
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Cell. Infect. Microbiol., 03 March 2021
Sec. Parasite and Host
Volume 11 - 2021 | https://doi.org/10.3389/fcimb.2021.614665
This article is part of the Research TopicAdvances in the Molecular Biology of Trypanosomatid PathogensView all 14 articles
 Carlos Talavera-López1,2*
Carlos Talavera-López1,2* Louisa A. Messenger3
Louisa A. Messenger3 Michael D. Lewis3
Michael D. Lewis3 Matthew Yeo3
Matthew Yeo3 João Luís Reis-Cunha4Gabriel Machado Matos5
João Luís Reis-Cunha4Gabriel Machado Matos5 Daniella C. Bartholomeu4
Daniella C. Bartholomeu4 José E. Calzada6
José E. Calzada6 Azael Saldaña6
Azael Saldaña6 Juan David Ramírez7
Juan David Ramírez7 Felipe Guhl8Sofía Ocaña-Mayorga9Jaime A. Costales9Rodion Gorchakov10
Felipe Guhl8Sofía Ocaña-Mayorga9Jaime A. Costales9Rodion Gorchakov10 Kathryn Jones10
Kathryn Jones10 Melissa S. Nolan10Santuza M. R. Teixeira11
Melissa S. Nolan10Santuza M. R. Teixeira11 Hernán José Carrasco12Maria Elena Bottazzi10Peter J. Hotez10
Hernán José Carrasco12Maria Elena Bottazzi10Peter J. Hotez10 Kristy O. Murray10
Kristy O. Murray10 Mario J. Grijalva9,13Barbara Burleigh14
Mario J. Grijalva9,13Barbara Burleigh14 Edmundo C. Grisard15
Edmundo C. Grisard15 Michael A. Miles3
Michael A. Miles3 Björn Andersson1*
Björn Andersson1*Trypanosoma cruzi, a zoonotic kinetoplastid protozoan parasite, is the causative agent of American trypanosomiasis (Chagas disease). Having a very plastic, repetitive and complex genome, the parasite displays a highly diverse repertoire of surface molecules, with pivotal roles in cell invasion, immune evasion and pathogenesis. Before 2016, the complexity of the genomic regions containing these genes impaired the assembly of a genome at chromosomal level, making it impossible to study the structure and function of the several thousand repetitive genes encoding the surface molecules of the parasite. We here describe the genome assembly of the Sylvio X10/1 genome sequence, which since 2016 has been used as a reference genome sequence for T. cruzi clade I (TcI), produced using high coverage PacBio single-molecule sequencing. It was used to analyze deep Illumina sequence data from 34 T. cruzi TcI isolates and clones from different geographic locations, sample sources and clinical outcomes. Resolution of the surface molecule gene distribution showed the unusual duality in the organization of the parasite genome, a synteny of the core genomic region with related protozoa flanked by unique and highly plastic multigene family clusters encoding surface antigens. The presence of abundant interspersed retrotransposons in these multigene family clusters suggests that these elements are involved in a recombination mechanism for the generation of antigenic variation and evasion of the host immune response on these TcI strains. The comparative genomic analysis of the cohort of TcI strains revealed multiple cases of such recombination events involving surface molecule genes and has provided new insights into T. cruzi population structure.
Trypanosoma cruzi is a kinetoplastid protozoan and the etiologic agent of Chagas disease, considered one of the most important human parasitic disease in Latin America. The Global Burden of Disease Study 2013 reported that almost 7 million people live with Chagas disease in the Western Hemisphere (GBD 2013 Mortality and Causes of Death Collaborators, 2015), with the expectation that up to one third will progress to develop chronic chagasic cardiomyopathy (CCC) or other life-threatening symptoms. In 2015, 5,742,167 people were estimated to be infected with T. cruzi in 21 Latin American countries and around 13% of the Latin American population is at risk of contracting T. cruzi infection due to domicile infestation of triatomine bugs or due to non-vectorial transmission via blood transfusion, organ transplant, oral, congenital or accidental infection (“WHO | 6 February 2015, Vol. 90, 6 (pp. 33–44)” 2015). Human Chagas disease is not restricted to Latin America. The migration of infected humans to non-endemic areas has made it a new public health threat in other geographic areas such as North America, Europe and Asia (Bern, 2015). Also, sylvatic T. cruzi transmission cycles, often associated with human disease, have been described in areas formerly considered as free from this disease such as in Texas (USA) (Bern, 2015).
The acute phase of the disease frequently lacks specific symptoms, is often undiagnosed and usually resolves in a few weeks in immunocompetent individuals but may be fatal in around 5% of diagnosed cases. Without successful treatment, a T. cruzi infection is normally carried for life. The disease progresses to either a chronic indeterminate phase that is asymptomatic, or to a chronic symptomatic phase with severe clinical syndromes such as cardiomyopathy, megaesophagus and/or megacolon (Rassi et al., 2010); meningoencephalitis may occur, especially in immunocompromised patients (Bern, 2015). The current prolonged chemotherapy (benznidazole or nifurtimox) is mostly effective only in the acute phase, particularly because severe side effects may interrupt treatment of adults in the chronic phase. There is currently no effective treatment for advanced chagasic cardiomyopathy (Morillo et al., 2015), and there is an urgent need to identify new potential drug and vaccine targets (Pecoul et al., 2016).
T. cruzi infection is a zoonosis, and the parasite has a complex life cycle; where transmission to humans occurs most frequently by contamination with infected feces from triatomine insect vectors (Subfamily Triatominae). The parasite evades the immune responses with the aid of multiple surface molecules from three large diverse gene families (Trans-Sialidases, Mucins and Mucin-Associated Surface Proteins - MASPs), which are also involved in cell invasion and possibly pathogenicity (Schenkman et al., 1994; Frasch, 2000; Yoshida and Cortez, 2008; Osorio et al., 2012).
Six distinct genetic clades of T. cruzi have been recognized, named TcI to TcVI (Discrete Typing Units or DTU-I to VI). The first genome sequence for T. cruzi was produced using Sanger sequencing technology from a hybrid, highly polymorphic, TcVI strain. The resultant genome sequence, while extremely useful for the core regions of the genome, was highly fragmented, especially in repetitive regions (El-Sayed et al., 2005). This sequence has been improved using enhanced scaffolding algorithms, but many repetitive regions remain unresolved (Weatherly et al., 2009). Subsequently, FLX 454 Titanium and Illumina sequencing were used to sequence a less polymorphic TcI strain (Sylvio X10/1), which allowed the first comparative genomic studies of T. cruzi, but correct assembly of repetitive regions was still impossible (Franzén et al., 2011; Franzén et al., 2012). The thousands of related genes that code for the surface proteins are generally located in large multigene family clusters of the T. cruzi genome (Kim et al., 2005), in the form of extremely repetitive segments with multiple gene copies and pseudogenes. These multigene family clusters are distinct from the core regions of the genome, defined as regions that share gene content and synteny with the genomes of other trypanosomatids (Llewellyn et al., 2009). The repetitive nature of the tandem arrays, and the length of the repeats, made correct assembly impossible using short and medium-sized sequence reads. The available T. cruzi genome sequences were therefore incomplete and inaccurate in these important regions, making it impossible to study the complex surface gene families in contrast to conserved core genomic regions (Berná et al., 2018).
We made a near-complete reconstruction of the majority (~98.5% of the estimated genome size) of the T. cruzi TcI Sylvio X10/1 genome available in Genbank and TriTrypDB in 2016, and it was described in a preprint made available in June 2018 (Talavera-López et al., 2018). This sequence has served as a main genome sequence for T. cruzi clade 1, since it was made public. We were able to decipher the majority of the organisation of T. cruzi surface protein coding gene repertoire from the TcI Sylvio X10/1 strain, revealing large numbers of evenly spaced retrotransposons, which may play a role in generating genomic structural diversity and antigenic variation. This study has been followed by several others using similar approaches and parasite strains (Callejas-Hernández et al., 2018; Reis-Cunha et al., 2018; Schwabl et al., 2019).
The population structure of T. cruzi is complex, and there is a high degree of genetic and phenotypic variation. The current TcI to TcVI clades are based on biochemical and molecular markers (Zingales et al., 2012), although there is substantial diversity even within these six groups (Llewellyn et al., 2009). The TcI clade is widespread and can be found across the American continent, and has been associated with CCC (Ramírez et al., 2010) and sudden death (Bern et al., 2011; Montgomery et al., 2014), among other clinical manifestations. In conjunction with the Sylvio X10/1 genome sequence, we generated Illumina whole-genome sequencing data for 34 T. cruzi TcI isolates and clones from different geographic locations for comparative analyses. These data was used to carry out population genetics studies, where strains from different environments and geographic locations were compared. We found patterns of active recombination possibly associated with the generation of new surface molecule variants. These studies contributed to answering longstanding questions on the biology of Chagas disease and host-parasite interaction in general. The availability of the close to complete repertoire of genes encoding surface molecules allows further research on virulence and pathogenesis, as well as the identification of drug targets and vaccine candidates, focused on shared and conserved motifs present within these variable families.
The Trypanosoma cruzi Sylvio X10/1 strain was isolated from an acute human case of Chagas disease in Brazil. Total genomic DNA of this TcI strain was obtained from culture epimastigotes as formerly described 11 and used to produce PacBio CCS data according to standard protocols from the Genomic Facility of Science for Life Laboratory (Sweden) and Pacific Biosciences (USA). Genomic DNA was sequenced to a depth of 210X using the PacBio platform, supplying raw reads with an average length of 5.8 Kb. These reads were corrected by means of the PBcR v8.3 pipeline with the MHAP algorithm (Berlin et al., 2015) using the auto-correction parameters described to merge haplotypes and skipping the assembly step, producing a total of 1,216 contigs (NG50 = 62 Kb). Illumina sequences at an average coverage of approximately 120X, with a mean read length of 101 bp were added. The reads were trimmed from adaptors and filtered using the Nesoni utility (https://github.com/Victorian-Bioinformatics-Consortium/nesoni), which is now part of Tail Tools (https://github.com/Monash-RNA-Systems-Biology-Laboratory/tail-tools) in order to remove bases with a quality score < 20 and length < 75.
Later, the assembly was scaffolded using the corrected PacBio reads with the SSPACE-Long scaffolder yielding 310 scaffolds (NG50 = 788 Kb); 118 gaps were filled using Illumina reads with GapFiller and corrected PacBio reads with PBJelly2. Finally, the core regions of these scaffolds were aligned against the core regions of the TcVI CL Brener reference genome using ABACAS (http://abacas.sourceforge.net), producing 47 scaffolds, henceforth designated as chromosomes. The quality of the new assembly was assessed with FRC_bam with the Illumina paired end reads generated at the Genomic Facility of Science for Life Laboratory (Sweden) using the same genomic DNA extraction used for PacBio sequencing. The final genome size was 41382871 bp in 47 scaffolds. The number of gaps was 1005, and they are indicated by rows of Ns in the sequence.
The genome sequence was annotated using a new kinetoplastid genome annotation pipeline combining homology-based gene model transfer with de novo gene prediction. To allow for the sensitive identification of partial genes, input sequences were split at stretches of undefined bases, effectively creating a set of ‘pseudocontigs’, each of which does not contain any gaps. Gene finding was then performed on both the original sequences and the pseudocontigs using AUGUSTUS, which also calls partial genes at the boundaries of each pseudocontig. The minimum ORF length that was considered for annotation was 50 amino acids to allow for the identification of short peptides that were supported by a contig at least twice the length of a read. AUGUSTUS models were trained on 800 genes randomly sampled from the 41 Esmeraldo-type (TcII) T. cruzi CL Brener chromosomes in GeneDB. Protein-DNA alignments of reference proteins against the new T. cruzi sequences, generated using Exonerate, were additionally used to improve the accuracy of the gene prediction. In addition, the RATT software was used to transfer highly conserved gene models from the T. cruzi CL Brener annotation to the target. A non-redundant set of gene models was obtained by merging the results of both RATT and AUGUSTUS and, for each maximal overlapping set of gene models, selecting the non-overlapping subset that maximizes the total length of the interval covered by the models, weighted by varying levels of a priori assigned confidence. Spurious low-confidence protein coding genes with a reading direction in disagreement with the directions of the polycistronic transcriptional units were removed automatically. The result of this integration process was then merged with ncRNA annotations produced by specific tools such as ARAGORN and Infernal. Finally, protein-DNA alignments with frame shifts produced by BLAST were used in a computational approach to identify potential pseudogenes in the remaining sequence.
Downstream of the structural annotation phase, gene models were automatically assigned IDs and further extended with product descriptions and GO terms, both transferred from CL Brener orthologs and inferred from Pfam protein domain hits and represented as feature attributes or Sequence Ontology-typed subfeatures tagged with appropriate evidence codes. This annotation pipeline has been implemented in the Companion web server. The assembled genome was scanned for small RNAs using INFERNAL against the curated RFAM database using cmsearch with a minimum e-value of 1 ×10-10, a GC-bias of 0 and a minimum alignment length of 10 nt. This annotation process has been implemented into the web-based annotation pipeline COMPANION (Steinbiss et al., 2016) from the Wellcome Trust Sanger Institute.
Repetitive sequences were annotated using RepeatMasker with the NCBI+ search engine and LTRHarvest. Using the genomic coordinates of the repetitive elements, the genome was split in windows of 10 Kb to identify VIPER and L1Tc retroelements adjacent to surface molecule genes (i.e: trans-sialidases, mucins and MASP). A one-sided Fisher’s exact test was used to evaluate if the retroelements were enriched in genomic segments containing surface molecule genes.
An improved short-read mapping strategy was used to assign the reads to their target sequences with high accuracy, especially in regions rich in simple and low complexity repeats, by taking advantage of the statistical read placement implemented in the Stampy read mapper to accurately call genomic variants from the mapped reads. Reads from all 34 T. cruzi TcI isolates (SRA BioProject accession number: PRJNA325924) were mapped against the assembled T. cruzi Sylvio X10/1 genome using a two-step mapping process to improve the mapping of Illumina data to highly repetitive regions: First, reads were mapped using BWA MEM with default parameters; later, the BAM file produced by BWA was remapped with Stampy (v1.23) using the –bamkeepgoodreads option. The final mapping file was sorted and filtered for PCR duplicates using Picard Tools v1.137. Variants were called using FreeBayes with a minimum per-base quality of 30, minimum mapping quality of 30 and minimum coverage of 15 bases. Variants that were found in a potentially misassembled region were excluded from the analysis. Additionally, genomic variants were called using FermiKit—which is an assembly-based variant caller—to validate the genomic variants observed in subtelomeric regions. A consensus of the two methods was used as a final set of variants for downstream analyses. Haplotypes were phased using Beagle r1399. The phased markers were used for downstream analyses with SNPrelate and VCFtools and the functional effect of the identified variants was predicted using SnpEff.
Genomic structural variants were identified within TcI isolates, using a consensus of different methods: Delly2, Lumpy, FermiKit and FindTranslocations (https://github.com/vezzi/FindTranslocations.git) using both raw reads and realigned BAM files. For each method, an SV must had a depth of coverage > 10 reads and a mapping quality of > 30. Later, a consensus was created with all the SV that were supported by all the methods. SVs that were supported by FermiKit and at least one of the mapping-based methods were also included but labeled as ‘Low Confidence’. SVs identified by only one method were not included. Breakpoint analysis was done with custom Python scripts and their functional effect was predicted using SNPeff. Analyses of copy number variation (CNV) were done using the BAM files for each sample with the Control-FREEC package. Determination of the fixation index (Fst) using VCFTools was carried out for the T. cruzi CG and FcHc clones obtained from human TcI isolates from Colombia (Supplementary Table 1). For each strain, replicate clones from the original sample were isolated and cultured under the same conditions, and five of the replicates from each sample were sequenced in the Illumina HiSeq 2500 run.
The final Sylvio X10/1 (TcI) genome assembly reconstructed 98.5% of the genome size, as previously estimated by analyzing gene content, and was contained in 47 chromosomes (Figure 1A) assembled from 210 X PacBio sequence data and a previous Illumina data set (Table 1). We tentatively refer to these as chromosomes in this paper, even though more verification of the complete karyotype is needed. Reads corresponding to mitochondrial DNA, kDNA, were removed by homology searches and were thus not included in the analysis. Comparison with the available short read assembly of the TcVI strain CL Brener revealed conserved core syntenic blocks composed of stretches of homologous sequences separated by non-syntenic regions (Figure 1B and Supplementary Table 2) that corresponded to regions that were in some cases initially not reconstructed in the hybrid TcVI strain, but have been partially resolved in later versions of this genome sequence. The non-syntenic regions mostly contained surface molecule gene arrays, and other repeated regions. In some cases, we found possible other gene-rich regions that were expanded in longer CL Brener chromosomes (Figure 1B). The length of the PacBio reads and the high coverage allowed the reconstruction of long stretches of repetitive sequences in the Sylvio X10/1 genome that could previously not be resolved using shorter read data for this genome.
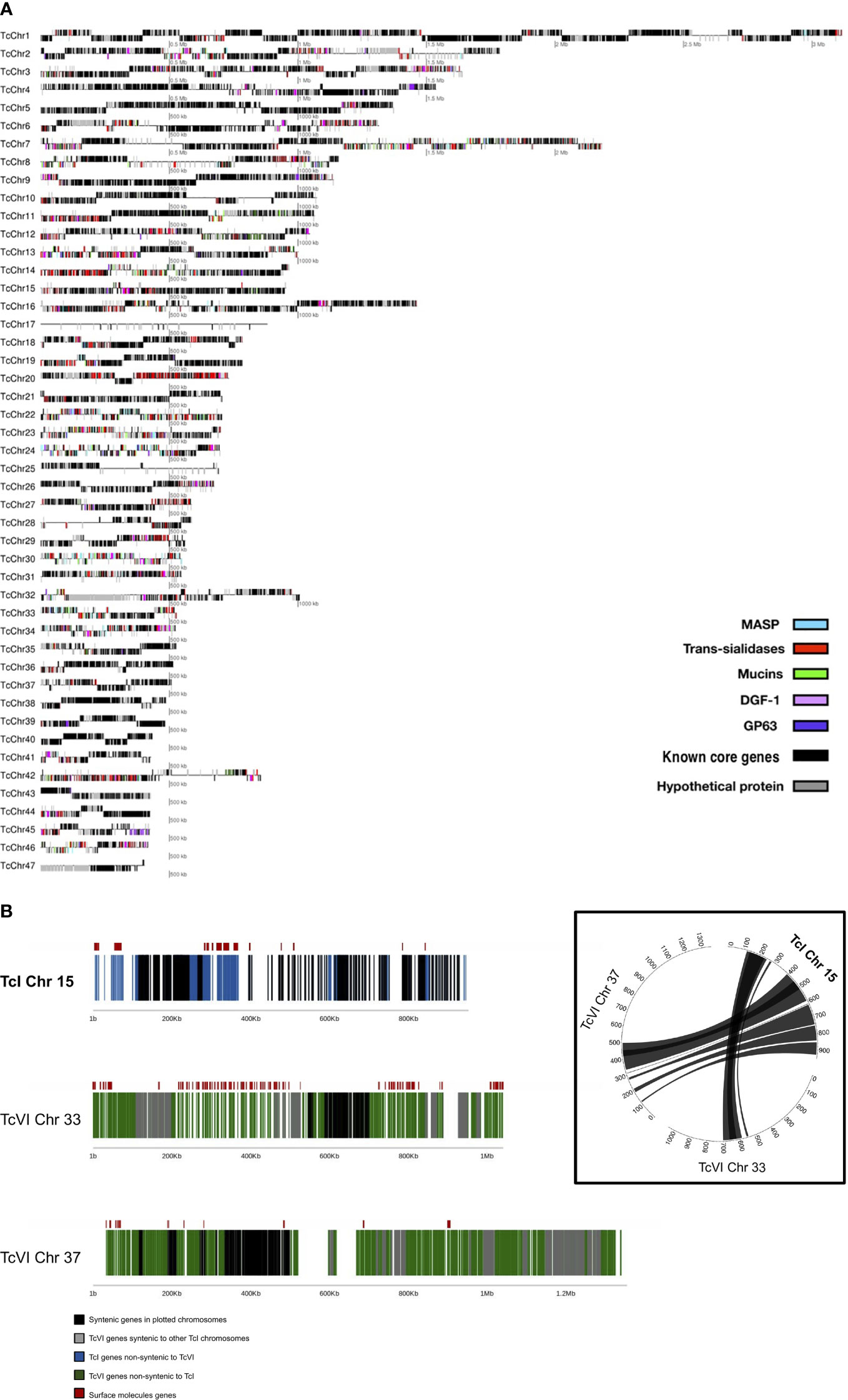
Figure 1 (A) Distribution of surface molecule gene tandem arrays in the 47 chromosomes of the TcI Sylvio X10/1 genome. In this image, each line corresponds to an assembled putative chromosome drawn in proportion to its size, where genes corresponding to the largest T. cruzi multigene families are represented by colored boxes, hypothetical genes are represented by gray boxes and known core genes as black boxes. The position of the gene boxes above or below the line corresponds to the direction of transcription. (B) Comparison of chromosome 15 from the TcI Sylvio X10/1 assembly with TcVI CL Brener chromosomes containing syntenic blocks. The TcI (Sylvio X10/1) chromosome is depicted with black boxes indicating genes that have a CL Brener homolog, and blue boxes showing genes with no synteny with CL Brener. For TcVI (CL Brener) green boxes indicate genes that have no Sylvio X10/1 homolog. The red boxes above these lines show the location of complete genes coding for large surface molecule gene family members. In the circle plot, the lines between chromosomes represent regions of synteny between orthologous genes.

Table 1 Trypanosoma cruzi Sylvio X10/1 strain (Tc-I) genome assembly.
The coverage of genomic regions coding for surface molecules was similar to that of known non-repetitive regions, which supported the correct reconstruction of these areas with a limited amount of assembly errors. To further investigate the quality of the new assembly, Illumina short reads were mapped and analyzed with FRC_bam, which revealed assembly artefacts related to low coverage, wrong paired-end read orientation, and higher than expected sequencing coverage in regions with long stretches of simple repeats. Coverage data based on mapping Illumina reads to the final genome sequence is presented in Supplementary Figures 1 and 2.
Repetitive elements comprised 18.43% of the TcI Sylvio X10/1 genome, 2.18% of which could not be classified using the repeat databases. LINE retroelements of the R1/Jockey group (3.63%) and VIPER LTRs (2.87%) were found to be the most prevalent types of retroelements, covering 6.89% of the genome, which is much higher than the 2.57% estimated from the previously published Sylvio X10/1 draft assembly using short reads (Franzén et al., 2012).
Although retrotransposons were found to be present throughout the genome, the frequency of VIPER and L1Tc elements was markedly higher in multigene family-rich regions and they were found within one kilobase of pseudogenes, hypothetical proteins and surface molecule gene tandem arrays (One-sided Fisher exact test, p-value < 1.32 × 10-16). This distribution indicates that these elements may contribute to increased recombination activity in the gene family clusters by providing a source of microhomology. We do not have experimental evidence for the activity of these retroelements in T. cruzi and it is unknown if they directly affect gene expression.
Simple and low complexity repeats were observed surrounding surface molecule coding sequences and were also more abundant in the multigene family regions (2.18%), extending up to 4 Kb, compared to core regions (0.98%) where they were much shorter (10–120 bp). The most prevalent type of simple repeat had the (C)n motif (11.7%), (TG)n repeat motif (5.6%) and (CA)n repeat motif (5.1%); each variable in length. The microhomology of these simple subtelomeric repeats may facilitate recombination resulting in new surface molecule variants, as described in other parasitic protozoa, including Trypanosoma brucei and Plasmodium falciparum (Hall et al., 2013; Claessens et al., 2014). However, it is noteworthy to mention that such subtelomeric regions are far less complex and shorter in T. brucei (African, virulent) and T. rangeli (Stoco et al., 2014) (American, non-virulent).
Based on our annotation approach, a total of 19,096 genes were identified in the TcI Sylvio X10/1 haploid genome sequence. The public CL Brener genome assembly has 11,106 annotated genes in one haplotype, 10, 596 in the other, and 3,397 in smaller contigs. We have previously estimated the total gene content of CL Brener, based on read coverage, to approximately 22,570 for the haploid genome (Arner et al., 2007). This is mostly due to the larger size of the multigene family clusters in the TcVI hybrid genome. The genome sequence was longer and less fragmented than the version generated previously using short-read sequencing of the same strain (Franzén et al., 2011), which indicated resolution of additional regions of the genome. About 24.1% (n = 4,602) of the total annotated genes were truncated, mostly due to the introduction of premature stop codons, and 67% of these were located within surface molecule gene arrays, sharing motifs of the complete genes.
The new assembly allowed an improved analysis of the T. cruzi surface molecule gene repertoire. While the regions can be described as large gene arrays that contain genes from different surface molecule gene families, the genes of each of the three major surface molecules families were mostly organized as multiple smaller groups or tandem arrays within the larger regions. After genome annotation, the total number of such smaller arrays were: trans-sialidases, 312, with 2,048 complete gene copies and 201 pseudogenes; mucins 98, with 2,466 complete copies and 111 pseudogenes; MASPs 264, with 1,888 complete copies and 245 pseudogenes. These three surface molecule gene families comprised 16.02 Mbp (39.04%) of the TcI Sylvio X10/1 genome and presented a high level of sequence diversity (Figure 1A). Sequence strand switches often delimited the surface molecule tandem arrays. Commonly, these arrays had two to four complete copies immediately followed by two or more truncated copies with motifs similar to the complete gene. The intergenic spaces between arrays were rich in simple and low complexity repeats with no identifiable regulatory elements. The VIPER and L1Tc retrotransposon elements, in clusters of two to four copies, were found in the proximity of, or inside, tandem arrays containing trans-sialidases, mucins and MASP genes. As the surface molecule genes are known to evolve rapidly and be highly variable (Andersson, 2011), the enrichment of VIPER and L1Tc elements in these regions supports the hypothesis that they may be involved in generating new surface molecule gene variants via recombination mediated by sequence homology.
Both Ser/Thr kinases and DEAD-box RNA helicase genes were found at both extremes of 34 (10.81%) trans-sialidase arrays located in chromosomes 1, 2, and 8. Searches against the RFAM database identified 1,618 small RNAs in the TcI Sylvio X10/1 genome. These were mostly ribosomal RNAs with the 5S rDNA subunit being the most common (31.9%) followed by ACA Box snoRNAs (30.9%), SSU rDNA (12.2%) and LSU rDNA (10.2%) subunits. We also found hits to telomerase RNA component (TERC), Catabolite Repression Control sequester (CrcZ), Protozoa Signal Recognition Particle RNAs, spliceosomal RNA subunits and miRNAs. The putative miRNAs identified in Sylvio X10/1 belong to the MIR2118 and MIR1023 families, previously not found in protozoan parasites. The functional relevance of these predicted small RNAs will need to be further validated in vitro. The miRNA segments were located in both strands within 1 Kb of genes coding for DEAD-box RNA helicases surrounding surface molecule gene tandem arrays.
Intra-TcI genomic diversity was examined among 34 samples from six countries: United States, Mexico, Panama, Colombia, Venezuela and Ecuador, derived from a range of triatomine vectors and human patients of different clinical stages (Table 2 and Supplementary Table 1). Our hybrid variant calling strategy, combined with removal of repeat elements and repeated genes, allowed us to identify genomic variants in the core and multigene family clusters in a reliable fashion (See methods).
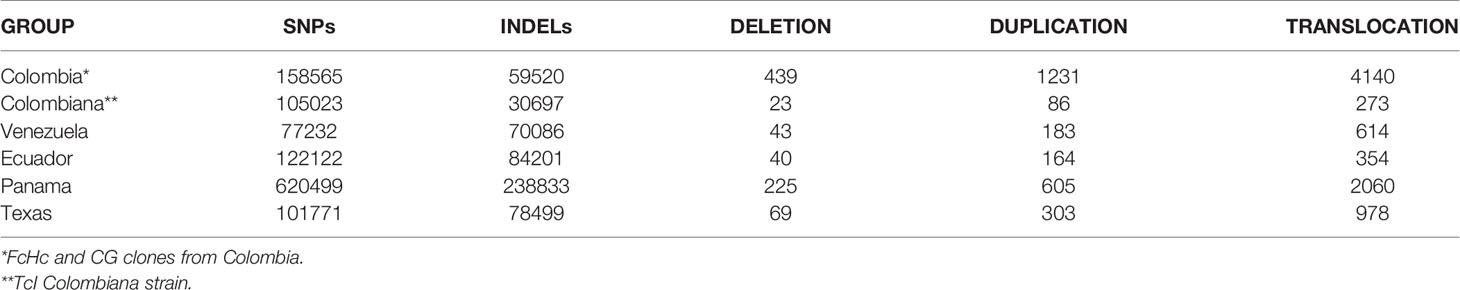
Table 2 Genomic variants identified among the Trypanosoma cruzi TcI isolates.
A total of 1,031,785 SNPs and 279,772 INDELs shorter than 50 bp were called for all sequenced TcI isolates relative to the Sylvio X10/1 genome. INDELs presented an average density of 5.3 variants per Kb and SNPs 24.1 variants per Kb. An individual T. cruzi TcI isolate was found to contain an average of 61,000 SNPs and 6,820 INDELs with a density of 31.8 variants per Kb. However, these measures fluctuated depending on the geographical and biological source of the sample. Core regions had an average SNP density of 0.4 variants per Kb, in contrast with surface molecule multigene family clusters where approx. 10 variants per Kb were found. It was not surprising that the bulk of the genomic variants were located in the multigenic family clusters regions in all the isolates, with fewer differences in the core regions. Although several studies using single gene markers have identified heterogeneity in the TcI clade (Llewellyn et al., 2009; Guhl and Ramírez, 2011), the extent of this variation had not previously been assessed genome-wide.
The majority of INDELs (96%) were found in intergenic or noncoding regions, and 81% of these were located in surface molecule multigene family regions. INDELs within coding sequences were exclusively found to cause frameshifts turning the affected coding sequence into a pseudogene. This distribution of INDELs is a genomic signature that has been associated with non-allelic homologous recombination due to unequal crossing over (Parks et al., 2015) or microhomology-mediated end joining (Sfeir and Symington, 2015; Weckselblatt et al., 2015) (Table 3). However, these repair mechanisms, or something similar, have not yet been shown to be present in T. cruzi. Short insertions were more prevalent than short deletions, a pattern common to all the analyzed TcI genomes when compared to Sylvio X10/1. In the subtelomeric regions, short insertions (1–3 bp) occurred within the upstream and downstream portions of the coding sequences and usually involved the addition of one or more cytosines or guanines. Single-base pair deletions of an adenine or thymine were also observed within these regions, but at a lower frequency. Longer deletions (5–20 bp) and insertions (8–10 bp) were observed within trans-sialidases, Retrotransposon Hot Spot (RHS), pseudogenes and, at a lower frequency, L1Tc retroelements.

Table 3 Patterns of INDELs and their associated mechanisms of origin.
We used the short genomic variants to analyze the population genomics of the T. cruzi TcI clade, and where possible taking into account the different sample sources (insect vector or human host), clinical outcome of the infected patients and geographic locations (Supplementary Table 1). This sampling strategy allowed comparison of parasite population structure in different environments. Interestingly, a Bayesian PCA analysis using INDELs and IBD-based hierarchical clustering using only SNPs from core regions for all the samples showed a mostly geography-specific population structure (Figures 2A, B).
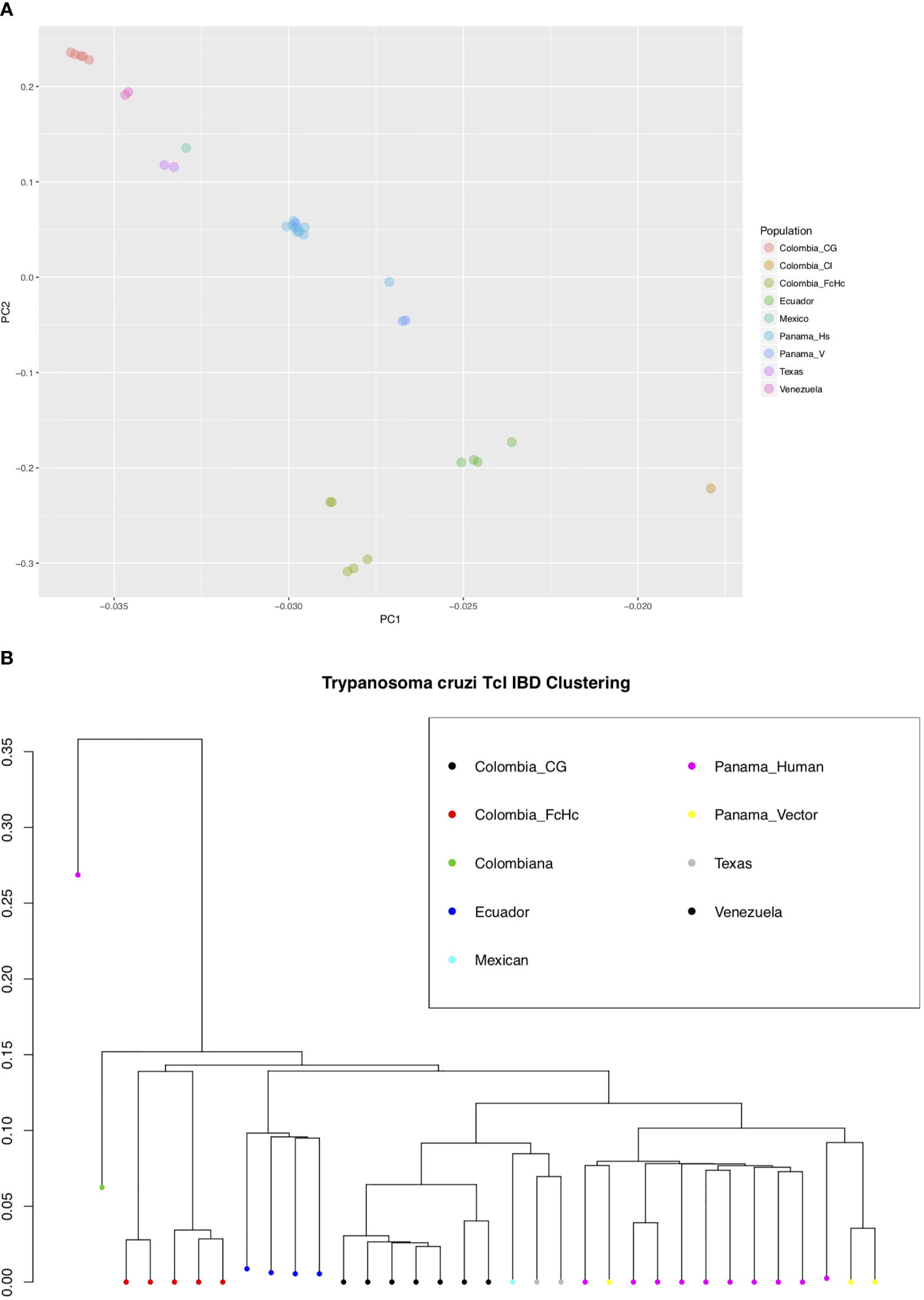
Figure 2 (A) Bayesian principal component analysis (PCA) of T. cruzi TcI strains using INDELs. The percentage of variance for PC1 was 46.2 and for PC2 28.6. (B) Identity by Descent (IBD) dendogram of T. cruzi TcI strains using SNPs, calculated using 1000 bootstraps. Both analyses, using different markers, support the population structure of the analyzed TcI samples. Notably, the highly virulent TcI Colombiana and the Panamanian TcI H1 from a chronic patient are presented as outliers [(B), far left].
The analysis of the variation between two Colombian TcI isolates made it possible to compare parasites from a HIV-positive patient with fatal cardiomyopathy (CG) and from an acute chagasic patient infected by oral transmission (FcHc). To increase accuracy, repeat elements, repeated genes and repeated surface molecule family genes were excluded from this analysis, while core regions and non-repeated, unique surface molecule genes and other genes were kept, in order to have markers outside the core regions. The latter were selected by lower sequence similarity to known surface molecule genes. A total of 158,565 well-supported SNPs, called by both GATK and FreeBayes, was selected from these clones, and used to calculate global and per-site population genetic statistics. These samples displayed distinctive behavior in a global analysis of genomic diversity by separating into two well-defined clusters, as can be seen in Figures 2A, B. Linkage Disequilibrium (LD) analyses were performed genome-wide for both groups using the r2 statistic; revealing a fluctuating pattern of LD across the entire genome with large blocks of low r2 values—implying a recombinatorial process—present at distinctive chromosomal locations that were specific to each group of clones. Particularly, CG clones had less genetic diversity than FcHc clones (Figure 3A) and displayed a trend toward LD, whereas FcHc clones presented a more dynamic LD pattern. Values of r2 near zero were more common in LD sliding windows containing genes coding for surface molecules and r2 values closer to one were present exclusively in core regions rich in housekeeping genes, indicating that these regions are more stable. For the CG and FcHc clones we calculated a global Fixation index (Fst) value of -0.9377958 and -0.1162212, respectively (Figure 3B). These values are consistent with genetic differentiation in recombination hotspots in the multigene family regions. The global Tajima’s D value for the CG clones was 1.373 and 0.9906 for FcHc clones, suggesting the presence of multiple alleles at variable frequencies in both populations (Figure 3C). This pattern was more evident in the multigene family regions, which is consistent with balancing selection of surface molecules. The values for each set of clones were calclulated separately, from different sets of SNPs, which makes a direct comparison in smaller regions difficult. We were only able to detect the larger patterns described above.
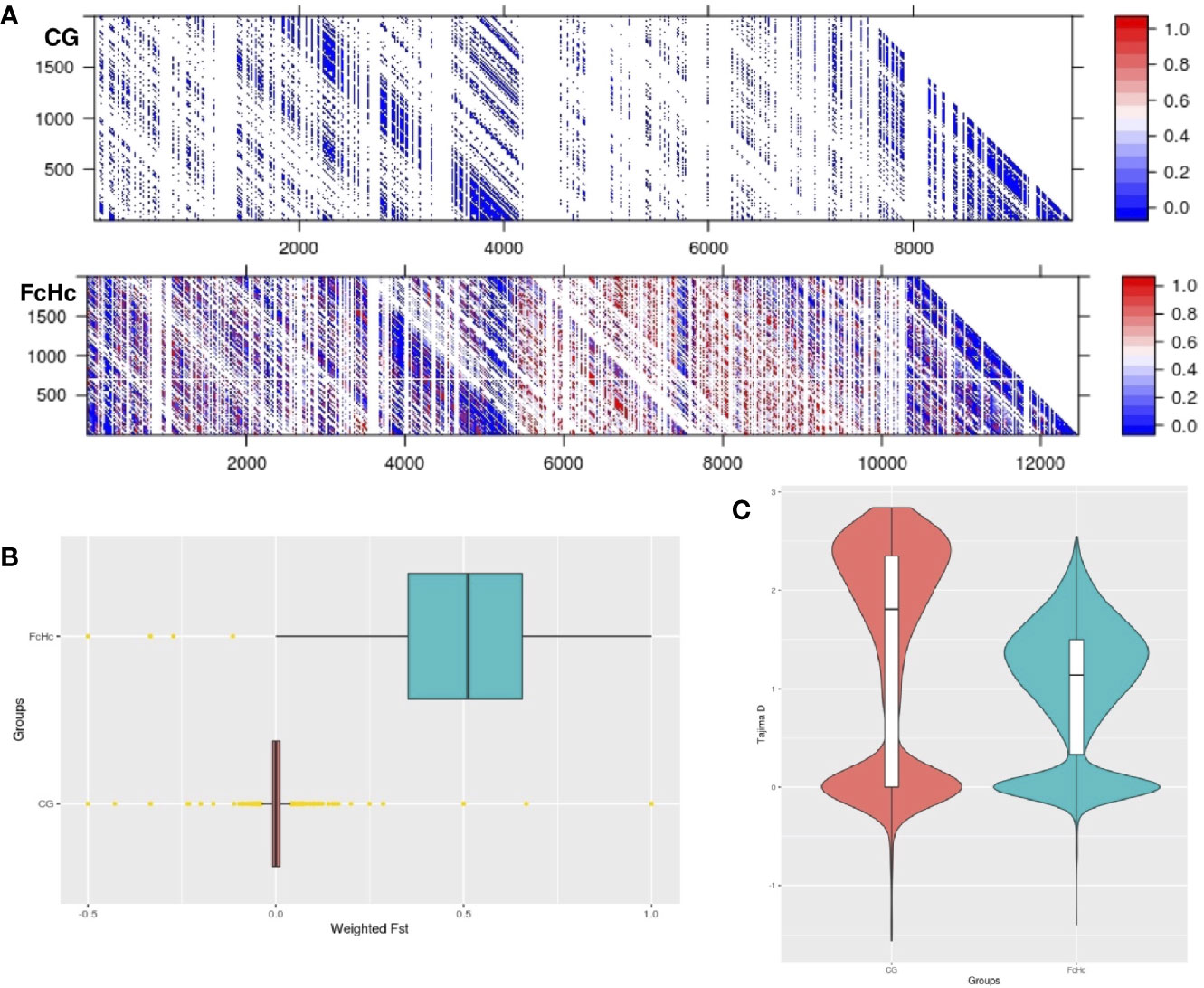
Figure 3 (A) Linkage disequilibrium matrix (r2) of chromosome 2 for the Colombian CG and FcHc clones. LD values range from 0 (recombination) to 1 (no recombination). (B) Genome-wide Fst distribution in 10 Kb bins displaying a state of panmixia for the CG clones and moderate genetic differentiation in the FcHc clones, yellow dots represent outlier bins. The differences in chromosome length are caused by missing sequences in these strains, that resulted in regions with no mapping that were removed from the analysis. (C) Distribution of subtelomeric Tajima’s D selection test in both groups displaying overall balancing selection (D > 0) in these regions for both clones.
Analyses of genomic variation between samples isolated from humans and vectors from Mexico, Panama and Ecuador revealed that the global genetic differentiation among samples isolated from vectors was Fst = 0.1289547 whereas for samples isolated from humans the observed was Fst = -0.05521983. The patterns of linkage disequilibrium between human and vector derived isolates were similar to those observed in the Colombian clones. Estimates of the Tajima’s D statistic revealed a distinctive pattern of selection between the two groups. Balancing selection was detected specifically in regions containing tandem gene arrays coding for surface molecules in all the samples derived from vectors, regardless of their geographical origin; whereas selective sweeps were present in the same regions in human-derived samples. Large genomic areas (> 50 Kb) containing surface molecule genes displayed negative Tajima’s D values in human-derived isolates, in contrast with the pattern observed in vector-derived isolates with long genomic stretches (> 70 Kb) of positive Tajima’s D values and short genomic blocks (< 5 Kb) with negative values. We speculate that these patterns may be caused by selection pressure from the immune system in human-derived strains, which is absent in strains that have grown in insects for extended times.
Genomic structural variants, such as deletions, tandem and interspersed duplications, genomic inversions and chromosomal break-ends, were observed ubiquitously throughout the genomes of the analyzed TcI strains. The most common type of intrachromosomal structural variant observed was tandem duplications followed by deletions larger than 50 Kb (Table 2). Chromosomal break-ends, similar to the unbalanced chromosomal translocations observed in many eumetazoans, were the most abundant type of structural rearrangement and they were only present in surface molecule multigene family regions that were statistically enriched with retroelements and simple repeats. The recombination breakpoints were found to occur at simple repeats and retrotransposons of the VIPER and L1Tc class.
The detected recombination events were found to involve fragments ranging between 20–150 Kb in length and in most cases contained fragments or even complete coding sequences for surface molecule genes, such as trans-sialidases, mucins and MASP genes and other surface glycoproteins (gp63/gp85). Housekeeping genes did not appear to have been affected by these genomic rearrangements. We detected several instances where rearrangements resulted in altered or longer coding sequences by superimposing fragments—or the entire coding sequence—on genes of the same family located in a different genomic location. This was found to have occurred both in the Colombian and the Texas isolates by recombination between trans-sialidases from different chromosomes. The biological relevance of the new altered gene sequences is not known.
Retroelements could be found within or near genomic regions containing surface molecule gene tandem arrays and L1Tc fragments or their entire sequence were also found near all the observed rearrangements, where they were inserted into regions containing simple repeats composed by AT dimers. Data on the genomic positions for repeat elements have been listed in Supplementary Tables 3–5.
Multiple such examples of the generation of possible new surface molecule gene variants were identified in TcI. It is a possibility that the parasite uses specific molecular mechanisms of recombination that can rapidly generate surface molecule diversity, allowing it to increase the genomic plasticity required to adapt to changing environments and evade immune responses during short and long-term infections in various host species.
The sizes of the tandem duplications ranged from 6 to 75 Kb and mainly involved tandem arrays coding for surface molecules, mostly trans-sialidases and mucins, but also Dispersed Gene Family 1 (DGF-1) and several hypothetical proteins. The breakpoints of these duplications were surrounded by simple repeats and retroelements in multigene family regions. A tandem duplication event could involve between four and 25 copies of a specific gene when in the surface multigene family regions, whereas in core regions the number was between two and eight. We observed that large deletions occurring in multigene family regions were surrounded by simple repeats of the type (T)n and (AT)n and retrotransposons of the L1Tc class, containing surface molecule gene tandem arrays. Deletions in these genomic regions tended to be shorter (4–12 Kb) and sample-specific.
CNV varied extensively between T. cruzi TcI strains. There have been previous attempts to assess CNV in the T. cruzi genome (Minning et al., 2011), but these studies were performed using DNA tiling microarrays with probes designed using the TcVI CL Brener strain assembly, in which multigenefamily regions are more difficult to study.
The distribution of CNV in the genomes of the studied TcI samples involved segments of an average size of 5 Kb. We observed blocks of segmental CNV within a chromosome with a pattern that was unique to each sample. Notably, the Colombian clones presented individual profiles of CNV (Figures 4A, B and Supplementary Figures 3–8) despite being derived from the same clinical isolates.
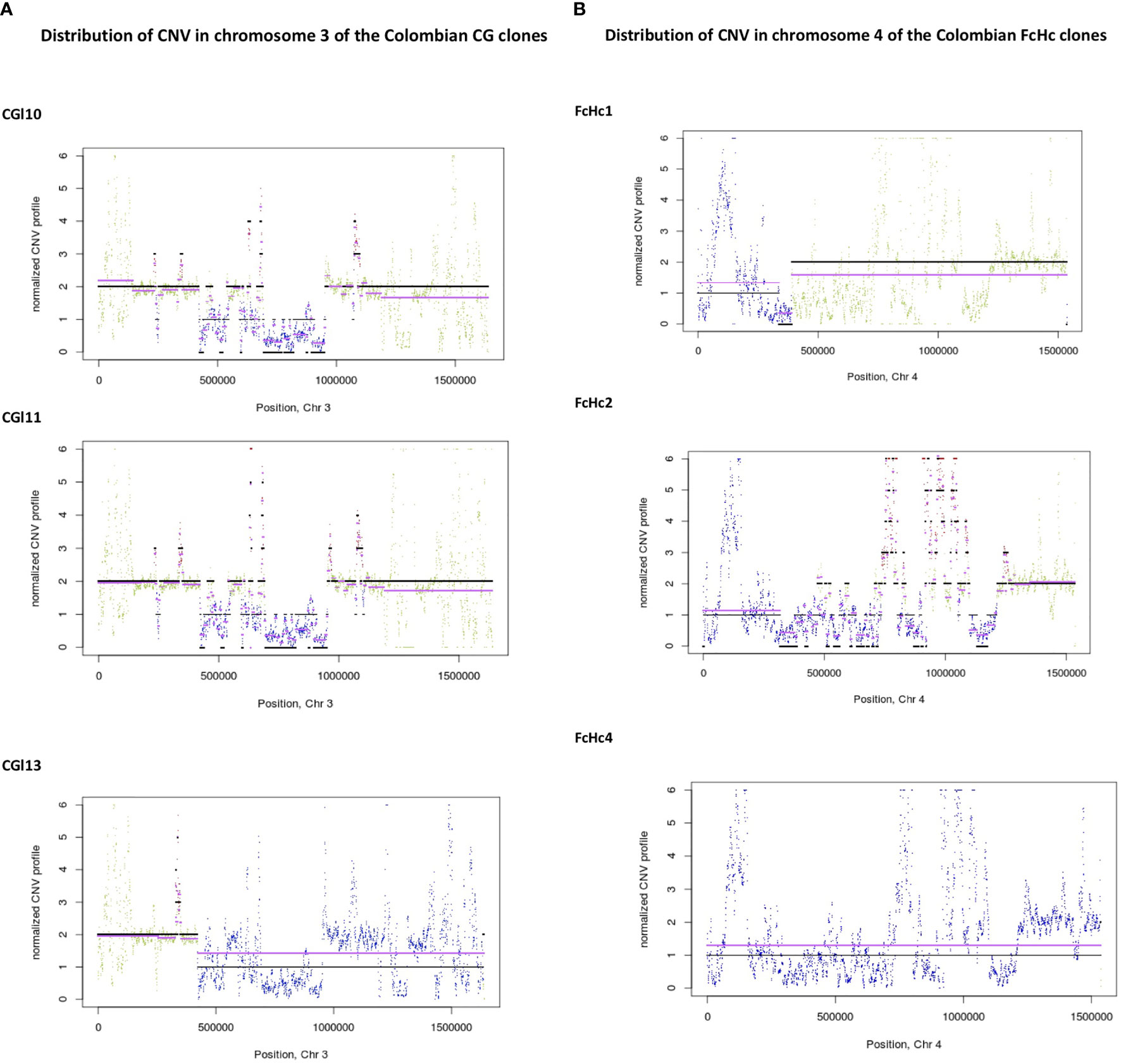
Figure 4 Distribution of CNV changes in chromosome 3 of the Colombian (A) CG clones and (B) FcHc clones. Black lines represent the reference genome sequence and cyan lines represent the sample under study. Each sliding window for CNV evaluation is represented as a dot. A drastic change in CNV can be noted in the FcHc clones whereas the CG clones seem to be less affected.
Sequence blocks affected by segmental CNVs contained retrotransposons of the VIPER and L1Tc class, as well as surface molecule genes surrounded by simple repeats. The isolate-specific nature of these CNV events demonstrates the high level of within-clade diversity of the TcI samples. The distribution of CNV across the T. cruzi genome reinforces the dynamic nature of the multigene family clusters and the surface molecule gene families.
Complete reconstruction of the T. cruzi genome to encompass the subtelomeric regions and surface molecule multigene family clusters proved to be difficult to achieve using short reads, due to sequencing library preparation biases and a genome architecture that is rich in long stretches of simple repeats, large repetitive gene families and multiple retrotransposons. In 2016, we used long PacBio sequencing reads to provide the most complete genome sequence of a T. cruzi strain to date and this reference genome was made public through Genbank and TriTrypDB. This allowed us to perform a detailed analysis of the repertoire of complex genes families that encode cell surface molecules, considered to be involved in cell invasion and evasion of the host immune response. We could clearly see the duality in the organisation of the parasite genome, comprised of a core genomic component with few repetitive elements and a slow evolutionary rate, resembling that of other related protozoa, and contrasting, highly plastic multigene family clusters encoding fast-evolving surface antigens, with abundant interspersed retrotransposons. The structural changes that generate and maintain diversity in T. cruzi surface molecules have certain mechanistic parallels in other protozoa such as those recently described in Plasmodium falciparum (Miles et al., 2016), but differing from the shorter, less repetitive genome of the non-virulent, human-infective Trypanosoma rangeli.
In order to overcome the limitations of short read mapping to a highly complex genome such as T. cruzi, we first mapped the reads against the repeat masked genome using `bwa-mem` with probabilistic read placement and multi-mapping probability assignment. The mapping results were subsequently submitted for statistical evaluation using Stampy (https://genome.cshlp.org/content/21/6/936). The Stampy algorithm assigns a probability for read-base misplacement and repeats. To evaluate the results we used two different variant callers (GATK + Freebayes) which also take into account these scores. For a variant to be considered, it needed to be reported by both variant calling methods and pass the filters. This strategy and the TcI reference genome made it possible to carry out the Tc1 population genomics study.
Early studies of the genetic diversity of T. cruzi using geographically disparate sampling and restricted comparisons of genetic diversity suggested a clonal population structure (Tibayrenc et al., 1990; Tibayrenc and Ayala, 1991); however, population genetics with an expanded set of markers have now challenged this view (Gaunt et al., 2003; Westenberger et al., 2005; Llewellyn et al., 2009). Nevertheless, there are still conflicting views as to which model best describes the population structure of T. cruzi (Messenger and Miles, 2015; Tibayrenc and Ayala, 2015). The newer Sylvio X10/1 genome sequence has enabled extensive genome-wide comparative population genomics analyses, which may shed light on this issue (Schwabl et al., 2019; Rose et al., 2020). Our comparative analyses of 34 T. cruzi isolates and clones from the TcI clade suggested many recombination events and population indices normally associated with genetic exchange between strains, which are more likely to be caused by the extensive repeat-driven recombination in the subtelomeric regions. The extent of variation in the multigene family clusters rich in surface antigen coding genes and the geographical clustering of strains based on geographic distribution indicates active, on-going adaptation to host and vectors. This need for phenotypic—and thus genomic—versatility may impel the active generation of sequence diversity in T. cruzi. Further analyses of the evolution of multigene family clusters will yield much more detailed understanding of diversity within and between the six currently recognised genetic lineages of T. cruzi (Andersson, 2011). We have shown how the genome architecture and dynamic multigene family clusters of T. cruzi may provide a mechanism to rapidly generate sequence diversity, required to escape the host immune response and adapt in response to new environments. It is the striking richness in simple repeat, retrotransposons and motif conservation in the multigene family clusters that renders these genomic areas susceptible to structural change, similar to yeast and other pathogens (Aksenova et al., 2013; de Jonge et al., 2013; Faino et al., 2016; Weatherly et al., 2016). Retrotransposons have been associated with the generation of complexity in genomic regions in mammals and plants and with control of gene expression (de Jonge et al., 2013; McConnell et al., 2013). In the case of T. cruzi, they appear to generate novel variants via mechanisms that exploit sequence homology. The presence of the simple repeats and retrotransposons near surface molecule coding genes provides the microhomology for both mechanisms to operate in such regions. Besides retrotransposons, the modular structure of the multigene families MASP and Trans-sialidase, where different genes share conserved motifs, could also provide microhomology needed for this homologous recombination (El-Sayed et al., 2005; Weatherly et al., 2016). Our analysis of INDELs and chromosomal breakpoints in the subtelomeric regions confirmed that a mechanism similar to NAHR or MMEJ operates as source of sequence diversity, for example by transposition of trans-sialidase genes or pseudogenes to produce new sequence mosaics. The required recombination machinery is conserved in T. cruzi (Ramesh et al., 2005). Furthermore, these mechanisms would explain the higher amount of pseudogenes observed in the surface molecule regions.
Retrotransposons were first reported from T. cruzi in 1991 (Villanueva et al., 1991). The presence of these elements may also partly account for the previously reported widespread observation of copy number variation in different T. cruzi strains (Minning et al., 2011). Thus, we find that repeats near the surface molecule genes appear to drive recombination in T. cruzi. The apparent inability of T. cruzi to condense chromatin may facilitate transposition in a stochastic fashion, facilitating generation of sequence diversity in exposed regions of the genome. A similar process has been described in the neurons of mammals and insects (Erwin et al., 2014) but not in any other unicellular organism. Retrotransposons may also have an important role as gene transcription regulators: they may either silence or promote gene expression, due to their susceptibility to DNA methylation or by providing potential binding sites respectively, as observed previously (Elbarbary et al., 2016). This lack of a well-defined transcriptional regulation machinery in the T. cruzi genome may suggest a link to the requirement for retrotransposon closely associated with gene tandem arrays.
Here we describe the sequencing and assembly of the complete genome of the Trypanosoma cruzi TcI strain Sylvio X10/1, which was made public in 2016. This genome sequence enabled the first resolution of the complex multiple gene families that encode T. cruzi surface molecules, and provided a basis for T. cruzi population genomics. We discovered an extraordinary concentration of retrotransposons among the multigene family clusters and indications of repeat-driven recombination and generation of antigenic diversity, providing the mechanisms for T. cruzi to evade the host immune response, and to facilitate the adaption to new host and vectors. This genome will provide an invaluable resource to facilitate the prospective discovery of novel drug targets and vaccine candidates for Chagas disease.
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found below: https://www.ncbi.nlm.nih.gov/genbank/, ADWP00000000 and https://www.ncbi.nlm.nih.gov/genbank/, SRP076682.
CT-L and BA conceived and designed the study. CT-L designed and executed computational analyses. MY prepared Sylvio X10/1 genomic DNA for PacBio sequencing and performed manual annotation of surface molecule genes. JEC, AS, JR, FG, SO-M, JAC, ST, HC, RG, KJ, MB, PH, KM, MJG, and BB provided genomic DNA for TcI isolates. JR-C and DB created chromosome maps for surface molecules. MM, LM, ML, JR-C, GM, and EG contributed to the interpretation of the results. CT-L, EG, MM, and BA wrote the manuscript. All authors contributed to the article and approved the submitted version.
This research was funded by grants from the Knut and Alice Wallenberg Foundation, The Swedish Research Council and the European FP7 program, Contract No. 223034. LM was funded by a grant from the NIH (5R01AI107028); DB, EG, and ST were funded by grants from CNPq (Brazilian Government Agency). GM was a recipient of a CAPES/PRINT Program Scholarship.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The authors wish to thank John Kuijpers and Lawrence Hon from Pacific Biosciences (PacBio) for providing the data for the Sylvio X10/1 genome and preliminary assembly results. We would also like to acknowledge Eric Dumonteil, University of the Yucatan, Mexico, and Ed Wozniak, Texas Department of State Health Services, for past work on parasite collection. This manuscript has been released as a preprint at Biorxiv (Talavera-Lopez et al., 2018).
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fcimb.2021.614665/full#supplementary-material
Supplementary Table 2 | A list of coordinates for the synteny analysis comparing the Sylvio X10/1 genome with CL Brener for all chromosomes.
Supplementary Table 3–5 | The tables list the genomic coordinates and identity of repeat elements of various types that have been identified in the Sylvio X10/1 genome sequence. The lists have been used for the annotation of the publically available genome sequence.
Supplementary Figure 1 | Results from mapping Illumina reads back to the completed genome sequence. The graphs show the overall coverage across the genome.
Supplementary Figure 2 | Results from mapping Illumina reads back to the completed genome sequence. The plots show coverage across each chromosome. Local variation in coverage indicate the presence of highly repeated regions.
Supplementary Figure 3–8 | Distribution of CNV changes in the Colombian CG clones and FcHc clones for all chromosomes.
Aksenova A. Y., Greenwell P. W., Dominska M., Shishkin A. A., Kim J. C., Petes T. D., et al. (2013). Genome Rearrangements Caused by Interstitial Telomeric Sequences in Yeast. Proc. Natl. Acad. Sci. U.S.A. 110 (49), 19866–19871. doi: 10.1073/pnas.1319313110
Andersson B. (2011). The Trypanosoma Cruzi Genome; Conserved Core Genes and Extremely Variable Surface Molecule Families. Res. Microbiol. 162 (6), 619–625. doi: 10.1016/j.resmic.2011.05.003
Arner E., Kindlund E., Nilsson D., Farzana F., Ferella M., Tammi M. T., et al. (2007). Database of Trypanosoma cruzi repeated genes: 20,000 additional gene variants. BMC Genomics 8, 391. doi: 10.1186/1471-2164-8-391
Berlin K., Koren S., Chin C.-S., Drake J. P., Landolin J. M., Phillippy A. M. (2015). Assembling Large Genomes with Single-Molecule Sequencing and Locality-Sensitive Hashing. Nat. Biotechnol. no August 2014, 1–11. doi: 10.1038/nbt.3238
Bern C., Kjos S., Yabsley M. J., Montgomery S. P. (2011). Trypanosoma Cruzi and Chagas’ Disease in the United States. Clin. Microbiol. Rev. 24 (4), 655–681. doi: 10.1128/CMR.00005-11
Berná L., Rodriguez M., Chiribao M. L., Parodi-Talice A., Pita S., Rijo G., et al. (2018). Expanding an expanded genome: long-read sequencing of Trypanosoma cruzi. Microbial. Genomics 4 (5), e000177. doi: 10.1099/mgen.0.000177
Callejas-Hernández F., Rastrojo A., Poveda C., Gironès N., Fresno M. (2018). Genomic Assemblies of Newly Sequenced Trypanosoma Cruzi Strains Reveal New Genomic Expansion and Greater Complexity. Sci. Rep. 8 (1), 14631. doi: 10.1038/s41598-018-32877-2
Claessens A., Hamilton W. L., Kekre M., Otto T. D., Faizullabhoy A., Rayner J. C., et al. (2014). Generation of Antigenic Diversity in Plasmodium Falciparum by Structured Rearrangement of Var Genes during Mitosis. PloS Genet. 10 (12), e1004812. doi: 10.1371/journal.pgen.1004812
de Jonge R., Bolton M. D., Kombrink A., van den Berg G. C.M., Yadeta K. A., et al. (2013). Extensive Chromosomal Reshuffling Drives Evolution of Virulence in an Asexual Pathogen. Genome Res. 23 (8), 1271–1282. doi: 10.1101/gr.152660.112
Elbarbary R. A., Lucas B. A., Maquat L. E. (2016). Retrotransposons as Regulators of Gene Expression. Science 351 (6274), aac7247. doi: 10.1126/science.aac7247
El-Sayed N. M., Myler P. J., Bartholomeu D. C., Nilsson D., Aggarwal G., Tran A.-N., et al. (2005). The Genome Sequence of Trypanosoma Cruzi, Etiologic Agent of Chagas Disease. Science 309 (5733), 409–415. doi: 10.1126/science.1112631
Erwin J. A., Marchetto M. C., Gage F. H. (2014). Mobile DNA Elements in the Generation of Diversity and Complexity in the Brain. Nat. Rev. Neurosci. 15 (8), 497–506. doi: 10.1038/nrn3730
Faino L., Seidl M. F., Shi-Kunne X., Pauper M., van den Berg G. C. M., Wittenberg A. H. J., et al. (2016). Transposons Passively and Actively Contribute to Evolution of the Two-Speed Genome of a Fungal Pathogen. Genome Res. 26 (8), 1091–1100. doi: 10.1101/gr.204974.116
Franzén O., Ochaya S., Sherwood E., Lewis M. D., Llewellyn M. S., Miles M. A., et al. (2011). Shotgun Sequencing Analysis of Trypanosoma Cruzi I Sylvio X10/1 andCruzi VI CL Brener. PloS Neglect. Trop. Dis. 5 (3), e984. doi: 10.1371/journal.pntd.0000984
Franzén O., Talavera-López C., Ochaya S., Butler C. E., Messenger L. A., Lewis M. D., et al. (2012). Comparative Genomic Analysis of Human Infective Trypanosoma Cruzi Lineages with the Bat-Restricted Subspecies T. Cruzi Marinkellei. BMC Genomics 13 (January), 531. doi: 10.1186/1471-2164-13-531
Frasch A. C. (2000). Functional diversity in the trans-sialidase and mucin families in Trypanosoma cruzi. Parasitol. Today 16 (7), 282–286. doi: 10.1016/S0169-4758(00)01698-7
Gaunt M. W., Yeo M., Frame I. A., Stothard J. R. (2003). Mechanism of Genetic Exchange in American Trypanosomes” Nature 421 (6926), 936–939. doi: 10.1038/nature01438
GBD 2013 Mortality and Causes of Death Collaborators (20159963). Global, Regional, and National Age-Sex Specific All-Cause and Cause-Specific Mortality for 240 Causes of Deat-2013: A Systematic Analysis for the Global Burden of Disease Study 2013. Lancet 385, 117–171. doi: 10.1016/S0140-6736(14)61682-2
Guhl F., Ramírez J. D. (2011). Trypanosoma Cruzi I Diversity: Towards the Need of Genetic Subdivision? Acta Tropica 119 (1), 1–4. doi: 10.1016/j.actatropica.2011.04.002
Hall J. P.J., Wang H., Barry J. D. (2013). Mosaic VSGs and the Scale of Trypanosoma Brucei Antigenic Variation. PloS Pathog. 9 (7), e1003502. doi: 10.1371/journal.ppat.1003502
Kim D., Chiurillo M. A., El-Sayed N., Jones K., Santos M. R.M., Porcile P. E., et al. (2005). Telomere and Subtelomere of Trypanosoma Cruzi Chromosomes Are Enriched in (pseudo)genes of Retrotransposon Hot Spot and Trans-Sialidase-like Gene Families: The Origins of T. Cruzi Telomeres. Gene 346 (February), 153–161. doi: 10.1016/j.gene.2004.10.014
Llewellyn M. S., Miles M. A., Carrasco H. J., Lewis M. D., Yeo M., Vargas J., et al. (2009). Genome-Scale Multilocus Microsatellite Typing of Trypanosoma Cruzi Discrete Typing Unit I Reveals Phylogeographic Structure and Specific Genotypes Linked to Human Infection. PloS Pathog. 5 (5), e1000410. doi: 10.1371/journal.ppat.1000410
McConnell M. J., Lindberg M. R., Brennand K. J., Piper J. C., Voet T., Cowing-Zitron C., et al. (2013). Mosaic Copy Number Variation in Human Neurons. Science 342 (6158), 632–637. doi: 10.1126/science.1243472
Messenger L. A., Miles M. A. (2015). Evidence and Importance of Genetic Exchange among Field Populations of Trypanosoma Cruzi. Acta Tropica 151 (November), 150–155. doi: 10.1016/j.actatropica.2015.05.007
Miles A., Iqbal Z., Vauterin P., Pearson R., Campino S., Theron M., et al. (2016). Indels, Structural Variation, and Recombination Drive Genomic Diversity in Plasmodium Falciparum. Genome Res. 26 (9), 1288–1299. doi: 10.1101/gr.203711.115
Minning T. A., Weatherly D. B., Flibotte S., Tarleton R. L. (2011). Widespread, Focal Copy Number Variations (CNV) and Whole Chromosome Aneuploidies in Trypanosoma Cruzi Strains Revealed by Array Comparative Genomic Hybridization. BMC Genomics 12 (1), 139. doi: 10.1186/1471-2164-12-139
Montgomery S. P., Starr M. C., Cantey P. T., Edwards M. S., Meymandi S. K. (2014). Neglected Parasitic Infections in the United States: Chagas Disease. Am. J. Trop. Med. Hygiene 90 (5), 814–818. doi: 10.4269/ajtmh.13-0726
Morillo C. A., Marin-Neto J. A., Avezum A., Sosa-Estani S., Rassi A. Jr, Rosas F., et al. (2015). Randomized Trial of Benznidazole for Chronic Chagas’ Cardiomyopathy. New Engl. J. Med. 373 (14), 1295–1306. doi: 10.1056/NEJMoa1507574
Osorio L., Ríos I., Gutiérrez B., González J. (2012). Virulence Factors of Trypanosoma Cruzi: Who Is Who? Microbes Infect. / Institut Pasteur 14 (15), 1390–1402. doi: 10.1016/j.micinf.2012.09.003
Parks M. M., Lawrence C. E., Raphael B. J. (2015). Detecting Non-Allelic Homologous Recombination from High-Throughput Sequencing Data. Genome Biol. 16 (April), 72. doi: 10.1186/s13059-015-0633-1
Pecoul B., Batista C., Stobbaerts E., Ribeiro I., Vilasanjuan R., Gascon J., et al. (2016). The BENEFIT Trial: Where Do We Go from Here? PloS Neglect. Trop. Dis. 10 (2), e0004343. doi: 10.1371/journal.pntd.0004343
Ramesh M. A., Malik S.-B., Logsdon J. M. Jr (2005). A Phylogenomic Inventory of Meiotic Genes: Evidence for Sex in Giardia and an Early Eukaryotic Origin of Meiosis. Curr. Biology: CB 15 (2), 185–191. doi: 10.1016/S0960-9822(05)00028-X
Ramírez J. D., Guhl F., Rendón L. M., Rosas F., Marin-Neto J. A., Morillo C. A. (2010). Chagas Cardiomyopathy Manifestations and Trypanosoma Cruzi Genotypes Circulating in Chronic Chagasic Patients. PloS Neglect. Trop. Dis. 4 (11), e899. doi: 10.1371/journal.pntd.0000899
Rassi A., Rassi A., Marin-Neto J. A. (2010). Chagas Disease. Lancet 375 (9723), 1388–1402. doi: 10.1016/S0140-6736(10)60061-X
Reis-Cunha J. L., Baptista R. P., Rodrigues-Luiz G. F., Coqueiro-Dos-Santos A., Valdivia H. O., de Almeida L. V., et al. (2018). Whole Genome Sequencing of Trypanosoma Cruzi Field Isolates Reveals Extensive Genomic Variability and Complex Aneuploidy Patterns within TcII DTU. BMC Genomics 19 (1), 816. doi: 10.1186/s12864-018-5198-4
Rose E., Carvalho J. L., Hecht M. (2020). Mechanisms of DNA Repair in Trypanosoma Cruzi: What Do We Know so Far? DNA Repair 91-92 (July), 102873. doi: 10.1016/j.dnarep.2020.102873
Schenkman S., Eichinger D., Pereira M. E., Nussenzweig V. (1994). Structural and functional properties of Trypanosoma trans-sialidase. Annu. Rev. Microbiol. 48, 499–523. doi: 10.1146/annurev.mi.48.100194.002435
Schwabl P., Imamura H., Van den Broeck F., Costales J. A., Maiguashca-Sánchez J., Miles M. A., et al. (2019). Meiotic Sex in Chagas Disease Parasite Trypanosoma Cruzi. Nat. Commun. 10 (1), 3972. doi: 10.1038/s41467-019-11771-z
Sfeir A., Symington L. S. (2015). Microhomology-Mediated End Joining: A Back-up Survival Mechanism or Dedicated Pathway? Trends Biochem. Sci. 40 (11), 701–714. doi: 10.1016/j.tibs.2015.08.006
Steinbiss S., Silva-Franco F., Brunk B., Foth B., Hertz-Fowler C., Berriman M., et al. (2016). Companion: A Web Server for Annotation and Analysis of Parasite Genomes. Nucleic Acids Res. 44 (W1), W29–W34. doi: 10.1093/nar/gkw292
Stoco P. H., Wagner G., Talavera-Lopez C., Gerber A., Zaha A., Thompson C. E., et al. (2014). Genome of the Avirulent Human-Infective Trypanosome–Trypanosoma Rangeli. PloS Neglect. Trop. Dis. 8 (9), e3176. doi: 10.1371/journal.pntd.0003176
Talavera-López C., Reis-Cunha J. L., Messenger L. A., Lewis M. D., Yeo M., Bartholomeu D. C., et al. (2018). Repeat-Driven Generation of Antigenic Diversity in a Major Human Pathogen, Trypanosoma Cruzi. BioRxiv 283531. doi: 10.1101/283531
Tibayrenc M., Ayala F. J. (1991). Towards a Population Genetics of Microorganisms: The Clonal Theory of Parasitic Protozoa. Parasitol. Today 7 (9), 228–232. doi: 10.1016/0169-4758(91)90234-F
Tibayrenc M., Ayala F. J. (2015). The Population Genetics of Trypanosoma Cruzi Revisited in the Light of the Predominant Clonal Evolution Model. Acta Tropica 151 (November), 156–165. doi: 10.1016/j.actatropica.2015.05.006
Tibayrenc M., Kjellberg F., Ayala F. J. (1990). A Clonal Theory of Parasitic Protozoa: The Population Structures of Entamoeba, Giardia, Leishmania, Naegleria, Plasmodium, Trichomonas, and Trypanosoma and Their Medical and Taxonomical Consequences. Proc. Natl. Acad. Sci. U. States America 87 (7), 2414–2418. doi: 10.1073/pnas.87.7.2414
Villanueva M. S., Williams S. P., Beard C. B., Richards F. F., Aksoy S. (1991). A New Member of a Family of Site-Specific Retrotransposons Is Present in the Spliced Leader RNA Genes of Trypanosoma Cruzi. Mol. Cell. Biol. 11 (12), 6139–6148. doi: 10.1128/MCB.11.12.6139
Weatherly D. B., Boehlke C., Tarleton R. L. (2009). Chromosome Level Assembly of the Hybrid Trypanosoma Cruzi Genome. BMC Genomics 10 (June), 255. doi: 10.1186/1471-2164-10-255
Weatherly D. B., Peng D., Tarleton R. L. (2016). Recombination-Driven Generation of the Largest Pathogen Repository of Antigen Variants in the Protozoan Trypanosoma Cruzi. BMC Genomics 17 (1), 729. doi: 10.1186/s12864-016-3037-z
Weckselblatt B., Hermetz K. E., Rudd M. K. (2015). Unbalanced Translocations Arise from Diverse Mutational Mechanisms Including Chromothripsis. Genome Res. 25 (7), 937–947. doi: 10.1101/gr.191247.115
Westenberger S. J., Barnabé C., Campbell D. A., Sturm N. R. (2005). Two Hybridization Events Define the Population Structure of Trypanosoma Cruzi. Genetics 171 (2), 527–543. doi: 10.1534/genetics.104.038745
WHO (2015). Available at: http://www.who.int/wer/2015/wer9006/en/ (Accessed 6 February 2015).
Yoshida N., Cortez M. (2008). Trypanosoma cruzi: parasite and host cell signaling during the invasion process. Subcellular Biochem. 47, 82–91. doi: 10.1007/978-0-387-78267-6_6
Zingales B., a. Miles M., Campbell D. A., Tibayrenc M., Macedo A. M., Teixeira M. M. G., et al. (2012). The Revised Trypanosoma Cruzi Subspecific Nomenclature: Rationale, Epidemiological Relevance and Research Applications. Infect. Genet. Evol.: J. Mol. Epidemiol. Evol. Genet. Infect. Dis. 12 (2), 240–253. doi: 10.1016/j.meegid.2011.12.009
Keywords: Trypanosoma cruzi, genome sequence, antigenic variation, population genetics, parasitology, microbial genomics, tropical medicine, pathology of infectious diseases
Citation: Talavera-López C, Messenger LA, Lewis MD, Yeo M, Reis-Cunha JL, Matos GM, Bartholomeu DC, Calzada JE, Saldaña A, Ramírez JD, Guhl F, Ocaña-Mayorga S, Costales JA, Gorchakov R, Jones K, Nolan MS, Teixeira SMR, Carrasco HJ, Bottazzi ME, Hotez PJ, Murray KO, Grijalva MJ, Burleigh B, Grisard EC, Miles MA and Andersson B (2021) Repeat-Driven Generation of Antigenic Diversity in a Major Human Pathogen, Trypanosoma cruzi. Front. Cell. Infect. Microbiol. 11:614665. doi: 10.3389/fcimb.2021.614665
Received: 06 October 2020; Accepted: 22 January 2021;
Published: 03 March 2021.
Edited by:
Julius Lukes, Academy of Sciences of the Czech Republic (ASCR), CzechiaReviewed by:
Carlos A. Buscaglia, Consejo Nacional de Investigaciones Científicas y Técnicas (CONICET), ArgentinaCopyright © 2021 Talavera-López, Messenger, Lewis, Yeo, Reis-Cunha, Matos, Bartholomeu, Calzada, Saldaña, Ramírez, Guhl, Ocaña-Mayorga, Costales, Gorchakov, Jones, Nolan, Teixeira, Carrasco, Bottazzi, Hotez, Murray, Grijalva, Burleigh, Grisard, Miles and Andersson. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Björn Andersson, Ympvcm4uYW5kZXJzc29uQGtpLnNl; Carlos Talavera-López, Y3Q1QHNhbmdlci5hYy51aw==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.