 Retsepile E. Maphasa
Retsepile E. Maphasa Mervin Meyer2
Mervin Meyer2 Admire Dube
Admire Dube
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Cell. Infect. Microbiol., 08 February 2021
Sec. Bacteria and Host
Volume 10 - 2020 | https://doi.org/10.3389/fcimb.2020.618414
This article is part of the Research TopicModulation of Macrophage Signaling Pathways during Bacterial InfectionsView all 20 articles
The major causative agent of tuberculosis (TB), i.e., Mycobacterium tuberculosis (Mtb), has developed mechanisms to evade host defense responses and persist within host cells for prolonged periods of time. Mtb is also increasingly resistant to existing anti-TB drugs. There is therefore an urgent need to develop new therapeutics for TB and host directed therapies (HDTs) hold potential as effective therapeutics for TB. There is growing interest in the induction of autophagy in Mtb host cells using autophagy inducing compounds (AICs). Nanoparticles (NPs) can enhance the effect of AICs, thus improving stability, enabling cell targeting and providing opportunities for multimodal therapy. In this review, we focus on the macrophage responses to Mtb infection, in particular, the mechanistic aspects of autophagy and the evasion of autophagy by intracellular Mtb. Due to the overlap between the onset of autophagy and apoptosis; we also focus on the relationship between apoptosis and autophagy. We will also review known AICs in the context of Mtb infection. Finally, we discuss the applications of NPs in inducing autophagy with the intention of sharing insights to encourage further research and development of nanomedicine HDTs for TB therapy.
TB caused by an infection from the bacillus Mycobacterium tuberculosis (Mtb) is among the top ten leading causes of death globally, and is the main cause of death from a single infectious agent. An estimated 1.7 billion people across the world are infected with Mtb and in 2018, approximately 10 million new cases were reported, corresponding to 130 cases per 100,000 population (World Health Organization, 2019). Geographically, the majority of TB cases are found mostly in South-East Asia, Africa, and the Western Pacific. The eight countries; India, China, Indonesia, the Philippines, Pakistan, Nigeria, Bangladesh and South Africa account for two thirds of all cases globally (World Health Organization, 2019).
TB commonly affects the lungs, where it is called pulmonary TB (PTB) but can also affect other organs throughout the body and in this case; it is known as extra-pulmonary TB (EPTB) (World Health Organization, 2018). It is estimated that between 5 and 15% of the estimated 1.7 billion people (about 20% of the world population) is infected with Mtb will develop TB during their lifetime (World Health Organization, 2018). However, the probability of developing TB is greater for people living with HIV and also higher for people who smoke, consume alcohol, have diabetes, and/or are under-nourished (World Health Organization, 2019). The current drug therapy for TB is a six-month regimen that consists of a two month intensive phase, where patients receive four first-line antibiotics [i.e., ethambutol hydrochloride (EMB), isoniazid (INH), rifampicin (RIF), and pyrazinamide (PZA)], followed by a four month continuous phase, where patients receive RIF and INH (Grange and Zumla, 2002; Nasiruddin et al., 2017; Bekale et al., 2018).
The lengthy multidrug therapy has been shown to overwhelm patients with high pill load and toxic side effects, resulting in a considerable reduction inpatient compliance and adherence to the medication (Dartois, 2014). This therefore increases the potential risk for the development of drug-resistant strains in noncompliant patients (Kimmey and Stallings, 2016; Kaufmann et al., 2018). As a result, drug-resistant TB remains a major public health hazard. In 2018, approximately 500,000 new cases of RIF-resistant TB (RR-TB) (of which 78% had multidrug-resistant TB) were reported (World Health Organization, 2019).
India, China and the Russian Federation were found to be the three countries with the largest share of the global burden of RR-TB, with each having 27%, 14% and 9%, respectively. In addition, 18% of treated cases in 2018, were either multidrug-resistant TB (MDR-TB) or RIF-resistant TB (RR-TB), with the highest proportions (>50%) of these cases being in countries of the former Soviet Union (World Health Organization, 2019). MDR-TB plus resistance to at least one of the fluoroquinolones and one of the injectable agents used in MDR-TB treatment regimens is known as extensively drug-resistant TB (XDR-TB). In 2018, at least one case of XDR-TB was reported by each of the 131 WHO Member States. However, over the past 15 years, the average proportion of MDR-TB cases with XDR-TB reported by 128 countries was 6.2% of all TB cases (World Health Organization, 2019).
Macrophages are most common host cells of Mtb in pulmonary TB. Therefore, the successful treatment of pulmonary TB depends on the efficiency of TB drugs to infiltrate complex lung lesions, penetrate the cell membrane of macrophages and other host cells and finally, for the drugs to be taken up by the intracellular Mtb (Dartois, 2014). A study by Gurumurthy et al., demonstrated that TB drugs are malabsorbed, rifampin in particular, was found to be malabsorbed in patients with advanced HIV and diarrhea and cryptosporidial infection. The bioavailability of INH was found to be more reduced in rapid acetylators, while the absorption of EMB and PZA was also reduced in the same patient group. These results suggest that the malabsorption of anti-TB drugs in TB patients with HIV, and diarrhea could be one of the factors affecting the success of anti-TB therapy in this patient cluster (Gurumurthy et al., 2004).
Anti-TB fixed dose combination (FDC) drugs and products have been described as unstable in formulations due to chemical interactions between the drugs, this incompatibility can also lead to a reduction in the bioavailability of the drugs during oral administration. Therefore, in a previous study by Aucamp et al., interactions as well as possible incompatibilities between the four commonly prescribed commercial anti-TB FDC drugs were investigated at a temperature of 50°C. The study revealed that INH and RIF are chemically incompatible (Aucamp et al., 2019). Anti-TB drugs should also be taken on an empty stomach to reduce interaction with various foods, which results in the absortion of these drugs being reduced. In addition, anti-TB drugs also interact with other drugs when taken as part of a combination treatment, this interaction with foods and/or other drugs has been shown to increase the side effect inducing capacity of anti-TB drugs e.g., interaction between isonizid with levodopa, or carbohydrate rich foods (Arbex et al., 2010).
First-line and second-line TB drugs have been shown to posess different intracellular uptake properties, and intracellular to extracellular (I/E) ratios ranging between 0.1 to >20. In both in vitro and in vivo studies, aminoglycosides and β-lactams (antibiotics that are used to treat XDR TB) were found to have I/E ratios that were below 1, the I/E ratio of INH was found to be approximately 1. On the other hand, the I/E ratio ofRIF ranged between two and five, while the I/E ratios of ETH and the macrolides were shown to range between 10 to >20 (Johnson et al., 1980; Hand et al., 1984). Fluoroquinolones have also been found to have variable intracellular penetration properties: the concentrations of moxifloxacin (MXF) have been shown to be 20–70 fold higher in pulmonary alveolar macrophages when compared to the concentrations in the plasma (Soman et al., 1999). The concentrations of levofloxacin were only marginally higher in alveolar cells when compared with concentrations in the plasma (Andrews et al., 1997). On the other hand, ciprofloxacin was found to have an alveolar macrophage to plasma ratio of 5–10, indicating an intermediate penetration capacity (Schüler et al., 1997).
The penetration and accumulation of TB drugs inside Mtb is the final step of the long journey the drugs have to take to reach the site of action. The reduced permeability of Mtb to small-molecule drugs has also been shown to contribute to the drug tolerance found in the dormant Mtb population (Dartois, 2014). Nutrient-starved non-replicating Mtb also display a significant reduction in intracellular accumulation of fluoroquinolones, which may help explain the resistence of quiescent Mtb against fluoroquinolone activity (Sarathy et al., 2013a). Polyamines decrease the ability of drugs to permeate Mtb, and have also been shown to prevent the uptake of fluoroquinolones by the mycobacteria (Sarathy et al., 2013b). These results, therefore suggest that the intracellular production of polyamines by macrophages might be partially responsible for the dormancy and phenotypic drug resistance of intracellular Mtb (Dartois, 2014).
There is therefore an urgent need to develop new therapeutic approaches for the treatment of TB (Kimmey and Stallings, 2016; Kaufmann et al., 2018; Paik et al., 2019). Host directed therapy (HDT) is a novel and promising concept in TB treatment, where small molecules, together with or without additional antibodies, are used to modify host responsesto better control the progression of TB. HDT drugs are distinct from antibiotics, as they directly modulate the functions of host cells without interacting with Mtb; thereby preventing the development of drug resistance by the infecting pathogen (Kolloli and Subbian, 2017; Paik et al., 2019). The applications of HDT-TB can possibly be effective in the treatment of MDR or XDR-TB through the therapeutic targeting of various clinically relevant biological pathways in hosts. Modulation of pathological or protective responses in hosts has revealed that various components of the immune system are central therapeutic targets for HDT against TB (Zumla et al., 2016; Kolloli and Subbian, 2017; Paik et al., 2019).
Autophagy is an intracellular catabolic process that assists in the maintainance of homeostasis or the removal of invading pathogens through a lysosomal degradation mechanism. The activation of autophagy in infected host cells, through the use of various drugs/compounds is a promising treatment strategy against Mtb infection, which may also work against drug-resistant strains with or without adjunctive agents (Gupta et al., 2016; Kimmey and Stallings, 2016; Paik et al., 2019). As autophagy and HDT are still relatively new approaches in TB, there is a scarcity of research focusing on the delivery of autophagy inducing compounds (AICs) and the treatment duration of autophagy therapy aspecially when combined with conventional antibiotic treatment. However, it is still necessary to directly deliver the AICs to the infected cells/tissues/organs to prevent unwanted side effects and delivery to non-target cells (Paik et al., 2019).
Nanoparticles (NPs) can be utilized as a delivery system to improve the activity of AICs against intracellular Mtb by transporting the encapsulated AICs to their target sites while protecting them from biodegradation and enhancing their absorption across biological barriers (Dube et al., 2013; Tukulula et al., 2018). NPs also allow for sustained drug release in the target tissue. This allows for the reduction of dosing frequency, thus lessening the drug-associated side effects in the process (Grabowski et al., 2013). In addition, the materials used to synthesize the NPs can possess autophagy inducing activity or the surface of the NPs can be functionalized with an AIC (Bekale et al., 2018).
In this review, we focus on the macrophage responses to Mtb infection, in particular, the mechanistic aspects of autophagy and the evasion of autophagy by intracellular Mtb. Due to the overlap between the onset of autophagy and apoptosis; we also focus on the relationship between apoptosis and autophagy. We will also review known AICs in the context of Mtb infection. Finally, we discuss the applications of NPs in inducing autophagy with the intention of sharing insights to encourage further research and development of nanomedicine HDTs for the treatment of TB.
TB can be spread when people with active pulmonary TB expel Mtb into the air, for example, by coughing. Aerosols of the pathogen can persist in the air for a prolonged period of time and once inhaled, the bacilli travel through the upper respiratory tract to reach the alveoli in the lungs, where they are taken-up primarily by alveolar macrophages, type 2 pneumocytes and polymorphonuclear neutrophils (PMNs). However, alveolar macrophages commonly act as the first line of defense against the pathogen (Silva Miranda et al., 2012; Dartois, 2014; Bekale et al., 2018). Mtb is internalized by macrophages through phagocytosis and subsequently located within phagosomes, which undergoa series of fusion events to mature and attain anti-microbial properties. The progression of phagosome maturation is actively regulated and driven by the network of Rab GTPases (Rab), which also play a role in the identity of the endosomal organelle (e.g., Rab5, early endosomes; Rab7, late endosome), sorting of protein and lipids through the recycling pathway as well as regulating membrane-fusion events (Gruenberg and van der Goot, 2006; Gutierrez, 2013; Prashar et al., 2017).
Therefore, the process of phagosome maturation is characterized by the recruitment of specific Rab GTPases, which ultimately facilitate the fusion between the phagosome and the lysosome to form the phagolysosome, a cytoplasmic body that contains a set of hydrolytic enzymes necessary for the clearance of pathogens. Rab20, Rab34 and the proneurotropin receptor sortilin have also been described as important regulators of phagosome maturation and are thus critical in the control and eradication of intracellular Mtb (Kasmapour et al., 2012; Vazquez et al., 2016; Schnettger et al., 2017). The completion of phagosome maturation is accompanied by a reduction in intraphagosomal pH from neutral to the more acidic pH 5, via a vesicular proton-pump ATPase (HC-V-ATPase)-dependent process (Russell et al., 2009). Phagosomal acidification creates a suitable environment for the production of reactive oxygen species (ROS) and for the optimal activity of lysosomal digestive enzymes; hence, the process is essential for the clearance of intracellular Mtb (Vieira et al., 2002; Sun-Wada et al., 2009).
The uptake of Mtb by phagocytic macrophages is facilitated by the recognition of Pathogen Associated Molecular Patterns (PAMP) present at the bacterial surface by various Pattern Recognition Receptors (PRRs) on the surface of macrophages such as Scavenger Receptors (SR), cytosolic DNA sensors,C-type Lectin Receptors (CLR/CTL), Fc Receptors (FcR) and Toll-Like Receptors (TLR), mannose receptors, surfactant protein A receptors, CD14, immunoglobulin receptors, and complement receptors (Songane et al., 2012; Nasiruddin et al., 2017; Queval et al., 2017). Phagocytosis, innate immune responses and various cellular processes such as antigen presentation, inflammasome activation, autophagy, phagosome maturation, and apoptosis are dependent on the stimulation of the PRRs (Queval et al., 2017). Mtb has been shown to activate phagocytosis in macrophages by interacting with TLRs. Recognition of Mtb is more specifically facilitated by TLR2 using a variety of microbial structures, such as the lipoproteins19 kDa, lipomannans (LM), lipoarabinomannan (LAM) and the glycolipid phosphatidyl-myo-inositolmannoside (PIM) (Quesniaux et al., 2004).
It has been noted that the loss of lipooligosaccharide (LOS) production in the evolution of TB causing mycobacteria is responsible for the rough colony morphology of these pathogens, further contributing to enhancing the recognition of Mtb by TLR2 (Boritsch et al., 2016). In addition, TLR9 has been reported to recognize unmethylated CpG motifs in bacterial DNA (Bafica et al., 2005). These events trigger a signaling cascade that stimulates Myeloid Differentiation primary response protein 88 (MyD88) resulting in the activation of pro-inflammatory cytokine production, activation and nuclear translocation of transcription factors, including nuclear factor kappa-light-chain-enhancer of activated B cells (NF-kB) (Nigou et al., 2001).
C-type lectin receptors (CLR/CTL) are a family of calcium dependent membrane bound receptors that recognize carbohydrate rich molecules. Among these receptors, the mannose Receptor (MR) is one of the most well-known, and recognizes mannose on the surface of Mtb. The stimulation of macrophages through the MR has been shown to induce the production of anti-inflammatory cytokines without activating oxidative responses (Nigou et al., 2001). The Dendritic Cell immune Activating Receptor (DCAR), Dectin-1, Dectin-2, C-type lectin superfamily member 8 (Clecsf8) and the Mincle receptor belong to a sub-family of the CLR and represent the most known CLR expressed by macrophages (Yonekawa et al., 2014; Marakalala and Ndlovu, 2017).
DCAR is an Fc receptor γ-chain (FcRγ) coupled receptor that induces Th1 responses against Mtb infections by recognizing PIM. T cells of DCAR-deficient mice have been shown to have weakened IFN-γ production resulting in elevated bacterial loads (Marakalala and Ndlovu, 2017). Dectin-1 is a transmembrane receptor with an extracellular C-type lectin-like domain (CRD) and a cytoplasmic immunoreceptor tyrosine-based activation motif (ITAM)-like domain (hemITAM). Dectin-1 is a known fungal β (1, 3)-glucan receptor but the precise PAMP for Mtb remains unknown. However, Dectin-1 triggers the production of IL-12p40 in a Syk-dependent mannerin splenic DCs infected with Mycobacterium Bovis (M. bovis) or pathogenic Mtb (Dinadayala et al., 2004; van de Veerdonk et al., 2010; Marakalala and Ndlovu, 2017).
Dectin-2 on the other hand, has been shown to recognize ManLAM to induce host immune responses against Mtb infection. The structure of Dectin-2 consists of a transmembrane domain, a CRD, and a short cytoplasmic tail. The short tail recruits an ITAM-linked FcRγ, which activates signaling through Syk and possibly the Caspase recruitment domain family member 9/B-Cell CLL/lymphoma 10/Mucosa-associated lymphoid tissue lymphoma translocation protein 1 (CARD9/BCL10/MALT1) complex. Dectin-2 has also been shown to recognize the virulent Mtb H37Rv strain, however, the protective capacity of this receptor against Mtb is yet to be demonstrated in vivo (Toyonaga et al., 2016; Marakalala and Ndlovu, 2017).
The Mincle receptor recognizes and specifically binds to what is thought to be likely the most abundant glycolipid in the mycobacterial cell wall, the mycobacterial cord factor Trehalose-6,6-dimycolate (TDM) (Ishikawa et al., 2009; Marakalala and Ndlovu, 2017). The ligation of TDM to Mincle triggers the generation of Th1/Th17 immune responses, production of pro-inflammatory cytokines and granuloma formation (Ishikawa et al., 2009; Marakalala and Ndlovu, 2017; Mishra et al., 2017). Clecsf8 also interacts with mycobacteria through the TDM glycolipid. This PRR is commonly expressed on macrophages, classical monocytes, peripheral blood neutrophils, and by a subsets of dendritic cells (DCs). Upon engaging with TDM, Clecsf8 stimulates Mincle expression through a protein to protein interaction, resulting in an increase in intracellular responses. Downstream signaling of this FcRγ-coupled receptor is mediated through Syk kinase and CARD9/BCL10/MALT1 pathways and induces various intracellular responses, including NF-κβ activation, pro-inflammatory cytokine production, phagocytosis, and respiratory burst. Clecsf8 interaction with TDM also induces DC maturation and T-cell priming (Marakalala and Ndlovu, 2017).
Complement Receptor (CR) and Fc receptors are also highly expressed on the surface of macrophages and CR3 has been shown to help facilitate the phagocytosis of Mtb by macrophages through the recognition of PIM and/or Mtb polysaccharides (Villeneuve et al., 2005). Mammalian monocytes and macrophages express scavenger receptors to recognize oxidized or acetylated lipoproteins. The Macrophage Receptor with Collagenous (MARCO) structure is the most studied scavenger receptor and has been found to cooperate with TLR2 to stimulate the secretion of proinflammatory cytokines and the activation of transcriptional factor NF-kB through the recognition of TDM (Bowdish et al., 2009). DCs and macrophages use Dendritic Cell Specific Intercellular adhesion molecule-3 Grabbing Non-integrin (DCSIGN/CD209) to recognize conserved sugar motifs on various parasites, viruses, and bacteria, including Mtb (Tanne and Neyrolles, 2010).
The failure to eradicate intracellular Mtb post-infection, results in either the active or latent form of TB. In the latent phase, an assortment of immune cells (more macrophages, lymphocytes and DCs) are recruited to the region of infection to keep the intracellular bacteria inactive. Together these cells may at times form a tissue structure known as a granuloma, which consists of a core composed of Mtb infected macrophages. In the presence of activated T cells, the granuloma becomes fully organized and the core becomes enclosed with lymphocytes (mainly γ/δ T Cells, CD4+ and CD8+) which initiate the secretion of IL-8, IL-12, TNF-α, and other pro-inflammatory cytokines by macrophages (Ehlers and Schaible, 2012; Hajipour et al., 2012; Nasiruddin et al., 2017). The end result consists of a continuous balance between necrotic infected cells and the recruitement of new phagocytes onto the granuloma is known as latency and is one of the hallmarks of latent TB (van Crevel et al., 2002; Hajipour et al., 2012; Nasiruddin et al., 2017).
However, this impasse between the host and pathogen, is dynamic and consists of an equilibrium between cell death and replenishment through cellular recruitment, as well as vascular and tissue remodeling (Ehlers and Schaible, 2012). The bacteria can persist within the granuloma in a latent state for prolonged periods of time (lasting up to the lifetime of the individual), however, as illustrated in Figure 1, a weakened immune system (e.g., due to malnutrition, HIV infection etc.) can lead to the reactivation of the Mtb. In this active phase, the bacteria can begin to proliferate, ultimately inciting the death of the infected macrophages through necrosis (Silva Miranda et al., 2012). Mtb can now exit the granuloma and further disseminate to form lesions in other regions of the lungs. The active Mtb can also be transmitted to other individuals through for example, coughing (Mueller and Pieters, 2006).
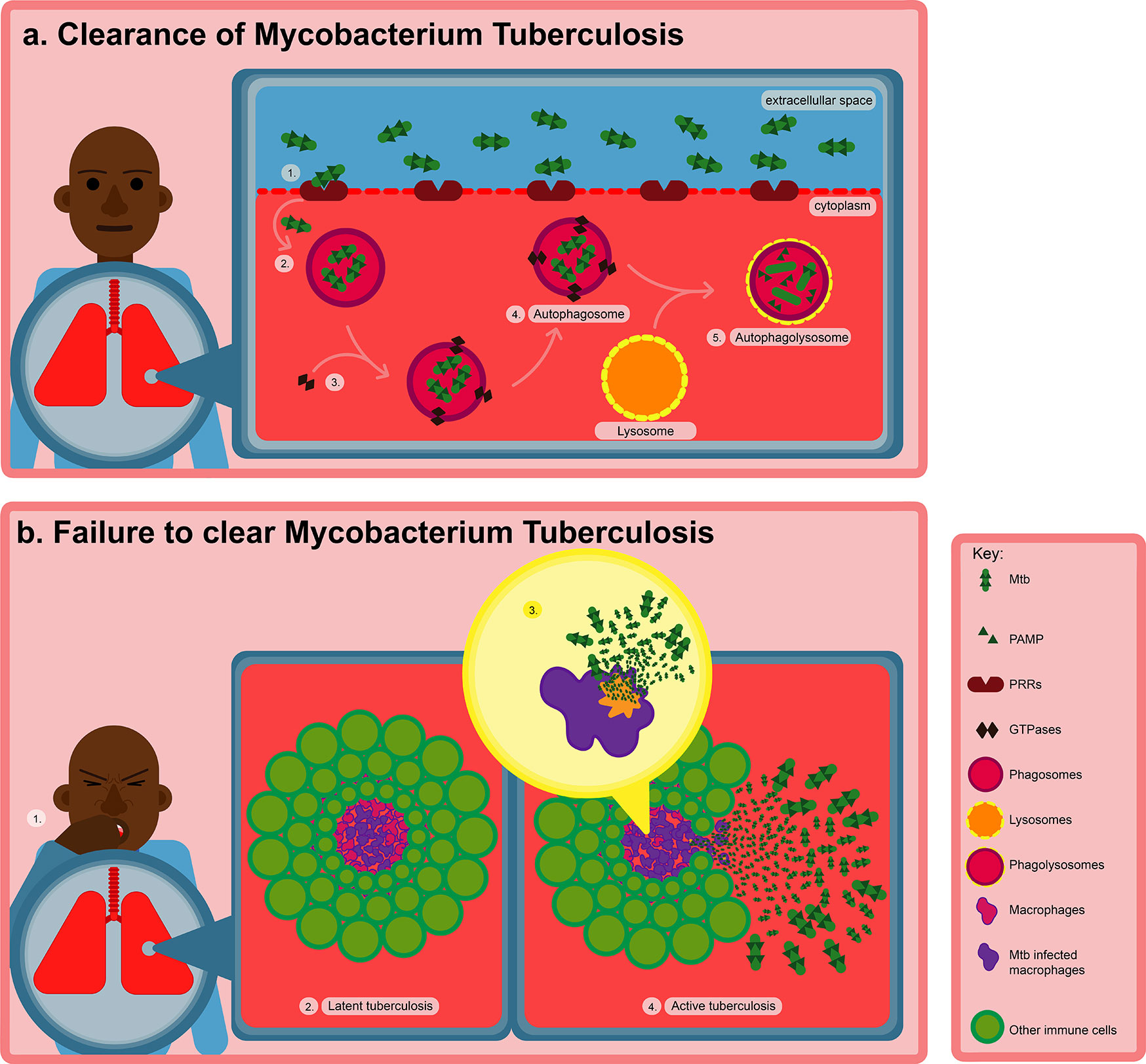
Figure 1 Transmission and pathogenesis of TB after Mtb infection. (A) Macrophages use PRRs to take up Mtb by recognizing various PAMPs on the surface of the bacteria. Recognition of Mtb is more precisely facilitated by TLR2, which triggers phagocytosis, innate immune responses and various cellular processes such as antigen presentation, inflammasome activation, autophagy, and apoptosis resulting in the eradication of intracellular Mtb. (B) However, failure to eradicate intracellular Mtb after infection, results in either the latent or active form of TB. In the latent phase, the Mtb can remain dormant within the granuloma for years. However, when the immune system becomes weakened, Mtb can become reactivated, subsequently inducing the death of the infected macrophages through necrosis.
Macrophages infected with Mtb commonly express two types of cell death, i.e., necrosis: a death modality that ends in cell lysis and apoptosis: a form of cell death that maintains an intact cell membrane (Figure 2) (Behar et al., 2011). Apoptosis can be instigated through various mechanisms; nonetheless, the process is defined using several molecular and morphological decisive factors. The common hallmarks of apoptosis are DNA fragmantation, exposure of phosphatidylserine on the outer leaflet of the plasma membrane, and lastly, containment of cellular components in membrane-bound blebs (Behar et al., 2011).
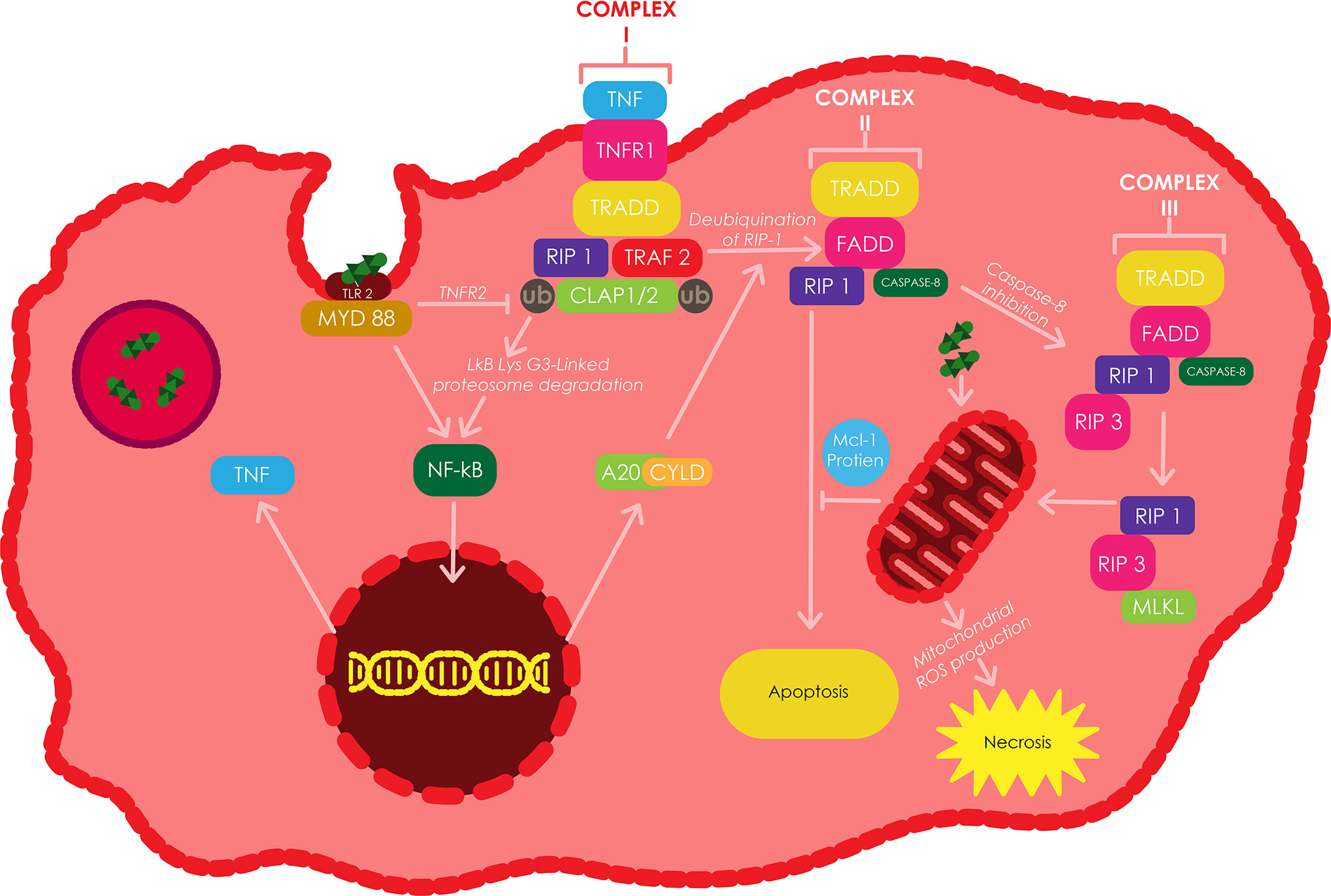
Figure 2 The induction of apoptosis and/or necroptosis by TNF in Mtb infected macrophages. TLR2 can be stimulated by different components of the Mtb cell wall, which triggers the translocation of NF-κB to the nucleus resulting in the silmultaneous expression of TNF through a myeloid differentiation factor 88 (MyD88) dependent approach. The binding of TNF to TNFR1, induces the binding of TRADD to RIP1 and TRAF2/5, which consecutively bind to cIAP1/2 to make up complex I. cIAPs facilitates the Lys-63-linked polyubiquitination of RIP1 or TRAF2 resulting in Lys 63-linked proteasomal degradation of IκB. The resulting NF-κB translocates into the nucleus and triggers the transcription of various genes including A20 and CYLD. RIP1 becomes deubiquitinated by A20 and CYLD, futher switching complex I into complex II, which then initiates apoptosis through the recruitment and activation of Caspase-8. Casepase 8 inactivation triggers the formation of complex III, which induces the recruitment of RIP3 by RIP1 and subsequently the recruitment of MLKL by RIP3, ultimately resulting in necroptosis. Adapted from (Xu et al., 2014).
Vertebrate cells generally undergo apoptosis by utilizing the intrinsic apoptotic pathway; relying on mitochondrial outer membrane permeabilization (MOMP) as the defining hallmark (Green and Kroemer, 2004). MOMP facilitates the release of apoptosis inducing factor, Smac-DIABLO, cytochrome c, and other factors from the mitochondrial intermembrane resulting in the activation of caspase-3/7 which are responsible for the induction of apoptosis. These events can also occur due to the opening of the mitochondrial inner membrane pore (permeability transition pore), resulting in the loss of the mitochondrial intermembrane potential (Δψm), mitochondrial permeability transition (MPT), and ultimately the induction of necrosis. Mtb infections have also been shown to induce these changes in mitochondrial membranes; therefore, these changes are believed to determine the mode of death in infected macrophages (Behar et al., 2011).
In the first scenario, MPT can cause the mitochondria to become permeable to water, resulting in swelling, dysfunction, and ultimately necrosis (Green and Kroemer, 2004). In the second scenario, MOMP can occur as a by-product of the outer mitochondrial membrane damage caused by irreversible MPT. Such a scenario can materialize when hepatocytes undergo apoptosis and/or necrosis due to oxidative stress, or other toxic treatments (Kim et al., 2003). However, the occurance of apoptosis and MOMP is not always dependent on MPT. In this scenario, members of the Bcl-2 family of apoptosis inducing proteins, for example, induce MOMP without affecting the mitochondrial inner membrane (Chipuk and Green, 2008). More specifically, processed Bcl-2 only protein BID activates the proapoptotic Bcl-2 family proteins BAX and BAK which subsequently induce MOMP and translocation of proapoptotic factorsinto the cytosol, resulting in the activation of caspase-9, caspase-3 and ultimately apoptosis (Behar et al., 2011).
A permeable mitochondrial outer membrane allows for effector molecules to damage the mitochondrial inner membrane by gaining access into the mitochondrial intermembrane space. This could be the same mechanism utilized by capase-3 to damage a component of the electron transport chain called Ndufs1 (Ricci et al., 2004). Ndufs1 damage induces ROS production by disrupting the electron transport chain in the inner membrane, resulting in necrosis. It is believed that caspase-3 gains access to the mitochondrial intermembrane space through a permeable mitochondrial outer membrane, which also facilitates the escape of proapoptotic factors including cytochrome c into the cytosol (Behar et al., 2011). Finally, there is a scenario where MPT can occur independently from the induction of MOMP. This is believed to be a mechanism utilized by the protease, granzyme A, to damage the components of the mitochondrial inner membrane (Martinvalet et al., 2008). Granzyme A enters the mitochondrial intermembrane space without damaging the mitochondrial outer membrane, due to the cytosolic chaperone activity of Hsp70 and Hsp90. Once inside, the protease cleaves Ndufs3 resulting in MPT (Martinvalet et al., 2008).
Various components of the mycobacterial cell wallcan stimulate the production of tumor necrosis factor (TNF) in Mtb infected macrophages through the TLR2-mediated signaling pathway. TNF induces apoptosis through TNF receptors, which can be found as either membrane-bound or in soluble form (Xu et al., 2014). TNF minimizes the spread of Mtb by activating the Caspase-8-mediated extrinsic cell death pathway (Behar et al., 2011). Avirulent or attenuated Mtb strains such as Mtb H37Ra have been shown to induce more macrophage apoptosis when compared to virulent strains such as Mtb H37Rv, which have been confirmed to elude TNF-dependent apoptosis by activating the release of membrane-bound TNFR2 in solution, through an IL-10 dependent pathway (Behar et al., 2011), and by increasing the expression of Mcl-1 protein, a member of the anti-apoptotic B-cell lymphoma/leukemia-2 (Bcl-2) family of proteins which are located in the outer membrane of mitochondria (Xu et al., 2014).
The virulent strain H37Rv has been shown to induce an enhanced release of IL-10 in Mtb infected macrophages, when compared to the attenuated strain H37Ra. This elevated IL-10 release triggers a greater build up of sTNFR2, which through the formation of a soluble receptor-ligand complex leads to the inhibition of TNF-α activity (Balcewicz-Sablinska et al., 1998). Conversely, increased production of TNF has been shown to induce mitochondrial ROS in infected macrophages via the receptor interacting protein 1 (RIP1) receptor interacting protein 3 (RIP3) mixed lineage kinase domain-like protein (MLKL) dependent pathways. The mitochondrial ROS induces a type of necrosis called necroptosis in Mtb infected macrophages, which leads to the release of mycobacteria into the extracellular environment (Wang et al., 2012). The repression of RIP3 or MLKL has been shown to switch Mtb infected macrophages from a state of TNF-induced necroptosis to a state of delayed RIP1-dependent apoptosis (Han et al., 2011). The stimulation of TNF receptor 1 (TNFR1) by TNF triggers the binding of TRADD (TNF receptor-associated death domain) to RIP1, TNF receptor-associated factor 2/5 (TRAF2/5) and cIAP1/2 (cellular inhibitor of apoptosis 1/2) to form complex 1 (Xu et al., 2014).
The Lys 63-linked polyubiquitination of RIP1 or TRAF2 by cIAPs induces the translocation of NF-κB into the nucleus to induce the transcription of A20 and cylindromatosis (CYLD), which can both deubiquitinate RIP1 (Vandenabeele et al., 2010). Inhibition of cIAP proteins or deubiquitination of RIP1 facilitates the conversion of complex I to complex II, which is comprised of FADD, RIP1, TRADD and Caspase-8 (Han et al., 2011). Complex II facilitates the activation of Caspase-8 which further instigates apoptosis. The inhibition of Caspase-8 activity triggers the recruitment of RIP3 by RIP1 to form complex III (also called necrosome), which also comprises of Caspase-8, FADD and possibly TRADD. RIP1 subsequently phosphorylates RIP3 to recruits MLKL, resulting in the onset of necroptosis. In addition, the presence of inhibitors of ubiquitinating enzymes, virulent strains of Mtb, and deubquitinating enzymes keep Caspase-8 inactive (Xu et al., 2014).
Therefore, avirulent Mtb strains tend to stimulate apoptosis, whereas virulent Mtb strains are inclined to trigger necroptosis, a type of cell death known to favourthe pathogen and facilitate the dissemination of Mtb (Jayaraman et al., 2013). These observations suggest that at some point during the infection, virulent Mtb strains stimulate higher expression of TNF when compared to avirulent strains resulting in necroptosis. In addition, virulent strains possibly secrete a biological agent that blocks the activity of Caspase-8, reslting in the inhibition of apoptosis. In contrast, avirulent strains might be lack this ability (Xu et al., 2014).
Macrophage apoptosis is associated with an increase in ROS production. During apoptosis, DCs engulf and facilitate the degradation of the Mtb or Mtb product containing apoptotic bodies by cross-presenting the bacterial antigens to CD8+ T cells through a process called efferocytosis (Lee et al., 2004; Ashida et al., 2011; Silva Miranda et al., 2012). The binding of apoptotic bodies to the phagocytes during efferocytosis triggers the expression of anti-inflammatory cytokines such as interleukin-10 (IL-10) and transforming growth factor-β. The suppression of inflammation by these cytokines is believed to assist in minimizing the tissue damage caused by the accidental release of intracellular contents, including degradative enzymes, to the extracellular space (Lee et al., 2009). As the bacteria are typically killed during this process, Mtb infected macrophages have ‘alternative activation’ which is characterized by the arrest of phagosome maturation, increased secretion of macrophage attractant chemokines and M2 cytokines, suppression of microautophagy and apoptosis inhibition (Lancellotti et al., 2006; Ashida et al., 2011).
Highly virulent Mtb strains are known to primarily induce necrosis; however, apoptosis has also been detected in macrophages infected with virulent Mtb (Figure 2). The induction of apoptosis in Mtb infected macrophages has been shown to be TNF-α dependent, although, the virulent Mtb strains induce extensively less apoptosis in host cells when compared to related attenuated strains (van Crevel et al., 2002; Behar et al., 2011). This difference in apoptosis induction has been explained using selective induction and release of neutralizing soluble TNF-α receptors by pathogenic strains. In turn, the release of TNF-α receptors is regulated by IL-10 production. Therefore, virulent strains of Mtb may selectively induce the production of IL-10, resulting inreduced TNF-α activity and decreased apoptosis of infected cells (van Crevel et al., 2002). However, it is still not clear whether virulent Mtb inhibits the induction of apoptosis or prevents its completion by locking its downstream events (Behar et al., 2011).
The TLR2 dependent activation of the NF-kB cell survival pathway and the enhanced production of the soluble TNF receptor 2 (sTNFR2), neutralize the activity of TNFR1 resulting in Mtb persisting inside the macrophage phagosomes in either an active form or a dormant form (Lancellotti et al., 2006; Ashida et al., 2011). Therefore, in order to investigate the role of apoptotic macrophages in adaptive immunity, a new adoptive transfer model has been developed where in vitro Mtb infected macrophages have been transferred from wild type or knockout mice and instilled into normal recipient mice via the trachea. This approach has been used to study the influence of macrophage genotypes on the T-cell response and control of infection (Divangahi et al., 2010).
Recent studies have investigatedhow the eicosanoid prostaglandin (PGE2) and lipoxins (LXs) regulate programmed celldeath in Mtb infected macrophages (Figure 3). Cyclooxygenases (COX), namely COX1 and COX2, are responsible for the production of PGs by metabolizing arachidonic acid into PGH (Kurtz et al., 2006; Behar et al., 2011). The various PGs produced comprise of PGD2, PGE2, PGF2α, PGI2, and thromboxane. Among these, PGE2 is the most studied PG, and has a variety of biological functions triggered by interacting withany of its four specific receptors EP1, EP2, EP3, and EP4 (Kurtz et al., 2006; Behar et al., 2011). The interaction between PGE2 and the EP2 receptor prevents bacteria induced damage to the mitochondria in macrophages. On the contrary, interacting with EP4 triggers the repair of the plasma membrane in infected macrophages through the recruitment of lysosomal membranes to the cell surface (Behar et al., 2011).
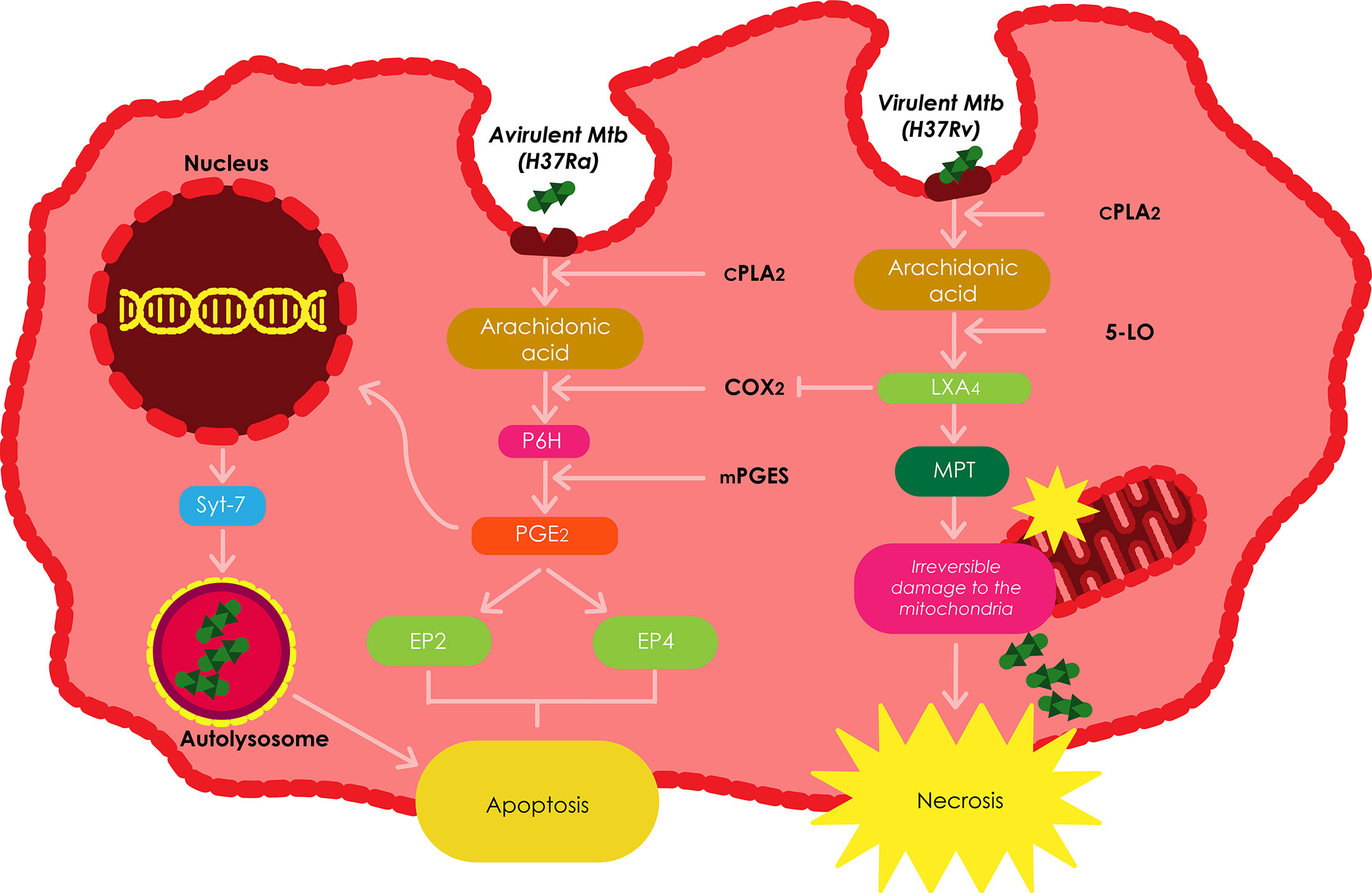
Figure 3 The roles played by eicosanoids and lipoxins in regulating the death of infected macrophages. The balance between prostanoid and lipoxin production is the dictator of the modality of death in Mtb infected macrophages. Increased production of LXA4 induces necrosis; whereas, excess production of PGE2 inhibits necrosis and favors apoptosis. Adapted from (Behar et al., 2011).
Mtb H37Ra has been shown to stimulate PGE2 in host macrophages; however, virulent Mtb has been demonstrated to stimulate the production of LXA4in macrophages (Bousso, 2008). In these cells, 5-lipoxygenase (5-LO) metabolizes arachidonic acid to produce LXA4. The ensuing LXA4 plays a key role in the onset of inflammation and has been shown to stimulate macrophages to engulf apoptotic neutrophils using phagocytosis (Behar et al., 2011). LXA4 inhibitsthe expression of COX2 and thus, the generation of PGE2, subsequently redirecting the fate of the infected macrophage to necrosis. Other pro-necrotic roles of LXA4 include inhibiting the stimulation ofapoptosis by lipopolysaccharide/interferon-γ, prevention of the secretion of pro-apoptotic mediators by the mitochondria, as well as inhibiting caspase activation and ROS generation. This data establishes LXA4 as the most important mediator of necrosis (Behar et al., 2011).
The fate of the macrophage following Mtb infection is a key component of the immune response against the pathogen. Several PGE2 dependant cellular pathways have been found to promote apoptosis while preventing necrosis in Mtb infected macrophages (Bousso, 2008). These pathways include the induction of prostaglandin H (PGH) synthase which is propelled by membrane associated prostaglandin E synthase-1 (mPGES) to produce PGE2, the production of COX2, transcription of synaptotagmin-7 (Syt-7), a lysosomal Ca 2+ sensor that facilitates the fusion of the plasma membrane with lysosomal vesicles requiredto induce apoptosis, prevention of irreversible damage of the mitochondrial inner membrane by virulent Mtb strains using MPT (Bousso, 2008), and the production of plasminogen activator inhibitor type 2 (serpin B2) (Winau et al., 2006).
In addition, PGE2 also induces the transcription of Syt-7, which facilitates the repair of the plasma membrane repair by means of lysosomal exocytosis (Pozzi et al., 2005). There are numerous PGE2 dependent mechanisms leading to apoptosis, and the balance between the production of PGE2 and LXA4 regulates the number of cells undergoing apoptosis or necrosis (Behar et al., 2011).
Autophagy is a homeostatic lysosomal process that involves the degradation of cellular components to their basic elements as a response to stress conditions (Lam et al., 2017; Paik et al., 2019). Based on how the cargo is targeted and delivered to the lysosomes, the autophagy pathway can be catagorized into three different types of autophagy processes, i.e., macroautophagy, microautophagy, and chaperone-mediated autophagy. Macroautophagy (generally referred to as autophagy) is triggered by numerous stress signals such as damaged intracellular organelles, hypoxia, starvation, and microbial infection (Lam et al., 2017; Paik et al., 2019). As illustrated in Figure 4, autophagy is initiated by the formation of the early phagophores (isolation membrane) from the endoplasmic reticulum–mitochondria contact sites, followed by the expansion of the phagophores around the intra-cytoplasmic cargo, and the growth of the phagophores into double membrane autophagosomes. Eventually, the autophagosomes become autolysosomes by coalescing with endosomes/lysosomes to facilitate the degradation of cytoplasmic cargo. Autophagy is also essentialfor the regulation of various immune responses, including: inflammation, innate immunity, and macrophage antibacterial defenses (Lam et al., 2017; Paik et al., 2019).
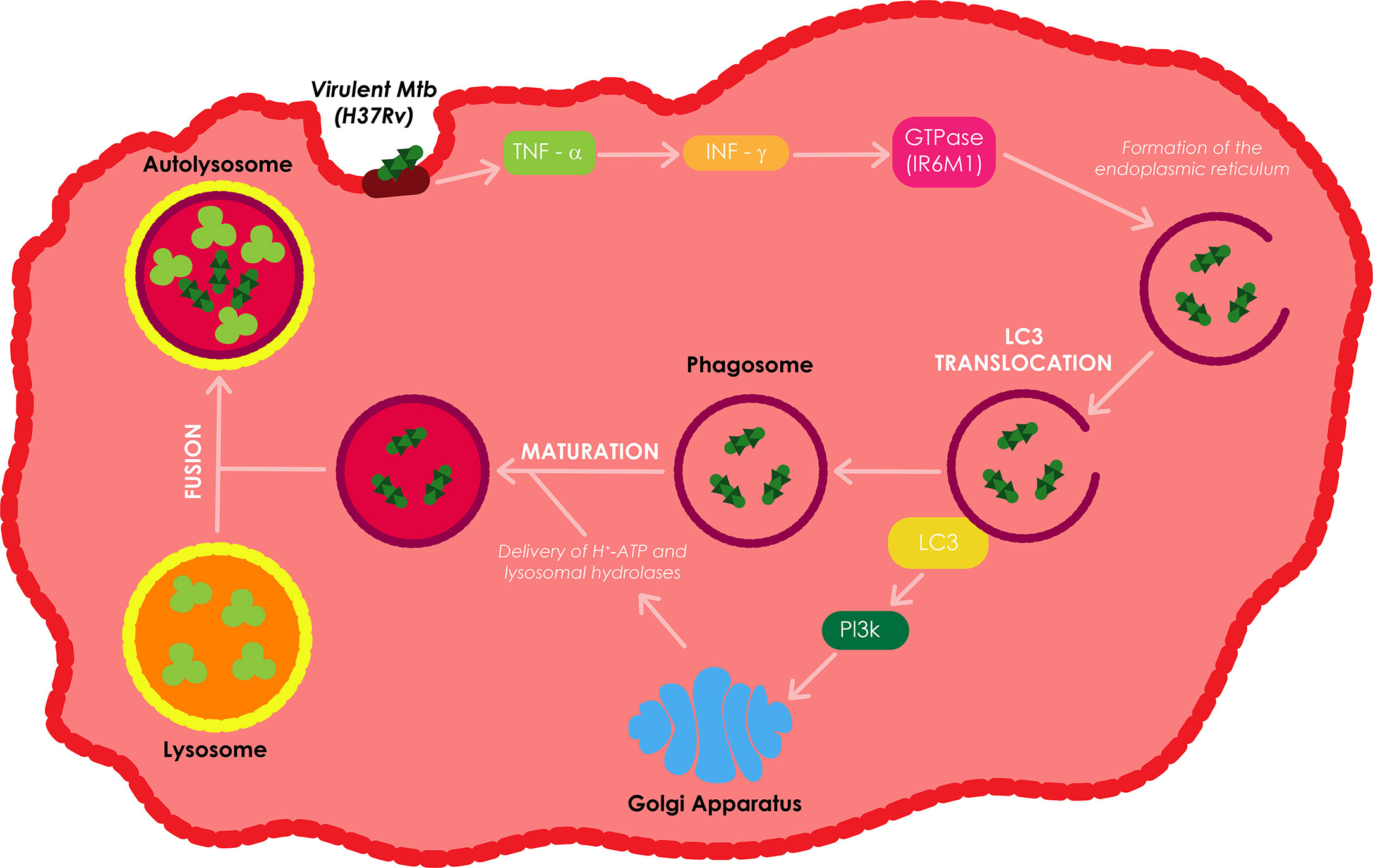
Figure 4 Overview of macroautophagy. Macroautophagy is one of autophagic process which is induced by various stimuli or stress including infection. Autophagy sequesters aggregated proteins, damaged organelles, and microbes from cytoplasm through formation of double-membraned structure (autophagosome). These autophagosomes fuse with lysosome or endosome that contains endocytic compartments to form amphisome or autolysosome. Finally, cargos are degraded in autolysosome for the maintenance of cellular homeostasis. Adapted from (Songane et al., 2012).
Bacteria mainly use pathogen induced damage-associated molecular patterns (DAMPs) and PAMPs to induce autophagy in host cells (Deretic et al., 2015; Shibutani et al., 2015). Cell surface recognition and cytosolic sensing of these molecules triggers signaling cascades that stimulatethe rapid and localized recruitment of the autophagy machinery (van der Vaart et al., 2014). Transcriptional factors such as NF-kB and transcription factor EB (TFEB) can further regulate autophagy by promoting the expression of diverse autophagy genes that extend the activation of autophagy (van der Vaart et al., 2014; Pastore et al., 2016).
The IL-12 and IL-18 dependent secretion of interferon gamma (IFN-γ), together with the production of tumor necrosis factor alpha (TNF-α) have all been shown to be vital in the defensive instigation of autophagy against Mtb infection (Salgame, 2005). IFN-γ induces autophagy in both human and murine macrophage cell lines; this type of autophagy reduces the survival of intracellular virulent Mtb H37Rv. IFN-γ enhances the translocation of LC3 and the proteolysis of long-lived proteins, which are both autophagy markers, and subsequently increases autophagosome formation (Shi and Kehrl, 2010). Upon IFN-γ stimulation, GTPase (IRGM1), an autophagy linked gene downstream of IFN-γ, controls Mtb replication through phagosome formation, facilitates the PI3K-dependent maturation of Mtb containing phagosomes, and autophagolysosome formation through the fusion of the phagosome to the lysosome (MacMicking et al., 2003).
The delivery of H+-ATP and lysosomal hydrolases from the trans-Golgi network to the phagosome as well as the fusion of the phagosome with the lysosome enhances the acidification of the internal environment of the autophagolysosome or autolysosome resulting in the killing of Mtb (Fratti et al., 2003; Songane et al., 2012). Treatment of Mtb infected macrophages with TNF-α leads to an increase inIFN-γ induced phagosome maturation in these macrophages (Songane et al., 2012). Autophagy can also produce and supply antimicrobial peptides to the bacterial compartment to improve the bacterial killing (Alonso et al., 2007). Depending on the nature and localization of PAMP/DAMP, autophagy can use a mechanism called xenophagy to selectively capture and eradicate bacteria, damaged organelles, and other signaling platforms activated during the infection process (Bah and Vergne, 2017).
Xenophagy refers to the process whereby cells use autophagy against pathogens and this cellular mechanism has been found to be an innate component of immune responses. Xenophagy is triggered against Mtb infection by the cytoplasmic release of the bacteria through the Mtb ESX-1 secretion system (Watson et al., 2012; Paik et al., 2019). The autophagic receptors p62 and NDP52, and the STING dependent cytosolic pathway play a crucial role in the elimination of Mtb by xenophagy (Watson et al., 2012; Franco et al., 2017). Mice possessing monocyte derived cells and neutrophils that are devoid of Atg5 have been shown to be more susceptible to Mtb infection. However, other Atgs such as Beclin-1 and Atg14 (Figure 5), were found to cause no susceptibility. These results therefore suggest that autophagy may not be involved in regulating the initial stages of infection, however these claims are yet to be confirmed (Castillo et al., 2012; Watson et al., 2012; Kimmey et al., 2015).
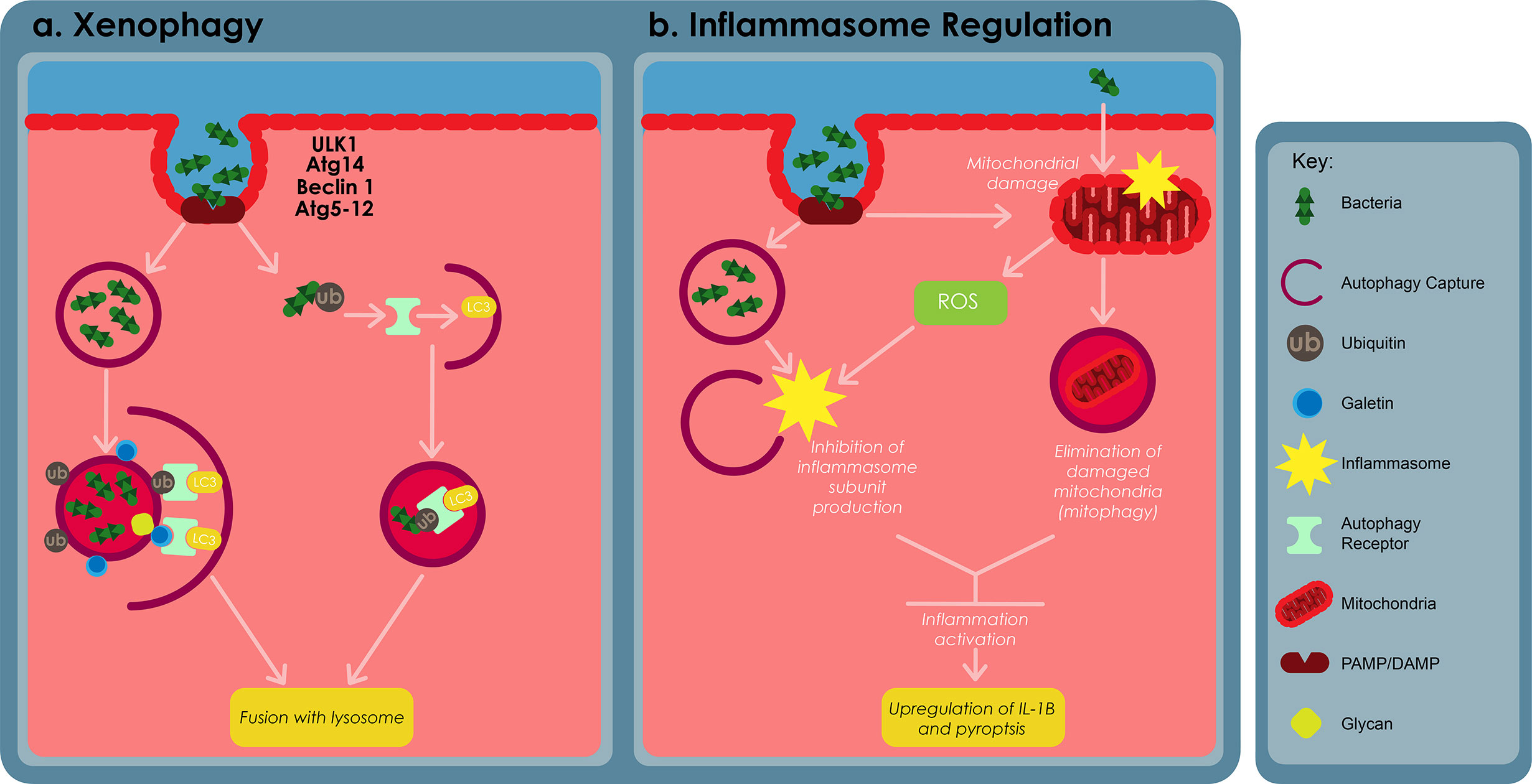
Figure 5 The roles of autophagy in macrophage defences against bacterial infections. (A) Cytosolic and vascular pathogens are selectively captured and degraded in the lysosome through a process called xenophagy. Xenophagy depends on ULK1, autophagy related (Atg) 14, Atg5-12, Beclin-1, autophagy receptor proteins, such as p62 and requires formation of an autophagosome. Autophagosomes capture bacterial compartments either through ubiquitination or through host glycan recognition by galectins. (B) Autophagy regulates inflammation by restraining inflammasome activation. This can be archieved by inhibiting the production of inflammasome subunits or through the elimination of damaged mitochondria, using a process called mitophagy. Adapted from (Bah and Vergne, 2017).
Mtb can also damage the phagosomal membrane and access the cytosol using its ESX-1 secretion system (Watson et al., 2012). Cytosolic sensor c-GAS can recognizethe bacteria’s DNA, thus inducing ubiquitination of the bacteria or its phagosome capture using ubiquitin ligases Parkin and Smurf1 (Watson et al., 2015; Franco et al., 2017). As a result, ubiquitin chains bind to autophagy adaptors, such as p62 and NDP52, recruitmicrotubule-associated-protein-1 light chain 3 (LC3) which subsequently delivers Mtb into an autophagosome. Cytosolic lectins of the galectin family also recognize the host glycan present on the lumen of damaged phagosomes, subsequently targeting these damage phagosomes for degradation by means of the autophagosome (Gomes and Dikic, 2014; Chauhan et al., 2015b).
LC3-associated phagocytosis (LAP) is a non-canonical autophagy process known to play a significant role in antibacterial defenses in macrophages (Galluzzi et al., 2017; Mitchell and Isberg, 2017). During LAP, pathogens are taken up by macrophages through phagocytosis. Binding of pathogens on surface receptors, such as the Fc gamma receptors, toll-like receptors, or Dectin-1, triggers the conjugation of LC3 onto the phagosomal membrane without requiring autophagosome formation (Sanjuan et al., 2007; Huang et al., 2009; Ma et al., 2012). Unlike canonical autophagy, LAP depends on the activation of Beclin-1/Rubicon complex and NADPH oxidase-2 (NOX2) and does not rely on ULK1 or Beclin-1/Atg14 complexes (Huang et al., 2009; Martinez et al., 2015). LC3 conjugation is triggered by the recruitment of two ubiquitin-like conjugation systems that are promoted by phosphatidylinositol-3-phosphate (PI3P) and ROS. The production of ROS has been shown to be PI3P dependent, whereas the synthesis of PI3P relies on Rubicon. The MORN2 protein has also been implicated in the LC3 conjugation pathway, though its role in the process remains unknown (Abnave et al., 2014).
A number of studies have shown that the conjugation of LC3 onto the phagosome improves the fusion between the phagosome and the lysosome. As a result, macrophages with a deficient LAP pathway are less effective at controlling intracellular growth of numerous bacteria including Mtb, Staphylococcus aureus, Legionella pneumophila, M. bovis BCG, and Listeria monocytogenes (Yang et al., 2012; Abnave et al., 2014). In addition, Rubicon-deficient mice have been shown to retain a larger bacterial load and are more vulnerable to L. Monocytogenes infection (Yang et al., 2012). LAP has also been shown to assist in antigen presentation process via MHC class II molecules, thus bridging the gap between innate and adaptive immunity (Ma et al., 2012; Romao et al., 2013).
However, it is important to note that LC3 conjugation at times delays or does not affect phagosome maturation. This indicates that LC3 alone is not enough to drive the fusion of the phagosome with the lysosome (Bah and Vergne, 2017). Autophagy can also indirectly regulate phagocytosis by varying the expression of phagocytic surface receptors. For example, suppression of the Atg 7 protein synthesis in macrophages promotes the upregulation of MARCO and MSR1, two class-A scavenger receptors that enable phagocytosis of M. bovis BCG and Mtb (Bonilla et al., 2013). Autophagy-deficient cells have also been shown to amass p62, thus promoting the dissociation of transcription factor nuclear erythroid-related factor 2 (NFE2L2) from Keap1 and its subsequent translocation into the nucleus, where it facilitates the expression of these receptors. In some cases, the lack of autophagy during infection can lead to reduced phagocytosis dependent on the bacteria (Jacquel et al., 2012).
ROS serve as signaling molecules that are involved in various cellular processes and play a key role in the killing of intracellular pathogens in macrophages (Lammas et al., 1997). Phagocytosis is a first-line innate immune defense mechanism. As illustrated on Figure 6, this process can be activated through the stimulation of specific membrane receptors, such as the β-glucan receptor, complement receptors, Fcγ receptors (FcγRs) or TLRs. Previous studies have revealed that the activation of TLR signaling during phagocytosis induces the recruitment of the LC3 autophagy protein to the phagosome, hence promoting phagosome maturation, plausibly by mediating membrane hemifusion/tethering, which results in microbial killing (Huang et al., 2009; Yang et al., 2012). FcγR and TLR signaling have been shown to induce the assembly of NOX2 NADPH oxidase on the nascent phagosomal membrane, which results in the production of superoxide through the transfer of electrons from the cytosolic NADPH to the oxygen in the phagosome lumen. Superoxide and other ROS generated during this process are highly potent and can directly kill intracellular microbes. The presence of NOX2-generated ROS has also been revealed to be compulsory for targeting LC3 to phagosomes (Huang et al., 2009; Yang et al., 2012).
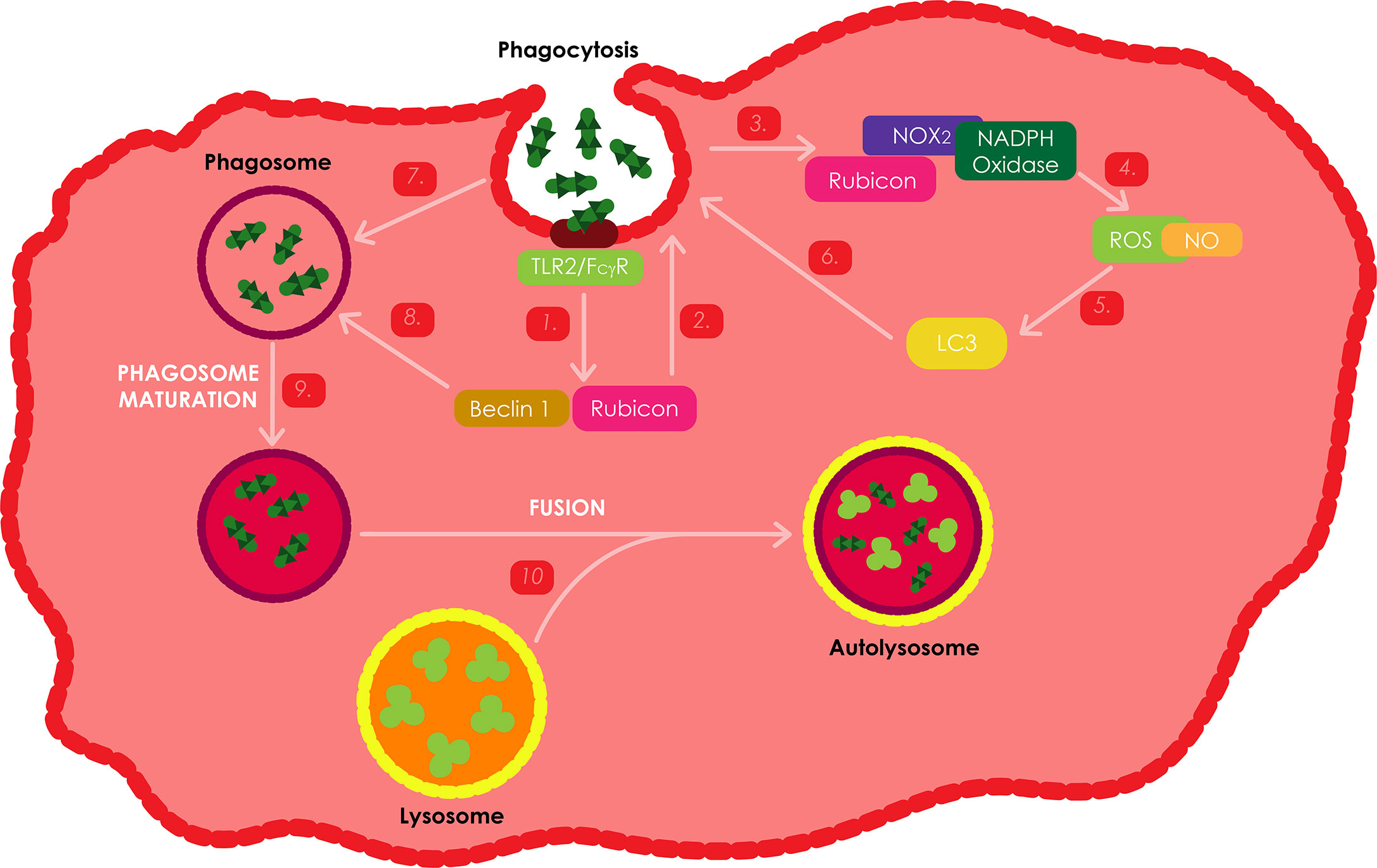
Figure 6 ROS production as an immune response against Mtb infections. Stimulation of either FcγRs or TLRs during phagocytosis activates similar kinetics to induce theactivation of ROS dependent autophagy. These two receptors activate the NOX2 NADPH oxidase, which is responsible for the production of ROS. Rubicon has also been shown to be an important positive regulator of the NADPH oxidase complex. During Mtb infection, the bacteria stimulate TLR2 resulting in the activation of Rubicon to interact with the p22phox subunit of the NADPH oxidase complex, enabling its phagosomal trafficking to induce the production of NOX2 dependent ROS, which leads to the recruitment of LC3 to the phagosome and ultimately the onset of autophagy and phagosome maturation through the assistance of Bectin-1.
Upon TLR stimulation or microbial infection (using Listeria monocytogenes), Rubicon was found to function as part of a Beclin-1 containing autophagy complex and has been shown to interact with the NADPH oxidase phagocytic complex, resulting in, firstly, the activation of NADPH oxidase which induces ROS-mediated antimicrobial oxidative activity; and secondly, the activation of Beclin-1-kinase complex relocate to phagosomes and trigger phagosomal maturation as a non-oxidative antimicrobial activity (Yang et al., 2012). IFN-γ inducible nitric oxide synthase (iNOS) pathways are an important antimicrobial alternative to the NADPH phagocyte oxidase pathways in phagocytic cells, which facilitates the production of nitric oxide (NO) radicals. Stimulation with zymosan, L. monocytogenes and a combination of the two, was found to produce significantly higher levels of ROS and NO in Raw264.7 Rubicon expressing macrophages. However, Rubicon expression as a result of IFN-γ stimulation was found to only induce minor differences in ROS and NO production. These results thus, suggest that Rubicon is primarily involved in the TRL2 dependent antimicrobial activity pathway, and that autophagy can either be ROS and NO dependent or non-dependent (Yang et al., 2012).
Inflammation is crucial to control bacterial infections; however, extreme inflammatory responses (cytokine storm) can result in injured host tissue, thus facilitating the progression of the disease. A number of studies have demonstrated that autophagy regulates inflammation in both infectious and non-infectious conditions (Shibutani et al., 2015; Cadwell, 2016). In cases of infections, autophagy uses multiple mechanisms to down-regulate the activation of inflammasomes (Saitoh and Akira, 2016; Harris et al., 2017). Pseudomonas aeruginosa infections damage the mitochondria, resulting in NLRC4 inflammasome. Autophagy eliminates the damaged mitochondria througha process called mitophagy, thus limiting inflammasome activation in both an in vitro and in vivo setting (Jabir et al., 2015). Atg7 deficient mice have been shown to have enhanced susceptibility to P. Aeruginosa infection resulting in increased neutrophil infiltration and severe lung damage. Atg7 deficiency in alveolar macrophages promotes up-regulation of IL-1β and pyroptosis (Pu et al., 2017). Aside frommitophagy, autophagy can also inhibit inflammasome activation by arresting the production of inflammasome subunits (Shi et al., 2012; Kimura et al., 2015).
It has also been shown that the mechanisms of autophagy are greatly manipulated by the micro-environment of the macrophage. Pro-inflammatory cytokines such as IFN-γ, IL-1β, and TNF-α are known to activate autophagy, whereas anti-inflammatory cytokines IL-4, IL-10, and IL-13 seem to inhibit autophagy (Harris, 2011). The recruitment of autophagy proteins p62 and Atg4B also promoted by the IFN-γ mediated xenophagy can only be accomplished through the help of ubiquilin-1 and guanylate binding proteins 1 and 7 (Kim et al., 2011; Sakowski et al., 2015). The presence of vitaminD in serum has been demonstrated to significantly enhance autophagy in macrophages by promoting the expression of cathelicidin, an antimicrobial peptide (Yuk et al., 2009). T cells have also been shownto induce autophagy in Mtb infected human macrophages (Petruccioli et al., 2012). In addition, the probiotic, Bacillus amyloliquefaciens, has also been shown to up-regulate autophagy genes in macrophages, resulting in enhanced Escherichia coli (E. coli) killing (Wu et al., 2017).
Intracellular Mtb have developed a variety of strategies to overcome the antibacterial defense of the macrophage, including autophagy (Bah and Vergne, 2017). A majority of the uncovered strategies presented in Figure 7, are mainly directed at and specifically target distinct steps of xenophagy. Mtb have been shown to target the JNK-dependent autophagy by secreting an N-acetyltransferase, Eis. However, this type of autophagy does not show sufficient influence on the intracellular growth of the pathogen (Shin et al., 2010; Kim et al., 2012). In addition, both in vitro and in vivo studies have demonstrated that Mtb inhibit the recruitment of NADPH oxidase onto phagosomes that contain the pathogen, thus favoring intracellular growth of Mtb (Koster et al., 2017).
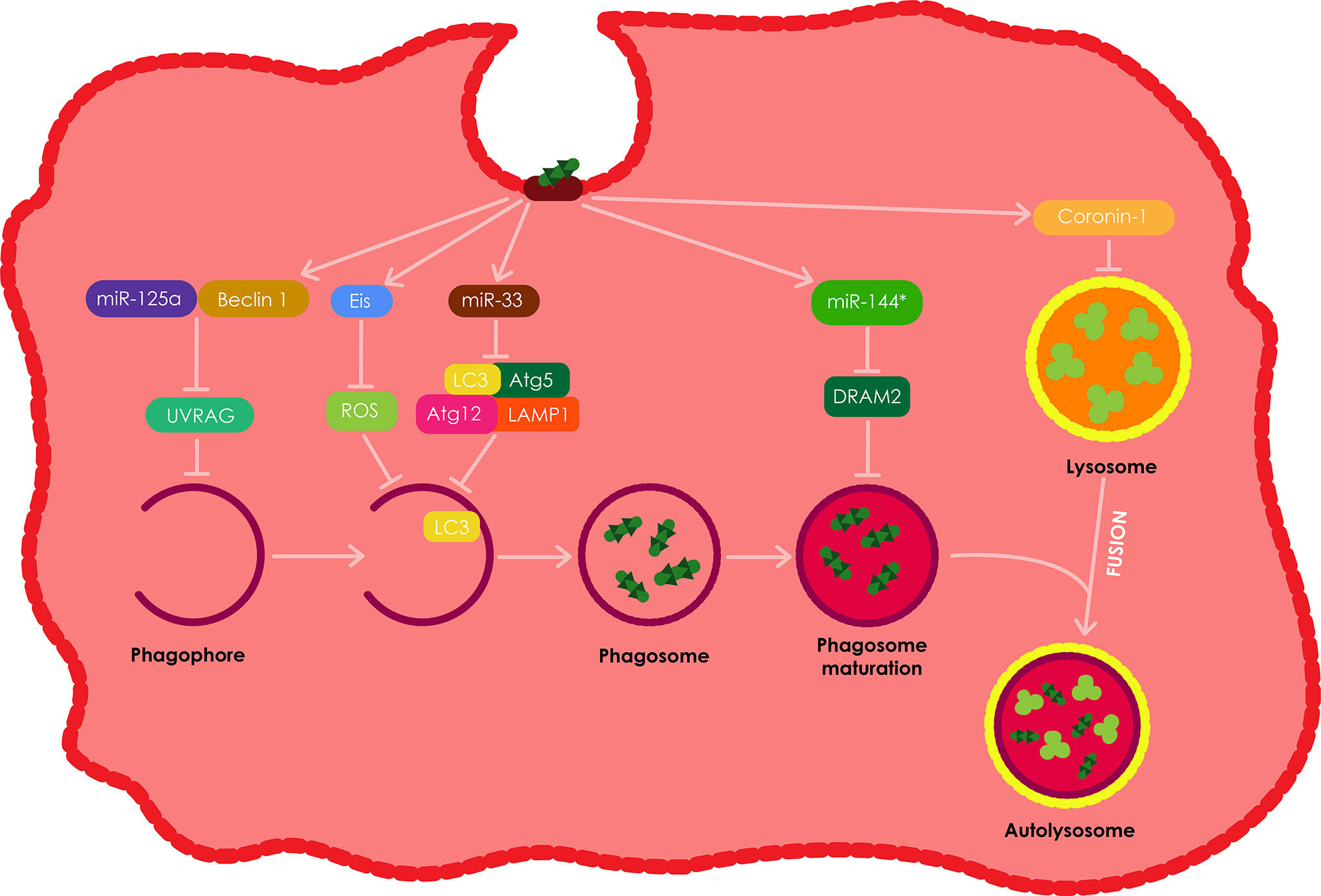
Figure 7 The evasion of autophagy by intracellular Mtb. Intracellular Mtb have been shown to employ various strategies to evade autophagy in macrophages. Among these, Mtb have been shown to induce the production of Eis, which results in the inhibition of ROS and ultimately autophagy. Mtb also impedes autophagy by inducing miR-33 expression which in turn supresses the expression of numerous autophagy proteins including; Atg5, Atg12, LC3, and LAMP1. Mtb also induces the expression of miR-125a which forms a complex with Beclin-1 resulting in the inhibition of UVRAG and ultimately the inhibition of autophagy in macrophages. Mtb also stimulates the production ofmiR-144*, which inhibits DRAM2 resulting in the inhibition of autophagosome maturation. Lastly, intracellular Mtb also induces the secretion of coronin1 by theinfected macrophages, which inhibits the fusion of phagosomes with lysosomes. All these autophagy evasion tactics therefore allow for the intracellular survival of Mtb in the infected phagocytes.
Autophagy is commonly modulated via post-translational modification. However, the process can also be modulated at a post-transcriptional level through the expression of numerous small non-coding RNAs (miR). Mtb impedes autophagy by down regulating the expression of several autophagy proteins such as Atg5, Atg12, LC3, and LAMP1 by means of inducing miR-33 expression (Ouimet et al., 2016). However, mice with hematopoietic miR-33 deficiency have been demonstrated to retain their ability to control Mtb infection. Mtb also targets UVRAG through the expression of miR-125a in complex with Beclin-1 resulting in the inhibition of autophagy in macrophages, therefore allowing for the intracellular survival of Mtb in the process (Kim et al., 2017).
However, other studies have demonstrated that miR-33 and miR-125a can be induced using PRR ligands separate from Mtb (Ouimet et al., 2016; Kim et al., 2017). These findings suggest that these miRs may be expressed as a normal negative feedback loop in host cells to avoid excessive autophagy, which Mtb takes advantage of. In addition, Mtb stimulates the up-regulation of miR-144* which it uses to decrease the expression of DRAM2, a newly discovered autophagy protein, responsible for promoting autophagosome maturation by activating a second Beclin-1 complex (Kim et al., 2017). Lastly, Mtb also promotes the expression of Mc1, which targets and inhibits Bectin-1 complex, by down-regulating the expression of miR-17, thus inhibiting autophagy (Kumar et al., 2016).
The Mtb secretes bacterial proteins ATP1/2 (secretion ATPase1/2, secreted secA1/2 protein) and early secretory antigen-6/culture filtrate protein to inhibit the accumulation of vacuolar ATP and GTP enzymes, therefore decreasing intracellular pH, which results in the suppression of phagocyte maturation (Rohde et al., 2007; Zhai et al., 2019). Mtb also supresses the formation of lysosomes by inducing an increase in the expression of the coronin 1 protein on the host phagocyte membrane; the extent of the recruitment process and the amount of coronin 1 are proportional to the amount of activated Mtb in the microsomes (Schuller et al., 2001; Flynn and Chan, 2003; Zhai et al., 2019).
The IFN cytokine interferon has also been shown to suppress phagocyte maturation through the induction of IL-10 production in a STAT1-dependent manner. IL-10 inhibits the caspase1-dependent IL-1 maturation of pleural fluid mononuclear cells by reducing excess IL-1 (Ma et al., 2014). Serine/threonine-protein kinase G (PKnG) has been shown to augment the virulence, metabolism, growth rate, and drug resistance of Mtb by increasing the infectivity of Mtb lowering the expression of GlpK and ALD, increasing the expression of Ag85A and Ag85C, and supressing the maturation of lysosomes. PKnG also hinder the fusion of phagosomes and lysosomes by increasing signal transduction in host cells (Walburger et al., 2004; Wong et al., 2013).
As described earlier, Mtb can inhibit autophagosome maturation using miR-144*, although, the bacterial factors involved in that process are still unidentified (Kumar et al., 2016). A number of studies have demonstrated that a vital Mtb virulence factor called Esat-6 is involved in the process of inhibiting autophagosome maturation in both macrophages and dendritic cells (Romagnoli et al., 2012; Chandra et al., 2015). The exact role of Esat-6 in the process is still unknown but it is estimated that it might be partially facilitated by miR modulation (Kumar et al., 2015). A number of other bacterial pathogens such as Chlamydia trachomatis, Yersinia pestis, and Y. Pseudotuberculosis can also block autophagosome maturation to insure their intracellular survival or growth in macrophages, and similar to Mtb their molecular mechanisms involved in the process remain unknown (Pujol et al., 2009; Moreau et al., 2010; Yasir et al., 2011). Mannosylated beads hinder phagosome maturation post phagocytosis, indicating the plausible influence of glycolipids on the survival of intracellular mycobacterium (Astarie-Dequeker et al., 1999; Kang et al., 2005).
The pro-inflammatory transcription factor NF-kB has been shown to control the release of lysosomal enzymes into phagosomes, therefore regulating the killing of intracellular pathogens. In addition, the fusion of phagolysosomes during infection is also regulated by NF-kB, by increasing the production of membrane transport molecules (Gutierrez et al., 2008). The fusion between phagosomes and lysosomes is also suppressed through the calmodulin dependent production of phosphatidylinositol-3-phosphate (PI3P), lower biosynthesis and via trafficking the toxin lipoarabinomannan (LAM) after Mtb infection (Vergne et al., 2003). As mentioned above, Mtb can survive and persist in macrophages by obstracting the fusion between the lysosome and phagosome (Figure 7). In DCs, DC-specific intercellular adhesion molecule-3 grabbing nonintegrin (DC-SIGN) facilitates the entry of Mtb by interacting with the mycobacterial cell wall component ManLAM. The interaction between DC-SIGN and ManLAM leads to the supression of DC maturation (Geijtenbeek et al., 2003; Tailleux et al., 2003).
Mtb inhibits phagosome acidification and persists by maintaining a relatively lower acidic intracellular environment (pH~6.2). Mtb initiates this process by changing the composition of the phagosome; as the structure and particular molecules on the cell wall function as a barrier that allows macrophages to retain a neutral pH (Chen et al., 2015). This step is followed by encoding the protein tyrosine kinase A (PtkA) in the same operon as PtpA. IFN-γ inducible nitric oxide synthase 2 has been shown to directly activate Mtb infected macrophages, resulting in the suppression of the replication of intracellular Mtb. LRG-47, a member of the 47-kD guanosine triphosphate familyhas also been demonstrated to play a protective nitric oxide synthase 2 independent role in macrophages against disease. As a result, LRG-47 defient macrophages have a reduced response to Mtb due to an inability to completely acidify (MacMicking et al., 2003).
Oxidative stress refers to a state of imbalance between the pro and antioxidants. Together with the inhibition of macrophage phagocytosis, lysosome maturation and acidification, inhibition of oxidative stress is mandatory process toward archieving Mtb latency in the host cells. Oxidative stress can induce lipid peroxidation, protein oxidation, and cause damage to DNA bases. The mitochondria and the NADPH oxidase (NOX) are the two main producers of ROS required to induce autophagy (Shin et al., 2010). NOX plays a key role in the elimination of pathogens and the NOX deficiency in mice has been shown to impair the mice’s ability to eradicate a number of pathogens (Fang, 2004).
However, Mtb has been shown to have the ability to deactivate NOX2-derived ROS (Miller et al., 2010), although it is still unclear if there is increased susceptibility to TB in patients with chronic granulomatous disease (CGD) that causes ROS defects as a result of genetic defects in NADPH oxidase. This is because patients with CGD are almost only identified and diagnosed in developed countries where there is a very low prevalence of TB, therefore it is unknown if these patients have increased susceptibility or show a flawed autophagy process against Mtb in countries with a high prevalence of TB (Songane et al., 2012). The presence of SigH, an Mtb alternative sigma factore induced by heat, oxidative and nitric oxide stresses, has been shown to becompulsory for long lasting Mtb infection. The interaction of host innate immune responses with Mtb is also regulated by a SigH-dependent regulon (Dutta et al., 2012).
Mycothiol (MSH), a major thiol in Mtb, has been shown to have antioxidant activity and helps Mtb persist in macrophages through the detoxification of various intracellular compounds. The gene mshD encodes for mycothiol synthase, the final enzyme in the biosynthesis of MSH. A strain of Mtb devoid of an mshD gene has been shown to be sensitive to H2O2 and also displays poor growth on catalase and oleic acid deficient agar plates (Buchmeier et al., 2006). Polymorphonuclear neutrophils (PMNs) eliminate pathogens and are key components in the first line of defense against Mtb through the induction of ROS production (Schuller et al., 2001). The enzymes KatG and TrxB2 have been shown to help Mtb with stand the oxidative environment and the expression of their related genes are notably increased by H2O2 and NO (Lamichhane, 2011).
In addition, Mtb also produces the protein Eis to enhance its intracellular survival through the blockade of ROS production, thus inhibiting macrophage inflammatory responses, cell death and autophagy (Shin et al., 2010). The Eis protein has been located in the cytoplasm and in Mtb containing phagosomes of infected macrophages (Samuel et al., 2007). In addition, traces of anti-Eis antibodies have also been detected in sera obtained from patients with pulmonary TB, thus signifying that Eis is produced during Mtb infection in humans (Dahl et al., 2001). Finally, Eis deficient Mtb infections have been shown to induce increased caspase-dependent cell death in the host cells, however, this cell death can be inhibited by blocking autophagy and c-Jun N-terminal kinase ROS signaling (Shin et al., 2010).
The stimulation of autophagy using carbamazepine, rapamycin, small molecule enhancers of rapamycin (SMERs), specifically SMERs 18 and 28, as well as vitamin D3 has been shown to induce Mtb killing in macrophages. In addition, apart from reducing bacterial burden, carbamazepine has also been shown to stimulate adaptive immunity and improve lung pathology in mice with MDR-TB (Floto et al., 2007; Schiebler et al., 2015; Paik et al., 2019). Using both chemical and RNAi screening, nortriptyline was revealed to be one of the majorcompounds that modulate intracellular mycobacteria and was shown to considerably reduce the survival of intracellular Mtb in macrophages through the induction of autophagy (Sundaramurthy et al., 2013). Nitazoxanide (antiprotozoan drug) and flubendazole (antihelminthic drug) were revealed to activate autophagy to limit both bacterial and HIV infections in macrophages (Lam et al., 2012; Chauhan et al., 2015a).
Trehalose has also been reported to induce bacterial killing by activating autophagy and xenophagy in Mtb infected macrophages (Sharma et al., 2017). In addition, trehalose has also been shown to be effective againstan HIV and Mtb co-infection and regulates the survival of Mtb by overriding the HIV mediated blockade of autophagy (Sharma et al., 2017). Vitamin D3 was also shown to induce autophagy to facilitate the killing of Mtb amidst an HIV co-infection (Campbell and Spector, 2012). Various host factors have been studied as potential anti-Mtb therapeutics and have been reported to regulate autophagy and xenophagy in host cells. For example, the compound saracatinib (AZD0530) was revealed to induce autophagy and lysosomal maturation to facilitate the killing of Mtb through inhibiting the Src kinase in host cells (Chandra et al., 2016a; Chandra et al., 2016b).
Fluoxetine, a selective serotonin re-uptake inhibitor, has been shown to induce autophagy to restrict the survival of intracellular Mtb, possibly by increasing the secretion of TNF-α in Mtb infected macrophages. Baicalin also induces anti-inflammatory and antimicrobial activity against Mtb infection, by activating autophagy through the PI3K/Akt/mTOR pathway in host cells (Paik et al., 2019). Antimycobacterial agents; loperamide and verapamil, have been revealed to induce autophagy, to facilitate the inhibition of intracellular Mtb survival in murine alveolar macrophages. Ambroxol has also been shown to induce autophagy in vitro and in vivo, resulting in the killing of Mtb in murine macrophages (Paik et al., 2019) Table 1.

Table 1 List of some known authophagy inducing compounds and their applications in TB treatment.
In addition, the interaction of zymosan with macrophage TLRs or the FcγRs during phagocytosis has been shown to promote the recruitment of the LC3 autophagy protein to phagosomes. Both TLRs and FcγRs activate NOX2 NADPH oxidase, which is known to induce the production of reactive oxygen species (ROS). NOX2-dependent ROS production has been shown to either induce or aid in LC3 recruitment to phagosomes, thus promoting phagosome maturation and ultimately microbial killing (Huang et al., 2009; Yang et al., 2012).
In this decade, NPs have received significant attention in disease therapy (Mallakpour, 2016). NPs are defined as engineered particles with a size range typically of 1–100 nm and also up to 1000 nm (Nagavarma et al., 2012). NPs can be classified into various categories as either organic (e.g. liposomes, polymeric NPs, lipid NPs or dendrimers), metallic (iron oxide Nps, gold NPs (GNPs), silver NPs, quantum dots) or ceramic (silica NPs, calcium carbonate NPs). The application of NPs toward drug delivery and diagnostics is known as nanomedicine. As drug delivery systems, drugs can either be encapsulated within the NP or covalently attached onto the surface of the NP (Figure 8). By so doing, the properties of these drugs can be improved, e.g. the aqeous solubility, chemical stability and translocation across biological barriers. The release of the drugs can also be controlled to occur over a desired time course or to occur at target sites following an external trigger (e.g. pH orenzymatic action) thus modulating plasma and intracellular pharmacokinetics of the drugs (Dube et al., 2013). Cells also take up NPs more efficiently making NPs a more auspicious transport and delivery system (Nasiruddin et al., 2017).
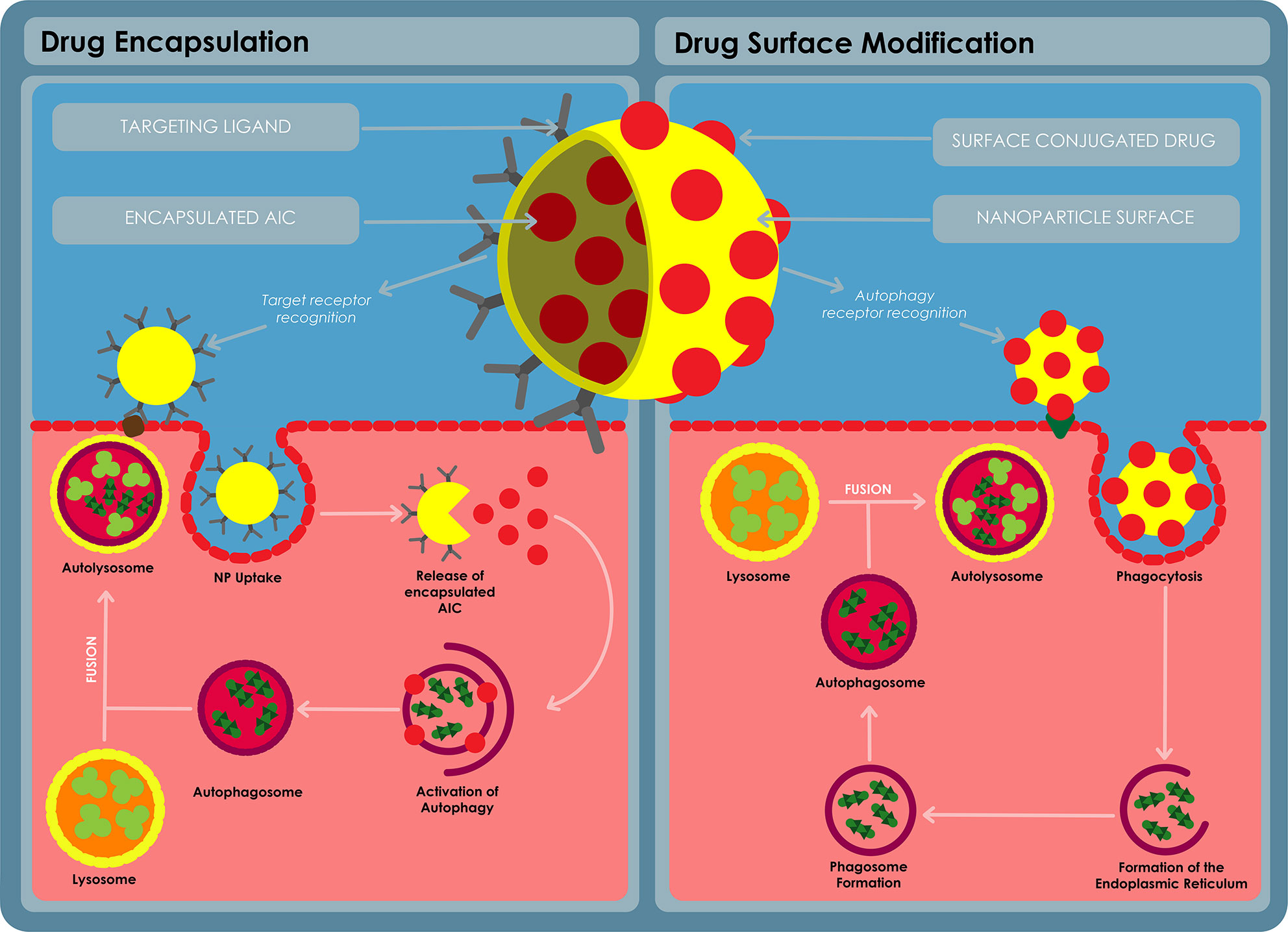
Figure 8 A diagram for autophagy inducing nanomedicine. The nanomedicine can be formulated in a variety of ways, the AIC can be encapsulated on the inside of a nanoparticle and released at the target site using a ligand to target the delivery nanosystem into the target cells. The AIC can also be conjugated on the surface of the nanoparticle and used to target the AIC to the target cells using receptor recognition. The AIC can stimulate autophagy via receptor stimulation or the AIC can stimulate specific autophagy pathways after being taken up by the cells.
NPs have been observed to induce ROS production in various cell types (Yu et al., 2014; Chiu et al., 2015; Guo et al., 2015). Spherical silica NPs of 60 nm in size were shown to stimulate ROS generation in (HepG2) cells at a final concentration of 100 μg/ml (Yu et al., 2014). Amine-modified but not pristine or carboxylate modified 60 nm polystyrene spheres were also observed to induce the generation of ROS in both BEAS-2B lung epithelial cells and RAW264.7 macrophages atconcentrations of 20 μg/ml. The amine-modified spheres also induced an increase of LC3 in both of the cells lines (Chiu et al., 2015). RAW264.7 macrophages were observed to use phagocytic pathways to take up elongated pristine iron oxide nanorods (1000 nm × 100 nm), which are then distributed in the cytosol by autophagosome-like vacuoles 24h post exposure to cells (Park et al., 2014). Other studies have revealed that these nanorods also induce an increase in ROS and nitrous oxidelevels in RAW264.7 macrophages. In addition, the nanorods also induced an increase in the expression levels of Atg5, Beclin1, Erk, p62, and LC3-II, while reducing JNK phosphorylation levels in the same cell line (Xia et al., 1995; Nordstrom et al., 2009).
GNPs of size 5 and 13 nm were observed to enhance ROS production in hypoxic HK-2 cells and increase autophagy in hypoxic cells. To study the relationship between apoptosis and autophagy, 3-methyladenine (3-MA), a well-known autophagy inhibitor was used. 3-MA significantly inhibited the GNP induced autophagy as expected. However, this treatment resulted in a 36% reduction in the viability and a 29% enhancement in the apoptosis of GNP treated cells under normoxic conditions. On the other hand, exposure to GNPs resulted in 61% increase in viability and a 52% decrease in the apoptosis of in HK-2 cells under hypoxic conditions (Ding et al., 2014).
Silica NPs at 500 μg/ml, were observed to induce the highest TNF-α release RAW264.7 macrophages, followed by poly(lactic-co-glycolic acid) (PLGA) NPs which induced the release of significantly lower TNF-α and finally, silk NPswhich induced the lowest release of TNF-α in RAW264.7 macrophages (Saborano et al., 2017). Unloaded Eudragit® RS NPs have been observed to be cytotoxic to NR8383 rat macrophages exposed to doses varying from 15 - 100 µg/mL, the internalized NPs were also shown to reach and alter the structure of the mitochondria. In addition, exposure to the NPs also triggered an increase in ROS production and autophagy in the macrophages (Eidi et al., 2012).
Particulate β-glucans (WGP) derived from the yeast Sacchromyces cerevisiae have also been shown to induce LC3 recruitment by increasing LC3-II expression. Formation of an autophagosome in WGP-induced DCs was also detected, indicating that the binding of WGP to dectin-1 in DCs, triggers both autophagy and LC3-associated phagocytosis. In addition, autophagy induced TNF-α production in WGP treated DCs, but had no influence on the secretion of IL-12, suggesting that the maturation of autophagy induced DCs may be archived using a unique pathway. Furthermore, the WGP also reduced iNOS expression in autophagy deficient DCs. WGP treated DCs also induced greater proliferation in CD4 +T-cells when compared to autophagy deficient DCs (Ding et al., 2019).
In a study demonstrating autophagy in Mtb infected macrophages (HDT-TB), Upadhyay et al. (2019) reported the capacity of yeast derived glucan particles (GP) loaded with high payload of rifabutin (RB) NPs [(RB-NPs)-GP] to induce anti-mycobacterial and cellular activation responses in Mtb infected J774 macrophage cells. The exposure to (RB-NPs)-GP was observed to trigger strong innate immune responses in Mtb infected macrophages including the induction of apoptosis, autophagy as well as reactive oxygen and nitrogen species. In addition, the (RB-NPs)-GP formulation was found to induce a 2.5 fold increase in the efficacy of the RB drug (Upadhyay et al., 2019). However, additional investigations in vivo as well as studies to target and deliver known AICs are required.
The interaction between Mtb and the host immunity has been shown to determine the outcome of the infection. The innate defense mechanisms of the host play a pivotal role in the process. An overlap was observed between the pathways that induce apoptosis and autophagy, for example Mtb has been shown to stimulate the production of TNF in Mtb infected macrophages through the TLR2-mediated signaling pathway. In turn, the induction of TNF-α activity leads to apoptosis in Mtb infected cell. On the other hand, the production of TNF-α has also been shown to be vital in the instigation of autophagy against Mtb infection. In addition, TRL2 has also been shown to induce mitochondrial ROS in infected macrophages resulting in both apoptosis and/or autophagy.
Nonetheless, Mtb has developed various mechanisms to evade the host’s protective immunity and the increase of Mtb’s resistance to a variety of existing antibiotics has become a threat to public health across the world. Therefore, the requirement for new therapeutic strategies has become a key priority. A promising approach is the use of AICs as part of HDT. This strategy can be coupled with nanotechnology, to improve the delivery of these compounds or to synthesize novel nanosized HDT-TB therapeutics, as studies have revealed that various NPs can induce autophagy in a variety of cell types. In addition, the introduction of NPs to HDT-TB can allow for targeted drug delivery, therefore minimizing the toxic side effects, while increasing the amount of the drugs that reach target cells.
However, despite the wide array of available AICs and NPs, there remains a very limited amount of research regarding the application of AICs and therapeutic NPs against Mtb. Mechanistic aspects of the macrophage response to Mtb and its suppression of autophagy, could be exploited to rationally design nanomedicines against this infection. The efficacy of nanomedicines to eradicate intracellular Mtb within the granuloma structureis also yet to be investigated. Macrophages have typically been the target in TB immunotherapy, however, DCs should also be targeted, as they are the bridge between the innate and adaptive immunity and can therefore be targeted for the purpose of both vaccination and therapy. The use of animal models is also required to determine efficacy of NP HDT in pre-clinical studies. Toxicity of the selected NPs is also a key concern and thus, we advocate for the use of biodegradable NPs as they have been shown to have minimum toxicity in vitro. Ultimately, we predict that greater understanding of both TB pathogenesis and the mode of action of AICs will reveal greater insights toward designing a superior treatment strategy against Mtb infections in the future.
RM wrote the full review paper. MM and AD co-supervised, guided, and helped shape the paper and also edited the paper before the paper was submitted. All authors contributed to the article and approved the submitted version.
Research reported in this publication was supported by the Fogarty International Center of the National Institutes of Health under Award Number K43TW010371 granted to AD and by National Research Foundation (NRF) of South Africa, award number UID 111887 awarded to RM. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The authors gratefully acknowledge Mr Prince Dlozi for producing all the images in this manuscript.
Abnave P., Mottola G., Gimenez G., Boucherit N., Trouplin V., Torre C., et al. (2014). Screening in planarians identifies MORN2 as a key component in LC3-associated phagocytosis and resistance to bacterial infection. Cell Host Microbe 16, 338–350. doi: 10.1016/j.chom.2014.08.002
Alonso S., Pethe K., Russell D. G., Purdy G. E. (2007). Lysosomal killing of Mycobacterium mediated by ubiquitin-derived peptides is enhanced by autophagy. Proc. Natl. Acad. Sci. U.S.A. 104, 6031–6036. doi: 10.1073/pnas.0700036104
Andrews J. M., Honeybourne D., Jevons G., Brenwald N. P., Cunningham B., Wise R. (1997). Concentrations of levofloxacin (HR 355) in the respiratory tract following a single oral dose in patients undergoing fibre-optic bronchoscopy. J. Antimicrob. Chemother. 40, 573–577. doi: 10.1093/jac/40.4.573
Arbex M. A., Varella Mde C., Siqueira H. R., Mello F. A. (2010). Antituberculosis drugs: drug interactions, adverse effects, and use in special situations. Part 1: first-line drugs. J. Bras. Pneumol. 36, 626–640. doi: 10.1590/s1806-37132010000500016
Ashida H., Mimuro H., Ogawa M., Kobayashi T., Sanada T., Kim M., et al. (2011). Cell death and infection: a double-edged sword for host and pathogen survival. J. Cell Biol. 195, 931–942. doi: 10.1083/jcb.201108081
Astarie-Dequeker C., N’diaye E. N., Le Cabec V., Rittig M. G., Prandi J., Maridonneau-Parini I. (1999). The mannose receptor mediates uptake of pathogenic and nonpathogenic mycobacteria and bypasses bactericidal responses in human macrophages. Infect. Immun. 67, 469–477. doi: 10.1128/IAI.67.2.469-477.1999
Aucamp M., Liebenberg W., Okaecwe T., Geldenhuys M., Stieger N. (2019). Compatibility Between Four Anti-TB Drugs and Tablet Excipients Determined By Microcalorimetry. Pharmazie 74, 350–351. doi: 10.1691/ph.2019.8189
Bafica A., Scanga C. A., Feng C. G., Leifer C., Cheever A., Sher A. (2005). TLR9 regulates Th1 responses and cooperates with TLR2 in mediating optimal resistance to Mycobacterium tuberculosis. J. Exp. Med. 202, 1715–1724. doi: 10.1084/jem.20051782
Bah A., Vergne I. (2017). Macrophage Autophagy and Bacterial Infections. Front. Immunol. 8, 1483. doi: 10.3389/fimmu.2017.01483
Balcewicz-Sablinska M., Keane J., Kornfeld H., Remold H. (1998). Pathogenic Mycobacterium tuberculosis evades apoptosis of host macrophages by release of TNF-R2, resulting in inactivation of TNF-alpha. J. Immunol. (Baltimore Md. 1950) 161, 2636–2641.
Behar S. M., Martin C. J., Nunes-Alves C., Divangahi M., Remold H. G. (2011). Lipids, apoptosis, and cross-presentation: links in the chain of host defense against Mycobacterium tuberculosis. Microbes Infect. 13, 749–756. doi: 10.1016/j.micinf.2011.03.002
Bekale R. B., Du Plessis S. M., Hsu N. J., Sharma J. R., Sampson S. L., Jacobs M., et al. (2018). Mycobacterium Tuberculosis and Interactions with the Host Immune System: Opportunities for Nanoparticle Based Immunotherapeutics and Vaccines. Pharm. Res. 36, 8. doi: 10.1007/s11095-018-2528-9
Bonilla D. L., Bhattacharya A., Sha Y., Xu Y., Xiang Q., Kan A., et al. (2013). Autophagy regulates phagocytosis by modulating the expression of scavenger receptors. Immunity 39, 537–547. doi: 10.1016/j.immuni.2013.08.026
Boritsch E. C., Frigui W., Cascioferro A., Malaga W., Etienne G., Laval F., et al. (2016). pks5-recombination-mediated surface remodelling in Mycobacterium tuberculosis emergence. Nat. Microbiol. 1, 15019. doi: 10.1038/nmicrobiol.2015.19
Bousso P. (2008). T-cell activation by dendritic cells in the lymph node: lessons from the movies. Nat. Rev. Immunol. 8, 675–684. doi: 10.1038/nri2379
Bowdish D. M., Sakamoto K., Kim M. J., Kroos M., Mukhopadhyay S., Leifer C. A., et al. (2009). MARCO, TLR2, and CD14 are required for macrophage cytokine responses to mycobacterial trehalose dimycolate and Mycobacterium tuberculosis. PloS Pathog. 5, e1000474. doi: 10.1371/journal.ppat.1000474
Buchmeier N. A., Newton G. L., Fahey R. C. (2006). A mycothiol synthase mutant of Mycobacterium tuberculosis has an altered thiol-disulfide content and limited tolerance to stress. J. Bacteriol. 188, 6245–6252. doi: 10.1128/JB.00393-06
Cadwell K. (2016). Crosstalk between autophagy and inflammatory signalling pathways: balancing defence and homeostasis. Nat. Rev. Immunol. 16, 661–675. doi: 10.1038/nri.2016.100
Campbell G. R., Spector S. A. (2012). Autophagy induction by vitamin D inhibits both Mycobacterium tuberculosis and human immunodeficiency virus type 1. Autophagy 8, 1523–1525. doi: 10.4161/auto.21154
Castillo E. F., Dekonenko A., Arko-Mensah J., Mandell M. A., Dupont N., Jiang S., et al. (2012). Autophagy protects against active tuberculosis by suppressing bacterial burden and inflammation. Proc. Natl. Acad. Sci. U.S.A. 109, E3168–E3176. doi: 10.1073/pnas.1210500109
Chandra P., Ghanwat S., Matta S. K., Yadav S. S., Mehta M., Siddiqui Z., et al. (2015). Mycobacterium tuberculosis Inhibits RAB7 Recruitment to Selectively Modulate Autophagy Flux in Macrophages. Sci. Rep. 5, 16320. doi: 10.1038/srep16320
Chandra P., Rajmani R. S., Verma G., Bhavesh N. S., Kumar D. (2016a). Targeting Drug-Sensitive and -Resistant Strains of Mycobacterium tuberculosis by Inhibition of Src Family Kinases Lowers Disease Burden and Pathology. mSphere 1, e00043–e00015. doi: 10.1128/mSphere.00043-15
Chandra P., Rajmani R. S., Verma G., Bhavesh N. S., Kumar D. (2016b). Targeting Drug-Sensitive and -Resistant Strains of Mycobacterium tuberculosis by Inhibition of Src Family Kinases Lowers Disease Burden and Pathology. mSphere 1, 1–13. doi: 10.1128/mSphere.00043-15
Chauhan S., Ahmed Z., Bradfute S. B., Arko-Mensah J., Mandell M. A., Won Choi S., et al. (2015a). Pharmaceutical screen identifies novel target processes for activation of autophagy with a broad translational potential. Nat. Commun. 6, 8620. doi: 10.1038/ncomms9620
Chauhan S., Mandell M. A., Deretic V. (2015b). IRGM governs the core autophagy machinery to conduct antimicrobial defense. Mol. Cell 58, 507–521. doi: 10.1016/j.molcel.2015.03.020
Chen Z., Wang T., Liu Z., Zhang G., Wang J., Feng S., et al. (2015). Inhibition of Autophagy by MiR-30A Induced by Mycobacteria tuberculosis as a Possible Mechanism of Immune Escape in Human Macrophages. Jpn. J. Infect. Dis. 68, 420–424. doi: 10.7883/yoken.JJID.2014.466
Chipuk J. E., Green D. R. (2008). How do BCL-2 proteins induce mitochondrial outer membrane permeabilization? Trends Cell Biol. 18, 157–164. doi: 10.1016/j.tcb.2008.01.007
Chiu H. W., Xia T., Lee Y. H., Chen C. W., Tsai J. C., Wang Y. J. (2015). Cationic polystyrene nanospheres induce autophagic cell death through the induction of endoplasmic reticulum stress. Nanoscale 7, 736–746. doi: 10.1039/C4NR05509H
Choi S. W., Gu Y., Peters R. S., Salgame P., Ellner J. J., Timmins G. S., et al. (2018). Ambroxol Induces Autophagy and Potentiates Rifampin Antimycobacterial Activity. Antimicrob. Agents Chemother. 62, e01019–e01018. doi: 10.1128/AAC.01019-18
Dahl J. L., Wei J., Moulder J. W., Laal S., Friedman R. L. (2001). Subcellular localization of the Iitracellular survival-enhancing Eis protein of Mycobacterium tuberculosis. Infect. Immun. 69, 4295–4302. doi: 10.1128/IAI.69.7.4295-4302.2001
Dartois V. (2014). The path of anti-tuberculosis drugs: from blood to lesions to mycobacterial cells. Nat. Rev. Microbiol. 12, 159–167. doi: 10.1038/nrmicro3200
Deretic V., Kimura T., Timmins G., Moseley P., Chauhan S., Mandell M. (2015). Immunologic manifestations of autophagy. J. Clin. Invest. 125, 75–84. doi: 10.1172/JCI73945
Dinadayala P., Lemassu A., Granovski P., Cerantola S., Winter N., Daffe M. (2004). Revisiting the structure of the anti-neoplastic glucans of Mycobacterium bovis Bacille Calmette-Guerin. Structural analysis of the extracellular and boiling water extract-derived glucans of the vaccine substrains. J. Biol. Chem. 279, 12369–12378. doi: 10.1074/jbc.M308908200
Ding F., Li Y., Liu J., Liu L., Yu W., Wang Z., et al. (2014). Overendocytosis of gold nanoparticles increases autophagy and apoptosis in hypoxic human renal proximal tubular cells. Int. J. Nanomed. 9, 4317–4330. doi: 10.2147/IJN.S68685
Ding J., Ning Y., Bai Y., Xu X., Sun X., Qi C. (2019). beta-Glucan induces autophagy in dendritic cells and influences T-cell differentiation. Med. Microbiol. Immunol. 208, 39–48. doi: 10.1007/s00430-018-0556-z
Divangahi M., Desjardins D., Nunes-Alves C., Remold H. G., Behar S. M. (2010). Eicosanoid pathways regulate adaptive immunity to Mycobacterium tuberculosis. Nat. Immunol. 11, 751–758. doi: 10.1038/ni.1904
Dube A., Lemmer Y., Hayeshi R., Balogun M., Labuschagne P., Swai H., et al. (2013). State of the art and future directions in nanomedicine for tuberculosis. Expert Opin. Drug Delivery 10, 1725–1734. doi: 10.1517/17425247.2014.846905
Dutta N. K., Mehra S., Martinez A. N., Alvarez X., Renner N. A., Morici L. A., et al. (2012). The stress-response factor SigH modulates the interaction between Mycobacterium tuberculosis and host phagocytes. PloS One 7, e28958. doi: 10.1371/journal.pone.0028958
Ehlers S., Schaible U. E. (2012). The granuloma in tuberculosis: dynamics of a host-pathogen collusion. Front. Immunol. 3, 411. doi: 10.3389/fimmu.2012.00411
Eidi H., Joubert O., Nemos C., Grandemange S., Mograbi B., Foliguet B., et al. (2012). Drug delivery by polymeric nanoparticles induces autophagy in macrophages. Int. J. Pharm. 422, 495–503. doi: 10.1016/j.ijpharm.2011.11.020
Fang F. C. (2004). Antimicrobial reactive oxygen and nitrogen species: concepts and controversies. Nat. Rev. Microbiol. 2, 820–832. doi: 10.1038/nrmicro1004
Floto R. A., Sarkar S., Perlstein E. O., Kampmann B., Schreiber S. L., Rubinsztein D. C. (2007). Small molecule enhancers of rapamycin-induced TOR inhibition promote autophagy, reduce toxicity in Huntington’s disease models and enhance killing of mycobacteria by macrophages. Autophagy 3, 620–622. doi: 10.4161/auto.4898
Flynn J. L., Chan J. (2003). Immune evasion by Mycobacterium tuberculosis: living with the enemy. Curr. Opin. Immunol. 15, 450–455. doi: 10.1016/S0952-7915(03)00075-X
Franco L. H., Nair V. R., Scharn C. R., Xavier R. J., Torrealba J. R., Shiloh M. U., et al. (2017). The Ubiquitin Ligase Smurf1 Functions in Selective Autophagy of Mycobacterium tuberculosis and Anti-tuberculous Host Defense. Cell Host Microbe 22, 421–423. doi: 10.1016/j.chom.2017.08.005
Fratti R. A., Chua J., Vergne I., Deretic V. (2003). Mycobacterium tuberculosis glycosylated phosphatidylinositol causes phagosome maturation arrest. Proc. Natl. Acad. Sci. U.S.A. 100, 5437–5442. doi: 10.1073/pnas.0737613100
Galluzzi L., Baehrecke E. H., Ballabio A., Boya P., Bravo-San Pedro J. M., Cecconi F., et al. (2017). Molecular definitions of autophagy and related processes. EMBO J. 36, 1811–1836. doi: 10.15252/embj.201796697
Geijtenbeek T. B., Van Vliet S. J., Koppel E. A., Sanchez-Hernandez M., Vandenbroucke-Grauls C. M., Appelmelk B., et al. (2003). Mycobacteria target DC-SIGN to suppress dendritic cell function. J. Exp. Med. 197, 7–17. doi: 10.1084/jem.20021229
Gomes L. C., Dikic I. (2014). Autophagy in antimicrobial immunity. Mol. Cell 54, 224–233. doi: 10.1016/j.molcel.2014.03.009
Grabowski N., Hillaireau H., Vergnaud J., Santiago L. A., Kerdine-Romer S., Pallardy M., et al. (2013). Toxicity of surface-modified PLGA nanoparticles toward lung alveolar epithelial cells. Int. J. Pharm. 454, 686–694. doi: 10.1016/j.ijpharm.2013.05.025
Grange J. M., Zumla A. (2002). The global emergency of tuberculosis: what is the cause? J. R. Soc. Promot. Health 122, 78–81. doi: 10.1177/146642400212200206
Green D. R., Kroemer G. (2004). The pathophysiology of mitochondrial cell death. Science 305, 626–629. doi: 10.1126/science.1099320
Gruenberg J., Van Der Goot F. G. (2006). Mechanisms of pathogen entry through the endosomal compartments. Nat. Rev. Mol. Cell Biol. 7, 495–504. doi: 10.1038/nrm1959
Guo C., Xia Y., Niu P., Jiang L., Duan J., Yu Y., et al. (2015). Silica nanoparticles induce oxidative stress, inflammation, and endothelial dysfunction in vitro via activation of the MAPK/Nrf2 pathway and nuclear factor-kappaB signaling. Int. J. Nanomed. 10, 1463–1477. doi: 10.2147/IJN.S76114
Gupta A., Misra A., Deretic V. (2016). Targeted pulmonary delivery of inducers of host macrophage autophagy as a potential host-directed chemotherapy of tuberculosis. Adv. Drug Delivery Rev. 102, 10–20. doi: 10.1016/j.addr.2016.01.016
Gurumurthy P., Ramachandran G., Hemanth Kumar A. K., Rajasekaran S., Padmapriyadarsini C., Swaminathan S., et al. (2004). Decreased bioavailability of rifampin and other antituberculosis drugs in patients with advanced human immunodeficiency virus disease. Antimicrob. Agents Chemother. 48, 4473–4475. doi: 10.1128/AAC.48.11.4473-4475.2004
Gutierrez M. G., Mishra B. B., Jordao L., Elliott E., Anes E., Griffiths G. (2008). NF-kappa B activation controls phagolysosome fusion-mediated killing of mycobacteria by macrophages. J. Immunol. 181, 2651–2663. doi: 10.4049/jimmunol.181.4.2651
Gutierrez M. G. (2013). Functional role(s) of phagosomal Rab GTPases. Small GTPases 4, 148–158. doi: 10.4161/sgtp.25604
Hajipour M. J., Fromm K. M., Akbar Ashkarran A., Jimenez De Aberasturi D., Larramendi I. R. D., Rojo T., et al. (2012). Antibacterial properties of nanoparticles. Trends Biotechnol. 30, 499–511. doi: 10.1016/j.tibtech.2012.06.004
Han J., Zhong C. Q., Zhang D. W. (2011). Programmed necrosis: backup to and competitor with apoptosis in the immune system. Nat. Immunol. 12, 1143–1149. doi: 10.1038/ni.2159
Hand W. L., Corwin R. W., Steinberg T. H., Grossman G. D. (1984). Uptake of antibiotics by human alveolar macrophages. Am. Rev. Respir. Dis. 129, 933–937. doi: 10.1164/arrd.1984.129.6.933
Harris J., Lang T., Thomas J. P. W., Sukkar M. B., Nabar N. R., Kehrl J. H. (2017). Autophagy and inflammasomes. Mol. Immunol. 86, 10–15. doi: 10.1016/j.molimm.2017.02.013
Huang J., Canadien V., Lam G. Y., Steinberg B. E., Dinauer M. C., Magalhaes M. A., et al. (2009). Activation of antibacterial autophagy by NADPH oxidases. Proc. Natl. Acad. Sci. U.S.A. 106, 6226–6231. doi: 10.1073/pnas.0811045106
Ishikawa E., Ishikawa T., Morita Y. S., Toyonaga K., Yamada H., Takeuchi O., et al. (2009). Direct recognition of the mycobacterial glycolipid, trehalose dimycolate, by C-type lectin Mincle. J. Exp. Med. 206, 2879–2888. doi: 10.1084/jem.20091750
Jabir M. S., Hopkins L., Ritchie N. D., Ullah I., Bayes H. K., Li D., et al. (2015). Mitochondrial damage contributes to Pseudomonas aeruginosa activation of the inflammasome and is downregulated by autophagy. Autophagy 11, 166–182. doi: 10.4161/15548627.2014.981915
Jacquel A., Obba S., Boyer L., Dufies M., Robert G., Gounon P., et al. (2012). Autophagy is required for CSF-1-induced macrophagic differentiation and acquisition of phagocytic functions. Blood 119, 4527–4531. doi: 10.1182/blood-2011-11-392167
Jayaraman P., Sada-Ovalle I., Nishimura T., Anderson A. C., Kuchroo V. K., Remold H. G., et al. (2013). IL-1β promotes antimicrobial immunity in macrophages by regulating TNFR signaling and caspase-3 activation. J. Immunol. 190, 4196–4204. doi: 10.4049/jimmunol.1202688
Johnson J. D., Hand W. L., Francis J. B., King-Thompson N., Corwin R. W. (1980). Antibiotic uptake by alveolar macrophages. J. Lab. Clin. Med. 95, 429–439.
Juárez E., Carranza C., Sánchez G., González M., Chávez J., Sarabia C., et al. (2016). Loperamide Restricts Intracellular Growth of Mycobacterium tuberculosis in Lung Macrophages. Am. J. Respir. Cell Mol. Biol. 55, 837–847. doi: 10.1165/rcmb.2015-0383OC
Kang P. B., Azad A. K., Torrelles J. B., Kaufman T. M., Beharka A., Tibesar E., et al. (2005). The human macrophage mannose receptor directs Mycobacterium tuberculosis lipoarabinomannan-mediated phagosome biogenesis. J. Exp. Med. 202, 987–999. doi: 10.1084/jem.20051239
Kasmapour B., Gronow A., Bleck C. K., Hong W., Gutierrez M. G. (2012). Size-dependent mechanism of cargo sorting during lysosome-phagosome fusion is controlled by Rab34. Proc. Natl. Acad. Sci. U.S.A. 109, 20485–20490. doi: 10.1073/pnas.1206811109
Kaufmann S. H. E., Dorhoi A., Hotchkiss R. S., Bartenschlager R. (2018). Host-directed therapies for bacterial and viral infections. Nat. Rev. Drug Discovery 17, 35–56. doi: 10.1038/nrd.2017.162
Kim J. S., He L., Lemasters J. J. (2003). Mitochondrial permeability transition: a common pathway to necrosis and apoptosis. Biochem. Biophys. Res. Commun. 304, 463–470. doi: 10.1016/S0006-291X(03)00618-1
Kim B. H., Shenoy A. R., Kumar P., Das R., Tiwari S., Macmicking J. D. (2011). A family of IFN-gamma-inducible 65-kD GTPases protects against bacterial infection. Science 332, 717–721. doi: 10.1126/science.1201711
Kim K. H., An D. R., Song J., Yoon J. Y., Kim H. S., Yoon H. J., et al. (2012). Mycobacterium tuberculosis Eis protein initiates suppression of host immune responses by acetylation of DUSP16/MKP-7. Proc. Natl. Acad. Sci. U.S.A. 109, 7729–7734. doi: 10.2210/pdb3sxo/pdb
Kim J. K., Lee H. M., Park K. S., Shin D. M., Kim T. S., Kim Y. S., et al. (2017). MIR144* inhibits antimicrobial responses against Mycobacterium tuberculosis in human monocytes and macrophages by targeting the autophagy protein DRAM2. Autophagy 13, 423–441. doi: 10.1080/15548627.2016.1241922
Kimmey J. M., Stallings C. L. (2016). Bacterial Pathogens versus Autophagy: Implications for Therapeutic Interventions. Trends Mol. Med. 22, 1060–1076. doi: 10.1016/j.molmed.2016.10.008
Kimmey J. M., Huynh J. P., Weiss L. A., Park S., Kambal A., Debnath J., et al. (2015). Unique role for ATG5 in neutrophil-mediated immunopathology during M. tuberculosis infection. Nature 528, 565–569. doi: 10.1038/nature16451
Kimura T., Jain A., Choi S. W., Mandell M. A., Schroder K., Johansen T., et al. (2015). TRIM-mediated precision autophagy targets cytoplasmic regulators of innate immunity. J. Cell Biol. 210, 973–989. doi: 10.1083/jcb.201503023
Kolloli A., Subbian S. (2017). Host-Directed Therapeutic Strategies for Tuberculosis. Front. Med. (Lausanne) 4, 171. doi: 10.3389/fmed.2017.00171
Koster S., Upadhyay S., Chandra P., Papavinasasundaram K., Yang G., Hassan A., et al. (2017). Mycobacterium tuberculosis is protected from NADPH oxidase and LC3-associated phagocytosis by the LCP protein CpsA. Proc. Natl. Acad. Sci. U.S.A. 114, E8711–e8720. doi: 10.1073/pnas.1707792114
Kumar M., Sahu S. K., Kumar R., Subuddhi A., Maji R. K., Jana K., et al. (2015). MicroRNA let-7 modulates the immune response to Mycobacterium tuberculosis infection via control of A20, an inhibitor of the NF-kappaB pathway. Cell Host Microbe 17, 345–356. doi: 10.1016/j.chom.2015.01.007
Kumar R., Sahu S. K., Kumar M., Jana K., Gupta P., Gupta U. D., et al. (2016). MicroRNA 17-5p regulates autophagy in Mycobacterium tuberculosis-infected macrophages by targeting Mcl-1 and STAT3. Cell Microbiol. 18, 679–691. doi: 10.1111/cmi.12540
Kurtz S., Mckinnon K. P., Runge M. S., Ting J. P., Braunstein M. (2006). The SecA2 secretion factor of Mycobacterium tuberculosis promotes growth in macrophages and inhibits the host immune response. Infect. Immun. 74, 6855–6864. doi: 10.1128/IAI.01022-06
Lam K. K., Zheng X., Forestieri R., Balgi A. D., Nodwell M., Vollett S., et al. (2012). Nitazoxanide stimulates autophagy and inhibits mTORC1 signaling and intracellular proliferation of Mycobacterium tuberculosis. PloS Pathog. 8, e1002691. doi: 10.1371/journal.ppat.1002691
Lam A., Prabhu R., Gross C. M., Riesenberg L. A., Singh V., Aggarwal S. (2017). Role of apoptosis and autophagy in tuberculosis. Am. J. Physiol. Lung Cell Mol. Physiol. 313, L218–l229. doi: 10.1152/ajplung.00162.2017
Lamichhane G. (2011). Mycobacterium tuberculosis response to stress from reactive oxygen and nitrogen species. Front. Microbiol. 2, 176. doi: 10.3389/fmicb.2011.00176
Lammas D. A., Stober C., Harvey C. J., Kendrick N., Panchalingam S., Kumararatne D. S. (1997). ATP-induced killing of mycobacteria by human macrophages is mediated by purinergic P2Z(P2X7) receptors. Immunity 7, 433–444. doi: 10.1016/S1074-7613(00)80364-7
Lancellotti M., Brocchi M., Dias Da Silveira W. (2006). Bacteria-induced apoptosis: An approach to bacterial pathogenesis. Braz. J. Morphol. Sci. 23, 75–86.
Lee J., Cho E. C., Cho K. (2004). Incorporation and release behavior of hydrophobic drug in functionalized poly(D,L-lactide)-block-poly(ethylene oxide) micelles. J. Control Rel. 94, 323–335. doi: 10.1016/j.jconrel.2003.10.012
Lee J., Hartman M., Kornfeld H. (2009). Macrophage apoptosis in tuberculosis. Yonsei Med. J. 50, 1–11. doi: 10.3349/ymj.2009.50.1.1
Ma J., Becker C., Lowell C. A., Underhill D. M. (2012). Dectin-1-triggered recruitment of light chain 3 protein to phagosomes facilitates major histocompatibility complex class II presentation of fungal-derived antigens. J. Biol. Chem. 287, 34149–34156. doi: 10.1074/jbc.M112.382812
Ma J., Yang B., Yu S., Zhang Y., Zhang X., Lao S., et al. (2014). Tuberculosis antigen-induced expression of IFN-alpha in tuberculosis patients inhibits production of IL-1beta. FASEB J. 28, 3238–3248. doi: 10.1096/fj.13-247056
Macmicking J. D., Taylor G. A., Mckinney J. D. (2003). Immune control of tuberculosis by IFN-gamma-inducible LRG-47. Science 302, 654–659. doi: 10.1126/science.1088063
Mallakpour S. (2016). Polymeric nanoparticles: Recent development in synthesis and application. Express Polymer. Lett. 10, 895–913. doi: 10.3144/expresspolymlett.2016.84
Marakalala M. J., Ndlovu H. (2017). Signaling C-type lectin receptors in antimycobacterial immunity. PloS Pathog. 13, e1006333. doi: 10.1371/journal.ppat.1006333
Martinez J., Malireddi R. K., Lu Q., Cunha L. D., Pelletier S., Gingras S., et al. (2015). Molecular characterization of LC3-associated phagocytosis reveals distinct roles for Rubicon, NOX2 and autophagy proteins. Nat. Cell Biol. 17, 893–906. doi: 10.1038/ncb3192
Martinvalet D., Dykxhoorn D. M., Ferrini R., Lieberman J. (2008). Granzyme A cleaves a mitochondrial complex I protein to initiate caspase-independent cell death. Cell 133, 681–692. doi: 10.1016/j.cell.2008.03.032
Miller J. L., Velmurugan K., Cowan M. J., Briken V. (2010). The type I NADH dehydrogenase of Mycobacterium tuberculosis counters phagosomal NOX2 activity to inhibit TNF-alpha-mediated host cell apoptosis. PloS Pathog. 6, e1000864. doi: 10.1371/journal.ppat.1000864
Mishra A., Akhtar S., Jagannath C., Khan A. (2017). Pattern recognition receptors and coordinated cellular pathways involved in tuberculosis immunopathogenesis: Emerging concepts and perspectives. Mol. Immunol. 87, 240–248. doi: 10.1016/j.molimm.2017.05.001
Mitchell G., Isberg R. R. (2017). Innate Immunity to Intracellular Pathogens: Balancing Microbial Elimination and Inflammation. Cell Host Microbe 22, 166–175. doi: 10.1016/j.chom.2017.07.005
Moreau K., Lacas-Gervais S., Fujita N., Sebbane F., Yoshimori T., Simonet M., et al. (2010). Autophagosomes can support Yersinia pseudotuberculosis replication in macrophages. Cell Microbiol. 12, 1108–1123. doi: 10.1111/j.1462-5822.2010.01456.x
Mueller P., Pieters J. (2006). Modulation of macrophage antimicrobial mechanisms by pathogenic mycobacteria. Immunobiology 211, 549–556. doi: 10.1016/j.imbio.2006.06.004
Nagavarma B. V. N., Yadav H., Ayaz A., Vasudha L., Shivakumar H. (2012). Different techniques for preparation of polymeric nanoparticles- A review. Asian J. Pharm. Clin. Res. 5, 16–23.
Nasiruddin M., Neyaz M. K., Das S. (2017). Nanotechnology-Based Approach in Tuberculosis Treatment. Tuberc. Res. Treat 2017, 4920209. doi: 10.1155/2017/4920209
Nigou J., Zelle-Rieser C., Gilleron M., Thurnher M., Puzo G. (2001). Mannosylated lipoarabinomannans inhibit IL-12 production by human dendritic cells: evidence for a negative signal delivered through the mannose receptor. J. Immunol. 166, 7477–7485. doi: 10.4049/jimmunol.166.12.7477
Nordstrom E., Fisone G., Kristensson K. (2009). Opposing effects of ERK and p38-JNK MAP kinase pathways on formation of prions in GT1-1 cells. FASEB J. 23, 613–622. doi: 10.1096/fj.08-115360
Ouimet M., Koster S., Sakowski E., Ramkhelawon B., Van Solingen C., Oldebeken S., et al. (2016). Mycobacterium tuberculosis induces the miR-33 locus to reprogram autophagy and host lipid metabolism. Nat. Immunol. 17, 677–686. doi: 10.1038/ni.3434
Paik S., Kim J. K., Chung C., Jo E. K. (2019). Autophagy: A new strategy for host-directed therapy of tuberculosis. Virulence 10, 448–459. doi: 10.1080/21505594.2018.1536598
Park E. J., Umh H. N., Kim S. W., Cho M. H., Kim J. H., Kim Y. (2014). ERK pathway is activated in bare-FeNPs-induced autophagy. Arch. Toxicol. 88, 323–336. doi: 10.1007/s00204-013-1134-1
Pastore N., Brady O. A., Diab H. II, Martina J. A., Sun L., Huynh T., et al. (2016). TFEB and TFE3 cooperate in the regulation of the innate immune response in activated macrophages. Autophagy 12, 1240–1258. doi: 10.1080/15548627.2016.1179405
Petruccioli E., Romagnoli A., Corazzari M., Coccia E. M., Butera O., Delogu G., et al. (2012). Specific T cells restore the autophagic flux inhibited by Mycobacterium tuberculosis in human primary macrophages. J. Infect. Dis. 205, 1425–1435. doi: 10.1093/infdis/jis226
Pozzi L. A., Maciaszek J. W., Rock K. L. (2005). Both dendritic cells and macrophages can stimulate naive CD8 T cells in vivo to proliferate, develop effector function, and differentiate into memory cells. J. Immunol. 175, 2071–2081. doi: 10.4049/jimmunol.175.4.2071
Prashar A., Schnettger L., Bernard E. M., Gutierrez M. G. (2017). Rab GTPases in Immunity and Inflammation. Front. Cell Infect. Microbiol. 7, 435. doi: 10.3389/fcimb.2017.00435
Pu Q., Gan C., Li R., Li Y., Tan S., Li X., et al. (2017). Atg7 Deficiency Intensifies Inflammasome Activation and Pyroptosis in Pseudomonas Sepsis. J. Immunol. 198, 3205–3213. doi: 10.4049/jimmunol.1601196
Pujol C., Klein K. A., Romanov G. A., Palmer L. E., Cirota C., Zhao Z., et al. (2009). Yersinia pestis can reside in autophagosomes and avoid xenophagy in murine macrophages by preventing vacuole acidification. Infect. Immun. 77, 2251–2261. doi: 10.1128/IAI.00068-09
Quesniaux V., Fremond C., Jacobs M., Parida S., Nicolle D., Yeremeev V., et al. (2004). Toll-like receptor pathways in the immune responses to mycobacteria. Microbes Infect. 6, 946–959. doi: 10.1016/j.micinf.2004.04.016
Queval C. J., Brosch R., Simeone R. (2017). The Macrophage: A Disputed Fortress in the Battle against Mycobacterium tuberculosis. Front. Microbiol. 8, 2284. doi: 10.3389/fmicb.2017.02284
Ricci J. E., Muñoz-Pinedo C., Fitzgerald P., Bailly-Maitre B., Perkins G. A., Yadava N., et al. (2004). Disruption of mitochondrial function during apoptosis is mediated by caspase cleavage of the p75 subunit of complex I of the electron transport chain. Cell 117, 773–786. doi: 10.1016/j.cell.2004.05.008
Rohde K., Yates R. M., Purdy G. E., Russell D. G. (2007). Mycobacterium tuberculosis and the environment within the phagosome. Immunol. Rev. 219, 37–54. doi: 10.1111/j.1600-065X.2007.00547.x
Romagnoli A., Etna M. P., Giacomini E., Pardini M., Remoli M. E., Corazzari M., et al. (2012). ESX-1 dependent impairment of autophagic flux by Mycobacterium tuberculosis in human dendritic cells. Autophagy 8, 1357–1370. doi: 10.4161/auto.20881
Romao S., Gasser N., Becker A. C., Guhl B., Bajagic M., Vanoaica D., et al. (2013). Autophagy proteins stabilize pathogen-containing phagosomes for prolonged MHC II antigen processing. J. Cell Biol. 203, 757–766. doi: 10.1083/jcb.201308173
Russell D. G., Cardona P. J., Kim M. J., Allain S., Altare F. (2009). Foamy macrophages and the progression of the human tuberculosis granuloma. Nat. Immunol. 10, 943–948. doi: 10.1038/ni.1781
Saborano R., Wongpinyochit T., Totten J. D., Johnston B. F., Seib F. P., Duarte I. F. (2017). Metabolic Reprogramming of Macrophages Exposed to Silk, Poly(lactic-co-glycolic acid), and Silica Nanoparticles. Adv. Healthc. Mater. 6, 1–13. doi: 10.1002/adhm.201601240
Saitoh T., Akira S. (2016). Regulation of inflammasomes by autophagy. J. Allergy Clin. Immunol. 138, 28–36. doi: 10.1016/j.jaci.2016.05.009
Sakowski E. T., Koster S., Portal Celhay C., Park H. S., Shrestha E., Hetzenecker S. E., et al. (2015). Ubiquilin 1 Promotes IFN-gamma-Induced Xenophagy of Mycobacterium tuberculosis. PloS Pathog. 11, e1005076. doi: 10.1371/journal.ppat.1005076
Salgame P. (2005). Host innate and Th1 responses and the bacterial factors that control Mycobacterium tuberculosis infection. Curr. Opin. Immunol. 17, 374–380. doi: 10.1016/j.coi.2005.06.006
Samuel L. P., Song C. H., Wei J., Roberts E. A., Dahl J. L., Barry C. E., et al. (2007). Expression, production and release of the Eis protein by Mycobacterium tuberculosis during infection of macrophages and its effect on cytokine secretion. Microbiology 153, 529–540. doi: 10.1099/mic.0.2006/002642-0
Sanjuan M. A., Dillon C. P., Tait S. W., Moshiach S., Dorsey F., Connell S., et al. (2007). Toll-like receptor signalling in macrophages links the autophagy pathway to phagocytosis. Nature 450, 1253–1257. doi: 10.1038/nature06421
Sarathy J., Dartois V., Dick T., Gengenbacher M. (2013a). Reduced drug uptake in phenotypically resistant nutrient-starved nonreplicating Mycobacterium tuberculosis. Antimicrob. Agents Chemother. 57, 1648–1653. doi: 10.1128/AAC.02202-12
Sarathy J. P., Lee E., Dartois V. (2013b). Polyamines inhibit porin-mediated fluoroquinolone uptake in mycobacteria. PloS One 8, e65806. doi: 10.1371/journal.pone.0065806
Schiebler M., Brown K., Hegyi K., Newton S. M., Renna M., Hepburn L., et al. (2015). Functional drug screening reveals anticonvulsants as enhancers of mTOR-independent autophagic killing of Mycobacterium tuberculosis through inositol depletion. EMBO Mol. Med. 7, 127–139. doi: 10.15252/emmm.201404137
Schnettger L., Rodgers A., Repnik U., Lai R. P., Pei G., Verdoes M., et al. (2017). A Rab20-Dependent Membrane Trafficking Pathway Controls M. tuberculosis Replication by Regulating Phagosome Spaciousness and Integrity. Cell Host Microbe 21, 619–628.e5. doi: 10.1016/j.chom.2017.04.004
Schüler P., Zemper K., Borner K., Koeppe P., Schaberg T., Lode H. (1997). Penetration of sparfloxacin and ciprofloxacin into alveolar macrophages, epithelial lining fluid, and polymorphonuclear leucocytes. Eur. Respir. J. 10, 1130–1136. doi: 10.1183/09031936.97.10051130
Schuller S., Neefjes J., Ottenhoff T., Thole J., Young D. (2001). Coronin is involved in uptake of Mycobacterium bovis BCG in human macrophages but not in phagosome maintenance. Cell Microbiol. 3, 785–793. doi: 10.1046/j.1462-5822.2001.00155.x
Sharma V., Makhdoomi M., Kumar P., Khan N., Singh S., Verma H. N., et al. (2017). Induction of autophagy by trehalose limits opportunistic mycobacterial infections in HIV-infected macrophages. bioRxiv 1, 202697. doi: 10.1101/202697
Sharma V., Makhdoomi M., Singh L., Kumar P., Khan N., Singh S., et al. (2020). Trehalose limits opportunistic mycobacterial survival during HIV co-infection by reversing HIV-mediated autophagy block. Autophagy, 1–20. doi: 10.1080/15548627.2020.1725374
Shi C. S., Kehrl J. H. (2010). TRAF6 and A20 regulate lysine 63-linked ubiquitination of Beclin-1 to control TLR4-induced autophagy. Sci. Signal 3, ra42. doi: 10.1126/scisignal.2000751
Shi C. S., Shenderov K., Huang N. N., Kabat J., Abu-Asab M., Fitzgerald K. A., et al. (2012). Activation of autophagy by inflammatory signals limits IL-1beta production by targeting ubiquitinated inflammasomes for destruction. Nat. Immunol. 13, 255–263. doi: 10.1038/ni.2215
Shibutani S. T., Saitoh T., Nowag H., Munz C., Yoshimori T. (2015). Autophagy and autophagy-related proteins in the immune system. Nat. Immunol. 16, 1014–1024. doi: 10.1038/ni.3273
Shin D. M., Jeon B. Y., Lee H. M., Jin H. S., Yuk J. M., Song C. H., et al. (2010). Mycobacterium tuberculosis eis regulates autophagy, inflammation, and cell death through redox-dependent signaling. PloS Pathog. 6, e1001230. doi: 10.1371/journal.ppat.1001230
Silva Miranda M., Breiman A., Allain S., Deknuydt F., Altare F. (2012). The tuberculous granuloma: an unsuccessful host defence mechanism providing a safety shelter for the bacteria? Clin. Dev. Immunol. 2012, 139127. doi: 10.1155/2012/139127
Soman A., Honeybourne D., Andrews J., Jevons G., WISE R. (1999). Concentrations of moxifloxacin in serum and pulmonary compartments following a single 400 mg oral dose in patients undergoing fibre-optic bronchoscopy. J. Antimicrob. Chemother. 44, 835–838. doi: 10.1093/jac/44.6.835
Songane M., Kleinnijenhuis J., Netea M. G., Van Crevel R. (2012). The role of autophagy in host defence against Mycobacterium tuberculosis infection. Tuberculosis (Edinb.) 92, 388–396. doi: 10.1016/j.tube.2012.05.004
Stanley S. A., Barczak A. K., Silvis M. R., Luo S. S., Sogi K., Vokes M., et al. (2014). Identification of Host-Targeted Small Molecules That Restrict Intracellular Mycobacterium tuberculosis Growth. PloS Pathog. 10, e1003946. doi: 10.1371/journal.ppat.1003946
Sundaramurthy V., Barsacchi R., Samusik N., Marsico G., Gilleron J., Kalaidzidis I., et al. (2013). Integration of chemical and RNAi multiparametric profiles identifies triggers of intracellular mycobacterial killing. Cell Host Microbe 13, 129–142. doi: 10.1016/j.chom.2013.01.008
Sun-Wada G. H., Tabata H., Kawamura N., Aoyama M., Wada Y. (2009). Direct recruitment of H+-ATPase from lysosomes for phagosomal acidification. J. Cell Sci. 122, 2504–2513. doi: 10.1242/jcs.050443
Tailleux L., Schwartz O., Herrmann J. L., Pivert E., Jackson M., Amara A., et al. (2003). DC-SIGN is the major Mycobacterium tuberculosis receptor on human dendritic cells. J. Exp. Med. 197, 121–127. doi: 10.1084/jem.20021468
Tanne A., Neyrolles O. (2010). C-type lectins in immune defense against pathogens: the murine DC-SIGN homologue SIGNR3 confers early protection against Mycobacterium tuberculosis infection. Virulence 1, 285–290. doi: 10.4161/viru.1.4.11967
Toyonaga K., Torigoe S., Motomura Y., Kamichi T., Hayashi J. M., Morita Y. S., et al. (2016). C-Type Lectin Receptor DCAR Recognizes Mycobacterial Phosphatidyl-Inositol Mannosides to Promote a Th1 Response during Infection. Immunity 45, 1245–1257. doi: 10.1016/j.immuni.2016.10.012
Tukulula M., Gouveia L., Paixao P., Hayeshi R., Naicker B., Dube A. (2018). Functionalization of PLGA Nanoparticles with 1,3-β-glucan Enhances the Intracellular Pharmacokinetics of Rifampicin in Macrophages. Pharm. Res. 35, 111. doi: 10.1007/s11095-018-2391-8
Upadhyay T. K., Fatima N., Sharma A., Sharma D., Sharma R. (2019). Nano-Rifabutin entrapment within glucan microparticles enhances protection against intracellular Mycobacterium tuberculosis. Artif. Cells Nanomed. Biotechnol. 47, 427–435. doi: 10.1080/21691401.2018.1559180
Van Crevel R., Ottenhoff T. H. M., Van Der Meer J. W. M. (2002). Innate Immunity to Mycobacterium tuberculosis. Clin. Microbiol. Rev. 15, 294. doi: 10.1128/CMR.15.2.294-309.2002
Van De Veerdonk F. L., Teirlinck A. C., Kleinnijenhuis J., Kullberg B. J., Van Crevel R., Van Der Meer J. W., et al. (2010). Mycobacterium tuberculosis induces IL-17A responses through TLR4 and dectin-1 and is critically dependent on endogenous IL-1. J. Leukoc. Biol. 88, 227–232. doi: 10.1189/jlb.0809550
Van Der Vaart M., Korbee C. J., Lamers G. E., Tengeler A. C., Hosseini R., Haks M. C., et al. (2014). The DNA damage-regulated autophagy modulator DRAM1 links mycobacterial recognition via TLR-MYD88 to autophagic defense [corrected]. Cell Host Microbe 15, 753–767. doi: 10.1016/j.chom.2014.05.005
Vandenabeele P., Declercq W., Van Herreweghe F., Vanden Berghe T. (2010). The role of the kinases RIP1 and RIP3 in TNF-induced necrosis. Sci. Signal 3, re4. doi: 10.1126/scisignal.3115re4
Vazquez C. L., Rodgers A., Herbst S., Coade S., Gronow A., Guzman C. A., et al. (2016). The proneurotrophin receptor sortilin is required for Mycobacterium tuberculosis control by macrophages. Sci. Rep. 6, 29332. doi: 10.1038/srep29332
Vergne I., Chua J., Deretic V. (2003). Tuberculosis toxin blocking phagosome maturation inhibits a novel Ca2+/calmodulin-PI3K hVPS34 cascade. J. Exp. Med. 198, 653–659. doi: 10.1084/jem.20030527
Vieira O. V., Botelho R. J., Grinstein S. (2002). Phagosome maturation: aging gracefully. Biochem. J. 366, 689–704. doi: 10.1042/bj20020691
Villeneuve C., Gilleron M., Maridonneau-Parini I., Daffe M., Astarie-Dequeker C., Etienne G. (2005). Mycobacteria use their surface-exposed glycolipids to infect human macrophages through a receptor-dependent process. J. Lipid Res. 46, 475–483. doi: 10.1194/jlr.M400308-JLR200
Walburger A., Koul A., Ferrari G., Nguyen L., Prescianotto-Baschong C., Huygen K., et al. (2004). Protein kinase G from pathogenic mycobacteria promotes survival within macrophages. Science 304, 1800–1804. doi: 10.1126/science.1099384
Wang Z., Jiang H., Chen S., Du F., Wang X. (2012). The mitochondrial phosphatase PGAM5 functions at the convergence point of multiple necrotic death pathways. Cell 148, 228–243. doi: 10.1016/j.cell.2011.11.030
Watson R. O., Manzanillo P. S., Cox J. S. (2012). Extracellular M. tuberculosis DNA targets bacteria for autophagy by activating the host DNA-sensing pathway. Cell 150, 803–815. doi: 10.1016/j.cell.2012.06.040
Watson R. O., Bell S. L., Macduff D. A., Kimmey J. M., Diner E. J., Olivas J., et al. (2015). The Cytosolic Sensor cGAS Detects Mycobacterium tuberculosis DNA to Induce Type I Interferons and Activate Autophagy. Cell Host Microbe 17, 811–819. doi: 10.1016/j.chom.2015.05.004
Winau F., Weber S., Sad S., De Diego J., Hoops S. L., Breiden B., et al. (2006). Apoptotic vesicles crossprime CD8 T cells and protect against tuberculosis. Immunity 24, 105–117. doi: 10.1016/j.immuni.2005.12.001
Wong D., Chao J. D., Av-Gay Y. (2013). Mycobacterium tuberculosis-secreted phosphatases: from pathogenesis to targets for TB drug development. Trends Microbiol. 21, 100–109. doi: 10.1016/j.tim.2012.09.002
World Health Organization (2018). Global Tuberculosis Report 2018 (Geneva: World Health Organization).
World Health Organization (2019). Global Tuberculosis Report 2019 (Geneva: World Health Organization).
Wu Y., Wang Y., Zou H., Wang B., Sun Q., Fu A., et al. (2017). Probiotic Bacillus amyloliquefaciens SC06 Induces Autophagy to Protect against Pathogens in Macrophages. Front. Microbiol. 8, 469. doi: 10.3389/fmicb.2017.00469
Xia Z., Dickens M., Raingeaud J., Davis R. J., Greenberg M. E. (1995). Opposing effects of ERK and JNK-p38 MAP kinases on apoptosis. Science 270, 1326–1331. doi: 10.1126/science.270.5240.1326
Xu G., Wang J., Gao G. F., Liu C. H. (2014). Insights into battles between Mycobacterium tuberculosis and macrophages. Protein Cell 5, 728–736. doi: 10.1007/s13238-014-0077-5
Yang C. S., Lee J. S., Rodgers M., Min C. K., Lee J. Y., Kim H. J., et al. (2012). Autophagy protein Rubicon mediates phagocytic NADPH oxidase activation in response to microbial infection or TLR stimulation. Cell Host Microbe 11, 264–276. doi: 10.1016/j.chom.2012.01.018
Yasir M., Pachikara N. D., Bao X., Pan Z., Fan H. (2011). Regulation of chlamydial infection by host autophagy and vacuolar ATPase-bearing organelles. Infect. Immun. 79, 4019–4028. doi: 10.1128/IAI.05308-11
Yonekawa A., Saijo S., Hoshino Y., Miyake Y., Ishikawa E., Suzukawa M., et al. (2014). Dectin-2 is a direct receptor for mannose-capped lipoarabinomannan of mycobacteria. Immunity 41, 402–413. doi: 10.1016/j.immuni.2014.08.005
Yu Y., Duan J., Yu Y., Li Y., Liu X., Zhou X., et al. (2014). Silica nanoparticles induce autophagy and autophagic cell death in HepG2 cells triggered by reactive oxygen species. J. Hazard. Mater. 270, 176–186. doi: 10.1016/j.jhazmat.2014.01.028
Yuk J. M., Shin D. M., Lee H. M., Yang C. S., Jin H. S., Kim K. K., et al. (2009). Vitamin D3 induces autophagy in human monocytes/macrophages via cathelicidin. Cell Host Microbe 6, 231–243. doi: 10.1016/j.chom.2009.08.004
Zhai W., Wu F., Zhang Y., Fu Y., Liu Z. (2019). The Immune Escape Mechanisms of Mycobacterium Tuberculosis. Int. J. Mol. Sci. 20, 1–18. doi: 10.3390/ijms20020340
Zhang Q., Sun J., Wang Y., He W., Wang L., Zheng Y., et al. (2017). Antimycobacterial and Anti-inflammatory Mechanisms of Baicalin via Induced Autophagy in Macrophages Infected with Mycobacterium tuberculosis. Front. Microbiol. 8, 2142. doi: 10.3389/fmicb.2017.02142
Keywords: Mycobacterium tuberculosis, innate immunity, autophagy and tuberculosis, host directed therapies, immunotherapeutic nanoparticles, xenophagy, LC3-associated phagocytosis, apoptosis and tuberculosis
Citation: Maphasa RE, Meyer M and Dube A (2021) The Macrophage Response to Mycobacterium tuberculosis and Opportunities for Autophagy Inducing Nanomedicines for Tuberculosis Therapy. Front. Cell. Infect. Microbiol. 10:618414. doi: 10.3389/fcimb.2020.618414
Received: 16 October 2020; Accepted: 18 December 2020;
Published: 08 February 2021.
Edited by:
Manisha Yadav, University of Delhi, IndiaReviewed by:
Chinnaswamy Jagannath, Weill Cornell Medical College of Cornell University, United StatesCopyright © 2021 Maphasa, Meyer and Dube. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Admire Dube, YWR1YmVAdXdjLmFjLnph
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.