been reported to be activated in response to different parasites (Bullen et al., 2003a; Zimmermann et al., 2006; Dalpke et al., 2008) and to play a role in regulating immune response in various infectious disease including leishmaniasis (Bullen et al., 2003b). Indeed, Leishmania LPG, an agonist of TLR2 (de Veer et al., 2003) and L. donovani parasites have been shown to induce SOCS3 and SOCS1 expression in macrophages (Bertholet et al., 2003). The early and transient expression of SOCS3 may suppress the reactive oxygen species-mediated apoptotic signaling cascade (Srivastav et al., 2014) while the stable and robust expression of SOCS1 could inhibit phosphorylation of STAT1 and induce the down-regulation the IL-12 cytokine level in infected macrophages (Chandrakar et al., 2020).
Hence, in response to Leishmania infection, several mechanisms, acting at various steps of the JAK-STAT pathway might potentially cooperate to coordinately shutdown JAK-STAT pathway, crucial for macrophage-T cells cross talk.
Transcription Factors Underlying Leishmania-Induced Inflammatory Response
Macrophages sense protozoan parasites using different surface receptors that participate in the course of infection and have different effect on macrophage-Leishmania interaction. Among these, the Toll like receptors (TLRs), activated by various parasites surface molecules, play a crucial role in initiating host innate immune responses and directing adaptive immune responses against invading pathogens. TLRs localize variously as TLR1, -2, -4, -5, and 6, are expressed on the cell membrane, whereas TLR3, -7, -8, -9, are expressed in the endosome. Once activated adaptor molecules, such as MyD88 (myeloid differentiation primary-response protein 88) shared by all TLRs except for TLR3, TRIF (TIR-domain-containing adaptor protein inducing beta interferon (IFN-β), unique to TLR3 and TLR4 signaling, TIRAP (TIR-domain-containing adaptor protein) that mediates the activation of signaling downstream of TLR2 and TLR4, or TRAM (TIR-domain-containing adaptor molecule) unique to TLR4, are recruited to the receptor (Satoh and Akira, 2016).
Different TLRs such as TLR2, TLR4 (Kropf et al., 2004) and TLR9 have been involved in providing protection against Leishmania spp infection (Liese et al., 2007; Gurung and Kanneganti, 2015). It was therefore not surprising that mice deficient for MyD88, the common adaptor acting downstream of almost all the TLRs, were highly susceptible to L. major infections (de Veer et al., 2003; Debus et al., 2003; Muraille et al., 2003).
The interaction between the Leishmania ligands and TLRs induce the recruitment of different adaptor molecules that transduce TLRs intracellular signals and lead to the activation of various transcription factors, including NF-κB and IRFs which play a key role in defining functional phenotype of macrophages (Colonna, 2007).
Indeed, MyD88 adaptor has been shown to form a multimolecular complex consisting of IRAKI, IRAK4, TRAF6, IRF-5, and IRF-7. IRF7 is a master regulator of type I IFN genes (Honda et al., 2005) whereas IRF5 is implicated in TLR-dependent induction of pro-inflammatory cytokines, such as interleukin-6 (L-6), IL-12p40 and tumour-necrosis factor-alpha (TNFα) as confirmed by IRF5 deficient mice (Takaoka et al., 2005). IRF-5 that play an essential role in M1 macrophages polarization, compete with IRF4 for binding to MyD88 (Ferrante and Leibovich, 2012). This negative regulator of MyD88 signaling is crucial for M2 polarization and IRF4 and IRF5 competition for binding to MyD88 plays a key role in choosing whether macrophage polarization goes toward M1 or M2 phenotype (Negishi et al., 2005).
The TRIF-dependent pathway, restricted to TLR-3 and TLR-4, recruits IRF3 to induce type I IFN responses, particularly IFN-beta (Sharma et al., 2003).
IRF1, highly induced by IFNγ, directly interacts with and is activated by MyD88 upon TLR9 activation (Schmitz et al., 2007) and the TLR9-mediated IRF1 activation, as confirmed in IRF1-deficient cells, is required for optimal regulation of a specific gene subset that includes IFNβ, inducible NO synthase (iNOS) and IL-12p35 (Negishi et al., 2006).
Hence multiple TLR pathways require either IRF-1, IRF-3 or IRF-7 for type I IFN-induction. IRF-1 and IRF-7 are recruited and activated through MyD88, which simultaneously triggers IRF-5 and a parallel signaling pathway for NF-κB activation leading to secretion of proinflammatory cytokines (Colonna, 2007).
IRF-1 (Lohoff et al., 1997), IRF-2 (Lohoff et al., 2000) and IRF-8 (Giese et al., 1997) knockout mice fail to mount a protective Th1 response and are thus highly susceptible to L. major infection. However, a defect in IRF1 expression is also observed in Leishmania infected macrophages treated with IFNγ (Matte and Descoteaux, 2010). This defect may be due to the inhibition of STAT1 activation by Leishmania infection. Indeed, IRF1 expression is mediated by STAT1 and NF-κB transcription factors, a finding confirmed by the data showing that IFN-induced expression of IRF-1 mRNA was completely abolished in STATl deficient cells (Meraz et al., 1996). Similarly, IRF5 was shown to be required for the development of host-protective Th1 responses in response to Leishmania donovani. However, in IRF5-/- mice (Paun et al., 2011) and in myeloïd CD11c specific IRF5-/- mice, Leishmania failed to induce splenomegaly which seems to be detrimental to the host (Mai et al., 2019).
Besides IRFs, the TLR MyD88/TIRAP signaling pathways culminate in activation of the transcription factor nuclear factor-kappaB (NF-κB) via either classical pathway engaging the IKK-related kinases TBK1 (TRAF family member-associated NF-κB activator (TANK) binding kinase-1) downstream of TRAF6 or alternative pathways (Kawai and Akira, 2007).
NF-κB transcription factor family has five different members: NF-κB1(p50), NF-κB2 (p52) (formed from a larger precursor, respectively p105 et p102), p65 relA, c-rel and rel-B. They are all able to operate as a homo- or hetero-dimers and potentially yield to 15 different NF-κB complexes. These NF-κB dimers bind an inhibitory protein “inhibitor of κB” (IκB) and are trapped in the cytoplasm. The phosphorylation of IκB induces its ubiquitination and degradation and result in the release and the nuclear translocation of this master transcription factor that regulates hundred of genes. A large bulk of these genes are involved in both immediate and delayed expression of inflammatory mediators making NF-κB transcription factor a key regulator of proinflammatory pathway (Reinhard et al., 2012), NADPH oxidase expression and mitochondrial activity.
NF-κB pathway plays a crucial role in host protection against Leishmania infection and the deficiencies in different NF-κB family members in mice result in high susceptibility to Leishmania infection. Indeed, a defect in relB induces a multi-organ inflammation in mice, underlying the inhibitory effect of relB protein (Weih et al., 1995). Similarly, NF-κB2 deficient mice, with resistant background, develop chronic non healing lesions associated with uncontrolled parasite replication as a result of a reduced CD40-induced IL-12 production by macrophages (Speirs et al., 2002). RelA deficiency is fatal and induces the death of mice during the fetal life. However, RelA-/- chimeric mice present defects in macrophage function and show a reduced production of NO and an inability to control parasite replication and to clear infection (Mise-Omata et al., 2009). While relA is implicated in the regulation of iNOS expression, c-rel seems to regulate IL-12 production and c-Rel-deficient mice were unable to resolve Leishmania infection in the absence of IL-12 recombinant treatment (Grigoriadis et al., 1996; Sanjabi et al., 2000; Abu-Dayyeh et al., 2010).
In macrophages, Leishmania parasite seems to be unable to activate p65 NF-κB subunit. Indeed, Leishmania Mexicana promastigotes induce the cleavage of p65 relA subunit and generate a smaller p35 protein whereas the amastigote form fully degrades relA (Abu-Dayyeh et al., 2010). Leishmania amazoniensis induces the p50/p50 repressor complex which leads to a reduced activation of nitric oxide synthase (iNOS) (Calegari-Silva et al., 2015). From his side, Leishmania major down-regulates relA containing complexes but induces a c-rel containing complexes that retain the ability to strengthen the innate immune response of the host through the regulation of proinflammatory cytokines expression (Guizani-Tabbane et al., 2004).
NF-κB regulates the expression of myriads of cytokines and chemokines including IL-1β that play crucial role in cellular defense. This cytokine is produced as pro-IL-1β which is processed and released by the NLRP3 inflammasome activated caspase1. Different species of Leishmania have been reported to induce NLRP3 inflammasome, that have been shown to play a key role in restricting Leishmania replication in macrophages (Lefèvre et al., 2013; Lima-Junior et al., 2013) although detrimental effects have been reported for certain Leishmania strains such L. major Seidman strain or L. braziliensis for which disease severity is associated to IL-1β production (Charmoy et al., 2016; Santos et al., 2018). It is therefore consistent that several Leishmania species actively inhibit inflammasome formation and have developed different strategies to inhibit NF-κB-dependent inflammatory response. This was achieved in part by targeting the cytosolic inducible protein A20, also known as TNFα-induced protein 3 that de-ubiquitinates TRAF6 to inhibit IκB degradation and hence NF-κB activation reducing the extend of NLRP3 inflammasome activation (Boone et al., 2004; Gupta et al., 2017). Recent study highlights a key role for epigenetic modifications at the promoter of NF-κB-regulated genes that dampen inflammasome activation (Lecoeur et al., 2020). Indeed, Leishmania amazoniensis amastigotes effector molecules, target histone H3 post-translational acetylation, to reduce expression of positive regulators of the NF-κB pathway and upregulate transcripts of anti-inflammatory molecules and known inhibitors of NF-κB activation, leading to the inhibition of NF-κB-NLRP3 axis.
Transcription Factor HIF-1α and Leishmania-Induced Metabolic Reprograming
Since a decade, the importance of metabolic reprogramming in macrophages fighting microbes has emerged (Chauhan and Saha, 2018; Wang et al., 2019). Indeed, low oxygen levels have been described in the areas of myeloid cell activity and site of inflammation. Studies extending back almost a century have demonstrated that macrophages in these areas are highly dependent on anaerobic glycolysis for the production of ATP. This glycolysis surge and reduced glucose mitochondrial oxidation have been shown to depend on HIF-α transcription factor, which plays a key role in assisting macrophage to adapt to hypoxic conditions and achieve their microbicidal function (Marín-Hernández et al., 2009).
Under hypoxia, the inactivation of the oxygen-sensitive enzymes prolylhydroxylases (PHDs) allows HIFα (one of the HIF-α isoforms: HIF-1α or HIF-2α) to escape proteosomal degradation. HIF-α accumulates and translocates to the nucleus where it forms a heterodimer with the constitutively expressed HIF-1ß subunit. The HIFα/β heterodimer binds to the hypoxia response elements (HREs) and drive then the expression of different genes (Ke and Costa, 2006). HIF-1α is a direct transcriptional activator of glucose transporters genes involved in glucose uptake as well as keys glycolytic and gluconeogenesis enzymes. It also decreases mitochondrial respiration and thus oxygen consumption by upregulating the expression of pyruvate dehydrogenase kinase (PDK) (Bishop and Ratcliffe, 2014) that limit the formation of acetyl-CoA. Glycolysis even less efficient than oxidative phosphorylation for ATP generation can be promptly ramped up to sustain ATP production and meet the rapid energy requirement of macrophages (Nagao et al., 2019).
Similarly to other site of infection, Leishmania infected skin has been demonstrated to be hypoxic. This low oxygen level has been described for L. amazonensis and L. major infected tissue (Araujo et al., 2013) and resolution of the wound was reported to correlate with reoxygenation (Mahnke et al., 2014).
HIF-lα expression can also occur in an oxygen-independent manner in myeloid cells fighting pathogens, driving metabolic reprogramming to modulate oxygen consumption and modulating innate immunity (Jantsch and Schödel, 2015; Devraj et al., 2017; Stothers et al., 2018; Knight and Stanley, 2019). Leishmania parasite also induces HIF-1α activation (Charpentier et al., 2016; Schatz et al., 2018). Indeed, the HIF-lα has been detected in the cutaneous lesions of Balb/c mice (Arrais-Silva et al., 2005) and in the nucleus of L. amazonensis infected mouse peritoneal macrophages and human MDM (Degrossoli et al., 2007). HIF-1α also accumulates in L. donovani infected macrophagic cell lines (Singh et al., 2012) under normoxic conditions. In our hand, L. major induces the accumulation of HIF-la in BMDM cells as assessed by confocal microscopy (unpublished data). A more recent study has however shown that L. major infection induces HIF-lα accumulation in the macrophage-rich cutaneous Leishmania lesions of mice and humans, but needs, in vitro, additional stimulation such IFNγ (Schatz et al., 2016).
Moreover, different HIF1α target genes including glucose transporters assuring glucose uptake, glycolytic enzymes and pyruvate dehydrogenase kinase 1 (PDK1) leading to inactivation of the TCA cycle are induced in Leishmania infected macrophages (Rabhi et al., 2012; Moreira et al., 2015). These allow the metabolic reprogramming of infected macrophages toward aerobic glycolysis and hence increased glucose uptake, activation of the pentose phosphate pathway (PPP) that fuels NADPH to activate NADPH oxidase for ROS generation, enhanced lactate production and decreased mitochondrial oxygen consumption.
Activation of HIF1α has contrasting effects on the pathogen survival. Unlike for bacterial and for some viral infections (Devraj et al., 2017), HIF-lα activation during several parasitic infections could promote the intracellular survival of parasites. Indeed, HIF-la stabilization is required for the survival of Toxoplasma gondii (Wiley et al., 2010); Similarly, Theileria annulata induces hif1-α transcription and drive host cell toward aerobic glycolysis limiting the production of H202 and hence protecting the parasites in infected macrophages (Metheni et al., 2014).
The impact of HIF1α on Leishmania parasite survival has been addressed in several studies and has yielded contrasting results. Indeed, Singh et al., using macrophagic cell lines, show that knockdown of HIF-lα inhibited intracellular growth whereas over expression of this TF promotes intracellular growth of L. donovani parasites (Singh et al., 2012). However, different other studies show that HIF-1 alpha is a host protective factor against infection by Leishmania parasites. Indeed, silencing of HIF-lα in macrophages, using RNA interference, inhibited LPS/IFNy-induced NO release in response to L. major and abolished the leishmanicidal activity of macrophages showing that HIF-lα expression by myeloid cells contributes to the control of L. major parasites in vitro and in vivo (Schatz et al., 2016). Finally, the viability of L. donovani is enhanced in myeloïd-restricted HIF-1-/- mice (mHIF-1-/-) and in macrophages from mHIF-1-/-. This increased intracellular growth both in vitro and in vivo, was the result of enhanced lipid synthesis induced by a defect in the expression of BNIP3 that allows the activation of the mTOR/SREBPlc axis (Mesquita et al., 2020).
Thus, besides carbohydrates metabolism regulation, HIF1α, by regulating mTOR signaling pathway, controls lipid metabolism.
HIF-1 is also linked to inflammatory response and microbicidal activities of myeloid cells. Indeed, the relationship between the hypoxic and inflammatory responses is tightly controlled and cross talk between HIF1α and NF-κB, the two main molecular players involved in hypoxia and innate immunity, has been demonstrated (Palazon et al., 2014; D’Ignazio et al., 2016). Different studies have underlined the importance of HIF-1 in regulating inflammatory cytokines. Mice with HIF-1α myeloid-specific deletion showed impaired inflammatory responses, underlining the importance of HIF1α and the high glycolytic rates and energy generation for basic myeloid cell activities (Cramer et al., 2003). Both HIFs are able to regulate a number of pro-inflammatory cytokines and chemokines directly, further contributing to the inflammation response (D’Ignazio et al., 2016) and NF-κB is critical transcriptional activator of HIF1α (Rius et al., 2008; van Uden et al., 2008).
Hypoxic response is also tightly connected to innate immune response and HIF1 and NF-κB synergistically respond against pathogens. Indeed, hypoxia and bacterial infections increased NF-kB activity in phagocytes, leading to the increase in HIF-1a mRNA transcription (Rius et al., 2008). Moreover, bacteria-infected macrophages are characterized by a defective HIF-1α expression following ablation of Iκκb, an essential regulator of NF-κB activity (Rius et al., 2008).
Hence, Leishmania infection, by inducing NF-κB and HIF1 transcription factor, provides a bridge for regulating immune response and ignite inflammation by glucose fueling (Figure 1).
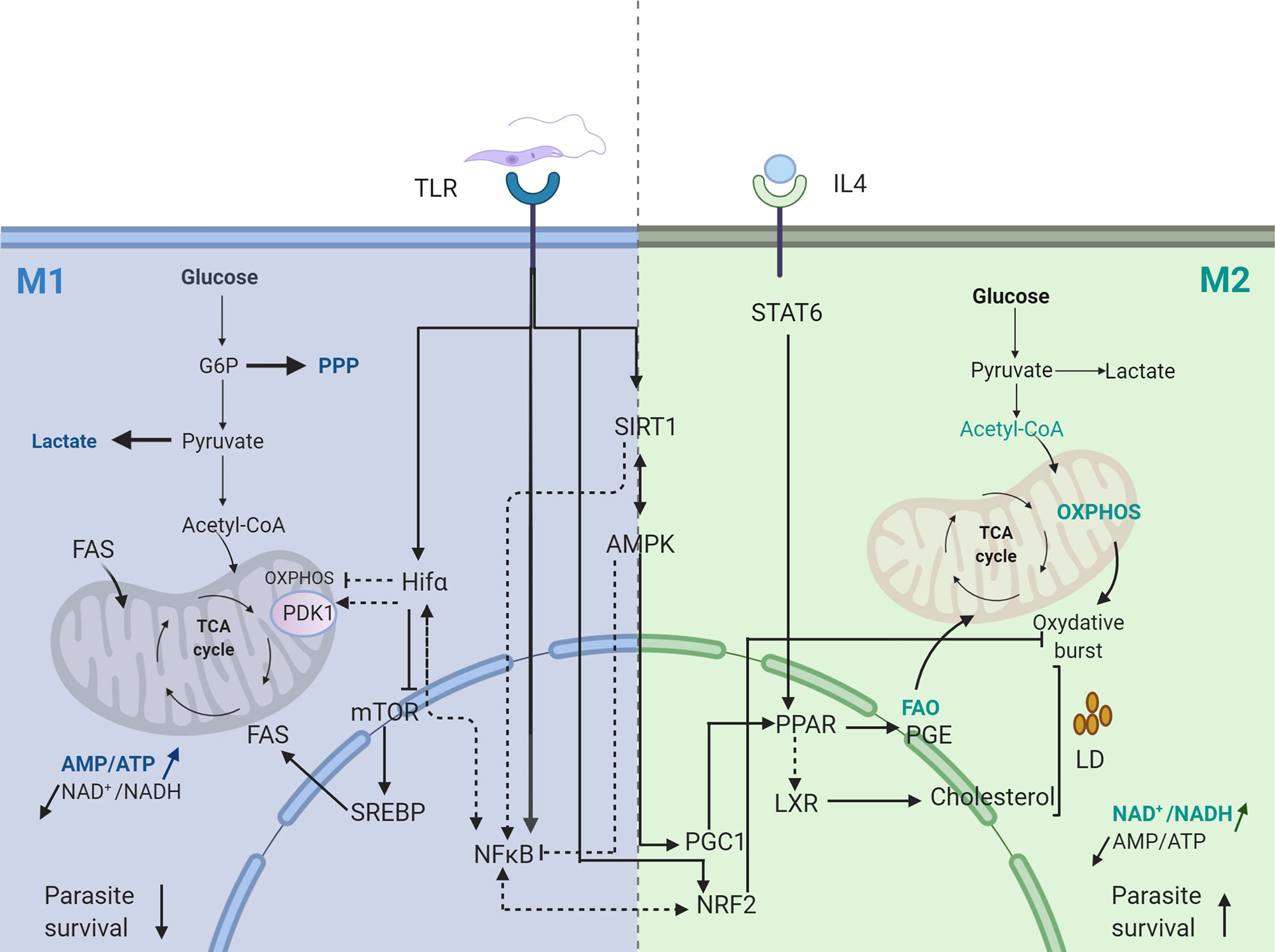
Figure 1 Transcriptional signature of Leishmania infected macrophage. Dotted arrows represent pathways not validated in Leishmania infected macrophages. Straight arrows represent pathways validated in Leishmania infected macrophages. FAS, Fatty Acid Synthesis; FAO, Fatty Acid Oxydation; PGE, Prostaglandine E; PPP, Pentose Phosphate Pathway; OXPHOS, Oxidative Phosphorylation. Created with BioRender.com.
Interplay Between Transcription Factors and Energy Sensors in Leishmania-Infected Macrophages
One target gene of NFκB is SIRT1 whose promoter sequence contains several putative NF-kB binding sites (Voelter-Mahlknecht and Mahlknecht, 2006; Mahlknecht and Voelter-Mahlknecht, 2009). Sirt1 belong to the Sirtuins family of nicotinamide nucleotide dinucleotide NAD(+)-dependent protein deacetylases that contain seven enzymatic activities in mammals (SIRT1–SIRT7) with different subcellular localisation that function to suppress gene transcription by epigenetic mechanisms. A second energy sensor, the adenosine 5’-monophosphate (AMP)-activated protein kinase (AMPK) is a key player in preserving cellular ATP.
During the early pro-inflammatory phase relying on glycolysis, decreases in ATP production and later on in NADH formation, increase the ratios of AMP/ATP, and NAD+/NADH. The increased AMP/ATP ratio induces AMPK while the enhanced cellular NAD+ activates SIRT proteins. These two pathways, can act in concert to reinforce one another in order to ensure the metabolic homeostasis of the cell. Indeed, On one hand SIRT1 protein has been described as an upstream activator of the AMPK through LKB1 (the kinase that mediates the phosphorylation of AMPK) deacetylation (Lan et al., 2008) and on the other hand has been considered as a downstream target of AMPK, becoming activated by the AMPK-induced increased levels of NAD+ (Cantó et al., 2010). Moreover, SIRT1 and AMPK proteins share several downstream targets such as PGC-1α (Rodgers et al., 2005) and PPARα (Purushotham et al., 2009).
SirT1 directly interacts with and deacetylates NFκB p65 (Yeung et al., 2004) inhibiting its transactivation capacity and suppressing the transcription of NF-κB dependent genes expression (Natoli, 2009). This antagonistic cross talk between NF-κB signaling pathway and SIRT1 pathways, bridge energy metabolism with innate immunity and have crucial role in the termination of NF-κB driven inflammatory response (Kauppinen et al., 2013; Vachharajani et al., 2016).
SIRT1 also deacetylates another key player in the regulation of energy metabolism/promoting mitochondrial biogenesis, the peroxisome proliferator activated receptor γ (PPARγ) coactivator 1 (PGC1) α, upregulating its activity which is further enhanced by AMK-mediated phosphorylation (Cantó et al., 2010). PGC-1α promotes the metabolic switch toward M2 profile. Indeed, the overexpression of PGC-1 reduces the transcriptional activity of NF-κB and suppressed the expression of proinflammatory cytokines and free fatty acids (Eisele et al., 2013). PGC-1α cooperates with the peroxisome proliferator-activated receptor alpha (PPARα) to control the transcription of genes encoding mitochondrial fatty acid oxidation enzymes shifting the fuel usage from glucose to fatty acids (Vega et al., 2000). Moreover, PGC1α is also a coactivator of a wide variety of transcription factors including Forkhead box-containing protein type O 1 (FOXO1), nuclear respiratory factors (NRF)1 and NRF2, many of which are involved in the transcriptional control of mitochondrial proteins and thus in mitochondrial biogenesis (Feige and Auwerx, 2007).
Hence, these energy sensors have emerged as key regulators of inflammation and metabolic switches, playing a crucial role in macrophage metabolic reprogramming toward M2 macrophages profile during the progression of inflammatory response.
These energy sensors have been shown to play an important role in the polarization of parasite infected macrophages. Indeed, the intracellular growth of Trypanosoma cruzi was recently shown to rely predominantly on energy production, nucleotide metabolism and fatty acid oxidation of the host and silencing of AMPK catalytic or regulatory subunits, favors intracellular T. cruzi growth (Caradonna et al., 2013). Similarly, activation of the host AMPK signaling inhibits the intracellular growth of Plasmodium falciparum parasite (Ruivo et al., 2016). The initial transient aerobic glycolytic phase induced by Leishmania parasites has been shown to be followed by a metabolic shift towards mitochondrial metabolism, allowing the metabolic recovery of the host cell (Moreira et al., 2015). This metabolic switch to oxidative phosphorylation and β-oxidation, is associated with an increase in PGC-1α levels and a significant increase in the AMP/ATP ratio. This metabolic reprogramming requires the activation of AMPK downstream to SIRT1 and LKB1 and ultimately contributes to parasite survival in vitro and in vivo (Moreira et al., 2015). Hence targeting the SIRT-AMPK axis may explain how the Leishmania parasite subverts the host metabolism to insure its persistence and replication (Figure 1).
Microenvironment Impact on Activity of PGC1 Co-Activator
Microenvironment play a role, in vivo, in the metabolic reprograming of Leishmania infected macrophages and their shift from M1to M2. Indeed, the significantly raised plasma levels of IL-4/IL-13 and IL-10 in VL patients suggested a microenvironment conducive for alternative activation of monocytes/macrophages (Roy et al., 2015). Indeed, both IL-4 and IL-13 cytokines induced the expression of the two metabolic regulators, Pparγ, Pparδ and of the coactivator protein PGC-1β, through the activation of STAT6.
The Peroxisome proliferator-activated receptors (PPARs) but also the Liver X Receptors (LXRs) nuclear receptors are two transcription factors families implicated in the regulation of macrophage lipids metabolism. Both ligand-dependent transcription factors heterodimerize with the retinoid X receptor (RXR).
The peroxisome proliferator-activated receptor-γ (PPARγ) is a master regulator that assures fatty acid homeostasis. It binds mono- and polyunsaturated fatty acids and derivatives such as eicosanoids to regulate the transcription of various genes that control lipid metabolism and fine-tune immune function. Indeed, a peroxisome proliferator response element (PRE) have been identified in all the genes that encode the rate limiting steps in the transport, synthesis, storage, mobilization, activation and oxidation of fatty acids but also in the promoter region of arginase I (Odegaard et al., 2007) and in the promoters region of phospholipase A2 (cPLA2) and cyclooxygenase-2 (COX-2), two enzymes regulating the prostaglandin production that have been often associated with anti-inflammatory activities. Phospholipase A2 (cPLA2) and cyclooxygenase-2 (COX-2) have been also reported to be activated upon TLR2 stimulation; whereas the increased macrophage cPLA2 activity induced by Leishmania infection is regulated at the post transcriptional level, Leishmania-driven COX2 induction was regulated by calcineurin and calcineurin-dependent NFAT transcription factor (Bhattacharjee et al., 2016). PPARγ inhibits the induction of a broad subset of inflammatory genes, suggesting that transrepression might be a primary mechanism responsible for the anti-inflammatory action of PPARγ.
PPARγ deficient macrophages show reduced rates of β-oxidation of fatty acids and blunted mitochondrial biogenic response and are hence unable to induce oxidative phosphorylation, showing that PPARy plays a crucial role in the differentiation of circulating monocytes toward anti-inflammatory M2 macrophages, both in humans and mice (Bouhlel et al., 2007; Odegaard et al., 2007). Similarly, the overexpression of the PPARγ transcriptional coactivators PGC-1β primes macrophages for alternative activation and prevents an M1 inflammatory response following stimulation with LPS and IFNγ (Vats et al., 2006). Indeed, the two PPAR subtypes expressed in macrophages have been implicated in the negative regulation of inflammatory responses as they inhibit pro-inflammatory gene expression including IL1β, IL6, TNFα, IL12 and iNOS (Delerive et al., 2001; Kostadinova et al., 2005).
PPARγ also activates a cholesterol efflux by inducing the Liver X receptor-alpha (LXR) expression either directly or through the induction of CYP27, which is capable of generating 27- hydroxycholesterol, an endogenous LXR activator.
Endogenous agonists binding to LXR induces conformational changes that trigger co-repressor release and co-activators recruitment, leading to transcriptional regulation of the target genes. These include those encoding the sterol regulatory element binding protein 1 (SREBPlc), the other master transcriptional regulator of fatty acid synthesis (Repa et al., 2000) but also the ATP binding cassette transporters ABCAl and ABCG1 as well as the gene encoding apolipoprotein E (APOE) (Repa et al., 2002), the cholesterol transporters that promote cholesterol efflux. LXR activation also leads to an increase in the levels of long chain polyunsaturated fatty acids (PUFAs) and triglyceride synthesis.
The Liver X Receptors, (LXRs) also play a crucial role in the macrophage anti inflammatory response promoting alternative (M2) macrophage activation. Two LXR subtypes, LXRα and LXRβ are expressed in hematopoietic cells with LXRα being restricted to the myeloid lineage. The deficient regulation of both isoforms of LXRs activity can lead to chronic inflammatory conditions. Indeed, several LXR regulated genes are targets of NF-κB and LXR agonist treatment can limit the transcriptional up-regulation of inflammatory genes such as tumor necrosis factor alpha (TNFα), cyclooxygenase 2 (COX2), inducible nitric oxide synthase (NOS2) and matrix metalloprotease 9 (MMP9) (Castrillo et al., 2003; Joseph et al., 2003).
Several mechanisms, have been proposed to explain LXR capacity to inhibit inflammation including the trans-repression of NF-κB (Ghisletti et al., 2009) and regulation of genes encoding enzymes involved in the synthesis of long chain PUFAS with known anti-inflammatory activity (Repa et al., 2000). Moreover, together with fatty acids, LXR regulates expression of LPCAT3 an enzyme that controls Fatty acyl-chain remodeling in phospholipid (Hashidate-Yoshida et al., 2015). These changes in phospholipid composition can reduce inflammation both by modulating kinase activation through changes in membrane composition and by affecting substrate availability for inflammatory mediator production (Rong et al., 2013).
Nuclear receptors (NRs) are emerging as key players in infectious diseases and targeting NRs as a novel host-directed treatment approach to infectious diseases appears to be a valid solution (Leopold Wager et al., 2019).
LXR deficiency in mice leads to reduced control of the intracellular Mycobacterium tuberculosis growth (Korf et al., 2009) and increased susceptibility to Listeria monocytogenes (Joseph et al., 2004). By contrast, mice deficient for LXR and challenged with the visceralizing species Leishmania chagasi/infantum had significantly decreased parasite burdens compared to C57Bl6 wild-type mice and enhanced NO production showing enhanced resistance of LXR-deficient mice to Leishmania infection (Ghisletti et al., 2009).
Regardless of PPARs subtypes induced by Trypanosoma cruzi (i.e., PPARy in murine peritoneal macrophage and PPARα in BMdM and Raw 264.7 macrophagic cell line), PPAR is involved in the in vitro M2 polarization of macrophages isolated from T. cruzi-infected mice and have a protective role in T. cruzi infection (Penas et al., 2015; Koo et al., 2018). In Leishmania infection the rôle of PPAR is controversial. PPAR agonists treatment of L. mexicana infected macrophages induced M1 polarization through IL-10 downregulation, cPLA2-COX-2 prostaglandins pathway deactivation and increased ROS production that control parasite burden in murine macrophages (Díaz-Gandarilla et al., 2013). By contrast, PPARy is activated by L. donovani both in vivo and in vitro and have been shown to promote the parasite survival, whereas the inhibition of PPAR-y facilitates Leishmania clearance in C57B16 peritoneal macrophages (Chan et al., 2012), suggesting that Leishmania parasites harness PPARγ to sustain the M2 phenotype and increase dissemination of the infection. PPARγ knockout Balb/c mice had significantly less footpad swelling 5-7 weeks after injection of L. major promastigotes than wild type mice, suggesting that absence of PPARγ delay disease progression in Balb/c mice (Odegaard et al., 2007). In lipid loaded Leishmania-infected BMDM, PPAR ligands promote L. major amastigote growth in an arginase-dependent manner (Gallardo-Soler et al., 2008).
Like other trypanosomatid parasites, Leishmania depend on their hosts for several nutrients necessary for their replication and survival. While carbohydrates have been proposed as the primary source of carbon for various parasites, including Leishmania (Naderer et al., 2010) various other sources have been suggested. Lipids would be a potential source especially since the infection induces the production of intracellular lipid droplets which rapidly localized in close association with the parasites (Rabhi et al., 2012; Rabhi et al., 2016).
Hence, like different other pathogens Leishmania parasite target metabolic pathway to scavenge nutrients to ensure its survival and to subvert the host immune response feeding two birds with one scone.
NRF2 Antioxidant Pathway
The metabolic reprogramming toward aerobic glycolysis attenuates the activities of the respiratory chain, allowing reactive-oxygen species (ROS) production. The respiratory burst or ROS, generated by the NADPH oxydase and the mitochondrial electron transport chain after the phagocytosis of pathogens, has been recognized since decades as an essential mechanism of microbial killing. To avoid detrimental side effects and oxidative damage, host cells produce antioxidants enzymes in order to deplete ROS and RNS. The transcription of phase II defense gene including detoxifying enzymes, such as genes involved in the regeneration of glutathione (GSH), genes encoding proteins involved in sequestration of iron ions (ferritine, heme oxygenase (HO-1), genes encoding proteins responsible for the production of NADPH and anti-oxidant genes (e.g., SODs, GPx, GSH reductase), is orchestrated by the master transcription (nuclear factor (erythroid-derived 2)-like 2 (NRF2) (Suzuki and Yamamoto, 2015).
Upon cell activation, NRF2 oxidative sensor is released from its inhibitor Kelch-like ECH-associated protein-1 (Keap-1), phosphorylated by different kinases including the MAPK, phosphatidylinositol-3 Kinase (Kang et al., 2002) and double-stranded RNA-dependent protein kinase R (PKR) (Vivarini et al., 2017) and translocated to the nucleus where it binds, once associated with small Maf proteins, to the antioxidant response elements (AREs) present in the promoter region of target genes (Jung and Kwak, 2010; Ding et al., 2019; Ingram et al., 2019). This master transcription factor orchestrates the regulation of about 200 genes, working in a network to regulate various functions.
NRF2 is a key player regulating inflammation; Indeed, Nrf2 knock-out mice shows increased expression of cytokines, chemokines, and adhesion molecules and sustains the development of different inflammatory disorders (Thimmulappa et al., 2006; Kim et al., 2010). Cross talk between NRF2 and NF-κB is essential to resolve inflammatory response of the cell. Indeed, NFκB can directly regulate the induction of Nrf2 transcription, but can also by interacting with Keap1 suppress the Nrf2-ARE pathway, resulting in the repression of various genes (Wardyn et al., 2015; Bellezza et al., 2018). This inhibitory effect on macrophage inflammatory response may be also indirect, induced by NRF2-activated anti-oxidant genes. Among the antioxidative stress genes whose expression is regulated by NRF2, the Heme oxygenase 1 (HO-1) has received considerable attention. HO-1 is the rate limiting enzyme that catalysis the degradation of heme into carbon monoxide (CO) and iron and biliverdin to bilirubin. NRF2-mediated HO-1 expression, inhibited NF-κB signaling preventing the expression of inflammasome components such IL-1β and nlrp3 induced by LPS (Vitali et al., 2020). Similarly to NRF2 knockout mouse, HO-1 knockout mouse displayed chronic inflammatory disorders, are highly vulnerable to experimental sepsis induced by the classical pro-inflammatory mediator endotoxin (Wiesel et al., 2000). Human cases of genetic HO-1 deficiency are similar to those observed in HO-1 knockout mice (Kawashima et al., 2002) confirming the anti-inflammatory role of HO-1 (Paine et al., 2010). Moreover, Nrf2 expression but also HO-1 induction have been identified as a key mediator of polarization to a novel macrophage phenotype (Mox), that develops in response to oxidative tissue damage (Kadl et al., 2010). This M2 phenotype has redox and antioxidant potential and induces the expression of the anti-inflammatory and antiapoptotic Cox2, IL1β, HO-1, VEGF, and Nrf2.
Thus, the complex network of protective mechanisms against the oxidative burst orchestrated by NRF2 and the NRF2-HO-1 axis implicated in the resolution of inflammation, modulates the function and phenotype of macrophages driving macrophages polarization towards M2 phenotype (Naito et al., 2014).
The effect of NRF2 on mitochondria biogenesis may also play a role during infection. Indeed, four AREs were identified in the NRF1 (nuclear respiratory factors 1) gene promoter and were capable of Nrf2 binding (Piantadosi et al., 2008). NRF1 through TFAM (transcription factor A mitochondrial), is involved in the mitochondrial DNA replication. Moreover, crosstalk between PGC1a and NRF2 has been also recently reported highlighting a link between cellular redox and mitochondrial homeostasis.
The NRF2 pathway orchestrates the expression of anti-oxidant and cytoprotective genes. Its activity is induced upon exposure to oxidative stress to protect cell from environmental insults and to adapt to endogenous stressors.
However, it is not clear to what extend oxidative stress is crucial for pathogens clearance as several microorganisms seem to be able to survive and even thrive in an oxidative environment (Paiva and Bozza, 2014). Indeed, different bacteria and parasites can thrive in oxidative environments while antioxidants promote their clearance. Trypanosoma cruzi infected mice treated with several Nrf2 inducers, including cobalt protoporphyrin (CoPP), sulforaphane, N-acetyl-cysteine (NAC), or pterostilbene, had a decreased parasite burden. The inhibition of Nrf2 activity with SnPP or the promotion of oxidative stress with paraquat or H202 treatment increased T. cruzi parasitism, suggesting that oxidative stress fuels Trypanosoma cruzi infection in mice (Paiva et al., 2012). During the malaria-induced inflammatory processes, the treatment of macrophages with NRF2 inducers enhances the CD36 expression and CD36-mediated Plasmodium phagocytosis and thus the control of severe malaria through parasite clearance (Olagnier et al., 2011). Nrf2 participates in infections by other protozoan microorganisms and different study show that various parasites such Toxoplasma gondii, activates Nrf2 signaling pathway that is required for parasites replication (Pang et al., 2019).
The potential role of NRF2 signaling in Leishmania infection outcomes has been recently reviewed (Vivarini and Lopes, 2019). NRF2 antioxidant pathway is activated in response to different Leishmania species even if differences are observed between species. Indeed, Leishmania amazoniensis but also Leishmania brasiliensis, induce in vitro, NRF2 expression, via protein kinase R signaling (Barreto-de-Souza et al., 2015; Vivarini et al., 2017), a result confirmed in skin biopsies from Leishmania infected patients. Treatment of L. major infected BALB/c mice with NAC, a Nrf2 inducer, reduces the parasitism in their footpads (Rocha-Vieira et al., 2003), whereas depletion of glutathione, increases the burden in the footpads of C57BL/6 mice infected with L. major (Cruz et al., 2008). However, for L. amazoniensis, the Nrf2 knockdown that promotes oxidative stress, impairs parasite survival in macrophages (Vivarini et al., 2017).
Whereas NRF2-induced anti-oxidant genes, HO-1, is required for protection against Toxoplasma gondii (Araujo et al., 2013), antioxidative enzymes such SOD1 and HO-1, favor the establishment of Leishmania infection. Indeed, L. pifanoi amastigotes and L. chagasi infection induce in macrophages an increased expression of HO-1 that promotes Leishmania persistence (Pham et al., 2005; Luz et al., 2012). Moreover, in mouse peritoneal and human macrophages lineages, infection by L. amazonensis leads to the increase of SOD1 expression in a PKR/Nrf2-dependent manner (Vivarini et al., 2017). SOD1 reduces the Leishmania oxidative stress and may promote the parasite survival and affect the outcome of the infection (Khouri et al., 2009; Khouri et al., 2010).
Conclusion and Perspectives
In recent years much progress has been made in immunometabolism and there is a growing understanding of how metabolic pathways are regulated to support or direct functional changes in immune cells, controlling immunity and inflammation. Metabolic signature of immune cells including macrophages is now known to change depending on stages of development, activation state and pathological conditions. A better understanding of the metabolic mechanisms and metabolic needs of immune cells, as well as the interactions that underlie the complex metabolic and immune networks, is essential to develop promising drugs with effective metabolic interventions that enhance immune cell response.
In the context of anticancer immunotherapy, drugs targeting cancer metabolism might synergistically enhance immunotherapy via metabolic reprogramming of the tumor microenvironment. Different combinations of metabolic agents and immunotherapies are already in clinical trials (Li et al., 2019). Deciphering the intricate interaction existent between metabolic reprogramming and immune response has also promising therapeutic application for inflammatory and autoimmune diseases. Different small molecules targeting metabolic processes in immune cells as a strategy to limit inflammation and change the outcome of inflammatory and autoimmune diseases are currently in use clinically (Pålsson-McDermott and O’Neill, 2020). Hence, the majority of available or under development anti-rheumatic drugs, target various metabolites in immune cells belonging to purine, pyrimidine or GSH metabolism (Piranavan et al., 2020). The host directed therapies includes different promising treatment strategies targeting macrophages’ polarization to the M2 phenotype that have been found associated with atherosclerosis regression and different compound are being tested to develop more efficient antiatherosclerosis therapy (Bi et al., 2019). Infectious diseases are more complex pathologies given the additional interactions that take place between the host cell and the pathogen. Host directed therapy (HDT) targeting metabolic pathway could represent new therapeutic strategies to fight infectious diseases in the face of emergence of antimicrobial resistance or compromised host immunity. Indeed, recent studies suggest that foamy macrophages, the major contributors to Tuberculosis pathogenesis, and lipid metabolism are promising targets for HDT against Tuberculosis (Shim et al., 2020). Macrophage polarization is also an important tool in the physiopathology of leishmaniasis (Tomiotto-Pellissier et al., 2018) and targeting metabolic pathway, once a better understanding of the balance required to develop a protective response is gained, might potentially change the outcome of the infection.
Targeting transcription factors could be another promising therapeutic strategy. Indeed, deregulation of transcription factors is a common feature to the majority of human cancers and is involved in a large number of human diseases (Papavassiliou and Papavassiliou, 2016). A better understanding of the pathologies, of the mechanisms of transcriptional regulation and their precise function as well as the development of new technologies (Hagenbuchner and Ausserlechner, 2016; Hecker and Wagner, 2017; Becskei, 2020), has allowed the development of novel therapeutic strategies that target transcription factors. Different small molecules that interact in various way with transcription factors to modulate their expression, degradation, interaction with DNA or other proteins, are actually used or evaluated for cancer treatment (Lambert et al., 2018). Agents targeting transcription factors for the treatment of chronic kidney disease are under clinical trial (Hishikawa et al., 2018). Transcription factors induced in response to Leishmania infection play crucial role in regulating immune and metabolic responses and hence macrophage polarization. Almost all the transcription factors involved in the macrophage immune and metabolic response to Leishmania infection have been described as therapeutic target in different diseases including infectious ones. These includes HIF that couple immunity with metabolism (Bhandari and Nizet, 2014; Santos and Andrade, 2017), nuclear receptors in metabolic, inflammatory and infectious diseases (Rudraiah et al., 2016; Wang et al., 2017; Mirza et al., 2019) and NRF2 in cancer (Lu et al., 2016). Despite the challenges that remain to be overcome (delivery methods, compensatory and pleiotropic effects), targeting transcription factors could represent new therapeutic strategies to reprogram the macrophage response in order to amplify the microbicidal activity that should allow the fight against infectious diseases while preventing an exacerbated immune response leading to a worsening of the lesions.
Author Contributions
All authors contributed to the article and approved the submitted version.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
Continuous support by the Tunisian Ministry of higher Education is gratefully acknowledged. Our warmest thanks to Dr. R. Herwig and Dr. M. Essefi for their critical reading of the manuscript.
References
Abu-Dayyeh I., Hassani K., Westra E. R., Mottram J. C., Olivier M. (2010). Comparative study of the ability of Leishmania mexicana promastigotes and amastigotes to alter macrophage signaling and functions. Infect. Immun. 78, 2438–2445. doi: 10.1128/IAI.00812-09
Araujo E. C. B., Barbosa B. F., Coutinho L. B., Barenco P. V. C., Sousa L. A., Milanezi C. M., et al. (2013). Heme oxygenase-1 activity is involved in the control of Toxoplasma gondii infection in the lung of BALB/c and C57BL/6 and in the small intestine of C57BL/6 mice. Vet. Res. 44, 89. doi: 10.1186/1297-9716-44-89
Arrais-Silva W. W., Paffaro V. A., Yamada A. T., Giorgio S. (2005). Expression of hypoxia-inducible factor-1alpha in the cutaneous lesions of BALB/c mice infected with Leishmania amazonensis. Exp. Mol. Pathol. 78, 49–54. doi: 10.1016/j.yexmp.2004.09.002
Barreto-de-Souza V., Ferreira P. L. C., Vivarini A. C., Calegari-Silva T., Soares D. C., Regis E. G., et al. (2015). IL-27 enhances Leishmania amazonensis infection via ds-RNA dependent kinase (PKR) and IL-10 signaling. Immunobiology 220, 437–444. doi: 10.1016/j.imbio.2014.11.006
Becskei A. (2020). Tuning up Transcription Factors for Therapy. Molecules Basel Switz 25, 1902. doi: 10.3390/molecules25081902
Bellezza I., Giambanco I., Minelli A., Donato R. (2018). Nrf2-Keap1 signaling in oxidative and reductive stress. Biochim. Biophys. Acta Mol. Cell Res. 1865, 721–733. doi: 10.1016/j.bbamcr.2018.02.010
Bertholet S., Dickensheets H. L., Sheikh F., Gam A. A., Donnelly R. P., Kenney R. T. (2003). Leishmania donovani-induced expression of suppressor of cytokine signaling 3 in human macrophages: a novel mechanism for intracellular parasite suppression of activation. Infect. Immun. 71, 2095–2101. doi: 10.1128/iai.71.4.2095-2101.2003
Bhandari T., Nizet V. (2014). Hypoxia-Inducible Factor (HIF) as a Pharmacological Target for Prevention and Treatment of Infectious Diseases. Infect. Dis. Ther. 3, 159–174. doi: 10.1007/s40121-014-0030-1
Bhattacharjee A., Majumder S., Das S., Ghosh S., Biswas S., Majumdar S. (2016). Leishmania donovani-Induced Prostaglandin E2 Generation Is Critically Dependent on Host Toll-Like Receptor 2-Cytosolic Phospholipase A2 Signaling. Infect. Immun. 84, 2963–2973. doi: 10.1128/IAI.00528-16
Bi Y., Chen J., Hu F., Liu J., Li M., Zhao L. (2019). M2 Macrophages as a Potential Target for Antiatherosclerosis Treatment. Neural Plast. 2019:6724903. doi: 10.1155/2019/6724903
Bishop T., Ratcliffe P. J. (2014). Signaling hypoxia by hypoxia-inducible factor protein hydroxylases: a historical overview and future perspectives. Hypoxia Auckl NZ 2, 197–213. doi: 10.2147/HP.S47598
Blanchette J., Racette N., Faure R., Siminovitch K. A., Olivier M. (1999). Leishmania-induced increases in activation of macrophage SHP-1 tyrosine phosphatase are associated with impaired IFN-gamma-triggered JAK2 activation. Eur. J. Immunol. 29, 3737–3744. doi: 10.1002/(SICI)1521-4141(199911)29:11<3737::AID-IMMU3737>3.0.CO;2-S
Boone D. L., Turer E. E., Lee E. G., Ahmad R.-C., Wheeler M. T., Tsui C., et al. (2004). The ubiquitin-modifying enzyme A20 is required for termination of Toll-like receptor responses. Nat. Immunol. 5, 1052–1060. doi: 10.1038/ni1110
Bouhlel M. A., Derudas B., Rigamonti E., Dièvart R., Brozek J., Haulon S., et al. (2007). PPARgamma activation primes human monocytes into alternative M2 macrophages with anti-inflammatory properties. Cell Metab. 6, 137–143. doi: 10.1016/j.cmet.2007.06.010
Buates S., Matlashewski G. (2001). General Suppression of Macrophage Gene Expression During Leishmania donovani Infection. J. Immunol. 166, 3416–3422. doi: 10.4049/jimmunol.166.5.3416
Bullen D. V. R., Hansen D. S., Siomos M.-A. V., Schofield L., Alexander W. S., Handman E. (2003a). The lack of suppressor of cytokine signalling-1 (SOCS1) protects mice from the development of cerebral malaria caused by Plasmodium berghei ANKA. Parasite Immunol. 25, 113–118. doi: 10.1046/j.1365-3024.2003.00616.x
Bullen D. V. R., Baldwin T. M., Curtis J. M., Alexander W. S., Handman E. (2003b). Persistence of lesions in suppressor of cytokine signaling-1-deficient mice infected with Leishmania major. J. Immunol. Baltim Md 1950 170, 4267–4272. doi: 10.4049/jimmunol.170.8.4267
Calegari-Silva T. C., Vivarini Á. C, Miqueline M., Dos Santos G. R. R. M., Teixeira K. L., Saliba A. M., et al. (2015). The human parasite Leishmania amazonensis downregulates iNOS expression via NF-κB p50/p50 homodimer: role of the PI3K/Akt pathway. Open Biol. 5, 150118. doi: 10.1098/rsob.150118
Cantó C., Jiang L. Q., Deshmukh A. S., Mataki C., Coste A., Lagouge M., et al. (2010). Interdependence of AMPK and SIRT1 for metabolic adaptation to fasting and exercise in skeletal muscle. Cell Metab. 11, 213–219. doi: 10.1016/j.cmet.2010.02.006
Caradonna K. L., Engel J. C., Jacobi D., Lee C.-H., Burleigh B. A. (2013). Host metabolism regulates intracellular growth of Trypanosoma cruzi. Cell Host Microbe 13, 108–117. doi: 10.1016/j.chom.2012.11.011
Castrillo A., Joseph S. B., Marathe C., Mangelsdorf D. J., Tontonoz P. (2003). Liver X receptor-dependent repression of matrix metalloproteinase-9 expression in macrophages. J. Biol. Chem. 278, 10443–10449. doi: 10.1074/jbc.M213071200
Chan M. M., Adapala N., Chen C. (2012). Peroxisome Proliferator-Activated Receptor-γ-Mediated Polarization of Macrophages in Leishmania Infection. PPAR Res. 2012, 796235. doi: 10.1155/2012/796235
Chandrakar P., Parmar N., Descoteaux A., Kar S. (2020). Differential Induction of SOCS Isoforms by Leishmania donovani Impairs Macrophage-T Cell Cross-Talk and Host Defense. J. Immunol. Baltim Md 1950 204, 596–610. doi: 10.4049/jimmunol.1900412
Charmoy M., Hurrell B. P., Romano A., Lee S. H., Ribeiro-Gomes F., Riteau N., et al. (2016). The Nlrp3 inflammasome, IL-1β, and neutrophil recruitment are required for susceptibility to a nonhealing strain of Leishmania major in C57BL/6 mice. Eur. J. Immunol. 46, 897–911. doi: 10.1002/eji.201546015
Charpentier T., Hammami A., Stäger S. (2016). Hypoxia inducible factor 1α: A critical factor for the immune response to pathogens and Leishmania. Cell Immunol. 309, 42–49. doi: 10.1016/j.cellimm.2016.06.002
Chauhan P., Saha B. (2018). Metabolic regulation of infection and inflammation. Cytokine 112, 1–11. doi: 10.1016/j.cyto.2018.11.016
Chaussabel D., Semnani R. T., McDowell M. A., Sacks D., Sher A., Nutman T. B. (2003). Unique gene expression profiles of human macrophages and dendritic cells to phylogenetically distinct parasites. Blood 102, 672–681. doi: 10.1182/blood-2002-10-3232
Colonna M. (2007). TLR pathways and IFN-regulatory factors: to each its own. Eur. J. Immunol. 37, 306–309. doi: 10.1002/eji.200637009
Cramer T., Yamanishi Y., Clausen B. E., Förster I., Pawlinski R., Mackman N., et al. (2003). HIF-1alpha is essential for myeloid cell-mediated inflammation. Cell 112, 645–657. doi: 10.1016/s0092-8674(03)00154-5
Cruz K. K., Fonseca S. G., Monteiro M. C., Silva O. S., Andrade V. M., Cunha F. Q., et al. (2008). The influence of glutathione modulators on the course of Leishmania major infection in susceptible and resistant mice. Parasite Immunol. 30, 171–174. doi: 10.1111/j.1365-3024.2007.01014.x
Dalpke A., Heeg K., Bartz H., Baetz A. (2008). Regulation of innate immunity by suppressor of cytokine signaling (SOCS) proteins. Immunobiology 213, 225–235. doi: 10.1016/j.imbio.2007.10.008
de Veer M. J., Curtis J. M., Baldwin T. M., DiDonato J. A., Sexton A., McConville M. J., et al. (2003). MyD88 is essential for clearance of Leishmania major: possible role for lipophosphoglycan and Toll-like receptor 2 signaling. Eur. J. Immunol. 33, 2822–2831. doi: 10.1002/eji.200324128
Debus A., Gläsner J., Röllinghoff M., Gessner A. (2003). High levels of susceptibility and T helper 2 response in MyD88-deficient mice infected with Leishmania major are interleukin-4 dependent. Infect. Immun. 71, 7215–7218. doi: 10.1128/iai.71.12.7215-7218.2003
Degrossoli A., Bosetto M. C., Lima C. B. C., Giorgio S. (2007). Expression of hypoxia-inducible factor 1alpha in mononuclear phagocytes infected with Leishmania amazonensis. Immunol. Lett. 114, 119–125. doi: 10.1016/j.imlet.2007.09.009
Delerive P., Fruchart J. C., Staels B. (2001). Peroxisome proliferator-activated receptors in inflammation control. J. Endocrinol. 169, 453–459. doi: 10.1677/joe.0.1690453
Devraj G., Beerlage C., Brüne B., Kempf V. A. J. (2017). Hypoxia and HIF-1 activation in bacterial infections. Microbes Infect. 19, 144–156. doi: 10.1016/j.micinf.2016.11.003
Díaz-Gandarilla J. A., Osorio-Trujillo C., Hernández-Ramírez V. I., Talamás-Rohana P. (2013). PPAR activation induces M1 macrophage polarization via cPLA₂-COX-2 inhibition, activating ROS production against Leishmania mexicana. BioMed. Res. Int. 2013:215283. doi: 10.1155/2013/215283
Ding L., Yuan X., Yan J., Huang Y., Xu M., Yang Z., et al. (2019). Nrf2 exerts mixed inflammation and glucose metabolism regulatory effects on murine RAW264.7 macrophages. Int. Immunopharmacol. 71, 198–204. doi: 10.1016/j.intimp.2019.03.023
D’Ignazio L., Bandarra D., Rocha S. (2016). NF-κB and HIF crosstalk in immune responses. FEBS J. 283, 413–424. doi: 10.1111/febs.13578
Eisele P. S., Salatino S., Sobek J., Hottiger M. O., Handschin C. (2013). The peroxisome proliferator-activated receptor γ coactivator 1α/β (PGC-1) coactivators repress the transcriptional activity of NF-κB in skeletal muscle cells. J. Biol. Chem. 288, 2246–2260. doi: 10.1074/jbc.M112.375253
Feige J. N., Auwerx J. (2007). Transcriptional coregulators in the control of energy homeostasis. Trends Cell Biol. 17, 292–301. doi: 10.1016/j.tcb.2007.04.001
Ferrante C. J., Leibovich S. J. (2012). Regulation of Macrophage Polarization and Wound Healing. Adv. Wound Care 1, 10–16. doi: 10.1089/wound.2011.0307
Forget G., Gregory D. J., Olivier M. (2005). Proteasome-mediated degradation of STAT1alpha following infection of macrophages with Leishmania donovani. J. Biol. Chem. 280, 30542–30549. doi: 10.1074/jbc.M414126200
Forget G., Gregory D. J., Whitcombe L. A., Olivier M. (2006). Role of host protein tyrosine phosphatase SHP-1 in Leishmania donovani-induced inhibition of nitric oxide production. Infect. Immun. 74, 6272–6279. doi: 10.1128/IAI.00853-05
Gallardo-Soler A., Gómez-Nieto C., Campo M. L., Marathe C., Tontonoz P., Castrillo A., et al. (2008). Arginase I induction by modified lipoproteins in macrophages: a peroxisome proliferator-activated receptor-gamma/delta-mediated effect that links lipid metabolism and immunity. Mol. Endocrinol. Baltim Md 22, 1394–1402. doi: 10.1210/me.2007-0525
Ghisletti S., Huang W., Jepsen K., Benner C., Hardiman G., Rosenfeld M. G., et al. (2009). Cooperative NCoR/SMRT interactions establish a corepressor-based strategy for integration of inflammatory and anti-inflammatory signaling pathways. Genes Dev. 23, 681–693. doi: 10.1101/gad.1773109
Giese N. A., Gabriele L., Doherty T. M., Klinman D. M., Tadesse-Heath L., Contursi C., et al. (1997). Interferon (IFN) consensus sequence-binding protein, a transcription factor of the IFN regulatory factor family, regulates immune responses in vivo through control of interleukin 12 expression. J. Exp. Med. 186, 1535–1546. doi: 10.1084/jem.186.9.1535
Grigoriadis G., Zhan Y., Grumont R. J., Metcalf D., Handman E., Cheers C., et al. (1996). The Rel subunit of NF-kappaB-like transcription factors is a positive and negative regulator of macrophage gene expression: distinct roles for Rel in different macrophage populations. EMBO J. 15, 7099–7107. doi: 10.1002/j.1460-2075.1996.tb01101.x
Guizani-Tabbane L., Ben-Aissa K., Belghith M., Sassi A., Dellagi K. (2004). Leishmania major amastigotes induce p50/c-Rel NF-kappa B transcription factor in human macrophages: involvement in cytokine synthesis. Infect. Immun. 72, 2582–2589. doi: 10.1128/iai.72.5.2582-2589.2004
Gupta A. K., Ghosh K., Palit S., Barua J., Das P. K., Ukil A. (2017). Leishmania donovani inhibits inflammasome-dependent macrophage activation by exploiting the negative regulatory proteins A20 and UCP2. FASEB J. 31, 5087–5101. doi: 10.1096/fj.201700407R
Gurung P., Kanneganti T.-D. (2015). Innate immunity against Leishmania infections. Cell Microbiol. 17, 1286–1294. doi: 10.1111/cmi.12484
Hagenbuchner J., Ausserlechner M. J. (2016). Targeting transcription factors by small compounds–Current strategies and future implications. Biochem. Pharmacol. 107, 1–13. doi: 10.1016/j.bcp.2015.12.006
Hashidate-Yoshida T., Harayama T., Hishikawa D., Morimoto R., Hamano F., Tokuoka S. M., et al. (2015). Fatty acid remodeling by LPCAT3 enriches arachidonate in phospholipid membranes and regulates triglyceride transport. eLife 4, e06328. doi: 10.7554/eLife.06328
Hecker M., Wagner A. H. (2017). Transcription factor decoy technology: A therapeutic update. Biochem. Pharmacol. 144, 29–34. doi: 10.1016/j.bcp.2017.06.122
Hishikawa A., Hayashi K., Itoh H. (2018). Transcription Factors as Therapeutic Targets in Chronic Kidney Disease. Molecules Basel Switz 23, 1123. doi: 10.3390/molecules23051123
Honda K., Yanai H., Negishi H., Asagiri M., Sato M., Mizutani T., et al. (2005). IRF-7 is the master regulator of type-I interferon-dependent immune responses. Nature 434, 772–777. doi: 10.1038/nature03464
Ingram S., Mengozzi M., Sacre S., Mullen L., Ghezzi P. (2019). Differential induction of nuclear factor-like 2 signature genes with toll-like receptor stimulation. Free Radic. Biol. Med. 135, 245–250. doi: 10.1016/j.freeradbiomed.2019.03.018
Jantsch J., Schödel J. (2015). Hypoxia and hypoxia-inducible factors in myeloid cell-driven host defense and tissue homeostasis. Immunobiology 220, 305–314. doi: 10.1016/j.imbio.2014.09.009
Jenner R. G., Young R. A. (2005). Insights into host responses against pathogens from transcriptional profiling. Nat. Rev. Microbiol. 3, 281–294. doi: 10.1038/nrmicro1126
Joseph S. B., Castrillo A., Laffitte B. A., Mangelsdorf D. J., Tontonoz P. (2003). Reciprocal regulation of inflammation and lipid metabolism by liver X receptors. Nat. Med. 9, 213–219. doi: 10.1038/nm820
Joseph S. B., Bradley M. N., Castrillo A., Bruhn K. W., Mak P. A., Pei L., et al. (2004). LXR-dependent gene expression is important for macrophage survival and the innate immune response. Cell 119, 299–309. doi: 10.1016/j.cell.2004.09.032
Jung K.-A., Kwak M.-K. (2010). The Nrf2 system as a potential target for the development of indirect antioxidants. Molecules Basel Switz 15, 7266–7291. doi: 10.3390/molecules15107266
Kadl A., Meher A. K., Sharma P. R., Lee M. Y., Doran A. C., Johnstone S. R., et al. (2010). Identification of a novel macrophage phenotype that develops in response to atherogenic phospholipids via Nrf2. Circ. Res. 107, 737–746. doi: 10.1161/CIRCRESAHA.109.215715
Kang K. W., Lee S. J., Park J. W., Kim S. G. (2002). Phosphatidylinositol 3-kinase regulates nuclear translocation of NF-E2-related factor 2 through actin rearrangement in response to oxidative stress. Mol. Pharmacol. 62, 1001–1010. doi: 10.1124/mol.62.5.1001
Kauppinen A., Suuronen T., Ojala J., Kaarniranta K., Salminen A. (2013). Antagonistic crosstalk between NF-κB and SIRT1 in the regulation of inflammation and metabolic disorders. Cell Signal 25, 1939–1948. doi: 10.1016/j.cellsig.2013.06.007
Kawai T., Akira S. (2007). TLR signaling. Semin. Immunol. 19, 24–32. doi: 10.1016/j.smim.2006.12.004
Kawashima A., Oda Y., Yachie A., Koizumi S., Nakanishi I. (2002). Heme oxygenase-1 deficiency: the first autopsy case. Hum. Pathol. 33, 125–130. doi: 10.1053/hupa.2002.30217
Ke Q., Costa M. (2006). Hypoxia-inducible factor-1 (HIF-1). Mol. Pharmacol. 70, 1469–1480. doi: 10.1124/mol.106.027029
Kelly B., O’Neill L. A. (2015). Metabolic reprogramming in macrophages and dendritic cells in innate immunity. Cell Res. 25, 771–784. doi: 10.1038/cr.2015.68
Khouri R., Bafica A., Silva M da P. P., Noronha A., Kolb J.-P., Wietzerbin J., et al. (2009). IFN-beta impairs superoxide-dependent parasite killing in human macrophages: evidence for a deleterious role of SOD1 in cutaneous leishmaniasis. J. Immunol. Baltim Md 1950 182, 2525–2531. doi: 10.4049/jimmunol.0802860
Khouri R., Novais F., Santana G., de Oliveira C. I., Vannier dos Santos M. A., Barral A., et al. (2010). DETC induces Leishmania parasite killing in human in vitro and murine in vivo models: a promising therapeutic alternative in Leishmaniasis. PloS One 5, e14394. doi: 10.1371/journal.pone.0014394
Kim J., Cha Y.-N., Surh Y.-J. (2010). A protective role of nuclear factor-erythroid 2-related factor-2 (Nrf2) in inflammatory disorders. Mutat. Res. 690, 12–23. doi: 10.1016/j.mrfmmm.2009.09.007
Knight M., Stanley S. (2019). HIF-1α as a central mediator of cellular resistance to intracellular pathogens. Curr. Opin. Immunol. 60, 111–116. doi: 10.1016/j.coi.2019.05.005
Koo S.-J., Szczesny B., Wan X., Putluri N., Garg N. J. (2018). Pentose Phosphate Shunt Modulates Reactive Oxygen Species and Nitric Oxide Production Controlling Trypanosoma cruzi in Macrophages. Front. Immunol. 9:202. doi: 10.3389/fimmu.2018.00202
Korf H., Vander Beken S., Romano M., Steffensen K. R., Stijlemans B., Gustafsson J.-A., et al. (2009). Liver X receptors contribute to the protective immune response against Mycobacterium tuberculosis in mice. J. Clin. Invest. 119, 1626–1637. doi: 10.1172/JCI35288
Kostadinova R., Wahli W., Michalik L. (2005). PPARs in diseases: control mechanisms of inflammation. Curr. Med. Chem. 12, 2995–3009. doi: 10.2174/092986705774462905
Kropf P., Freudenberg N., Kalis C., Modolell M., Herath S., Galanos C., et al. (2004). Infection of C57BL/10ScCr and C57BL/10ScNCr mice with Leishmania major reveals a role for Toll-like receptor 4 in the control of parasite replication. J. Leukoc. Biol. 76, 48–57. doi: 10.1189/jlb.1003484
Lambert M., Jambon S., Depauw S., David-Cordonnier M.-H. (2018). Targeting Transcription Factors for Cancer Treatment. Molecules Basel Switz 23, 1479. doi: 10.3390/molecules23061479
Lan F., Cacicedo J. M., Ruderman N., Ido Y. (2008). SIRT1 modulation of the acetylation status, cytosolic localization, and activity of LKB1. Possible role in AMP-activated protein kinase activation. J. Biol. Chem. 283, 27628–27635. doi: 10.1074/jbc.M805711200
Lecoeur H., Prina E., Rosazza T., Kokou K., N’Diaye P., Aulner N., et al. (2020). Targeting Macrophage Histone H3 Modification as a Leishmania Strategy to Dampen the NF-κB/NLRP3-Mediated Inflammatory Response. Cell Rep. 30, 1870–1882.e4. doi: 10.1016/j.celrep.2020.01.030
Lefèvre L., Lugo-Villarino G., Meunier E., Valentin A., Olagnier D., Authier H., et al. (2013). The C-type lectin receptors dectin-1, MR, and SIGNR3 contribute both positively and negatively to the macrophage response to Leishmania infantum. Immunity 38, 1038–1049. doi: 10.1016/j.immuni.2013.04.010
Leopold Wager C. M., Arnett E., Schlesinger L. S. (2019). Macrophage nuclear receptors: Emerging key players in infectious diseases. PloS Pathog. 15, e1007585. doi: 10.1371/journal.ppat.1007585
Li X., Wenes M., Romero P., Huang S. C.-C., Fendt S.-M., Ho P.-C. (2019). Navigating metabolic pathways to enhance antitumour immunity and immunotherapy. Nat. Rev. Clin. Oncol. 16, 425–441. doi: 10.1038/s41571-019-0203-7
Liese J., Schleicher U., Bogdan C. (2007). TLR9 signaling is essential for the innate NK cell response in murine cutaneous leishmaniasis. Eur. J. Immunol. 37, 3424–3434. doi: 10.1002/eji.200737182
Lima-Junior D. S., Costa D. L., Carregaro V., Cunha L. D., Silva A. L. N., Mineo T. W. P., et al. (2013). Inflammasome-derived IL-1β production induces nitric oxide-mediated resistance to Leishmania. Nat. Med. 19, 909–915. doi: 10.1038/nm.3221
Lohoff M., Ferrick D., Mittrucker H. W., Duncan G. S., Bischof S., Rollinghoff M., et al. (1997). Interferon regulatory factor-1 is required for a T helper 1 immune response in vivo. Immunity 6, 681–689. doi: 10.1016/s1074-7613(00)80444-6
Lohoff M., Duncan G. S., Ferrick D., Mittrücker H. W., Bischof S., Prechtl S., et al. (2000). Deficiency in the transcription factor interferon regulatory factor (IRF)-2 leads to severely compromised development of natural killer and T helper type 1 cells. J. Exp. Med. 192, 325–336. doi: 10.1084/jem.192.3.325
Lu M.-C., Ji J.-A., Jiang Z.-Y., You Q.-D. (2016). The Keap1-Nrf2-ARE Pathway As a Potential Preventive and Therapeutic Target: An Update. Med. Res. Rev. 36, 924–963. doi: 10.1002/med.21396
Luz N. F., Andrade B. B., Feijó D. F., Araújo-Santos T., Carvalho G. Q., Andrade D., et al. (2012). Heme oxygenase-1 promotes the persistence of Leishmania chagasi infection. J. Immunol. Baltim Md 1950 188, 4460–4467. doi: 10.4049/jimmunol.1103072
Mahlknecht U., Voelter-Mahlknecht S. (2009). Chromosomal characterization and localization of the NAD+-dependent histone deacetylase gene sirtuin 1 in the mouse. Int. J. Mol. Med. 23, 245–252. doi: 10.3892/ijmm_00000123
Mahnke A., Meier R. J., Schatz V., Hofmann J., Castiglione K., Schleicher U., et al. (2014). Hypoxia in Leishmania major skin lesions impairs the NO-dependent leishmanicidal activity of macrophages. J. Invest. Dermatol. 134, 2339–2346. doi: 10.1038/jid.2014.121
Mai L. T., Smans M., Silva-Barrios S., Fabié A., Stäger S. (2019). IRF-5 Expression in Myeloid Cells Is Required for Splenomegaly in L. donovani Infected Mice. Front. Immunol. 10:3071. doi: 10.3389/fimmu.2019.03071
Marín-Hernández A., Gallardo-Pérez J. C., Ralph S. J., Rodríguez-Enríquez S., Moreno-Sánchez R. (2009). HIF-1alpha modulates energy metabolism in cancer cells by inducing over-expression of specific glycolytic isoforms. Mini Rev. Med. Chem. 9, 1084–1101. doi: 10.2174/138955709788922610
Matte C., Descoteaux A. (2010). Leishmania donovani Amastigotes Impair Gamma Interferon-Induced STAT1α Nuclear Translocation by Blocking the Interaction between STAT1α and Importin-α5. Infect. Immun. 78, 3736–3743. doi: 10.1128/IAI.00046-10
Meraz M. A., White J. M., Sheehan K. C., Bach E. A., Rodig S. J., Dighe A. S., et al. (1996). Targeted disruption of the Stat1 gene in mice reveals unexpected physiologic specificity in the JAK-STAT signaling pathway. Cell 84, 431–442. doi: 10.1016/s0092-8674(00)81288-x
Mesquita I., Ferreira C., Moreira D., Kluck G. E. G., Barbosa A. M., Torrado E., et al. (2020). The Absence of HIF-1α Increases Susceptibility to Leishmania donovani Infection via Activation of BNIP3/mTOR/SREBP-1c Axis. Cell Rep. 30, 4052–4064.e7. doi: 10.1016/j.celrep.2020.02.098
Metheni M., Echebli N., Chaussepied M., Ransy C., Chéreau C., Jensen K., et al. (2014). The level of H₂O₂ type oxidative stress regulates virulence of Theileria-transformed leukocytes. Cell Microbiol. 16, 269–279. doi: 10.1111/cmi.12218
Mirza A. Z., Althagafi I. I., Shamshad H. (2019). Role of PPAR receptor in different diseases and their ligands: Physiological importance and clinical implications. Eur. J. Med. Chem. 166, 502–513. doi: 10.1016/j.ejmech.2019.01.067
Mise-Omata S., Kuroda E., Sugiura T., Yamashita U., Obata Y., Doi T. S. (2009). The NF-kappaB RelA subunit confers resistance to Leishmania major by inducing nitric oxide synthase 2 and Fas expression but not Th1 differentiation. J. Immunol. Baltim Md 1950 182, 4910–4916. doi: 10.4049/jimmunol.0800967
Moreira D., Rodrigues V., Abengozar M., Rivas L., Rial E., Laforge M., et al. (2015). Leishmania infantum modulates host macrophage mitochondrial metabolism by hijacking the SIRT1-AMPK axis. PloS Pathog. 11, e1004684. doi: 10.1371/journal.ppat.1004684
Muraille E., De Trez C., Brait M., De Baetselier P., Leo O., Carlier Y. (2003). Genetically resistant mice lacking MyD88-adapter protein display a high susceptibility to Leishmania major infection associated with a polarized Th2 response. J. Immunol. Baltim Md 1950 170, 4237–4241. doi: 10.4049/jimmunol.170.8.4237
Murray P. J. (2017). Macrophage Polarization. Annu. Rev. Physiol. 79, 541–566. doi: 10.1146/annurev-physiol-022516-034339
Muxel S. M., Aoki J. I., Fernandes J. C. R., Laranjeira-Silva M. F., Zampieri R. A., Acuña S. M., et al. (2017). Arginine and Polyamines Fate in Leishmania Infection. Front. Microbiol. 8:2682. doi: 10.3389/fmicb.2017.02682
Naderer T., Heng J., McConville M. J. (2010). Evidence that intracellular stages of Leishmania major utilize amino sugars as a major carbon source. PloS Pathog. 6, e1001245. doi: 10.1371/journal.ppat.1001245
Nagao A., Kobayashi M., Koyasu S., Chow C. C. T., Harada H. (2019). HIF-1-Dependent Reprogramming of Glucose Metabolic Pathway of Cancer Cells and Its Therapeutic Significance. Int. J. Mol. Sci. 20, 238. doi: 10.3390/ijms20020238
Naito Y., Takagi T., Higashimura Y. (2014). Heme oxygenase-1 and anti-inflammatory M2 macrophages. Arch. Biochem. Biophys. 564, 83–88. doi: 10.1016/j.abb.2014.09.005
Nandan D., Reiner N. E. (1995). Attenuation of gamma interferon-induced tyrosine phosphorylation in mononuclear phagocytes infected with Leishmania donovani: selective inhibition of signaling through Janus kinases and Stat1. Infect. Immun. 63, 4495–4500. doi: 10.1128/IAI.63.11.4495-4500.1995
Natoli G. (2009). When sirtuins and NF-kappaB collide. Cell 136, 19–21. doi: 10.1016/j.cell.2008.12.034
Negishi H., Ohba Y., Yanai H., Takaoka A., Honma K., Yui K., et al. (2005). Negative regulation of Toll-like-receptor signaling by IRF-4. Proc. Natl. Acad. Sci. U.S.A. 102, 15989–15994. doi: 10.1073/pnas.0508327102
Negishi H., Fujita Y., Yanai H., Sakaguchi S., Ouyang X., Shinohara M., et al. (2006). Evidence for licensing of IFN-gamma-induced IFN regulatory factor 1 transcription factor by MyD88 in Toll-like receptor-dependent gene induction program. Proc. Natl. Acad. Sci. U.S.A. 103, 15136–15141. doi: 10.1073/pnas.0607181103
Odegaard J. I., Ricardo-Gonzalez R. R., Goforth M. H., Morel C. R., Subramanian V., Mukundan L., et al. (2007). Macrophage-specific PPARgamma controls alternative activation and improves insulin resistance. Nature 447, 1116–1120. doi: 10.1038/nature05894
Olagnier D., Lavergne R.-A., Meunier E., Lefèvre L., Dardenne C., Aubouy A., et al. (2011). Nrf2, a PPARγ alternative pathway to promote CD36 expression on inflammatory macrophages: implication for malaria. PloS Pathog. 7, e1002254. doi: 10.1371/journal.ppat.1002254
Osorio y Fortea J., de La Llave E., Regnault B., Coppee J.-Y., Milon G., Lang T., et al. (2009). Transcriptional signatures of BALB/c mouse macrophages housing multiplying Leishmania amazonensis amastigotes. BMC Genomics 10:119. doi: 10.1186/1471-2164-10-119
Paine A., Eiz-Vesper B., Blasczyk R., Immenschuh S. (2010). Signaling to heme oxygenase-1 and its anti-inflammatory therapeutic potential. Biochem. Pharmacol. 80, 1895–1903. doi: 10.1016/j.bcp.2010.07.014
Paiva C. N., Bozza M. T. (2014). Are reactive oxygen species always detrimental to pathogens? Antioxid. Redox Signal 20, 1000–1037. doi: 10.1089/ars.2013.5447
Paiva C. N., Feijó D. F., Dutra F. F., Carneiro V. C., Freitas G. B., Alves L. S., et al. (2012). Oxidative stress fuels Trypanosoma cruzi infection in mice. J. Clin. Invest. 122, 2531–2542. doi: 10.1172/JCI58525
Palazon A., Goldrath A. W., Nizet V., Johnson R. S. (2014). HIF Transcription Factors, Inflammation, and Immunity. Immunity 41, 518–528. doi: 10.1016/j.immuni.2014.09.008
Pålsson-McDermott E. M., O’Neill L. A. J. (2020). Targeting immunometabolism as an anti-inflammatory strategy. Cell Res. 30, 300–314. doi: 10.1038/s41422-020-0291-z
Pang Y., Zhang Z., Chen Y., Cao S., Yang X., Jia H. (2019). The Nrf2 pathway is required for intracellular replication of Toxoplasma gondii in activated macrophages. Parasite Immunol. 41, e12621. doi: 10.1111/pim.12621
Papavassiliou K. A., Papavassiliou A. G. (2016). Transcription Factor Drug Targets. J. Cell Biochem. 117, 2693–2696. doi: 10.1002/jcb.25605
Paun A., Bankoti R., Joshi T., Pitha P. M., Stäger S. (2011). Critical role of IRF-5 in the development of T helper 1 responses to Leishmania donovani infection. PloS Pathog. 7, e1001246. doi: 10.1371/journal.ppat.1001246
Penas F., Mirkin G. A., Vera M., Cevey Á., González C. D., Gómez M. I., et al. (2015). Treatment in vitro with PPARα and PPARγ ligands drives M1-to-M2 polarization of macrophages from T. cruzi-infected mice. Biochim. Biophys. Acta 1852, 893–904. doi: 10.1016/j.bbadis.2014.12.019
Pham N.-K., Mouriz J., Kima P. E. (2005). Leishmania pifanoi amastigotes avoid macrophage production of superoxide by inducing heme degradation. Infect. Immun. 73, 8322–8333. doi: 10.1128/IAI.73.12.8322-8333.2005
Piantadosi C. A., Carraway M. S., Babiker A., Suliman H. B. (2008). Heme oxygenase-1 regulates cardiac mitochondrial biogenesis via Nrf2-mediated transcriptional control of nuclear respiratory factor-1. Circ. Res. 103, 1232–1240. doi: 10.1161/01.RES.0000338597.71702.ad
Piranavan P., Bhamra M., Perl A. (2020). Metabolic Targets for Treatment of Autoimmune Diseases. Immunometabolism 2, e200012. doi: 10.20900/immunometab20200012
Purushotham A., Schug T. T., Xu Q., Surapureddi S., Guo X., Li X. (2009). Hepatocyte-specific deletion of SIRT1 alters fatty acid metabolism and results in hepatic steatosis and inflammation. Cell Metab. 9, 327–338. doi: 10.1016/j.cmet.2009.02.006
Rabhi I., Rabhi S., Ben-Othman R., Rasche A., Daskalaki A., Trentin B., et al. (2012). Transcriptomic signature of Leishmania infected mice macrophages: a metabolic point of view. PloS Negl. Trop. Dis. 6, e1763. doi: 10.1371/journal.pntd.0001763
Rabhi S., Rabhi I., Trentin B., Piquemal D., Regnault B., Goyard S., et al. (2016). Lipid Droplet Formation, Their Localization and Dynamics during Leishmania major Macrophage Infection. PloS One 11, e0148640. doi: 10.1371/journal.pone.0148640
Ray M., Gam A. A., Boykins R. A., Kenney R. T. (2000). Inhibition of Interferon-γ Signaling by Leishmania donovani. J. Infect. Dis. 181, 1121–1128. doi: 10.1086/315330
Reinhard K., Huber M., Lohoff M., Visekruna A. (2012). The role of NF-κB activation during protection against Leishmania infection. Int. J. Med. Microbiol. 302, 230–235. doi: 10.1016/j.ijmm.2012.07.006
Repa J. J., Liang G., Ou J., Bashmakov Y., Lobaccaro J. M., Shimomura I., et al. (2000). Regulation of mouse sterol regulatory element-binding protein-1c gene (SREBP-1c) by oxysterol receptors, LXRalpha and LXRbeta. Genes Dev. 14, 2819–2830. doi: 10.1101/gad.844900
Repa J. J., Berge K. E., Pomajzl C., Richardson J. A., Hobbs H., Mangelsdorf D. J. (2002). Regulation of ATP-binding cassette sterol transporters ABCG5 and ABCG8 by the liver X receptors alpha and beta. J. Biol. Chem. 277, 18793–18800. doi: 10.1074/jbc.M109927200
Rius J., Guma M., Schachtrup C., Akassoglou K., Zinkernagel A. S., Nizet V., et al. (2008). NF-kappaB links innate immunity to the hypoxic response through transcriptional regulation of HIF-1alpha. Nature 453, 807–811. doi: 10.1038/nature06905
Rocha-Vieira E., Ferreira E., Vianna P., De Faria D. R., Gaze S. T., Dutra W. O., et al. (2003). Histopathological outcome of Leishmania major-infected BALB/c mice is improved by oral treatment with N-acetyl-l-cysteine. Immunology 108, 401–408. doi: 10.1046/j.1365-2567.2003.01582.x
Rodgers J. T., Lerin C., Haas W., Gygi S. P., Spiegelman B. M., Puigserver P. (2005). Nutrient control of glucose homeostasis through a complex of PGC-1alpha and SIRT1. Nature 434, 113–118. doi: 10.1038/nature03354
Rodriguez N. E., Chang H. K., Wilson M. E. (2004). Novel program of macrophage gene expression induced by phagocytosis of Leishmania chagasi. Infect. Immun. 72, 2111–2122. doi: 10.1128/iai.72.4.2111-2122.2004
Rong X., Albert C. J., Hong C., Duerr M. A., Chamberlain B. T., Tarling E. J., et al. (2013). LXRs regulate ER stress and inflammation through dynamic modulation of membrane phospholipid composition. Cell Metab. 18, 685–697. doi: 10.1016/j.cmet.2013.10.002
Roy S., Mukhopadhyay D., Mukherjee S., Ghosh S., Kumar S., Sarkar K., et al. (2015). A Defective Oxidative Burst and Impaired Antigen Presentation are Hallmarks of Human Visceral Leishmaniasis. J. Clin. Immunol. 35, 56–67. doi: 10.1007/s10875-014-0115-3
Rudraiah S., Zhang X., Wang L. (2016). Nuclear Receptors as Therapeutic Targets in Liver Disease: Are We There Yet? Annu. Rev. Pharmacol. Toxicol. 56, 605–626. doi: 10.1146/annurev-pharmtox-010715-103209
Ruivo M. T. G., Vera I. M., Sales-Dias J., Meireles P., Gural N., Bhatia S. N., et al. (2016). Host AMPK Is a Modulator of Plasmodium Liver Infection. Cell Rep. 16, 2539–2545. doi: 10.1016/j.celrep.2016.08.001
Sanjabi S., Hoffmann A., Liou H. C., Baltimore D., Smale S. T. (2000). Selective requirement for c-Rel during IL-12 P40 gene induction in macrophages. Proc. Natl. Acad. Sci. U.S.A. 97, 12705–12710. doi: 10.1073/pnas.230436397
Santos D., Campos T. M., Saldanha M., Oliveira S. C., Nascimento M., Zamboni D. S., et al. (2018). IL-1β Production by Intermediate Monocytes Is Associated with Immunopathology in Cutaneous Leishmaniasis. J. Invest. Dermatol. 138, 1107–1115. doi: 10.1016/j.jid.2017.11.029
Santos S. A. D., Andrade DR de (2017). HIF-1alpha and infectious diseases: a new frontier for the development of new therapies. Rev. Inst. Med. Trop. Sao Paulo 59, e92. doi: 10.1590/S1678-9946201759092
Satoh T., Akira S. (2016). Toll-Like Receptor Signaling and Its Inducible Proteins. Microbiol. Spectr. 4, 6. doi: 10.1128/microbiolspec.MCHD-0040-2016
Schatz V., Strüssmann Y., Mahnke A., Schley G., Waldner M., Ritter U., et al. (2016). Myeloid Cell-Derived HIF-1α Promotes Control of Leishmania major. J. Immunol. Baltim Md 1950 197, 4034–4041. doi: 10.4049/jimmunol.1601080
Schatz V., Neubert P., Rieger F., Jantsch J. (2018). Hypoxia, Hypoxia-Inducible Factor-1α, and Innate Antileishmanial Immune Responses. Front. Immunol. 9:216. doi: 10.3389/fimmu.2018.00216
Schmitz F., Heit A., Guggemoos S., Krug A., Mages J., Schiemann M., et al. (2007). Interferon-regulatory-factor 1 controls Toll-like receptor 9-mediated IFN-beta production in myeloid dendritic cells. Eur. J. Immunol. 37, 315–327. doi: 10.1002/eji.200636767
Sharma S., tenOever B. R., Grandvaux N., Zhou G.-P., Lin R., Hiscott J. (2003). Triggering the interferon antiviral response through an IKK-related pathway. Science 300, 1148–1151. doi: 10.1126/science.1081315
Shim D., Kim H., Shin S. J. (2020). Mycobacterium tuberculosis Infection-Driven Foamy Macrophages and Their Implications in Tuberculosis Control as Targets for Host-Directed Therapy. Front. Immunol. 11:910. doi: 10.3389/fimmu.2020.00910
Singh A. K., Mukhopadhyay C., Biswas S., Singh V. K., Mukhopadhyay C. K. (2012). Intracellular pathogen Leishmania donovani activates hypoxia inducible factor-1 by dual mechanism for survival advantage within macrophage. PloS One 7, e38489. doi: 10.1371/journal.pone.0038489
Speirs K., Caamano J., Goldschmidt M. H., Hunter C. A., Scott P. (2002). NF-kappa B2 is required for optimal CD40-induced IL-12 production but dispensable for Th1 cell Differentiation. J. Immunol. Baltim Md 1950 168, 4406–4413. doi: 10.4049/jimmunol.168.9.4406
Srivastav S., Basu Ball W., Gupta P., Giri J., Ukil A., Das P. K. (2014). Leishmania donovani prevents oxidative burst-mediated apoptosis of host macrophages through selective induction of suppressors of cytokine signaling (SOCS) proteins. J. Biol. Chem. 289, 1092–1105. doi: 10.1074/jbc.M113.496323
Stothers C. L., Luan L., Fensterheim B. A., Bohannon J. K. (2018). Hypoxia-inducible factor-1α regulation of myeloid cells. J. Mol. Med. Berl Ger 96, 1293–1306. doi: 10.1007/s00109-018-1710-1
Suzuki T., Yamamoto M. (2015). Molecular basis of the Keap1-Nrf2 system. Free Radic. Biol. Med. 88, 93–100. doi: 10.1016/j.freeradbiomed.2015.06.006
Takaoka A., Yanai H., Kondo S., Duncan G., Negishi H., Mizutani T., et al. (2005). Integral role of IRF-5 in the gene induction programme activated by Toll-like receptors. Nature 434, 243–249. doi: 10.1038/nature03308
Thimmulappa R. K., Lee H., Rangasamy T., Reddy S. P., Yamamoto M., Kensler T. W., et al. (2006). Nrf2 is a critical regulator of the innate immune response and survival during experimental sepsis. J. Clin. Invest. 116, 984–995. doi: 10.1172/JCI25790
Tomiotto-Pellissier F., Bortoleti BT da S., Assolini J. P., Gonçalves M. D., Carloto A. C. M., Miranda-Sapla M. M., et al. (2018). Macrophage Polarization in Leishmaniasis: Broadening Horizons. Front. Immunol. 9:2529. doi: 10.3389/fimmu.2018.02529
Vachharajani V. T., Liu T., Wang X., Hoth J. J., Yoza B. K., McCall C. E. (2016). Sirtuins Link Inflammation and Metabolism. J. Immunol. Res. 2016:8167273. doi: 10.1155/2016/8167273
van Uden P., Kenneth N. S., Rocha S. (2008). Regulation of hypoxia-inducible factor-1alpha by NF-kappaB. Biochem. J. 412, 477–484. doi: 10.1042/BJ20080476
Vats D., Mukundan L., Odegaard J. I., Zhang L., Smith K. L., Morel C. R., et al. (2006). Oxidative metabolism and PGC-1beta attenuate macrophage-mediated inflammation. Cell Metab. 4, 13–24. doi: 10.1016/j.cmet.2006.05.011
Vega R. B., Huss J. M., Kelly D. P. (2000). The coactivator PGC-1 cooperates with peroxisome proliferator-activated receptor alpha in transcriptional control of nuclear genes encoding mitochondrial fatty acid oxidation enzymes. Mol. Cell Biol. 20, 1868–1876. doi: 10.1128/mcb.20.5.1868-1876.2000
Vitali S. H., Fernandez-Gonzalez A., Nadkarni J., Kwong A., Rose C., Mitsialis S. A., et al. (2020). Heme oxygenase-1 dampens the macrophage sterile inflammasome response and regulates its components in the hypoxic lung. Am. J. Physiol. Lung Cell Mol. Physiol. 318, L125–L134. doi: 10.1152/ajplung.00074.2019
Vivarini A. C., Lopes U. G. (2019). The Potential Role of Nrf2 Signaling in Leishmania Infection Outcomes. Front. Cell Infect. Microbiol. 9:453. doi: 10.3389/fcimb.2019.00453
Vivarini Á. C., Calegari-Silva T. C., Saliba A. M., Boaventura V. S., França-Costa J., Khouri R., et al. (2017). Systems Approach Reveals Nuclear Factor Erythroid 2-Related Factor 2/Protein Kinase R Crosstalk in Human Cutaneous Leishmaniasis. Front. Immunol. 8:1127. doi: 10.3389/fimmu.2017.01127
Voelter-Mahlknecht S., Mahlknecht U. (2006). Cloning, chromosomal characterization and mapping of the NAD-dependent histone deacetylases gene sirtuin 1. Int. J. Mol. Med. 17, 59–67. doi: 10.3892/ijmm.17.1.59
Wang Z., Schaffer N. E., Kliewer S. A., Mangelsdorf D. J. (2017). Nuclear receptors: emerging drug targets for parasitic diseases. J. Clin. Invest. 127, 1165–1171. doi: 10.1172/JCI88890
Wang S., Liu R., Yu Q., Dong L., Bi Y., Liu G. (2019). Metabolic reprogramming of macrophages during infections and cancer. Cancer Lett. 452, 14–22. doi: 10.1016/j.canlet.2019.03.015
Wardyn J. D., Ponsford A. H., Sanderson C. M. (2015). Dissecting molecular cross-talk between Nrf2 and NF-κB response pathways. Biochem. Soc. Trans. 43, 621–626. doi: 10.1042/BST20150014
Weih F., Carrasco D., Durham S. K., Barton D. S., Rizzo C. A., Ryseck R. P., et al. (1995). Multiorgan inflammation and hematopoietic abnormalities in mice with a targeted disruption of RelB, a member of the NF-kappa B/Rel family. Cell 80, 331–340. doi: 10.1016/0092-8674(95)90416-6
Wiesel P., Patel A. P., DiFonzo N., Marria P. B., Sim C. U., Pellacani A., et al. (2000). Endotoxin-induced mortality is related to increased oxidative stress and end-organ dysfunction, not refractory hypotension, in heme oxygenase-1-deficient mice. Circulation 102, 3015–3022. doi: 10.1161/01.cir.102.24.3015
Wiley M., Sweeney K. R., Chan D. A., Brown K. M., McMurtrey C., Howard E. W., et al. (2010). Toxoplasma gondii activates hypoxia-inducible factor (HIF) by stabilizing the HIF-1alpha subunit via type I activin-like receptor kinase receptor signaling. J. Biol. Chem. 285, 26852–26860. doi: 10.1074/jbc.M110.147041
Xue J., Schmidt S. V., Sander J., Draffehn A., Krebs W., Quester I., et al. (2014). Transcriptome-based network analysis reveals a spectrum model of human macrophage activation. Immunity 40, 274–288. doi: 10.1016/j.immuni.2014.01.006
Yeung F., Hoberg J. E., Ramsey C. S., Keller M. D., Jones D. R., Frye R. A., et al. (2004). Modulation of NF-kappaB-dependent transcription and cell survival by the SIRT1 deacetylase. EMBO J. 23, 2369–2380. doi: 10.1038/sj.emboj.7600244
Zhu L., Zhao Q., Yang T., Ding W., Zhao Y. (2015). Cellular Metabolism and Macrophage Functional Polarization. Int. Rev. Immunol. 34, 82–100. doi: 10.3109/08830185.2014.969421
Keywords: Leishmania, macrophages, transcription factor, immune response, metabolic response
Citation: Bichiou H, Bouabid C, Rabhi I and Guizani-Tabbane L (2021) Transcription Factors Interplay Orchestrates the Immune-Metabolic Response of Leishmania Infected Macrophages. Front. Cell. Infect. Microbiol. 11:660415. doi: 10.3389/fcimb.2021.660415
Received: 29 January 2021; Accepted: 22 March 2021;
Published: 07 April 2021.
Edited by:
Yongliang Zhang, National University of Singapore, SingaporeReviewed by:
Saikat Majumder, University of Pittsburgh, United StatesLucas P. Carvalho, Gonçalo Moniz Institute (IGM), Brazil
Copyright © 2021 Bichiou, Bouabid, Rabhi and Guizani-Tabbane. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Lamia Guizani-Tabbane, bGFtaWEuZ3VpemFuaUBQYXN0ZXVyLnJucy50bg==