
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Cell. Infect. Microbiol. , 18 May 2021
Sec. Microbiome in Health and Disease
Volume 11 - 2021 | https://doi.org/10.3389/fcimb.2021.646467
 Prince Kofi Parbie1,2,3
Prince Kofi Parbie1,2,3 Taketoshi Mizutani4Aya Ishizaka4
Taketoshi Mizutani4Aya Ishizaka4 Ai Kawana-Tachikawa1,3,4Lucky Ronald Runtuwene1
Ai Kawana-Tachikawa1,3,4Lucky Ronald Runtuwene1 Sayuri Seki1Christopher Zaab-Yen Abana2Dennis Kushitor2
Sayuri Seki1Christopher Zaab-Yen Abana2Dennis Kushitor2 Evelyn Yayra Bonney2Sampson Badu Ofori5
Evelyn Yayra Bonney2Sampson Badu Ofori5 Satoshi Uematsu4,6,7Seiya Imoto4,7Yasumasa Kimura4
Satoshi Uematsu4,6,7Seiya Imoto4,7Yasumasa Kimura4 Hiroshi Kiyono4,8,9
Hiroshi Kiyono4,8,9 Koichi Ishikawa1
Koichi Ishikawa1 William Kwabena Ampofo2*
William Kwabena Ampofo2* Tetsuro Matano1,3,4*
Tetsuro Matano1,3,4*HIV-1 infected individuals under antiretroviral therapy can control viremia but often develop non-AIDS diseases such as cardiovascular and metabolic disorders. Gut microbiome dysbiosis has been indicated to be associated with progression of these diseases. Analyses of gut/fecal microbiome in individual regions are important for our understanding of pathogenesis in HIV-1 infections. However, data on gut/fecal microbiome has not yet been accumulated in West Africa. In the present study, we examined fecal microbiome compositions in HIV-1 infected adults in Ghana, where approximately two-thirds of infected adults are females. In a cross-sectional case-control study, age- and gender-matched HIV-1 infected adults (HIV+; n = 55) and seronegative controls (HIV-; n = 55) were enrolled. Alpha diversity of fecal microbiome in HIV+ was significantly reduced compared to HIV- and associated with CD4 counts. HIV+ showed reduction in varieties of bacteria including Faecalibacterium, the most abundant in seronegative controls, but enrichment of Proteobacteria. Ghanaian HIV+ exhibited enrichment of Dorea and Blautia; bacteria groups whose depletion has been reported in HIV-1 infected individuals in several other cohorts. Furthermore, HIV+ in our cohort exhibited a depletion of Prevotella, a genus whose enrichment has recently been shown in men having sex with men (MSM) regardless of HIV-1 status. The present study revealed the characteristics of dysbiotic fecal microbiome in HIV-1 infected adults in Ghana, a representative of West African populations.
Antiretroviral therapy (ART) can inhibit HIV-1 replication and prevent AIDS progression but is not able to eliminate the viruses in HIV-1 infected individuals (Dieffenbach and Fauci, 2011). HIV-1 infected individuals on ART are likely to develop non-AIDS diseases such as cardiovascular and metabolic disorders (Antiretroviral Therapy Cohort Collaboration, 2010). Gut dysbiosis in HIV-1 infected individuals has been indicated to be associated with progression of these non-AIDS diseases (Vujkovic-Cvijin et al., 2013; Nwosu et al., 2014; West et al., 2015; Kehrmann et al., 2019). Severe gastrointestinal barrier impairment resulting in translocation of microbial components from intestinal mucosa into blood stream during the acute phase of HIV-1 infection has been reported as a major driver of systemic inflammation and immune activation with subsequent detrimental disease outcomes (Brenchley et al., 2006; Marchetti et al., 2011; Sandler and Douek, 2012; Mudd and Brenchley, 2016). Circulating lipopolysaccharide (LPS) levels are considered to be a strong predictor of disease progression in HIV-1 infection (Marchetti et al., 2011), and association of circulating microbial product levels with systemic immune activation has been indicated in simian immunodeficiency virus (SIV) infected macaques as well as HIV-1 infected individuals (Brenchley et al., 2006).
Gut microbiome composition is different all over the world, and individual populations show varieties of biological interaction between host factors and microbiome. For instance, it has been suggested that diet and gut microbial compositions are determinant for higher risk of colorectal carcinogenesis in African Americans compared to native Africans and Caucasians (O’Keefe et al., 2007). On the other hand, influence of host genetics (HLA-B27 and HLA-DRB1) on gut microbial dysbiosis has been indicated in ankylosing spondylitis and rheumatoid arthritis (Gill et al., 2018; Asquith et al., 2019). Analysis of gut/fecal microbiome is thus important for our understanding of pathogenesis in HIV-1 infections in individual regions. Therapeutic interventions targeting gut microbiota have currently been explored (Irvine et al., 2011; Klatt et al., 2013; D’Ettorre et al., 2015; Ortiz et al., 2016), but lacking in data on gut/fecal microbiome may be an obstacle for application of these attempts. However, studies on gut microbiome in people living with HIV (PLWH) in sub-Saharan Africa, a population with the vast majority of PLWH, are limited (Gootenberg et al., 2017). Indeed, reports comparing microbial compositions of HIV-1 infected and uninfected groups adequately matched on age, gender, and residence are quite limited. To address the issues described above, we started analyzing fecal microbiome in HIV-1 infected individuals in Ghana, West Africa, where data on enteric microbiome has not yet been accumulated. Approximately two-thirds of HIV-1-infected Ghanaian adults are females (UNAIDS, 2020), while data on gut microbiome in HIV-1 infected individuals has been accumulated mostly by analyzing cohorts including larger number of men having sex with men (MSM) in developed countries (Lozupone et al., 2013; McHardy et al., 2013; Vujkovic-Cvijin et al., 2013; Dillon et al., 2014; Mutlu et al., 2014; Vujkovic-Cvijin and Somsouk, 2019). In the present study, we compared fecal microbiome in HIV-1 infected and uninfected groups matched on gender, age, and community of residence in Ghana. Analysis revealed the characteristics of dysbiotic fecal microbiome in HIV-1 infected Ghanaians.
In a cross-sectional case-control study, we enrolled matched pairs of HIV-1 infected (HIV+) and uninfected (seronegative; HIV-) individuals. HIV-1 infected individuals routinely attending an HIV/AIDS clinic at the Eastern Regional Hospital, Koforidua (RHK), Ghana, were enrolled into the study. HIV-1 infected participants were identified to reside in 7 communities (Tafo, Suhum, Koforidua, Nkurakan, Jumapo, Oyoko, and Akwadum) in the Eastern Region of Ghana (Supplementary Information 1). HIV-1 seronegative individuals were recruited during a community health screening in these communities. Seronegative individuals matched by age (± 2 years), gender and community of residence were enrolled as controls. Only adults above 18 years old were enrolled in this study. Seronegative participants who took antibiotics within 4 weeks prior to sample collection were not enrolled. Data on HIV- was described previously (Parbie et al., 2020).
Venous blood and stool samples were collected from enrolled participants. All biological samples were transported to Noguchi Memorial Institute for Medical Research (NMIMR), University of Ghana and processed for storage within 24 hours of sample collection. Plasma and peripheral blood mononuclear cells (PBMC) were prepared from venous blood. Stool samples were collected and stored at -80°C until DNA extraction.
Bacterial pellets were prepared from frozen fecal samples as previously described (Morita et al., 2007; Parbie et al., 2020) with minor modifications. Briefly, 1 g of stool was washed three times with 3 ml of SM-plus buffer (100 mM NaCl, 50 mM Tris-HCl [pH7.4], 8 mM MgSO4-7H2O, 5 mM CaCl2-2H2O, 0.01% [w/v] Gelatin) and centrifuged at 6,000 x g for 5 min. Then, pellets were resuspended in 20 ml of SM-plus buffer and filtered through a 100-μm cell strainer (Corning, Tokyo, Japan). One ml out of the filtrated 20 ml of bacterial suspension was used for DNA extraction.
DNA was extracted from fecal sample-derived bacteria fraction as previously described (Kim et al., 2013). Gene libraries for the hypervariable V3-V4 region of 16S rRNA was prepared as previously described (Parbie et al., 2020) according to the 16S Metagenomic Sequencing Library Preparation guide (Illumina, San Diego, USA; Part # 15044223 Rev. B). Sequencing was performed on the Illumina MiSeq using MiSeq Reagent Kit v3 (600-cycle) with a 20% PhiX (Illumina) spike-in at NMIMR.
Sequences were quality filtered, denoised and analyzed with the Quantitative Insights Into Microbial Ecology 2 (QIIME 2™ version 2019.4) (Bolyen et al., 2019) (Supplementary Information 2). Briefly, paired-end reads were denoised into amplicon sequence variants (ASVs) with DADA2 (Callahan et al., 2016). Taxonomy was assigned to the resulting ASVs against the SILVA database (release 132) (Quast et al., 2012), trimmed to the V3-V4 region of the 16S rRNA gene, using Naive Bayesian classifier (Fabian et al., 2011). To remove low abundant or rare taxa prior to differential abundance analysis, sequences were preprocessed based on methodology described in ANCOM-II (Kaul et al., 2017). Differentially abundant taxa by HIV-1 status were identified using the analysis of composition of microbiome (ANCOM) and linear discriminant analysis (LDA) effect size (LEfSe) methods (Segata et al., 2011; Mandal et al., 2015).
Plasma lipopolysaccharide binding protein (LBP), soluble CD14 (sCD14), and intestinal fatty acid-binding protein (I-FABP) levels were measured by enzyme-linked immunosorbent assay (ELISA) according to manufacturer specifications (R&D Systems, Minneapolis, MN, USA).
Data were obtained from 2017 to 2019. GraphPad Prism version 7.04 and R 3.6.0 packages were used for statistical analyses. Comparison between categorical variables between groups were performed with Fisher’s exact test. Continuous variables were assessed using Wilcoxon rank-sum test, Mann-Whitney test, or Kruskal Wallis test. Multiple test correction was performed when assessing differences between groups using Benjamini, Krieger and Yekutieli FDR correction. p values less than 0.05 were considered significant. To test associations involving bacteria taxa showing significant difference in abundance by HIV-1 status and clinical and immunological markers, correlation analysis was performed using R package Hmisc v4.5. Multivariate analysis of phenotypes with significant correlation was performed to ascertain association using Microbiome Multivariable Associations with Linear Models, MaAsLin2 (Mallick et al., 2021).
A total of 110 participants consisting of 55 HIV-1 infected (HIV+) and 55 uninfected (HIV-) adults were enrolled in the present study. The median age of all participants was 45.5 years (IQR, 34-51) and 86 (78%) were females. Fifty-two of the HIV-1 infected participants (95%) had been on ART for a median duration of 62 months (IQR, 26-105) (Table 1) and 32 of them were controlling viremia (< 1,000 copies/mL). The ART regimen consisted of two kinds of nucleoside reverse transcriptase inhibitors and a non-nucleoside reverse transcriptase inhibitor (either efavirenz [n = 42] or nevirapine [n = 10]). Out of the 3 ART naïve HIV-1 infected participants, 2 had undetectable plasma viral RNA (< 20 copies/mL). The median duration since last co-trimoxazole treatment was 72 months (IQR, 24-108) in the HIV-1 infected individuals.
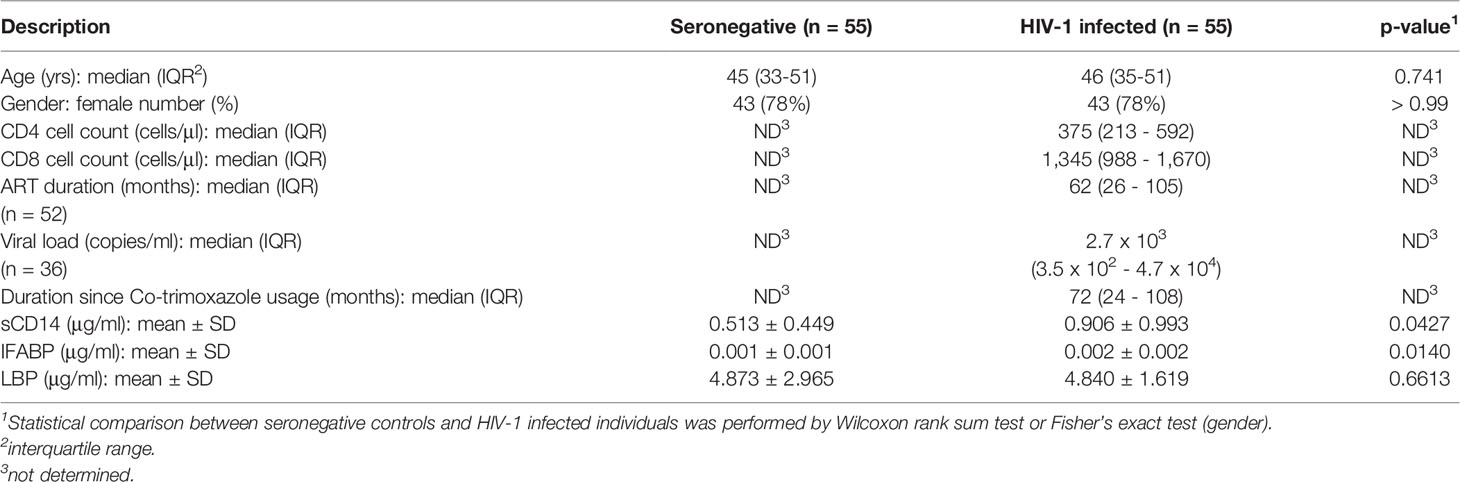
Table 1 Clinical and demographic characteristics.
The 16S rRNA gene libraries were prepared from fecal sample-derived bacteria fraction and subjected to next generation sequencing. Fecal microbiome compositions in the healthy seronegative Ghanaian adults were described previously (Parbie et al., 2020). Mean relative abundance showed that Firmicutes is the most abundant at the phylum level in the entire study cohort (Supplementary Figure 1). Proteobacteria, Bacteroidetes, and Actinobacteria were also abundant in HIV-, while more abundant Proteobacteria but less Bacteroidetes were observed in HIV+ individuals. At the genus level, Faecalibacterium and Subdoligranulum belonging to Firmicutes were dominant in HIV-, whereas Faecalibacterium was depleted and Stenotrophomonas and Achromobacter belonging to Proteobacteria as well as Subdoligranulum looked abundant in HIV+ (Figure 1).
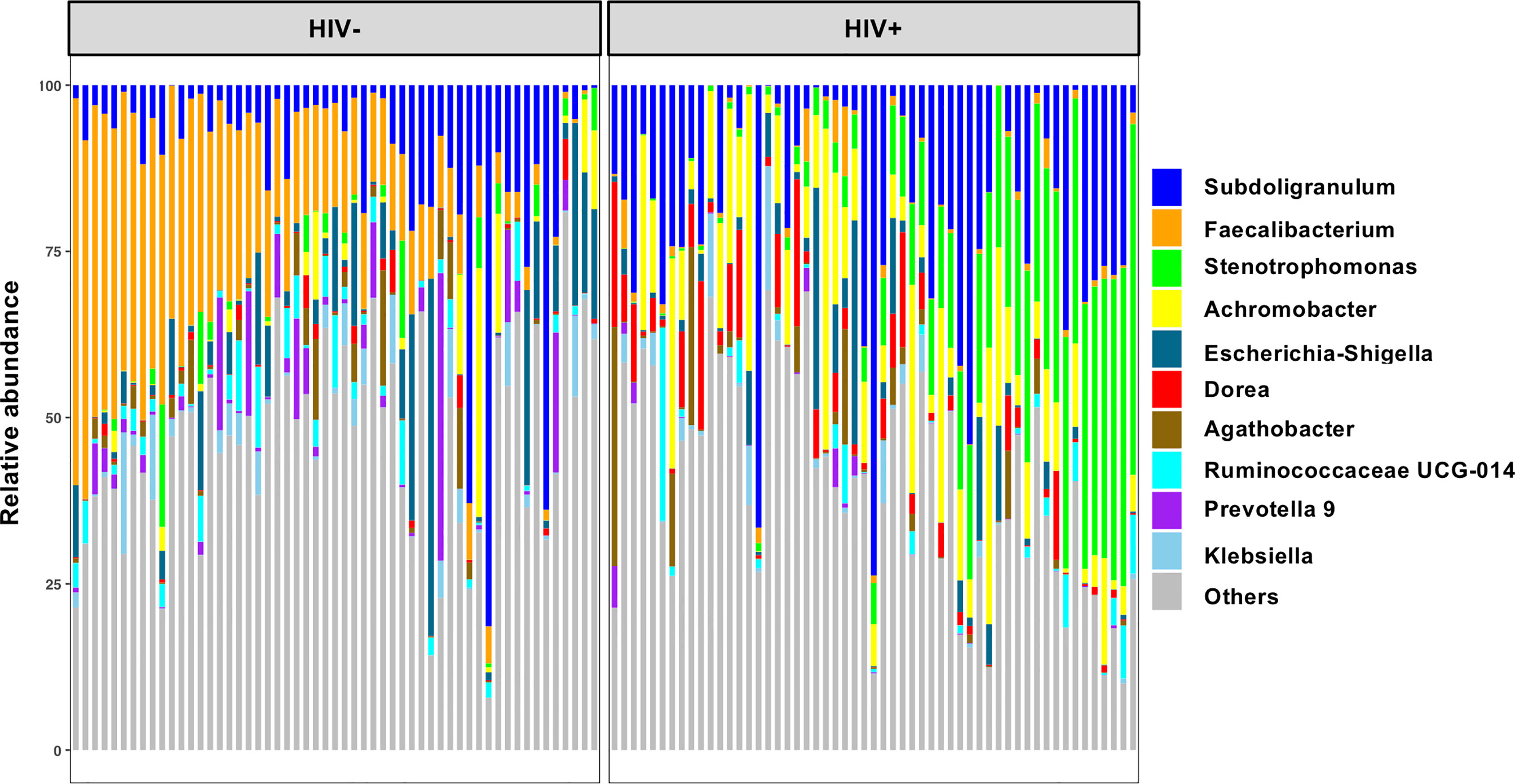
Figure 1 Top 10 abundant genera in fecal microbiome in all participants in the present study. Taxa bar plots showing the top 10 abundant genera in all participants (n = 110) are shown. Each bar represents frequencies of the genera in fecal microbiome of an individual. HIV-, HIV-1 uninfected individuals (n = 55); HIV+, HIV-1 infected individuals (n = 55).
Analyses of Chao’s richness and Fisher’s richness (Fisher et al., 1943; Chao, 1984) showed significantly lower richness in fecal microbiome of HIV+ than HIV- (Supplementary Figures 2A, B). Analyses of Shannon’s index and Faith’s phylogenic diversity (Shannon and Weaver, 1949; Faith, 1992) indicated significant reduction in alpha diversity in HIV+ compared to HIV- (Figures 2A, B). Comparison in alpha diversity between groups with low (< 200) and high (> 200) CD4 counts, short (< 2 years) and long (> 2 years) period of ART, or low (< 1,000 copies/ml) and high (> 1,000 copies/ml) plasma viral loads showed no significant difference in HIV-1 infected individuals (data not shown). However, a tendency of higher microbial richness in ART-treated HIV-1 non-controllers with > 1,000 copies/ml of plasma viral load than ART-treated HIV-1 controllers was observed (Figures 2C, D and Supplementary Figures 2C, D). While it has been indicated that different ART regimens may have different impacts on gut microbiome (Pinto-Cardoso et al., 2017; Ray et al., 2020; Imahashi et al., 2021), no significant difference in fecal microbiome richness or diversity was observed between individuals on ART with efavirenz and nevirapine in our cohort (Figures 2E, F and Supplementary Figures 2E, F).
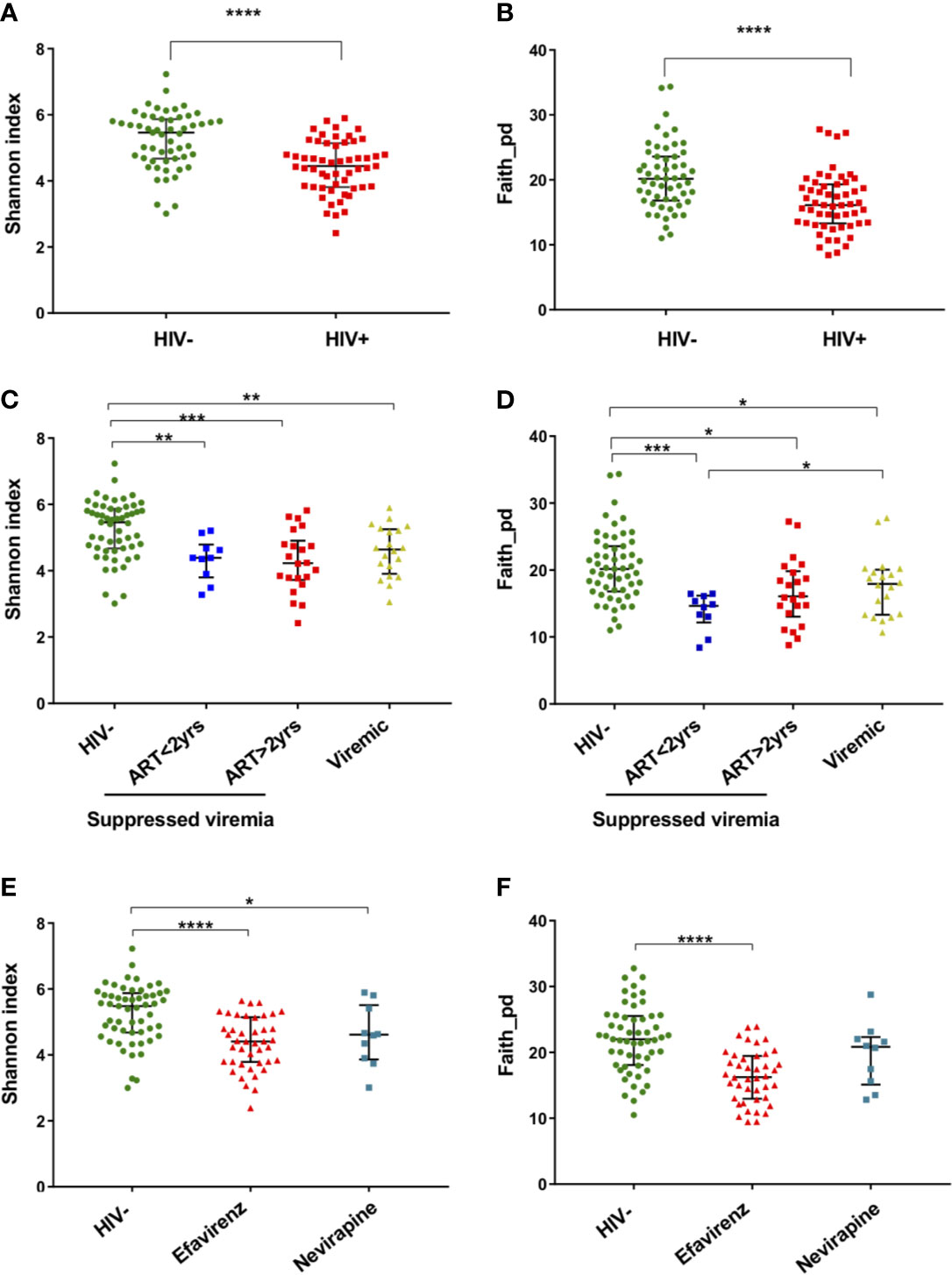
Figure 2 Alpha diversity of fecal microbiome in HIV-1 uninfected and infected Ghanaian adults. (A) Comparison of Shannon diversities of fecal microbiome between HIV- (n = 55) and HIV+ (n = 55). HIV- showed significantly higher diversity than HIV+ (p < 0.001). (B) Comparison of Faith phylogenetic diversities between HIV- and HIV+. HIV- showed significantly higher diversity than HIV+ (p < 0.001). (C) Comparison of Shannon diversities among HIV-, HIV-1 infected controllers (viral load < 1,000 copies/ml) under ART for less than 2 years (ART<2 yrs, n = 10), HIV-1 infected controllers under ART for more than 2 years (ART>2 yrs, n = 22), and HIV-1 infected non-controllers (viral load > 1,000 copies/ml) under ART (Viremic, n = 20). HIV- showed significantly higher diversity than ART<2yrs (p < 0.01), ART>2yrs (p < 0.005), and Viremic (p < 0.01), respectively. (D) Comparison of Faith phylogenetic diversities among HIV-, ART<2 yrs, ART>2 yrs, and Viremic. HIV- showed significantly higher diversity than ART<2yrs (p < 0.005), ART>2yrs (p < 0.05), and Viremic (p < 0.05), respectively. ART<2yrs showed significantly lower diversity than Viremic (p < 0.05). (E) Comparison of Shannon diversities among HIV-, HIV+ on ART with Efavirenz (n = 42), and HIV+ on ART with Nevirapine (n = 10). (F) Comparison of Faith phylogenetic diversities among HIV-, HIV+ with Efavirenz, and HIV+ with Nevirapine. Significant difference was determined by Wilcoxon rank sum test (A, B) or Kruskal Wallis test with Benjamini, Krieger and Yekutieli FDR correction (C–F); *, **, ***, and **** indicate significant differences with p < 0.05, p < 0.01, p < 0.005, and p < 0.001, respectively.
Beta-diversity (similarity/dissimilarity) in fecal microbiome was assessed by weighted Unifrac, Bray-Curtis, and robust Aitchison metrics (Sorensen, 1948; Lozupone et al., 2007; Martino et al., 2019). Principal coordinates analyses (PCoA) using these matrices revealed segregation of fecal microbiome compositions between HIV- and HIV+ (Figure 3). This was confirmed by ADONIS permutation-based statistical test (Anderson, 2001) showing significant differences based on HIV status with r2 of approximately 0.32, 0.11, and 0.16 in Aitchison, Bray-Curtis, and Weighted Unifrac PCoA, respectively (Table 2). The impact of difference in gender, age, CD4 counts or viral loads on beta-diversity in fecal microbiome was not clear (Table 2).
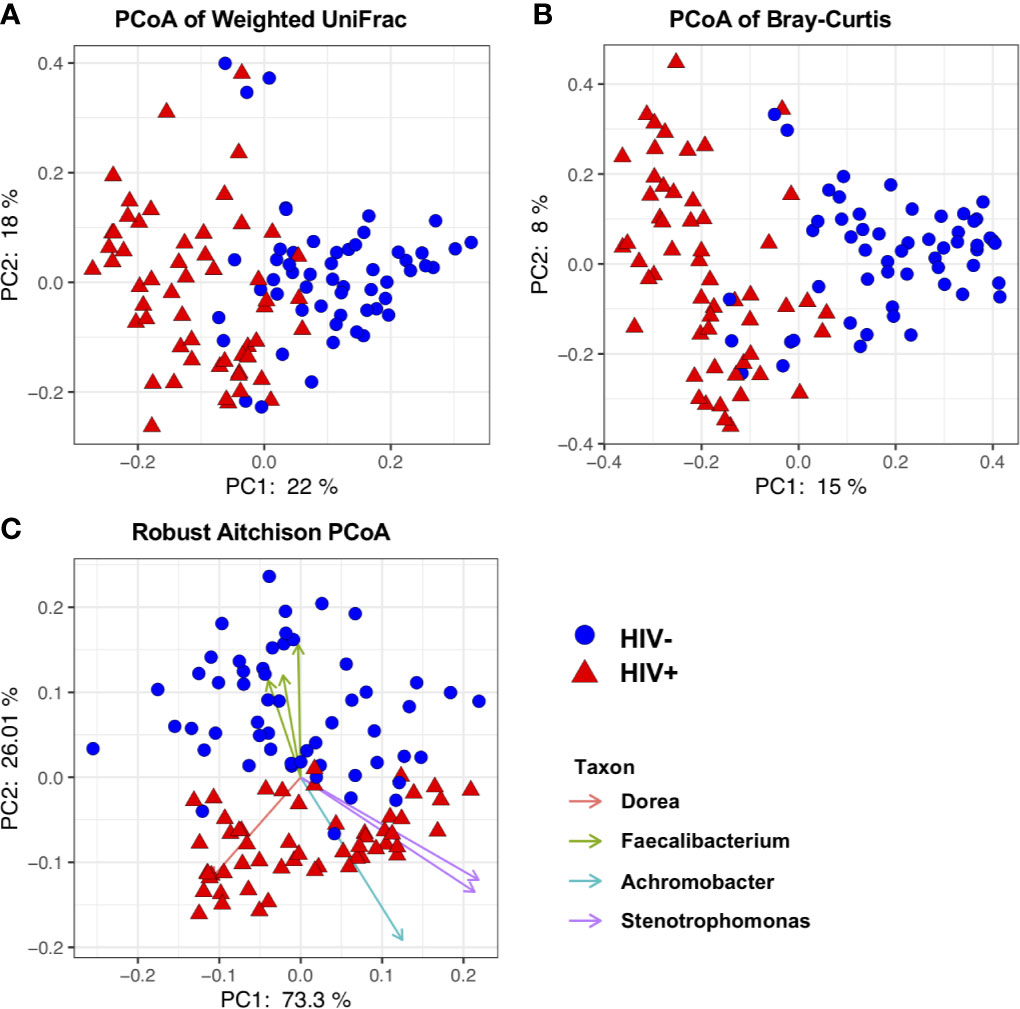
Figure 3 Beta diversity of fecal microbiome in HIV-1 uninfected and infected Ghanaian adults. (A) Principal Coordinates Analysis (PCoA) of weighted Unifrac distances in fecal microbiome in HIV- and HIV+ individuals. (B) PCoA of Bray-Curtis distances in HIV- and HIV+. (C) PCoA of Aitchison distances in HIV- and HIV+. Seven representative amplicon sequence variants (ASV) influencing the observed segregation are shown. These analyses indicate segregation between HIV- and HIV+.

Table 2 Summary of ADONIS permutation-based statistical test on beta diversity metrices.
Differences in taxa in fecal microbiome between HIV- and HIV+ were first determined by linear discriminant analysis (LDA) effect size (LEfSe) (Segata et al., 2011) (Figure 4). Firmicutes and Bacteroidetes were significantly more abundant in HIV- compared to HIV+. The abundance of Firmicutes was driven by the families of Ruminococcaceae, Veillonellaceae, and Clostridiaceae_1, while the abundance of Bacteroidetes was driven by the families of Prevotellaceae and Bacteroidaceae in HIV-. In contrast, Proteobacteria was significantly more abundant in fecal microbiome of HIV+ compared to HIV-. The abundance of Proteobacteria in HIV+ was driven by the families of Xanthomonadaceae and Burkholderiaceae. In addition, Streptococcaceae belonging to Firmicutes was abundant in HIV+.
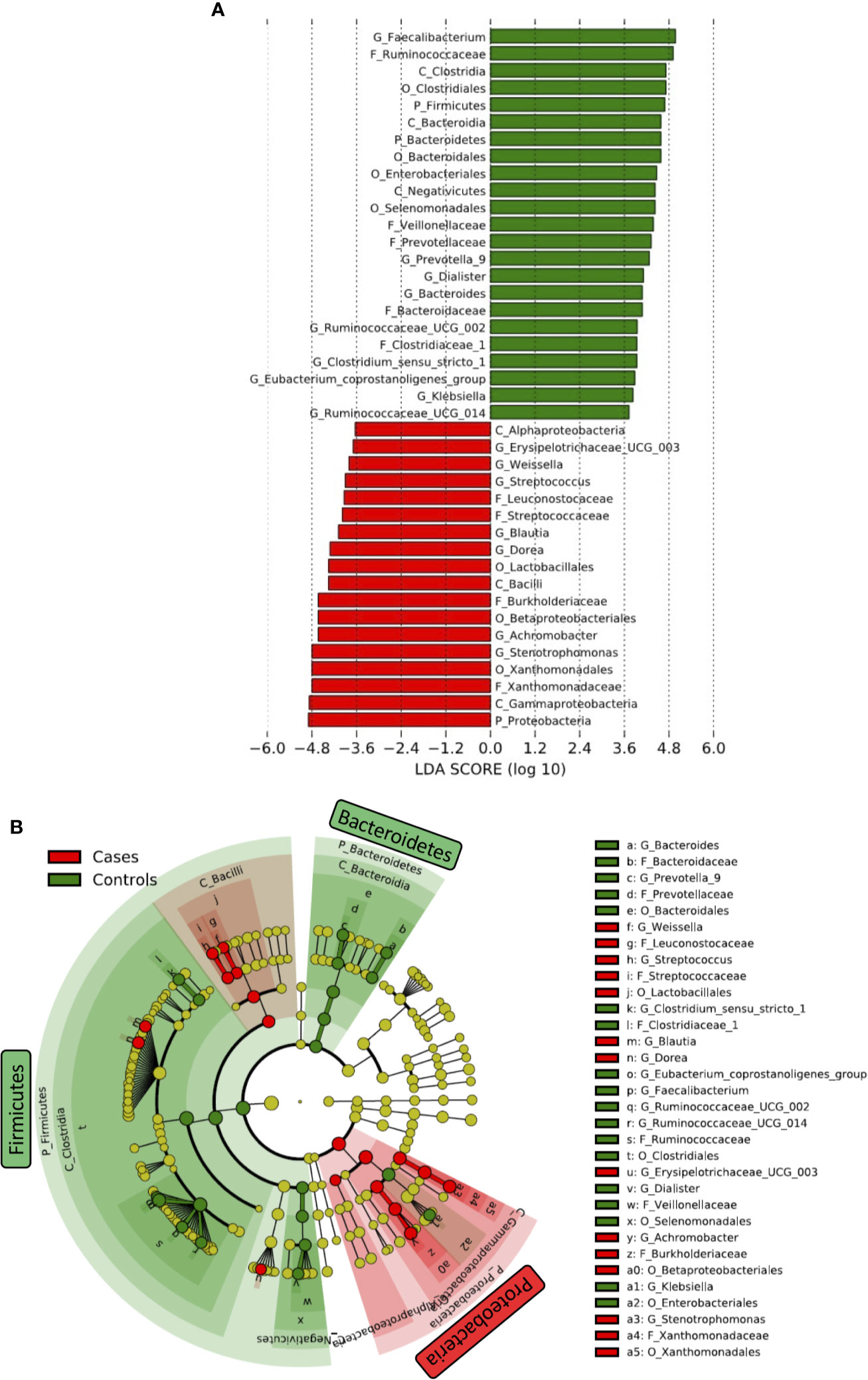
Figure 4 Difference in abundant taxa in fecal microbiome between HIV-1 uninfected and infected Ghanaian adults. (A) Data determined by linear discriminant analysis (LDA) effect size (LEfSe). Alpha value of 0.01 and 3.6 threshold on the logarithmic LDA score for discriminative features was used. P_, C_, O_, F_, G_ represent Phylum, Class, Order, Family, and Genus, respectively. (B) Cladogram. Green and Red indicate HIV- (Controls) and HIV+ (Cases), respectively.
We also determined several significant discriminating taxa at the genus level by LEfSe and analysis of composition of microbiome (ANCOM) (Mandal et al., 2015; Kaul et al., 2017) comparing HIV- and HIV+ (Figure 4 and Supplementary Figure 3). Faecalibacterium, [Eubacterium] coprostanoligenes group, Ruminococcaceae UCG-002, Bacteroides, Prevotella_9, Butyricimonas, Dialister, Phascolarctobacterium, Ruminococcaceae UCG-014, Parabacteroides, Ruminococcaceae UCG-005, Clostridium sensu_stricto_1, and Klebsiella were more abundant in HIV-. In contrast, Stenotrophomonas, Dorea, Achromobacter, Blautia, Streptococcus, Subdoligranulum, Erysipelotrichaceae UCG-003, Ochrobactrum, Weissella, and Gemella were more abundant in HIV+.
Plasma levels of I-FABP, sCD14, and LBP, markers for microbial translocation (Younas et al., 2019), were also examined. Analysis revealed significantly higher levels of I-FABP and sCD14 in HIV+ than in HIV- (Figure 5). However, there was no significant difference in plasma LBP levels between HIV- and HIV+. Correlation analysis found a strong positive correlation between plasma I-FABP and sCD14 levels (p < 0.0001, r = 0.55) (Figure 6). Plasma LBP levels did not show significant correlation with I-FABP or sCD14 but were correlated negatively with CD4 counts (p < 0.005, r = -0.38) and CD8 counts (p < 0.05, r = -0.32) and positively with plasma viral load (p < 0.05, r = 0.39) (Figure 6 and Supplementary Figure 4). These microbial translocation markers exhibited no significant correlation with alpha diversity of fecal microbiome (Figure 6). However, the alpha diversity showed a moderate but significant positive correlation with CD4 count (Faith_pd: p < 0.05, r = 0.28; Shannon: p < 0.05, r = 0.27) (Figure 6 and Supplementary Figure 4).
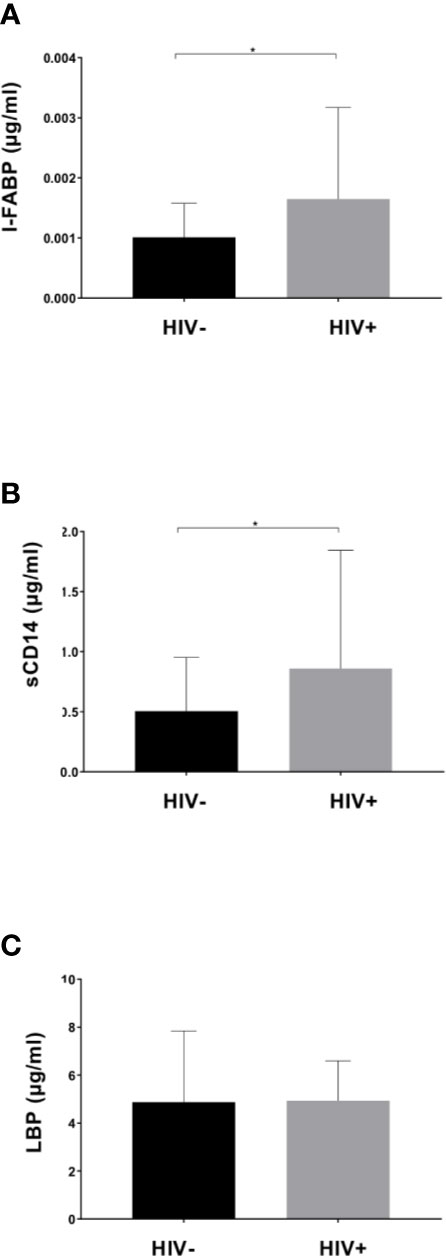
Figure 5 Comparison of plasma markers between HIV-1 uninfected and infected Ghanaian adults. Comparisons of plasma I-FABP (A), sCD14 (B), and LBP (C) levels are shown. HIV+ (n = 55) showed significantly higher I-FABP and sCD14 levels than uninfected HIV- (n = 55) (p < 0.05 [*]; Wilcoxon rank sum test).
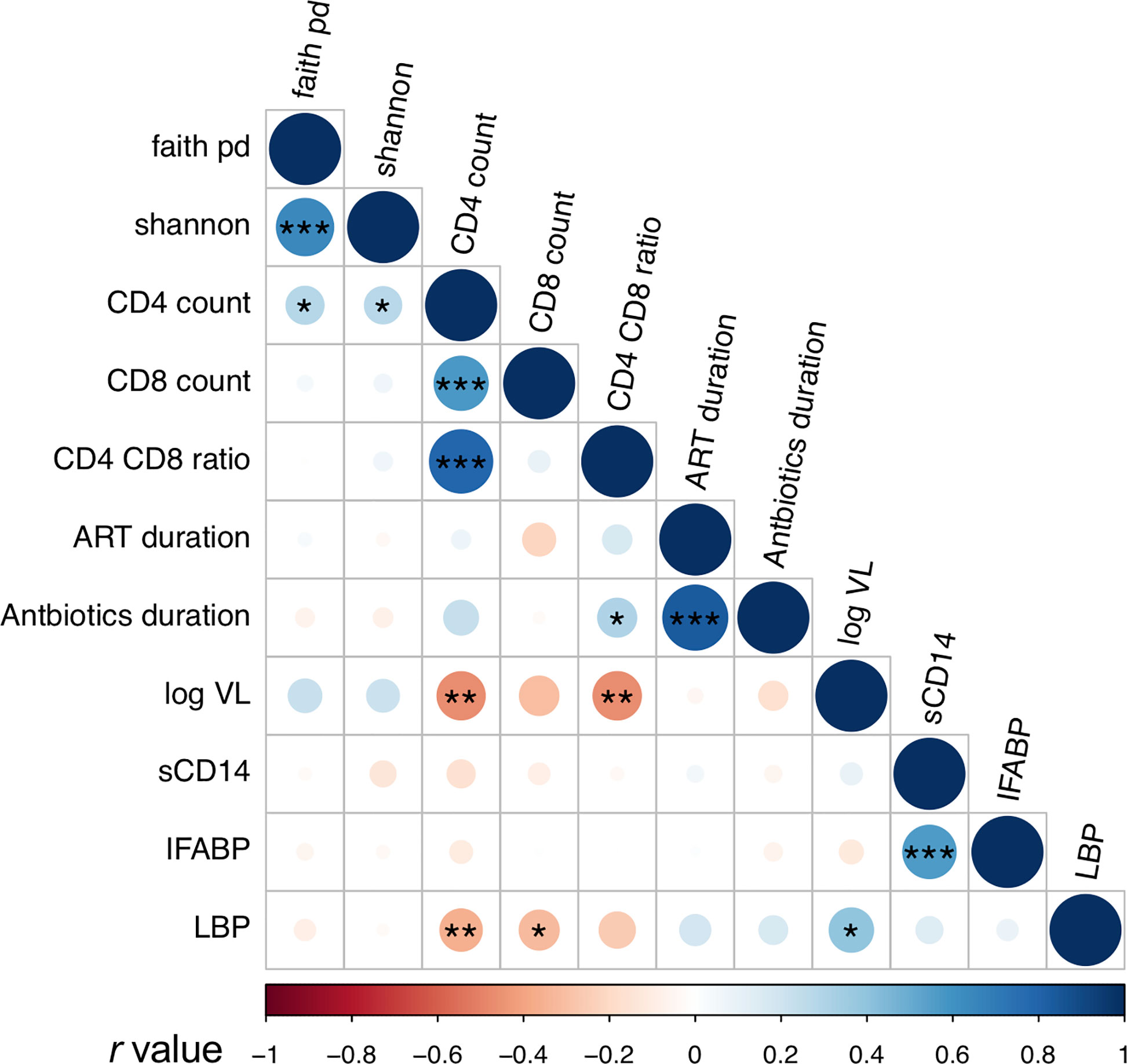
Figure 6 Analyses of correlation between fecal microbiome diversity and clinical markers. Asterisks indicate significant correlation determined by Spearman’s test; *, **, and *** indicate significant differences with p < 0.05, p < 0.01 and p < 0.005, respectively. Circle sizes are proportional to strength of correlation. Faith_pd, Faith’s phylogenetic diversity; shannon, Shannon’s index; ART_duration, duration under ART; Antibiotics_duration, duration since last Co-trimoxazole treatment; log_VL, viral load; sCD14, plasma soluble CD14 levels; IFABP, plasma intestinal fatty acid-binding protein levels; LBP, plasma lipopolysaccharide binding protein levels. n = 110 in faith_pd, shannon, sCD14, IFABP, and LBP; n = 55 (HIV+) in CD4 count, CD8 count, and CD4_CD8_ratio; n = 52 in ART_duration; n = 54 in Antibiotics_duration, n = 36 in log_VL (> 20 copies/ml).
In all the genera identified to be more abundant in HIV+, the relative abundance showed a trend of negative correlation with fecal microbiome alpha diversity and CD4 counts (Figure 7). In particular, strong inverse correlation was observed between relative abundance in Streptococcus and Gemella and CD4 counts (p < 0.001, r < -0.4) (Supplementary Figures 5 and 9). In contrast, the relative abundance in the genera identified to be more abundant in HIV- showed a trend of positive correlation with fecal microbiome alpha diversity and CD4 counts (Figure 7). In particular, relative abundance in Phascolarctobacterium, Faecalibacterium, Butyricimonas, and [Eubacterium] coprostanoligenes group showed strong positive correlation with fecal microbiome alpha diversity and CD4 counts (p < 0.001, r > 0.4) (Supplementary Figures 6–9).
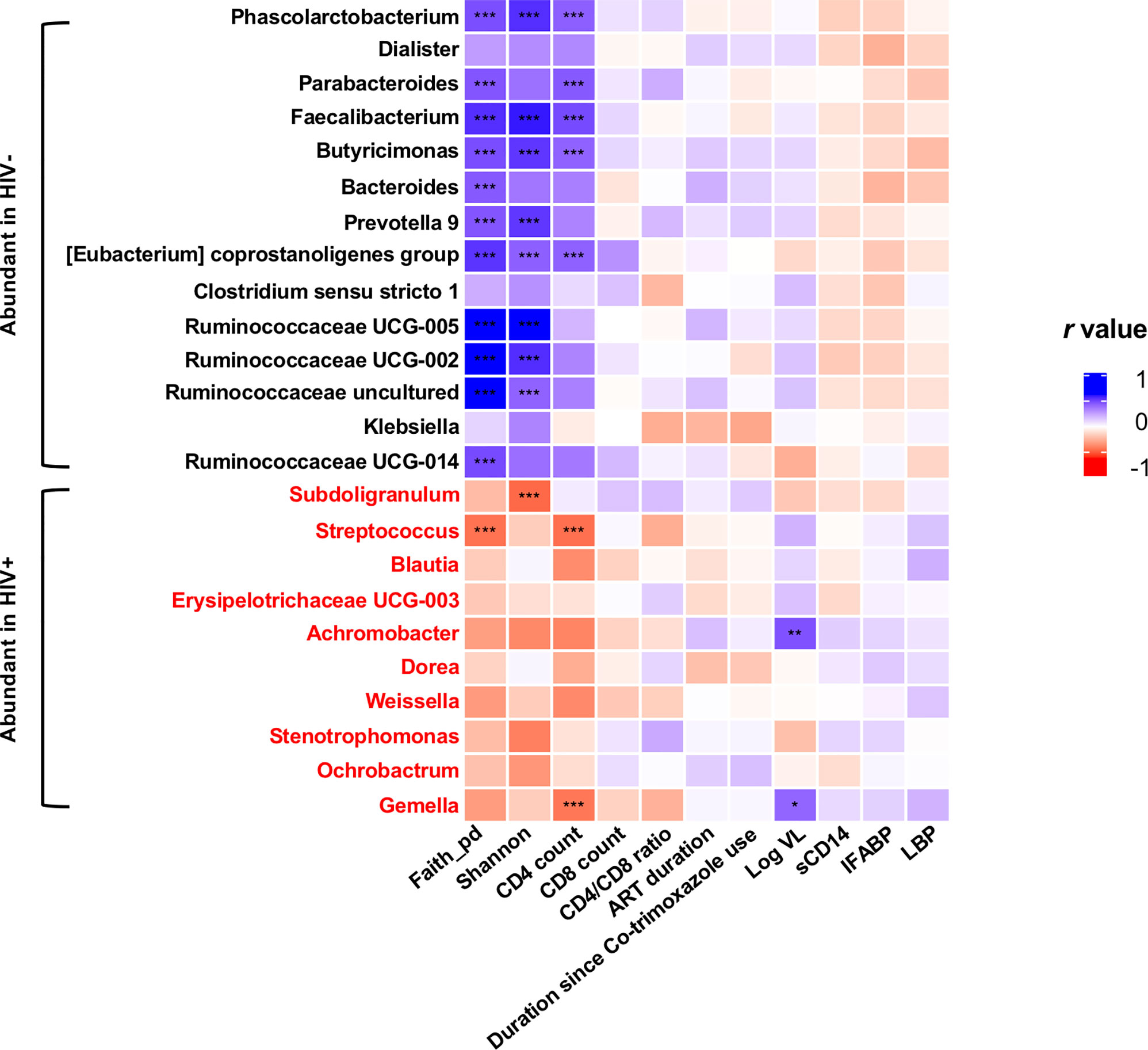
Figure 7 Analyses of correlation of genera showing significant difference in abundance in HIV- and HIV+ with clinical and immunological markers and microbiome diversities. Faith_pd, Faith’s phylogenetic diversity (n = 110); shannon, Shannon’s index (n = 110); CD4 count (n = 55); CD8 count (n = 55); CD4_CD8_ratio (n = 55); ART_duration, duration under ART (n = 52); Duration since Co-trimoxazole use (n = 54); Log VL, viral load (> 20 copies/ml) (n = 36); sCD14, plasma soluble CD14 levels (n = 110); IFABP, plasma intestinal fatty acid-binding protein levels (n = 110); LBP, plasma lipopolysaccharide binding protein levels (n = 110). Asterisks indicate significant correlation determined by Spearman’s test; *, **, and *** indicate significant differences with p < 0.05, p < 0.01, and p < 0.005, respectively. Significantly abundant genera were determined by ANCOM and LEfSE. Genera significantly abundant in HIV+ are shown by red.
Gut microbiota are known to be influenced by factors such as dietary behavior linked with socio-cultural and socio-economic status, which are different among populations (O’Keefe et al., 2007; Yatsunenko et al., 2012; Senghor et al., 2018; Hansen et al., 2019). It is thus important to obtain data on microbiome in individual populations. In sub-Saharan Africa, studies on gut microbiome in rural populations have indicated abundance in Prevotella which is associated with high fiber-carbohydrate diet (Yatsunenko et al., 2012; Schnorr et al., 2014; Morton et al., 2015; Monaco et al., 2016; Nowak et al., 2017; Senghor et al., 2018; Hansen et al., 2019). In industrialized populations, however, abundance in Bacteroides associated with high animal fat and protein has been indicated (Schnorr et al., 2014; Brewster et al., 2019). Individuals from suburban communities show abundance in Prevotella and Bacteroides, exhibiting both rural and urban features (Brewster et al., 2019). Our cohort consisting of individuals from peri-urban communities exhibited abundance in Prevotella and Bacteroides in fecal microbiome, suggesting the pattern of dietary habit in transition from rural to industrialized area (Parbie et al., 2020). The present study describes the characteristics of fecal microbiome of HIV-1 infected Ghanaians in comparison with the healthy seronegative Ghanaian control. Faecalibacterium (the most abundant genus) as well as Prevotella and Bacteroides were not included in the top 10 abundant taxa in HIV+, indicating loss of critical commensals in HIV-1 infected adults in our cohort.
Consistent with previous reports (Monaco et al., 2016; Nowak et al., 2017; Flygel et al., 2020), our analyses showed significant reduction in alpha diversity in HIV+ compared to HIV- (Figure 2). There was also a significant difference in beta diversity of fecal microbiome between HIV- and HIV+ (Figure 3 and Table 2). The difference indicated by weighted Unifrac distances (which incorporates phylogeny) was considered to be mainly attributed by enrichment of Proteobacteria and loss of Firmicutes and Bacteroidetes in HIV+ (Figure 4).
Several reports have indicated that microbiome diversity decreases after ART initiation but is recovered by prolonged ART treatment (Klase et al., 2015; Nowak et al., 2017; Flygel et al., 2020). This can explain our observation of a trend of higher alpha diversity in HIV-1 controllers under longer ART (> 2 years) compared to those under shorter ART (< 2 years) in the present study (Figures 2C, D and Supplementary Figures 2C, D). Unexpectedly, microbial richness in HIV-1 non-controllers under ART looked higher than HIV-1 controllers under ART (Figures 2C, D). One possible speculation is poor drug compliance in HIV-1 non-controllers.
The present study confirmed differences in fecal microbiome compositions between HIV+ and HIV- in Ghana. Our analysis showed significant increase in Proteobacteria but decrease in Firmicutes and Bacteroidetes at the phylum level in fecal microbiome of HIV-1 infected individuals in Ghana (Supplementary Figure 1), similarly with the previous observations in other countries (McHardy et al., 2013; Vujkovic-Cvijin et al., 2013; McHardy et al., 2013; Monaco et al., 2016; Vujkovic-Cvijin and Somsouk, 2019). Depletion of these bacteria groups can be associated with non-AIDS disease progression (Vujkovic-Cvijin et al., 2013). However, some bacteria belonging to Firmicutes such as Streptococcus were enriched in HIV+ (Figure 4). Although depletion of Lachnospiraceae including Dorea and Blautia in HIV-1 infected individuals have been reported in several cohorts (Mutlu et al., 2014; Vázquez-Castellanos et al., 2015; Vujkovic-Cvijin and Somsouk, 2019), Ghanaian HIV+ individuals exhibited enrichment of Dorea and Blautia (Figure 4). Furthermore, we found a depletion of Prevotella in HIV+ individuals in Ghana, whereas previous studies on gut microbiome have described enrichment of Prevotella in HIV-1-infected populations (Lozupone et al., 2013; Dillon et al., 2014; Mutlu et al., 2014; Vujkovic-Cvijin and Somsouk, 2019). Recently, enrichment of Prevotella has been attributed to sexual practice in MSM independent of HIV status (Armstrong et al., 2018; Vujkovic-Cvijin and Somsouk, 2019; Vujkovic-Cvijin et al., 2020). Since females are 78% of our cohorts, our results suggest a decrease in Prevotella in HIV+ non-MSM Ghanaians.
The alpha diversity of fecal microbiome exhibited a significant correlation with CD4 counts (Figure 7 and Supplementary Figures 4D, E). Relative abundance in the genera identified to be more abundant in HIV+ showed a trend of negative correlation with fecal microbiome alpha diversity and CD4 counts (Figure 7). In contrast, the relative abundance in the genera identified to be more abundant in HIV- showed a trend of positive correlation with fecal microbiome alpha diversity and CD4 counts. Enrichment of Streptococcus in fecal microbiome of HIV-1 infected individuals with low CD4 counts has been reported previously (Monaco et al., 2016; Shenoy et al., 2019). The inverse correlation of relative abundance in Gemella and Streptococcus with CD4 counts in the present study (Figure 7 and Supplementary Figures 5C, D) may imply association of these bacteria with immunosuppression (Shenoy et al., 2019; Ueberroth and Roxas, 2019). Furthermore, this study showed reduction of several butyrate producing bacteria including Faecalibacterium, Butyricimonas, and other members of Ruminococcaceae, known for their role in maintenance of gut homeostasis (Furusawa et al., 2013), in HIV+ as previously reported (Dillon et al., 2017). These results describing genera that exhibit increase or decrease associated with disease progression could be a fundamental data for our understanding of the pathogenesis in HIV-1 infection.
Potential confounders in gut microbiome studies include nutrition, body mass index (BMI) and antibiotic usage. Unfortunately, in our cohort, information on nutrition and BMI was unavailable, while HIV- participants who took antibiotics within 4 weeks prior to sample collection were excluded. Only one HIV+ participant had taken co-trimoxazole within 1 month prior to sample collection.
Most non-African cohorts in previous reports largely included MSM, but 86 of 110 individuals are female in our cohort (Lozupone et al., 2013; McHardy et al., 2013; Vujkovic-Cvijin et al., 2013; Dillon et al., 2014; Mutlu et al., 2014; Vujkovic-Cvijin and Somsouk, 2019). Hence, we looked for available data on a cohort including substantial number of females and chose a cohort in the Netherlands (Vujkovic-Cvijin et al., 2020) as a representative non-African cohort for comparison. We used data on 37 non-MSM males (HIV- = 20, HIV+ = 17) and 39 females (HIV- = 20, HIV+ = 19) from the Dutch cohort. Comparison of Shannon’s index of fecal microbiome in Ghana and Netherlands indicated that both HIV- and HIV+ groups in our study had significantly lower alpha diversity compared to their respective counterparts from Netherlands (Supplementary Figure 10A). These differences were not observed in Faith’s phylogenic diversity (Supplementary Figure 10B). However, reduction in diversity in HIV+ compared to HIV- was more distinct in our cohort than in the Netherlands in both Shannon’s index and Faith’s phylogenic diversity. Principal coordinates analyses of Weighted Unifrac distances revealed significant differences in beta diversity between Ghanaian and Dutch HIV- and between Ghanaian and Dutch HIV+ (Supplementary Figure 10C).
This is the first study, although descriptive, that analyzes the fecal microbiome in HIV-1 infected Ghanaian adults using high throughput metagenomic tools. Analysis revealed significant difference in fecal microbiome diversity and compositions between HIV-1 infected and uninfected individuals. Our results describing the feature of fecal microbiome in Ghana provide valuable data in West Africa, where data on enteric microbiome has not yet been systematically accumulated, and thus contribute to understanding of the interaction between HIV-1 and enteric microbiota in a population specific manner.
The data and materials supporting the finding in this study are available within the publication or can be obtained upon request to the corresponding author. The sequences obtained by NGS were deposited in the DNA Data Bank of Japan (DDBJ) (https://ddbj.nig.ac.jp/DRASearch; accession number: DRA010770).
The studies involving human participants were reviewed and approved by The Institutional Review Board of Noguchi Memorial Institute for Medical Research (NMIMR) and the Ethical Committee of National Institute of Infectious Diseases (NIID). The patients/participants provided their written informed consent to participate in this study.
PP, TMi, KI, WA, and TMa conceived and designed the experiments. SO and WA contributed to sample collection. PP, TMi, AI, AK-T, SS, CA, DK, and EB performed the experiments. PP, TMi, AI, AK-T, LR, SU, SI, YK, WA, and TMa analyzed the data. PP, TMi, HK, KI, WA, and TMa wrote the paper. All authors contributed to the article and approved the submitted version.
This study was supported by Japan Agency for Medical Research & Development (AMED) (grant number: JP18fk0410003, JP20fk0410011, JP20fk0108125, JP20fk0108139, JP19kk0205024, and JP20jk0210002), AMED-JICA (the Science and Technology Research Partnership for Sustainable Development [SATREPS]; JP20jm0110012), and the Ministry of Education, Culture, Sports, Science, and Technology in Japan (18H02666). The Kumamoto University International Scholarship Program generously provided travel support during this study.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
We thank the staff of NMIMR-Virology Department, RHK, and NIID-ARC for their administrative and technical support. We are sincerely grateful to all the healthy uninfected and HIV-1 infected individuals who consented to participate in this study.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fcimb.2021.646467/full#supplementary-material
Anderson, M. J. (2001). A New Method for non-Parametric Multivariate Analysis of Variance. Austral Ecology 26, 32–46. doi: 10.1111/j.1442-9993.2001.01070.pp.x
Antiretroviral Therapy Cohort Collaboration (2010). Causes of Death in HIV-1 Infected Patients Treated With Antiretroviral Therapy 1996-2006: Collaborative Analysis of 13 HIV Cohort Studies. Clin. Infect. Dis. 50, 1387–1396. doi: 10.1086/652283
Armstrong, A. J., Shaffer, M., Nusbacher, N. M., Griesmer, C., Fiorillo, S., Schneider, J. M., et al. (2018). An Exploration of Prevotella-rich Microbiomes in HIV and Men Who Have Sex With Men. Microbiome 6, 198. doi: 10.1186/s40168-018-0580-7
Asquith, M., Sternes, P. R., Costello, M. E., Karstens, L., Diamond, S., Martin, T. M., et al. (2019). Hla Alleles Associated With Risk of Ankylosing Spondylitis and Rheumatoid Arthritis Influence the Gut Microbiome. Arthritis Rheumatol. 71, 1642–1650. doi: 10.1002/art.40917
Bolyen, E., Rideout, J. R., Dillon, M. R., Bokulich, N. A., Abnet, C. C., Al-Ghalith, G. A., et al. (2019). Author Correction: Reproducible, Interactive, Scalable and Extensible Microbiome Data Science Using Qiime 2. Nat. Biotechnol. 37, 1091. doi: 10.1038/s41587-019-0252-6
Brenchley, J. M., Price, D. A., Schacker, T. W., Asher, T. E., Silvestri, G., Rao, S., et al. (2006). Microbial Translocation is a Cause of Systemic Immune Activation in Chronic Hiv Infection. Nat. Med. 12, 1365–1371. doi: 10.1038/nm1511
Brewster, R., Tamburini, F. B., Asiimwe, E., Oduaran, O., Hazelhurst, S., Bhatt, A. S. (2019). Surveying Gut Microbiome Research in Africans: Toward Improved Diversity and Representation. Trends Microbiol. 27, 824–835. doi: 10.1016/j.tim.2019.05.006
Callahan, B. J., McMurdie, P. J., Rosen, M. J., Han, A. W., Johnson, A. J. A., Holmes, S. P. (2016). Dada2: High-resolution Sample Inference From Illumina Amplicon Data. Nat. Methods 13, 581–583. doi: 10.1038/nmeth.3869
Chao, A. (1984). Nonparametric Estimation of the Number of Classes in a Population. Scand. J. Stat. 1, 265–270. doi: 10.2307/4615964
D’Ettorre, G., Ceccarelli, G., Giustini, N., Serafino, S., Calantone, N., De Girolamo, G., et al. (2015). Probiotics Reduce Inflammation in Antiretroviral Treated, HIV-infected Individuals: Results of the “Probio-Hiv” clinical trial. PloS One 10, e0137200. doi: 10.1371/journal.pone.0137200
Dieffenbach, C. W., Fauci, A. S. (2011). Thirty Years of HIV and AIDS: Future Challenges and Opportunities. Ann. Intern. Med. 154, 766–771. doi: 10.7326/0003-4819-154-11-201106070-00345
Dillon, S. M., Kibbie, J., Lee, E. J., Guo, K., Santiago, M. L., Austin, G. L., et al. (2017). Low Abundance of Colonic Butyrate-Producing Bacteria in HIV Infection is Associated With Microbial Translocation and Immune Activation. AIDS 31, 511–521. doi: 10.1097/QAD.0000000000001366
Dillon, S. M., Lee, E. J., Kotter, C. V., Austin, G. L., Dong, Z., Hecht, D. K., et al. (2014). An Altered Intestinal Mucosal Microbiome in HIV-1 Infection is Associated With Mucosal and Systemic Immune Activation and Endotoxemia. Mucosal Immunol. 7, 983–994. doi: 10.1038/mi.2013.116
Fabian, P., Varoquaux, G., Gramfort, A., Michel, V., Thirion, B., Olivier, G., et al. (2011). Scikit-Learn: Machine Learning in Python. J. Mach. Learn Res. 12, 2825–2830. Available at: http://jmlr.org/papers/v12/pedregosa11a.html.
Faith, D. P. (1992). Conservation Evaluation and Phylogenetic Diversity. Biol. Conserv. 61, 1–10. doi: 10.1016/0006-3207(92)91201-3
Fisher, R. A., Corbet, A. S., Williams, C. B. (1943). The Relation Between the Number of Species and the Number of Individuals in a Random Sample of an Animal Population. J. Anim. Ecol. 1, 42–58. doi: 10.2307/1411
Flygel, T. T., Sovershaeva, E., Classen-Weitz, S., Hjerde, E., Mwaikono, K. S., Odland, J.Ø., et al. (2020). Composition of Gut Microbiota of Children and Adolescents With Perinatal Hiv Infection Taking Antiretroviral Therapy in Zimbabwe. J. Infect. Dis. 221, 483s–492. doi: 10.1093/infdis/jiz473
Furusawa, Y., Obata, Y., Fukuda, S., Endo, T. A., Nakato, G., Takahashi, D., et al. (2013). Commensal Microbe-Derived Butyrate Induces the Differentiation of Colonic Regulatory T Cells. Nature 504, 446–450. doi: 10.1038/nature12721
Gill, T., Asquith, M., Brooks, S. R., Rosenbaum, J. T., Colbert, R. A. (2018). Effects of HLA–B27 on Gut Microbiota in Experimental Spondyloarthritis Implicate an Ecological Model of Dysbiosis. Arthritis Rheumatol. 70, 555–565. doi: 10.1002/art.40405
Gootenberg, D. B., Paer, J. M., Luevano, J. M., Kwon, D. S. (2017). Hiv-Associated Changes in the Enteric Microbial Community: Potential Role in Loss of Homeostasis and Development of Systemic Inflammation. Curr. Opin. Infect. Dis. 30, 31–43. doi: 10.1097/QCO.0000000000000341
Hansen, M. E., Rubel, M. A., Bailey, A. G., Ranciaro, A., Thompson, S. R., Campbell, M. C., et al. (2019). Population Structure of Human Gut Bacteria in a Diverse Cohort From Rural Tanzania and Botswana. Genome Biol. 20, 16. doi: 10.1186/s13059-018-1616-9
Imahashi, M., Ode, H., Kobayashi, A., Nemoto, M., Matsuda, M., Hashiba, C., et al. (2021). Impact of Long-Term Antiretroviral Therapy on Gut and Oral Microbiotas in HIV-1-infected Patients. Sci. Rep. 11, 960. doi: 10.1038/s41598-020-80247-8
Irvine, S. L., Hummelen, R., Hekmat, S. (2011). Probiotic Yogurt Consumption may Improve Gastrointestinal Symptoms, Productivity, and Nutritional Intake of People Living With Human Immunodeficiency Virus in Mwanza, Tanzania. Nutr. Res. 31, 875–881. doi: 10.1016/j.nutres.2011.10.005
Kaul, A., Mandal, S., Davidov, O., Peddada, S. D. (2017). Analysis of Microbiome Data in the Presence of Excess Zeros. Front. Microbiol. 8, 2114. doi: 10.3389/fmicb.2017.02114
Kehrmann, J., Menzel, J., Saeedghalati, M., Obeid, R., Schulze, C., Holzendorf, V., et al. (2019). Gut Microbiota in Human Immunodeficiency Virus-Infected Individuals Linked to Coronary Heart Disease. J. Infect. Dis. 219, 497–508. doi: 10.1093/infdis/jiy524
Kim, S. W., Suda, W., Kim, S., Oshima, K., Fukuda, S., Ohno, H., et al. (2013). Robustness of Gut Microbiota of Healthy Adults in Response to Probiotic Intervention Revealed by High-Throughput Pyrosequencing. DNA Res. 20, 241–253. doi: 10.1093/dnares/dst006
Klase, Z., Ortiz, A., Deleage, C., Mudd, J., Quiñones, M., Schwartzman, E., et al. (2015). Dysbiotic Bacteria Translocate in Progressive Siv Infection. Mucosal Immunol. 8, 1009–1020. doi: 10.1038/mi.2014.128
Klatt, N. R., Canary, L. A., Sun, X., Vinton, C. L., Funderburg, N. T., Morcock, D. R., et al. (2013). Probiotic/Prebiotic Supplementation of Antiretrovirals Improves Gastrointestinal Immunity in SIV-infected Macaques. J. Clin. Invest. 123, 903–907. doi: 10.1172/JCI66227
Lozupone, C. A., Hamady, M., Kelley, S. T., Knight, R. (2007). Quantitative and Qualitative β Diversity Measures Lead to Different Insights Into Factors That Structure Microbial Communities. Appl. Environ. Microbiol. 73, 1576–1585. doi: 10.1128/AEM.01996-06
Lozupone, C. A., Li, M., Campbell, T. B., Flores, S. C., Linderman, D., Gebert, M. J., et al. (2013). Alterations in the Gut Microbiota Associated With HIV-1 Infection. Cell Host Microbe 14, 329–339. doi: 10.1016/j.chom.2013.08.006
Mallick, H., McIver, L. J., Rahnavard, A., Ma, S., Zhang, Y., Nguyen, L. H., et al. (2021) Multivariable Association in Population-scale Meta-Omics Studies. doi: 10.1101/2021.01.20.427420
Mandal, S., Van Treuren, W., White, R. A., Eggesbø, M., Knight, R., Peddada, S. D. (2015). Analysis of Composition of Microbiomes: A Novel Method for Studying Microbial Composition. Microb. Ecol. Health Dis. 26, 27663. doi: 10.3402/mehd.v26.27663
Marchetti, G., Cozzi-Lepri, A., Merlini, E., Bellistrì, G. M., Castagna, A., Galli, M., et al. (2011). Microbial Translocation Predicts Disease Progression of HIV-infected Antiretroviral-Naïve Patients With High Cd4+ Cell Count. AIDS. 25, 1385–1394. doi: 10.1097/QAD.0b013e3283471d10
Martino, C., Morton, J. T., Marotz, C. A., Thompson, L. R., Tripathi, A., Knight, R., et al. (2019). A Novel Sparse Compositional Technique Reveals Microbial Perturbations. MSystems. 4, e00016–e00019. doi: 10.1128/mSystems.00016-19
McHardy, I. H., Li, X., Tong, M., Ruegger, P., Jacobs, J., Borneman, J., et al. (2013). Hiv Infection is Associated With Compositional and Functional Shifts in the Rectal Mucosal Microbiota. Microbiome 1, 26. doi: 10.1186/2049-2618-1-26
Monaco, C. L., Gootenberg, D. B., Zhao, G., Handley, S. A., Ghebremichael, M. S., Lim, E. S., et al. (2016). Altered Virome and Bacterial Microbiome in Human Immunodeficiency Virus-Associated Acquired Immunodeficiency Syndrome. Cell Host Microbe 19, 311–322. doi: 10.1016/j.chom.2016.02.011
Morita, H., Kuwahara, T., Ohshima, K., Sasamoto, H., Itoh, K., Hattori, M., et al. (2007). An Improved Dna Isolation Method for Metagenomic Analysis of the Microbial Flora of the Human Intestine. Microbes Environ. 22, 214–222. doi: 10.1264/jsme2.22.214
Morton, E. R., Lynch, J., Froment, A., Lafosse, S., Heyer, E., Przeworski, M., et al. (2015). Variation in Rural African Gut Microbiota is Strongly Correlated With Colonization by Entamoeba and Subsistence. PloS Genet. 11, e1005658. doi: 10.1371/journal.pgen.1005658
Mudd, J. C., Brenchley, J. M. (2016). Gut Mucosal Barrier Dysfunction, Microbial Dysbiosis, and Their Role in HIV-1 Disease Progression. J. Infect. Dis. 214, S58–S66. doi: 10.1093/infdis/jiw258
Mutlu, E. A., Keshavarzian, A., Losurdo, J., Swanson, G., Siewe, B., Forsyth, C., et al. (2014). A Compositional Look At the Human Gastrointestinal Microbiome and Immune Activation Parameters in HIV Infected Subjects. PloS Pathog. 10, e1003829. doi: 10.1371/journal.ppat.1003829
Nowak, R. G., Bentzen, S. M., Ravel, J., Crowell, T. A., Dauda, W., Ma, B., et al. (2017). Rectal Microbiota Among HIV-uninfected, Untreated HIV, and Treated HIV-Infected in Nigeria. AIDS. 31, 857–862. doi: 10.1097/QAD.0000000000001409
Nwosu, F. C., Avershina, E., Wilson, R., Rudi, K. (2014). Gut Microbiota in HIV Infection: Implication for Disease Progression and Management. Gastroenterol. Res. Pract. 2014, 1–6. doi: 10.1155/2014/803185
O’Keefe, S. J., Chung, D., Mahmoud, N., Sepulveda, A. R., Manafe, M., Arch, J., et al. (2007). Why do African Americans Get More Colon Cancer Than Native Africans? J. Nutr. 137, 175S–182S. doi: 10.1093/jn/137.1.175S
Ortiz, A. M., Klase, Z. A., Dinapoli, S. R., Vujkovic-Cvijin, I., Carmack, K., Perkins, M. R., et al. (2016). Il-21 and Probiotic Therapy Improve Th17 Frequencies, Microbial Translocation, and Microbiome in ARV-treated, Siv-Infected Macaques. Mucosal Immunol. 9, 458–467. doi: 10.1038/mi.2015.75
Parbie, P. K., Mizutani, T., Ishizaka, A., Kawana-Tachikawa, A., Runtuwene, L. R., Seki, S., et al. (2020). Fecal Microbiome Composition in Healthy Adults in Ghana. Jpn J. Infect. Dis. 74, 42–47. doi: 10.7883/yoken.JJID.2020.469
Pinto-Cardoso, S., Lozupone, C., Briceño, O., Alva-Hernández, S., Téllez, N., Adriana, A., et al. (2017). Fecal Bacterial Communities in Treated Hiv Infected Individuals on Two Antiretroviral Regimens. Sci. Rep. 7, 43741. doi: 10.1038/srep43741
Quast, C., Pruesse, E., Yilmaz, P., Gerken, J., Schweer, T., Yarza, P., et al. (2012). The SILVA Ribosomal RNA Gene Database Project: Improved Data Processing and Web-Based Tools. Nucleic Acids Res. 41, D590–D596. doi: 10.1093/nar/gks1219
Ray, S., Narayanan, A., Giske, C. G., Neogi, U., Sönnerborg, A., Nowak, P. (2020). Altered Gut Microbiome Under Antiretroviral Therapy: Impact of Efavirenz and Zidovudine. ACS Infect. Dis. doi: 10.1021/acsinfecdis.0c00536
Sandler, N. G., Douek, D. C. (2012). Microbial Translocation in HIV Infection: Causes, Consequences and Treatment Opportunities. Nat. Rev. Microbiol. 10, 655–666. doi: 10.1038/nrmicro2848
Schnorr, S. L., Candela, M., Rampelli, S., Centanni, M., Consolandi, C., Basaglia, G., et al. (2014). Gut Microbiome of the Hadza Hunter-Gatherers. Nat. Commun. 5, 3654. doi: 10.1038/ncomms4654
Segata, N., Izard, J., Waldron, L., Gevers, D., Miropolsky, L., Garrett, W. S., et al. (2011). Metagenomic Biomarker Discovery and Explanation. Genome Biol. 12, R60. doi: 10.1186/gb-2011-12-6-r60
Senghor, B., Sokhna, C., Ruimy, R., Lagier, J. C. (2018). Gut Microbiota Diversity According to Dietary Habits and Geographical Provenance. Hum. Microbiom J. 7, 1–9. doi: 10.1016/j.humic.2018.01.001
Shannon, C. E., Weaver, W. (1949). The Mathematical Theory of Communication (Illonois: University of Illonois Press).
Shenoy, M. K., Fadrosh, D. W., Lin, D. L., Worodria, W., Byanyima, P., Musisi, E., et al. (2019). Gut Microbiota in HIV–pneumonia Patients is Related to Peripheral Cd4 Counts, Lung Microbiota, and In Vitro Macrophage Dysfunction. Microbiome. 7, 37. doi: 10.1186/s40168-019-0651-4
Sorensen, T. (1948). A Method of Establishing Groups of Equal Amplitude in Plant Sociology Based on Similarity of Species Content. Det Kongelige Danske Videnskabernes Selskab Biologiske Skrifter 5, 1–34.
Ueberroth, B. E., Roxas, R. (2019). Gemella Morbillorum Isolated From a Pelvic Abscess in an HIV-positive Patient With Squamous Cell Carcinoma of the Perianal Region. BMJ Case Rep. 12, e227352. doi: 10.1136/bcr-2018-227352
UNAIDS (2020)Ghana 2019. In: Country Fact Sheets. Available at: https://www.unaids.org/en/regionscountries/countries/ghana (Accessed December 07, 2020).
Vázquez-Castellanos, J. F., Serrano-Villar, S., Latorre, A., Artacho, A., Ferrus, M. L., Madrid, N., et al. (2015). Altered Metabolism of Gut Microbiota Contributes to Chronic Immune Activation in HIV-infected Individuals. Mucosal Immunol. 8, 760–772. doi: 10.1038/mi.2014.107
Vujkovic-Cvijin, I., Dunham, R. M., Iwai, S., Maher, M. C., Albright, R. G., Broadhurst, M. J., et al. (2013). Dysbiosis of the Gut Microbiota is Associated With HIV Disease Progression and Tryptophan Catabolism. Sci. Transl. Med. 5, 193ra91. doi: 10.1126/scitranslmed.3006438
Vujkovic-Cvijin, I., Somsouk, M. (2019). HIV and the Gut Microbiota: Composition, Consequences, and Avenues for Amelioration. Curr. HIV/AIDS Rep. 16, 204–123. doi: 10.1007/s11904-019-00441-w
Vujkovic-Cvijin, I., Sortino, O., Verheij, E., Sklar, J., Wit, F. W., Kootstra, N. A., et al. (2020). Hiv-Associated Gut Dysbiosis is Independent of Sexual Practice and Correlates With Noncommunicable Diseases. Nat. Commun. 11, 2448. doi: 10.1038/s41467-020-16222-8
West, C. E., Renz, H., Jenmalm, M. C., Kozyrskyj, A. L., Allen, K. J., Vuillermin, P., et al. (2015). The Gut Microbiota and Inflammatory Noncommunicable Diseases: Associations and Potentials for Gut Microbiota Therapies. J. Allergy Clin. Immunol. 135, 3–13. doi: 10.1016/j.jaci.2014.11.012
Yatsunenko, T., Rey, F. E., Manary, M. J., Trehan, I., Dominguez-Bello, M. G., Contreras, M., et al. (2012). Human Gut Microbiome Viewed Across Age and Geography. Nature. 486, 222–227. doi: 10.1038/nature11053
Keywords: HIV-1, gut microbiome, dysbiosis, sub‐Saharan Africa, Ghana
Citation: Parbie PK, Mizutani T, Ishizaka A, Kawana-Tachikawa A, Runtuwene LR, Seki S, Abana CZ-Y, Kushitor D, Bonney EY, Ofori SB, Uematsu S, Imoto S, Kimura Y, Kiyono H, Ishikawa K, Ampofo WK and Matano T (2021) Dysbiotic Fecal Microbiome in HIV-1 Infected Individuals in Ghana. Front. Cell. Infect. Microbiol. 11:646467. doi: 10.3389/fcimb.2021.646467
Received: 27 December 2020; Accepted: 19 April 2021;
Published: 18 May 2021.
Edited by:
Wayne Young, AgResearch Ltd, New ZealandReviewed by:
Scott Hazelhurst, University of the Witwatersrand, South AfricaCopyright © 2021 Parbie, Mizutani, Ishizaka, Kawana-Tachikawa, Runtuwene, Seki, Abana, Kushitor, Bonney, Ofori, Uematsu, Imoto, Kimura, Kiyono, Ishikawa, Ampofo and Matano. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: William K. Ampofo, V0FtcG9mb0Bub2d1Y2hpLnVnLmVkdS5naA==; Tetsuro Matano, dG1hdGFub0BuaWguZ28uanA=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.