 Grzegorz Czerwonka
Grzegorz Czerwonka Dawid Gmiter
Dawid Gmiter- Institute of Biology, Jan Kochanowski University in Kielce, Kielce, Poland
Proteus mirabilis is a pathogenic, Gram-negative, rod-shaped bacterium that causes ascending urinary tract infections. Swarming motility, urease production, biofilm formation, and the properties of its lipopolysaccharide (LPS) are all factors that contribute to the virulence of this bacterium. Uniquely, members of the O18 serogroup elaborate LPS molecules capped with O antigen polymers built of pentasaccharide repeats; these repeats are modified with a phosphocholine (ChoP) moiety attached to the proximal sugar of each O unit. Decoration of the LPS with ChoP is an important surface modification of many pathogenic and commensal bacteria. The presence of ChoP on the bacterial envelope is correlated with pathogenicity, as decoration with ChoP plays a role in bacterial adhesion to mucosal surfaces, resistance to antimicrobial peptides and sensitivity to complement-mediated killing in several species. The genome of P. mirabilis O18 is 3.98 Mb in size, containing 3,762 protein-coding sequences and an overall GC content of 38.7%. Annotation performed using the RAST Annotation Server revealed genes associated with choline phosphorylation, uptake and transfer. Moreover, amino acid sequence alignment of the translated licC gene revealed it to be homologous to LicC from Streptococcus pneumoniae encoding CTP:phosphocholine cytidylyltransferase. Recognized homologs are located in the O antigen gene clusters of Proteus species, near the wzx gene encoding the O antigen flippase, which translocates lipid-linked O units across the inner membrane. This study reveals the genes potentially engaged in LPS decoration with ChoP in P. mirabilis O18.
Introduction
Proteus mirabilis is an opportunistic Gram-negative bacterial pathogen that swarms across solid surfaces, which often leads to catheter-associated urinary tract infections. The hindered eradication of P. mirabilis results in recurrent urinary tract infections (Schaffer and Pearson, 2015). Flagella-based swarming motility and biofilm formation, as well as the production of urease, hemolysin, and lipopolysaccharide (LPS) endotoxin, all contribute to the virulence of P. mirabilis. The presence of a long-chain O antigen is important for resistance to normal serum, and is involved in swarming motility as well as the formation of the cell-surface glycocalyx (Knirel et al., 2011). The O antigen consists of repeating polysaccharide units (O units) that typically contain two to eight sugar residues that define the serological specificity of Gram-negative bacteria (Liu et al., 2014). Due to the high structural diversity of P. mirabilis O antigens, infections by different serotypes may activate different host immune responses. To date, 83 serogroups within the genus Proteus have been identified (Siwińska et al., 2020). An interesting and unique modification of the P. mirabilis O antigen is its decoration with phosphocholine (ChoP), which occurs in strains belonging to the O18 serogroup (Fudala et al., 2003). Decoration of glycans with ChoP protects bacteria from innate and adaptive immune system responses, and modifies interactions with host proteins engaged in bacterial pathogenesis (Young et al., 2013).
Biosynthesis of ChoP and decoration of LPS involves four enzymes: choline kinase (LicA), choline transmembrane transporter (LicB), CTP:phosphocholine cytidylyltransferase (LicC) and lipopolysaccharide cholinephosphotransferase (LicD). The first steps in this process is choline uptake from the cell surroundings, involving choline permease LicB and phosphorylation of free choline to ChoP by choline kinase LicA (Young et al., 2013). Choline uptake is carried by both LicB and BetT (a high-affinity choline transporter). The BetT permease contributes to the utilisation of choline in the osmoprotection system, in which choline is oxidized to form glycine betaine that acts as an osmoprotectant, as demonstrated for Escherichia coli (Fan et al., 2003). In the LPS decoration pathway, phosphorylated choline is modified via the CDP-choline pathway to CDP-ChoP by LicC. This enzyme catalyzes the transfer of a cytidine monophosphate from CTP to phosphocholine to form CDP-choline (Kwak et al., 2002). The final event in the decoration process is CDP-ChoP transfer to a target glycan, mediated by LicD (Young et al., 2013). This enzyme decorates cell surface structures such as LPS in Gram-negative species, e.g. H. influenzae and commensal Neisseria, the pili of Neisseria meningitidis, and lipoteichoic acids and teichoic acids of S. pneumoniae (Geiger et al., 2013). Importantly, only in P. mirabilis and Morganella morganii, the O antigens are decorated with ChoP (Clark and Weiser, 2013).
Our studies revealed the homologs of licABCD genes presumably responsible for LPS decoration with ChoP. The biosynthesis of O antigen is regulated by genes which are generally organized in an O antigen gene cluster (Yu et al., 2017). Certain O antigens may contain unique modifications, and genes engaged in their processing are also located within this cluster (Yu et al., 2017). In the Wzx/Wzy-dependent assembly pathway the O antigen repeating units are linked to a lipid carrier (undecaprenyl pyrophosphate) and engage a transmembrane flippase (Wzx) that flips O units from the cytoplasm to the periplasm, where O antigen subunits are polymerized by Wzy. The length of the O antigen chain is regulated by the action of Wzz, a polysaccharide copolymerase (PCP) (Islam and Lam, 2014). The conservation of the O antigen gene cluster among bacteria is low, and heterogeneity of this region results in the synthesis of different O antigens (Islam and Lam, 2014). The low conservation between wzy genes of different serotypes enables the use of this gene as a molecular marker for molecular serotyping (Wang et al., 2017).
The first complete genome of P. mirabilis was characterised for the widely-studied strain HI4320 (serogroup O28) isolated from the urine of a patient with a long-term indwelling urinary catheter. Genome annotation revealed the coding sequences and genomic locations of previously-characterized virulence determinants. From this annotation, genes predicted to be involved in LPS biosynthesis were identified (Pearson et al., 2008). Subsequent studies have reported that Proteus serogroups can be genetically distinguished based on the sequences of their respective O antigen biosynthesis clusters; however, information about the O18 serogroup was incomplete (Yu et al., 2017). Here we report the draft genome sequence of P. mirabilis strain PrK 34/57 belonging to the O18 serogroup, including the fully-sequenced and annotated O antigen biosynthesis gene cluster.
Materials and Methods
Bacterial Strains and DNA Extraction
P. mirabilis strain PrK 34/57 (O18) was obtained from the Czech National Collection of Type Cultures in Prague, Czech Republic. Cells were grown overnight in 50 ml of LB (Biocorp, Warsaw, Poland), diluted in ratio 1:100 with fresh LB medium and cultivated for following tests at 37°C with shaking (160 rpm) for 12–18 h in an Ecotron incubator (Infors HT, Basel, Switzerland). Genomic DNA was isolated with a GenElute™ Bacterial Genomic DNA Kit (Sigma-Aldrich, Saint Louis, MO, USA) according to manufacturer’s protocol from 1.5 ml of overnight culture. Final elution was performed with 100 μl of nuclease-free water. DNA quality was assessed using a NanoDrop 2000 Spectrophotometer (Thermo Fisher Scientific, Waltham, MA, USA).
Sequencing
Libraries were prepared using the Nextera XT kit (Illumina Inc., San Diego, CA, USA) according to the manufacturer’s protocol. Libraries were sequenced on the NextSeq machine (Illumina) with 2 × 150 bp paired-end reads with a depth of 200-fold coverage. Over 91.00% of bases of sequencing reads had quality scores ≥Q30.
Genome ‘De Novo’ Assembly
The trimmed Illumina reads were assembled using Unicycler, an assembly pipeline for bacterial genomes (Wick et al., 2017). The Unicycler Version 0.4.8.0 was available online on the Galaxy web Server (https://usegalaxy.org/). The Unicycler pipeline functions as a SPAdes-optimizer. For the assembly, the default options of SPADes were selected, that includes turned on error correction, the k-mer in a range of 0.2 to 0.95 (expressed as a fraction of the read length). Contigs with a fraction of the chromosomal depth lower than 0.25 were filtered out. In terms of Unicycler options, the Normal Bridge mode (moderate contigs size and moderate misassembly rate) was selected. Contigs shorter than 500 bp were excluded from the final assembly.
Genome Annotation
The obtained contigs were reordered relative to the P. mirabilis HI4320 reference genome using Mauve Contig Mover (MCM) of Mauve 2.4.0 software to facilitate the study (Darling et al., 2004; Rissman et al., 2009). The default parameters were used. Genome sequence of PrK 34/57 was primarily functionally annotated by Rapid Annotation Subsystems Technology (RAST) server using the ClassicRAST annotation scheme, FIGfams version 70, automatic error correction, and automatic frame shift correction (Aziz et al., 2008; Gmiter et al., 2019). Further, during WGS (Whole Genome Shotgun) submission to NCBI, it was annotated using the NCBI Prokaryotic Genome Annotation Pipeline (PGAP) (Tatusova et al., 2016) to ensure better insight into the genomic features.
Variant Calling Analysis
For variant calling analysis of PrK 34/57, the Illumina reads were tested using Snippy (Galaxy Version 4.4.5+galaxy2) with the default parameters and HI4320 as a reference genome (https://github.com/tseemann/snippy) (Bush et al., 2020).
Phylogenetic Analysis
In order to perform the P. mirabilis PrK 34/57 comparative study, selected P. mirabilis, both complete and WGS type, genomes were downloaded from NCBI and presented in Table 1. For WGS genomes, the reordering of contigs was performed using MCM as above. The comparative genomics include the phylogenomic analysis based on average nucleotide identities (ANI) using FastANI algorithm (Jain et al., 2018) and single nucleotide polymorphisms (SNPs) using CSI Phylogeny webserver (Kaas et al., 2014). The CSI Phylogeny webserver was used with the default options, including minimum depth at SNP positions = 10×, minimum relative depth at SNP positions = 10%, minimum distance between SNPs = 10 bp, minimum SNP quality = 30, minimum read mapping quality = 25 and minimum Z-score = 1.96. The obtained phylogeny tree includes the P. mirabilis HI4320 as a reference genome. The tree resulted from the CSI Phylogeny webserver was visualized using Interactive Tree Of Life (iTOL) version 5 (Letunic and Bork, 2019). To determine global rearrangement structure between P. mirabilis PrK 34/57 and other genomes, the progressiveMauve option of Mauve was used (Darling et al., 2004; Darling et al., 2010). The backbone file, an output of Mauve, was used to visualize the genomes comparison with the R package genoPlotR (Guy et al., 2011).
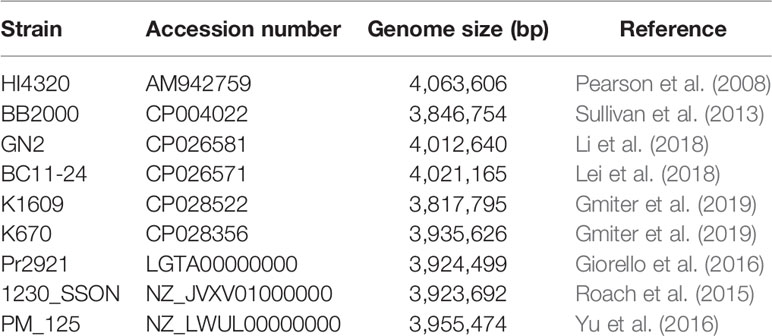
Table 1 Proteus mirabilis genomes used in the study.
KEGG Annotation
Analysis of KEGG pathways in PrK 34/57 was conducted by GhostKOALA, an automated metagenome annotation server that characterizes gene functions and pathways based on KEGG Orthology sequence assignments (Kanehisa et al., 2016). As an input file, the Amino-Acid FASTA file generated by RAST was used.
The Virulome Investigation
The presence of virulence driving genes was investigated using a local data base created with the makeblastdb option of BLAST+ (Camacho et al., 2009). Genes for data base construction were selected based on previously-described genome of P. mirabilis HI4320 strain. The database included genes responsible for ureolitic, proteolitic and hemolytic activity, motility (flagellum synthesis and chemotaxis), and fimbriae synthesis.
Resistance Gene Identifier
The resistome of P. mirabilis PrK 34/57 was predicted from nucleotide data based on homology and SNP models using the Resistance Gene Identifier (RGI) based on The Comprehensive Antibiotic Resistance Database (CARD; https://card.mcmaster.ca) (Alcock et al., 2020). The DNA was used as a data type, selection criteria was selected as perfect and strict hits only, the nudges above 95% was excluded, and the sequence quality was selected as a high coverage.
Genome Accession Number
This Whole Genome Shotgun Project of P. mirabilis PrK 34/57 has been deposited at GenBank (http://www.ncbi.nlm.nih.gov) under the accession JAAMPE000000000. The version described in this paper is version JAAMPE010000000.
Results
Genome Overview
The genome of P. mirabilis strain PrK 34/57 was determined to be 3,970,593 bp in length, with a GC content of 38.7%. It was assembled into 60 contigs (>500 bp). Based on PGAP annotation, there are 3,722 genes, of which 3,594 (96.6%) are protein-coding genes, 56 (1.5%) are pseudogenes and the remaining 72 are predicted RNA-coding genes, including 66 tRNAs, 2 rRNAs and 4 ncRNAs. The general features of P. mirabilis PrK 34/57 assembly identified by RAST are presented in Table 2. In contrast to PGAP, 3,620 genes were annotated using RAST, which were assigned to 497 subsystems. Importantly, among virulence, disease and defence subsystem categories, we identified genes involved in bacteriocin tolerance and production, as well as tolerance to antibiotics and heavy metals (including efflux pumps). Moreover, PrK 34/57 possesses 56 genes related to iron acquisition and metabolism. Additionally, there is a set of 57 genes related to motility and chemotaxis, both of which are important for the ability of P. mirabilis strains to swarm over solid surfaces. We also identified genes encoding urease subunits (nitrogen metabolism category), an important enzyme for P. mirabilis survival in the urinary tract. Furthermore, genes involved in choline transport and metabolism were identified (miscellaneous category). The detailed subsystems present in PrK 34/57 are summarised in Table 3. Variant calling is a useful comparative genomic method that provides insight into organismal differences at the nucleotide level (Fariq et al., 2019). A set of variants are identified from tested and reference genomes, including small polymorphisms, specifically single-nucleotide polymorphisms (SNPs), insertions and deletions (indels), multi-nucleotide polymorphisms (MNPs) and complex events (composite insertion and substitution events) smaller than the length of short-read sequencing alignment. During our current analysis, 18,240 variants were identified between PrK 34/57 and HI4320. For all variants, their FILTERs status was PASS, indicating that variants in the raw data are true calls and not false positives resulting from low coverage depth. Observed variants were divided into five categories, and numbers and frequencies are summarized in Table 4. Using GhostKOALA annotation server totally 2342 entries (64.7%) was annotated in the genome of PrK 34/57. The distribution and frequency of KEGG pathways was presented on Figure 1.

Table 2 Proteus mirabilis PrK 34/57 genome assembly statistics based on RAST annotation server.

Table 3 Subsystem distribution of Proteus mirabilis PrK 34/57 via RAST server-based annotation.
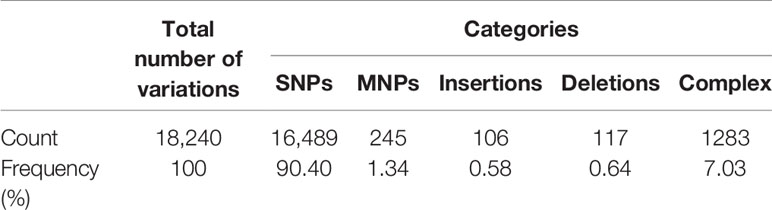
Table 4 Summary of variants in Proteus mirabilis PrK 34/57 genome against P. mirabilis HI4320 reference strain.
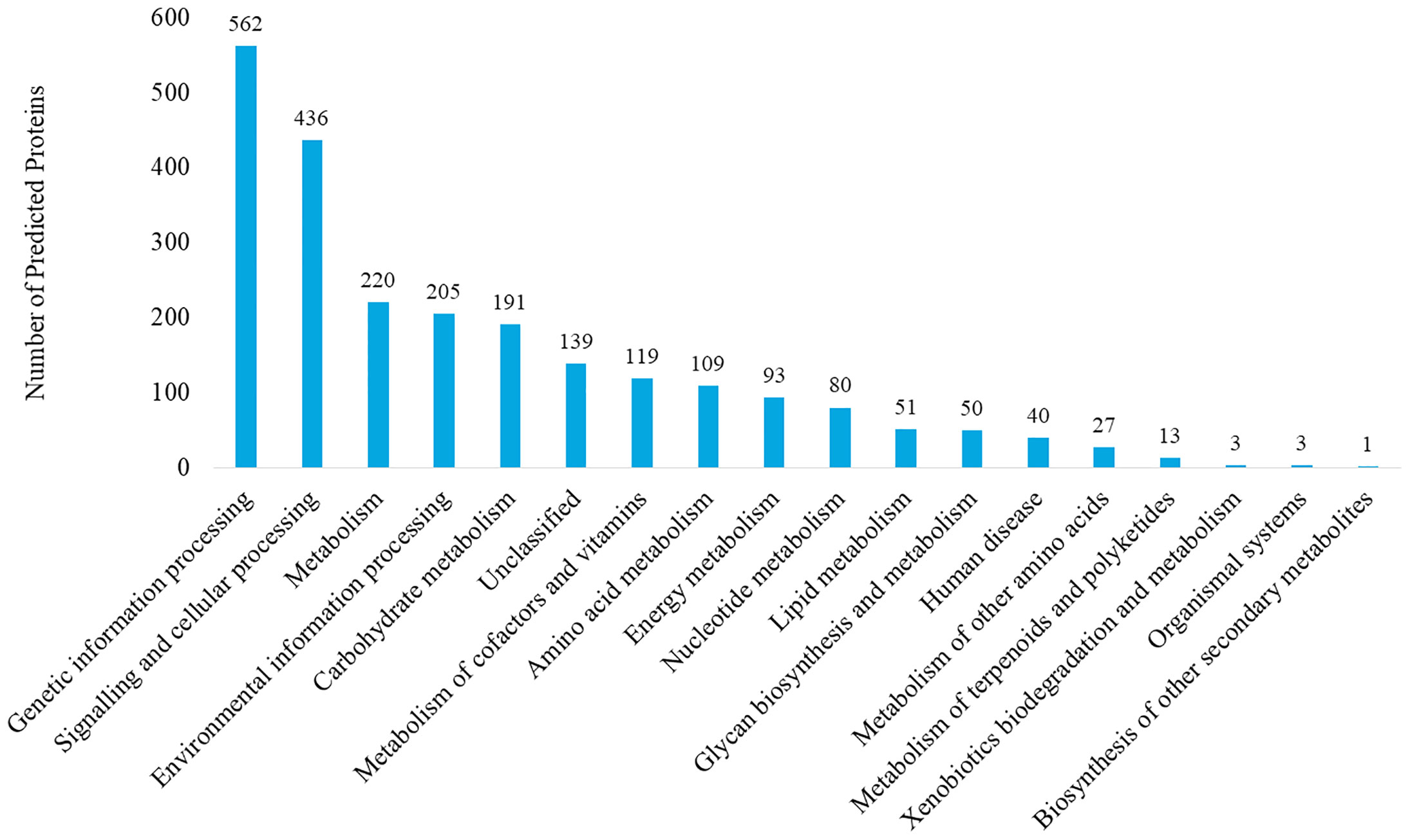
Figure 1 KEGG pathway analysis of Proteus mirabilis PrK 34/57 strain. Proteins were identified and categorized using the GhostKOALA tool against the Amino-Acid file generated by RAST server.
Phylogenetic Analysis
The fastANI analysis of the P. mirabilis PrK 34/57 genome revealed high similarity with other genomes. ANI values were 98.6077 to 99.1747% compared with BC11-24 and K670 genomes, respectively. These values far exceed the generally accepted 95% cut-off level for taxonomy affiliation of newly sequenced genomes (Figueras et al., 2014). The results of detailed comparison of the genomes are presented in Table 5.
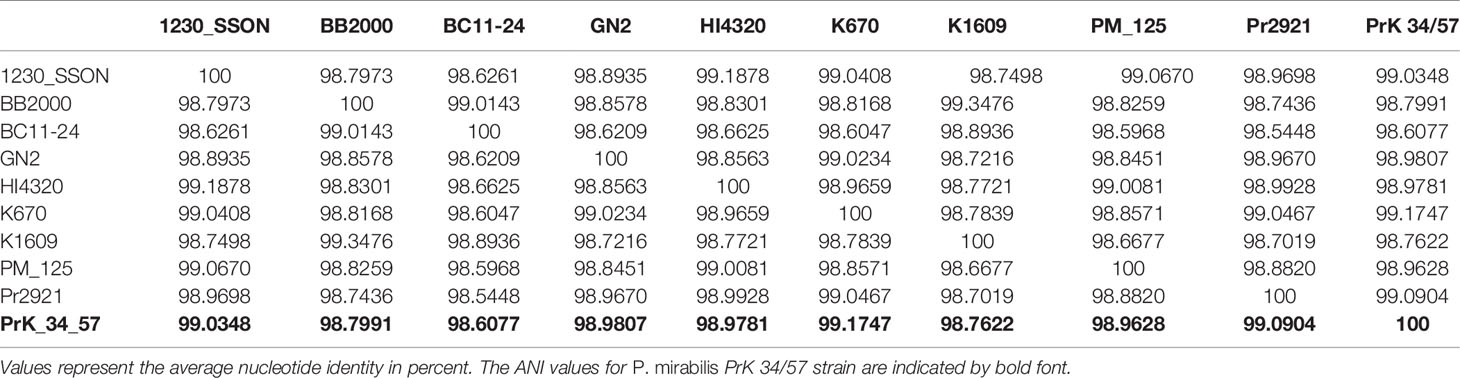
Table 5 ANI similarity matrix between studied Proteus mirabilis genomes.
A phylogenetic tree based on SNPs identified from whole genome comparisons (Figure 2) resulted in three major clades: PrK 34/57; the HI4320 reference genome; and BB2000, another frequently studied P. mirabilis strain (Sullivan et al., 2013). The PrK 34/57 genome is most similar to the previously reported genome of K670 (Gmiter et al., 2019), and shares greater similarity with HI43200 than BB2000. These observations correspond to the results of ANI-based analysis. Similar results were obtained following analysis of whole genome phylogeny of P. mirabilis strains based on the Mauve comparison (data not shown). The global rearrangement structure of the studied P. mirabilis genomes is visualized in Figure 3. Significant similarity between genomes is evident, with only a few genome rearrangement events. These occasional events are illustrated by the crossing lines that link Locally Collinear Blocks (LCBs). LCBs were calculated by Mauve to identify conserved segments that appear to be internally free from genome rearrangements. However, the PrK 34/57 genome displays blocks below the genome’s center line, which suggests the presence of inverted regions in PrK 34/57 (regions that align in the reverse complement (inverse) orientation) compared with other strains (Darling et al., 2004).
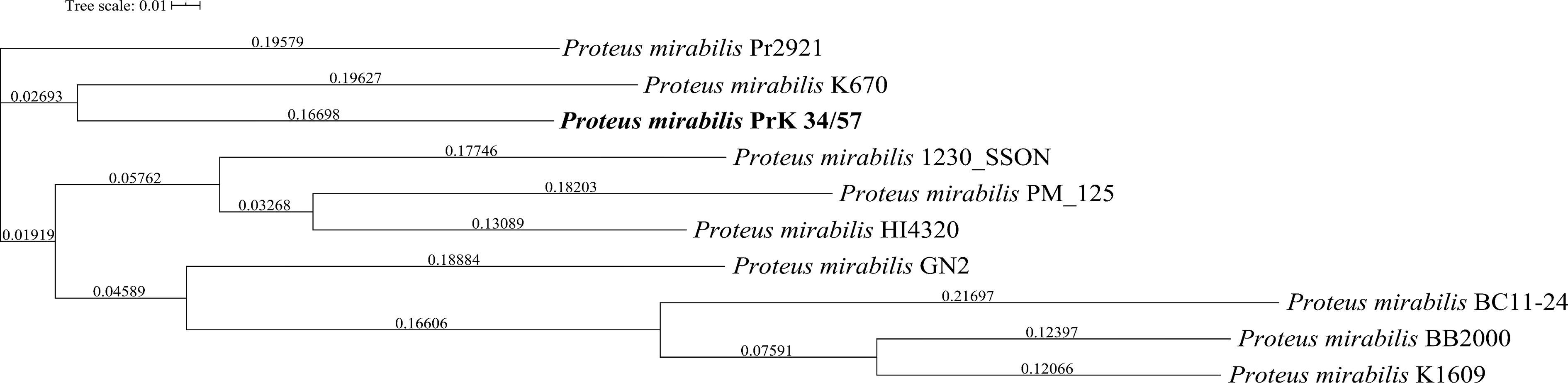
Figure 2 Whole genome phylogeny of Proteus mirabilis strains based on single nucleotide polymorphisms (SNPs) using CSI Phylogeny webserver. The sequence of studied P. mirabilis PrK 34/57 strain is indicated by bold font. The obtained Newick file was visualized using Interactive Tree Of Life (iTOL) version 5.

Figure 3 Whole genome comparison of Proteus mirabilis genomes using progressiveMauve option of Mauve software. Genomes are represented in blue, and blocks with borders of different colors are homologous between genomes The backbone file was visualized using the R package genoPlotR.
The Virulome Investigation
To better explore virulence potential of P. mirabilis strain PrK 34/57 the presence of genes involved in expression of most important virulence factors was tested using local virulence factor databases. From this analysis, genes associated with all major virulence factors were detected in PrK 34/57. The sequence similarity with HI4320 was up to 100% depending on gene class. Interestingly, though previously detected in strain HI4320, genes encoding fimbriae 3 (fim3A) were not detected in PrK 34/57 (Table 6).
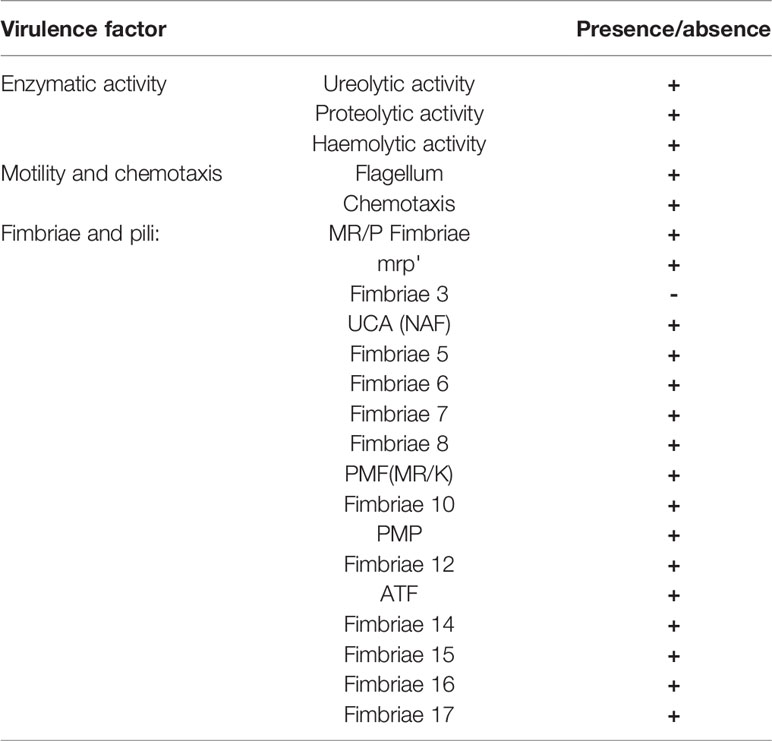
Table 6 Presence of genes related to Proteus mirabilis virulence in PrK 34/57.
Identification of Putative Antibiotic Resistance Genes
Based on the Comprehensive Antibiotic Research Database (CARD), the Resistance Gene Identifier (RGI) tool identified homologs of genes responsible for resistance to aminoglycosides (amikacin, tobramycin) and beta-lactams (ampicillin, piperacillin, amoxicillin), and the results are presented in Table 7. Antimicrobial peptides resistance in this strain is presumably supported by homologs of Klebsiella pneumoniae KpnFH efflux pumps (Poirel et al., 2017; Aghapour et al., 2019).
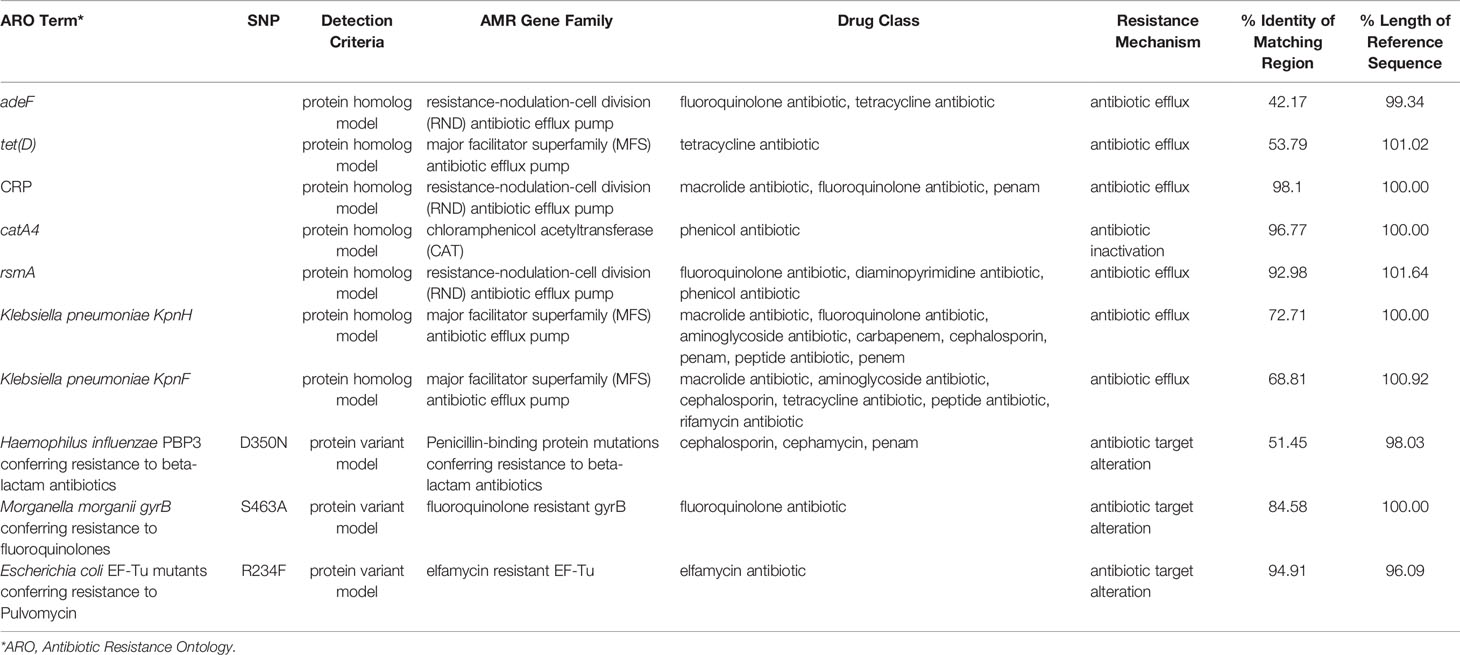
Table 7 The putative antibiotic resistance genes identified in genome of P. mirabilis PrK 34/57 by the Resistance Gene Identifier (RGI) based on the Comprehensive Antibiotic Research Database (CARD).
Organization of the P. mirabilis O18 Antigen Gene Cluster
Most O antigen gene clusters are located at fixed positions on bacterial chromosomes, and genetic variation in these gene clusters is largely responsible for the diversity of O antigen forms (Liu et al., 2008). There are three types of molecular biosynthesis pathways for O antigens: Wzx/Wzy-dependent, synthase-dependent, and ABC transporter-dependent pathways (Bertani and Ruiz, 2018). The Wzx/Wzy-dependent pathway is the most common, and the main components are the integral inner membrane flippase Wzx, the polymerase Wzy, and the chain-length regulator protein Wzz (Islam and Lam, 2014; Yu et al., 2017). The P. mirabilis O antigen gene cluster is located between cpxA and secB genes, and contains both the wzx and wzy genes (Wang et al., 2010; Yu et al., 2017). Genome annotation using the RAST server identified the genes involved in choline transport and metabolism. Three of the genes were licA located at open reading frame (ORF) ORF04, licB located at ORF05, and licD located at ORF08 based on the cluster scheme proposed by Yu and colleagues (Yu et al., 2017). The O antigen cluster contained one additional ORF (ORF06); amino acid sequence analysis by Phyre2 software (Kelley et al., 2015) revealed 98% coverage, and a 224 amino acid polypeptide was modelled with 100.0% confidence with ctp:phosphocholine cytidylytransferase (LicC) as the single highest scoring template. The licABC genes overlap with each other, suggesting they are co-transcribed during the O antigen assembly process. The sequence of the licD gene overlaps with that of the wzx gene. Both regions are divided by a short intergenic sequence. The licABC genes are localized between the wemA gene, encoding a UDP-galactofuranosyl transferase according to Phyre2 (confidence 98.1%, coverage 49%), separated from licD by wzx encoding a flippase. The complete O antigen biosynthesis cluster for the P. mirabilis O18 serogroup is presented on Figure 4.
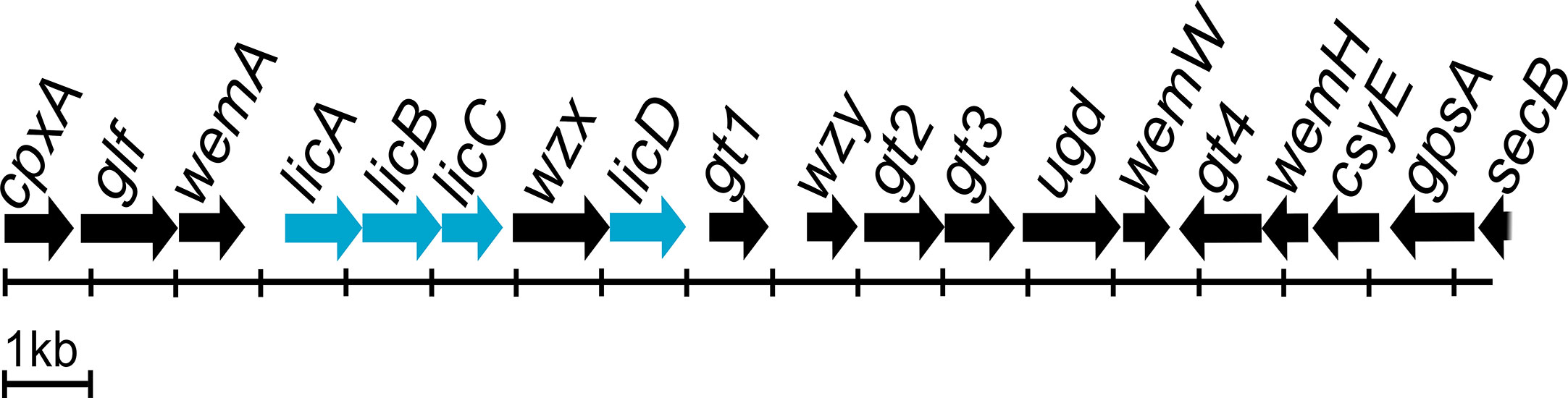
Figure 4 Proteus mirabilis O18 O antigen gene cluster arrangement. Highlighted genes represent licABCD genes and its organisation in the Wzx/Wzy-dependent O antigen biosynthesis gene cluster. Scheme modified from Yu et al. (2017).
Discussion
P. mirabilis is a Gram-negative, commensal bacterium, which is frequently responsible for catheter-associated urinary tract infections. In this work, we determined the draft genome of P. mirabilis strain PrK 34/57 (O18) containing the unique ChoP modification of O antigen. Furthermore, the obtained data allowed us to investigate the general features of the studied genome. PGAP and RAST annotation revealed the presence of important genes involved in P. mirabilis pathogenicity, including genes responsible for swarming motility, ureolytic activity and iron acquisition (Różalski et al., 1997). The structure and organisation of the PrK 34/57 genome was compared with other P. mirabilis strains, and the genome of PrK 34/57 is generally similar in terms of features such as genome size, GC ratio and the number of coding sequences (Pearson et al., 2008; Sullivan et al., 2013; Shi et al., 2016; Gmiter et al., 2019). In the present study, the HI4320 genome was used as a reference, since this strain has been studied extensively (Armbruster et al., 2018). Other complete and WGS genomes were also included to better understand the general position of PrK 34/57 among P. mirabilis isolates from around the world. P. mirabilis PrK 34/57 shares high similarity with strain HI4320, but it is most closely related to the previously studied K670 strain. Simple variant calling analysis was performed using HI4320 and PrK 34/57. The results indicated few P. mirabilis PrK 34/57 polymorphisms (<20,000 variants), especially considering the high level of polymorphisms revealed in previously studied Acidithiobacillus ferrooxidans strains (>70,000 variants) (Fariq et al., 2019). These variant calling analyses may be relevant for future studies on PrK 34/57. Similar conclusions about the high genetic relatedness between PrK 34/57 and other strains can be drawn from the ANI results, which were in agreement with previous observations (Armbruster et al., 2018).
The obtained draft genome sequence of ChoP-containing P. mirabilis O18 strain PrK 34/57 is important in a serological context and will be beneficial during subsequent studies of the pathogenicity of this bacterium. Choline uptake and LPS decoration protects bacteria from osmotic pressure, and enables avoiding the immune system recognition. Choline uptake is provided in P. mirabilis O18 by a high-affinity choline transport protein BetT. Choline gained in this way is involved in the glycine betaine biosynthesis pathway, which acts as an osmoprotectant (Deole and Hoff, 2020). The other way for choline recruitment into the cell is the ChoP decoration pathway, where licABC genes products (kinase, permease, and cytidylyltransferase, respectively) incorporate, and modify choline molecule. In the next step, CDP-choline is attached to the target glycans by the transferase LicD (Young et al., 2013). In P. mirabilis O18 antigen gene cluster genes licABC overlaps, which suggest that they are organised in a separate cluster, and expressed simultaneously. The action of this cluster may result in incorporation choline into the cell and its modification to CDP-choline. The licD encoding transferase overlaps with the sequence of the wzx flippase, which suggests that both genes are co-transcribed, and expression of licD depends on expression of wzx and is regulated with the same factors. This finding reveals that ChoP attachment to O antigen repeating unit in P. mirabilis is probably regulated by two independent regulatory regions, upstream the licABC and wzx/licD clusters, and it occurs as two independently controlled events. The decoration of a single O antigen unit presumably occurs before flipping of the undecaprenyl phosphate-linked O units to the periplasmic face of the inner membrane by the Wzx flippase.
In conclusion, determination of the P. mirabilis O18 genome sequence revealed the presence and localisation of genes licABCD potentially involved in choline uptake, modification and decoration of LPS. Enzymes involved in ChoP decoration, a choline kinase, choline permease, and ctp:phosphocholine cytidylytransferase are encoded by the licABC cluster. This cluster is separated from licD homolog (encoding lipopolysaccharide cholinephosphotransferase) by the wzx gene encoding a crucial LPS biosynthesis transmembrane flippase. The results allowed us to present the provisional organisation of the O antigen gene cluster in the P. mirabilis O18 serogroup.
Data Availability Statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/supplementary material.
Author Contributions
GC—carried out the experiments with DNA isolation, and genome analysis in order to investigate the presence of licABCD gene and organisation the O antigen gene cluster as well as antibiotic resistance identification. Contributed to the interpretation of the results, and took the lead in writing the manuscript. DG—carried out DNA isolation, genome annotation and deposition in GenBank. Carried out the bioinformatic analysis of general features of reported genome, interpretation of the results, and writing the manuscript. KD-P—Contributed to the interpretation of the results, and contributed in writing the manuscript. All authors provided critical feedback and helped shape the research, analysis, and manuscript. All authors contributed to the article and approved the submitted version.
Funding
This research was supported by the Polish National Science Centre (Grant No. 2017/01/X/NZ6/01141), and Jan Kochanowski University Rector’s Grant 2019 (no SIGR.RN.20.061.604). DG received the PhD scholarship from the NSC Grant No. 2019/32/T/NZ1/00515.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We would like to thank Prof. Wiesław Kaca for his contribution of a discussion to our paper.
References
Aghapour Z., Gholizadeh P., Ganbarov K., Bialvaei A. Z., Mahmood S. S., Tanomand A., et al. (2019). Molecular mechanisms related to colistin resistance in Enterobacteriaceae.. Infect Drug Resist. 12, 965–975. doi: 10.2147/IDR.S199844
Alcock B. P., Raphenya A. R., Lau T. T. Y., Tsang K. K., Bouchard M., Edalatmand A., et al. (2020). CARD 2020: Antibiotic resistome surveillance with the comprehensive antibiotic resistance database. Nucleic Acids Res. 48, D517–D525. doi: 10.1093/nar/gkz935
Armbruster C. E., Mobley H. L. T., Pearson M. M. (2018). Pathogenesis of Proteus mirabilis Infection. EcoSal Plus 8:1–73. doi: 10.1128/ecosalplus.ESP-0009-2017
Aziz R. K., Bartels D., Best A., DeJongh M., Disz T., Edwards R. A., et al. (2008). The RAST Server: Rapid annotations using subsystems technology. BMC Genomics 9, 1–15. doi: 10.1186/1471-2164-9-75
Bertani B., Ruiz N. (2018). Function and Biogenesis of Lipopolysaccharides. EcoSal Plus 8, 1–33. doi: 10.1128/ecosalplus.esp-0001-2018
Bush S. J., Foster D., Eyre D. W., Clark E. L., De Maio N., Shaw L. P., et al. (2020). Genomic diversity affects the accuracy of bacterial single-nucleotide polymorphism-calling pipelines. GigaScience 9, 1–21. doi: 10.1093/gigascience/giaa007
Camacho C., Coulouris G., Avagyan V., Ma N., Papadopoulos J., Bealer K., et al. (2009). BLAST+: Architecture and applications. BMC Bioinf. 10, 1–9. doi: 10.1186/1471-2105-10-421
Clark S. E., Weiser J. N. (2013). Microbial modulation of host immunity with the small molecule phosphorylcholine. Infect. Immun. 81, 392–401. doi: 10.1128/IAI.01168-12
Darling A. C. E., Mau B., Blattner F. R., Perna N. T. (2004). Mauve: Multiple Alignment of Conserved Genomic Sequence With Rearrangements Mauve. Genome Res. 14 (7), 1394–1403. doi: 10.1101/gr.2289704
Darling A. E., Mau B., Perna N. T. (2010). Progressivemauve: Multiple genome alignment with gene gain, loss and rearrangement. PloS One 5 (6), e11147. doi: 10.1371/journal.pone.0011147
Deole R., Hoff W. D. (2020). A potassium chloride to glycine betaine osmoprotectant switch in the extreme halophile Halorhodospira halophila. Sci. Rep. 10, 3383. doi: 10.1038/s41598-020-59231-9
Fan X., Pericone C. D., Lysenko E., Goldfine H., Weiser J. N. (2003). Multiple mechanisms for choline transport and utilization in Haemophilus influenzae. Mol. Microbiol. 50, 537–548. doi: 10.1046/j.1365-2958.2003.03703.x
Fariq A., Blazier J. C., Yasmin A., Gentry T. J., Deng Y. (2019). Whole genome sequence analysis reveals high genetic variation of newly isolated Acidithiobacillus ferrooxidans IO-2C. Sci. Rep. 9, 1–10. doi: 10.1038/s41598-019-49213-x
Figueras M. J., Beaz-Hidalgo R., Hossain M. J., Liles M. R. (2014). Taxonomic Affiliation of New Genomes Should Be Verified Using Average Nucleotide Identity and Multilocus Phylogenetic Analysis. Genome Announc. 2, e00927–14-e00927-14. doi: 10.1128/genomeA.00927-14
Fudala R., Kondakova A. N., Bednarska K., Senchenkova S. N., Shashkov A. S., Knirel Y. A., et al. (2003). Structure and serological characterization of the O-antigen of Proteus mirabilis O18 with a phosphocholine-containing oligosaccharide phosphate repeating unit. Carbohydr. Res. 338, 1835–1842. doi: 10.1016/S0008-6215(03)00274-X
Geiger O., López-Lara I. M., Sohlenkamp C. (2013). Phosphatidylcholine biosynthesis and function in bacteria. Biochim. Biophys. Acta - Mol. Cell Biol. Lipids 1831, 503–513. doi: 10.1016/j.bbalip.2012.08.009
Giorello F. M., Romero V., Farias J., Scavone P., Umpiérrez A., Zunino P., et al. (2016). Draft genome sequence and gene annotation of the uropathogenic bacterium Proteus mirabilis Pr2921. Genome Announc. 4, 3–4. doi: 10.1128/genomeA.00564-16
Gmiter D., Czerwonka G., Drewnowska J. M., Swiecicka I., Kaca W. (2019). Draft Genome Sequences of Proteus mirabilis K1609 and K670: A Model Strains for Territoriality Examination. Curr. Microbiol. 76, 144–152. doi: 10.1007/s00284-018-1598-6
Guy L., Kultima J. R., Andersson S. G. E., Quackenbush J. (2011). GenoPlotR: comparative gene and genome visualization in R. Bioinformatics 27, 2334–2335. doi: 10.1093/bioinformatics/btq413
Islam S. T., Lam J. S. (2014). Synthesis of bacterial polysaccharides via the Wzx/Wzy-dependent pathway. Can. J. Microbiol. 60, 697–716. doi: 10.1139/cjm-2014-0595
Jain C., Rodriguez-R L. M., Phillippy A. M., Konstantinidis K. T., Aluru S. (2018). High throughput ANI analysis of 90K prokaryotic genomes reveals clear species boundaries. Nat. Commun. 9, 1–8. doi: 10.1038/s41467-018-07641-9
Kaas R. S., Leekitcharoenphon P., Aarestrup F. M., Lund O. (2014). Solving the problem of comparing whole bacterial genomes across different sequencing platforms. PloS One 9, 1–8. doi: 10.1371/journal.pone.0104984
Kanehisa M., Sato Y., Morishima K. (2016). BlastKOALA and GhostKOALA: KEGG Tools for Functional Characterization of Genome and Metagenome Sequences. J. Mol. Biol. 428, 726–731. doi: 10.1016/j.jmb.2015.11.006
Kelley L. A., Mezulis S., Yates C. M., Wass M. N., Sternberg M. J. (2015). The Phyre2 web portal for protein modeling, prediction and analysis. Nat. Protoc. 10, 845–858. doi: 10.1038/nprot.2015-053
Knirel Y. A., Perepelov A. V., Kondakova A. N., Senchenkova S. N., Sidorczyk Z., Rozalski A., et al. (2011). Structure and serology of O-antigens as the basis for classification of Proteus strains. Innate Immun. 17, 70–96. doi: 10.1177/1753425909360668
Kwak B. Y., Zhang Y. M., Yun M., Heath R. J., Rock C. O., Jackowski S., et al. (2002). Structure and mechanism of CTP:Phosphocholine cytidylyltransferase (LicC) from Streptococcus pneumoniae. J. Biol. Chem. 277, 4343–4350. doi: 10.1074/jbc.M109163200
Lei C.-W., Chen Y.-P., Kong L.-H., Zeng J., Wang Y.-X., Zhang A.-Y., et al. (2018). PGI2 Is a Novel SGI1-Relative Multidrug-Resistant Genomic Island Characterized in Proteus mirabilis. Antimicrob. Agents Chemother. 62, 1–4. doi: 10.1128/AAC.00019-18
Letunic I., Bork P. (2019). Interactive Tree Of Life (iTOL) v4: recent updates and new developments. Nucleic Acids Res. 47, W256–W259. doi: 10.1093/nar/gkz239
Li B., Feng J., Zhan Z., Yin Z., Jiang Q., Wei P., et al. (2018). Dissemination of KPC-2-encoding IncX6 plasmids among multiple Enterobacteriaceae Species in a single Chinese hospital. Front. Microbiol. 9, 478. doi: 10.3389/fmicb.2018.00478
Liu B., Knirel Y. A., Feng L., Perepelov A. V., Senchenkova S. N., Wang Q., et al. (2008). Structure and genetics of Shigella O antigens. FEMS Microbiol. Rev. 32, 627–653. doi: 10.1111/j.1574-6976.2008.00114.x
Liu B., Knirel Y. A., Feng L., Perepelov A. V., Senchenkova S. N., Reeves P. R., et al. (2014). Structural diversity in Salmonella O antigens and its genetic basis. FEMS Microbiol. Rev. 38, 56–89. doi: 10.1111/1574-6976.12034
Pearson M. M., Sebaihia M., Churcher C., Quail M. A., Seshasayee A. S., Luscombe N. M., et al. (2008). Complete genome sequence of uropathogenic Proteus mirabilis, a master of both adherence and motility. J. Bacteriol. 190, 4027–4037. doi: 10.1128/JB.01981-07
Poirel L., Jayol A., Nordmann P. (2017). Polymyxins: Antibacterial activity, susceptibility testing, and resistance mechanisms encoded by plasmids or chromosomes. Clin. Microbiol. Rev. 30, 557–596. doi: 10.1128/CMR.00064-16
Rissman A. I., Mau B., Biehl B. S., Darling A. E., Glasner J. D., Perna N. T. (2009). Reordering contigs of draft genomes using the Mauve Aligner. Bioinformatics 25, 2071–2073. doi: 10.1093/bioinformatics/btp356
Roach D. J., Burton J. N., Lee C., Stackhouse B., Butler-Wu S. M., Cookson B. T., et al. (2015). A Year of Infection in the Intensive Care Unit: Prospective Whole Genome Sequencing of Bacterial Clinical Isolates Reveals Cryptic Transmissions and Novel Microbiota. PloS Genet. 11, 1–21. doi: 10.1371/journal.pgen.1005413
Różalski A., Sidorczyk Z., Kotełko K. (1997). Potential virulence factors of Proteus bacilli. Microbiol. Mol. Biol. Rev. 61 (1), 65–89.
Schaffer J. N., Pearson M. M. (2015). Proteus mirabilis and Urinary Tract Infections. Microbiol. Spectr. 3, 212–263. doi: 10.1128/microbiolspec.uti-0017-2013
Shi X., Lin Y., Qiu Y., Li Y., Jiang M., Chen Q., et al. (2016). Comparative screening of digestion tract toxic genes in Proteus mirabilis. PloS One 11, 1–12. doi: 10.1371/journal.pone.0151873
Siwińska M., Zabłotni A., Levina E. A., Shashkov A. S., Ovchinnikova O. G., Różalski A., et al. (2020). The unique structure of bacterial polysaccharides - Immunochemical studies on the O-antigen of Proteus penneri 4034-85 clinical strain classified into a new O83 Proteus serogroup. Int. J. Biol. Macromol. 163, 1168–1174. doi: 10.1016/j.ijbiomac.2020.07.012
Sullivan N. L., Septer A. N., Fields A. T., Wenren L. M., Gibbs K. A. (2013). The Complete Genome Sequence of Proteus mirabilis Strain BB2000 Reveals Differences from the P. mirabilis Reference Strain. Genome Announc. 1, e00024–13-e00024-13. doi: 10.1128/genomeA.00024-13
Tatusova T., Dicuccio M., Badretdin A., Chetvernin V., Nawrocki E. P., Zaslavsky L., et al. (2016). NCBI prokaryotic genome annotation pipeline. Nucleic Acids Res. 44, 6614–6624. doi: 10.1093/nar/gkw569
Wang Q., Torzewska A., Ruan X., Wang X., Rozalski A., Shao Z., et al. (2010). Molecular and genetic analyses of the putative Proteus O antigen gene locus. Appl. Environ. Microbiol. 76, 5471–5478. doi: 10.1128/AEM.02946-09
Wang H., Zheng H., Li Q., Xu Y., Wang J., Du P., et al. (2017). Defining the genetic features of O-antigen biosynthesis gene cluster and performance of an O-antigen serotyping scheme for Escherichia albertii. Front. Microbiol. 8, 1857. doi: 10.3389/fmicb.2017.01857
Wick R. R., Judd L. M., Gorrie C. L., Holt K. E. (2017). Unicycler: Resolving bacterial genome assemblies from short and long sequencing reads. PloS Comput. Biol. 13, 1–22. doi: 10.1371/journal.pcbi.1005595
Young N. M., Foote S. J., Wakarchuk W. W. (2013). Review of phosphocholine substituents on bacterial pathogen glycans: Synthesis, structures and interactions with host proteins. Mol. Immunol. 56, 563–573. doi: 10.1016/j.molimm.2013.05.237
Yu C. Y., Ang G. Y., Ngeow Y. F., Tee K. K., Yin W. F., Chan K. G. (2016). Genome sequences of two multidrug-resistant Proteus mirabilis strains harboring CTX-M-65 isolated from Malaysia. Genome Announc. 4, 2015–2016. doi: 10.1128/genomeA.01301-16
Keywords: Proteus mirabilis, phosphocholine, lipopolysaccharide, urinary tract infection, genome
Citation: Czerwonka G, Gmiter D and Durlik-Popińska K (2021) Draft Genome of Proteus mirabilis Serogroup O18 Elaborating Phosphocholine-Decorated O Antigen. Front. Cell. Infect. Microbiol. 11:620010. doi: 10.3389/fcimb.2021.620010
Received: 21 October 2020; Accepted: 02 March 2021;
Published: 25 March 2021.
Edited by:
Andres Felipe Opazo-Capurro, University of Concepcion, ChileReviewed by:
Michael Marceau, Université Lille Nord de France, FranceSalim T. Islam, Université du Québec, Canada
Copyright © 2021 Czerwonka, Gmiter and Durlik-Popińska. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Grzegorz Czerwonka, Z2N6ZXJ3b25rYUB1amsuZWR1LnBs