 Honghong Liu1
Honghong Liu1 Siqin Feng
Siqin Feng Shuyang Zhang
Shuyang Zhang Chenhong Zhang
Chenhong Zhang- 1Department of Cardiology, Peking Union Medical College Hospital, Peking Union Medical College & Chinese Academy of Medical Sciences, Beijing, China
- 2State Key Laboratory of Microbial Metabolism, School of Life Sciences and Biotechnology, Shanghai Jiao Tong University, Shanghai, China
Familial hypercholesterolemia (FH) is an inherited rare disease leading to markedly elevated low-density lipoprotein cholesterol (LDL-C) levels and increased risk for cardiovascular event. Gut microbiota has been implicated as a pivotal contributing factor in hyperlipidemia, however, its role in FH remains elusive. We performed whole-exome and metagenomics sequencing on a family with 22 members in which myocardial infarctions occurred at a young age with unclear etiology. We confirmed the missense mutation of LDLR c.1723C>T accounted for the abnormal cholesterol metabolism in the family through co-segregation analysis. In addition, Prevotella dentalis was found elevated and strongly associated with LDL-C level in FH family members with mutation of LDLR c.1723C>T compared to unaffected members with hyperlipidemia. Overall, our work suggests that whole-exome sequencing can facilitate identification of disease-causing variants and enable preventive treatment of FH. Our metagenomics analysis provides early insights into potential contributions of host-microbe interactions in genetic and common hypercholesterolemia.
Introduction
Familial hypercholesterolemia (MIM#143890), an inherited rare disease mainly caused by the mutation of low-density lipoprotein receptor (LDLR) gene or apolipoprotein B (APOB) gene in autosomal dominant pattern, which is typically associated with premature coronary artery disease (CAD) (Defesche et al., 2017). Based on the presence of FH-causing alleles, the prevalence of the disease has been estimated worldwide to be 1/1,000,000 for homozygotes (Santos et al., 2016). The risk for the development of CAD in patients with untreated heterozygous familial hypercholesterolemia (HeFH) is 20 times higher than that in treated patients (Khera et al., 2016). Although PCSK9 inhibitors such as evolocumab emerged as a new LDL-C lowering therapy and showed significant reductions in cardiovascular outcomes (Sabatine et al., 2015), FH remains underdiagnosed and undertreated in most countries, which need further effort to better manage (Nordestgaard et al., 2013). In recent years, emerged evidence shows that the gut microbiota, as an internalized environmental factor, plays an essential role in human cardiovascular disease (Brown and Hazen, 2018). Moreover, compelling evidence from animal and human studies suggests that genotype also affects the composition of gut microbiome (Goodrich et al., 2014). Thus, the effect and interaction of the host genotype and gut microbiota in lipid metabolic phenotypes remain elusive, which might provide important information for treatment of hyperlipidemia and CAD.
Here, we reported on an extended myocardial infarction (MI) family, which remained without diagnosis with FH until whole-exome sequencing (WES) was performed. We identified the pathogenic LDLR variant accounting for the abnormally elevated LDL-C level. In particular, we investigated metagenomic profile of this family by comparing FH members with unaffected members to explore the role of gut microbiota in genetically predisposed hypercholesterolemia. The current study illustrates the need for a more systematic employment of multi-omics analysis of FH to increase the awareness and timely initiation of medical treatment.
Materials and Methods
Study Subjects
We recruited a three-generation Chinese family with FH from the Peking Union Medical College Hospital in 2019. All subjects analyzed in this study gave written informed consent before participating. Venous fasting blood was drawn in the morning the day after recruitment. Then, the laboratory data including lipid profile (total cholesterol, low-density lipoprotein cholesterol, high-density lipoprotein cholesterol, and triglycerides) were measured immediately. Stool samples freshly collected from each participant under instruction were immediately transported to the laboratory and frozen at −80°C immediately. The local Ethical Committee (JS-1233, Peking Union Medical College Hospital) approved the study.
Whole-Exome Sequencing Analysis and Mutation Screening
Genomic DNA was extracted with QIAamp DNA Blood Mini kit (Qiagen, Hilden, Germany) following the manufacturer’s protocol. The extracted DNA was fragmented randomly and then purified using magnetic particle method. DNA fragments were ligated with adaptors and captured by probes of the IDT XGen Exome Research Panel (IDT, Lowa, USA) targeting about 19,396 genes. The DNA libraries after enrichment and purification were sequenced on the NovaSeq 6000 sequencer according to the manufacturer’s instructions (Illumina, San Diego, USA).
All reads were aligned to the reference human genome (UCSC hg19) by Burrows-Wheeler Aligner (BWA, v.0.5.9-r16) (Li and Durbin, 2010). Picard (v2.5.0) was used to mark duplicate and low‐quality reads. Genome Analysis Toolkit (GATK, v4.0.6.0) was used for variant calling (McKenna et al., 2010). Resulting variants were annotated with Annovar (2018Apr16) to evaluate their effect on coding sequences, allele frequency in the general population, and the predicted level of pathogenicity (Wang et al., 2010). Allele frequencies were reported using the GnomAD, TOPMED, ExAC, and G1000. Minor allele frequencies, prior evidence of disease causality, and pathogenicity scores predicted by SIFT (Ng and Henikoff, 2003) and Polyphen‐2 (Adzhubei et al., 2013) were used to prioritize variants during the curation of the filtered variant list. We put emphasis on variants in genes with strong clinical evidence. The chosen variants were then verified using Sanger sequencing.
Metagenomics Sequencing and Analysis
DNA extraction was performed using the Qiagen QIAamp DNA Stool Mini Kit (Qiagen) according to the manufacturer’s instructions. DNA library construction was performed following the manufacturer’s instruction (Illumina). All samples were sequenced based on the Illumina HiSeq X Ten platform. High-quality paired-end reads from each sample were used for de novo assembly with IDBA_UD v1.1.0 (Peng et al., 2012) into contigs longer than 500 bp. The high-quality reads from each sample were aligned against the gene catalogue by SOAP2 v2.22 (Li et al., 2009) using the criterion of “identity > 90%.” Prediction of genes from each sample was processed based on Meta-GeneMark v.2.1 (Zhu et al., 2010). A non-redundant gene catalog of 1,048,575 microbial genes was constructed with CD-HIT v4.6.4. The high-quality reads distribution of different samples was aligned with Bowtie2 (Langmead and Salzberg, 2012).
Putative sequences were translated from the gene catalog and aligned against the KEGG databases (release 89.1). MEGAN v4.6 (Afshinnekoo et al., 2015) was used for taxonomy annotation. Redundancy analyses was computed on transformed bray-curtis distances, the significance was assessed by permutational multivariate analysis of variance (PerMANOVA, 999 permutations) in R (3.6.0). All differential abundances of genes, species and KO screening were tested using the Wilcoxon rank sum test. Wherever mentioned, the Benjamini-Hochberg method was used to control the FDR.
Results
Clinical Case Description
The proband was a 33-years-old male (FM 1), who was diagnosed with acute ST-elevated MI 5 months ago. Emergency coronary angiography showed that 60 to 80% obstruction of left anterior descending artery (LAD) and 50% stenosis of left circumflex artery (LCX) (Figure 1A). The patient was presented to our hospital due to chest pain and fatigue during physical exercise recurrently. Electrocardiogram and laboratory cardiac enzyme test were all normal. The myocardial perfusion tomographic imaging (adenosine) revealed transverse, coronal, and sagittal planes showing areas of reduced radioactivity distribution in the basement of the posterior lateral wall of the left ventricle. His hyperlipidemia was first diagnosed at age 28, he had taken atorvastatin (10 mg, q.d.) irregularly since then for 4 years, and the LDL-C level ranged between 2.6 to 3.8 mmol/L. Unexpectedly, his LDL-C level rose to 6.18 mmol/L 1 month before MI owing to drug withdrawal (Figure 1B).
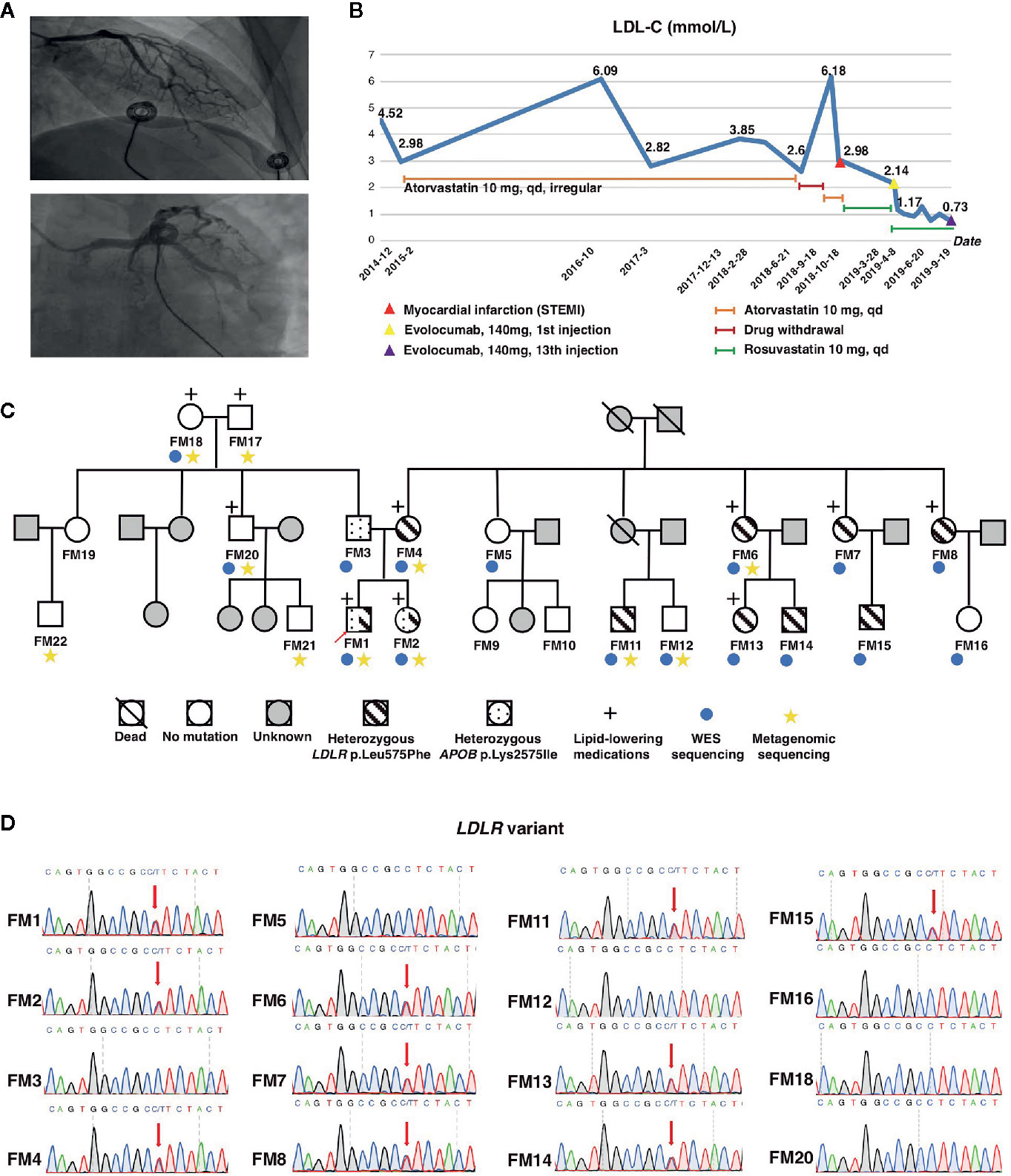
Figure 1 Pedigree of the HeFH family and mutation screening. (A) Coronary artery angiography showing obstruction in the left anterior descending artery (LAD) and left circumflex artery (LCX) of the proband. (B) LDL-C changes of the proband during recent 5 years. (C) Pedigree of the family members and their corresponding phenotypes. (D) Sequencing results of the LDLR c.1723C>T variant. Red arrow indicates heterozygous mutations. LDL-C, low-density lipoprotein cholesterol.
His mother experienced lethal MI at the age 50. She was also diagnosed with type 2 diabetes mellitus (T2DM) and hypertension. His several maternal aunties had hyperlipidemia and one of them was died of MI at the age 41. His father had T2DM at age 50. His sister was diagnosed of hypertension, T2DM, and hyperlipidemia at age of 29.
Identification of Heterozygote Low-Density Lipoprotein Receptor Mutant in this Familial Hypercholesterolemia Family
Since the patient had a family history of early-onset coronary heart disease, we highly suspect that his dyslipidemia was genetically related. We recruited the remaining 21 family members to investigate the genetic mechanism. The basic demographic data were summarized in Table 1. We performed WES analysis on this proband and other 15 family members (Figure 1C). We found the proband carried two deleterious mutations including LDLR (c.1723C>T, p.Leu575Phe) and APOB (c.7724A>T, p.Lys2575Ile) (Table 2). We identified that pattern of inheritance of heterozygous LDLR variants was fully co-segregated with the elevated levels of LDL‐C in this family and the causality was further verified by Sanger sequencing. The APOB c.7724A>T variant carried by the proband was inherited from the father who had normal cholesterol level. Meanwhile, the APOB mutation was not identified in FM18 and FM 20 who had elevated LDL-C level. Therefore, according to the principle of co-segregation, we exclude the pathogenicity of APOB mutation (Figures 1C, D). Finally, the proband and other nine family members were diagnosed with heterozygote familial hypercholesterolemia according DLCN (Dutch Lipid Clinic Network) criteria after genomic screening (Umans-Eckenhausen et al., 2001). After being diagnosed with FH, the proband received evolocumab treatment (140 mg) for 5 months and the LDL-C level dropped from 2.14 to 0.73 mmol/L immediately (Figure 1B).
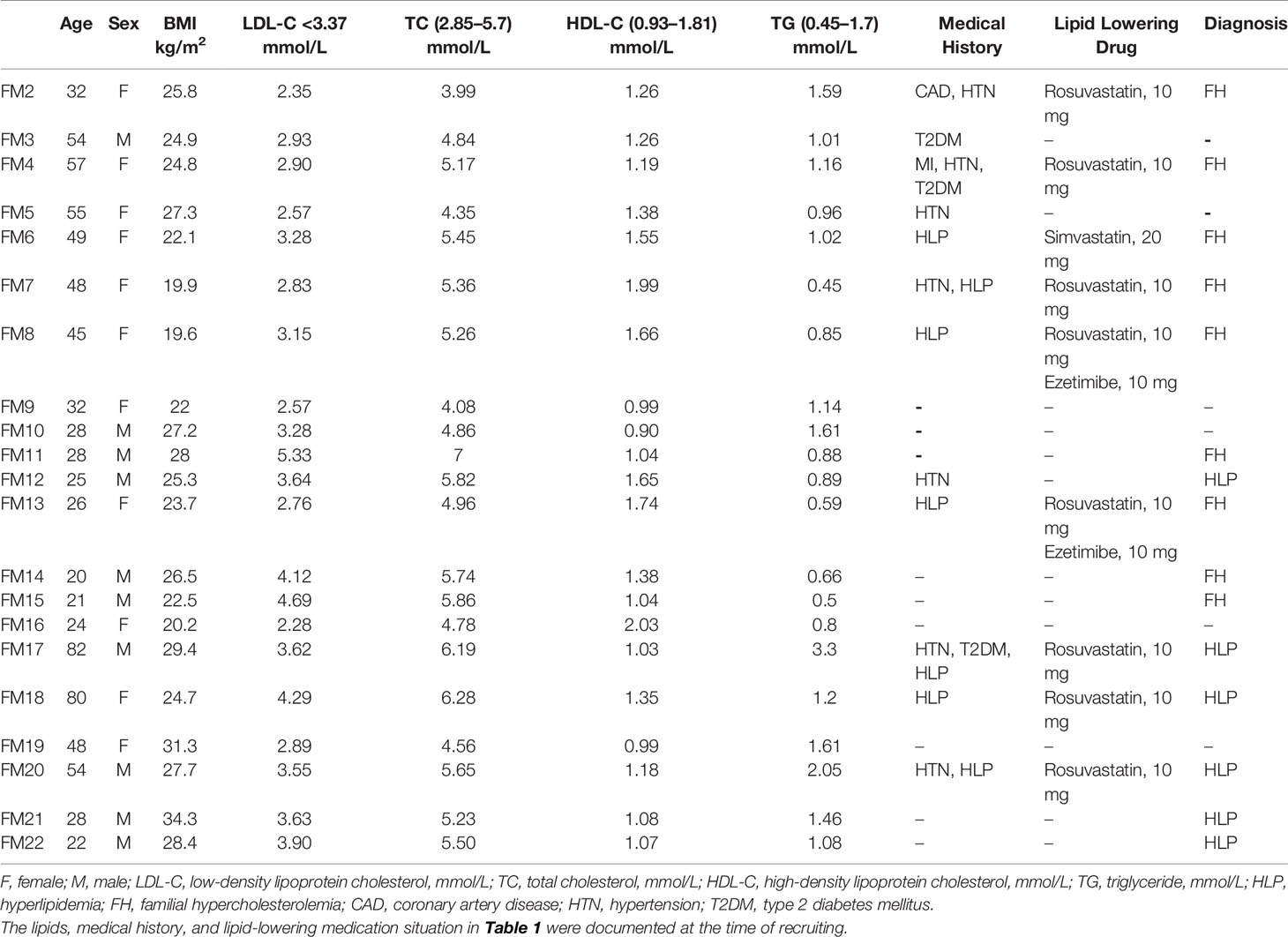
Table 1 Clinical characteristics of enrolled FH subjects.
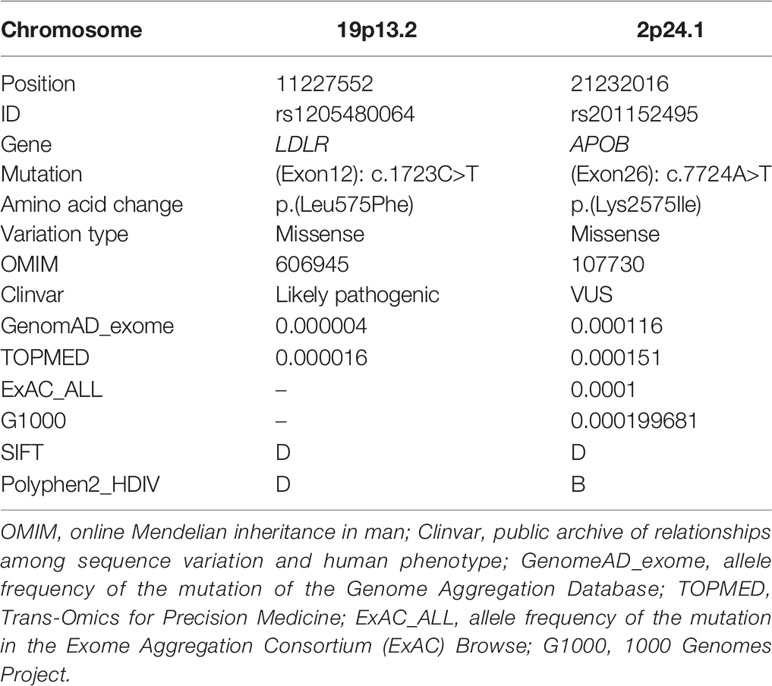
Table 2 Damaging mutations found for the proband using WES.
Metagenomics Analysis Comparing Familial Hypercholesterolemia and Unaffected Hyperlipidemia Subjects in This Family
Since several members without mutation of LDLR c.1723C>T in this family were also present with dyslipidemia, we then investigated the changes in the gut microbiome in the FH members with mutation of LDLR c.1723C>T (n = 5, FM1, FM2, FM4, FM6, and FM11) and unaffected members with hyperlipidemia (HLP, n = 6, FM12, FM17, FM18, FM20, FM21, and FM22) by metagenomics sequencing, to further explore the contribution of microbial communities to cholesterol homeostasis. Redundancy analysis indicated that age, statins therapy, and LDL-C level were the determinant factor for explaining the variations in the bacterial compositions (P = 0.046, PerMANOVA test with 999 permutations) (Figure 2A). Fourteen bacterial species were observed to be significantly altered (adjusted P < 0.05 in Wilcoxon rank sum test and |log2 fold change|> 1). Compared to HLP, the gut microbiota in FH patients harbored increased Veillonella sp. and Prevotella dentalis (Figure 2B). Moreover, we found that Prevotella dentalis showed significantly stronger positive correlation with LDL-C level only in FH individuals (Figure 2C). Clostridium clariflavum also had trendy positive correlation with LDL-C level in FH individuals (Figure 2C). FH- depleted microbial function included degradation of module of fatty acid, whereas increase of fatty acid initiation and elongation biosynthesis (Figures 2D, E). The bacterial fatty acid synthesis pathway has significant potential as a target for novel drugs and β-hydroxyacyl-ACP of the proper length is used for lipopolysaccharide (LPS) synthesis in Gram-negative bacteria (Heath and Rock, 2004).
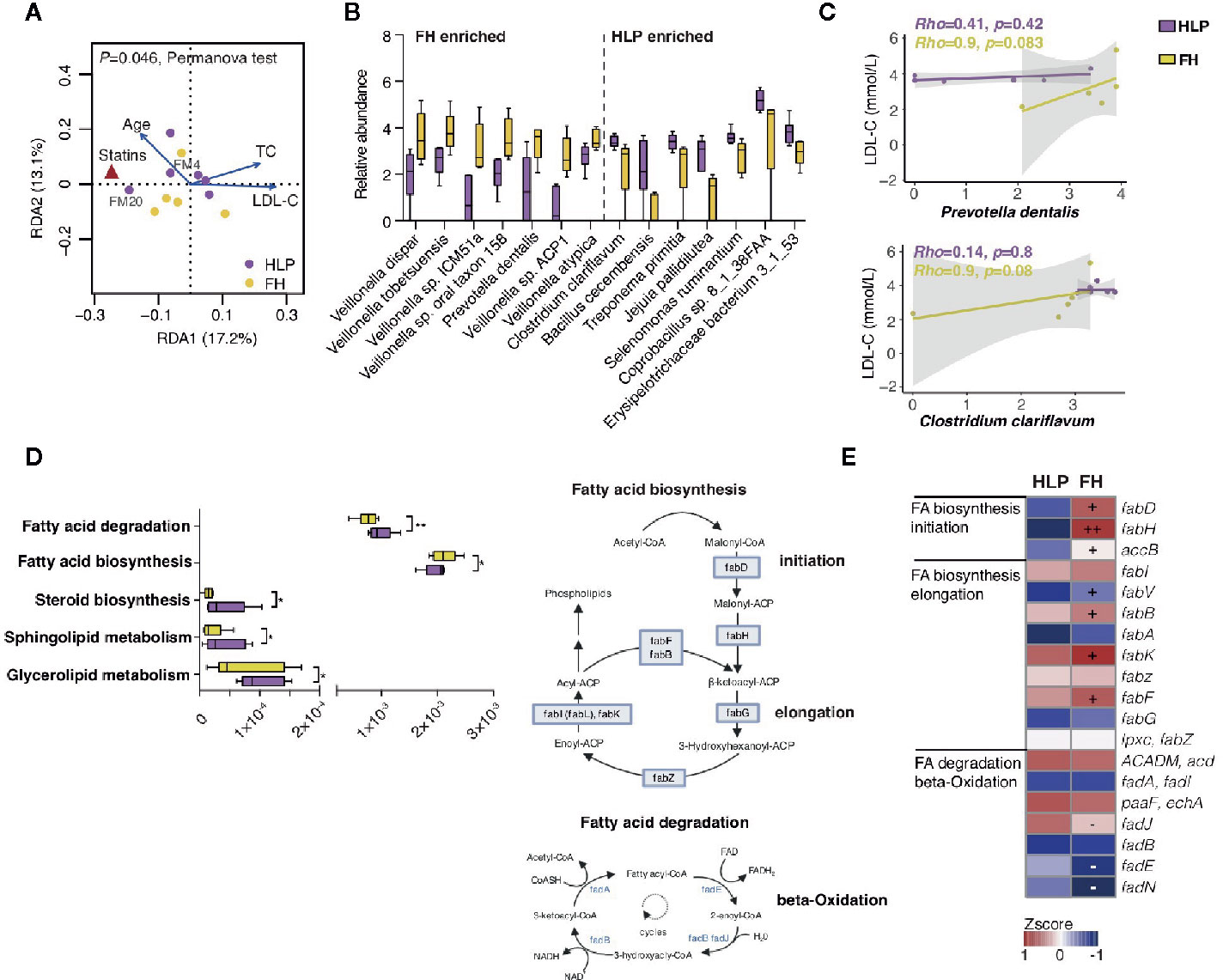
Figure 2 Metagenomics profile of this HeFH family. (A) Distance-based redundancy analysis (dbRDA) plot based on clinical parameters fitted to the variation in gut microbiota. (B) Relative abundance of significantly differed species between FH and HLP individuals (P < 0.05, Wilcoxon rank sum test). Boxes represent the inter-quartile ranges, and lines inside the boxes denote medians. (C) Spearman correlations between bacterial relative abundance and LDL-C levels. (D) Alterations in gut microbial lipid functional modules. *P < 0.05; **P < 0.01. (E) KO genes significantly changed between FH and HLP individuals are shown in the heat map. Significant changes (elevation and depletion) are denoted as follows: +, elevation with P < 0.05; −, depletion with P < 0.05.
Discussion
Here, we unmasked the genetic cause of a young male patient with early-onset MI through detailed clinical assessment and diagnosed other nine family members with HeFH in the extended family through WES analysis. The LDLR c.1723C>T variant was classified as likely pathogenic based on family co-segregation analyses. It was once detected in a young Chinese patient with FH in 2016 (Jiang et al., 2016).This mutation is in the exon 12 which is within the EGF precursor homology domain (i.e. exon 7–14), and is involved in the dissociation of the receptor and the lipoprotein in the endocytosis machinery (Zhang et al., 2007). Our study revealed that genomic test along with family screening, is confirmatory and clinically relevant, especially for individuals with borderline-high LDL-C levels.
In the current Chinese family cohort, the specific genetic relatedness help dissect the gut microbial variation between FH and hypercholesterolemia. We discovered that microbiota was affected by statins therapy in accordance with other research that higher gut biodiversity was associated with statin sensitive response (Sun et al., 2018). Statins therapy has been reported as a key covariate of modulating the prevalence of Prevotella sp. (Vieira-Silva et al., 2020). Similarly, the Veillonella sp. enriched in FH individuals are often present in the oral cavity and is identified to be abundant in hyperlipidemia cohorts (Mashima et al., 2016; Liu et al., 2019). Recent study indicated that microbiota is implicated in human genetic adaptations and host mechanisms can replace or recruit beneficial microbiota functions during local adaptation (Suzuki and Ley, 2020). We speculated that host gene-microbe interactions and lipids metabolic disorders caused by LDLR mutations may disturb bacteria community homeostasis in FH individuals, resulting in positive correlation between specific bacteria and LDL-C level. Prevotella sp. is regarded as potential pathogens for CVD (Liu et al., 2019), and Clostridiaceae was found associated with lipid profile in previously large cohort (Fu et al., 2015). In addition, bacterial fatty acid synthesis is a vital facet of bacterial physiology, whereas the intermediate metabolite could catalyze the initial step in the biosynthesis of LPS (Li et al., 2003; Yao and Rock, 2015). The gut microbiota in FH individuals may exhibit stronger function of pro-inflammatory potential which facilitate lipometabolic disturbance. However, more FH family trios are necessary in order to decipher the microbial predisposition and the mechanistic participation of genotype-microbiota interaction remain to be further biologically elucidated.
Overall, we showed a potential contribution of dysbiotic gut microbiota to the cholesterol disorder associated with genetically predisposed hyperlipidemia, as with simple hyperlipidemia although CAD risk factors and medications might imprint their effects microbiota. Whole exome sequencing in combination with clinical criteria facilitate early diagnosis of FH which can lead to preventive treatment that reduce cardiovascular risk. Our results characterize complex genetic and microbial variation in the regulation of cholesterol biological processes. This study provides conceptual insights that lay important groundwork for future applications in rare diseases, which will have to take into account both the genome and metagenome.
Data Availability Statement
The datasets presented in this study can be found in online repositories. The metagenomics raw sequence data have been deposited in the Genome Sequence Archive under accession number CRA002675 at https://bigd.big.ac.cn/gsa.
Ethics Statement
The studies involving human participants were reviewed and approved by the Peking Union Medical College Hospital. Written informed consent was obtained from the individuals for the publication of any potentially identifiable data included in this article.
Author Contributions
HL and YJ conceived and designed the analysis. RT and SF collected the samples and data. HL and CZ performed the analysis. HL, CZ, and SZ wrote the manuscript. YFW and YYW revised the manuscript. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by the National Natural Science Foundation of China (81670329), Center for Rare Diseases Research, Chinese Academy of Medical Sciences, Beijing, China (Grant No. 2016ZX310174–4), CAMS Innovation Fund for Medical Sciences (CIFMS) (No. 2016-I2M-1-011 and No. 2017-I2M-2-001).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We would like to thank the patients and their families for their participation and the KingMed Diagnostics association for their support. We thank Yifei Wang and Yiyang Wang for their technical assistance.
References
Adzhubei I., Jordan D. M., Sunyaev S. R. (2013). Predicting functional effect of human missense mutations using PolyPhen-2. Curr. Protoc. Hum. Genet. 7, 7.20. doi: 10.1002/0471142905.hg0720s76
Afshinnekoo E., Meydan C., Chowdhury S., Jaroudi D., Boyer C., Bernstein N., et al. (2015). Geospatial Resolution of Human and Bacterial Diversity with City-Scale Metagenomics. Cell Syst. 1 (1), 97–97.e93. doi: 10.1016/j.cels.2015.07.006
Brown J. M., Hazen S. L. (2018). Microbial modulation of cardiovascular disease. Nat. Rev. Microbiol. 16 (3), 171–181. doi: 10.1038/nrmicro.2017.149
Defesche J. C., Gidding S. S., Harada-Shiba M., Hegele R. A., Santos R. D., Wierzbicki A. S. (2017). Familial hypercholesterolaemia. Nat. Rev. Dis. Primers 3, 17093. doi: 10.1038/nrdp.2017.93
Fu J., Bonder M. J., Cenit M. C., Tigchelaar E. F., Maatman A., Dekens J. A., et al. (2015). The Gut Microbiome Contributes to a Substantial Proportion of the Variation in Blood Lipids. Circ. Res. 117 (9), 817–824. doi: 10.1161/CIRCRESAHA.115.306807
Goodrich J. K., Waters J. L., Poole A. C., Sutter J. L., Koren O., Blekhman R., et al. (2014). Human genetics shape the gut microbiome. Cell 159 (4), 789–799. doi: 10.1016/j.cell.2014.09.053
Heath R. J., Rock C. O. (2004). Fatty acid biosynthesis as a target for novel antibacterials. Curr. Opin. Investig. Drugs 5 (2), 146–153.
Jiang L., Wu W. F., Sun L. Y., Chen P. P., Wang W., Benito-Vicente A., et al. (2016). The use of targeted exome sequencing in genetic diagnosis of young patients with severe hypercholesterolemia. Sci. Rep. 6, 36823. doi: 10.1038/srep36823
Khera A. V., Won H. H., Peloso G. M., Lawson K. S., Bartz T. M., Deng X., et al. (2016). Diagnostic Yield and Clinical Utility of Sequencing Familial Hypercholesterolemia Genes in Patients With Severe Hypercholesterolemia. J. Am. Coll. Cardiol. 67 (22), 2578–2589. doi: 10.1016/j.jacc.2016.03.520
Langmead B., Salzberg S. L. (2012). Fast gapped-read alignment with Bowtie 2. Nat. Methods 9 (4), 357–359. doi: 10.1038/nmeth.1923
Li H., Durbin R. (2010). Fast and accurate long-read alignment with Burrows-Wheeler transform. Bioinformatics 26 (5), 589–595. doi: 10.1093/bioinformatics/btp698
Li X., Uchiyama T., Raetz C. R., Hindsgaul O. (2003). Synthesis of a carbohydrate-derived hydroxamic acid inhibitor of the bacterial enzyme (LpxC) involved in lipid A biosynthesis. Org. Lett. 5 (4), 539–541. doi: 10.1021/ol027458l
Li R., Yu C., Li Y., Lam T. W., Yiu S. M., Kristiansen K., et al. (2009). SOAP2: an improved ultrafast tool for short read alignment. Bioinformatics 25 (15), 1966–1967. doi: 10.1093/bioinformatics/btp336
Liu H., Pan L. L., Lv S., Yang Q., Zhang H., Chen W., et al. (2019). Alterations of Gut Microbiota and Blood Lipidome in Gestational Diabetes Mellitus With Hyperlipidemia. Front. Physiol. 10, 1015. doi: 10.3389/fphys.2019.01015
Liu Z., Li J., Liu H., Tang Y., Zhan Q., Lai W., et al. (2019). The intestinal microbiota associated with cardiac valve calcification differs from that of coronary artery disease. Atherosclerosis 284, 121–128. doi: 10.1016/j.atherosclerosis.2018.11.038
Mashima I., Theodorea C. F., Thaweboon B., Thaweboon S., Nakazawa F. (2016). Identification of Veillonella Species in the Tongue Biofilm by Using a Novel One-Step Polymerase Chain Reaction Method. PloS One 11 (6), e0157516. doi: 10.1371/journal.pone.0157516
McKenna A., Hanna M., Banks E., Sivachenko A., Cibulskis K., Kernytsky A., et al. (2010). The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 20 (9), 1297–1303. doi: 10.1101/gr.107524.110
Ng P. C., Henikoff S. (2003). SIFT: Predicting amino acid changes that affect protein function. Nucleic Acids Res. 31 (13), 3812–3814. doi: 10.1093/nar/gkg509
Nordestgaard B. G., Chapman M. J., Humphries S. E., Ginsberg H. N., Masana L., Descamps O. S., et al. (2013). Familial hypercholesterolaemia is underdiagnosed and undertreated in the general population: guidance for clinicians to prevent coronary heart disease: consensus statement of the European Atherosclerosis Society. Eur. Heart J. 34 (45), 3478–3490a. doi: 10.1093/eurheartj/eht273
Peng Y., Leung H. C., Yiu S. M., Chin F. Y. (2012). IDBA-UD: a de novo assembler for single-cell and metagenomic sequencing data with highly uneven depth. Bioinformatics 28 (11), 1420–1428. doi: 10.1093/bioinformatics/bts174
Sabatine M. S., Giugliano R. P., Wiviott S. D., Raal F. J., Blom D. J., Robinson J., et al. (2015). Efficacy and safety of evolocumab in reducing lipids and cardiovascular events. N. Engl. J. Med. 372 (16), 1500–1509. doi: 10.1056/NEJMoa1500858
Santos R. D., Gidding S. S., Hegele R. A., Cuchel M. A., Barter P. J., Watts G. F., et al. (2016). Defining severe familial hypercholesterolaemia and the implications for clinical management: a consensus statement from the International Atherosclerosis Society Severe Familial Hypercholesterolemia Panel. Lancet Diabetes Endocrinol. 4 (10), 850–861. doi: 10.1016/S2213-8587(16)30041-9
Sun B., Li L., Zhou X. (2018). Comparative analysis of the gut microbiota in distinct statin response patients in East China. J. Microbiol. 56 (12), 886–892. doi: 10.1007/s12275-018-8152-x
Suzuki T. A., Ley R. E. (2020). The role of the microbiota in human genetic adaptation. Science 370 (6521), eaaz6827. doi: 10.1126/science.aaz6827
Umans-Eckenhausen M. A., Defesche J. C., Sijbrands E. J., Scheerder R. L., Kastelein J. J. (2001). Review of first 5 years of screening for familial hypercholesterolaemia in the Netherlands. Lancet 357 (9251), 165–168. doi: 10.1016/S0140-6736(00)03587-X
Vieira-Silva S., Falony G., Belda E., Nielsen T., Aron-Wisnewsky J., Chakaroun R., et al. (2020). Statin therapy is associated with lower prevalence of gut microbiota dysbiosis. Nature 581 (7808), 310–315. doi: 10.1038/s41586-020-2269-x
Wang K., Li M., Hakonarson H. (2010). ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 38 (16), e164. doi: 10.1093/nar/gkq603
Yao J., Rock C. O. (2015). How bacterial pathogens eat host lipids: implications for the development of fatty acid synthesis therapeutics. J. Biol. Chem. 290 (10), 5940–5946. doi: 10.1074/jbc.R114.636241
Zhang D. W., Lagace T. A., Garuti R., Zhao Z., McDonald M., Horton J. D., et al. (2007). Binding of proprotein convertase subtilisin/kexin type 9 to epidermal growth factor-like repeat A of low density lipoprotein receptor decreases receptor recycling and increases degradation. J. Biol. Chem. 282 (25), 18602–18612. doi: 10.1074/jbc.M702027200
Keywords: familial hypercholesterolemia, LDLR gene mutation, whole exome sequencing, gut microbiota, Prevotella dentalis
Citation: Liu H, Jin Y, Tian R, Feng S, Zhang S and Zhang C (2021) A Comprehensive Analysis of Genomics and Metagenomics in a Heterozygote Familial Hypercholesterolemia Family. Front. Cell. Infect. Microbiol. 11:605954. doi: 10.3389/fcimb.2021.605954
Received: 14 September 2020; Accepted: 05 January 2021;
Published: 02 March 2021.
Edited by:
Marloes Dekker Nitert, The University of Queensland, AustraliaReviewed by:
Lei Dai, Shenzhen Institutes of Advanced Technology (CAS), ChinaXiaomin Yao, New York University, United States
Zhenjiang (Zech) Xu, Nanchang University, China
Copyright © 2021 Liu, Jin, Tian, Feng, Zhang and Zhang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Shuyang Zhang, c2h1eWFuZ3poYW5nMTAzQG5yZHJzLm9yZw==; Chenhong Zhang, emhhbmdjaGVuaG9uZ0BzanR1LmVkdS5jbg==