
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Cell. Infect. Microbiol., 16 March 2021
Sec. Bacteria and Host
Volume 11 - 2021 | https://doi.org/10.3389/fcimb.2021.615959
This article is part of the Research TopicHost-pathogen Interaction During Pneumococcal InfectionsView all 14 articles
 Hisanori Domon1,2*
Hisanori Domon1,2* Yutaka Terao1,2
Yutaka Terao1,2Streptococcus pneumoniae, also known as pneumococcus, is a Gram-positive diplococcus and a major human pathogen. This bacterium is a leading cause of bacterial pneumonia, otitis media, meningitis, and septicemia, and is a major cause of morbidity and mortality worldwide. To date, studies on S. pneumoniae have mainly focused on the role of its virulence factors including toxins, cell surface proteins, and capsules. However, accumulating evidence indicates that in addition to these studies, knowledge of host factors and host-pathogen interactions is essential for understanding the pathogenesis of pneumococcal diseases. Recent studies have demonstrated that neutrophil accumulation, which is generally considered to play a critical role in host defense during bacterial infections, can significantly contribute to lung injury and immune subversion, leading to pneumococcal invasion of the bloodstream. Here, we review bacterial and host factors, focusing on the role of neutrophils and their elastase, which contribute to the progression of pneumococcal pneumonia.
Pneumonia is a common and serious infectious disease and has been a significant cause of morbidity and mortality worldwide, accounting for approximately three million deaths annually. The World Health Organization (WHO) placed lower respiratory infections as the fourth most common cause of death in 2016. Among a number of infectious agents, Streptococcus pneumoniae, also known as pneumococcus, is the most common cause of pneumonia in all age groups. In addition to localized infections such as pneumonia and otitis media, pneumococcus may cause invasive diseases, including meningitis and septicemia. Furthermore, an increase in antimicrobial resistance among pneumococci has raised concerns about the effectiveness of empiric antimicrobial therapy for pneumococcal pneumonia (Feldman, 2004; Ferrara, 2005; Nagai et al., 2019).
S. pneumoniae is a Gram-positive diplococcus that colonizes the mucosal surfaces of the human nasopharynx. Nasopharyngeal aerosolization of S. pneumoniae is considered to be the primary mode of population transmission. The molecular interaction of pneumococcal virulence factors and host proteins with respect to nasopharyngeal colonization has been thoroughly reviewed elsewhere (Kadioglu et al., 2008; Weiser et al., 2018). It has been reported that 18–92% of children are carriers of S. pneumoniae (Le Polain de Waroux et al., 2014); thus, they are considered the main reservoirs and transmission vectors of pneumonia (Smith et al., 2019). The aspiration of nasopharyngeal secretions leads to the invasion and propagation of S. pneumoniae in the lung parenchyma at the alveolar level, which leads to pulmonary infection (Liu et al., 2015). It has been reported that bacterial virulence factors directly damage human tissues or cause malfunctioning of the human immune system, resulting in an excessive inflammatory response. This excessive or inappropriate host inflammatory response is considered to result in the clinical syndrome of pneumonia. In this review, we discuss the bacterial and host factors that contribute to the progression of pneumococcal pneumonia, specifically focusing on the role of neutrophils and their elastase.
Upon pneumococcal colonization or infection, the respiratory epithelium controls the bacterium through antimicrobial peptides, such as LL-37 and defensins (Bals and Hiemstra, 2004). However, S. pneumoniae can survive by removing its capsule from the surface (also see section 4) (Kietzman et al., 2016), which allows the organism to adhere to and invade the epithelium (Hammerschmidt et al., 2005). Pneumococcal interactions with other innate immune molecules, such as complements and surfactant protein-D, have been reviewed elsewhere (Kadioglu and Andrew, 2004). Following invasion of epithelial cells, the host innate immune system, which includes respiratory epithelial cells, alveolar macrophages, and dendritic cells, recognizes invading S. pneumoniae using pattern recognition receptors (PRRs) (Hartl et al., 2018). Different classes of PRRs include toll-like receptors (TLRs), nucleotide-binding oligomerization domain (NOD)-like receptors (NLRs), retinoic acid-inducible gene I-like receptors, and C-type lectin receptors (Takeuchi and Akira, 2010). These receptors are activated by conserved microbial molecules and bacterial virulence factors. Among the various TLRs, TLR2 recognizes several components of the pneumococcal cell wall, such as lipoteichoic acid, lipoproteins, and peptidoglycan (Yoshimura et al., 1999; Tomlinson et al., 2014), whereas TLR9 recognizes pneumococcal genomic DNA (Mogensen et al., 2006). Although TLR4 is known for its ability to detect lipopolysaccharide (LPS) from Gram-negative bacteria, it has been suggested that TLR4 might additionally recognize pneumolysin (Ply) (Malley et al., 2003), a pneumococcal pore-forming toxin. Additionally, we have demonstrated that pneumococcal cytosolic components, such as the chaperone protein DnaK, elongation factor Tu, and glyceraldehyde-3-phosphate dehydrogenase induce the production of proinflammatory cytokines via TLR4 (Nagai et al., 2018). NOD2 recognizes lysosome-digested peptidoglycan fragments of phagocytized S. pneumoniae (Davis et al., 2011). Additionally, Ply activates the NLRP3 inflammasome and promotes proinflammatory cytokine secretion by dendritic cells (McNeela et al., 2010). Intracellular signaling cascades triggered by PRRs lead to the transcriptional activation of inflammatory mediators, such as proinflammatory cytokines and chemokines. These mediators stimulate neighboring immune and non‐immune cells, activate the acute‐phase response, and recruit neutrophils (Koppe et al., 2012) (Figure 1).
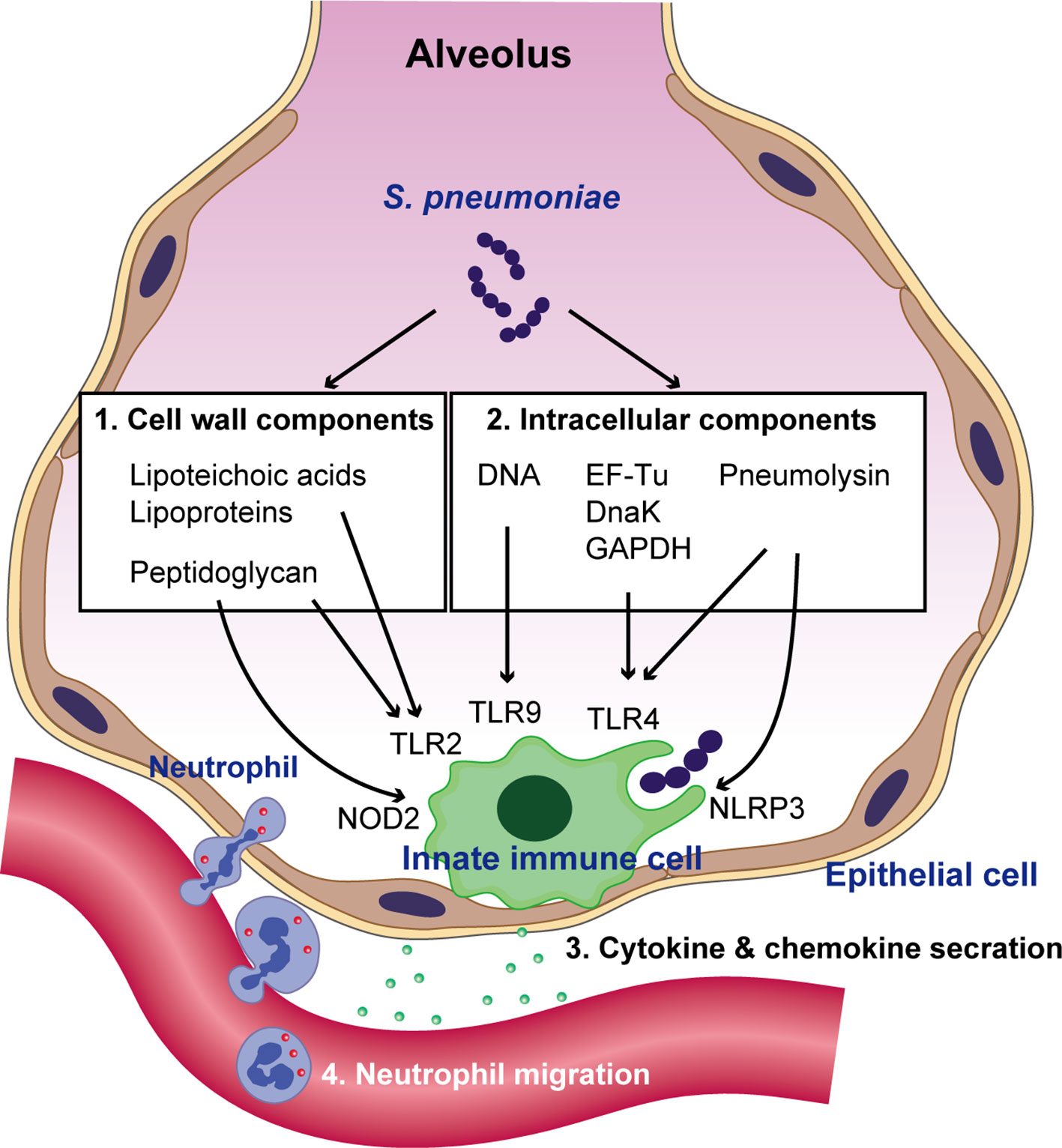
Figure 1 Recognition of S. pneumoniae by the innate immune system and neutrophil migration. Pneumococcal cell wall components such as lipoteichoic acids, lipoproteins, and peptidoglycan are recognized by TLR2. Following bacterial uptake by phagocytes and their degradation in phagosomes, pneumococcal peptidoglycan is recognized by NOD2. Pattern recognition receptors (PRRs) also sense pneumococcal intracellular molecules such as pneumolysin, genomic DNA, the chaperone protein DnaK, elongation factor Tu (EF-Tu), and glyceraldehyde-3-phosphate dehydrogenase (GAPDH). DNA is recognized by TLR9 within endosomes, whereas pneumolysin, DnaK, EF-Tu, and GAPDH are recognized by TLR4. Additionally, pneumolysin activates the NLRP3 inflammasome. The activation of PRR signaling leads to the transcriptional activation of cytokines and chemokines, which subsequently augments neutrophil migration.
When infectious agents invade the respiratory tract, immune cells and epithelial cells secrete chemokines and cytokines, as described above, promoting neutrophil migration into the lung through the pulmonary capillary walls (Maas et al., 2018). Neutrophils phagocytose and kill infectious agents with the help of reactive oxygen species, antimicrobial proteins, and serine proteases (Teng et al., 2017). An in vitro study demonstrated that neutrophils degrade phagocytized S. pneumoniae via serine proteases such as neutrophil elastase (NE) and cathepsin G (CG), which are stored in azurophilic granules (Standish and Weiser, 2009). NE- and CG-deficient mice exhibit impaired antibacterial defense against S. pneumoniae and decrease murine survival without affecting neutrophil recruitment (Hahn et al., 2011). Furthermore, neutrophil depletion results in profound defects in the clearance of S. pneumoniae in a murine model of pneumonia (Garvy and Harmsen, 1996). This converging evidence indicates that phagocytic function and phagolysosomal degradation of bacteria by neutrophils are crucial strategies for controlling pneumococcal infection.
In addition to their phagocytic function, previous studies have reported that neutrophils release chromatin DNA decorated with granule-derived antimicrobial peptides and enzymes, including NE, CG, α-defensins, and myeloperoxidase (Brinkmann et al., 2004; Papayannopoulos, 2018). These chromatin structures are termed neutrophil extracellular traps (NETs), which degrade virulence factors and kill multiple microbial genera (Brinkmann et al., 2004). The trapping of microbes by NETs may provide several benefits, including reducing the spread of infection by concentrating host antimicrobial agents at infection sites. In bacterial pneumonia, animal and human studies have indicated that NETs are increased in alveolar spaces (Lefrançais et al., 2018; Mikacenic et al., 2018). Furthermore, an in vitro study demonstrated that NETs exhibit significant antibacterial activity against S. pneumoniae (Mori et al., 2012). These findings suggest the functional importance of NETs in pneumococcal pneumonia. However, higher concentrations of NETs have been reported to be associated with reduced hazards of clinical stability and increased mortality in pneumonia (Ebrahimi et al., 2018). In this context, excess NETs released by activated neutrophils have been implicated in promoting tissue damage, including sepsis (Czaikoski et al., 2016), and lung injury (Narasaraju et al., 2011). The mechanisms responsible for NET-induced tissue damage involve NET components such as NE and other proteases that induce cell death in multiple cell types (Yang et al., 1996; Hou et al., 2014; Grechowa et al., 2017; Daniel et al., 2019; Hiyoshi et al., 2019).
A variety of pneumococcal virulence factors have been identified (Brooks and Mias, 2018; Feldman and Anderson, 2020). In the present review, we discuss the virulence factors associated with immune evasion.
The autolytic enzyme, autolysin, is known to be responsible for the characteristic autolytic behavior associated with pneumococci. The major autolysin of S. pneumoniae is N-acetylmuramyl-L-alanine amidase (LytA), which breaks down peptidoglycan (Höltje and Tomasz, 1976). Although the exact in vivo function of autolysis in pneumococcal pathogenesis is unclear, animal studies have demonstrated that pneumococcal strains deficient in LytA are less virulent than wild-type pneumococci (Berry et al., 1989a; Hirst et al., 2008). Recently, Kietzman et al. identified a novel physiological function of LytA. This enzyme was shown to drive rapid capsule shedding in response to antimicrobial peptides in the initial phases of infection (Kietzman et al., 2016). This response increases bacterial resistance to peptides, as well as invasion of the alveolar epithelium. LytA may also contribute to pneumococcal pathogenesis by catalyzing the release of the intracellular toxin Ply (Martner et al., 2008; Domon et al., 2016; Domon et al., 2018a), cell wall degradation products (Tuomanen et al., 1985), and cytosolic proteins (Nagai et al., 2018), which induce immune responses. Additionally, fragments from autolyzed bacteria inhibit phagocytosis of intact bacteria by peripheral blood mononuclear cells (Martner et al., 2009).
Ply is a potent intracellular toxin possessing multiple functions that augment pneumococcal virulence. Ply-deficient mutant strains of S. pneumoniae showed a significant reduction in virulence related to both intranasal and systemic infection (Berry et al., 1989b). Ply toxicity is mainly associated with its ability to induce ring-shaped pores in cholesterol-containing membranes (Tilley et al., 2005). In this regard, Ply has cytotoxic effects on various cell types, including alveolar epithelial cells (Rubins et al., 1993), microvascular endothelial cells (Zysk et al., 2001), and monocytes (Hirst et al., 2002). Thus, the direct cytotoxicity of Ply towards lung tissue is considered to play a primary role in lung injury in pneumococcal pneumonia. However, several studies have demonstrated that in vivo lung injury could be due to inflammation and microvascular leakage caused by Ply, rather than its cytotoxic activity (Witzenrath et al., 2006; García-Suárez et al., 2007; Witzenrath et al., 2007). Although neutrophils are required for the clearance of S. pneumoniae, intranasal or intratracheal infection of mice with wild-type S. pneumoniae demonstrated an increased neutrophil recruitment, increased bacterial burden in the lungs, and a higher prevalence of bacteremia compared to infection with Ply-negative mutant strains (Rubins et al., 1995; Kadioglu et al., 2000). Therefore, the proinflammatory interactions between Ply and neutrophils are considered to play a role in the aggravation of pneumococcal pneumonia. Indeed, Ply is cytotoxic to neutrophils (Cockeran et al., 2001). We demonstrated that Ply induces neutrophil cell death through specific interactions with the P2X7 receptor; whereas Ply is less cytotoxic against P2X7 receptor-negative alveolar epithelial cells and macrophages. This suggests that neutrophils are the primary target cells of Ply (Domon et al., 2016). The subsequent leakage of NE from dead neutrophils disrupts the pulmonary epithelial barrier. Another study demonstrated that Ply induces NET formation, which contains high levels of NE (Nel et al., 2016). Several others have reported pneumococcal evasion of NETs. Pneumococcal surface protein A plays a role in the resistance to NET-mediated killing (Martinez et al., 2019). Meanwhile pneumococcal endonucleases, EndA and TatD, allow the bacterium to degrade the DNA scaffolds of NETs and escape, followed by the release of NE from the NETs (Beiter et al., 2006; Jhelum et al., 2018).
A comprehensive review of the capsule, including its regulation in pathogenesis, capsule synthesis, and the genetic basis for serotype differences, has been published elsewhere (Paton and Trappetti, 2019). Accordingly, current mini reviews mainly focus on immune evasion related to virulence factors. The capsule, which confers protection against phagocytosis, has been extensively studied in the context of pneumococcal virulence (Jonsson et al., 1985; Andre et al., 2017). The capsule impairs bacterial opsonization with C3b/iC3b by the classical and alternative complement pathways and also inhibits the conversion of C3b, which bound to the bacterial surface, to iC3b, thus resulting in a profound inhibition of opsonophagocytosis by neutrophils (Hyams et al., 2010). Additionally, the capsule plays a role in bacterial adherence, colonization of the nasopharynx, and entry into alveolar epithelial cells (Hammerschmidt et al., 2005). Although capsules protect S. pneumoniae against trapping by NETs (Wartha et al., 2007), it has recently been observed that capsules of virulent pneumococcal serotypes enhance the formation of NETs during pneumonia (Moorthy et al., 2016). Moreover, NETs and neutrophil activity in the lungs generally correspond to disease severity after pneumococcal infection (Moorthy et al., 2016).
Neutrophil serine proteases, including NE, CG, and proteinase 3, are critical for the effective functioning of neutrophils, and contribute to immune protection (Pham, 2006). Among these proteases, NE has been well studied in both basic and clinical research. Although NE is a protease that degrades elastin (Janoff and Scherer, 1968), the degradation of foreign organic molecules phagocytosed by neutrophils is considered its main function (Kawabata et al., 2002). NE degrades outer membrane protein localized on the surface of Gram-negative bacteria to exert antimicrobial effects (Belaaouaj et al., 2000). NE-deficient mice are more susceptible to sepsis and death following infection with Gram-negative Klebsiella pneumoniae and Escherichia coli (Belaaouaj et al., 1998). However, the role of NE in Gram-positive bacterial infections remains controversial. It has been reported that NE does not contribute to neutrophil-mediated killing of Gram-positive Staphylococcus aureus (Belaaouaj et al., 1998). Specifically, in S. pneumoniae, NE plays an important role in degrading pneumococcal cell wall-localized aminopeptidase N, and mediates opsonophagocytic killing by neutrophils (Standish and Weiser, 2009; Nganje et al., 2019). However, some pneumococcal strains exhibit resistance to extracellular NE-mediated killing (Van der Windt et al., 2012; Domon et al., 2016).
Despite its fundamental importance in innate immunity, excessive neutrophil activation causes the release of NE, which contributes to tissue damage (Fox et al., 2013; Kovtun et al., 2018). In general, NE exerts potent catalytic effects against a broad array of host extracellular matrix components, including elastin, proteoglycan, fibronectin, and several collagen types (Janusz and Doherty, 1991; Taylor et al., 2018). The cross-linking of collagen and elastin imparts stability and functionality to the lung extracellular matrix, which plays an important role in the formation of alveolar gas exchange units (Mižíková et al., 2015). Many studies have indicated that increased NE activity in the lung is involved in the pathogenesis of various lung diseases such as pneumonia, acute lung injury, exacerbated chronic obstructive pulmonary disease, and cystic fibrosis (Polverino et al., 2017). Indeed, it has been reported that NE-deficient mice are protected to a significantly greater extent from the development of emphysema than wild-type mice (Shapiro et al., 2003). Additionally, N-formyl-methionyl-leucyl-phenylalanine-induced neutrophil influx in alveolar spaces results in decreased lung elastin content and the development of emphysema in mice (Cavarra et al., 1996). Moreover, instillation of NE into the lungs results in the destruction of alveolar walls in animals (Campbell, 2000). One possible mechanism could be that NE cleaves E-cadherin in alveolar epithelial cells which interferes with cell-cell adhesion (Boxio et al., 2016). As for bacterial infection, patients with bacterial pneumonia exhibit increased levels of NE in bronchoalveolar lavage fluid (BALF) (Boutten et al., 1996; Wilkinson et al., 2012), which may result in excessive proteolytic damage and worse clinical outcomes. Generally, NE is inhibited by serum α1-antitrypsin. However, neutrophils also release matrix metalloproteinases (MMPs) that inactivate α1-antitrypsin (Michaelis et al., 1990). Thus, the proteinase inhibitory capacity is decreased in the BALF in patients with bacterial pneumonia compared to that of healthy controls (Abrams et al., 1984).
In animal models, intratracheal pneumococcal infection causes acute lung injury, characterized by an increase in neutrophil accumulation and NE activity in the BALF (Yanagihara et al., 2007; Hagio et al., 2008). Subsequently, extracellular NE impairs the phagocytic activity of macrophages (Domon et al., 2016). Furthermore, NE cleaves extracellular matrix proteins, and proteins associated with the host immune response. In a murine model of bacterial pneumonia, NE, CG, and proteinase 3 cleave surfactant protein D, reducing the ability of the protein to promote bacterial aggregation (Hirche et al., 2004). NE also cleaves multiple cell surface receptors such as TLR2, TLR4, CD14, tumor necrosis factor receptor, and the C5a receptor, leading to an inhibition of downstream signaling (Wiedow and Meyer-Hoffert, 2005; Van den Berg et al., 2014; Domon et al., 2018b). Additionally, multiple cytokines and chemokines, such as interleukin (IL)-1β, IL-2, IL-6, IL-8, IL-12p40, IL-12p70, and tumor necrosis factor, are degraded and inactivated by NE (Wiedow and Meyer-Hoffert, 2005; Clancy et al., 2018; Domon et al., 2018b). Furthermore, we recently reported that NE cleaves human leukocyte antigen class II molecules in both cultured macrophages and in vivo mouse models, indicating that NE may disrupt antigen presentation and T-cell activation (Domon et al., 2021). In contrast, NE cleaves and activates MMP-9, which may also have a destructive role in lung diseases (Jackson et al., 2010). Collectively, these data imply that NE cleaves a variety of host immune proteins, induces lung injury, and may assist pneumococci in evading the immune system during pneumonia (Figure 2).
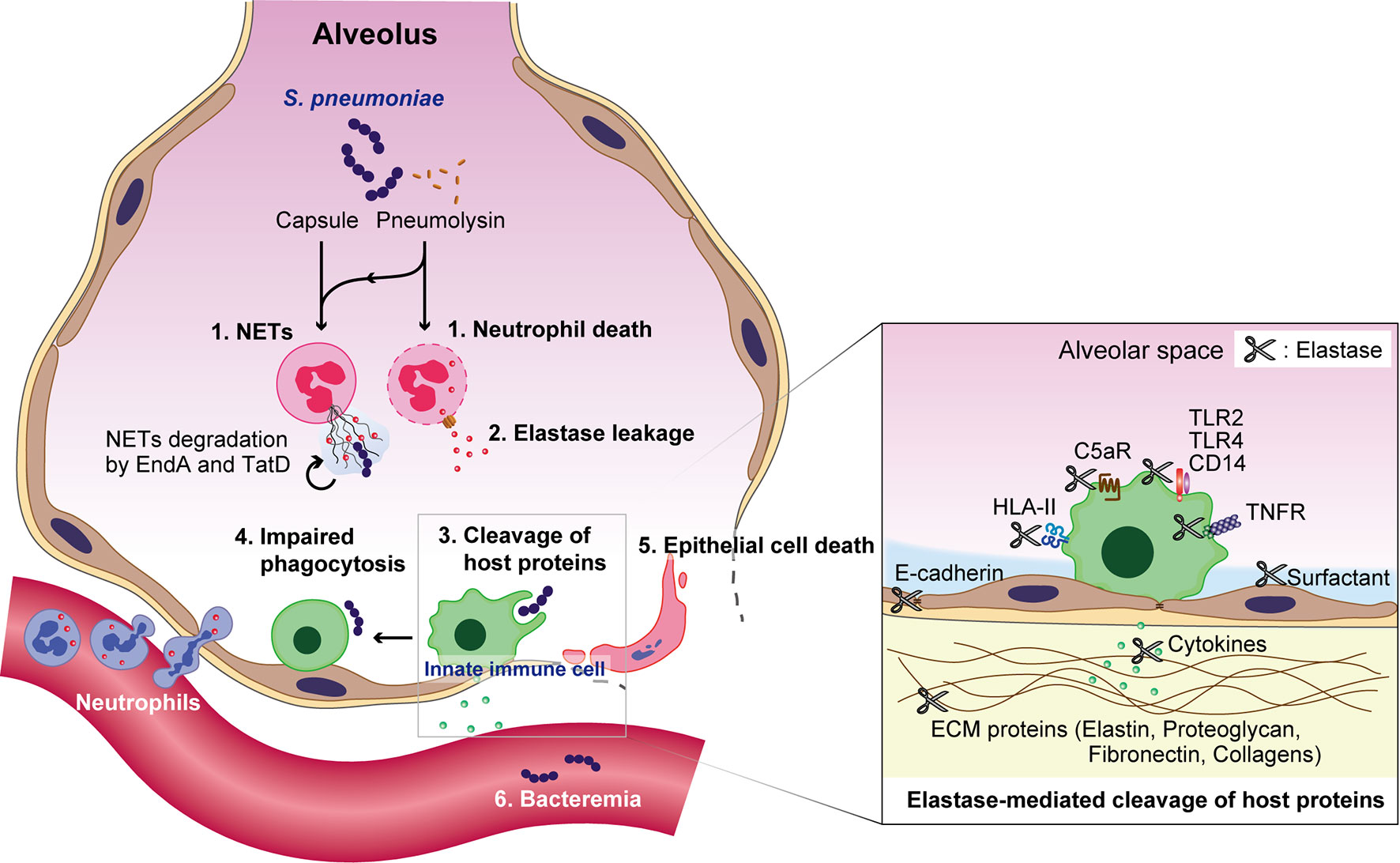
Figure 2 Overview of S. pneumoniae-induced immune subversion by the exploitation of neutrophils during pneumonia. Pneumococcal capsules and pneumolysin enhance the formation of neutrophil extracellular traps (NETs), which are subsequently degraded by the pneumococcal endonucleases EndA and TatD. Pneumolysin also exerts cytotoxicity against neutrophils. The subsequent leakage of neutrophil elastase induces the degradation of surfactant protein D, cell-cell adhesion molecule E-cadherin, and extracellular matrix components, such as elastin, proteoglycan, fibronectin, and several collagen types. Additionally, neutrophil elastase impairs the phagocytic activity of macrophages, induces the death of alveolar epithelial cells, and diminishes the pulmonary epithelial barrier. Furthermore, neutrophil elastase cleaves multiple cell surface proteins, such as toll-like receptor (TLR)2, TLR4, CD14, tumor necrosis factor receptor (TNFR), the C5a receptor (C5aR), and human leukocyte antigen class II (HLA-II); followed by the degradation of multiple cytokines and chemokines, such as interleukin (IL)-1β, IL-2, IL-6, IL-8, IL-12p40, IL-12p70, and tumor necrosis factor, which eventually disrupts the pulmonary immune defense.
Multiple studies have investigated the potential role of various NE inhibitors, such as sivelestat (ONO-5046), AZD9668, EPI-hNE-4, KRP-109, and pre-elafin in different lung diseases (Polverino et al., 2017). In animal models of LPS- or chemical-induced non-infectious acute lung injury, symptoms have been observed to be ameliorated upon treatment with EPI-hNE-4 (Honoré et al., 2004), sivelestat (Sakamaki et al., 1996; Inoue et al., 2005; Iba et al., 2006), or pre-elafin (Vachon et al., 2002). Preclinical and clinical studies have also demonstrated the efficacy of sivelestat and AZD9668 in treating acute lung injury and bronchiectasis, respectively (Tamakuma et al., 2004; Inoue et al., 2006; Fujii et al., 2010; Aikawa et al., 2011; Stockley et al., 2013). In animal models of pneumococcal pneumonia, the administration of sivelestat resulted in higher survival rates and decreased bacterial counts in the blood (Yanagihara et al., 2007; Domon et al., 2018b), suggesting that NE-induced lung injury and immune subversion cause bacterial invasion of the bloodstream followed by death. Mice treated with KRP-109 showed lower neutrophil infiltration and inflammation than control mice, with no effects on viable pneumococcal numbers in the lungs (Yamada et al., 2011). These findings suggest that NE contributes, at least in part, to the pathogenesis of pneumococcal pneumonia. Although only a few studies have investigated the effects of NE inhibitors in patients with bacterial pneumonia, a retrospective study suggested that the early administration of sivelestat improves patient survival rate (Nakamura et al., 2008). Although this finding provides convincing evidence of NE-induced tissue destruction in pneumonia in humans, since neutrophils are the first phagocytic cells recruited to the lung infection site, further randomized controlled trials are required to examine the efficacy of NE inhibitors against bacterial pneumonia.
Accumulating evidence indicates that both pneumococcal virulence factors and host proteases, including NE, are major mediators of lung injury during severe pneumococcal infections. Although the activation of PRRs in response to pneumococcal stimuli, followed by neutrophil infiltration, is key to the initiation of the innate immune response, this host defense strategy can be exploited by pneumococcus in lung tissues. S. pneumoniae targets infiltrated neutrophils and promotes the formation of NETs and cell lysis by utilizing Ply and other virulence factors, which in turn could increase the local NE concentration (Mori et al., 2012; Domon et al., 2016; Nel et al., 2016). Subsequently, elastase-induced proteolysis of extracellular matrix components (Taylor et al., 2018), cell-cell adhesion molecules (Boxio et al., 2016), and host immune molecules (Hirche et al., 2004; Wiedow and Meyer-Hoffert, 2005; Van den Berg et al., 2014; Clancy et al., 2018; Domon et al., 2018b) results in disruption of the alveolar epithelial barrier, which may allow pneumococci to invade the bloodstream. Additionally, several host proteases, including MMPs (Davey et al., 2011), CG, and proteinase 3 (Guyot et al., 2014), may contribute to lung injury. Thus, further basic research is still needed to understand the mechanisms of disease initiation, and to develop novel therapies for lung injury during bacterial pneumonia.
HD wrote the paper. HD and YT further developed and edited the manuscript. All authors contributed to the article and approved the submitted version.
This work was supported by the Japan Society for the Promotion of Science (JSPS) KAKENHI Grant (JP20K09903, JP20K21671, and JP20H03858), the Takeda Science Foundation, and the Kobayashi International Scholarship Foundation.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Abrams, W. R., Fein, A. M., Kucich, U., Kueppers, F., Yamada, H., Kuzmowycz, T., et al. (1984). Proteinase inhibitory function in inflammatory lung disease. I. Acute bacterial pneumonia. Am. Rev. Respir. Dis. 129, 735–741. doi: 10.1164/arrd.1984.129.5.735
Aikawa, N., Ishizaka, A., Hirasawa, H., Shimazaki, S., Yamamoto, Y., Sugimoto, H., et al. (2011). Reevaluation of the efficacy and safety of the neutrophil elastase inhibitor, Sivelestat, for the treatment of acute lung injury associated with systemic inflammatory response syndrome; a phase IV study. Pulm. Pharmacol. Ther. 24, 549–554. doi: 10.1016/j.pupt.2011.03.001
Andre, G. O., Converso, T. R., Politano, W. R., Ferraz, L. F., Ribeiro, M. L., Leite, L. C., et al. (2017). Role of Streptococcus pneumoniae Proteins in Evasion of Complement-Mediated Immunity. Front. Microbiol. 8, 224. doi: 10.3389/fmicb.2017.00224
Bals, R., Hiemstra, P. S. (2004). Innate immunity in the lung: how epithelial cells fight against respiratory pathogens. Eur. Respir. J. 23, 327–333. doi: 10.1183/09031936.03.00098803
Beiter, K., Wartha, F., Albiger, B., Normark, S., Zychlinsky, A., Henriques-Normark, B. (2006). An endonuclease allows Streptococcus pneumoniae to escape from neutrophil extracellular traps. Curr. Biol. 16, 401–407. doi: 10.1016/j.cub.2006.01.056
Belaaouaj, A., Mccarthy, R., Baumann, M., Gao, Z., Ley, T. J., Abraham, S. N., et al. (1998). Mice lacking neutrophil elastase reveal impaired host defense against gram negative bacterial sepsis. Nat. Med. 4, 615–618. doi: 10.1038/nm0598-615
Belaaouaj, A., Kim, K. S., Shapiro, S. D. (2000). Degradation of outer membrane protein A in Escherichia coli killing by neutrophil elastase. Science 289, 1185–1188. doi: 10.1126/science.289.5482.1185
Berry, A. M., Lock, R. A., Hansman, D., Paton, J. C. (1989a). Contribution of autolysin to virulence of Streptococcus pneumoniae. Infect. Immun. 57, 2324–2330. doi: 10.1128/IAI.57.8.2324-2330.1989
Berry, A. M., Yother, J., Briles, D. E., Hansman, D., Paton, J. C. (1989b). Reduced virulence of a defined pneumolysin-negative mutant of Streptococcus pneumoniae. Infect. Immun. 57, 2037–2042. doi: 10.1128/IAI.57.7.2037-2042.1989
Boutten, A., Dehoux, M. S., Seta, N., Ostinelli, J., Venembre, P., Crestani, B., et al. (1996). Compartmentalized IL-8 and elastase release within the human lung in unilateral pneumonia. Am. J. Respir. Crit. Care Med. 153, 336–342. doi: 10.1164/ajrccm.153.1.8542140
Boxio, R., Wartelle, J., Nawrocki-Raby, B., Lagrange, B., Malleret, L., Hirche, T., et al. (2016). Neutrophil elastase cleaves epithelial cadherin in acutely injured lung epithelium. Respir. Res. 17:129. doi: 10.1186/s12931-016-0449-x
Brinkmann, V., Reichard, U., Goosmann, C., Fauler, B., Uhlemann, Y., Weiss, D. S., et al. (2004). Neutrophil extracellular traps kill bacteria. Science 303, 1532–1535. doi: 10.1126/science.1092385
Brooks, L. R. K., Mias, G. I. (2018). Streptococcus pneumoniae’s Virulence and Host Immunity: Aging, Diagnostics, and Prevention. Front. Immunol. 9, 1366. doi: 10.3389/fimmu.2018.01366
Campbell, E. J. (2000). Animal models of emphysema: the next generations. J. Clin. Invest. 106, 1445–1446. doi: 10.1172/JCI11791
Cavarra, E., Martorana, P. A., Gambelli, F., Desanti, M., Vaneven, P., Lungarella, G. (1996). Neutrophil recruitment into the lungs is associated with increased lung elastase burden, decreased lung elastin, and emphysema in alpha1 proteinase inhibitor-deficient mice. Lab. Invest. 75, 273–280.
Clancy, D. M., Sullivan, G. P., Moran, H. B. T., Henry, C. M., Reeves, E. P., Mcelvaney, N. G., et al. (2018). Extracellular Neutrophil Proteases Are Efficient Regulators of IL-1, IL-33, and IL-36 Cytokine Activity but Poor Effectors of Microbial Killing. Cell Rep. 22, 2937–2950. doi: 10.1016/j.celrep.2018.02.062
Cockeran, R., Theron, A. J., Steel, H. C., Matlola, N. M., Mitchell, T. J., Feldman, C., et al. (2001). Proinflammatory interactions of pneumolysin with human neutrophils. J. Infect. Dis. 183, 604–611. doi: 10.1086/318536
Czaikoski, P. G., Mota, J. M., Nascimento, D. C., Sônego, F., Castanheira, F. V., Melo, P. H., et al. (2016). Neutrophil Extracellular Traps Induce Organ Damage during Experimental and Clinical Sepsis. PloS One 11, e0148142. doi: 10.1371/journal.pone.0148142
Daniel, C., Leppkes, M., Muñoz, L. E., Schley, G., Schett, G., Herrmann, M. (2019). Extracellular DNA traps in inflammation, injury and healing. Nat. Rev. Nephrol. 15, 559–575. doi: 10.1038/s41581-019-0163-2
Davey, A., Mcauley, D. F., O’kane, C. M. (2011). Matrix metalloproteinases in acute lung injury: mediators of injury and drivers of repair. Eur. Respir. J. 38, 959–970. doi: 10.1183/09031936.00032111
Davis, K. M., Nakamura, S., Weiser, J. N. (2011). Nod2 sensing of lysozyme-digested peptidoglycan promotes macrophage recruitment and clearance of S. pneumoniae colonization in mice. J. Clin. Invest. 121, 3666–3676. doi: 10.1172/JCI57761
Domon, H., Oda, M., Maekawa, T., Nagai, K., Takeda, W., Terao, Y. (2016). Streptococcus pneumoniae disrupts pulmonary immune defence via elastase release following pneumolysin-dependent neutrophil lysis. Sci. Rep. 6, 38013. doi: 10.1038/srep38013
Domon, H., Maekawa, T., Yonezawa, D., Nagai, K., Oda, M., Yanagihara, K., et al. (2018a). Mechanism of Macrolide-Induced Inhibition of Pneumolysin Release Involves Impairment of Autolysin Release in Macrolide-Resistant Streptococcus pneumoniae. Antimicrob. Agents Chemother. 62, e00161–e00118. doi: 10.1128/AAC.00161-18
Domon, H., Nagai, K., Maekawa, T., Oda, M., Yonezawa, D., Takeda, W., et al. (2018b). Neutrophil Elastase Subverts the Immune Response by Cleaving Toll-Like Receptors and Cytokines in Pneumococcal Pneumonia. Front. Immunol. 9, 732. doi: 10.3389/fimmu.2018.00732
Domon, H., Maekawa, T., Isono, T., Furuta, K., Kaito, C., Terao, Y. (2021). Proteolytic cleavage of HLA class II by human neutrophil elastase in pneumococcal pneumonia. Sci. Rep. 11, 2432. doi: 10.1038/s41598-021-82212-5
Ebrahimi, F., Giaglis, S., Hahn, S., Blum, C. A., Baumgartner, C., Kutz, A., et al. (2018). Markers of neutrophil extracellular traps predict adverse outcome in community-acquired pneumonia: secondary analysis of a randomised controlled trial. Eur. Respir. J. 51, 1701389. doi: 10.1183/13993003.01389-2017
Feldman, C., Anderson, R. (2020). Pneumococcal virulence factors in community-acquired pneumonia. Curr. Opin. Pulm. Med. 26, 222–231. doi: 10.1097/MCP.0000000000000674
Feldman, C. (2004). Clinical relevance of antimicrobial resistance in the management of pneumococcal community-acquired pneumonia. J. Lab. Clin. Med. 143, 269–283. doi: 10.1016/j.lab.2004.02.002
Ferrara, A. M. (2005). New fluoroquinolones in lower respiratory tract infections and emerging patterns of pneumococcal resistance. Infection 33, 106–114. doi: 10.1007/s15010-005-4102-8
Fox, E. D., Heffernan, D. S., Cioffi, W. G., Reichner, J. S. (2013). Neutrophils from critically ill septic patients mediate profound loss of endothelial barrier integrity. Crit. Care 17, R226. doi: 10.1186/cc13049
Fujii, M., Miyagi, Y., Bessho, R., Nitta, T., Ochi, M., Shimizu, K. (2010). Effect of a neutrophil elastase inhibitor on acute lung injury after cardiopulmonary bypass. Interact. Cardiovasc. Thorac Surg. 10, 859–862. doi: 10.1510/icvts.2009.225243
García-Suárez, M. D., Flórez, N., Astudillo, A., Vázquez, F., Villaverde, R., Fabrizio, K., et al. (2007). The role of pneumolysin in mediating lung damage in a lethal pneumococcal pneumonia murine model. Respir. Res. 8, 3. doi: 10.1186/1465-9921-8-3
Garvy, B. A., Harmsen, A. G. (1996). The importance of neutrophils in resistance to pneumococcal pneumonia in adult and neonatal mice. Inflammation 20, 499–512. doi: 10.1007/BF01487042
Grechowa, I., Horke, S., Wallrath, A., Vahl, C. F., Dorweiler, B. (2017). Human neutrophil elastase induces endothelial cell apoptosis by activating the PERK-CHOP branch of the unfolded protein response. FASEB J. 31, 3868–3881. doi: 10.1096/fj.201700012R
Guyot, N., Wartelle, J., Malleret, L., Todorov, A. A., Devouassoux, G., Pacheco, Y., et al. (2014). Unopposed cathepsin G, neutrophil elastase, and proteinase 3 cause severe lung damage and emphysema. Am. J. Pathol. 184, 2197–2210. doi: 10.1016/j.ajpath.2014.04.015
Hagio, T., Kishikawa, K., Kawabata, K., Tasaka, S., Hashimoto, S., Hasegawa, N., et al. (2008). Inhibition of neutrophil elastase reduces lung injury and bacterial count in hamsters. Pulm. Pharmacol. Ther. 21, 884–891. doi: 10.1016/j.pupt.2008.10.002
Hahn, I., Klaus, A., Janze, A. K., Steinwede, K., Ding, N., Bohling, J., et al. (2011). Cathepsin G and neutrophil elastase play critical and nonredundant roles in lung-protective immunity against Streptococcus pneumoniae in mice. Infect. Immun. 79, 4893–4901. doi: 10.1128/IAI.05593-11
Hammerschmidt, S., Wolff, S., Hocke, A., Rosseau, S., Müller, E., Rohde, M. (2005). Illustration of pneumococcal polysaccharide capsule during adherence and invasion of epithelial cells. Infect. Immun. 73, 4653–4667. doi: 10.1128/IAI.73.8.4653-4667.2005
Hartl, D., Tirouvanziam, R., Laval, J., Greene, C. M., Habiel, D., Sharma, L., et al. (2018). Innate Immunity of the Lung: From Basic Mechanisms to Translational Medicine. J. Innate Immun. 10, 487–501. doi: 10.1159/000487057
Hirche, T. O., Crouch, E. C., Espinola, M., Brokelman, T. J., Mecham, R. P., Desilva, N., et al. (2004). Neutrophil serine proteinases inactivate surfactant protein D by cleaving within a conserved subregion of the carbohydrate recognition domain. J. Biol. Chem. 279, 27688–27698. doi: 10.1074/jbc.M402936200
Hirst, R. A., Yesilkaya, H., Clitheroe, E., Rutman, A., Dufty, N., Mitchell, T. J., et al. (2002). Sensitivities of human monocytes and epithelial cells to pneumolysin are different. Infect. Immun. 70, 1017–1022. doi: 10.1128/iai.70.2.1017-1022.2002
Hirst, R. A., Gosai, B., Rutman, A., Guerin, C. J., Nicotera, P., Andrew, P. W., et al. (2008). Streptococcus pneumoniae deficient in pneumolysin or autolysin has reduced virulence in meningitis. J. Infect. Dis. 197, 744–751. doi: 10.1086/527322
Hiyoshi, T., Domon, H., Maekawa, T., Nagai, K., Tamura, H., Takahashi, N., et al. (2019). Aggregatibacter actinomycetemcomitans induces detachment and death of human gingival epithelial cells and fibroblasts via elastase release following leukotoxin-dependent neutrophil lysis. Microbiol. Immunol. 63, 100–110. doi: 10.1111/1348-0421.12672
Höltje, J. V., Tomasz, A. (1976). Purification of the pneumococcal N-acetylmuramyl-L-alanine amidase to biochemical homogeneity. J. Biol. Chem. 251, 4199–4207. doi: 10.1016/S0021-9258(17)33281-7
Honoré, S., Attalah, H. L., Azoulay, E., Soussy, C. J., Saudubray, F., Harf, A., et al. (2004). Beneficial effect of an inhibitor of leukocyte elastase (EPI-hNE-4) in presence of repeated lung injuries. Shock 22, 131–136. doi: 10.1097/01.shk.0000126861.77543.d0
Hou, H. H., Cheng, S. L., Chung, K. P., Wei, S. C., Tsao, P. N., Lu, H. H., et al. (2014). PlGF mediates neutrophil elastase-induced airway epithelial cell apoptosis and emphysema. Respir. Res. 15, 106. doi: 10.1186/s12931-014-0106-1
Hyams, C., Camberlein, E., Cohen, J. M., Bax, K., Brown, J. S. (2010). The Streptococcus pneumoniae capsule inhibits complement activity and neutrophil phagocytosis by multiple mechanisms. Infect. Immun. 78, 704–715. doi: 10.1128/IAI.00881-09
Iba, T., Kidokoro, A., Fukunaga, M., Takuhiro, K., Yoshikawa, S., Sugimotoa, K. (2006). Pretreatment of sivelestat sodium hydrate improves the lung microcirculation and alveolar damage in lipopolysaccharide-induced acute lung inflammation in hamsters. Shock 26, 95–98. doi: 10.1097/01.shk.0000223126.34017.d9
Inoue, Y., Seiyama, A., Tanaka, H., Ukai, I., Akimau, P., Nishino, M., et al. (2005). Protective effects of a selective neutrophil elastase inhibitor (sivelestat) on lipopolysaccharide-induced acute dysfunction of the pulmonary microcirculation. Crit. Care Med. 33, 1814–1822. doi: 10.1097/01.ccm.0000172547.54086.ad
Inoue, Y., Tanaka, H., Ogura, H., Ukai, I., Fujita, K., Hosotsubo, H., et al. (2006). A neutrophil elastase inhibitor, sivelestat, improves leukocyte deformability in patients with acute lung injury. J. Trauma 60, 936–943. doi: 10.1097/01.ta.0000217271.25809.a0
Jackson, P. L., Xu, X., Wilson, L., Weathington, N. M., Clancy, J. P., Blalock, J. E., et al. (2010). Human Neutrophil Elastase-Mediated Cleavage Sites of MMP-9 and TIMP-1: Implications to Cystic Fibrosis Proteolytic Dysfunction. Mol. Med. 16, 159–166. doi: 10.2119/molmed.2009.00109
Janoff, A., Scherer, J. (1968). Mediators of inflammation in leukocyte lysosomes. IX. Elastinolytic activity in granules of human polymorphonuclear leukocytes. J. Exp. Med. 128, 1137–1155. doi: 10.1084/jem.128.5.1137
Janusz, M. J., Doherty, N. S. (1991). Degradation of cartilage matrix proteoglycan by human neutrophils involves both elastase and cathepsin G. J. Immunol. 146, 3922–3928.
Jhelum, H., Sori, H., Sehgal, D. (2018). A novel extracellular vesicle-associated endodeoxyribonuclease helps Streptococcus pneumoniae evade neutrophil extracellular traps and is required for full virulence. Sci. Rep. 8, 7985. doi: 10.1038/s41598-018-25865-z
Jonsson, S., Musher, D. M., Chapman, A., Goree, A., Lawrence, E. C. (1985). Phagocytosis and killing of common bacterial pathogens of the lung by human alveolar macrophages. J. Infect. Dis. 152, 4–13. doi: 10.1093/infdis/152.1.4
Kadioglu, A., Andrew, P. W. (2004). The innate immune response to pneumococcal lung infection: the untold story. Trends Immunol. 25, 143–149. doi: 10.1016/j.it.2003.12.006
Kadioglu, A., Gingles, N. A., Grattan, K., Kerr, A., Mitchell, T. J., Andrew, P. W. (2000). Host cellular immune response to pneumococcal lung infection in mice. Infect. Immun. 68, 492–501. doi: 10.1128/iai.68.2.492-501.2000
Kadioglu, A., Weiser, J. N., Paton, J. C., Andrew, P. W. (2008). The role of Streptococcus pneumoniae virulence factors in host respiratory colonization and disease. Nat. Rev. Microbiol. 6, 288–301. doi: 10.1038/nrmicro1871
Kawabata, K., Hagio, T., Matsuoka, S. (2002). The role of neutrophil elastase in acute lung injury. Eur. J. Pharmacol. 451, 1–10. doi: 10.1016/s0014-2999(02)02182-9
Kietzman, C. C., Gao, G., Mann, B., Myers, L., Tuomanen, E. I. (2016). Dynamic capsule restructuring by the main pneumococcal autolysin LytA in response to the epithelium. Nat. Commun. 7, 10859. doi: 10.1038/ncomms10859
Koppe, U., Suttorp, N., Opitz, B. (2012). Recognition of Streptococcus pneumoniae by the innate immune system. Cell. Microbiol. 14, 460–466. doi: 10.1111/j.1462-5822.2011.01746.x
Kovtun, A., Messerer, D. A. C., Scharffetter-Kochanek, K., Huber-Lang, M., Ignatius, A. (2018). Neutrophils in Tissue Trauma of the Skin, Bone, and Lung: Two Sides of the Same Coin. J. Immunol. Res. 2018, 8173983. doi: 10.1155/2018/8173983
Le Polain De Waroux, O., Flasche, S., Prieto-Merino, D., Edmunds, W. J. (2014). Age-dependent prevalence of nasopharyngeal carriage of Streptococcus pneumoniae before conjugate vaccine introduction: a prediction model based on a meta-analysis. PloS One 9, e86136. doi: 10.1371/journal.pone.0086136
Lefrançais, E., Mallavia, B., Zhuo, H., Calfee, C. S., Looney, M. R. (2018). Maladaptive role of neutrophil extracellular traps in pathogen-induced lung injury. JCI Insight 3, e98178. doi: 10.1172/jci.insight.98178
Liu, N., Zheng, Z., Chen, P., Hou, P., Wang, X., Li, H., et al. (2015). [Detection of aspiration of nasopharyngeal secretion and the relationship between the aspiration of nasopharyngeal secretion and the incidence of pneumonia]. Zhonghua Jie He He Hu Xi Za Zhi 38, 511–515.
Maas, S. L., Soehnlein, O., Viola, J. R. (2018). Organ-Specific Mechanisms of Transendothelial Neutrophil Migration in the Lung, Liver, Kidney, and Aorta. Front. Immunol. 9, 2739. doi: 10.3389/fimmu.2018.02739
Malley, R., Henneke, P., Morse, S. C., Cieslewicz, M. J., Lipsitch, M., Thompson, C. M., et al. (2003). Recognition of pneumolysin by toll-like receptor 4 confers resistance to pneumococcal infection. Proc. Natl. Acad. Sci. U.S.A. 100, 1966–1971. doi: 10.1073/pnas.0435928100
Martinez, P. J., Farhan, A., Mustafa, M., Javaid, N., Darkoh, C., Garrido-Sanabria, E., et al. (2019). PspA facilitates evasion of pneumococci from bactericidal activity of neutrophil extracellular traps (NETs). Microb. Pathog. 136, 103653. doi: 10.1016/j.micpath.2019.103653
Martner, A., Dahlgren, C., Paton, J. C., Wold, A. E. (2008). Pneumolysin released during Streptococcus pneumoniae autolysis is a potent activator of intracellular oxygen radical production in neutrophils. Infect. Immun. 76, 4079–4087. doi: 10.1128/IAI.01747-07
Martner, A., Skovbjerg, S., Paton, J. C., Wold, A. E. (2009). Streptococcus pneumoniae autolysis prevents phagocytosis and production of phagocyte-activating cytokines. Infect. Immun. 77, 3826–3837. doi: 10.1128/IAI.00290-09
McNeela, E. A., Burke, A., Neill, D. R., Baxter, C., Fernandes, V. E., Ferreira, D., et al. (2010). Pneumolysin Activates the NLRP3 Inflammasome and Promotes Proinflammatory Cytokines Independently of TLR4. PloS Pathog. 6, e1001191. doi: 10.1371/journal.ppat.1001191
Michaelis, J., Vissers, M. C., Winterbourn, C. C. (1990). Human neutrophil collagenase cleaves α1-antitrypsin. Biochem. J. 270, 809–814. doi: 10.1042/bj2700809
Mikacenic, C., Moore, R., Dmyterko, V., West, T. E., Altemeier, W. A., Liles, W. C., et al. (2018). Neutrophil extracellular traps (NETs) are increased in the alveolar spaces of patients with ventilator-associated pneumonia. Crit. Care 22, 358. doi: 10.1186/s13054-018-2290-8
Mižíková, I., Ruiz-Camp, J., Steenbock, H., Madurga, A., Vadász, I., Herold, S., et al. (2015). Collagen and elastin cross-linking is altered during aberrant late lung development associated with hyperoxia. Am. J. Physiol. Lung Cell. Mol. Physiol. 308, L1145–L1158. doi: 10.1152/ajplung.00039.2015
Mogensen, T. H., Paludan, S. R., Kilian, M., Ostergaard, L. (2006). Live Streptococcus pneumoniae, Haemophilus influenzae, and Neisseria meningitidis activate the inflammatory response through Toll-like receptors 2, 4, and 9 in species-specific patterns. J. Leukoc. Biol. 80, 267–277. doi: 10.1189/jlb.1105626
Moorthy, A. N., Rai, P., Jiao, H., Wang, S., Tan, K. B., Qin, L., et al. (2016). Capsules of virulent pneumococcal serotypes enhance formation of neutrophil extracellular traps during in vivo pathogenesis of pneumonia. Oncotarget 7, 19327–19340. doi: 10.18632/oncotarget.8451
Mori, Y., Yamaguchi, M., Terao, Y., Hamada, S., Ooshima, T., Kawabata, S. (2012). α-Enolase of Streptococcus pneumoniae induces formation of neutrophil extracellular traps. J. Biol. Chem. 287, 10472–10481. doi: 10.1074/jbc.M111.280321
Nagai, K., Domon, H., Maekawa, T., Oda, M., Hiyoshi, T., Tamura, H., et al. (2018). Pneumococcal DNA-binding proteins released through autolysis induce the production of proinflammatory cytokines via toll-like receptor 4. Cell. Immunol. 325, 14–22. doi: 10.1016/j.cellimm.2018.01.006
Nagai, K., Kimura, O., Domon, H., Maekawa, T., Yonezawa, D., Terao, Y. (2019). Antimicrobial susceptibility of Streptococcus pneumoniae, Haemophilus influenzae, and Moraxella catarrhalis clinical isolates from children with acute otitis media in Japan from 2014 to 2017. J. Infect. Chemother. 25, 229–232. doi: 10.1016/j.jiac.2018.08.018
Nakamura, S., Yanagihara, K., Izumikawa, K., Seki, M., Kakeya, H., Yamamoto, Y., et al. (2008). [Efficacy of sivelestat for acute lung injury due to severe bacterial pneumonia with systemic inflammatory response syndrome]. Nihon Kokyuki Gakkai Zasshi 46, 793–797.
Narasaraju, T., Yang, E., Samy, R. P., Ng, H. H., Poh, W. P., Liew, A. A., et al. (2011). Excessive neutrophils and neutrophil extracellular traps contribute to acute lung injury of influenza pneumonitis. Am. J. Pathol. 179, 199–210. doi: 10.1016/j.ajpath.2011.03.013
Nel, J. G., Theron, A. J., Durandt, C., Tintinger, G. R., Pool, R., Mitchell, T. J., et al. (2016). Pneumolysin activates neutrophil extracellular trap formation. Clin. Exp. Immunol. 184, 358–367. doi: 10.1111/cei.12766
Nganje, C. N., Haynes, S. A., Qabar, C. M., Lent, R. C., Ghanem, E. N. B., Shainheit, M. G. (2019). PepN is a non-essential, cell wall-localized protein that contributes to neutrophil elastase-mediated killing of Streptococcus pneumoniae. PloS One 14, e0211632. doi: 10.1371/journal.pone.0211632
Papayannopoulos, V. (2018). Neutrophil extracellular traps in immunity and disease. Nat. Rev. Immunol. 18, 134–147. doi: 10.1038/nri.2017.105
Paton, J. C., Trappetti, C. (2019). Streptococcus pneumoniae Capsular Polysaccharide. Microbiol. Spectr. 7. doi: 10.1128/microbiolspec.GPP3-0019-2018
Pham, C. T. N. (2006). Neutrophil serine proteases: specific regulators of inflammation. Nat. Rev. Immunol. 6, 541–550. doi: 10.1038/nri1841
Polverino, E., Rosales-Mayor, E., Dale, G. E., Dembowsky, K., Torres, A. (2017). The role of neutrophil elastase inhibitors in lung diseases. Chest 152, 249–262. doi: 10.1016/j.chest.2017.03.056
Rubins, J. B., Duane, P. G., Clawson, D., Charboneau, D., Young, J. A., Niewoehner, D. E. (1993). Toxicity of Pneumolysin to Pulmonary Alveolar Epithelial Cells. Infect. Immun. 61, 1352–1358. doi: 10.1128/IAI.61.4.1352-1358.1993
Rubins, J. B., Charboneau, D., Paton, J. C., Mitchell, T. J., Andrew, P. W., Janoff, E. N. (1995). Dual function of pneumolysin in the early pathogenesis of murine pneumococcal pneumonia. J. Clin. Invest 95, 142–150. doi: 10.1172/JCI117631
Sakamaki, F., Ishizaka, A., Urano, T., Sayama, K., Nakamura, H., Terashima, T., et al. (1996). Effect of a specific neutrophil elastase inhibitor, ONO-5046, on endotoxin-induced acute lung injury. Am. J. Respir. Crit. Care Med. 153, 391–397. doi: 10.1164/ajrccm.153.1.8542148
Shapiro, S. D., Goldstein, N. M., Houghton, A. M., Kobayashi, D. K., Kelley, D., Belaaouaj, A. (2003). Neutrophil elastase contributes to cigarette smoke-induced emphysema in mice. Am. J. Pathol. 163, 2329–2335. doi: 10.1016/S0002-9440(10)63589-4
Smith, H. C., German, E., Ferreira, D. M., Rylance, J. (2019). Nasopharyngeal colonisation with Streptococcus pneumoniae in malnourished children: a systematic review and meta-analysis of prevalence. Trans. R. Soc. Trop. Med. Hyg. 113, 227–233. doi: 10.1093/trstmh/try139
Standish, A. J., Weiser, J. N. (2009). Human neutrophils kill Streptococcus pneumoniae via serine proteases. J. Immunol. 183, 2602–2609. doi: 10.4049/jimmunol.0900688
Stockley, R., De Soyza, A., Gunawardena, K., Perrett, J., Forsman-Semb, K., Entwistle, N., et al. (2013). Phase II study of a neutrophil elastase inhibitor (AZD9668) in patients with bronchiectasis. Respir. Med. 107, 524–533. doi: 10.1016/j.rmed.2012.12.009
Takeuchi, O., Akira, S. (2010). Pattern recognition receptors and inflammation. Cell 140, 805–820. doi: 10.1016/j.cell.2010.01.022
Tamakuma, S., Ogawa, M., Aikawa, N., Kubota, T., Hirasawa, H., Ishizaka, A., et al. (2004). Relationship between neutrophil elastase and acute lung injury in humans. Pulm. Pharmacol. Ther. 17, 271–279. doi: 10.1016/j.pupt.2004.05.003
Taylor, S., Dirir, O., Zamanian, R. T., Rabinovitch, M., Thompson, A. A. R. (2018). The Role of Neutrophils and Neutrophil Elastase in Pulmonary Arterial Hypertension. Front. Med. 5, 217. doi: 10.3389/fmed.2018.00217
Teng, T. S., Ji, A. L., Ji, X. Y., Li, Y. Z. (2017). Neutrophils and Immunity: From Bactericidal Action to Being Conquered. J. Immunol. Res. 2017, 9671604. doi: 10.1155/2017/9671604
Tilley, S. J., Orlova, E. V., Gilbert, R. J., Andrew, P. W., Saibil, H. R. (2005). Structural basis of pore formation by the bacterial toxin pneumolysin. Cell 121, 247–256. doi: 10.1016/j.cell.2005.02.033
Tomlinson, G., Chimalapati, S., Pollard, T., Lapp, T., Cohen, J., Camberlein, E., et al. (2014). TLR-mediated inflammatory responses to Streptococcus pneumoniae are highly dependent on surface expression of bacterial lipoproteins. J. Immunol. 193, 3736–3745. doi: 10.4049/jimmunol.1401413
Tuomanen, E., Liu, H., Hengstler, B., Zak, O., Tomasz, A. (1985). The induction of meningeal inflammation by components of the pneumococcal cell wall. J. Infect. Dis. 151, 859–868. doi: 10.1093/infdis/151.5.859
Vachon, E., Bourbonnais, Y., Bingle, C. D., Rowe, S. J., Janelle, M. F., Tremblay, G. M. (2002). Anti-inflammatory effect of pre-elafin in lipopolysaccharide-induced acute lung inflammation. Biol. Chem. 383, 1249–1256. doi: 10.1515/BC.2002.138
Van Den Berg, C. W., Tambourgi, D. V., Clark, H. W., Hoong, S. J., Spiller, O. B., Mcgreal, E. P. (2014). Mechanism of neutrophil dysfunction: neutrophil serine proteases cleave and inactivate the C5a receptor. J. Immunol. 192, 1787–1795. doi: 10.4049/jimmunol.1301920
Van Der Windt, D., Bootsma, H. J., Burghout, P., Van Der Gaast-De Jongh, C. E., Hermans, P. W., Van Der Flier, M. (2012). Nonencapsulated Streptococcus pneumoniae resists extracellular human neutrophil elastase- and cathepsin G-mediated killing. FEMS Immunol. Med. Microbiol. 66, 445–448. doi: 10.1111/j.1574-695X.2012.01028.x
Wartha, F., Beiter, K., Albiger, B., Fernebro, J., Zychlinsky, A., Normark, S., et al. (2007). Capsule and D-alanylated lipoteichoic acids protect Streptococcus pneumoniae against neutrophil extracellular traps. Cell. Microbiol. 9, 1162–1171. doi: 10.1111/j.1462-5822.2006.00857.x
Weiser, J. N., Ferreira, D. M., Paton, J. C. (2018). Streptococcus pneumoniae: transmission, colonization and invasion. Nat. Rev. Microbiol. 16, 355–367. doi: 10.1038/s41579-018-0001-8
Wiedow, O., Meyer-Hoffert, U. (2005). Neutrophil serine proteases: potential key regulators of cell signalling during inflammation. J. Intern. Med. 257, 319–328. doi: 10.1111/j.1365-2796.2005.01476.x
Wilkinson, T. S., Morris, A. C., Kefala, K., O’kane, C. M., Moore, N. R., Booth, N. A., et al. (2012). Ventilator-Associated Pneumonia Is Characterized by Excessive Release of Neutrophil Proteases in the Lung. Chest 142, 1425–1432. doi: 10.1378/chest.11-3273
Witzenrath, M., Gutbier, B., Hocke, A. C., Schmeck, B., Hippenstiel, S., Berger, K., et al. (2006). Role of pneumolysin for the development of acute lung injury in pneumococcal pneumonia. Crit. Care Med. 34, 1947–1954. doi: 10.1097/01.CCM.0000220496.48295.A9
Witzenrath, M., Gutbier, B., Owen, J. S., Schmeck, B., Mitchell, T. J., Mayer, K., et al. (2007). Role of platelet-activating factor in pneumolysin-induced acute lung injury. Crit. Care Med. 35, 1756–1762. doi: 10.1097/01.CCM.0000269212.84709.23
Yamada, K., Yanagihara, K., Araki, N., Harada, Y., Morinaga, Y., Izumikawa, K., et al. (2011). In vivo efficacy of KRP-109, a novel elastase inhibitor, in a murine model of severe pneumococcal pneumonia. Pulm. Pharmacol. Ther. 24, 660–665. doi: 10.1016/j.pupt.2011.08.001
Yanagihara, K., Fukuda, Y., Seki, M., Izumikawa, K., Miyazaki, Y., Hirakata, Y., et al. (2007). Effects of specific neutrophil elastase inhibitor, sivelestat sodium hydrate, in murine model of severe pneumococcal pneumonia. Exp. Lung Res. 33, 71–80. doi: 10.1080/01902140701198500
Yang, J. J., Kettritz, R., Falk, R. J., Jennette, J. C., Gaido, M. L. (1996). Apoptosis of endothelial cells induced by the neutrophil serine proteases proteinase 3 and elastase. Am. J. Pathol. 149, 1617–1626.
Yoshimura, A., Lien, E., Ingalls, R. R., Tuomanen, E., Dziarski, R., Golenbock, D. (1999). Cutting edge: recognition of Gram-positive bacterial cell wall components by the innate immune system occurs via Toll-like receptor 2. J. Immunol. 163, 1–5.
Keywords: innate immunity, neutrophil, pneumonia, pneumolysin, neutrophil elastase, Streptococcus pneumoniae, virulence factor
Citation: Domon H and Terao Y (2021) The Role of Neutrophils and Neutrophil Elastase in Pneumococcal Pneumonia. Front. Cell. Infect. Microbiol. 11:615959. doi: 10.3389/fcimb.2021.615959
Received: 10 October 2020; Accepted: 01 March 2021;
Published: 16 March 2021.
Edited by:
Victor Nizet, University of California, San Diego, United StatesReviewed by:
Elaine I. Tuomanen, St. Jude Children’s Research Hospital, United StatesCopyright © 2021 Domon and Terao. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Hisanori Domon, aGlzYS1kb21vbkBkZW50Lm5paWdhdGEtdS5hYy5qcA==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.