 Diana L. Rodriguez
Diana L. Rodriguez Morgan M. Quail1,2†
Morgan M. Quail1,2† Aaron D. Hernday
Aaron D. Hernday Clarissa J. Nobile
Clarissa J. Nobile
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Cell. Infect. Microbiol. , 03 December 2020
Sec. Fungal Pathogenesis
Volume 10 - 2020 | https://doi.org/10.3389/fcimb.2020.605711
This article is part of the Research Topic Functional Genomics of Transcriptional Regulation in Pathogenic Fungi View all 7 articles
Candida albicans is a commensal member of the human microbiota that colonizes multiple niches in the body including the skin, oral cavity, and gastrointestinal and genitourinary tracts of healthy individuals. It is also the most common human fungal pathogen isolated from patients in clinical settings. C. albicans can cause a number of superficial and invasive infections, especially in immunocompromised individuals. The ability of C. albicans to succeed as both a commensal and a pathogen, and to thrive in a wide range of environmental niches within the host, requires sophisticated transcriptional regulatory programs that can integrate and respond to host specific environmental signals. Identifying and characterizing the transcriptional regulatory networks that control important developmental processes in C. albicans will shed new light on the strategies used by C. albicans to colonize and infect its host. Here, we discuss the transcriptional regulatory circuits controlling three major developmental processes in C. albicans: biofilm formation, the white-opaque phenotypic switch, and the commensal-pathogen transition. Each of these three circuits are tightly knit and, through our analyses, we show that they are integrated together by extensive regulatory crosstalk between the core regulators that comprise each circuit.
C. albicans is a common human commensal that asymptomatically colonizes the skin, oral cavity, and gastrointestinal and genitourinary tracts of healthy individuals (Kennedy and Volz, 1985; Kumamoto, 2002; Achkar and Fries, 2010; Spiliopoulou et al., 2010; Kumamoto, 2011; Nobile and Johnson, 2015; Kan et al., 2020). It is also an opportunistic pathogen that is capable of causing superficial mucosal and life-threatening disseminated infections, especially in immunocompromised individuals (Wenzel, 1995; Calderone and Fonzi, 2001; Hube, 2004; Pappas et al., 2004; Mayer et al., 2013), such as in AIDS, chemotherapy and organ transplant patients, as well as in individuals with implanted medical devices (Wenzel, 1995; Nobile and Johnson, 2015). Multiple regulatory pathways controlling important C. albicans developmental processes allow this opportunistic fungal pathogen to adapt to and proliferate in distinct environmental niches in the host. In this review, we discuss the “core” transcriptional circuits controlling three major developmental processes in C. albicans: biofilm formation, the white-opaque phenotypic switch, and the commensal-pathogen transition. The core circuitry is defined as the direct physical interactions between transcriptional regulators that control these developmental processes and their respective upstream intergenic regions, where at least one direct binding interaction with other members of the circuit has been experimentally observed. These three circuits were chosen because they regulate persistent phenotypic changes in C. albicans that have been characterized using genome-wide transcriptional profiling (RNA-sequencing and/or microarray) and binding (chromatin immunoprecipitation) approaches. In our discussion of these circuits we focus largely on transcription factors (TFs) that bind to DNA in a sequence-specific manner; however, we also include some discussion of important cofactors for which genome-wide transcriptional profiling and binding data are available. In addition, we include information on “auxiliary” transcriptional regulators of these three developmental processes that we define as those that are known to regulate these processes, but that lack direct binding interactions with the core transcriptional regulators or binding data is not available for these transcriptional regulators under the growth condition of interest.
Biofilms are communities of adherent microbial cells encased in protective extracellular matrices (Kolter and Greenberg, 2006; Nobile and Johnson, 2015; Gulati and Nobile, 2016). Biofilms are ubiquitous in nature and are typically associated with interfaces, such as solid-liquid, liquid-gas, and liquid-liquid interfaces (Davey and O’toole, 2000; Kolter and Greenberg, 2006; Wilking et al., 2011; Desai and Ardekani, 2020). They are problematic when they form in industrial settings, such as in water distribution systems and on food preparation settings, and even more so when they form inside a host on tissues and on implanted medical devices. C. albicans biofilms are composed of several cell types, including round budding yeast-form cells, oval pseudohyphal cells, and elongated hyphal cells, encased in a protective extracellular matrix (Chandra et al., 2001; Desai and Mitchell, 2015). C. albicans biofilm formation occurs in four basic temporal stages: i) adherence of yeast-form cells to a surface; ii) growth and proliferation of yeast-form cells forming a basal layer of anchoring cells; iii) differentiation of a proportion of yeast-form cells into hyphal cells and production of the extracellular matrix; and iv) dispersion of yeast-form cells out of the biofilm to cause bloodstream infections or to colonize new sites for biofilm formation (Figure 1) (Desai and Mitchell, 2015; Nobile and Johnson, 2015; Gulati and Nobile, 2016). Indeed, C. albicans is a common cause of bloodstream infections worldwide, which often originate from biofilms (Edmond et al., 1999; Richards et al., 1999; Pfaller and Diekema, 2007). Given that cells within C. albicans biofilms are inherently resistant and tolerant to most antifungal drug treatments compared to planktonic (free-floating) cells, biofilm infections are particularly challenging to treat in the clinic. Understanding the genetic regulatory mechanisms that control C. albicans biofilm formation could lead to the development of novel therapeutic strategies effective in treating biofilm infections.
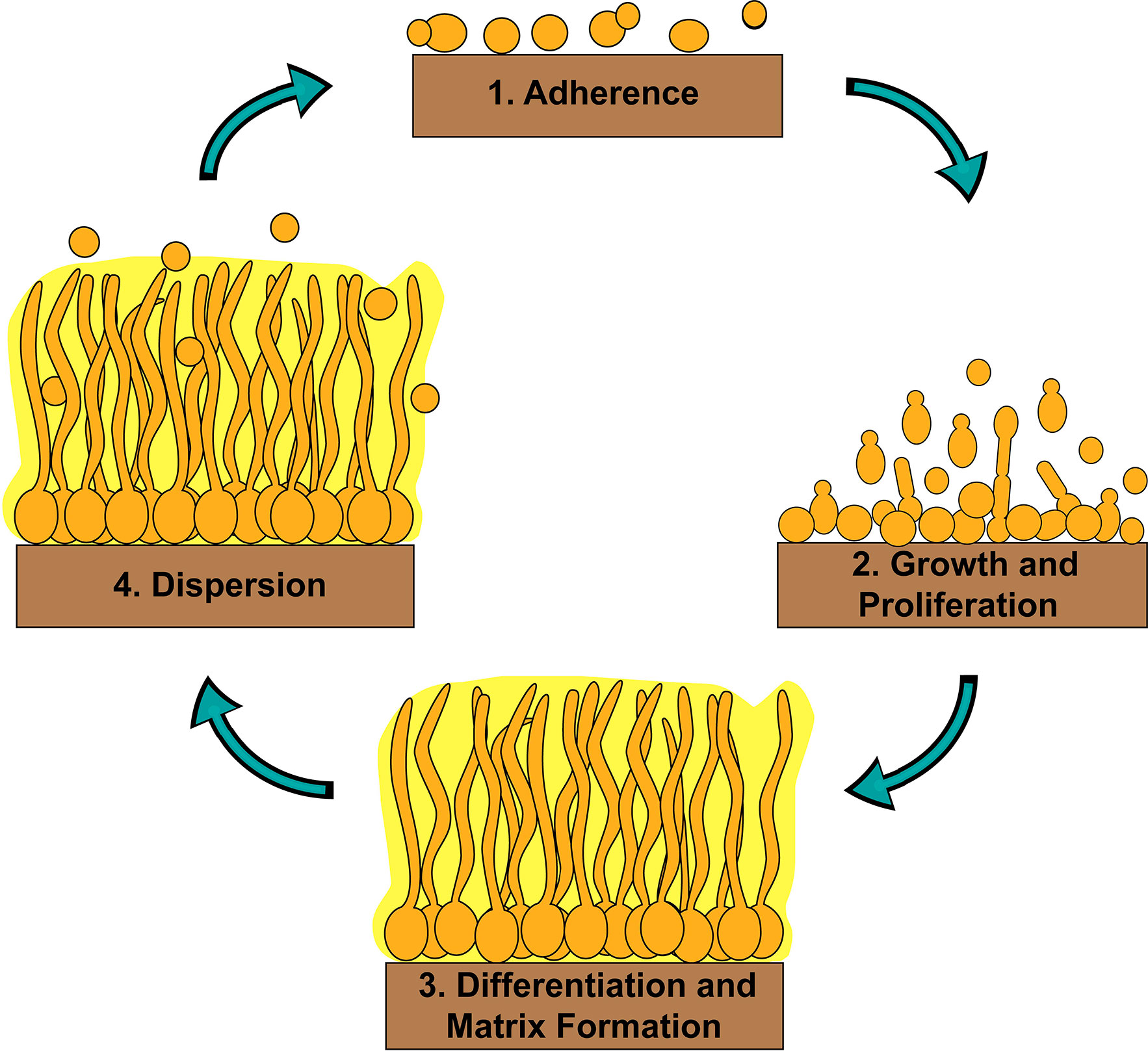
Figure 1 Stages of C. albicans biofilm formation. C. albicans biofilm formation occurs in four basic temporal stages: 1) adherence of yeast-form cells to a surface; 2) growth and proliferation of yeast-form cells forming a basal layer of anchoring cells; 3) differentiation of a proportion of yeast-form cells into hyphal cells and production of the extracellular matrix; and 4) dispersion of yeast-form cells out of the biofilm to cause bloodstream infections or to colonize new sites for biofilm formation.
The C. albicans transcriptional network controlling biofilm formation was first described eight years ago (Nobile et al., 2012). Six “master” biofilm transcriptional regulators (Bcr1, Tec1, Efg1, Ndt80, Rob1, and Brg1) were identified by screening a library of 165 transcription factor (TF) mutant strains (Homann et al., 2009) for defects in biofilm formation under standard in vitro biofilm growth conditions (Nobile et al., 2012). Here, we define a master biofilm transcriptional regulator as one whose deletion impairs biofilm formation throughout a 48-h period of biofilm growth under these standard conditions. All six TF mutant strains identified additionally had clear defects in biofilm formation in at least one of two in vivo animal models for biofilm formation (Nobile et al., 2012). Using genome-wide transcriptional profiling and chromatin immunoprecipitation techniques to study mature 48-h biofilms, a complex interconnected transcriptional network was discovered consisting of those six master transcriptional regulators, along with 1,061 downstream “target” genes (Nobile et al., 2012). These six master transcriptional regulators directly bound to the upstream intergenic regions and positively regulated the expression of each other, forming a tightly knit core biofilm circuit (Fox and Nobile, 2012; Nobile et al., 2012). Additionally, with the exception of Tec1, all of the six master biofilm transcriptional regulators acted as both repressors and activators of their directly bound biofilm target genes; Tec1, on the other hand, primarily acted as an activator (Nobile et al., 2012). Each of the six master biofilm transcriptional regulators controlled target genes that were in common with the other core transcriptional regulators in the circuit, as well as target genes that were unique to each transcriptional regulator. These findings suggest that each master biofilm transcriptional regulator in the circuit controls certain elements of biofilm formation independently, but that they also work together to coordinate concerted efforts important for biofilm formation. For example, Ndt80 regulates the expression of drug transporters independent of the other master biofilm transcriptional regulators in the circuit (such as, CDR4), and some in common with several of the other master biofilm transcriptional regulators in the circuit (such as, CDR3) (Nobile et al., 2012). Additionally, each master biofilm transcriptional regulator likely responds to unique environmental inputs, such as oxygen and nutrient availability, pH, temperature, and waste products. How different environmental inputs influence the biofilm transcriptional circuit is an intriguing area of future research. For example, we know that the six master biofilm transcriptional regulators discovered using in vitro biofilm assays are still required for in vivo biofilm formation in at least one of two in vivo biofilm models (Nobile et al., 2012). The majority (four) of the master biofilm transcriptional regulators discovered in this study were essential for biofilm formation in both in vivo biofilm models used; however, two of the master biofilm transcriptional regulators played different roles depending on the in vivo biofilm model (Nobile et al., 2012). Specifically, Bcr1 was essential for biofilm formation in a rat catheter biofilm model but was dispensable in a rat denture biofilm model (Nobile et al., 2012). Similarly, Brg1 was essential for biofilm formation in a rat denture biofilm model but was dispensable in a rat catheter biofilm model (Nobile et al., 2012). Future work on these master transcriptional regulators will determine their unique influences on biofilm formation dependent on the environmental inputs present.
In a subsequent study, three additional transcriptional regulators, Gal4, Rfx2, and Flo8, were added to the core biofilm transcriptional circuit (Fox et al., 2015). Gal4, Rfx2, and Flo8 were found to directly bind to the upstream intergenic regions of one or more of the previously identified six master biofilm transcriptional regulators and vice versa during biofilm development (Nobile et al., 2012; Fox et al., 2015). Gal4, Rfx2, and Flo8 were identified (in addition to the six previously identified transcriptional regulators) by screening a TF mutant library containing 192 TF mutant strains (Fox et al., 2015). This TF library contained the same 165 TF mutants (Homann et al., 2009) from the Nobile et al., 2012 study (Nobile et al., 2012) plus 27 additional newly constructed TF mutant strains. The TF mutants in this larger library were screened for their abilities to form biofilms over time at 90 min, 8, 24, and 48 h of biofilm growth (Fox et al., 2015). Flo8, like the other six previously identified master biofilm transcriptional regulators, was required for biofilm formation throughout a 48-h course of biofilm growth, and thus was deemed to be a master biofilm transcriptional regulator; Gal4 and Rfx2 were only required for normal biofilm formation at specific intermediate time points (Fox et al., 2015). Given that the initial biofilm circuit consisting of six master transcriptional regulators was discovered by assessing biofilm formation at a single mature time point (48 h) (Nobile et al., 2012), performing the genetic screen as a biofilm develops over time, with the additional TF mutant strains, contributed to the expansion of the core biofilm circuit (Fox et al., 2015). Genome-wide binding data was not performed for Gal4, Rfx2, and Flo8 as part of this study; however, directed chromatin immunoprecipitation followed by quantitative PCR was performed to determine that these three new transcriptional regulators are integrated into the core biofilm circuit, which now consists of nine core transcriptional regulators, seven of which are considered to be master biofilm transcriptional regulators (Figure 2) (Nobile et al., 2012; Fox et al., 2015). We note that although genome-wide binding experiments have been performed for Gal4 and Flo8 (Askew et al., 2009; Polvi et al., 2019), these experiments were not performed under biofilm conditions and thus the resulting data cannot be integrated into the biofilm transcriptional circuit. Overall, although the logic of the biofilm transcriptional circuit (defined as how each transcriptional regulator contributes to the regulatory dynamics of the circuit) has yet to be fully elucidated, the high degree of interconnectivity between the core biofilm transcriptional regulators likely contributes to the robustness, yet reversibility, of the biofilm state.
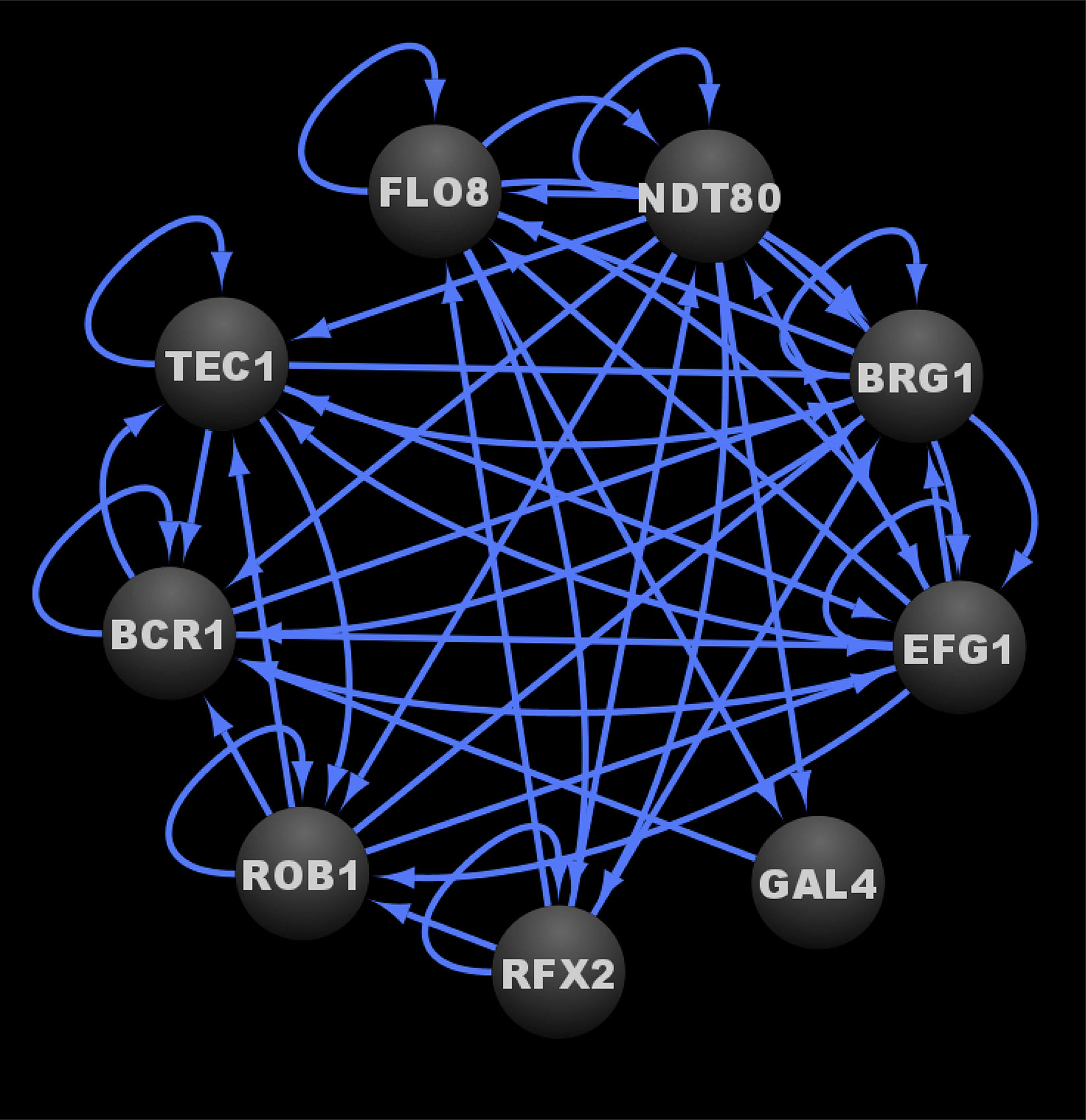
Figure 2 Transcriptional circuit controlling C. albicans biofilm formation. Ovals indicate each of the core biofilm transcriptional regulators with their respective names. Arrows indicate direct binding events. See Data Sheet S1, Tab4 for binding interactions. Data were derived from (Nobile et al., 2012; Fox et al., 2015). Figure was generated using Cytoscape (Shannon et al., 2003).
Although the nine core biofilm transcriptional regulators are known to be important for biofilm formation, how each one specifically contributes to biofilm processes (e.g. adhesion, filamentation, antifungal drug resistance, etc.), through detailed analyses of their mutant strains, has not been systematically determined. Table 1 summarizes the current knowledge of the roles of all known transcriptional regulators in known biofilm-related processes. Eight of the nine core biofilm transcriptional regulators (Bcr1, Brg1, Efg1, Flo8, Ndt80, Rfx2, Rob1, and Tec1) have been implicated in regulating filamentation (Schweizer et al., 2000; Bockmüh and Ernst, 2001; Cao et al., 2006; Elson et al., 2009; Hao et al., 2009; Sellam et al., 2010; Vandeputte et al., 2011; Du et al., 2012b; Nobile et al., 2012), which is a critical process necessary for maintaining the architectural stability of the biofilm structure. Four of the nine core biofilm transcriptional regulators (Bcr1, Efg1, Rfx2, and Tec1) have been implicated in regulating adhesion (Dieterich et al., 2002; Hao et al., 2009; Sahni et al., 2010; Finkel et al., 2012), including both cell-cell and cell-substrate adhesion, which is an essential process for both the initiation of biofilm formation as well as for the maintenance of a mature biofilm. Three of the nine core biofilm transcriptional regulators (Bcr1, Efg1, and Ndt80) are known to be involved in the regulation of antifungal drug resistance and/or tolerance (Chen et al., 2004; Sellam et al., 2009; Prasad et al., 2010; Desai et al., 2013), an important feature that contributes to the overall recalcitrance of established biofilms to antimicrobial compounds. Of the nine core biofilm transcriptional regulators, we know the least about the biofilm specific roles of Gal4, and only that it contributes to the structure of a biofilm at intermediate stages of biofilm development (Fox et al., 2015). In the future, additional roles of the nine core biofilm transcriptional regulators during biofilm formation will certainly be elucidated. For example, it seems likely that some of the core biofilm transcriptional regulators would be involved in the formation of the extracellular matrix; however, this role has not been examined to date in the mutant strains of the core biofilm transcriptional regulators. In addition, the ability of cells within biofilms to communicate with one another, called quorum sensing, is an important process for coordinating biofilm formation of many microorganisms; however, this role has yet to be examined in the mutant strains of the core biofilm transcriptional regulators. In fact, little is known in general on the regulation of quorum sensing during C. albicans biofilm development.
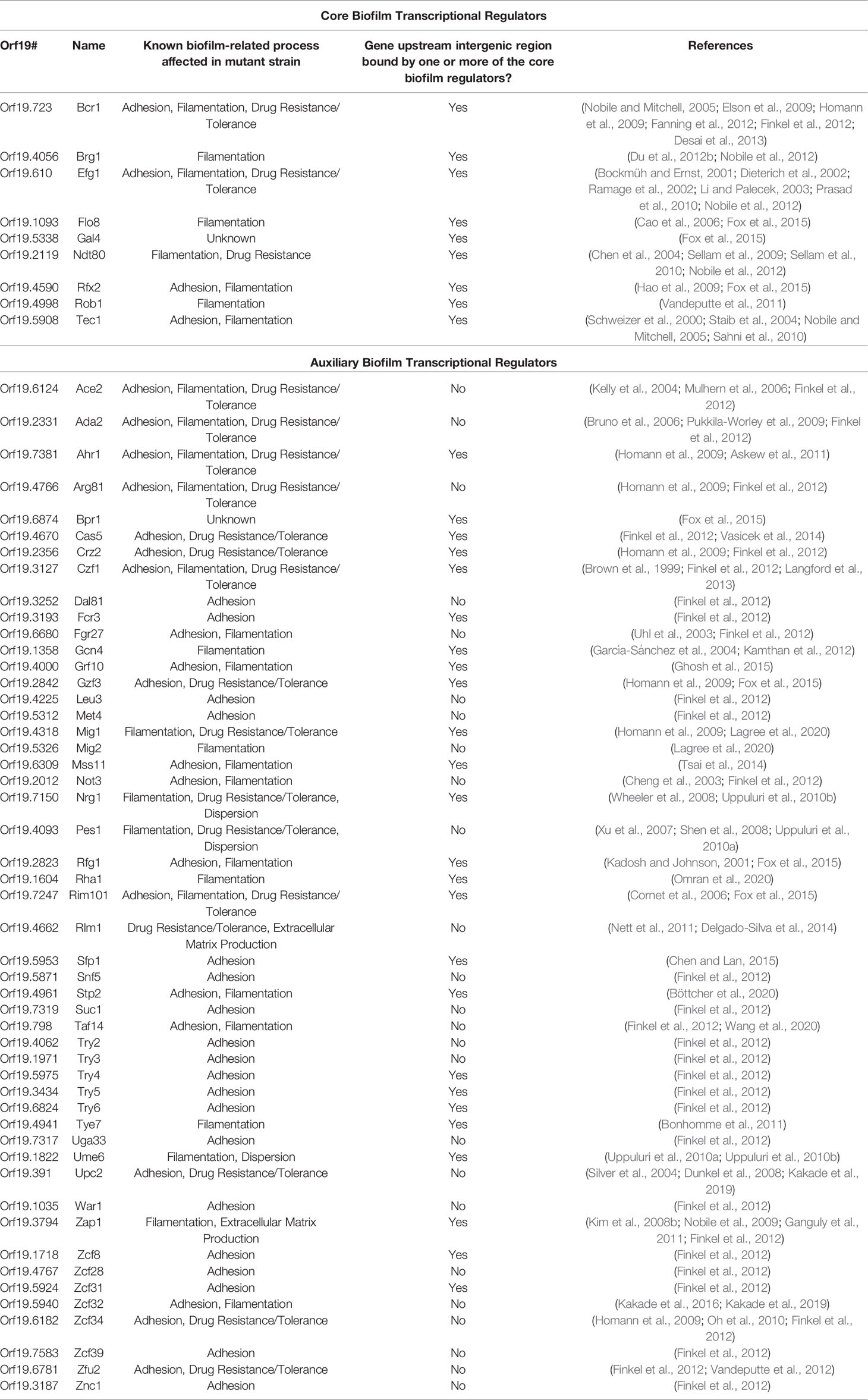
Table 1 Known transcriptional regulators with roles in C. albicans biofilm formation.
In addition to these nine transcriptional regulators that make up the core biofilm circuit, there are 50 “auxiliary” transcriptional regulators that have been implicated in biofilm formation (Table 1). The majority of these auxiliary biofilm transcriptional regulators are also bound in their upstream intergenic regions by at least one of the initial six master biofilm transcriptional regulators (Bcr1, Tec1, Efg1, Ndt80, Rob1, or Brg1; note that of the nine core biofilm transcriptional regulators, there is not genome-wide chromatin immunoprecipitation data available for Gal4, Rfx2, and Flo8, and thus we do not know whether they bind to the auxiliary biofilm transcriptional regulators) (Table 1) (Nobile et al., 2012). As such, several of the 50 auxiliary transcriptional regulators are integrated into the larger biofilm network that includes the core nine transcriptional regulators and all of their directly bound target genes (Nobile et al., 2012). Based on existing phenotypic analyses of the mutant strains of the auxiliary biofilm transcriptional regulators, the majority (48) are implicated in the regulation of adhesion and/or filamentation (Brown et al., 1999; Kadosh and Johnson, 2001; Cheng et al., 2003; Uhl et al., 2003; García-Sánchez et al., 2004; Kelly et al., 2004; Mulhern et al., 2006; Kim et al., 2008b; Shen et al., 2008; Wheeler et al., 2008; Homann et al., 2009; Nobile et al., 2009; Pukkila-Worley et al., 2009; Uppuluri et al., 2010a; Uppuluri et al., 2010b; Askew et al., 2011; Bonhomme et al., 2011; Ganguly et al., 2011; Finkel et al., 2012; Kamthan et al., 2012; Langford et al., 2013; Delgado-Silva et al., 2014; Tsai et al., 2014; Chen and Lan, 2015; Fox et al., 2015; Ghosh et al., 2015; Kakade et al., 2016; Kakade et al., 2019; Böttcher et al., 2020; Lagree et al., 2020; Omran et al., 2020; Wang et al., 2020); 16 are implicated in drug resistance and/or tolerance (Bruno et al., 2006; Cornet et al., 2006; Mulhern et al., 2006; Xu et al., 2007; Dunkel et al., 2008; Wheeler et al., 2008; Homann et al., 2009; Prasad et al., 2010; Nett et al., 2011; Vandeputte et al., 2012; Langford et al., 2013; Vasicek et al., 2014); two are implicated in the production of the extracellular matrix (Finkel et al., 2012; Delgado-Silva et al., 2014); and two are implicated in dispersion (Uppuluri et al., 2010b; Uppuluri et al., 2010a). Similar to the core biofilm transcriptional regulators, detailed analyses of the mutant strains of the auxiliary biofilm transcriptional regulators have not been systemically studied for known biofilm processes. Rather, most of their roles in biofilm processes have been determined through large-scale genetic screens. Of the auxiliary biofilm transcriptional regulators, we understand the least about the biofilm specific roles of Bpr1/Orf19.6874, which is only known to contribute to biofilm biomass throughout biofilm development (Fox et al., 2015). Future detailed phenotypic analyses of the auxiliary transcriptional regulator mutant strains in biofilm specific processes will certainly reveal new and additional roles for these transcriptional regulators in biofilm development.
The white-opaque switch in C. albicans is a form of phenotypic switching that gives rise to two distinct cell types called “white” and “opaque” that display distinct phenotypic characteristics at the single cell and colony levels (Anderson and Soll, 1987; Slutsky et al., 1987; Rikkerink et al., 1988; Bergen et al., 1990; Soll, 1992; Soll et al., 1993). White cells represent the standard budding yeast form of C. albicans, forming shiny, white, dome-shaped colonies on solid media plates, while opaque cells are larger and more elongated than white cells and form dull, off-white, flattened colonies on solid media plates (Slutsky et al., 1987; Soll et al., 1993; Lohse and Johnson, 2009; Noble et al., 2017). White and opaque cells differ in their virulence characteristics, metabolic preferences, mating competencies, interactions with the host innate immune system, and responses to environmental stimuli (Kolotila and Diamond, 1990; Lan et al., 2002; Lockhart et al., 2002; Miller and Johnson, 2002; Bennett et al., 2003; Geiger et al., 2004; Dumitru et al., 2007; Lohse and Johnson, 2008; Ramírez-Zavala et al., 2008; Huang et al., 2009; Huang et al., 2010; Lohse et al., 2013; Xie et al., 2013; Lohse et al., 2016; Du and Huang, 2016; Ene et al., 2016; Dalal et al., 2019). In total, nearly 20% of the transcriptome is differentially expressed, by at least twofold, between the two cell types, highlighting that the white-opaque switch involves major transcriptional rewiring (Tuch et al., 2010; Hernday et al., 2013). Under standard switch permissive growth conditions, switching between the white cell type, considered the “ground” state, and the opaque cell type, considered the “excited” state, occurs stochastically at a frequency of roughly one switch event per 1,000-10,000 cell divisions (Rikkerink et al., 1988; Bergen et al., 1990; Ramírez-Zavala et al., 2008; Alby and Bennett, 2009b). Each cell type is heritably maintained without any change to the primary sequence of the genome, thus fitting the classic definition of an epigenetic switch (Slutsky et al., 1987; Soll et al., 1993; Zordan et al., 2006; Zordan et al., 2007). The switch is responsive to the combined effects of environmental signals, such as carbon source, pH, CO2 levels, and temperature, which can differentially bias the cell population towards one of the two cell types (Dumitru et al., 2007; Ramírez-Zavala et al., 2008; Alby and Bennett, 2009a; Huang et al., 2009; Huang, 2012; Lohse et al., 2013; Du and Huang, 2016; Ene et al., 2016; Dalal et al., 2019). Mating type can also influence the ability of the cells to undergo white-opaque switching, where MTL heterozygous (a/α) cells are typically “locked” in the white state, while MTL hemizygous (a/Δ, α/Δ), homozygous (a/a, or α/α), and haploid (a or α) cells are capable of undergoing stochastic white-opaque switching (Hull and Johnson, 1999; Lockhart et al., 2002; Miller and Johnson, 2002). This mating type dependency, however, is not exclusive to all strains; in fact, a significant fraction of MTL heterozygous clinical isolates can be induced to form opaque cells under specific growth conditions that promote white to opaque switching in MTL hemizygous, homozygous, or haploid cells (Xie et al., 2013).
Through a combination of forward and reverse genetic approaches, a total of 112 transcriptional regulators and one protein binding cofactor (Ssn6) have been identified which, when deleted, significantly impact the frequency of white-opaque switching (Table 2) (Huang et al., 2006; Srikantha et al., 2006; Zordan et al., 2006; Zordan et al., 2007; Hernday et al., 2013; Lohse et al., 2013; Du et al., 2015; Hernday et al., 2016; Lohse et al., 2016; Lohse and Johnson, 2016). Of these 113 switch regulating proteins, eight (Wor1, Wor2, Wor3, Wor4, Czf1, Efg1, Ahr1, and Ssn6) are considered to be core switch regulators, and have been extensively characterized by genome-wide transcriptional profiling and chromatin immunoprecipitation approaches in white and opaque cell types; the remaining 105 switch regulating proteins are considered to be auxiliary switch regulators (Table 2) (Sonneborn et al., 1999; Srikantha et al., 2000; Huang et al., 2006; Zordan et al., 2006; Zordan et al., 2007; Vinces and Kumamoto, 2007; Lohse and Johnson, 2010; Wang et al., 2011; Hernday et al., 2013; Lohse et al., 2013; Lohse and Johnson, 2016; Hernday et al., 2016). Together, these eight core switch regulators form complex cell type specific networks, with 203 bound target genes in white cells and 756 bound target genes in opaque cells (Hernday et al., 2013; Lohse et al., 2013; Hernday et al., 2016; Lohse and Johnson, 2016). At the center of the white and opaque specific regulatory networks are two distinct transcriptional circuits (see Figure 3A for the white circuit, Figure 3B for the opaque circuit, and Figure 3C for the combined white and opaque overlayed circuits) that consist of interconnected positive and negative feedback loops that govern the cell fate and heritable maintenance of the white and opaque cell types (Vinces et al., 2006; Vinces and Kumamoto, 2007; Zordan et al., 2007; Hernday et al., 2013; Hernday et al., 2016; Lohse and Johnson, 2016). Although several groups have identified kinases, chromatin modifiers, and other proteins that also affect white-opaque switching (Hnisz et al., 2009; Noble et al., 2017; Rai et al., 2018); here, we focus on the eight core switch regulators (TFs: Wor1, Wor2, Wor3, Wor4, Czf1, Efg1, Ahr1; and cofactor: Ssn6) for which genome-wide transcriptional profiling and chromatin immunoprecipitation data are available.
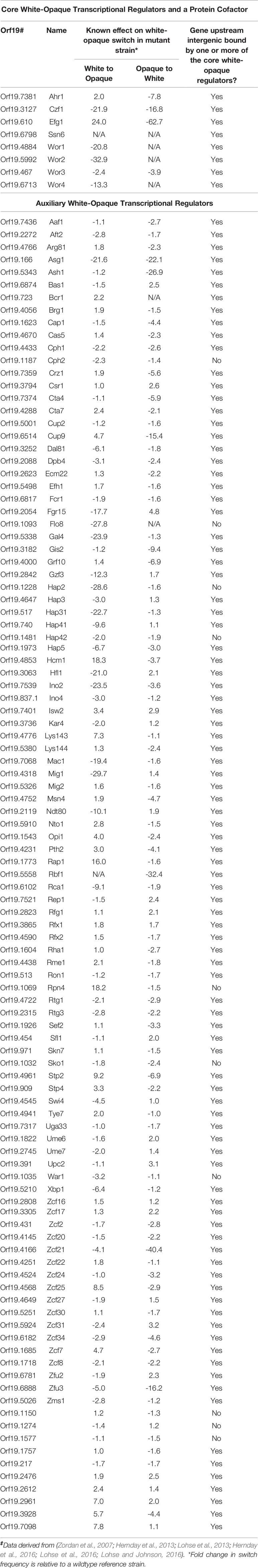
Table 2 Known transcriptional regulators and a protein cofactor with roles in the C. albicans white-opaque switch‡.
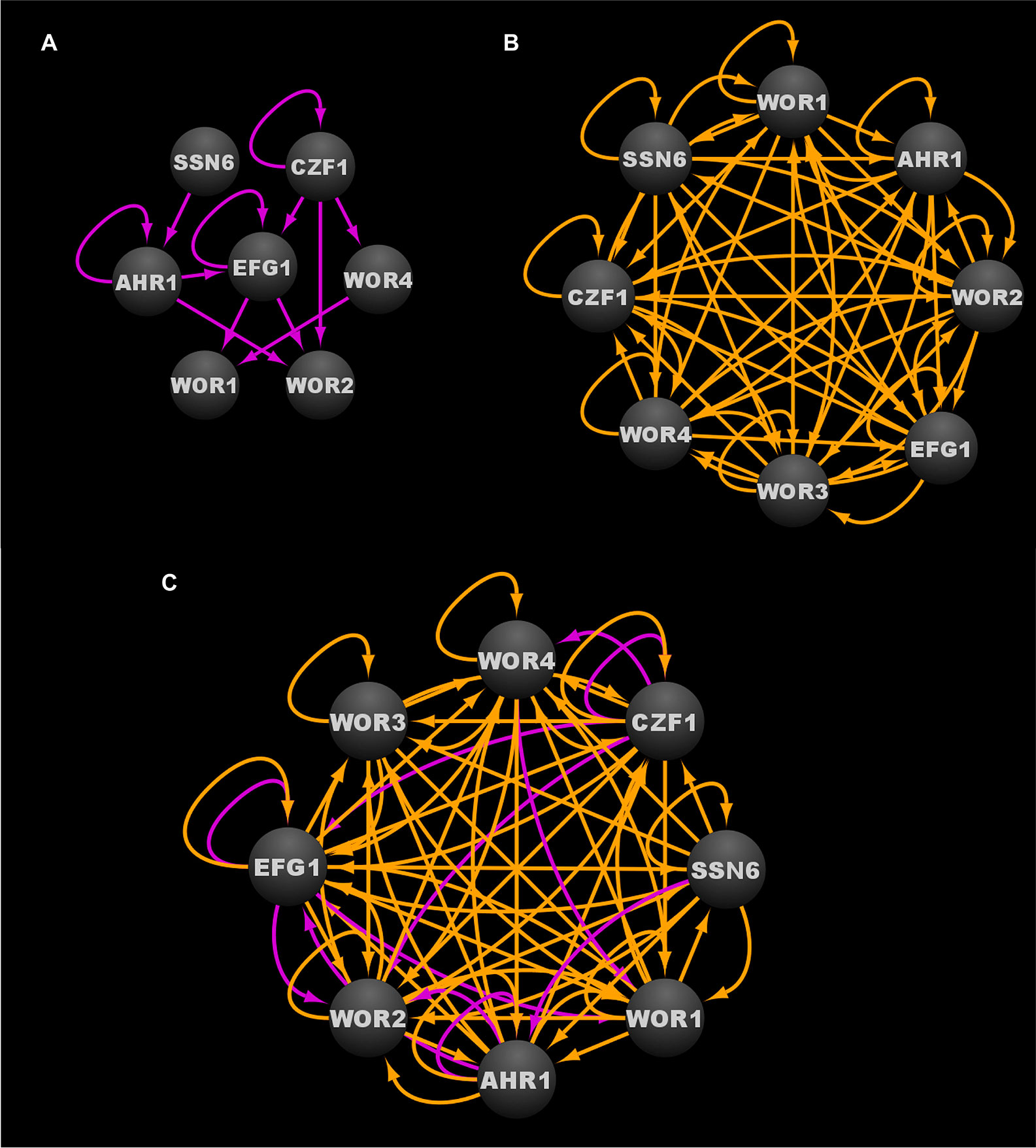
Figure 3 Transcriptional circuits controlling the C. albicans white-opaque phenotypic switch. (A) Transcriptional circuit of the white state. (B) Transcriptional circuit of the opaque state. (C) Overlayed transcriptional circuits regulating the white and opaque states. Ovals indicate each of the core regulators with their respective names. Arrows indicate direct binding events. See Data Sheet S1, Tab4 for binding interactions. Data were derived from (Zordan et al., 2007; Hernday et al., 2013; Lohse et al., 2013; Hernday et al., 2016; Lohse and Johnson, 2016). Figure was generated using Cytoscape (Shannon et al., 2003).
Wor1 is considered to be the master regulator of the white-opaque switch, as it is the only switch regulator that is known to be required for both the transition to, and heritable maintenance of, the opaque cell type (Huang et al., 2006; Srikantha et al., 2006; Zordan et al., 2006; Zordan et al., 2007; Hernday et al., 2013; Lohse et al., 2013; Hernday et al., 2016; Lohse and Johnson, 2016). Furthermore, ectopic WOR1 expression can rescue opaque cell formation in all known mutant backgrounds that fail to spontaneously switch to the opaque cell type (Zordan et al., 2007; Du et al., 2012a; Lohse and Johnson, 2016). WOR1 expression is repressed in white cells, where Wor1 protein levels have been found to be nearly undetectable (Huang et al., 2006; Srikantha et al., 2006; Zordan et al., 2006; Zordan et al., 2007; Lohse and Johnson, 2010). In opaque cells, WOR1 is highly transcribed, and Wor1 protein levels have been found to accumulate to elevated levels (Huang et al., 2006; Srikantha et al., 2006; Zordan et al., 2006; Zordan et al., 2007; Lohse and Johnson, 2010). Stochastic white to opaque switching is thought to be the result of transcriptional noise within the white cell circuit that occasionally allows Wor1 levels to surpass a critical threshold necessary to induce the transition to the opaque state (Srikantha et al., 2006; Hernday et al., 2010; Lohse and Johnson, 2010; Nobile et al., 2012; Guan and Liu, 2015; Horwitz et al., 2015; Lohse and Johnson, 2016; Lohse et al., 2016a; Tandonnet and Torres, 2017). Once established, the excited opaque cell circuit is stably maintained by a series of nested feedback loops, including a positive autoregulatory feedback loop generated by Wor1 binding to the upstream intergenic region of WOR1 (Zordan et al., 2007; Hernday et al., 2013). This Wor1-induced positive feedback loop, along with other opaque specific binding interactions between the white and opaque regulators and their respective upstream intergenic regions, is proposed to be a central mechanism that mediates the epigenetic heritability of the opaque cell type (Huang et al., 2006; Srikantha et al., 2006; Zordan et al., 2006; Zordan et al., 2007; Lohse and Johnson, 2010; Wang et al., 2011; Hernday et al., 2013; Lohse et al., 2013; Lohse and Johnson, 2016; Hernday et al., 2016). Stochastic opaque to white switching is believed to occur when transcriptional noise causes Wor1 levels to drop below a critical threshold, thus leading to a collapse of the excited opaque cell transcriptional program and a return to the ground white cell transcriptional program (Srikantha et al., 2006; Zordan et al., 2006; Lohse and Johnson, 2010).
The core transcriptional circuit in white cells consists of a series of feed-forward loops that ultimately repress the expression of WOR1 and WOR2, both of which are key players in the establishment and/or maintenance of the opaque cell type (Zordan et al., 2007). Efg1, Ahr1, and Ssn6 all contribute to the stability of the white cell circuit and are believed to directly or indirectly repress the expression of WOR1 and WOR2 (Zordan et al., 2007; Tuch et al., 2010; Hernday et al., 2013; Hernday et al., 2016). Deletion of EFG1, AHR1, or SSN6 destabilizes the white cell circuit such that most, if not all, of the cells in the population transition to the opaque state (Sonneborn et al., 1999; Srikantha et al., 2000; Vinces et al., 2006; Vinces and Kumamoto, 2007; Zordan et al., 2007; Wang et al., 2011; Hernday et al., 2016). Czf1, Wor3, and Wor4 are capable of destabilizing the white cell circuit, and induced expression of CZF1, WOR3, or WOR4 in white cells can promote white to opaque switching in a Wor1 dependent manner (Zordan et al., 2007; Hernday et al., 2013; Lohse et al., 2013; Lohse and Johnson, 2016). Interestingly, neither Czf1 nor Wor3 is required for the heritable maintenance of the opaque state once switching has occurred (Zordan et al., 2007; Lohse et al., 2013). Based on these results and the structure of the white cell regulatory circuit (Figure 3A), Czf1 and Wor4 are thought to destabilize the white cell type by directly and indirectly antagonizing the white cell stabilizing activities of Ssn6, Ahr1, and Efg1, and by inducing opaque promoting factors such as WOR3, thus introducing the transcriptional noise that leads to the stochastic activation of the WOR1 positive feedback loop and the transition to the opaque state. In addition to repression of WOR1 and WOR2, the white cell transcriptional program results in repression of opaque enriched transcripts (e.g. WOR3 and CZF1) as well as the activation of white enriched transcripts (e.g. EFG1), thus creating a series of feed-forward loops that act to stabilize the white cell circuit and prevent activation of the opaque state (Zordan et al., 2007; Hernday et al., 2013; Lohse et al., 2013).
In contrast to the core transcriptional circuit of the white cell type (Figure 3A), the core transcriptional circuit of the opaque cell type is extensively intertwined (Figure 3B). All of the core switch regulators are active in opaque cells, and they are each found to bind to their own upstream intergenic regions, along with the upstream intergenic regions of most, if not all, of the other core switch regulators (Figure 3B) (Huang et al., 2006; Srikantha et al., 2006; Zordan et al., 2006; Zordan et al., 2007; Wang et al., 2011; Hernday et al., 2013; Lohse et al., 2013; Hernday et al., 2016; Lohse and Johnson, 2016). To highlight this point, 58 of the 64 possible binding interactions between the core switch regulators and their respective upstream intergenic regions are observed in opaque cells (Data Sheet S1, Tab1). Although the logic of the opaque transcriptional circuit has yet to be fully elucidated, the high degree of interconnectivity between the core opaque regulators likely contributes to the robustness, yet reversibility, of the opaque cell state. Similar to the white cell circuit, Wor1 is a critical player in the opaque cell circuit; however, it is the sustained high levels of WOR1 expression, rather than its repression, that is required for the formation and stable maintenance of the opaque cell type (Huang et al., 2006; Srikantha et al., 2006; Zordan et al., 2006). Although not strictly required for the formation of an opaque cell, Wor2 and Wor4 also play important roles in the heritable maintenance of the opaque transcriptional program (Zordan et al., 2007; Lohse and Johnson, 2016). Strains lacking WOR2 or WOR4 are locked in the white cell type and fail to undergo spontaneous white to opaque switching, yet can be induced to form opaque cells by ectopic expression of WOR1 (Frazer et al., 2020). These induced opaque cells, however, are unstable, and quickly revert to the white cell type when ectopic WOR1 expression is repressed, indicating that Wor2 and Wor4 play essential roles in the heritability of opaque cells (Zordan et al., 2007). Interestingly, with the exception of Ahr1, all switch regulators discovered to date have been found to contain prion-like domains that enable liquid-liquid demixing and the formation of phase-separated condensates (Frazer et al., 2020). Several of the switch regulators, including Wor1 and Wor4, have been shown to undergo phase separation in vitro, and to form condensates at genomic loci in vivo, in a manner similar to the formation of mammalian super-enhancers (Frazer et al., 2020). Combined with the observation that many of the target genes bound by the switch regulators are flanked by unusually large upstream intergenic regions (Zordan et al., 2007; Hernday et al., 2013), and the discovery that specific residues within the Wor1 prion-like domain are required for condensate formation and white to opaque switching, it seems likely that these phase-separated condensates formed by the core switch regulators in opaque cells are critical factors that contribute to the formation and heritable maintenance of the opaque cell type.
C. albicans typically exists as a commensal member of the healthy human microbiota. It can also transition into a pathogen in response to specific host environmental cues. In its pathogenic state, C. albicans can cause a wide range of infections, from acute to chronic superficial mucosal infections to severe and life-threatening disseminated bloodstream infections (Wenzel, 1995; Hube, 2004; Pappas et al., 2004). Although immunocompetent individuals with healthy and balanced microbiota are typically not adversely affected by C. albicans, immunocompromised individuals can suffer severe infections with significant morbidity and mortality (Wenzel, 1995; Nobile and Johnson, 2015). Understanding the genetic regulatory mechanisms that control the C. albicans commensal-pathogen transition has the potential to lead to the development of targeted therapeutic strategies against C. albicans in its pathogenic state, without affecting its commensal state and the delicate balance of the microbiota.
Two distinct C. albicans transcriptional networks controlling the commensal-pathogen transition were described in 2011 and 2013, one governing iron homeostasis, and the other governing proliferation in the host, respectively (see Figure 4A for the iron homeostasis circuit, Figure 4B for the proliferation in the host circuit, and Figure 4C for the combined commensal-pathogen overlayed circuits) (Chen et al., 2011; Pérez et al., 2013). As a commensal of the gastrointestinal (GI) tract, C. albicans is exposed to varying and often abundant levels of iron from food, and thus a tightly regulated transcriptional response is important for C. albicans to control iron assimilation and to avoid iron toxicity in the GI tract (McCance and Widdowson, 1938; Martin et al., 1987; Miret et al., 2003). On the other hand, when C. albicans causes a disseminated bloodstream infection, iron is extremely limiting, and to survive, C. albicans must conserve and scavenge iron from the bloodstream. Three transcriptional regulators, Sef1, Sfu1, and Hap43, were found to form a tightly knit transcriptional network, encompassing 214 downstream target genes (Chen et al., 2011). These three transcriptional regulators control iron homeostasis and were found to be essential for C. albicans to survive as both a commensal and as a pathogen within the mammalian host (Chen et al., 2011). Iron homeostasis in many other fungi (such as in other ascomycetes and the basidiomycete, Cryptococcus neoformans) is commonly regulated by a bipartite regulatory circuit composed of orthologs of Sfu1 and Hap43, where Sfu1 orthologs repress iron acquisition genes and HAP43 orthologs, while Hap43 orthologs repress nonessential iron utilization genes and SFU1 orthologs. This mutually repressive regulatory interaction between orthologs of Sfu1 and Hap43 in other fungi is significantly altered in C. albicans by the intercalation of Sef1 as a third player within this circuit (Figure 4A) (Chen et al., 2011). In C. albicans, Sfu1 directly represses SEF1 and iron acquisition genes under iron replete conditions (Chen et al., 2011). In response to iron limitation, Sef1 serves to directly activate HAP43 and iron uptake genes, while Hap43 directly represses SFU1 and iron utilization genes (Chen et al., 2011). Although the roles for Hap43 in C. albicans are similar to those of other fungi, the reciprocal interaction between Sfu1 and HAP43 is altered in C. albicans by the inclusion of Sef1, which serves as an intermediary between Sfu1 and HAP43. C. albicans SEF1 and SFU1 are differentially expressed between growth in the GI tract versus growth in the bloodstream (Chen et al., 2011), thus providing dual inputs into the circuit controlling iron acquisition and utilization. While both Sef1 and Sfu1 serve to promote commensalism in a mouse GI commensal model, only Sef1 is required for virulence in a mouse disseminated infection model (Chen et al., 2011). Interestingly, deletion of SFU1 conferred a significant competitive advantage over wildtype cells in the disseminated infection model (Chen et al., 2011), indicating that Sfu1 serves not only to promote commensalism in the GI tract, but also to attenuate virulence in the bloodstream. (See Table 3 for information on these three core transcriptional regulators in the commensal-pathogen transition.) Ultimately the C. albicans iron homeostasis circuit produces a well conserved transcriptional output consisting of increased iron uptake and reduced iron utilization in iron limited environments, and decreased iron uptake and increased iron utilization in iron replete conditions. Despite being well conserved in its transcriptional output, the iron homeostasis circuit appears to be uniquely evolved in C. albicans to control the delicate balance between its commensal and pathogenic growth states.
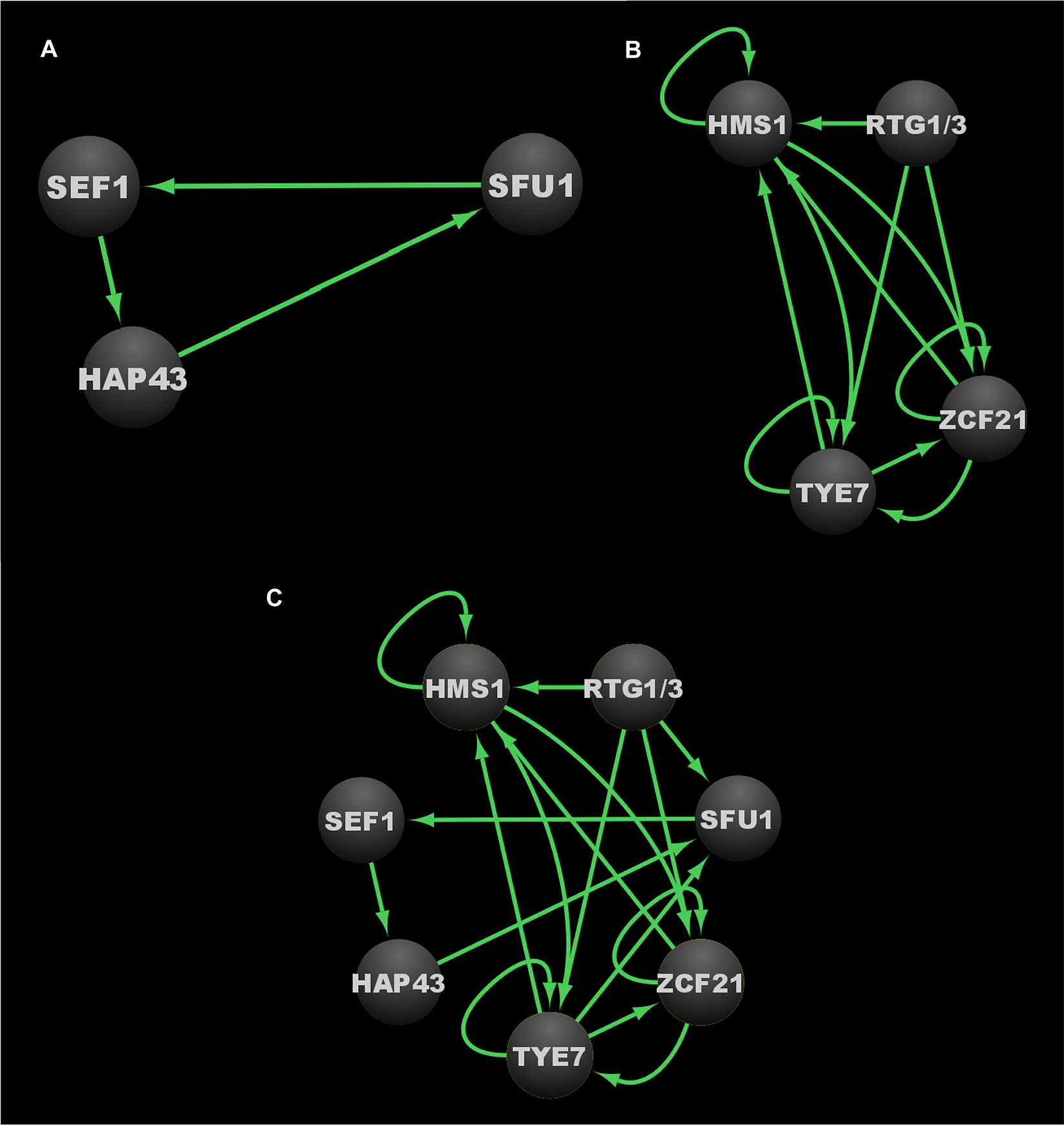
Figure 4 Transcriptional circuits controlling the C. albicans commensal-pathogen transition. (A) Transcriptional circuit controlling iron homeostasis. (B) Transcriptional circuit controlling proliferation in the host. (C) Overlayed transcriptional circuits controlling the commensal-pathogen transition. Ovals indicate each of the core regulators with their respective names. Arrows indicate direct binding events. Note that since Rtg1 and Rtg3 function as a heterodimer, and do not appear to bind DNA independently, they are represented as a single node in these regulatory circuit diagrams. See Data Sheet S1, Tab4 for binding interactions. Data were derived from (Chen et al., 2011; Pérez et al., 2013). Figure was generated using Cytoscape (Shannon et al., 2003).
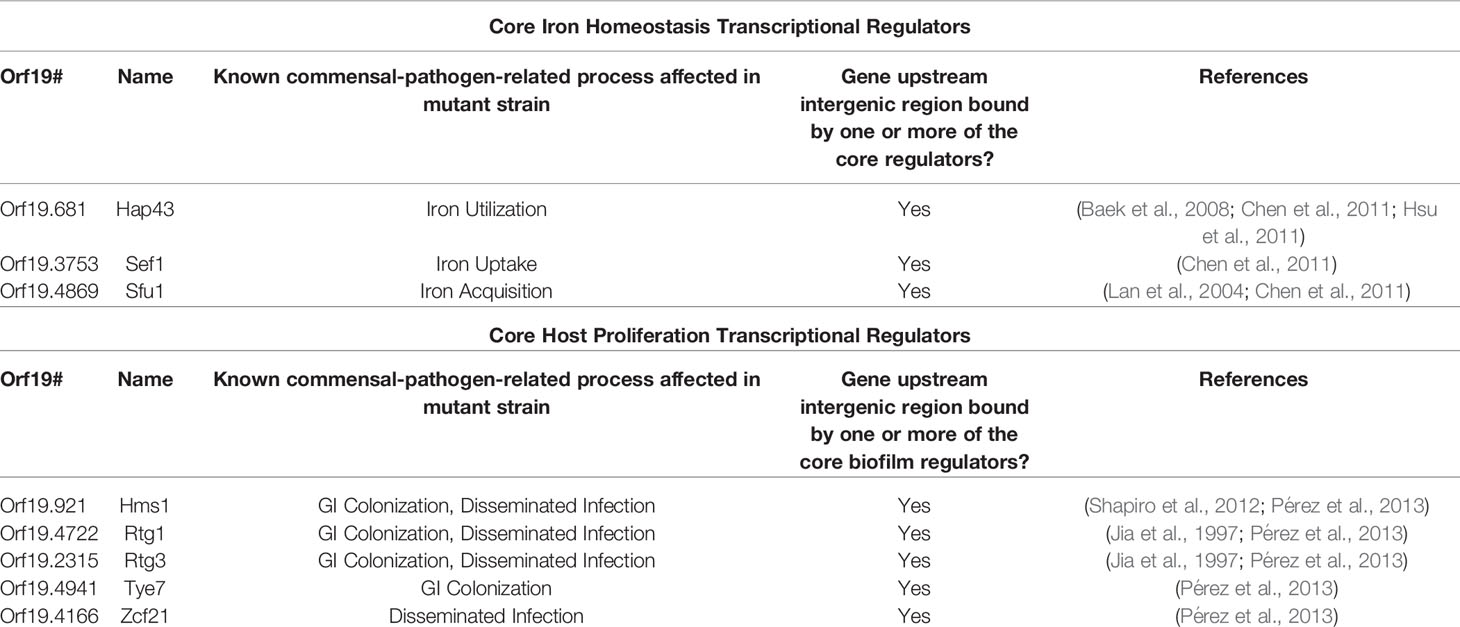
Table 3 Known transcriptional regulators with roles in the C. albicans commensal-pathogen transition.
A subsequent study identified eight transcriptional regulators (Tye7, Orf19.3625, Lys144, Zcf21, Lys14, Hsm1, Rtg1, and Rtg3) that influence C. albicans proliferation in the commensal and/or pathogenic growth states (Pérez et al., 2013). These regulators were identified by screening a subset of the commonly used C. albicans TF mutant library (Homann et al., 2009) for defects in a commensal (GI colonization) mouse model and a pathogenic (disseminated infection) mouse model. This subset of the TF mutant library consisted of those mutant strains that revealed no phenotypes in a diverse panel of in vitro growth conditions, and was screened to identify transcriptional regulators that were specifically required for normal (wildtype) levels of growth in either of the two mouse models (Homann et al., 2009; Pérez et al., 2013). Of the eight regulators that were identified, six (Rtg1, Rtg3, Tye7, Hms1, Orf19.3625, and Lys144) were required for GI colonization, while five (Rtg1, Rtg3, Hms1, Lys14, and Zcf21) were required for robust growth in the disseminated infection model (Pérez et al., 2013). Overall, Tye7, Orf19.3625, and Lys144 were found to be specific to commensal colonization of the GI tract; Zcf21 and Lys14 were found to be specific to disseminated infections; and Rtg1, Rtg3, and Hms1 were found to be associated generally with growth in the host (Pérez et al., 2013). Based on genome-wide transcriptional profiling and chromatin immunoprecipitation data, seven of these regulators (Tye7, Lys144, Zcf21, Lys14, Hsm1, Rtg1, and Rtg3) were found to form a transcriptional network consisting of 808 directly bound target genes. Significant overlap was observed between the bound target genes of this network and those genes that were upregulated in the mouse GI model compared to growth in vitro. Orf19.3625 was excluded from this analysis as it is a predicted subunit of a histone remodeling complex, and thus was not considered to be a specific regulator within the commensal-pathogen network. In contrast to the transcriptional network defined by Sef1, Sfu1, and Hap43, which is primarily responsible for regulating genes involved in iron homeostasis (Chen et al., 2011), the transcriptional network defined by Tye7, Lys144, Zcf21, Lys14, Hsm1, Rtg1, and Rtg3 appears to primarily regulate genes involved in the acquisition and metabolism of carbon and nitrogen, as well as genes that encode transporters and cell surface proteins (Pérez et al., 2013). The binding profiles for Rtg1 and Rtg3 were observed to be identical, and thus they likely function as a heterodimer to bind DNA (Pérez et al., 2013), which is consistent with their orthologs in Saccharomyces cerevisiae (Liu and Butow, 2006). Of the 153 direct target genes in this network that are upregulated during GI colonization and disseminated infection, 108 of them are bound by the Rtg1/3 heterodimer (Pérez et al., 2013), highlighting the central role that Rtg1/3 plays in this network. We note that a subsequent study by the same group identified five transcriptional regulators that influence fitness in an oropharyngeal candidiasis model (Cup9, Zcf8, Zcf21, Zcf27, and Orf19.217), and identified a set of genes that are differentially regulated in response to deletion of CUP9 (Meir et al., 2018). We did not include these data in our analyses since binding experiments that would be necessary to integrate these additional regulators into the commensal-pathogen transcriptional circuit have not been performed.
At the core of this commensal-pathogen transcriptional network lies a tightly interwoven regulatory circuit defined by the binding interactions between five of these transcriptional regulators (Hms1, Zcf21, Tye7, Rtg1, and Rtg3) and their respective upstream intergenic regions (Figure 4B). While Lys14 and Lys144 are clearly important for pathogenic and commensal growth, respectively, they are not integrated into the core transcriptional circuit and instead appear to function as auxiliary regulators. Interestingly, RTG1 and RTG3 are not regulated at the transcriptional level in response to growth in the GI tract and are not direct targets of any of the members of this commensal-pathogen transcriptional circuit (Pérez et al., 2013). Instead, Rtg1/3 seems to function as a major regulatory input into, rather than target of, this commensal-pathogen circuit. In S. cerevisiae, the Rtg1/3 heterodimer is known to be post-translationally modified and translocated into the nucleus in response to growth on poor nitrogen sources or mitochondrial dysfunction, suggesting that nitrogen assimilation and metabolic adaptation could be critical factors for the proliferation of C. albicans in the host (Liao and Butow, 1993; Jia et al., 1997; Liu and Butow, 2006). Hms1, which is also required for both commensal and pathogenic growth in the host, is known to be activated in response to elevated temperatures (Shapiro et al., 2012), indicating that temperature, along with nitrogen source(s), represent two critical environmental signals that influence the commensal and pathogenic growth programs of C. albicans. Zcf21 represses a variety of genes that encode key virulence factors, and plays a major role in pathogenesis by balancing the positive effects of these virulence factors during disseminated infection against the increased susceptibility to host immune system recognition and clearance that is correlated with their expression (Böhm et al., 2016). Finally, Tye7 has been implicated in the metabolism of carbohydrates, such as oligosaccharides and polysaccharides, as well as in the regulation of hyphal growth and biofilm formation (Askew et al., 2009; Bonhomme et al., 2011). (See Table 3 for information on these five core transcriptional regulators in the commensal-pathogen transition.) Although both the iron homeostasis and the host proliferation transcriptional networks are critical to the ability of C. albicans to grow as a commensal and as a pathogen, there is limited interconnectivity between these networks at the level of the core regulators of each circuit (Figure 4C). SFU1 serves as the sole point of integration between the two circuits, being bound by Rtg1/3 and Tye7. There are no binding interactions observed between the iron homeostasis regulators (Sef1, Sfu1, and Hap43) and the genes encoding the host proliferation regulators, suggesting that under certain growth conditions which alter the binding of Rtg1/3 and/or Tye7, the iron homeostasis circuit may function as a sub-circuit of the host proliferation circuit. Together, the transcriptional regulators involved in iron homeostasis and acquisition, and host proliferation, confer C. albicans with the ability to proliferate in different niches of the host as well as to transition between commensal and pathogenic states in response to changes in the host environment.
In total, the three larger regulatory networks, consisting of the core regulators and all of their directly bound target genes involved in biofilm formation, the white-opaque phenotypic switch, and the commensal-pathogen transition in C. albicans encompass at least 1657 directly bound individual target genes, making up a little over 25% of genes in the entire genome (note that Flo8, Gal4, and Rfx2 were excluded from this analysis since there is not genome-wide chromatin immunoprecipitation data available for them) (Data Sheet S1, Tab2) (Zordan et al., 2007; Chen et al., 2011; Nobile et al., 2012; Hernday et al., 2013; Lohse et al., 2013; Pérez et al., 2013; Fox et al., 2015; Hernday et al., 2016; Lohse and Johnson, 2016). These three networks are highly intertwined, with 40% (667/1657) of the target genes shared between at least two of the networks, and 11% (188/1657) of the target genes shared between all three networks (Data Sheet S1, Tab2). This high degree of interconnectivity is even more pronounced at the level of the core transcriptional circuits that control these three networks, as is evident by the extensive binding interactions present between the core regulators themselves (Figure 5 and Data Sheet S1, Tab1). Together, the twenty transcriptional regulators for which we have genome-wide chromatin immunoprecipitation data available form a total of 225 binding interactions within and between their core circuits, distributed roughly evenly between intra-circuit (49%) and inter-circuit (51%) interactions (note that the Rtg1/3 heterodimer is counted as a single regulator since neither subunit is known to bind independently) (Data Sheet S1, Tab3). The commensal-pathogen circuit and the biofilm circuit are highly intertwined with the regulators in the other circuits, with 66% and 59% inter-circuit interactions, respectively, while the opaque cell circuit appears to be much more isolated, with the majority (64%) of its interactions being intra-circuit (Data Sheet S1, Tab3). Perhaps the most striking example of integration between the circuits is exemplified by Ndt80 in the biofilm circuit, which binds to the upstream intergenic regions of 22 out of 24 of the core regulators (all but the upstream intergenic regions of RTG1 and RTG3) (Data Sheet S1, Tab1). The percentage of inter-circuit binding events is highest for Tye7 (79%), Zcf21 (75%), Bcr1 (71%), Brg1 (67%), and Rtg1/3 (67%), accounting for at least two out of three binding events for each of these regulators within the three core circuits (Data Sheet S1, Tab3). At the opposite end of the spectrum, Hap43, Hms1, and Sfu1 are exclusive to the commensal-pathogen circuit. In addition, at least two thirds of the binding events observed for Wor3 (88%), Czf1 (75%), Rob1 (71%), Ahr1 (70%), and Wor4 (70%) within the three core circuits occur within their respective core circuits (Data Sheet S1, Tab3). Interestingly, the degree of Efg1 inter-circuit interaction is unique to the circuit within which it lies, where 61% inter-circuit interactions are observed for Efg1 in the biofilm circuit, while only 42% inter-circuit interactions are observed for Efg1 in the white-opaque circuit (Data Sheet S1, Tab3). BRG1 is the most highly integrated target within the three circuits, where it is bound by seventeen of the twenty core regulators evaluated (leaving out Gal4, Rfx2, and Flo8, and considering Rtg1 and Rtg3 as a single regulator) (Data Sheet S1, Tab1). Overall, more than half (thirteen out of twenty-four) of the regulators that make up the three core circuits are bound by at least half (eleven or more) of the twenty core regulators evaluated (Data Sheet S1, Tab1). These rather striking numbers highlight the degree to which these circuits are intertwined, and these numbers are only likely to increase as additional core regulators are identified and incorporated into the three transcriptional circuits.
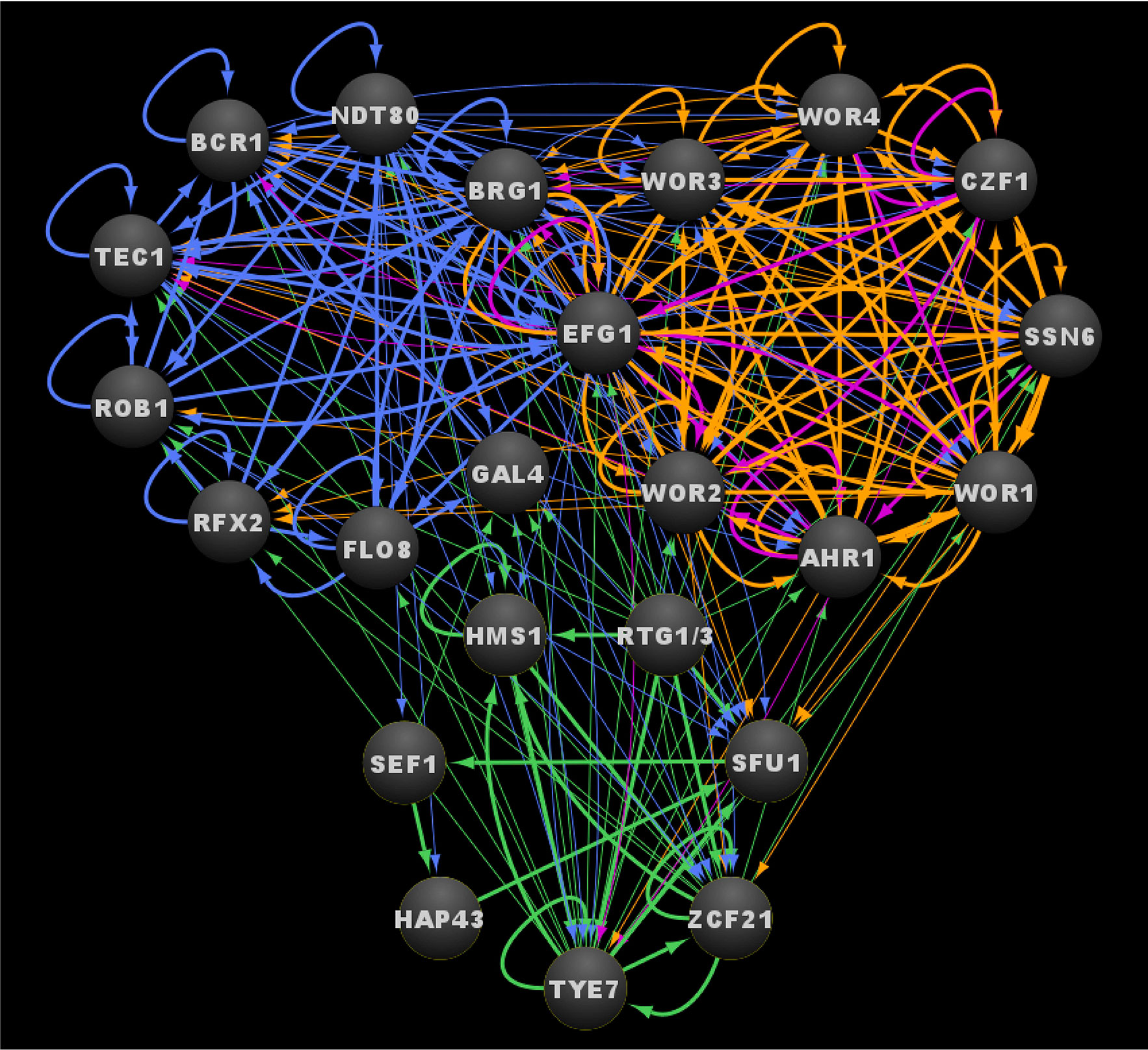
Figure 5 Integrated transcriptional circuits of C. albicans biofilm formation, the white-opaque switch and the commensal-pathogen transition. Ovals indicate each of the core regulators with their respective names. Arrows indicate direct binding events. See Data Sheet S1, Tab4 for binding interactions. Data were derived from (Zordan et al., 2007; Chen et al., 2011; Nobile et al., 2012; Hernday et al., 2013; Lohse et al., 2013; Pérez et al., 2013; Fox et al., 2015; Hernday et al., 2016; Lohse and Johnson, 2016). Figure was generated using Cytoscape (Shannon et al., 2003).
The extensive integration between these core transcriptional circuits appears to have significant functional relevance. For example, 14 of the 24 regulator genes discussed (AHR1, BCR1, BRG1, CZF1, GAL4, HAP43, HMS1, RFX2, SEF1, SFU1, TEC1, WOR1, WOR3, ZCF21) are differentially expressed by at least twofold between planktonic and biofilm growth conditions; of these fourteen genes, all but GAL4 are upregulated in biofilms (Data Sheet S1, Tab1) (Nobile et al., 2012). A similar trend is observed during white-opaque switching, where eleven of the twenty-four regulator genes (BRG1, CZF1, EFG1, GAL4, HMS1, RFX2, ROB1, TYE7, WOR1, WOR2, WOR3) are differentially expressed by at least twofold between white and opaque cell types (Data Sheet S1, Tab1) (Tuch et al., 2010). The interactions between the biofilm circuit and the white-opaque circuit are particularly striking. All eight of the core white-opaque regulator genes are bound by at least four of the six core biofilm regulators, and six of the eight white-opaque regulator genes (all but EFG1 and WOR4) are differentially expressed by twofold or more between planktonic and biofilm conditions (WOR1, AHR1, CZF1, and WOR3 are upregulated by 3-, 5-, 8-, and 32-fold, respectively, while WOR2 and SSN6 are both downregulated by 2-fold) (Data Sheet S1, Tab1). Conversely, five of the nine core biofilm regulator genes are bound by at least four of the eight white-opaque regulators in opaque cells (EFG1, BRG1, BCR1, TEC1, and RFX2 are bound by eight, eight, five, five, and four white-opaque regulators, respectively), and five of the nine biofilm regulator genes are differentially expressed by at least 2-fold between white and opaque cells (BRG1 and RFX2 are upregulated in opaque cells, while EFG1, GAL4, and ROB1 are upregulated in white cells) (Data Sheet S1, Tab1). The commensal-pathogen circuit regulators are closely intertwined with the biofilm circuit; however, there is relatively little overlap between the overlayed white-opaque circuit and the overlayed commensal-pathogen circuit. Six of the eight commensal-pathogen regulator genes (all but RTG1 and RTG3) are bound by at least one biofilm core regulator, half of which (SFU1, TYE7, and ZCF21) are bound by at least four of the biofilm regulators (Data Sheet S1, Tab1). All six of the commensal-pathogen regulator genes that are bound by biofilm regulators are differentially expressed by twofold or more between planktonic and biofilm conditions, with all but TYE7 being upregulated in biofilms (Data Sheet S1, Tab1). In contrast to the high degree of functional interaction between the biofilm circuit and the overlayed commensal-pathogen circuit, only three of the eight commensal-pathogen regulator genes (SFU1, TYE7, and ZCF21) are bound by any of the white-opaque regulators, and of the three target genes, only TYE7 is differentially expressed between white and opaque cells (upregulated twentyfold in opaque cells). The effect of growth under conditions relevant to the overlayed commensal-pathogen circuit (i.e. low iron or growth in the GI tract) is relatively limited when compared to the effects of biofilm formation and white-opaque switching. Upon growth in low iron, only the three regulator genes involved in iron homeostasis (HAP43, SEF1, SFU1) are differentially expressed (Data Sheet S1, Tab1) (Chen et al., 2011). While growth in the GI tract does affect the expression of core regulator genes in the other circuits, the impact of this expression is relatively limited, where AHR1 and TEC1 are upregulated and ROB1 is downregulated in the GI tract versus growth in vitro (Rosenbach et al., 2010).
The C. albicans transcriptional regulatory circuits controlling the developmental processes of biofilm formation, the white-opaque phenotypic switch, and the commensal-pathogen transition are individually tightly knit and we show that they are integrated together by extensive regulatory crosstalk between the core regulators that comprise each circuit. If we take into consideration all of the target genes in each of the larger transcriptional networks, each regulator controls individual subsets of target genes regulating distinct functions as well as subsets of target genes with functions in common with the other core regulators in each network. Strikingly, these three major transcriptional networks, together, encompass a little over 25% of genes in the entire genome, indicating that there is a high degree of functional redundancy across the networks. The complexity and functional redundancy of these network structures often make dissecting the logic of each network extremely challenging. The networks we discuss here in this review are overall structurally very similar to networks controlling complex transcriptional developmental processes in higher eukaryotes, such as the mammalian embryonic stem cell state (pluripotency) network (Boyer et al., 2005; Kim et al., 2008a). Given that mammals and C. albicans diverged from a common ancestor around 1.5 million years ago (Wang et al., 1999), it is notable that the structures of these independently evolved transcriptional networks are so similar. There are a couple hypotheses as to how these transcriptional networks could appear so structurally similar (Sorrells and Johnson, 2015). The first hypothesis is that these complex transcriptional networks represent the optimal solutions for organizing the biological processes they control (François and Hakim, 2004; Prill et al., 2005). The second hypothesis is that these transcriptional networks are not optimal solutions but are rather non-adaptive structures that have been retained over evolutionary time scales by purifying selection and are thus the result of high-probability evolutionary trajectories (Sorrells and Johnson, 2015). As we begin to discover and deconvolute complex transcriptional networks, we will begin to test these hypotheses and shed new light on the logic of these complex network structures.
Conceptualization: DR, MQ, AH, and CN. Formal Analysis: DR, MQ, AH, and CN. Investigation: DR, MQ, AH, and CN. Resources: AH, and CN. Data Curation: DR, MQ, AH, and CN. Writing—Original Draft Preparation: DR, MQ, AH, and CN. Writing—Review and Editing: DR, MQ, AH, and CN. Visualization: DR, AH, and CN. Supervision: AH and CN. Project Administration: AH and CN. Funding Acquisition: AH and CN. All authors contributed to the article and approved the submitted version.
This work was supported by the National Institutes of Health (NIH) National Institute of Allergy and Infectious Diseases (NIAID) and National Institute of General Medical Sciences (NIGMS) awards R21AI125801 and R35GM124594, respectively, by a Pew Biomedical Scholar Award from the Pew Charitable Trusts, and by the Kamangar family in the form of an endowed chair to CN. This work was also supported by NIH NIAID award R15AI37975 to AH. DR was supported by the National Science Foundation (NSF) Graduate Research Fellowship Program (GRFP) award 1744620. The funders had no role in the study design, data collection and interpretation, or the decision to submit the work for publication.
CN is a cofounder of BioSynesis, Inc., a company developing inhibitors and diagnostics of biofilms.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The reviewer AS declared a past co-authorship with one of the authors AH to the handling editor.
We thank all members of the Nobile and Hernday labs for insightful discussions on the topic of this manuscript.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fcimb.2020.605711/full#supplementary-material
Supplementary Data Sheet 1 | Compilation and analysis of regulator binding interactions and target gene expression. Tab1 labeled “Tab1_Combined Core Circuits” contains compiled genome-wide chromatin immunoprecipitation (ChIP-seq or ChIP-chip) and expression profiling (RNA-seq or microarray) data for the core circuit regulators and their respective target genes. ChIP data and RNA-seq data values are in log2 format. Biofilm regulators ChIP data and differential gene expression data were derived from (Nobile et al., 2012). White-opaque regulators ChIP data were derived from (Zordan et al., 2007; Hernday et al., 2013; Lohse et al., 2013; Hernday et al., 2016; Lohse and Johnson, 2016). White-opaque differential gene expression data were derived from (Tuch et al., 2010). Iron homeostasis regulators ChIP data and differential gene expression data were derived from (Chen et al., 2011). Host proliferation ChIP data were derived from (Pérez et al., 2013). Host proliferation differential gene expression data were derived from (Rosenbach et al., 2010). Tab2 labeled “Tab2_Combined Networks” contains compiled genome-wide chromatin immunoprecipitation (ChIP-seq or ChIP-chip) and expression profiling (RNAseq or microarray) data for the core circuit regulators and all possible target genes in the C. albicans genome. ChIP data and RNA-seq data values are in log2 format. Biofilm regulators ChIP data and differential gene expression data were derived from (Nobile et al., 2012). White-opaque regulators ChIP data were derived from (Zordan et al., 2007; Hernday et al., 2013; Lohse et al., 2013; Hernday et al., 2016; Lohse and Johnson, 2016). White-opaque differential gene expression data were derived from (Tuch et al., 2010). Iron homeostasis regulators ChIP data and differential gene expression data were derived from (Chen et al., 2011). Host proliferation ChIP data were derived from (Pérez et al., 2013). Host proliferation differential gene expression data were derived from (Rosenbach et al., 2010). Tab3 labeled “Tab3_Inter- vs Intra-circuit” contains an analysis of the genome-wide ChIP data from Tab1, tabulating the total number of bound targets for each of the regulators for which genome-wide binding data is available, and calculating the percentage of binding events that represent inter- versus intra-circuit binding interactions. Tab4 labeled “Tab4_Cytoscape Interactions” contains a representation of the genome-wide ChIP data from Tab1 in an interaction table format for visualization in Cytoscape (Shannon et al., 2003). This dataset was used to generate all of the circuit diagrams shown in Figures 2–5.
Achkar J. M., Fries B. C. (2010). Candida infections of the genitourinary tract. Clin. Microbiol. Rev. 23, 253–273. doi: 10.1128/CMR.00076-09
Alby K., Bennett R. J. (2009a). Stress-induced phenotypic switching in Candida albicans. Mol. Biol. Cell 20, 3178–3191. doi: 10.1091/mbc.e09-01-0040
Alby K., Bennett R. J. (2009b). To switch or not to switch?: Phenotypic switching is sensitive to multiple inputs in a pathogenic fungus. Commun. Integr. Biol. 2, 509–511. doi: 10.4161/cib.2.6.9487
Anderson J. M., Soll D. R. (1987). Unique phenotype of opaque cells in the white-opaque transition of Candida albicans. J. Bacteriol. 169, 5579–5588. doi: 10.1128/JB.169.12.5579-5588.1987
Askew C., Sellam A., Epp E., Hogues H., Mullick A., Nantel A., et al. (2009). Transcriptional regulation of carbohydrate metabolism in the human pathogen Candida albicans. PloS Pathog. 5, e1000612. doi: 10.1371/journal.ppat.1000612
Askew C., Sellam A., Epp E., Mallick J., Hogues H., Mullick A., et al. (2011). The zinc cluster transcription factor Ahr1p directs Mcm1p regulation of Candida albicans adhesion. Mol. Microbiol. 79, 940–953. doi: 10.1111/j.1365-2958.2010.07504.x
Baek Y. U., Li M., Davis D. A. (2008). Candida albicans ferric reductases are differentially regulated in response to distinct forms of iron limitation by the Rim101 and CBF transcription factors. Eukaryot. Cell 7, 1168–1179. doi: 10.1128/EC.00108-08
Bennett R. J., Uhl M. A., Miller M. G., Johnson A. D. (2003). Identification and characterization of a Candida albicans mating pheromone. Mol. Cell. Biol. 23, 8189–8201. doi: 10.1128/MCB.23.22.8189-8201.2003
Bergen M. S., Voss E., Soll D. R. (1990). Switching at the cellular level in the white-opaque transition of Candida albicans. J. Gen. Microbiol. 136, 1925–1936. doi: 10.1099/00221287-136-10-1925
Bockmüh D. P., Ernst J. F. (2001). A potential phosphorylation site for an A-Type kinase in the Efg1 regulator protein contributes to hyphal morphogenesis of Candida albicans. Genetics 157, 1523–1530.
Böhm L., Muralidhara P., Pérez J. C. (2016). A Candida albicans regulator of disseminated infection operates primarily as a repressor and governs cell surface remodeling. Mol. Microbiol. 100, 328–344. doi: 10.1111/mmi.13320
Bonhomme J., Chauvel M., Goyard S., Roux P., Rossignol T., D’Enfert C. (2011). Contribution of the glycolytic flux and hypoxia adaptation to efficient biofilm formation by Candida albicans. Mol. Microbiol. 80, 995–1013. doi: 10.1111/j.1365-2958.2011.07626.x
Böttcher B., Hoffmann B., Garbe E., Weise T., Cseresnyés Z., Brandt P., et al. (2020). The transcription factor Stp2 is important for Candida albicans biofilm establishment and sustainability. Front. Microbiol. 11, 794. doi: 10.3389/fmicb.2020.00794
Boyer L. A., Tong I. L., Cole M. F., Johnstone S. E., Levine S. S., Zucker J. P., et al. (2005). Core transcriptional regulatory circuitry in human embryonic stem cells. Cell 122, 947–956. doi: 10.1016/j.cell.2005.08.020
Brown D. H., Giusani A. D., Chen X., Kumamoto C. A. (1999). Filamentous growth of Candida albicans in response to physical environmental cues and its regulation by the unique CZF1 gene. Mol. Microbiol. 34, 651–662. doi: 10.1046/j.1365-2958.1999.01619.x
Bruno V. M., Kalachikov S., Subaran R., Nobile C. J., Kyratsous C., Mitchell A. P. (2006). Control of the C. albicans cell wall damage response by transcriptional regulator Cas5. PloS Pathog. 2, e21. doi: 10.1371/journal.ppat.0020021
Calderone R. A., Fonzi W. A. (2001). Virulence factors of Candida albicans. Trends Microbiol. 9, 327–335. doi: 10.1016/S0966-842X(01)02094-7
Cao F., Lane S., Raniga P. P., Lu Y., Zhou Z., Ramon K., et al. (2006). The Flo8 transcription factor is essential for hyphal development and virulence in Candida albicans. Mol. Biol. Cell 17, 295–307. doi: 10.1091/mbc.e05-06-0502
Chandra J., Kuhn D. M., Mukherjee P. K., Hoyer L. L., McCormick T., Ghannoum M. A. (2001). Biofilm formation by the fungal pathogen Candida albicans: development, architecture, and drug resistance. J. Bacteriol. 183, 5385–5394. doi: 10.1128/JB.183.18.5385-5394.2001
Chen C. G., Yang Y. L., Shih H. II, Su C. L., Lo H. J. (2004). CaNdt80 is involved in drug resistance in Candida albicans by regulating CDR1. Antimicrob. Agents Chemother. 48, 4505–4512. doi: 10.1128/AAC.48.12.4505-4512.2004
Chen C., Pande K., French S. D., Tuch B. B., Noble S. M. (2011). An iron homeostasis regulatory circuit with reciprocal roles in Candida albicans commensalism and pathogenesis. Cell Host Microbe 10, 118–135. doi: 10.1016/j.chom.2011.07.005
Chen H. F., Lan C. Y. (2015). Role of SFP1 in the regulation of Candida albicans biofilm formation. PloS One 10, e0129903. doi: 10.1371/journal.pone.0129903
Cheng S., Nguyen M. H., Zhang Z., Jia H., Handfield M., Clancy C. J. (2003). Evaluation of the roles of four Candida albicans genes in virulence by using gene disruption strains that express URA3 from the native locus. Infect. Immun. 71, 6101–6103. doi: 10.1128/IAI.71.10.6101-6103.2003
Cornet M., Gaillardin C., Richard M. L. (2006). Deletions of the endocytic components VPS28 and VPS32 in Candida albicans lead to echinocandin and azole hypersensitivity. Antimicrob. Agents Chemother. 50, 3492–3495. doi: 10.1128/AAC.00391-06
Dalal C. K., Zuleta I. A., Lohse M. B., Zordan R. E., El-Samad H., Johnson A. D. (2019). A population shift between two heritable cell types of the pathogen Candida albicans is based both on switching and selective proliferation. Proc. Natl. Acad. Sci. U.S.A. 116, 26918–26924. doi: 10.1073/pnas.1908986116
Davey M. E., O’toole G. A. (2000). Microbial biofilms: from ecology to molecular genetics. Microbiol. Mol. Biol. Rev. 64, 847–867. doi: 10.1128/MMBR.64.4.847-867.2000
Delgado-Silva Y., Vaz C., Carvalho-Pereira J., Carneiro C., Nogueira E., Correia A., et al. (2014). Participation of Candida albicans transcription factor RLM1 in cell wall biogenesis and virulence. PloS One 9, e86270. doi: 10.1371/journal.pone.0086270
Desai J. V., Mitchell A. P. (2015). Candida albicans biofilm development and its genetic control. Microbiol. Spectr. 5, 99–114. doi: 10.1128/microbiolspec.MB-0005-2014
Desai J. V., Bruno V. M., Ganguly S., Stamper R. J., Mitchell K. F., Solis N., et al. (2013). Regulatory role of glycerol in Candida albicans biofilm formation. mBio 4, 637–649. doi: 10.1128/mBio.00637-12
Desai N., Ardekani A. M. (2020). Biofilms at interfaces: microbial distribution in floating films. Soft Matter 16, 1731–1750. doi: 10.1039/C9SM02038A
Dieterich C., Schandar M., Noll M., Johannes F. J., Brunner H., Graeve T., et al. (2002). In vitro reconstructed human epithelia reveal contributions of Candida albicans EFG1 and CPH1 to adhesion and invasion. Microbiology 148, 497–506. doi: 10.1099/00221287-148-2-497
Du H., Guan G., Xie J., Cottier F., Sun Y., Jia W., et al. (2012a). The transcription factor Flo8 mediates CO2 sensing in the human fungal pathogen Candida albicans. Mol. Biol. Cell 23, 2692–2701. doi: 10.1091/mbc.e12-02-0094
Du H., Guan G., Xie J., Sun Y., Tong Y., Zhang L., et al. (2012b). Roles of Candida albicans Gat2, a GATA-Type zinc finger transcription factor in biofilm formation, filamentous growth and virulence. PloS One 7, e29707. doi: 10.1371/journal.pone.0029707
Du H., Li X., Huang G., Kang Y., Zhu L. (2015). The zinc-finger transcription factor, Ofi1, regulates white-opaque switching and filamentation in the yeast Candida albicans. Acta Biochim. Biophys. Sin. 47, 335–341. doi: 10.1093/abbs/gmv011
Du H., Huang G. (2016). Environmental pH adaption and morphological transitions in Candida albicans. Curr. Genet. 62, 283–286. doi: 10.1007/s00294-015-0540-8
Dumitru R., Navarathna D. H. M. L. P., Semighini C. P., Elowsky C. G., Dumitru R. V., Dignard D., et al. (2007). In vivo and in vitro anaerobic mating in Candida albicans. Eukaryot. Cell 6, 465–472. doi: 10.1128/EC.00316-06
Dunkel N., Liu T. T., Barker K. S., Homayouni R., Morschhäuser J., Rogers P. D. (2008). A gain-of-function mutation in the transcription factor Upc2p causes upregulation of ergosterol biosynthesis genes and increased fluconazole resistance in a clinical Candida albicans isolate. Eukaryot. Cell 7, 1180–1190. doi: 10.1128/EC.00103-08
Edmond M. B., Wallace S. E., McClish D. K., Pfaller M. A., Jones R. N., Wenzel R. P. (1999). Nosocomial bloodstream infections in United States hospitals: a three-year analysis. Clin. Infect. Dis. 29, 239–244. doi: 10.1086/520192
Elson S. L., Noble S. M., Solis N. V., Filler S. G., Johnson A. D. (2009). An RNA transport system in Candida albicans regulates hyphal morphology and invasive growth. PloS Genet. 5, e1000664. doi: 10.1371/journal.pgen.1000664
Ene I. V., Lohse M. B., Vladu A. V., Morschhäuser J., Johnson A. D., Bennett R. J. (2016). Phenotypic profiling reveals that Candida albicans opaque cells represent a metabolically specialized cell state compared to default white cells. mBio 7, e01269–16. doi: 10.1128/mBio.01269-16
Fanning S., Xu W., Solis N., Woolford C. A., Filler S. G., Mitchell A. P. (2012). Divergent targets of Candida albicans biofilm regulator Bcr1 in vitro and in vivo. Eukaryot. Cell 11, 896–904. doi: 10.1128/EC.00103-12
Finkel J. S., Xu W., Huang D., Hill E. M., Desai J. V., Woolford C. A., et al. (2012). Portrait of Candida albicans adherence regulators. PloS Pathog. 8, e1002525. doi: 10.1371/journal.ppat.1002525
Fox E. P., Bui C. K., Nett J. E., Hartooni N., Mui M. C., Andes D. R., et al. (2015). An expanded regulatory network temporally controls Candida albicans biofilm formation. Mol. Microbiol. 96, 1226–1239. doi: 10.1111/mmi.13002
Fox E. P., Nobile C. J. (2012). A sticky situation: untangling the transcriptional network controlling biofilm development in Candida albicans. Transcription 3, 315–322. doi: 10.4161/trns.22281
François P., Hakim V. (2004). Design of genetic networks with specified functions by evolution in silico. Proc. Natl. Acad. Sci. U.S.A. 101, 580–585. doi: 10.1073/pnas.0304532101
Frazer C., Staples M. II, Kim Y., Hirakawa M., Dowell M. A., Johnson N. V., et al. (2020). Epigenetic cell fate in Candida albicans is controlled by transcription factor condensates acting at super-enhancer-like elements. Nat. Microbiol. 5, 1374–1389 doi: 10.1038/s41564-020-0760-7
Ganguly S., Bishop A. C., Xu W., Ghosh S., Nickerson K. W., Lanni F., et al. (2011). Zap1 control of cell-cell signaling in Candida albicans biofilms. Eukaryot. Cell 10, 1448–1454. doi: 10.1128/EC.05196-11
García-Sánchez S., Aubert S., Iraqui I., Janbon G., Ghigo J. M., D’Enfert C. (2004). Candida albicans biofilms: A developmental state associated with specific and stable gene expression patterns. Eukaryot. Cell 3, 536–545. doi: 10.1128/EC.3.2.536-545.2004
Geiger J., Wessels D., Lockhart S. R., Soll D. R. (2004). Release of a potent polymorphonuclear leukocyte chemoattractant is regulated by white-opaque switching in Candida albicans. Infect. Immun. 72, 667–677. doi: 10.1128/IAI.72.2.667-677.2004
Ghosh A. K., Wangsanut T., Fonzi W. A., Rolfes R. J. (2015). The GRF10 homeobox gene regulates filamentous growth in the human fungal pathogen Candida albicans. FEMS Yeast Res. 15, fov093. doi: 10.1093/femsyr/fov093
Guan Z., Liu H. (2015). The WOR1 5’ untranslated region regulates white-opaque switching in Candida albicans by reducing translational efficiency. Mol. Microbiol. 97, 125–138. doi: 10.1111/mmi.13014
Gulati M., Nobile C. J. (2016). Candida albicans biofilms: development, regulation, and molecular mechanisms. Microbes Infect. 18, 310–321. doi: 10.1016/j.micinf.2016.01.002
Hao B., Clancy C. J., Cheng S., Raman S. B., Iczkowski K. A., Nguyen M. H. (2009). Candida albicans RFX2 encodes a DNA binding protein involved in DNA damage responses, morphogenesis, and virulence. Eukaryot. Cell 8, 627–639. doi: 10.1128/EC.00246-08
Hernday A. D., Noble S. M., Mitrovich Q. M., Johnson A. D. (2010). Genetics and molecular biology in Candida albicans. Methods Enzymol. 470, 737–758. doi: 10.1016/S0076-6879(10)70031-8
Hernday A. D., Lohse M. B., Fordyce P. M., Nobile C. J., DeRisi J. L., Johnson A. D. (2013). Structure of the transcriptional network controlling white-opaque switching in Candida albicans. Mol. Microbiol. 90, 22–35. doi: 10.1111/mmi.12329
Hernday A. D., Lohse M. B., Nobile C. J., Noiman L., Laksana C. N., Johnson A. D. (2016). Ssn6 Defines a new level of regulation of white-opaque switching in Candida albicans and is required for the stochasticity of the switch. mBio 7, e01565–e01515. doi: 10.1128/mBio.01565-15
Hnisz D., Sehwarzmüller T., Kuchler K. (2009). Transcriptional loops meet chromatin: a dual-layer network controls white-opaque switching in Candida albicans. Mol. Microbiol. 74, 1–15. doi: 10.1111/j.1365-2958.2009.06772.x
Homann O. R., Dea J., Noble S. M., Johnson A. D. (2009). A phenotypic profile of the Candida albicans regulatory network. PloS Genet. 5, e1000783. doi: 10.1371/journal.pgen.1000783
Horwitz A. A., Walter J. M., Schubert M. G., Kung S. H., Hawkins K., Platt D. M., et al. (2015). Efficient multiplexed integration of synergistic alleles and metabolic pathways in yeasts via CRISPR-Cas. Cell Syst. 1, 88–96. doi: 10.1016/j.cels.2015.02.001
Hsu P. C., Yang C. Y., Lan C. Y. (2011). Candida albicans Hap43 is a repressor induced under low-iron conditions and is essential for iron-responsive transcriptional regulation and virulence. Eukaryot. Cell 10, 207–225. doi: 10.1128/EC.00158-10
Huang G., Wang H., Chou S., Nie X., Chen J., Liu H. (2006). Bistable expression of WOR1, a master regulator of white-opaque switching in Candida albicans. Proc. Natl. Acad. Sci. 103, 12813–12818. doi: 10.1073/pnas.0605270103
Huang G., Srikantha T., Sahni N., Yi S., Soll D. R. (2009). CO2 regulates white-to-opaque switching in Candida albicans. Curr. Biol. 19, 330–334. doi: 10.1016/j.cub.2009.01.018
Huang G., Yi S., Sahni N., Daniels K. J., Srikantha T., Soll D. R. (2010). N-acetylglucosamine induces white to opaque switching, a mating prerequisite in Candida albicans. PloS Pathog. 6, e1000806. doi: 10.1371/journal.ppat.1000806
Huang G. (2012). Regulation of phenotypic transitions in the fungal pathogen Candida albicans. Virulence 3, 251–261. doi: 10.4161/viru.20010
Hube B. (2004). From commensal to pathogen: stage- and tissue-specific gene expression of Candida albicans. Curr. Opin. Microbiol. 7, 336–341. doi: 10.1016/j.mib.2004.06.003
Hull C. M., Johnson A. D. (1999). Identification of a mating type-like locus in the asexual pathogenic yeast Candida albicans. Science 285, 1271–1275. doi: 10.1126/science.285.5431.1271
Jia Y., Rothermel B., Thornton J., Butow R. A. (1997). A basic helix-loop-helix-leucine zipper transcription complex in yeast functions in a signaling pathway from mitochondria to the nucleus. Mol. Cell. Biol. 17, 1110–1117. doi: 10.1128/MCB.17.3.1110
Kadosh D., Johnson A. D. (2001). Rfg1, a protein related to the Saccharomyces cerevisiae hypoxic regulator Rox1, controls filamentous growth and virulence in Candida albicans. Mol. Cell. Biol. 21, 2496–2505. doi: 10.1128/MCB.21.7.2496-2505.2001
Kakade P., Sadhale P., Sanyal K., Nagaraja V. (2016). ZCF32, a fungus specific Zn(II)2 Cys6 transcription factor, is a repressor of the biofilm development in the human pathogen Candida albicans. Sci. Rep. 6, 1–15. doi: 10.1038/srep31124
Kakade P., Mahadik K., Balaji K. N., Sanyal K., Nagaraja V. (2019). Two negative regulators of biofilm development exhibit functional divergence in conferring virulence potential to Candida albicans. FEMS Yeast Res. 19, foy078. doi: 10.1093/femsyr/foy078
Kamthan M., Mukhopadhyay G., Chakraborty N., Chakraborty S., Datta A. (2012). Quantitative proteomics and metabolomics approaches to demonstrate N-acetyl-d-glucosamine inducible amino acid deprivation response as morphological switch in Candida albicans. Fungal Genet. Biol. 49, 369–378. doi: 10.1016/j.fgb.2012.02.006
Kan S., Pang Q., Song N., Mei H., Zheng H., Li D., et al. (2020). Study on vulvovaginal candidiasis: clinical epidemiology and in vitro susceptibility of pathogenic yeasts in China. SSRN. doi: 10.2139/ssrn.3521422
Kelly M. T., MacCallum D. M., Clancy S. D., Odds F. C., Brown A. J. P., Butler G. (2004). The Candida albicans CaACE2 gene affects morphogenesis, adherence and virulence. Mol. Microbiol. 53, 969–983. doi: 10.1111/j.1365-2958.2004.04185.x
Kennedy M. J., Volz P. A. (1985). Ecology of Candida albicans gut colonization: inhibition of Candida adhesion, colonization, and dissemination from the gastrointestinal tract by bacterial antagonism. Infect. Immun. 49, 654–663. doi: 10.1128/IAI.49.3.654-663.1985
Kim J., Chu J., Shen X., Wang J., Orkin S. H. (2008a). An extended transcriptional network for pluripotency of embryonic stem cells. Cell 132, 1049–1061. doi: 10.1016/j.cell.2008.02.039
Kim M. J., Kil M., Jung J. H., Kim J. (2008b). Roles of zinc-responsive transcription factor Csr1 in filamentous growth of the pathogenic yeast Candida albicans. J. Microbiol. Biotechnol. 18, 242–247.
Kolotila M. P., Diamond R. D. (1990). Effects of neutrophils and in vitro oxidants on survival and phenotypic switching of Candida albicans WO-1. Infect. Immun. 58, 1174–1179. doi: 10.1128/IAI.58.5.1174-1179.1990
Kolter R., Greenberg E. P. (2006). Microbial sciences: the superficial life of microbes. Nature 441, 300–302. doi: 10.1038/441300a
Kumamoto C. A. (2002). Candida biofilms. Curr. Opin. Microbiol. 5, 608–611. doi: 10.1016/S1369-5274(02)00371-5
Kumamoto C. A. (2011). Inflammation and gastrointestinal Candida colonization. Curr. Opin. Microbiol. 14, 386–391. doi: 10.1016/j.mib.2011.07.015
Lagree K., Woolford C. A., Huang M. Y., May G., McManus C. J., Solis N. V., et al. (2020). Roles of Candida albicans Mig1 and Mig2 in glucose repression, pathogenicity traits, and SNF1 essentiality. PloS Genet. 16, e1008582. doi: 10.1371/journal.pgen.1008582
Lan C. Y., Newport G., Murillo L. A., Jones T., Scherer S., Davis R. W., et al. (2002). Metabolic specialization associated with phenotypic switching in Candida albicans. Proc. Natl. Acad. Sci. U.S.A. 99, 14907–14912. doi: 10.1073/pnas.232566499
Lan C. Y., Rodarte G., Murillo L. A., Jones T., Davis R. W., Dungan J., et al. (2004). Regulatory networks affected by iron availability in Candida albicans. Mol. Microbiol. 53, 1451–1469. doi: 10.1111/j.1365-2958.2004.04214.x
Langford M. L., Hargarten J. C., Patefield K. D., Marta E., Blankenship J. R., Fanning S., et al. (2013). Candida albicans Czf1 and Efg1 coordinate the response to farnesol during quorum sensing, white-opaque thermal dimorphism, and cell death. Eukaryot. Cell 12, 1281–1292. doi: 10.1128/EC.00311-12
Li F., Palecek S. P. (2003). EAP1, a Candida albicans gene involved in binding human epithelial cells. Eukaryot. Cell 2, 1266–1273. doi: 10.1128/EC.2.6.1266-1273.2003
Liao X., Butow R. A. (1993). RTG1 and RTG2: two yeast genes required for a novel path of communication from mitochondria to the nucleus. Cell 72, 61–71. doi: 10.1016/0092-8674(93)90050-Z
Liu Z., Butow R. A. (2006). Mitochondrial retrograde signaling. Annu. Rev. Genet. 40, 159–185. doi: 10.1146/annurev.genet.40.110405.090613
Lockhart S. R., Pujol C., Daniels K. J., Miller M. G., Johnson A. D., Pfaller M. A., et al. (2002). In Candida albicans, white-opaque switchers are homozygous for mating type. Genetics 162, 737–745.
Lohse M. B., Hernday A. D., Fordyce P. M., Noiman L., Sorrells T. R., Hanson-Smith V., et al. (2013). Identification and characterization of a previously undescribed family of sequence-specific DNA-binding domains. Proc. Natl. Acad. Sci. U.S.A. 110, 7660–7665. doi: 10.1073/pnas.1221734110
Lohse M. B., Ene I. V., Craik V. B., Hernday A. D., Mancera E., Morschhäuser J., et al. (2016). Systematic genetic screen for transcriptional regulators of the Candida albicans white-opaque switch. Genetics 203, 1679–1692. doi: 10.1534/genetics.116.190645
Lohse M. B., Johnson A. D. (2008). Differential phagocytosis of white versus opaque Candida albicans by Drosophila and mouse phagocytes. PloS One 3, e1473. doi: 10.1371/journal.pone.0001473
Lohse M. B., Johnson A. D. (2009). White-opaque switching in Candida albicans. Curr. Opin. Microbiol. 12, 650–654. doi: 10.1016/j.mib.2009.09.010
Lohse M. B., Johnson A. D. (2010). Temporal anatomy of an epigenetic switch in cell programming: the white-opaque transition of C. albicans. Mol. Microbiol. 78, 331–343. doi: 10.1111/j.1365-2958.2010.07331.x
Lohse M. B., Johnson A. D. (2016). Identification and characterization of Wor4, a new transcriptional regulator of white-opaque switching. G3 (Bethesda Md.) 6, 721–729. doi: 10.1534/g3.115.024885
Martin R. B., Savory J., Brown S., Bertholf R. L., Wills M. R. (1987). Transferrin binding of Al3+ and Fe3+. Clin. Chem. 33, 405–407. doi: 10.1093/clinchem/33.3.405
Mayer F. L., Wilson D., Hube B. (2013). Candida albicans pathogenicity mechanisms. Virulence 4, 119–128. doi: 10.4161/viru.22913
McCance R. A., Widdowson E. M. (1938). The absorption and excretion of iron following oral and intravenous administration. J. Physiol. 94, 148–154. doi: 10.1113/jphysiol.1938.sp003669
Meir J., Hartmann E., Eckstein M. T., Guiducci E., Kirchner F., Rosenwald A., et al. (2018). Identification of Candida albicans regulatory genes governing mucosal infection. Cell. Microbiol. 20, e12841. doi: 10.1111/cmi.12841
Miller M. G., Johnson A. D. (2002). White-opaque switching in Candida albicans is controlled by mating-type locus homeodomain proteins and allows efficient mating. Cell 110, 293–302. doi: 10.1016/S0092-8674(02)00837-1
Miret S., Simpson R. J., McKie A. T. (2003). Physiology and molecular biology of dietary iron absorption. Annu. Rev. Nutr. 23, 283–301. doi: 10.1146/annurev.nutr.23.011702.073139
Mulhern S. M., Logue M. E., Butler G. (2006). Candida albicans transcription factor Ace2 regulates metabolism and is required for filamentation in hypoxic conditions. Eukaryot. Cell 5, 2001–2013. doi: 10.1128/EC.00155-06
Nett J. E., Sanchez H., Cain M. T., Ross K. M., Andes D. R. (2011). Interface of Candida albicans biofilm matrix-associated drug resistance and cell wall integrity regulation. Eukaryot. Cell 10, 1660–1669. doi: 10.1128/EC.05126-11
Nobile C. J., Nett J. E., Hernday A. D., Homann O. R., Deneault J. S., Nantel A., et al. (2009). Biofilm matrix regulation by Candida albicans Zap1. PloS Biol. 7, e1000133. doi: 10.1371/journal.pbio.1000133
Nobile C. J., Fox E. P., Nett J. E., Sorrells T. R., Mitrovich Q. M., Hernday A. D., et al. (2012). A recently evolved transcriptional network controls biofilm development in Candida albicans. Cell 148, 126–138. doi: 10.1016/j.cell.2011.10.048
Nobile C. J., Johnson A. D. (2015). Candida albicans biofilms and human disease. Annu. Rev. Microbiol. 69, 71–92. doi: 10.1146/annurev-micro-091014-104330
Nobile C. J., Mitchell A. P. (2005). Regulation of cell-surface genes and biofilm formation by the C. albicans transcription factor Bcr1p. Curr. Biol. 15, 1150–1155. doi: 10.1016/j.cub.2005.05.047
Noble S. M., Gianetti B. A., Witchley J. N. (2017). Candida albicans cell-type switching and functional plasticity in the mammalian host. Nat. Rev. Microbiol. 15, 96–108. doi: 10.1038/nrmicro.2016.157
Oh J., Fung E., Price M. N., Dehal P. S., Davis R. W., Giaever G., et al. (2010). A universal TagModule collection for parallel genetic analysis of microorganisms. Nucleic Acids Res. 38, e146. doi: 10.1093/nar/gkq419
Omran R. P., Law C., Dumeaux V., Morschhäuser J., Whiteway M. (2020). The zinc cluster transcription factor Rha1 is a positive filamentation regulator in Candida albicans. bioRxiv 2020.01.21.901744. doi: 10.1101/2020.01.21.901744
Pappas P. G., Rex J. H., Sobel J. D., Filler S. G., Dismukes W. E., Walsh T. J., et al. (2004). Guidelines for treatment of candidiasis. Clin. Infect. Dis. 38, 161–189. doi: 10.1086/380796
Pérez J. C., Kumamoto C. A., Johnson A. D. (2013). Candida albicans commensalism and pathogenicity are intertwined traits directed by a tightly knit transcriptional regulatory circuit. PloS Biol. 11, e1001510. doi: 10.1371/journal.pbio.1001510
Pfaller M. A., Diekema D. J. (2007). Epidemiology of invasive candidiasis: a persistent public health problem. Clin. Microbiol. Rev. 20, 133–163. doi: 10.1128/CMR.00029-06
Polvi E. J., Veri A. O., Liu Z., Hossain S., Hyde S., Kim S. H., et al. (2019). Functional divergence of a global regulatory complex governing fungal filamentation. PloS Genet. 15, e1007901. doi: 10.1371/journal.pgen.1007901
Prasad T., Hameed S., Manoharlal R., Biswas S., Mukhopadhyay C. K., Goswami S. K., et al. (2010). Morphogenic regulator EFG1 affects the drug susceptibilities of pathogenic Candida albicans. FEMS Yeast Res. 10, 587–596. doi: 10.1111/j.1567-1364.2010.00639.x
Prill R. J., Iglesias P. A., Levchenko A. (2005). Dynamic properties of network motifs contribute to biological network organization. PloS Biol. 3, e343. doi: 10.1371/journal.pbio.0030343
Pukkila-Worley R., Peleg A. Y., Tampakakis E., Mylonakis E. (2009). Candida albicans hyphal formation and virulence assessed using a Caenorhabditis elegans infection model. Eukaryot. Cell 8, 1750–1758. doi: 10.1128/EC.00163-09
Rai L. S., Singha R., Brahma P., Sanyal K. (2018). Epigenetic determinants of phenotypic plasticity in Candida albicans. Fungal Biol. Rev. 32, 10–19. doi: 10.1016/j.fbr.2017.07.002
Ramage G., VandeWalle K., López-Ribot J. L., Wickes B. L. (2002). The filamentation pathway controlled by the Efg1 regulator protein is required for normal biofilm formation and development in Candida albicans. FEMS Microbiol. Lett. 214, 95–100. doi: 10.1111/j.1574-6968.2002.tb11330.x
Ramírez-Zavala B., Reuß O., Park Y. N., Ohlsen K., Morschhäuser J. (2008). Environmental induction of white-opaque switching in Candida albicans. PloS Pathog. 4, e1000089. doi: 10.1371/journal.ppat.1000089
Richards M. J., Edwards J. R., Culver D. H., Gaynes R. P. (1999). Nosocomial infections in medical intensive care units in the United States. National Nosocomial Infections Surveillance System. Crit. Care Med. 27, 887–892. doi: 10.1097/00003246-199905000-00020
Rikkerink E. H., Magee B. B., Magee P. T. (1988). Opaque-white phenotype transition: a programmed morphological transition in Candida albicans. J. Bacteriol. 170, 895–899. doi: 10.1128/JB.170.2.895-899.1988
Rosenbach A., Dignard D., Pierce J. V., Whiteway M., Kumamoto C. A. (2010). Adaptations of Candida albicans for growth in the mammalian intestinal tract. Eukaryot. Cell 9, 1075–1086. doi: 10.1128/EC.00034-10
Sahni N., Yi S., Daniels K. J., Huang G., Srikantha T., Soll D. R. (2010). Tec1 mediates the pheromone response of the white phenotype of Candida albicans: insights into the evolution of new signal transduction pathways. PloS Biol. 8, e1000363. doi: 10.1371/journal.pbio.1000363
Schweizer A., Rupp S., Taylor B. N., Röllinghoff M., Schröppel K. (2000). The TEA/ATTS transcription factor CaTec1p regulates hyphal development and virulence in Candida albicans. Mol. Microbiol. 38, 435–445. doi: 10.1046/j.1365-2958.2000.02132.x
Sellam A., Tebbji F., Nantel A. (2009). Role of Ndt80p in sterol metabolism regulation and azole resistance in Candida albicans. Eukaryot. Cell 8, 1174–1183. doi: 10.1128/EC.00074-09
Sellam A., Askew C., Epp E., Tebbji F., Mullick A., Whiteway M., et al. (2010). Role of transcription factor CaNdt80p in cell separation, hyphal growth, and virulence in Candida albicans. Eukaryot. Cell 9, 634–644. doi: 10.1128/EC.00325-09
Shannon P., Markiel A., Ozier O., Baliga N. S., Wang J. T., Ramage D., et al. (2003). Cytoscape: A software Environment for integrated models of biomolecular interaction networks. Genome Res. 13, 2498–2504. doi: 10.1101/gr.1239303
Shapiro R. S., Sellam A., Tebbji F., Whiteway M., Nantel A., Cowen L. E. (2012). Pho85, Pcl1, and Hms1 signaling governs Candida albicans morphogenesis induced by high temperature or Hsp90 compromise. Curr. Biol. 22, 461–470. doi: 10.1016/j.cub.2012.01.062
Shen J., Cowen L. E., Griffin A. M., Chan L., Köhler J. R. (2008). The Candida albicans pescadillo homolog is required for normal hypha-to-yeast morphogenesis and yeast proliferation. Proc. Natl. Acad. Sci. U.S.A. 105, 20918–20923. doi: 10.1073/pnas.0809147105
Silver P. M., Oliver B. G., White T. C. (2004). Role of Candida albicans transcription factor Upc2p in drug resistance and sterol metabolism. Eukaryot. Cell 3, 1391–1397. doi: 10.1128/EC.3.6.1391-1397.2004
Slutsky B., Staebell M., Anderson J., Risen L., Pfaller M., Soll D. R. (1987). White-opaque transition: a second high-frequency switching system in Candida albicans. J. Bacteriol. 169, 189–197. doi: 10.1128/JB.169.1.189-197.1987
Soll D. R., Morrow B., Srikantha T. (1993). High-frequency phenotypic switching in Candida albicans. Trends Genet. 9, 61–65. doi: 10.1016/0168-9525(93)90189-O
Soll D. R. (1992). High-frequency switching in Candida albicans. Clin. Microbiol. Rev. 5, 183–203. doi: 10.1128/CMR.5.2.183
Sonneborn A., Tebarth B., Ernst J. F. (1999). Control of white-opaque phenotypic switching in Candida albicans by the Efg1p morphogenetic regulator. Infect. Immun. 67, 4655–4660. doi: 10.1128/IAI.67.9.4655-4660.1999
Sorrells T. R., Johnson A. D. (2015). Making sense of transcription networks. Cell 161, 714–723. doi: 10.1016/j.cell.2015.04.014
Spiliopoulou A., Vamvakopoulou S., Bartzavali C., Dimitracopoulos G., Anastassiou E. D., Christofidou M. (2010). Eleven-year retrospective survey of candidaemia in a university hospital in southwestern Greece. Clin. Microbiol. Infect. 16, 1378–1381. doi: 10.1111/j.1469-0691.2010.03193.x
Srikantha T., Tsai L. K., Daniels K., Soll D. R. (2000). EFG1 null mutants of Candida albicans switch but cannot express the complete phenotype of white-phase budding cells. J. Bacteriol. 182, 1580–1591. doi: 10.1128/JB.182.6.1580-1591.2000
Srikantha T., Borneman A. R., Daniels K. J., Pujol C., Wu W., Seringhaus M. R., et al. (2006). TOS9 regulates white-opaque switching in Candida albicans. Eukaryot. Cell 5, 1674–1687. doi: 10.1128/EC.00252-06
Staib P., Binder A., Kretschmar M., Nichterlein T., Schröppel K., Morschhäuser J. (2004). Tec1p-independent activation of a hypha-associated Candida albicans virulence gene during infection. Infect. Immun. 72, 2386–2389. doi: 10.1128/IAI.72.4.2386-2389.2004
Tandonnet S., Torres T. T. (2017). Traditional versus 3′ RNA-seq in a non-model species. Genomics Data 11, 9–16. doi: 10.1016/j.gdata.2016.11.002
Tsai P. W., Chen Y. T., Yang C. Y., Chen H. F., Tan T. S., Lin T. W., et al. (2014). The role of Mss11 in Candida albicans biofilm formation. Mol. Genet. Genomics 289, 807–819. doi: 10.1007/s00438-014-0846-0
Tuch B. B., Mitrovich Q. M., Homann O. R., Hernday A. D., Monighetti C. K., de La Vega F. M., et al. (2010). The transcriptomes of two heritable cell types illuminate the circuit governing their differentiation. PloS Genet. 6, e1001070. doi: 10.1371/journal.pgen.1001070
Uhl M. A., Biery M., Craig N., Johnson A. D. (2003). Haploinsufficiency-based large-scale forward genetic analysis of filamentous growth in the diploid human fungal pathogen C. albicans. EMBO J. 22, 2668–2678. doi: 10.1093/emboj/cdg256
Uppuluri P., Chaturvedi A. K., Srinivasan A., Banerjee M., Ramasubramaniam A. K., Köhler J. R., et al. (2010a). Dispersion as an important step in the Candida albicans biofilm developmental cycle. PloS Pathog. 6, e1000828. doi: 10.1371/journal.ppat.1000828
Uppuluri P., Pierce C. G., Thomas D. P., Bubeck S. S., Saville S. P., Lopez-Ribot J. L. (2010b). The transcriptional regulator Nrg1p controls Candida albicans biofilm formation and dispersion. Eukaryot. Cell 9, 1531–1537. doi: 10.1128/EC.00111-10
Vandeputte P., Ischer F., Sanglard D., Coste A. T. (2011). In vivo systematic analysis of Candida albicans Zn2-Cys6 transcription factors mutants for mice organ colonization. PloS One 6, e26962. doi: 10.1371/journal.pone.0026962
Vandeputte P., Pradervand S., Ischer F., Coste A. T., Ferrari S., Harshman K., et al. (2012). Identification and functional characterization of Rca1, a transcription factor involved in both antifungal susceptibility and host response in Candida albicans. Eukaryot. Cell 11, 916–931. doi: 10.1128/EC.00134-12
Vasicek E. M., Berkow E. L., Bruno V. M., Mitchell A. P., Wiederhold N. P., Barker K. S., et al. (2014). Disruption of the transcriptional regulator Cas5 results in enhanced killing of Candida albicans by fluconazole. Antimicrob. Agents Chemother. 58, 6807–6818. doi: 10.1128/AAC.00064-14
Vinces M. D., Haas C., Kumamoto C. A. (2006). Expression of the Candida albicans morphogenesis regulator gene CZF1 and its regulation by Efg1p and Czf1p. Eukaryot. Cell 5, 825–835. doi: 10.1128/EC.5.5.825-835.2006
Vinces M. D., Kumamoto C. A. (2007). The morphogenetic regulator Czf1p is a DNA-binding protein that regulates white–opaque switching in Candida albicans. Microbiology 153, 2877–2884. doi: 10.1099/mic.0.2007/005983-0
Wang D. Y. C., Kumar S., Hedges S. B. (1999). Divergence time estimates for the early history of animal phyla and the origin of plants, animals and fungi. Proc. R. Soc. B: Biol. Sci. 266, 163–171. doi: 10.1098/rspb.1999.0617
Wang H., Song W., Huang G., Zhou Z., Ding Y., Chen J. (2011). Candida albicans Zcf37, a zinc finger protein, is required for stabilization of the white state. FEBS Lett. 585, 797–802. doi: 10.1016/j.febslet.2011.02.005
Wang Q., Verma J., Vidan N., Wang Y., Tucey T. M., Lo T. L., et al. (2020). The YEATS domain histone crotonylation readers control virulence-related biology of a major human pathogen. Cell Rep. 31, 107528. doi: 10.1016/j.celrep.2020.107528
Wenzel R. P. (1995). Nosocomial candidemia: risk factors and attributable mortality. Clin. Infect. Dis. 20, 1531–1534. doi: 10.1093/clinids/20.6.1531
Wheeler R. T., Kombe D., Agarwala S. D., Fink G. R. (2008). Dynamic, morphotype-specific Candida albicans β-Glucan exposure during infection and drug treatment. PloS Pathog. 4, e1000227. doi: 10.1371/journal.ppat.1000227
Wilking J. N., Angelini T. E., Seminara A., Brenner M. P., Weitz D. A. (2011). Biofilms as complex fluids. MRS Bull. 36, 385–391. doi: 10.1557/mrs.2011.71
Xie J., Tao L., Nobile C. J., Tong Y., Guan G., Sun Y., et al. (2013). White-opaque switching in natural MTLa/α isolates of Candida albicans: evolutionary implications for roles in host adaptation, pathogenesis, and sex. PloS Biol. 11, e1001525. doi: 10.1371/journal.pbio.1001525
Xu D., Jiang B., Ketela T., Lemieux S., Veillette K., Martel N., et al. (2007). Genome-wide fitness test and mechanism-of-action studies of inhibitory compounds in Candida albicans. PloS Pathog. 3, e92. doi: 10.1371/journal.ppat.0030092
Zordan R. E., Galgoczy D. J., Johnson A. D. (2006). Epigenetic properties of white-opaque switching in Candida albicans are based on a self-sustaining transcriptional feedback loop. Proc. Natl. Acad. Sci. U.S.A. 103, 12807–12812. doi: 10.1073/pnas.0605138103
Keywords: Candida albicans, biofilms, commensal-pathogen transition, transcriptional regulation, transcriptional networks, transcriptional rewiring, white-opaque switching, transcriptional circuits
Citation: Rodriguez DL, Quail MM, Hernday AD and Nobile CJ (2020) Transcriptional Circuits Regulating Developmental Processes in Candida albicans. Front. Cell. Infect. Microbiol. 10:605711. doi: 10.3389/fcimb.2020.605711
Received: 13 September 2020; Accepted: 04 November 2020;
Published: 03 December 2020.
Edited by:
Bernard Turcotte, McGill University, CanadaReviewed by:
Adnane Sellam, Université de Montréal, CanadaCopyright © 2020 Rodriguez, Quail, Hernday and Nobile. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Aaron D. Hernday, YWhlcm5kYXlAdWNtZXJjZWQuZWR1; Clarissa J. Nobile, Y25vYmlsZUB1Y21lcmNlZC5lZHU=
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.