 Solanka E. Ledwaba1*†
Solanka E. Ledwaba1*† Deiziane V. S. Costa2†
Deiziane V. S. Costa2† David T. Bolick3
David T. Bolick3 Natasa Giallourou4
Natasa Giallourou4 Pedro H. Q. S. Medeiros5
Pedro H. Q. S. Medeiros5 Jonathan R. Swann4
Jonathan R. Swann4 Afsatou N. Traore1
Afsatou N. Traore1 Natasha Potgieter1
Natasha Potgieter1 James P. Nataro6Richard L. Guerrant3
James P. Nataro6Richard L. Guerrant3- 1Department of Microbiology, University of Venda, Thohoyandou, South Africa
- 2Department of Physiology and Pharmacology, Federal University of Ceará, Fortaleza, Brazil
- 3Center for Global Health, Division of Infectious Disease and International Health, University of Virginia School of Medicine, Charlottesville, VA, United States
- 4Faculty of Medicine, Department of Metabolism, Digestion and Reproduction, Imperial College, London, England
- 5Institute of Biomedicine, Federal University of Ceará, Fortaleza, Brazil
- 6Department of Pediatrics, University of Virginia School of Medicine, Charlottesville, VA, United States
Enteropathogenic E. coli (EPEC) are recognized as one of the leading bacterial causes of infantile diarrhea worldwide. Weaned C57BL/6 mice pretreated with antibiotics were challenged orally with wild-type EPEC or escN mutant (lacking type 3 secretion system) to determine colonization, inflammatory responses and clinical outcomes during infection. Antibiotic disruption of intestinal microbiota enabled efficient colonization by wild-type EPEC resulting in growth impairment and diarrhea. Increase in inflammatory biomarkers, chemokines, cellular recruitment and pro-inflammatory cytokines were observed in intestinal tissues. Metabolomic changes were also observed in EPEC infected mice with changes in tricarboxylic acid (TCA) cycle intermediates, increased creatine excretion and shifts in gut microbial metabolite levels. In addition, by 7 days after infection, although weights were recovering, EPEC-infected mice had increased intestinal permeability and decreased colonic claudin-1 levels. The escN mutant colonized the mice with no weight loss or increased inflammatory biomarkers, showing the importance of the T3SS in EPEC virulence in this model. In conclusion, a murine infection model treated with antibiotics has been developed to mimic clinical outcomes seen in children with EPEC infection and to examine potential roles of selected virulence traits. This model can help in further understanding mechanisms involved in the pathogenesis of EPEC infections and potential outcomes and thus assist in the development of potential preventive or therapeutic interventions.
Introduction
Gastroenteritis remains a major cause of morbidity and mortality in young children especially in developing countries (Liu et al., 2015). Enteropathogenic E. coli (EPEC) has been recognized by the Global Enteric Multicenter Study (GEMS) and Malnutrition and Enteric Disease (MAL-ED) studies as one of the major causes of moderate to severe diarrhea in children (Kotloff et al., 2013; Platts-Mills et al., 2015). Infection results in acute watery diarrhea accompanied by fever, vomiting and dehydration (Kaper et al., 2004; Guerrant et al., 2011).
EPEC contains the locus of enterocyte effacement regulator (Ler) gene, a major transcriptional activator of LEE pathogenicity island, comprising around 41 open reading frames (Frankel et al., 1998; Friedberg et al., 1999). EPEC virulence is mediated by the Type 3 secretion system (T3SS), characterized by attaching and effacing (A/E) lesions (Frankel et al., 1998). The T3SS consists of EPEC secreted components (Esc) and EPEC secretion proteins (Esp), encoded in the LEE pathogenicity island. In addition, EscN is the main driving force assisting in the ATPase response to enable activation of the T3SS, for efficient transportation of effector proteins into the enterocytes of the host (Andrade et al., 2007). During infection, EPEC attaches to epithelial cells via bundle forming pili (bfp) (Giron et al., 1991) followed by intimate adherence with the aid of the translocated intimin receptor (tir) and intimin (eae), which results in actin accumulation and formation of pedestal structures (Knutton et al., 1989; Guerrant et al., 2011). EPEC is characterized by the presence (typical EPEC) or absence (atypical EPEC) of bfp. Typical EPEC are characterized by Localized Adherence (LA) in vitro (Kaper et al., 2004) and have been reported to cause severe diarrhea in children under 12 months of age and in certain cases results in death (Kotloff et al., 2013; Platts-Mills et al., 2015). Atypical EPEC is characterized by LA-like (Scaletsky et al., 2010), aggregative adherence or diffuse adherence in vitro (Pelayo et al., 1999; Mora et al., 2009) and are increasingly being detected in children worldwide (Abe et al., 2009; Hu and Torres, 2015).
Pathogens such as EPEC compete with the resident microbiota for nutrients in order to colonize the intestinal environment. According to Freter’s nutrient niche, in order for microbes to be successful, they must have the capacity to grow fast in the intestine compared to its competitors (Freter et al., 1983). These pathogens require the same carbon pathways which commensal E. coli uses, such as mannose and galactose in vivo (Fabich et al., 2008).
EPEC have been studied extensively in vitro, which enables studies of localization traits, A/E lesions and expression of the T3SS effector proteins (Giron et al., 1991; Knutton et al., 1999; Leverton and Kaper, 2005). In vivo studies have shown that a complete intestinal environment helps further determine the specific roles of EPEC traits involved in infections in animals and humans (Law et al., 2013). Animal models such as Caenorhabditis elegans, rabbits, pigs, and cattle have been used to study EPEC infections (Moon et al., 1983; Larsen et al., 1995; Dean-Nystrom et al., 2002; Misyurina et al., 2010). Infections induced by EPEC in C57BL/6 mice have also been reported (Savkovic et al., 2005; Royan et al., 2010; Zhang et al., 2010; Rhee et al., 2011; Manthey et al., 2014; Dupont et al., 2016), showing colonization of EPEC in the intestinal epithelial microvilli (Savkovic et al., 2005), changes in tight junction morphology and epithelial barrier function accompanied by inflammatory responses (Zhang et al., 2010; Zhang et al., 2012).
Most of the previous EPEC infection murine models have used streptomycin in order to disrupt the intestinal microbiota and promote colonization in mice (Mundy et al., 2006; Zhang et al., 2010; Rhee et al., 2011; Zhang et al., 2012; Dupont et al., 2016). Recently, a study demonstrated that mice are susceptible to colonization with EPEC in an age and microbiota disruption-dependent manner, with infant (preweaning) mice being more susceptible (Dupont et al., 2016). These animal models have provided insights into the understanding of potential pathogenetic mechanisms of EPEC infection in humans. However, these models have not been able to replicate clinical outcomes observed in humans (growth decrement and diarrhea).
A murine EPEC infection model able to induce changes in body weight and diarrhea, which are important outcomes in children infected by EPEC, had been needed and was the focus of our current study (Law et al., 2013). We have previously shown that disruption of intestinal microbiota using a broad-spectrum antibiotic cocktail enabled colonization of bacterial pathogens such as Enterotoxigenic E. coli (Bolick et al., 2018), Campylobacter jejuni (Giallourou et al., 2018) and Shigella flexneri (Medeiros PH et al., 2019) resulting in diarrhea in C57BL/6 mice. This current study also used the same antibiotic cocktail to enable the assessment of disease outcomes associated with EPEC infection. We therefore, describe a weaned murine model in which the microbiota have been disrupted via broad-spectrum antibiotics to enable efficient colonization and clinical outcomes of EPEC infection in mice resulting in growth impairment, diarrhea, intestinal damage, metabolic alterations, and increased intestinal permeability.
Materials and Methods
Ethics Statement
The mice used in the study have been handled with strict accordance with the recommendations in the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health. The protocol has been approved by the Committee on the Ethics of Animal Experiments of the University of Virginia (Protocol Number: 3315). All efforts were made to minimize suffering. This is also in accordance with the Institutional Animal Care and Use Committee policies of the University of Virginia. The University of Virginia is accredited by the Association for the Assessment and Accreditation of Laboratory Animal Care, International (AAALAC).
Mice
Mice used in this study were male, 22 days old, C57BL/6 strain, ordered from Jackson Laboratories (Bar Harbor, ME). Mice weighed approximately 10 grams on arrival and were co-housed in groups of up to 4 animals per cage. The vivarium was kept at a temperature of between 20 and 23°C with a 14-h light and 10-h dark cycle. Mice were allowed to acclimate for 3 days upon arrival. Mice were fed standard rodent house chow diet from arrival and throughout the infection challenge.
EPEC Inoculum Preparation
Bacterial strains used included: wild type EPEC E2348/69 [(serotype O127:H6) first isolated in 1969 during infantile diarrhea outbreak in Taunton, United Kingdom, it belongs to phylogroup B2 with full length chromosomal nucleotide (Accession number: FM180568)] (Levine et al., 1985; Iguchi et al., 2009) and EPEC E2348/69 ΔescN CVD425 (Jarvis et al., 1995). Bacterial cultures were prepared from glycerol stocks maintained at -80°C. Cultures were grown in 20 ml Dulbecco’s modified Eagle’s medium containing phenol red (DMEM) at 37°C in a shaking incubator until cultures turned orange indicating optimal growth, OD600 ~ 0.6. Cultures were centrifuged at 3500 x g for 10 min at 4°C. The bacterial pellet was resuspended in DMEM high glucose in order to obtain 1010 CFU/ml.
Enteropathogenic Escherichia coli Infection Model
Four days prior to challenge with EPEC, mice were given an antibiotic cocktail of gentamicin (35 mg/L), vancomycin (45 mg/L), metronidazole (215 mg/L), and colistin (850 U/ml) in drinking water for 3 days in order to disrupt resident microbiota, followed by 1 day on normal water in order to clear antibiotics (Bolick et al., 2018). Then, mice were administered 100 µl of 1010 CFU/ml (1010 bacteria per mouse) bacterial culture in DMEM high glucose orally using 22-gauge feeding needles. Uninfected control mice were administered only 100 µl DMEM high glucose.
After infection, all mice were weighed and stools were collected daily until 8-days post infection (p.i.). Mice were euthanized on days 3, 7, and 8 p.i. Figure 1A shows the schematic presentation summarizing the experimental procedure.
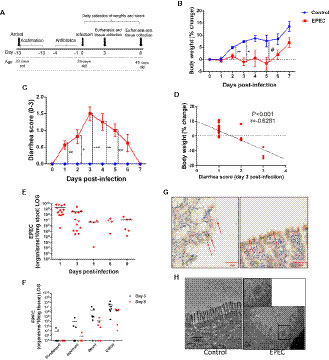
Figure 1 EPEC infection impairs weight gain, induces diarrhea and colon colonization in C57BL/6 mice. (A) Experimental timeline of EPEC infection model. Weaned C57BL/6 mice pretreated with antibiotic cocktail were orally infected with EPEC (1010 CFU). (B) Change in body weight of C57BL/6 mice infected with EPEC (EPEC) or uninfected (Control) (n=12/group). Line graphs represents mean ± SEM. **p< 0.009, *p<0.001 and #p< 0.04 using multiple Student’s t-test. (C) Diarrhea score of EPEC and control mice. Line graphs represent median ± SEM. *p<0.01, **p<0.006 and ***p<0.0001 using Kruskal-Wallis test followed by Dunn’s test. (D) Significant negative correlation between diarrhea score and change in body weight at day 3 p.i (Spearman rank test). (E). Quantification of EPEC shedding in stools. (F) Quantification of EPEC tissue burden in the intestinal tissues (duodenum, jejunum, ileum and colon) at days 3 and 8 p.i. (G) Immunohistochemistry staining of intimin (red arrows), in ileal tissue of EPEC infected mice at day 3 p.i. (H) TEM at day 3 p.i., showing disruption of the microvilli of mice infected with EPEC.
Analysis of Clinical Outcomes
The clinical outcomes, body weight and diarrhea were assessed daily. Body weight was measured for 9 days (starting before infection, day 0) and percentage of changes in body weight was measured based on each individual mouse weight from day 0 before infection. Diarrhea scores were measured until day 7 p.i. Diarrhea score were based on the following 0–4: 0-well-formed pellets; 1- stick stools adhering in microtubes wall; 2-pasty stools with or without mucus; 3-watery stools with or without mucus; and 4-Stools with blood.
Tissue Burden and Stool Shedding
For stool shedding, DNA was extracted from stools using the QIAamp DNA Stool Mini kit (Qiagen) according to the manufacturer’s instructions. For tissue burden, tissues were homogenized using the beat-beater and DNA was extracted using DNeasy Kit (Qiagen) according to the manufacturer’s instructions. The eae (intimin) gene was used as a specific target for detecting EPEC in stools and tissues. Primer sequences included eae 5’-CCCGAATTCGGCACAAGCATAAGC-3’ (sense) and 5’-CCCGGATCCGTCTCGCCAGTATTCG-3’ (antisense) (Zhang et al., 2002). Real-time PCR was performed using Bio-Rad CFX under the following conditions: 95°C for 3 min, followed by 40 cycles of 15 s at 95°C, 60 s at 55°C, and lastly 20 s at 72°C.
EPEC Adherence on the Intestine
Ileal tissue segments from EPEC-infected mice on day 3 p.i. were fixed in 4% formalin, embedded in paraffin, the slides were stained with rabbit anti-intimin at the University of Virginia Histology core, and viewed using light microscope.
Transmission Electron Microscopy
For TEM, the ileal tissues from EPEC-infected mice on day 3 p.i. and uninfected (control) were fixed with 4% glutaraldehyde. The samples were washed with 1X cacodylate buffer for 10 min and placed in 2% osmium tetroxide for 1 h. Then washed for 10 min with cacodylate buffer and distilled water. Followed by dehydration with 30% ethanol for 10 min and concentrations of 50%, 70%, 95%, and 100% ethanol all for 10 min each. About 1:1 ethanol/propylene oxide was used for 10 min followed by 100% propylene oxide (PO) for 10 min. The samples were then placed in 1:1 of PO/epoxy resin (EPON) overnight followed by 1:2 PO/EPON for 2 h, then 1:4 PO/EPON for 4 h, and lastly 100% EPON overnight. The samples were then embedded in fresh 100% EPON and allowed to bake in a 65°C oven. Ultra-thin sections were cut at 75 nm and picked up on 200 mesh copper grids. Sections were stained with 0.25% lead citrate and 2% uranyl acetate. The slides were viewed using JEOL 1230 microscope, with 4k x 4k CCD camera.
Histology Analysis
Mice intestinal samples (ileum and colon) from day 3 or day 7 p.i. were fixed in 10% neutral buffered formalin for 20 h, dehydrated and embedded in paraffin. Ileal and colonic sections (5 µm) were stained with hematoxylin and eosin staining (H&E) and were examined using light microscopy. Histopathological scores were performed by a blinded investigator, using previously described methods (Wong et al., 2015). Histological damage scores were determined by quantifying the intensity of epithelial tissue damage (0–3, 0-no damage, 1-mild, 2-moderate, 3-extensive), edema in submucosa layer (0-3), and neutrophil infiltration (0-3).
Intestinal Inflammatory Response by Enzyme-Linked Immunosorbent Assay
Protein lysates were extracted from the stools, ileum, colon and cecal contents using radioimmunoprecipitation assay (RIPA) buffer (20 mM Tris [pH 7.5], 150 mM NaCl, 1% Nonidet P-40, 0.5% sodium deoxycholate, 1 mM EDTA, 0.1% SDS) containing protease inhibitor cocktail (Roche) and phosphatase inhibitors (1 mM sodium orthovanadate, 5 mM sodium fluoride, 1 mM microcystin LR, and 5 mM β-glycerophosphate). Lysates were centrifuged at 13,000 rpm for 15 min and the supernatant was used to perform the protein assay using the bicinchoninic acid assay (Thermo Fisher Scientific). Inflammatory biomarkers (LCN-2, MPO, IL-23, IL-22, IL-17, GMCSF, IL-33, and IL-10) were measured using a commercial ELISA kit (R&D Systems) according to the manufacturer instructions. Interleukin-6 (IL-6), IL-1β, interferon gamma (INF-γ), TNF-α, KC (analogue to human IL-8), and IL-18 and CRP levels were measured using a ProcartPlex multiplex immunoassay (Invitrogen) by Luminex (Biorad). Biomarkers levels were measured as picograms per milligram of protein.
Phosphorylated-Signal Transducer and Activator of Transcription-3, Phosphorylated-Cyclic AMP Response Element-Binding Protein and Cleaved Caspase-3 Measurement
Colonic samples from EPEC-infected and control mice collected at day 3 p.i. were homogenized in ice-cold using RIPA buffer containing protease and phosphatase inhibitors. The colonic levels of phosphorylated STAT3 3 (pSTAT3), phosphorylated CREB (pCREB) and cleaved caspase-3 were measured using ELISA kits (R&D Systems) according to the manufacturer’s instructions.
TaqMan- Real Time Polymerase Chain Reaction
The isolation of total RNA from colon tissues of EPEC-infected (presenting moderate or severe diarrhea) and control mice were performed by using a Qiagen RNeasy mini kit and QIAcube. cDNA was synthetized from 1µg of total RNA, quantified by Qubit 3 fluorometer 3000 (Invitrogen) and purified by deoxyribonuclease I (Invitrogen) treatment, with the iScript cDNA (Bio-Rad) as described by manufacturer instructions. qPCR was performed with 50 ng of cDNA in each well and SensiFAST probe no-ROX mix (Bioline) using a CFX Connect system (Bio-Rad) with the following conditions: 95°C for 2 min, 40 cycles of 95°C for 10 s and 60°C for 50 s. A pre-designed TaqMan array mouse immune fast 96-well plates (Applied Biosystems) was used to assess the expression of 92 genes listed in Supplementary Table 1. Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) was used as a reference gene. All fold changes were determined using the ΔΔCt method (Livak and Schmittgen, 2001).
In Vivo Intestinal Permeability Assay
For assessing in vivo intestinal permeability fluorescein isothiocyanate (FITC)-labeled dextran assay (4kDa, Sigma Aldrich) was used. Mice were deprived food, with free access to water, for 4 h. Then, 200 µl of FITC-dextran solution (80 mg/ml in water) was administered by oral gavage for each mouse. After 4 h of FITC administration, mice were anesthetized to collect blood using cardiac puncture. Then, the blood samples were centrifuged (5 min, 8,000 rpm, 4°C) and plasma was obtained. Fluorescence intensity in 100 µl of plasma placed on Qubit 0.5 ml-microtubes (Life Technologies) was measured using Qubit 3fluorometer (Life Technologies) using an excitation wavelength of 470 nm. A plasma sample from mice not receiving FITC-dextran solution was used as a blank.
1H Nuclear Magnetic Resonance Spectroscopy-Based Metabolic Profiling
Urine specimen were collected in a sterile 1.5 ml eppendorf tube and placed at -80°C until further analysis. The metabolic profiling was performed on all urine samples using 1H nuclear magnetic resonance (NMR) spectroscopy. A 30 µl urine aliquot was combined with 30 µl of phosphate buffer (pH 7.4, 100% D2O, 0.2 M Na2HPO4/NaH2PO4) containing 1 mM of the internal standard, 3-(trimethylsilyl)-[2,2,3,3-2H4]-propionic acid (TSP), and 2 mM sodium azide (NaN3) as a bacteriocide. Samples were vortexed and spun at 13,000 x g for 10 min and 50 µl of the supernatant was then transferred to 1.7 mm NMR tubes. Spectroscopic analysis was performed on a 600 MHz Bruker Avance™ NMR spectrometer at 300 K using a Bruker BBI probe and an automated SampleJet for tube handling (Bruker, Germany). 1H NMR spectra of the urine samples were acquired using a standard one-dimensional pulse sequence [recycle delay (RD) -90°-t1–90°-tm-90°-acquire free induction decay (FID)]. The water signal was suppressed through irradiation during the RD of 4 s and a mixing time of (tm) 100 ms was used. For each spectrum, 64 scans were obtained into 64 K data points using a spectral width of 12.001 ppm. The NMR spectra were calibrated to the TSP resonance at 0 ppm using TopSpin 3.5 NMR software (Bruker, Germany) and imported into MATLAB (R2018a, Mathworks Inc, Natwick, MA) using in-house scripts. Regions containing the TSP, water and urea resonances were removed from the urinary spectra. 1H NMR spectra were manually aligned and normalized to the unit area.
Western Blotting
Colon tissues from EPEC-infected and control mice at day 7 p.i. were collected, lysed using RIPA lysis buffer containing complete EDTA-free protease inhibitor cocktail (Roche) and phosStop (Roche) and centrifuged (17 min, 4°C, 13,000 rpm). Then the supernatant was collected for extracting protein. Protein concentrations were determined through the bicinchoninic acid assay according to the manufacturer`s protocol (Thermo Fisher Scientific). Reduced 60 µg protein samples (previously prepared with sample reducing agent- Invitrogen- and protein loading buffer-LI-COR) were denatured at 95°C for 5 min, separated on NuPAGE 4%–12% BIS-Tris gel (Invitrogen) and transferred to nitrocellulose membranes (Life Technologies) for 2 h. The membranes were then immersed in iBind fluorescent detection solution (Life technologies) and placed in a iBIND automated Western device (Life Technologies) overnight at 4°C for blocking, incubating with primary antibodies (rabbit anti-β-actin, 1:1000, Thermo Fisher Scientific, PA1-183; mouse anti-claudin-2, 3:500, Invitrogen, 325600; rabbit anti-claudin-1, 1:1000, Novus biological, NBP1-77036) and secondary antibodies (Cy3-conjugated AffiniPure donkey anti-rabbit, 711-165-152, 1:1000, Jackson Immuno-Research and Cy5-conjugated AffiniPure donkey anti-mouse, 715-175-150, 1:1000, Jackson ImmunoResearch). Then, the membranes were immersed in ultrapure water and fluorescent signal was detected using the Typhoon system (GE healthcare). Densitometric quantification of bands was performed using ImageJ software (NIH, Bethesda, MD, USA).
Systemic Inflammation Analysis
Blood collected at day 3 p.i. was centrifuged at 8,000 rpm for 5 min at 4°C in order to obtain the plasma, to measure the levels of SAA as a marker of systemic inflammation. The levels of SAA were measured using a commercial ELISA kit (R&D Systems) according to the manufacturer’s instructions. The results were expressed as picograms per milliliter.
Statistical Analysis
All data were analyzed using GraphPad Prism 7 software (GraphPad Software). Data are presented as the mean ± standard error of the mean (SEM) or as medians when appropriate. Student’s t test and one-way Analysis of Variance (ANOVA) followed by Tukey’s test were used to compare means, and the Kruskal–Wallis and Dunn tests were used to compare medians. Spearman rank test was used to correlation analyses. Differences were considered significant when p< 0.05. Experiments were repeated at least two times.
For metabolomics data analysis multivariate statistical modelling was performed including principal component analysis (PCA) using the Imperial Metabolic Profiling and Chemometrics Toolbox (https://github.com/csmsoftware/IMPaCTS) in MATLAB (Version 2018a, Mathworks Inc) and unsupervised hierarchical clustering analysis (HCA) to unveil metabolic differences between EPEC infected and control mice. Unsupervised clustering for all samples was done with the use of the normalized abundance of metabolites that were identified through the PCA models. Hierarchical clusters were generated with the use of an average-linkage method by means of the pdist and linkage functions in the MATLAB bioinformatics toolbox. Heat maps and dendrograms after the HCA were generated with MATLAB imagesc and dendrogram functions, respectively. Pathway analysis was performed using the MetaboAnalyst 4.0 platform (https://www.metaboanalyst.ca/). The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
Results
EPEC Infection Leads to Growth Impairment and Diarrhea
Depletion of intestinal microbiota by antibiotics has been shown to be effective in promoting colonization by bacterial pathogens (Bolick et al., 2018; Giallourou et al., 2018; Medeiros PH et al., 2019). We therefore, tested whether pretreatment with antibiotics could enable the study of body weight and diarrhea in mice infected with EPEC (1010 CFU) (Figure 1A). EPEC inhibited the growth of mice when compared to the control group from days 2 to 5 post infection (p.i.) (p<0.05, Figure 1B). EPEC infection also induced moderate (days 1, 2, and 6 p.i.) to severe diarrhea at days 3 to 5 p.i. (p<0.0001, p<0.006) (Figure 1C). The changes in body weight exhibited by individual EPEC-infected mice were correlated with their individual diarrhea scores at the overall peak of diarrhea and weight decrements on day 3 (Figure 1D), showing that higher diarrhea scores tended to associate with greater weight shortfalls.
EPEC Colonizes the Ileum and Colon in Mice
In order to confirm that growth impairment and diarrhea were promoted by EPEC infection, DNA was extracted from stools of EPEC-infected mice and quantitative PCR was used to measure shedding. As shown in Figure 1E, most of the EPEC-infected mice exhibited 108-1010 organisms/10 mg stool at day 1 p.i. and less shedding was observed from days 5 to 8 p.i.
Given that EPEC is an intestinal pathogen, we further investigated which intestinal sections were predominantly colonized by EPEC using quantitative PCR to measure tissue burdens. EPEC was found to predominantly colonize the ileum (<107 organisms/mg tissue) and colon (<108 organisms/mg tissue) of mice at day 3 pi (Figure 1F), and the same trend was also observed at day 8 p.i. Figure 1G shows intimin-stained EPEC adherence on blunted ileal mucosa, with disruption of the microvilli shown by TEM (Figure 1H).
These findings indicate that EPEC infection promotes a self-limited symptomatic acute disease in antibiotic-pretreated mice. Fecal shedding of EPEC, and tissue burdens were detected up to day 8 p.i. in infected mice.
EPEC Infection Promotes Acute Intestinal Tissue Damage and Inflammation
Given that our EPEC infection model resulted in a significant colonization in the ileum and colon, we next investigated whether EPEC infection promotes ileal and colonic damage. The ileal and colonic histological damage in EPEC-infected mice was characterized by the loss of epithelial integrity, moderate edema in the submucosa and infiltration of inflammatory cells into the lamina propria and submucosa, with significant histology score differences from controls in both ileum and colon at day 3, and persistent significant differences in the ileum extending to day 7. The damage was found to be greater in colon compared to control mice (Figure 2A) at day 3 p.i., as confirmed by measurement of histologic damage score (p<0.0001, Figure 2B). On day 7 p.i., the damage in the ileum of EPEC-infected mice was higher when compared to the control mice (p<0.0001, Figures 2A, C).
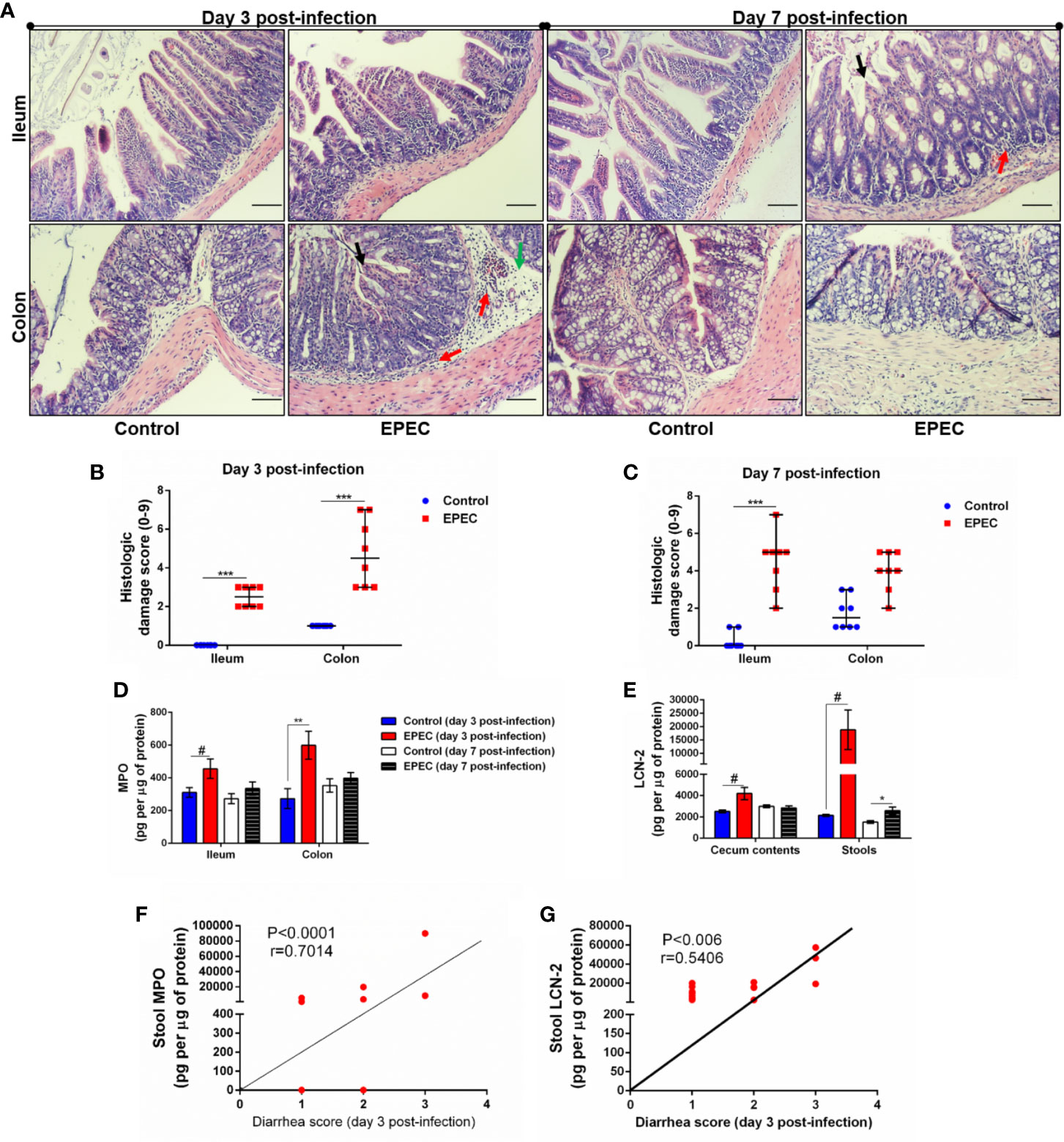
Figure 2 EPEC infection model induces acute colonic damage and inflammation followed by a positive correlation between diarrhea and inflammation markers in stools. (A) Representative H&E stains of ileal and colonic tissue collected from control and EPEC infected mice at days 3 and 7 p.i. Scale bars, 100 μm. (B, C) Histologic damage score based on epithelial damage (black arrow), mucosal edema (green arrow) and inflammatory cell infiltrate (red arrow) in the ileal and colonic tissue of uninfected (control group) and EPEC-infected (EPEC group) (B) mice at day 3 and (C) mice at day 7 p.i. Bars represent median ± SEM (n=8). ***p<0.0001 using Kruskal-Wallis test followed by Dunn’s test. (D) MPO levels measured in intestinal tissue lysates (ileum and colon) collected at days 3 and 7 p.i. (E) LCN-2 levels measured in the cecal and stool lysates collected at days 3 and 7 p.i. Bars represent mean ± SEM (n=8). #p<0.05, *p<0.03 and **p<0.007 using multiple Student’s t-test. (F) Significant positive correlation between diarrhea score and MPO levels in stools of EPEC-infected mice at day 3 p.i (Spearman rank test). (G) Significant positive correlation between diarrhea score and LCN-2 levels in stools of EPEC-infected mice at day 3 p.i (Spearman rank test).
Myeloperoxidase (MPO), a marker of neutrophil activity in intestinal mucosa, and lipocalin-2 (LCN-2), a glycoprotein upregulated in tissue damage under infection conditions, have been considered as biomarkers of environmental enteric dysfunction, including EPEC infection in children (Prata et al., 2016; Kosek et al., 2017). To ensure that our EPEC infection model mimics the alterations of these biomarkers as observed in children, we measured MPO and LCN-2 in the ileal and colonic tissues, as well as in cecal contents and stools. We found increased MPO levels in ileum and colon tissues of EPEC-infected mice at day 3 p.i. when compared to the control group (p<0.05 and p<0.03 respectively, Figure 2D). Of note, a trend of increase in MPO levels was also observed in the cecal contents and stools at day 3p.i. (Supplementary Figure 1), however no statistical significance was found. On day 7 p.i., MPO levels were reduced in the intestinal (ileum and colon), cecal contents and stools of EPEC infected mice compared to controls (Figure 2D, Supplementary Figure 1). However, increased LCN-2 levels were found in cecal contents (day 3 p.i.) and stools (days 3 and 7 p.i.) of EPEC-infected mice when compared to control mice (p<0.05-day 3 p.i. or p<0.03-day 7 p.i., Figure 2E).
We found that the diarrhea score correlated with MPO or LCN-2 levels in stools samples of EPEC-infected mice at day 3 p.i., when mice exhibited higher diarrhea score, a positive correlation was found between diarrhea score and MPO levels in stools (p<0.0001, r=0.7014, Figure 2F). Positive correlation was also seen of diarrhea score with LCN-2 levels at day 3 p.i. (p<0.006, r=0.5406, Figure 2G). These data indicated that high diarrhea scores are associated with increased MPO and LCN-2 levels.
EPEC Infection Alters Pro-Inflammatory and Anti-Inflammatory Cytokine Synthesis in Ileum and Colon in a Stage Disease-Dependent Manner in Mice
Next, we further analyzed which pro-inflammatory [IL-6, IL-1β, INF-γ, IL-23, tumor necrosis factor-α (TNF-α), IL-17, IL-18] and anti-inflammatory (IL-22, IL-33, IL-10) cytokines were affected by EPEC infection during acute (day 3 p.i.) and later (day 7 p.i.) stage of the disease. As shown in Figure 3, higher levels of IL-6 (p<0.05), IL-1β (p<0.05), INF-γ (p<0.05), IL-23 (p<0.01), and IL-22 (p<0.05) were detected in colonic tissues of EPEC-infected mice at day 3 p.i. and were significantly different when compared to controls (Figures 3A–D, F). EPEC infection in the ileal tissue resulted in significant increase in IL-6, IL-1β, and IL-22 at day 3 p.i. when compared to control mice (p<0.05, Figures 3A, B, F). In the later stage of disease (day 7 p.i.), TNF-α (ileal and colonic tissues) and IL-22 (colonic tissues) were significantly increased in EPEC-infected mice when compared to control mice (p<0.01, Figures 3E, G). In addition, increase of KC in the ileal and colonic tissues was observed, however, not statistically significant (Figure 3F).
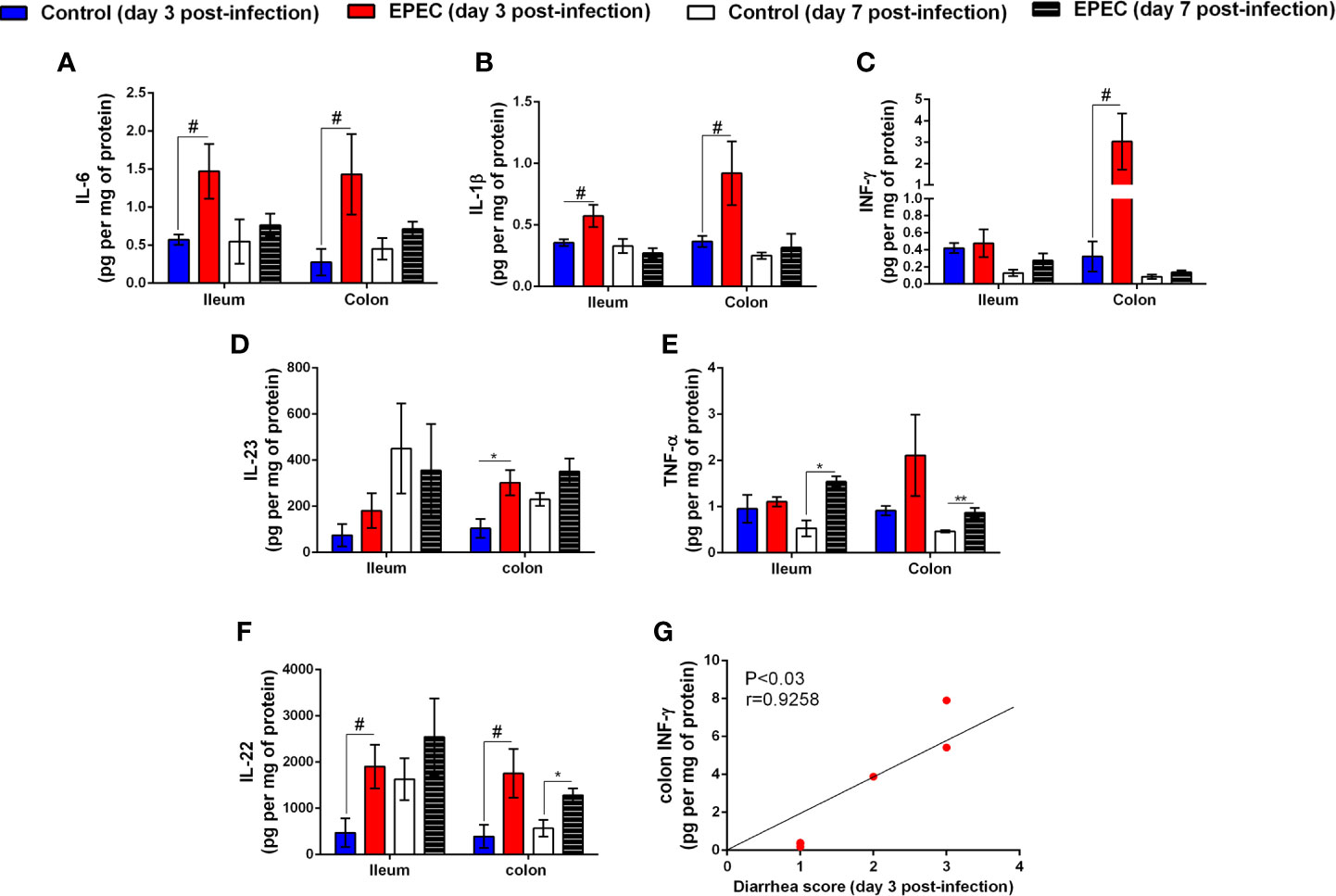
Figure 3 EPEC infection increases pro-inflammatory mediators, and diarrhea scores correlates positively with INF-γ and KC levels in colonic tissues of EPEC infected mice. (A) Levels of IL-6, (B) IL-1β, (C) INF-γ, (D) IL-23, (E) TNF-α, and (F) IL-22 in the ileal and colonic tissues of control and EPEC infected mice at days 3 and 7 p.i. were measured by ELISA. Bars represent mean ± SEM (n=8). #p<0.05, *p<0.01 and **p<0.001 using multiple Student’s t-test. (G) Significant positive correlation between diarrhea score and INF-γ levels in colonic tissues of EPEC infected mice at day 3 p.i (Spearman rank test).
We analyzed the correlation of diarrhea severity with the levels of INF-γ in colonic tissues of EPEC-infected mice and found a strong positive correlation between diarrhea score and colonic INF-γ levels (p<0.03, r=0.9258, Figure 3H). We did not find any change in the levels of IL-17, IL-18, granulocyte-macrophage colony–stimulating factor (GMCSF), IL-33, and IL-10 in ileal and colonic tissues samples of EPEC-infected mice when compared to the controls (Supplementary Figures 2A–E).
Taken together, these data indicate that EPEC infection promotes two-phase of disease in mice; an acute stage characterized by growth impairment, intestinal damage, presence of diarrhea and increased of MPO, LCN-2, IL-6, IL-1β, INF-γ, IL-23, and IL-22; and a later stage with an increase in LCN-2 and TNF-α levels in an absence of diarrhea. In addition, the data suggests that the colon is more affected by EPEC infection in mice.
EPEC-Infected Mice With Diarrhea Demonstrates Upregulation of Pro-Inflammatory Cytokines, Inflammatory Markers, STAT, and Apoptosis Markers in Colon
Due to an increase in diarrhea severity and colonic INF-γ levels that were positively correlated on day 3 p.i., we evaluated the profile of gene expression from the colonic tissues of EPEC-infected mice with diarrhea and controls using Taqman assay. In total, 92 genes were evaluated, among these 37 were upregulated and four were downregulated (p<0.05, Figure 4A). INF-γ, GZMB, CXCL10, IL-6, and IL-1β were the most upregulated genes, showing approximately 85, 30, 27, 23, and 16-fold-change in relation to controls (p<0.05, Figure 4A). In mice with diarrhea, EPEC infection resulted in upregulation of pro-inflammatory mediators (INF-γ, TNF-α, TNFRS, IL-1α, IL-1β, IL-2Rα, IL-5, IL-6, and IL-12b, Figure 4B), chemokines (CCL2, CCL5, CCL19, CXCL10, and CXCL11, Figure 4C), chemokine receptors (CCR2 and CCR7, Figure 4C), cellular recruitment (VCAM1, ICAM1, and SELP, Figure 4D), CD68 (a macrophage marker, Figure 4E), CD3ϵ (marker of T-cell activation, Figure 4E), inflammation markers (C3, CD38, CD40, CSF2, GZMB, and MD2, Figure 4F), transcription factors [phosphorylated-signal transducer and activator of transcription-1 (STAT1), STAT3, and STAT4, Figure 4G] and apoptosis markers (Fas and Bax, Figure 4H) in colon of mice at day 3 p.i. In addition, EPEC-infection also upregulated gene expression of anti-inflammatory mediators, such as TGF-β1, HMOX1, PTPRC, SOCS1, and LIF when compared to control mice (p<0.05, Figure 4I).
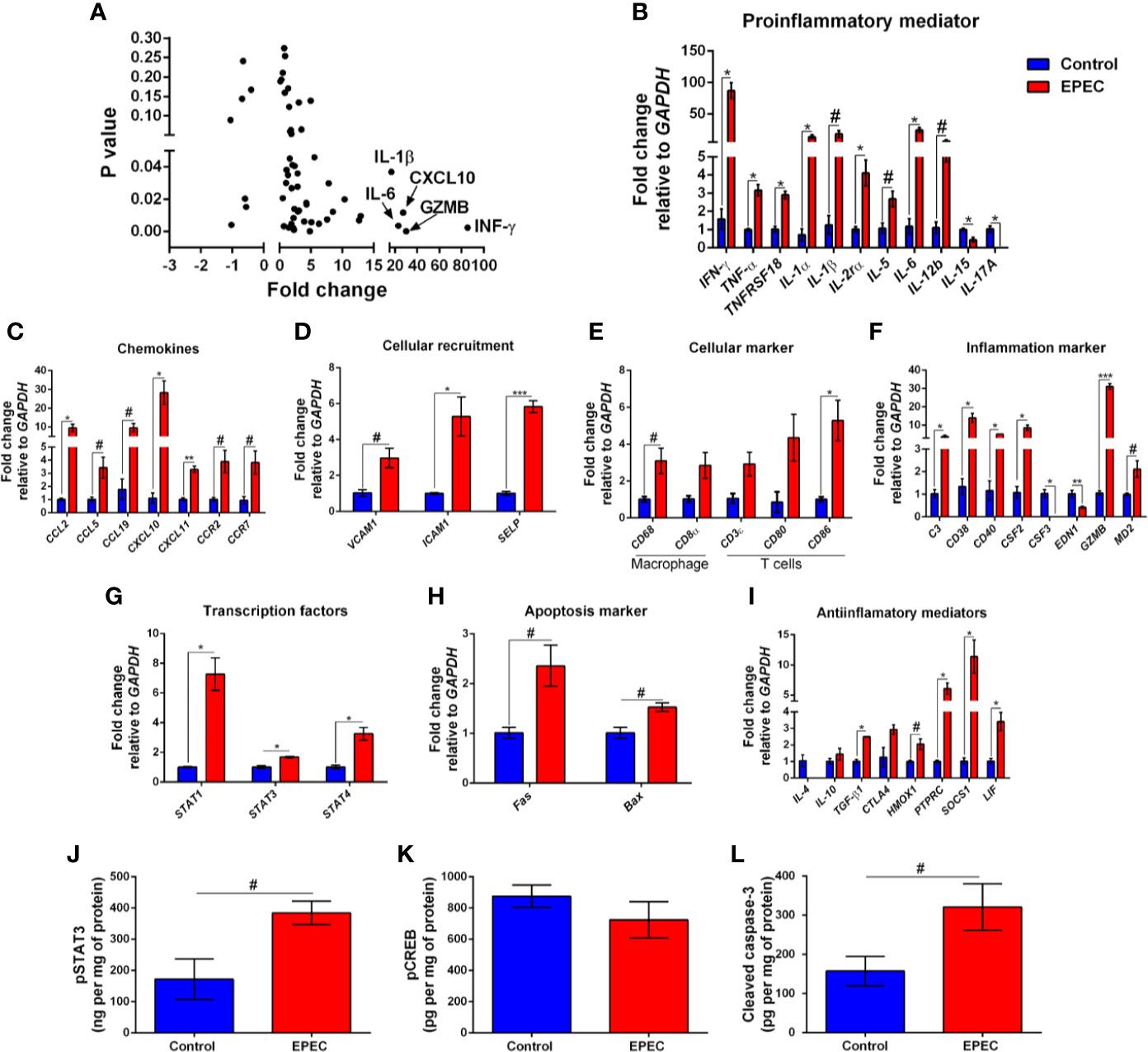
Figure 4 EPEC-infected mice with soft or unformed stools exhibit higher expression of mRNA for pro-inflammatory mediators, chemokines, and other inflammatory markers. (A) mRNA expression fold changes (on the x axis) and the corresponding p-values (on the y axis). Gene names are labeled next to highlighted significant results. Taqman qPCR analysis of (B) pro-inflammatory, (C) chemokines, (D) cellular recruitment, (E) cellular marker, (F) inflammation marker, (G) transcription factors, (H) apoptosis marker, and (I) anti-inflammatory genes in the colonic tissues of control and EPEC-infected mice with moderate (score=2) or severe (score=3) diarrhea scores at day 3 p.i. (A–I) Expression levels were normalized with GAPDH, as an internal housekeeping gene. Bars represent mean ± SEM (n=3). #p<0.05, *p<0.01, **p<0.001 and ***p<0.0001 using multiple Student’s t-test. Levels of (J) pSTAT3, (K) pCREB and (L) cleaved caspase-3 in colonic tissue lysates of control and EPEC-infected mice at day 3 p.i. measured using ELISA. Bars represent mean ± SEM (n=8). #p<0.05 using Student’s t-test.
Signal transducer and activator of transcription 3 (STAT3) is a transcription factor involved in response of cytokines such as IL-6, and its activation results in expression of target genes involved in inflammatory and anti-inflammatory responses (Griesinger et al., 2015; Aden et al., 2016). To investigate the levels of phosphorylated STAT3 (pSTAT3), its active form, we found increased levels of pSTAT3 in the colon of mice infected with EPEC on day 3 p.i. (p<0.05, Figure 4J). Cyclic AMP Response Element-Binding protein (CREB) is another transcription factor involved on the transcription of inflammatory (such as IL-6, IL-2, and TNF-α) and anti-inflammatory (IL-4, IL-10 and IL-13) mediators (Westbom et al., 2014; Zhang et al., 2019). To determine the levels of phosphorylated CREB (pCREB), its active form, we examined, but did not find a difference between EPEC-infected mice and controls on day 3 p.i. (Figure 4K).
Given that EPEC infection increased the gene expression of apoptosis markers, we further evaluated the levels of cleaved caspase-3 using ELISA. We found that EPEC infection induced an increase in cleaved caspase-3 in the colonic tissues when compared to control mice at day 3 p.i. (p<0.05, Figure 4L), confirming an apoptosis process during EPEC infection.
EPEC Infection Model Induces Metabolic Perturbations
Metabolic perturbations following EPEC infection were further analyzed using Principal Component analysis (PCA). Urinary metabolic profiles of each of the mice infected with EPEC were compared to the age-matched uninfected mice at days 1 and 3 p.i. No differences were observed between the controls and EPEC infected mice on the day 1 p.i. (Figure 5A).
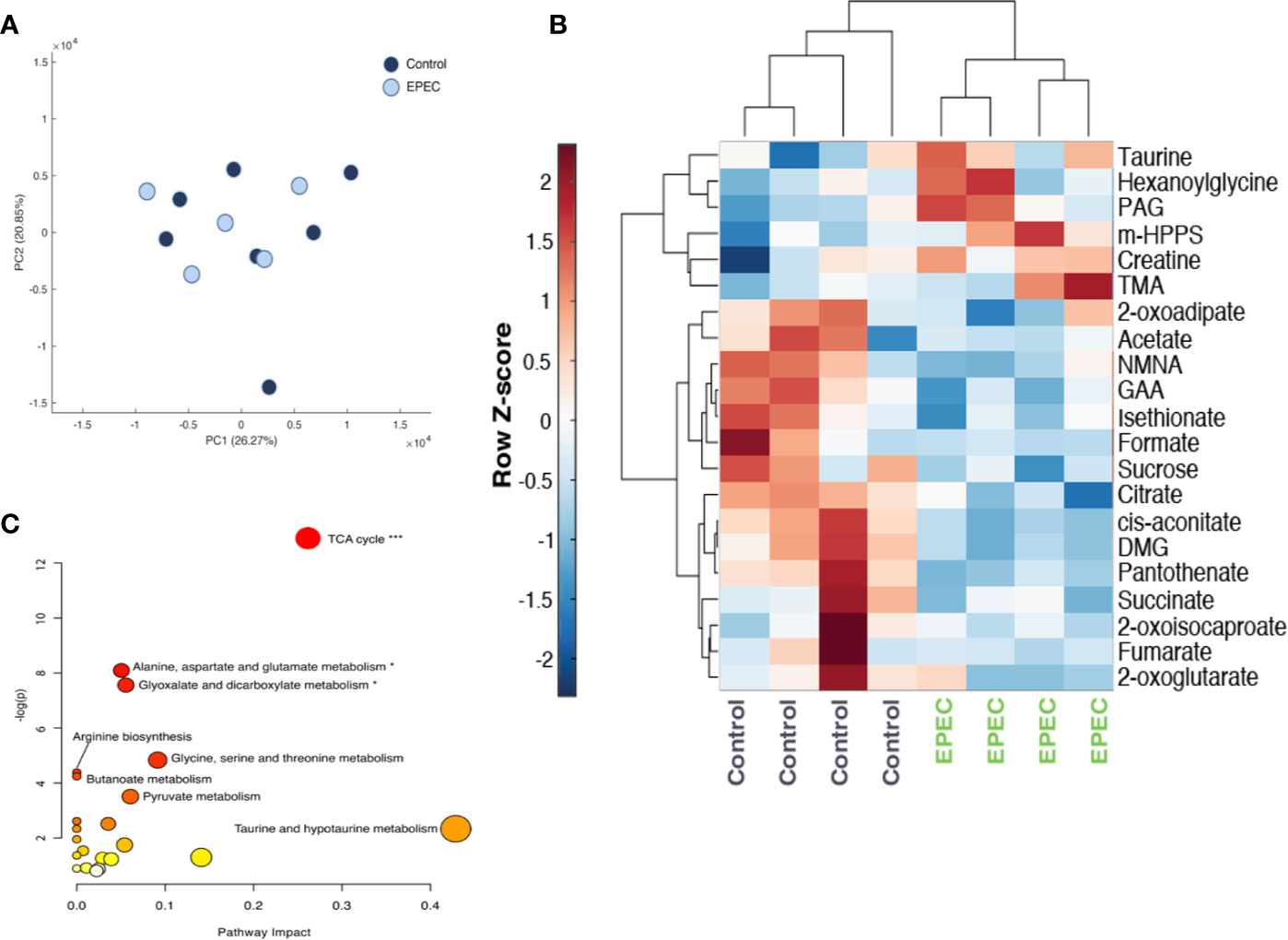
Figure 5 Metabolomic analysis of urinary specimens collected from age-matched, control, and EPEC infected mice. (A) Principal component analysis (PCA) scores plot at day 1 p.i. showing no difference in the metabolic profiles of between infected and uninfected mice (control n=7, EPEC n=5). (B) Unsupervised hierarchical clustering and heat-map of urinary metabolites from control and EPEC-infected mice at day 3 p.i (n=4). Each row represents a metabolite and each column represents a mouse sample. The row Z-score (scaled expression value) of each metabolite is plotted in red-blue color. The red color indicates metabolites, which are high in abundance and blue indicates metabolites low in abundance. DMG, dimethylglycine; GAA, guanidinoacetic acid; m-HPPS, m-hydroxyphenylpropionylsulfate; NMNA, N-methyl-nicotinic acid; PAG, phenylacetylglycine; TMA, trimethylamine. (C) Pathway analysis plot created using MetaboAnalyst 4.0 showing metabolic pathway alterations induced by EPEC. Each pathway is represented as a circle. Darker colors indicate more significant changes of metabolites annotated on the corresponding pathway. The x-axis represents the pathway impact value computed from pathway topological analysis, and the y-axis is the-log of the p-value obtained from pathway enrichment analysis. The pathways that were most significantly changed are characterized by both a high-log(p) value and high impact value (top right region). FDR adjusted p-values, *p<0.05, ***p<0.0001.
EPEC infection-driven metabolic variation was, however, observed at day 3 pi (PCA model: Q2 = 0.25, R2 = 0.59 (1000 permutations). EPEC infection resulted in reduced excretion of TCA cycle metabolites (succinate, cis-aconitate, citrate, 2-oxoglutarate and fumarate) and choline related metabolite, dimethylglycine (DMG) and increased trimethylamine (TMA) (Figure 5B). Lower urinary excretion of the tryptophan catabolite N-methylnicotinic acid (NMNA), the creatinine precursor guanidinoacetic acid (GAA) and the amino acid catabolites 2-oxoisocaproate, 2-oxoadipate were also observed. Isethionate, formate, pantothenate, and sucrose were also excreted in lower amounts by EPEC infected mice. Increases in the excretion of gut microbial-derived metabolites [acetate, phenylacetylglycine (PAG), m-hydroxyphenylpropionylsulfate (m-HPPS)], were observed in infected mice. Urinary excretion of taurine, creatine, and b-oxidation product hexanoylglycine were also elevated at day 3 p.i (Figure 5B).
Pathway analysis revealed that the TCA cycle was the biochemical pathway most influenced by the infection (Impact: 0.26, p-value: 2.5E-6, FDR adjusted p-value: 2.1E-4) (Figure 5C).
EPEC Infection Increases Intestinal Permeability and Decreases Colonic Claudin-1 Expression in Mice
Alteration on intestinal permeability related to EPEC infection has been reported in children (Johansen et al., 1989). Given that the intestinal tissues of EPEC-infected mice showed increase in inflammatory markers on later stage of disease, we investigated whether intestinal permeability was altered by using a FITC dextran assay in our experimental model. As shown in Figure 6A, EPEC infection resulted in increased levels of plasma 4kDa FITC dextran, indicating higher intestinal permeability when compared to control mice at day 7 p.i. (p<0.006). A strong positive correlation was found between the levels of plasma 4kDa FITC dextran and colonic INF-γ at day 7 p.i. in EPEC-infected mice (p<0.003, r=0.9219, Figure 6B).
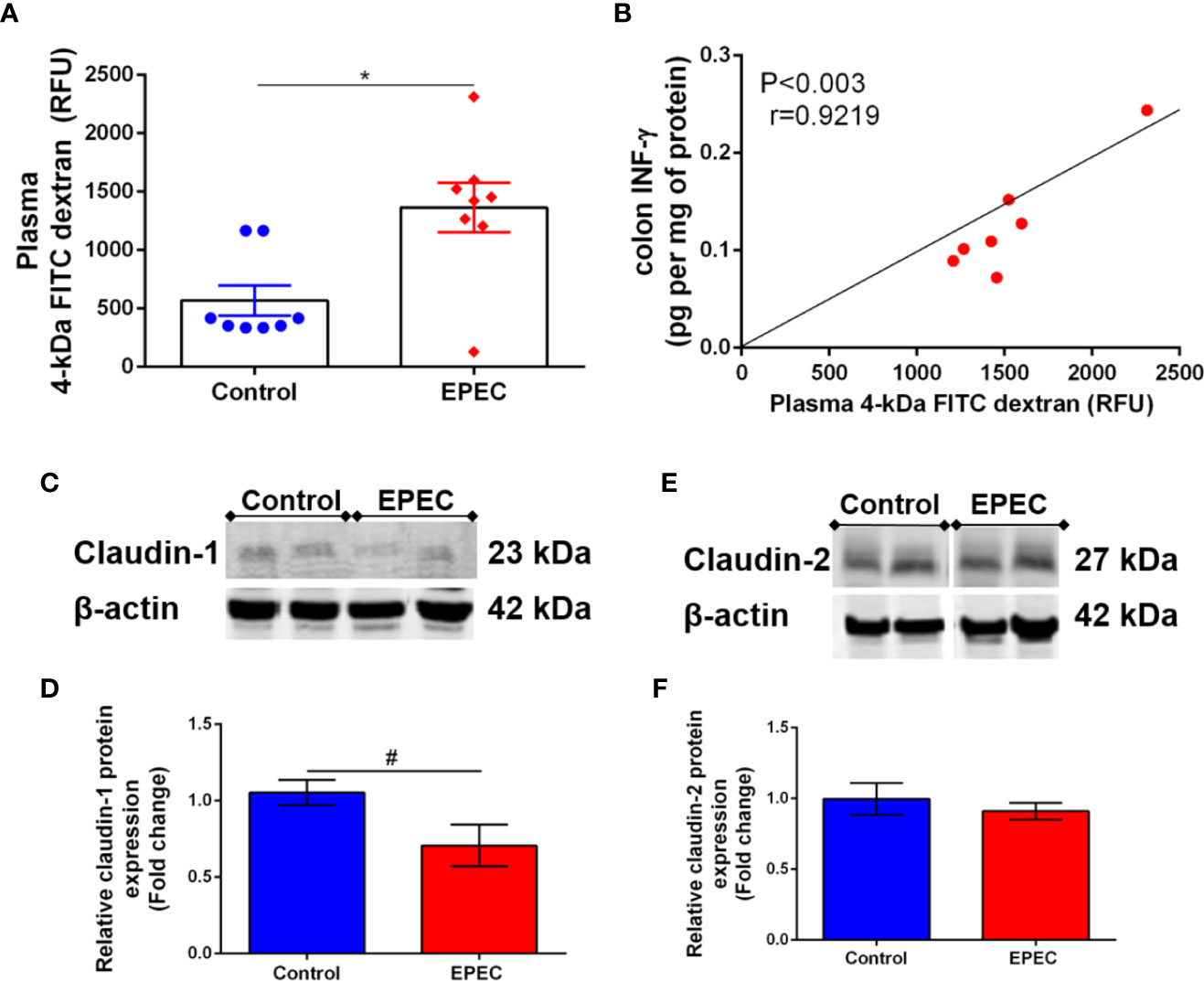
Figure 6 EPEC infection model increases intestinal permeability and decreases claudin-1 expression in colon of mice. (A) Intestinal permeability was determined in the plasma of control and EPEC-infected mice at day 7 p.i. using FITC dextran assay. Each point represents a mouse. Lines represent mean ± SEM (n=8). *p < 0.006 using Student’s t-test. (B) Significant positive correlation between diarrhea score and INF-γ levels in colon tissues of EPEC-infected mice at day 7 p.i (Spearman rank test). (C). Representative western blots of levels of claudin-1 and β-actin in colonic tissues of control and EPEC-infected mice at day 7 p.i. (D) Quantification of western blot bands of claudin-1 and β-actin. Bars represent mean ± SEM (n=3). #p < 0.03 using Student’s t-test. (E) Representative western blots of levels of claudin-2 and β-actin in colonic tissues of control and EPEC-infected mice at day 7 p.i. (F) Quantification of western blot bands of Claudin-2 and β-actin. Bars represent mean ± SEM (n=3).
Tight junctions play a crucial role in regulating intestinal permeability, we therefore, investigated if EPEC infection alters claudin-1 and claudin-2 expression in the colon of mice, and found that EPEC infection decreased claudin-1 (p<0.03, Figures 6C, D), but not claudin 2 (Figures 6E, F) in the colon when compared to control mice. Uncropped versions of Figures 6C, E are available in Supplementary Data (Supplementary Figures 3A, B).
Loss of escN Expression in EPEC Inhibits Intestinal and Systemic Inflammation Induced by Wild-Type Enteropathogenic Escherichia coli Infection Model Without Changes in Stools Shedding in Mice
The T3SS is essential for EPEC pathogenesis, and disruption of the escN gene (ATPase energizer), can lead to inefficient injection of EPEC effectors into the host cell (Andrade et al., 2007). We therefore, tested whether inactivation of escN in EPEC strain could affect changes in body weight, intestinal and systemic inflammation in our EPEC infection model. The ΔescN EPEC-infected mice exhibited weight gain when compared to wild-type (WT) EPEC-infected mice on days 1 and 3 p.i (p<0.05 and p<0.001, respectively, Supplementary Figure 4). Deletion of ΔescN did not affect EPEC shedding in the stools (Figure 7A) and colonization in the intestinal tissues (Figure 7B). However, at day 8 p.i., tissue burden of ΔescN EPEC infected mice was detected only in the colonic tissue (p<0.01 Figure 7C). No histological changes were observed in the colon of mice infected with ΔescN EPEC when compared to mice infected with WT EPEC (Figure 7D). Furthermore, ΔescN EPEC infection resulted in decreased LCN-2 in the stools (days 2 and 7 p.i., p<0.01 and p<0.05, respectively) and cecal contents (day 3 p.i., p<0.05) (Figure 7E); and MPO in stools (day 2 p.i., p<0.01) (Figure 7F), as well as IL-6 in ileum and colon (day 3 p.i., p<0.01) (Figure 7G).
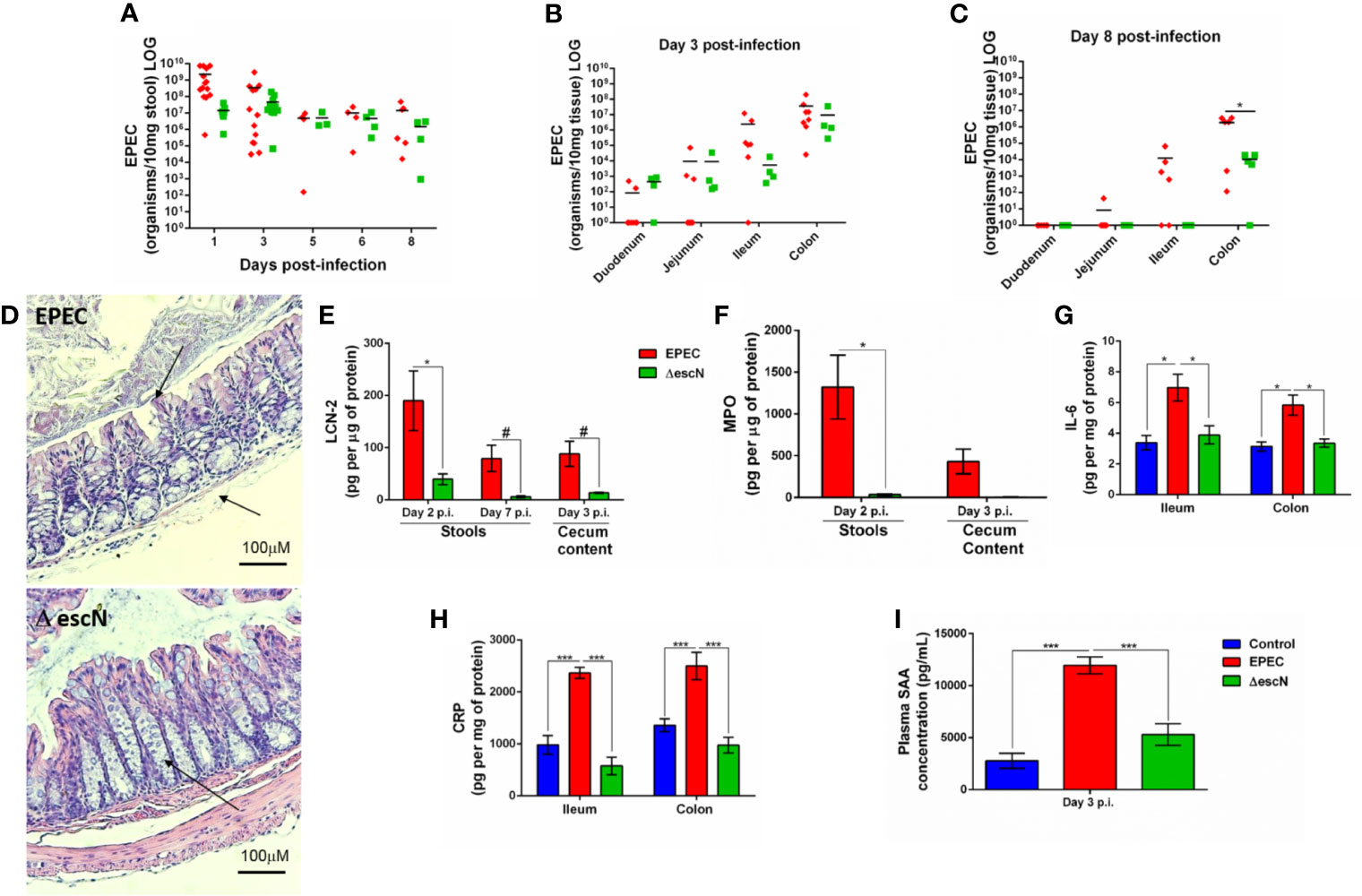
Figure 7 Inactivation of escN in EPEC decreases colonization in colon and reduces intestinal and systemic inflammation promoted by EPEC infection in mice. (A) Quantification of EPEC shedding in stools of WT EPEC (EPEC) or ΔescN EPEC infected mice. (B, C) Quantification of EPEC tissue burden in intestinal tissues (duodenum, jejunum, ileum and colon) at day 3 and 8 p.i. Bars represent mean ± SEM. *p < 0.01 using multiple Student’s t-test. (D) Representative H&E stains of colon from WT EPEC (EPEC) or ΔescN infected mice at day 3 p.i. Scale bars, 100 μm. (E) LCN-2 levels measured in stools (days 2 and 7 p.i.) and cecal contents (day 3 p.i.) of WT EPEC (EPEC) or ΔescN infected mice. (F) MPO levels measured in stools (day 2 p.i.) and cecal contents (day 3 p.i.). (G) IL-6 levels measured in the ileal and colonic tissue lysates at day 3 p.i. using ELISA (H) CRP levels measured in the ileal and colonic tissue lysates of uninfected (control, in blue), WT EPEC (in red) or ΔescN EPEC (in green) infected mice at day 3 p.i (I). Concentration of SAA in plasma at day 3 p.i. was determined using ELISA. Bars represent mean ± SEM (n=8). #p < 0.05, *p < 0.01 and ***p < 0.0001 using multiple Student’s t-test (E, F) or One-way ANOVA followed by Turkey´s test (G–I).
To assess whether WT EPEC infection induces systemic inflammation, as well as the role of the T3SS using ΔescN during infection, C-reactive protein (CRP) in intestinal tissues (ileum and colon) and serum amyloid A (SAA) in plasma of mice collected at day 3 p.i. were measured. WT EPEC infection resulted in increased the levels of CRP in the intestinal tissues (p<0.0001, Figure 7H) and increased SAA (p<0.0001, Figure 7I) when compared to control mice. Whereas deletion of ΔescN in EPEC led to a significant decrease in levels of these markers of systemic inflammation at day 3 p.i. when compared to WT EPEC-infected mice (p<0.0001, Figures 7H, I).
Taken together, these findings showed that EPEC induces later colonic colonization, intestinal and systemic inflammation, in an escN-encoded T3SS-dependent manner, confirming the effectivity of this new EPEC infection model in mice.
Discussion
Typical EPEC infections have been suggested to be associated with inflammatory enteropathy and/or diarrhea in resource-limited populations (Platts-Mills et al., 2015; Rogawski et al., 2018). Infections caused by EPEC have been widely reported in in vitro studies to demonstrate the effects of adherence traits (Knutton et al., 1999) and the T3SS (Andrade et al., 2007). However, there was still a need for a suitable in vivo model that clearly shows the effects of human EPEC infection in an intestinal environment. Citrobacter rodentium is a natural murine pathogen that has also been shown to cause A/E lesions, with the formation of pedestal structures and polarized actin accumulation at the site of infection analogous to that seen in human EPEC and Enterohemorrhagic E. coli (EHEC) infections (Luperchio and Schauer, 2001; Mundy et al., 2006).
Herein an EPEC murine infection model is reported using mice pretreated with antibiotic cocktail (vancomycin, gentamicin, metronidazole and colistin) to enable colonization, and induce growth impairment, acute diarrhea, intestinal damage and inflammation, as well as metabolomic perturbations and intestinal permeability alterations. Efforts to develop an EPEC model that mimics clinical outcomes, such as growth impairment and diarrhea as observed in humans have been a challenge for in vivo studies (Johansen et al., 1989; Savkovic et al., 2005; Law et al., 2013; Dupont et al., 2016). We also identified a two-phase disease promoted by the EPEC infection model, an acute symptomatic and a later asymptomatic phase. In the acute symptomatic phase of the disease, EPEC-infected mice had growth impairment and diarrhea as clinical outcomes, and this was accompanied with ileal and colonic damage (loss of integrity of epithelial cells, edema in submucosa and intense infiltrate of inflammatory cells) and intense inflammation. Similar findings showing disruption of colonic damage following EPEC infection has also been previously reported (Zhang et al., 2012).
In humans, EPEC infection promotes watery diarrhea and dehydration (Guerrant et al., 2011). Most of the EPEC infected mice developed moderate to severe diarrhea at day 3 p.i. Previously, C57BL/6 mice infected with EPEC have been reported to develop semi-solid stools in the proximal colon with no apparent diarrhea (Savkovic et al., 2005).
MPO has been used as biomarker of enteropathy in clinical studies (Guerrant et al., 2016; McCormick et al., 2017), also exhibiting inflammation, growth and development decrements in children infected with different enteropathogens in low-income countries. In our present study, MPO was increased during acute phase of disease, in the intestinal tissues (ileum and colon); MPO was detected mainly in stools of mice that developed soft or unformed stools or diarrhea. In addition, LCN-2 which is known as a neutrophil gelatinase-associated protein expressed by intestinal epithelial cells (Makhezer et al., 2019), was detected in higher concentrations in stools of all the mice that were infected with EPEC. Particularly impressive in our model was the striking inflammatory enteropathy (as evidenced by fecal LCN-2 and MPO during acute infection) with EPEC infected mice developing watery mucoid stools.
EPEC infection in infant C57BL/6 mice has been previously reported to colonize the small intestine and colon for 3 days as a result of human milk oligosaccharides administration (Manthey et al., 2014). Here, the ileum and colon were markedly colonized by EPEC during infection challenge. Similar to our findings, EPEC infection in other mouse models have also been reported to colonize the ileal and colonic tissues (Savkovic et al., 2005; Dupont et al., 2016). Moreover, the colon in our model was most affected by EPEC infection with higher colonization by EPEC; and similar high EPEC colonization has been previously reported in the colon of germ-free mice albeit without mention of diarrhea (Dupont et al., 2016).
The pro-inflammatory cytokines such as IL-6, IL-1β, IL-23, IL-22, and INF-γ were increased in colon of mice infected with EPEC. IL-6 is a pleiotropic cytokine showing a pro-inflammatory phenotype and is protective against infection. For instance, deficiency of IL-6 in C57BL/6 mice has been reported to cause colonic damage, increase infiltrate of inflammatory cells and apoptosis during infection with C. rodentium (Dann et al., 2008). Mice lacking IL-1β are more susceptible to C. rodentium-induced colonic inflammation (Liu et al., 2012), in contrast blocking IL-1β in EPEC-infected mice with persistent IL-1β response decreased the colonic damage (Sham et al., 2013), suggesting the role of IL-1β during intestinal infection in a concentration- and timing-dependent manner. Binding of IL-1β, to IL-1 receptor type I (IL-1RI) and activation of nuclear factor κB (NF-κB), promotes the recruitment of inflammatory cells at the site of inflammation by inducing the expression of adhesion molecules on endothelial cells and the release of chemokines (Bolick et al., 2007; Gabay et al., 2010). In our model, EPEC infection increased the expression of adhesion molecules (VCAM1, ICAM1, and SELP), as well as chemokines (CCL2, CCL5, CCL19, CXCL10, and CXCL11) and the chemokine receptors CCR2 (activated by CCL2 and expressed by macrophage and lymphocytes) and CCR7 (activated by CCL19, promoting migration of dendritic cells, monocytes and T cells) (Pezoldt et al., 2017), contributing to the intense recruitment of inflammatory cells in the colon, but not in the ileum, of EPEC-infected mice.
Similar to IL-6, INF-γ is a pleiotropic protein that promotes the transcription of pro-inflammatory mediators and CXCL10 (by binding to CXCR3 in order to promote the recruitment of monocytes/macrophages and T cells at the site of infection) (Lee et al., 2017), and were both upregulated in the colon of mice following challenge with EPEC, likely activating STAT1 by binding to INF-γ receptor (INFGR) (Green et al., 2017). In fecal samples from children with symptomatically EPEC infection, intermediate levels of INF-γ have been associated with an increase in infection duration (Long et al., 2010). INF-γ levels were increased in colonic tissues in our study, and severity of diarrhea was associated with higher levels of INF-γ in EPEC-infected mice at day 3 p.i.
IL-23, was also increased following EPEC infection, and has been shown to be required to promote IL-22 expression, a cytokine involved in promoting tissue regeneration and regulating inflammation, and also to negatively control the potentially deleterious production of IL-12 (Aychek et al., 2015). The data therefore, suggests that an increase in these cytokines during EPEC-infection in our model is protective, but not enough to prevent the intestinal damage promoted by EPEC. In C. rodentium infection, lack of IL-23 in macrophages led to increased mortality in mice (Aychek et al., 2015). Here, we also provided data suggesting that our EPEC infection model was able to activate NF-κB via IL-1β, STAT1 via INF-γ, and STAT3 via IL-22 and IL-6, but not CREB. STAT1 and STAT3 contribute to the expression of pro-apoptotic and anti-apoptotic genes respectively (Wittkopf et al., 2015; Lee et al., 2017). However, in the present study, it seemed that the response mediated by STAT1 (whose expression was higher than STAT3 in colon of EPEC-infected mice) prevailed over anti-apoptotic response promoted by STAT3, once increased cleaved caspase, a marker of apoptosis, were detected. Moreover, because our EPEC infection model exhibits evident diarrhea more investigation of how these cytokines contribute to its pathogeneses is needed.
Furthermore, during acute phase of infection, EPEC infection resulted in perturbations of multiple biochemical pathways, with the TCA cycle intermediates appearing to be the most sensitive to EPEC infection. The TCA cycle in E. coli is linked to energy metabolism in which CO2 concomitant is oxidized from pyruvate leading to production of NADH and FADH2 (Alteri and Mobley, 2012). In our model, the TCA cycle metabolites were excreted in lower quantities following EPEC infection, suggesting that energy production was reduced or conserved in the infected host. A shut-down of the TCA cycle during infection suggests that the energy requirements of the host were not met, potentially explaining in part the significant weight loss in the infected mice. C. jejuni infection in zinc deficient mice have also been reported to perturb the TCA cycle, affecting amino acid and muscle catabolism as a result of increased creatine excretion (Giallourou et al., 2018). Pantothenate is the key precursor of the fundamental TCA cycle cofactor, coenzyme A (Leonardi et al.). Reduced pantothenate excretion following EPEC colonization further adds to the TCA cycle disruption by infection. Interestingly, excretion of creatine which is a source for energy production in the form of ATP was also increased during infection. Sugiharto and colleagues reported on post-weaning pigs infected with E. coli F18 and found that there was a reduction in creatine and betaine which was due to inhibition of antioxidant system that resulted in piglets developing diarrhea (Sugiharto et al., 2014). Taurine has been shown to possess antioxidant properties and its concentrations are elevated in inflamed tissues where oxidants are abundant (Jeon et al., 2009; Oliveira et al., 2010). EPEC infection in this study was characterized by elevated urinary taurine excretion. As we have previously observed (Swann et al., 2011), treating rodents with antibiotics suppresses the bacterial metabolism of taurine thus increasing taurine bioavailability and uptake in the host reflected by greater urinary taurine excretion. Metabolites derived from bacteria in the gut were excreted in greater amounts following infection suggesting gut microbial metabolism was altered by EPEC infection. These findings help to understand host metabolism during infection, suggesting potential pathways to be further explored and targeted in future studies.
In relation to later phase of EPEC infection, we observed an increase in TNF-α in the colon and ileum, as well as increased LCN-2 in stools samples and increased intestinal permeability and decreased claudin-1. Despite TNF-α gene expression, as well as its receptor TNFRS being increased during the acute phase of disease, the TNF-α protein levels were increased in intestinal tissues of EPEC-infected mice only during the later phase. TNF-α synthesis is promoted by NF-κB activation which in turn can be promoted by IL-1β (Kalliolias and Ivashkiv, 2016). The biological effects of TNF-α mediated by binding to TNFRS include inflammation, apoptosis and tissue regeneration via activation of NF-κB, caspase-8 and AKT respectively (Kalliolias and Ivashkiv, 2016). Similar to our findings, later increases of TNF-α in the ileum and colon has been observed by others in EPEC-infected mice at day 5 p.i (Shifflett et al., 2005), findings that we showed at day 7 p.i. This increase in TNF-α was associated with increased LCN-2 in stools, indicating the presence of intestinal damage and inflammation, despite partial recovery from the acute phase of disease. TNF-α has been shown to induce LCN-2 expression by activating NF-κB (Makhezer et al., 2019). TNF-α and INF-γ have been associated with a loss of integrity of the intestinal epithelial barrier (Chelakkot et al., 2018). Despite TNF-α, but not INF-γ, being increased in colonic tissues in EPEC-infected mice, only INF-γ levels were associated positively with an increase in intestinal permeability; a similar association has also been reported in an in vitro study using T84 epithelial cell monolayer (Smyth et al., 2011). The permeability of the intestinal barrier is regulated by the tight junction proteins (Wang et al., 2017). EPEC infection has been reported to impair tight junction barrier function of ileal and colonic mucosa (Mundy et al., 2006; Guttman et al., 2006; Zhang et al., 2010; Zhang et al., 2012; Ugalde-Silva et al., 2016). Claudin-1, a component of tight junction expressed by epithelial cells from small and large intestine, is responsible for increasing barrier tightness (Luissint et al., 2016). In our model, a decrease of claudin-1 in the colon of EPEC-infected mice was associated with an increase in intestinal permeability. Although we detected alterations in intestinal permeability at the later stage of EPEC infection, this might have been due in part to an increase in systemic markers (SAA and CRP) that were detected at day 3 p.i. during the acute phase leading to disruption of intestinal tight junctions. SAA has been a biomarker of enteropathy in clinical studies also associated with inflammation, and with growth and developmental impairment in children infected with multiple enteropathogens in low-income countries (Guerrant et al., 2016).
The T3SS is essential for EPEC pathogenesis and requires an effective ATPase energizer, escN (Gauthier et al., 2003; Andrade et al., 2007; Zarivach et al., 2007). Here, we also demonstrated that mice infected with escN deletion mutant resulted in diminished growth impairment and inflammation. Even without an effective T3SS, escN mutant was able to colonize all sections of the intestinal tissue (due to the presence of functional bfp), albeit at much lower levels to day 8 p.i., as shown by our results. These findings reinforce the importance of a functional T3SS in the virulence of EPEC in this model (Frankel et al., 1998; Andrade et al., 2007).
In conclusion, our findings showed that EPEC infection causes growth impairment, diarrhea and increased inflammatory responses in weaned antibiotic pretreated mice. These effects were also dependent on an intact EPEC T3SS. In addition, metabolic perturbations and intestinal permeability were also observed in mice with EPEC infection, suggesting relevant biochemical pathways involved. Further, the findings presented here suggest that EPEC infections leads to an increase in intestinal and systemic inflammatory responses and transient overt diarrhea and growth impairment, as is often seen in children with EPEC infections. This EPEC infection model also presents two phases of diseases: an acute symptomatic and a later asymptomatic phase (Figure 8). This model can help further explore mechanisms involved in EPEC pathogenesis and perhaps facilitate the development of vaccines or therapeutic interventions.
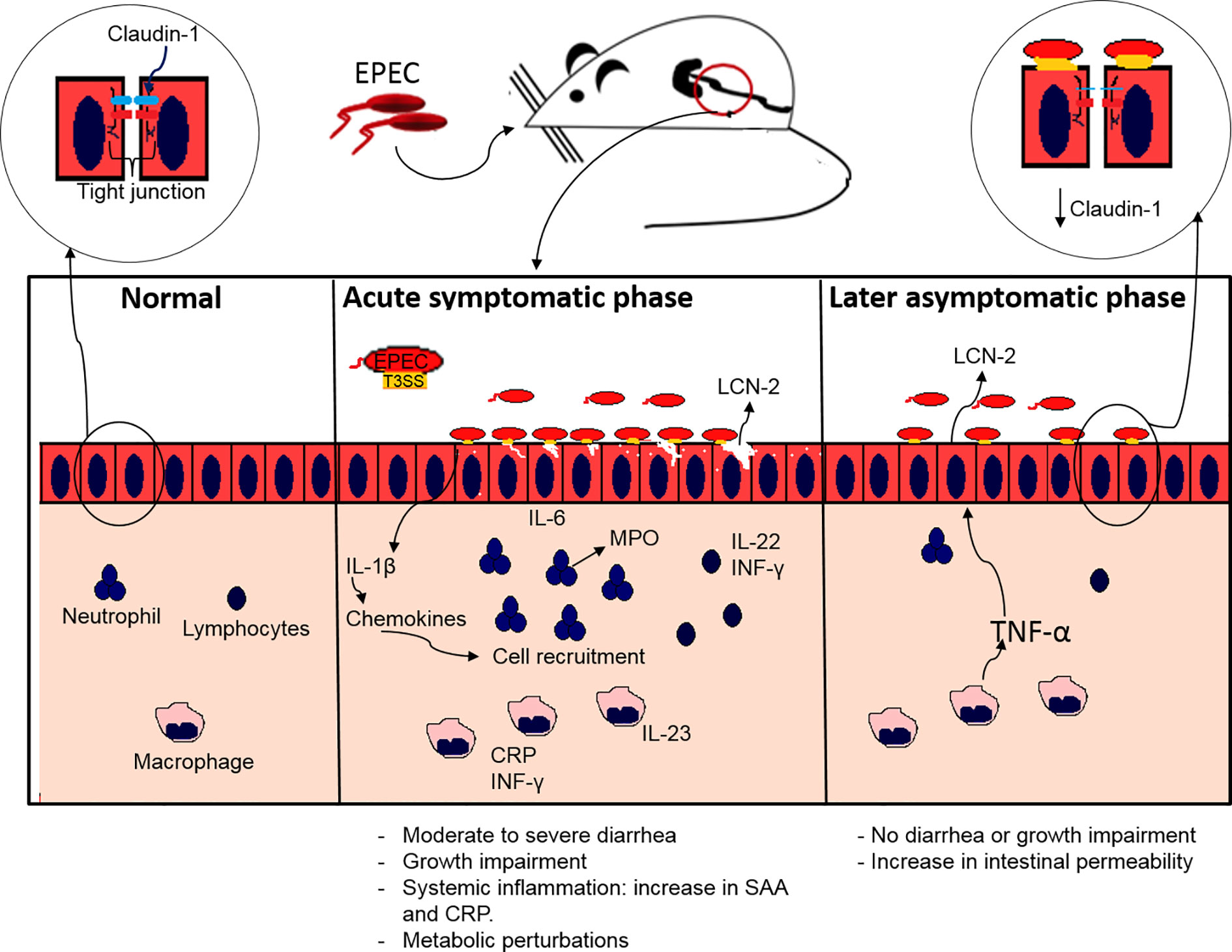
Figure 8 Proposed model of EPEC infection antibiotic pretreated mice displaying acute symptomatic and late asymptomatic phase. During acute symptomatic phase, colonization by EPEC leads to growth impairment, accompanied with moderate to severe diarrhea. Adherence of EPEC on the intestinal epithelial cells leads to increased IL-1β, which in turn stimulates chemokines synthesis, and consequently recruitment of neutrophils, macrophages and lymphocytes, resulting in increased release of MPO by neutrophils and LCN-2 by epithelial cells, as well as pro-inflammatory (IL-6, IL-23, INF-γ) and IL-22 cytokines indicating intestinal inflammation. A further increase in CRP and SAA indicates acute systemic inflammation. During the later asymptomatic phase an increase in LCN-2 inflammatory biomarker and an increase in TNF-α leads to increased intestinal permeability affecting the tight junction integrity by decreasing claudin-1 with no signs of diarrhea and growth impairment.
Author’s Note
This manuscript has been released as a pre-print at bioRxiv 2020.06.12.148593 (Ledwaba et al., 2020).
Data Availability Statement
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
Ethics Statement
The animal study was reviewed and approved by the Committee on the Ethics of Animal Experiments of the University of Virginia (Protocol Number: 3315). The mice used in the study have been handled with strict accordance with the recommendations in the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health. All efforts were made to minimize suffering. This is also in accordance with the Institutional Animal Care and Use Committee policies of the University of Virginia. The University of Virginia is accredited by the Association for the Assessment and Accreditation of Laboratory Animal Care, International (AAALAC).
Author Contributions
SL, DB, RG, JN, NP, and AT designed the project. SL, DC, and DB performed the experiments. SL, DC, DB, and RG analyzed the data and wrote the manuscript. PM assisted in analysis of results. NG and JS performed and assisted with metabolome analysis. All authors revised the manuscript. NP, AT, RG, and JN supervised the project. All authors contributed to the article and approved the submitted version.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
Research reported in this publication was supported by the Bill & Melinda Gates Foundation Opportunity ID: OPP1137923, the NIH’s Fogarty International Center award number D43TW006578, NIAID award number U19AI109776 and National Research Foundation (NRF). The content is solely the responsibility of the authors and does not necessarily represent the official views of the funders.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fcimb.2020.595266/full#supplementary-material
References
Abe C. M., Trabulsi L. R., Blanco J., Blanco M., Dahbi G., Blanco J. E., et al. (2009). Virulence features of atypical enteropathogenic Escherichia coli identified by the eae+ EAF-negative stx– genetic profile. Diagn. Microbiol. Infect. Dis. 64 (4), 357–365. doi: 10.1016/j.diagmicrobio.2009.03.025
Aden K., Rehman A., Falk-Paulsen M., Secher T., Kuiper J., Tran F., et al. (2016). Epithelial IL-23R signaling licenses protective IL-22 responses in intestinal inflammation. Cell Rep. 16 (8), 2208–2218. doi: 10.1016/j.celrep.2016.07.054
Alteri C. J., Mobley H. L. (2012). Escherichia coli physiology and metabolism dictates adaptation to diverse host microenvironments. Curr. Opin. Microbiol. 15 (1), 3–9. doi: 10.1016/j.mib.2011.12.004
Andrade A., Pardo J. P., Espinosa N., Pérez-Hernández G., González-Pedrajo B. (2007). Enzymatic characterization of the enteropathogenic Escherichia coli type III secretion ATPase EscN. Arch. Biochem. Biophys. 468 (1), 121–127. doi: 10.1016/j.abb.2007.09.020
Aychek T., Mildner A., Yona S., Kim K. W., Lampl N., Reich-Zeliger S., et al. (2015). IL-23-mediated mononuclear phagocyte crosstalk protects mice from Citrobacter rodentium-induced colon immunopathology. Nat. Commun. 6, 6525. doi: 10.1038/ncomms7525
Bolick D. T., Whetzel A. M., Skaflen M., Deem T. L., Lee J., Hedrick C. C. (2007). Absence of the G Protein–Coupled Receptor G2A in Mice Promotes Monocyte/Endothelial Interactions in Aorta. Circ. Res. 100 (4), 572–580. doi: 10.1161/01.RES.0000258877.57836.d2
Bolick D. T., Medeiros P. H., Ledwaba S. E., Lima A. A., Nataro J. P., Barry E. M., et al. (2018). Critical Role of Zinc in a new murine model of enterotoxigenic Escherichia coli Diarrhea. Infect. Immun. 86 (7). doi: 10.1128/IAI.00183-18
Chelakkot C., Ghim J., Ryu S. H. (2018). Mechanisms regulating intestinal barrier integrity and its pathological implications. Exp. Mol. Med. 50 (8):103. doi: 10.1038/s12276-018-0126-x
Dann S. M., Spehlmann M. E., Hammond D. C., Iimura M., Hase K., Choi L. J., et al. (2008). IL-6-dependent mucosal protection prevents establishment of a microbial niche for attaching/effacing lesion-forming enteric bacterial pathogens. J. Immunol. 180 (10), 6816–6826. doi: 10.4049/jimmunol.180.10.6816
Dean-Nystrom E. A., Gansheroff L. J., Mills M., Moon H. W., O’Brien A. D. (2002). Vaccination of pregnant dams with intiminO157 protects suckling piglets from Escherichia coli O157: H7 infection. Infect. Immun. 70 (5), 2414–2418. doi: 10.1128/iai.70.5.2414-2418.2002
Dupont A., Sommer F., Zhang K., Repnik U., Basic M., Bleich A., et al. (2016). Age-dependent susceptibility to enteropathogenic Escherichia coli (EPEC) infection in mice. PloS Pathog. 12 (5), e1005616. doi: 10.1371/journal.ppat.1005616
Fabich A. J., Jones S. A., Chowdhury F. Z., Cernosek A., Anderson A., Smalley D., et al. (2008). Comparison of carbon nutrition for pathogenic and commensal Escherichia coli strains in the mouse intestine. Infect. Immun. 76 (3), 1143–1152. doi: 10.1128/IAI.01386-07
Frankel G., Phillips A. D., Rosenshine I., Dougan G., Kaper J. B., Knutton S. (1998). Enteropathogenic and enterohaemorrhagic Escherichia coli: more subversive elements. Mol. Microbiol. 30 (5), 911–921. doi: 10.1046/j.1365-2958.1998.01144.x
Freter R., Brickner H., Fekete J., Vickerman M. M., Carey K. E. (1983). Survival and implantation of Escherichia coli in the intestinal tract. Infect. Immun. 39 (2), 686–703. doi: 10.1128/IAI.39.2.686-703.1983
Friedberg D., Umanski T., Fang Y., Rosenshine I. (1999). Hierarchy in the expression of the locus of enterocyte effacement genes of enteropathogenic Escherichia coli. Mol. Microbiol. 34 (5), 941–952. doi: 10.1046/j.1365-2958.1999.01655.x
Gabay C., Lamacchia C., Palmer G. (2010). IL-1 pathways in inflammation and human diseases. Nat. Rev. Rheumatol. 6 (4), 232–241. doi: 10.1038/nrrheum.2010.4
Gauthier A., Puente J. L., Finlay B. B. (2003). Secretin of the enteropathogenic Escherichia coli type III secretion system requires components of the type III apparatus for assembly and localization. Infect. Immun. 71 (6), 3310–3319. doi: 10.1128/iai.71.6.3310-3319.2003
Giallourou N., Medlock G. L., Bolick D. T., Medeiros P. H., Ledwaba S. E., Kolling G. L., et al. (2018). A novel mouse model of Campylobacter jejuni enteropathy and diarrhea. PloS Pathog. 14 (3), e1007083. doi: 10.1371/journal.ppat.1007083
Giron J. A., Ho A. S., Schoolnik G. K. (1991). An inducible bundle-forming pilus of enteropathogenic Escherichia coli. Science 254 (5032), 710–713. doi: 10.1126/science.1683004
Green D. S., Young H. A., Valencia J. C. (2017). Current prospects of type II interferon γ signaling and autoimmunity. J. Biol. Chem. 292 (34), 13925–13933. doi: 10.1074/jbc.R116.774745
Griesinger A. M., Josephson R. J., Donson A. M., Levy J. M., Amani V., Birks D. K., et al. (2015). Interleukin-6/STAT3 pathway signaling drives an inflammatory phenotype in Group A ependymoma. Cancer Immunol. Res. 3 (10), 1165–1174. doi: 10.1158/2326-6066.CIR-15-0061
Guerrant R. L., Walker D. H., Weller P. F. (2011). Tropical Infectious Diseases: Principles, Pathogens and Practice E-Book. Elsevier Health Sci. 3, 110–120.
Guerrant R. L., Leite A. M., Pinkerton R., Medeiros P. H., Cavalcante P. A., DeBoer M., et al. (2016). Biomarkers of environmental enteropathy, inflammation, stunting, and impaired growth in children in northeast Brazil. PloS One 11 (9), e0158772. doi: 10.1371/journal.pone.0158772
Guttman J. A., Samji F. N., Li Y., Vogl A. W., Finlay B. B. (2006). Evidence that tight junctions are disrupted due to intimate bacterial contact and not inflammation during attaching and effacing pathogen infection in vivo. Infect. Immun. 74 (11), 6075–6084. doi: 10.1128/IAI.00721-06
Hu J., Torres A. G. (2015). Enteropathogenic Escherichia coli: foe or innocent bystander? Clin. Microbiol. Infect. 21 (8), 729–734. doi: 10.1016/j.cmi.2015.01.015
Iguchi A., Thomson N. R., Ogura Y., Saunders D., Ooka T., Henderson I. R., et al. (2009). Complete genome sequence and comparative genome analysis of enteropathogenic Escherichia coli O127: H6 strain E2348/69. J. Bacteriol. 191 (1), 347–354. doi: 10.1128/JB.01238-08
Jarvis K. G., Giron J. A., Jerse A. E., McDaniel T. K., Donnenberg M. S., Kaper J. B. (1995). Enteropathogenic Escherichia coli contains a putative type III secretion system necessary for the export of proteins involved in attaching and effacing lesion formation. Proc. Natl. Acad. Sci. U.S.A. 92 (17), 7996–8000. doi: 10.1073/pnas.92.17.7996
Jeon S. H., Lee M. Y., Rahman M. M., Kim S. J., Kim G. B., Park S. Y., et al. (2009). The antioxidant, taurine reduced lipopolysaccharide (LPS)-induced generation of ROS, and activation of MAPKs and Bax in cultured pneumocytes. Pulm. Pharmacol. Ther. 22 (6), 562–566. doi: 10.1016/j.pupt.2009.07.004
Johansen K., Stintzing G., Magnusson K. E., Sundqvist T., Jalil F., Murtaza A., et al. (1989). Intestinal permeability assessed with polyethylene glycols in children with diarrhea due to rotavirus and common bacterial pathogens in a developing community. J. Pediatr. Gastroenterol. Nutr. J. Pediatr. Gastroenterol. Nutr. 9 (3), 307–313. doi: 10.1097/00005176-198910000-00008
Kalliolias G. D., Ivashkiv L. B. (2016). TNF biology, pathogenic mechanisms and emerging therapeutic strategies. Nat. Rev. Rheumatol. 12 (1), 49–62. doi: 10.1038/nrrheum.2015.169
Kaper J. B., Nataro J. P., Mobley H. L. (2004). Pathogenic Escherichia coli. Nat. Rev. Microbiol. 2 (2), 123–140. doi: 10.1038/nrmicro818
Knutton S., Baldwin T., Williams P. H., McNeish A. S. (1989). Actin accumulation at sites of bacterial adhesion to tissue culture cells: basis of a new diagnostic test for enteropathogenic and enterohemorrhagic Escherichia coli. Infect. Immun. 57 (4), 1290–1298. doi: 10.1128/IAI.57.4.1290-1298.1989
Knutton S., Shaw R. K., Anantha R. P., Donnenberg M. S., Zorgani A. A. (1999). The type IV bundle-forming pilus of enteropathogenic Escherichia coli undergoes dramatic alterations in structure associated with bacterial adherence, aggregation and dispersal. Mol. Microbiol. 33 (3), 499–509. doi: 10.1046/j.1365-2958.1999.01495.x
Kosek M. N., Ahmed T., Bhutta Z., Caulfield L., Guerrant R., Houpt E., et al. (2017). Causal pathways from enteropathogens to environmental enteropathy: findings from the MAL-ED birth cohort study. EBioMedicine 18, 109–117. doi: 10.1016/j.ebiom.2017.02.024
Kotloff K. L., Nataro J. P., Blackwelder W. C., Nasrin D., Farag T. H., Panchalingam S., et al. (2013). Burden and aetiology of diarrhoeal disease in infants and young children in developing countries (the Global Enteric Multicenter Study, GEMS): a prospective, case-control study. Lancet 382 (9888), 209–222. doi: 10.1016/S0140-6736(13)60844-2
Larsen P. L., Albert P. S., Riddle D. L. (1995). Genes that regulate both development and longevity in Caenorhabditis elegans. Genetics 139 (4), 1567–1583.
Law R. J., Gur-Arie L., Rosenshine I., Finlay B. B. (2013). In vitro and in vivo model systems for studying enteropathogenic Escherichia coli infections. Cold Spring Harb. Perspect. Med. 3 (3), a009977. doi: 10.1101/cshperspect.a009977
Ledwaba E., Costa D., Bolick D. T., Giallourou N., Medeiros P. H., Swann J., et al. (2020). Enteropathogenic Escherichia coli (EPEC) infection induces diarrhea, intestinal damage, metabolic alterations and increased intestinal permeability in a murine model. bioRxiv. doi: 10.1101/2020.06.12.148593
Lee J. H., Kim B., Jin W. J., Kim H. H., Ha H., Lee Z. H. (2017). Pathogenic roles of CXCL10 signaling through CXCR3 and TLR4 in macrophages and T cells: relevance for arthritis. Arthritis Res. Ther. 19 (1), 163. doi: 10.1186/s13075-017-1353-6
Leonardi R., Zhang Y. M., Lykidis A., Rock C. O., Jackowski S. (2007). Localization and regulation of mouse pantothenate kinase 2. FEBS Lett. 581, 4639–4644. doi: 10.1016/j.febslet.2007.08.056
Leverton L. Q., Kaper J. B. (2005). Temporal expression of enteropathogenic Escherichia coli virulence genes in an in vitro model of infection. Infect. Immun. 73 (2), 1034–1043. doi: 10.1128/IAI.73.2.1034-1043.2005
Levine M. M., Nataro J. P., Karch H., Baldini M. M., Kaper J. B., Black R. E., et al. (1985). The diarrheal response of humans to some classic serotypes of enteropathogenic Escherichia coli is dependent on a plasmid encoding an enteroadhesiveness factor. J. Infect. Dis. 152 (3), 550–559. doi: 10.1093/infdis/152.3.550
Liu Z., Zaki M. H., Vogel P., Gurung P., Finlay B. B., Deng W., et al. (2012). Role of inflammasomes in host defense against Citrobacter rodentium infection. J. Biol. Chem. 287 (20), 16955–16964. doi: 10.1074/jbc.M112.358705
Liu L., Oza S., Hogan D., Perin J., Rudan I., Lawn J. E., et al. (2015). Global, regional, and national causes of child mortality in 2000–13, with projections to inform post-2015 priorities: an updated systematic analysis. Lancet 385 (9966), 430–440. doi: 10.1016/S0140-6736(14)61698-6
Livak K. J., Schmittgen T. D. (2001). Analysis of relative gene expression data using real-time quantitative PCR and the 2– ΔΔCT method. Methods 25 (4), 402–408. doi: 10.1006/meth.2001.1262
Long K. Z., Rosado J. L., Santos J. I., Haas M., Al Mamun A., DuPont H. L., et al. (2010). Associations between mucosal innate and adaptive immune responses and resolution of diarrheal pathogen infections. Infect. Immun. 78 (3), 1221–1228. doi: 10.1128/IAI.00767-09
Luissint A. C., Parkos C. A., Nusrat A. (2016). Inflammation and the intestinal barrier: leukocyte–epithelial cell interactions, cell junction remodeling, and mucosal repair. Gastroenterol 151 (4), 616–632. doi: 10.1053/j.gastro.2016.07.008
Luperchio S. A., Schauer D. B. (2001). Molecular pathogenesis of Citrobacter rodentium and transmissible murine colonic hyperplasia. Microbes Infect. 3 (4), 333–340. doi: 10.1016/s1286-4579(01)01387-9
Makhezer N., Khemis M. B., Liu D., Khichane Y., Marzaioli V., Tlili A., et al. (2019). NOX1-derived ROS drive the expression of Lipocalin-2 in colonic epithelial cells in inflammatory conditions. Mucosal Immunol. 12 (1), 117–131. doi: 10.1038/s41385-018-0086-4
Manthey C. F., Autran C. A., Eckmann L., Bode L. (2014). Human milk oligosaccharides protect against enteropathogenic Escherichia coli attachment in vitro and EPEC colonization in suckling mice. J. Pediatr. Gastroenterol. Nutr. 58 (2), 165–168. doi: 10.1097/MPG.0000000000000172
McCormick B. J., Lee G. O., Seidman J. C., Haque R., Mondal D., Quetz J., et al. (2017). Dynamics and trends in fecal biomarkers of gut function in children from 1–24 months in the MAL-ED study. Am. J. Trop. Med. Hyg. 96 (2), 465–472. doi: 10.4269/ajtmh.16-0496
Medeiros PH Q. S., Ledwaba S. E., Bolick D. T., Giallourou N., Yum L. K., Costa D. V., et al. (2019). A murine model of diarrhea, growth impairment and metabolic disturbances with Shigella flexneri infection and the role of zinc deficiency. Gut Microbes 10 (5), 615–630. doi: 10.1080/19490976.2018.1564430
Misyurina O., Asper D. J., Deng W., Finlay B. B., Rogan D., Potter A. A. (2010). The role of Tir, EspA, and NleB in the colonization of cattle by Shiga toxin producing Escherichia coli O26: H11. Can. J. Microbiol. 56 (9), 739–747. doi: 10.1139/w10-059
Moon H. W., Whipp S. C., Argenzio R. A., Levine M. M., Giannella R. A. (1983). Attaching and effacing activities of rabbit and human enteropathogenic Escherichia coli in pig and rabbit intestines. Infect. Immun. 41 (3), 1340–1351. doi: 10.1128/IAI.41.3.1340-1351.1983
Mora A., Blanco M., Yamamoto D., Dahbi G., Blanco J. E., López C., et al. (2009). HeLa-cell adherence patterns and actin aggregation of enteropathogenic Escherichia coli (EPEC) and Shiga-toxin-producing E. coli (STEC) strains carrying different eae and tir alleles. Int. Microbiol. 12 (4), 243–251.
Mundy R., Girard F., FitzGerald A. J., Frankel G. (2006). Comparison of colonization dynamics and pathology of mice infected with enteropathogenic Escherichia coli, enterohaemorrhagic E. coli and Citrobacter rodentium. FEMS Microbiol. Lett. 265 (1), 126–132. doi: 10.1111/j.1574-6968.2006.00481.x
Oliveira M. W., Minotto J. B., de Oliveira M. R., Zanotto-Filho A., Behr G. A., Rocha R. F., et al. (2010). Scavenging and antioxidant potential of physiological taurine concentrations against different reactive oxygen/nitrogen species. Pharmacol. Rep. 62 (1), 185–193. doi: 10.1016/s1734-1140(10)70256-5
Pelayo J. S., Scaletsky I. C., Pedroso M. Z., Sperandio V., Giron J. A., Frankel G., et al. (1999). Virulence properties of atypical EPEC strains. J. Med. Microbiol. 48 (1), 41–49. doi: 10.1099/00222615-48-1-41
Pezoldt J., Pisano F., Heine W., Pasztoi M., Rosenheinrich M., Nuss A. M., et al. (2017). Impact of CCR7 on T-Cell Response and Susceptibility to Yersinia pseudotuberculosis Infection. J. Infect. Dis. 216 (6), 752–760. doi: 10.1093/infdis/jix037
Platts-Mills J. A., Babji S., Bodhidatta L., Gratz J., Haque R., Havt A., et al. (2015). Pathogen-specific burdens of community diarrhoea in developing countries: a multisite birth cohort study (MAL-ED). Lancet Glob. Health 3 (9), e564–e575. doi: 10.1016/S2214-109X(15)00151-5
Prata M. D., Havt A., Bolick D. T., Pinkerton R., Lima A. A., Guerrant R. L. (2016). Comparisons between myeloperoxidase, lactoferrin, calprotectin and lipocalin-2, as fecal biomarkers of intestinal inflammation in malnourished children. J. Trasl. Sci. 2 (2), 134. doi: 10.15761/JTS.1000130
Rhee K. J., Cheng H., Harris A., Morin C., Kaper J. B., Hecht G. (2011). Determination of spatial and temporal colonization of enteropathogenic E. coli and enterohemorrhagic E. coli in mice using bioluminescent in vivo imaging. Gut Microbes 2 (1), 34–41. doi: 10.4161/gmic.2.1.14882
Rogawski E. T., Liu J., Platts-Mills J. A., Kabir F., Lertsethtakarn P., Siguas M., et al. (2018). Use of quantitative molecular diagnostic methods to investigate the effect of enteropathogen infections on linear growth in children in low-resource settings: longitudinal analysis of results from the MAL-ED cohort study. Lancet Glob. Health 6 (12), e1319–e1328. doi: 10.1016/S2214-109X(18)30351-6
Royan S. V., Jones R. M., Koutsouris A., Roxas J. L., Falzari K., Weflen A. W., et al. (2010). Enteropathogenic E. coli non-LEE encoded effectors NleH1 and NleH2 attenuate NF-κB activation. Mol. Microbiol. 78 (5), 1232–1245. doi: 10.1111/j.1365-2958.2010.07400.x
Savkovic S. D., Villanueva J., Turner J. R., Matkowskyj K. A., Hecht G. (2005). Mouse model of enteropathogenic Escherichia coli infection. Infect. Immun. 73 (2), 1161–1170. doi: 10.1128/IAI.73.2.1161-1170.2005
Scaletsky I. C., Aranda K. R., Souza T. B., Silva N. P. (2010). Adherence factors in atypical enteropathogenic Escherichia coli strains expressing the localized adherence-like pattern in HEp-2 cells. J. Clin. Microbiol. 48 (1), 302–306. doi: 10.1128/JCM.01980-09
Sham H. P., Yu E. Y., Gulen M. F., Bhinder G., Stahl M., Chan J. M., et al. (2013). SIGIRR, a negative regulator of TLR/IL-1R signalling promotes Microbiota dependent resistance to colonization by enteric bacterial pathogens. PloS Pathog. 9 (8), e1003539. doi: 10.1371/journal.ppat.1003539
Shifflett D. E., Clayburgh D. R., Koutsouris A., Turner J. R., Hecht G. A. (2005). Enteropathogenic E. coli disrupts tight junction barrier function and structure in vivo. Lab. Invest. 85 (10), 1308–1324. doi: 10.1038/labinvest.3700330
Smyth D., Phan V., Wang A., McKay D. M. (2011). Interferon-γ-induced increases in intestinal epithelial macromolecular permeability requires the Src kinase Fyn. Lab. Invest. 91 (5), 764–777. doi: 10.1038/labinvest.2010.208
Sugiharto S., Hedemann M. S., Lauridsen C. (2014). Plasma metabolomic profiles and immune responses of piglets after weaning and challenge with E. coli. J. Anim. Sci. Biotechnol. 5 (1):17. doi: 10.1186/2049-1891-5-17
Swann J. R., Tuohy K. M., Lindfors P., Brown D. T., Gibson G. R., Wilson I. D., et al. (2011). Variation in antibiotic-induced microbial recolonization impacts on the host metabolic phenotypes of rats. J. Proteome Res. 10 (8), 3590–3603. doi: 10.1021/pr200243t
Ugalde-Silva P., Gonzalez-Lugo O., Navarro-Garcia F. (2016). Tight junction disruption induced by type 3 secretion system effectors injected by enteropathogenic and enterohemorrhagic Escherichia coli. Front. Cell Infect. Microbiol. 6, 87. doi: 10.3389/fcimb.2016.00087
Wang Y., Mumm J. B., Herbst R., Kolbeck R., Wang Y. (2017). IL-22 increases permeability of intestinal epithelial tight junctions by enhancing claudin-2 expression. J. Immunol. 199 (9), 3316–3325. doi: 10.4049/jimmunol.1700152
Westbom C. M., Shukla A., MacPherson M. B., Yasewicz E. C., Miller J. M., Beuschel S. L., et al. (2014). CREB-induced inflammation is important for malignant mesothelioma growth. Am. J. Pathol. 184 (10), 2816–2827. doi: 10.1016/j.ajpath.2014.06.008
Wittkopf N., Pickert G., Billmeier U., Mahapatro M., Wirtz S., Martini E., et al. (2015). Activation of intestinal epithelial Stat3 orchestrates tissue defense during gastrointestinal infection. PloS One 10 (3), e0118401. doi: 10.1371/journal.pone.0118401
Wong D. V., Lima-Júnior R. C., Carvalho C. B., Borges V. F., Wanderley C. W., Bem A. X., et al. (2015). The adaptor protein Myd88 is a key signaling molecule in the pathogenesis of irinotecan-induced intestinal mucositis. PloS One 10 (10), e0139985. doi: 10.1371/journal.pone.0139985
Zarivach R., Vuckovic M., Deng W., Finlay B. B., Strynadka N. C. (2007). Structural analysis of a prototypical ATPase from the type III secretion system. Nat. Struct. Mol. Biol. 14 (2), 131–137. doi: 10.1038/nsmb1196
Zhang W. L., Köhler B., Oswald E., Beutin L., Karch H., Morabito S., et al. (2002). Genetic diversity of intimin genes of attaching and effacing Escherichia coli strains. J. Clin. Microbiol. 40 (12), 4486–4492. doi: 10.1128/jcm.40.12.4486-4492.2002
Zhang Q., Li Q., Wang C., Liu X., Li N., Li J. (2010). Enteropathogenic Escherichia coli changes distribution of occludin and ZO-1 in tight junction membrane microdomains in vivo. Microb. Pathog. 48 (1), 28–34. doi: 10.1016/j.micpath.2009.10.002
Zhang Q., Li Q., Wang C., Li N., Li J. (2012). Redistribution of tight junction proteins during EPEC infection in vivo. Inflammation 35 (1), 23–32. doi: 10.1007/s10753-010-9285-1
Keywords: enteropathogenic Escherichia coli, murine model, diarrhea, enteropathy, antibiotics, inflammation
Citation: Ledwaba SE, Costa DVS, Bolick DT, Giallourou N, Medeiros PHQS, Swann JR, Traore AN, Potgieter N, Nataro JP and Guerrant RL (2020) Enteropathogenic Escherichia coli Infection Induces Diarrhea, Intestinal Damage, Metabolic Alterations, and Increased Intestinal Permeability in a Murine Model. Front. Cell. Infect. Microbiol. 10:595266. doi: 10.3389/fcimb.2020.595266
Received: 15 August 2020; Accepted: 11 November 2020;
Published: 17 December 2020.
Edited by:
Dongyan Niu, University of Calgary, CanadaReviewed by:
Fernando Navarro-Garcia, National Polytechnic Institute of Mexico (CINVESTAV), MexicoJorge Blanco, University of Santiago de Compostela, Spain
Copyright © 2020 Ledwaba, Costa, Bolick, Giallourou, Medeiros, Swann, Traore, Potgieter, Nataro and Guerrant. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Solanka E. Ledwaba, ZWxsZW5sZWR3YWJhQGdtYWlsLmNvbQ==
†These authors have contributed equally to this work