
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Cell. Infect. Microbiol. , 10 September 2020
Sec. Microbiome in Health and Disease
Volume 10 - 2020 | https://doi.org/10.3389/fcimb.2020.00481
This article is part of the Research Topic Viruses: Drivers or Passengers of the Human Gut Microbiome? View all 8 articles
 Emma Guerin1,2
Emma Guerin1,2 Colin Hill1,2*
Colin Hill1,2*The human gut is a complex environment that contains a multitude of microorganisms that are collectively termed the microbiome. Multiple factors have a role to play in driving the composition of human gut bacterial communities either toward homeostasis or the instability that is associated with many disease states. One of the most important forces are likely to be bacteriophages, bacteria-infecting viruses that constitute by far the largest portion of the human gut virome. Despite this, bacteriophages (phages) are the one of the least studied residents of the gut. This is largely due to the challenges associated with studying these difficult to culture entities. Modern high throughput sequencing technologies have played an important role in improving our understanding of the human gut phageome but much of the generated sequencing data remains uncharacterised. Overcoming this requires database-independent bioinformatic pipelines and even those phages that are successfully characterized only provide limited insight into their associated biological properties, and thus most viral sequences have been characterized as “viral dark matter.” Fundamental to understanding the role of phages in shaping the human gut microbiome, and in turn perhaps influencing human health, is how they interact with their bacterial hosts. An essential aspect is the isolation of novel phage-bacteria host pairs by direct isolation through various screening methods, which can transform in silico phages into a biological reality. However, this is also beset with multiple challenges including culturing difficulties and the use of traditional methods, such as plaquing, which may bias which phage-host pairs that can be successfully isolated. Phage-bacteria interactions may be influenced by many aspects of complex human gut biology which can be difficult to reproduce under laboratory conditions. Here we discuss some of the main findings associated with the human gut phageome to date including composition, our understanding of phage-host interactions, particularly the observed persistence of virulent phages and their hosts, as well as factors that may influence these highly intricate relationships. We also discuss current methodologies and bottlenecks hindering progression in this field and identify potential steps that may be useful in overcoming these hurdles.
The human microbiome is collectively comprised of trillions of microbial cells originating from all domains of cellular life (Bacteria, Archaea, Eukarya) as well as viruses (Ursell et al., 2012). The microbiome has received significant attention in recent decades, and has been considered as an organ in its own right due to its important role in multiple aspects of human health (O'Hara and Shanahan, 2006; Baquero and Nombela, 2012). A major fraction of these microorganisms reside in the gastrointestinal tract (GIT) where they partake in essential metabolic and physiological processes that impact human biology (Turnbaugh et al., 2007; Sender et al., 2016). Thus, the GIT forms a complex ecosystem where biotic factors and abiotic factors, such as immune components and physiochemical parameters, are intertwined (Mirzaei and Maurice, 2017; Shkoporov and Hill, 2019). A healthy human gut is populated by more than 1,000 bacterial species with up to 90% resolving into two phyla, the Firmicutes and Bacteroidetes (Turnbaugh et al., 2007; Lozupone et al., 2012). These bacterial compositions will generally be stable over years and perhaps even decades (Faith et al., 2013). Perturbations and shifts in the composition, abundance, and diversity of bacterial communities has been associated with multiple disease states (Lozupone et al., 2012). These include inflammatory bowel disease, obesity, diabetes, eczema, cancer and neurological disorders (Giongo et al., 2011; Karlsson et al., 2011; Gkouskou et al., 2014; Goodrich et al., 2014; Casén et al., 2015; Hartstra et al., 2015; Halfvarson et al., 2017; Vogt et al., 2017; Helmink et al., 2019). The driving forces behind these shifts remains a vital question in the field of human gut microbiome research. One of the most understudied aspects of the gut microbiome are viruses of bacteria, bacteriophages (phages). It is well known that phages have important roles in shaping bacterial communities in different environments both by predation or horizontal gene transfer through transduction (Koskella and Meaden, 2013).
Together bacteria and their viruses form the most abundant biological entities in the human gut (Cani, 2018). The virome includes both bacteriophages and eukaryotic viruses; however, phages (phageome) are significantly more abundant (Gregory et al., 2020). Furthermore, recent studies have shown that microbiome composition is driven by phages and not by the eukaryotic viruses which are generally disease or diet associated (Moreno-Gallego et al., 2019). Phage-driven alterations of the bacteriome by direct interactions or potentially via the human immune system have been linked to a number of disease states including inflammatory bowel disease (IBD), diabetes, malnutrition, Parkinson's disease (Norman et al., 2015; Reyes et al., 2015; Manrique et al., 2016; Belleghem et al., 2017; Nguyen et al., 2017; Zhao et al., 2017; Kieser et al., 2018; Ma et al., 2018; Tetz et al., 2018; Clooney et al., 2019; Gogokhia et al., 2019; Zuo et al., 2019). Fecal filtrate transfer (FFT) studies further indicate that the phageome has the potential to shape the microbiome. For example, the administration of a bacteria-free fecal filtrate from healthy donors to patients with Clostridioides difficile infection (CDI) was demonstrated to prevent disease recurrence, albeit in only five subjects (Ott et al., 2017). The engraftment of the donor phageome has also been observed following fecal microbiota transplantation (FMT) in CDI patients (Draper et al., 2018).
Modern sequencing technology has been critical in providing insights that would have been impossible with culture-based methods (Shkoporov et al., 2019). However, this methodology is also limited because most viral sequences lack homology with phages in current databases. Up to 86–99% of reads in a dataset can remain uncharacterised and are sometimes referred to as “viral dark matter” (Aggarwala et al., 2017). Many bioinformatic pipelines are database-dependent and this results in the exclusion of the majority of viral sequences from subsequent analyses, potentially resulting in skewed findings (Clooney et al., 2019; Sutton and Hill, 2019; Gregory et al., 2020). In terms of laboratory-based research there are also multiple challenges. Many gut bacteria are extremely difficult to culture, thus making isolation of their associated phages difficult (Browne et al., 2016). Furthermore, many phages are strain specific, mimicking gut conditions in vitro is problematic, and host factors such as phase variable expression of host receptors and the existence of multiple phage resistance mechanisms can add another level of complexity (Porter et al., 2020).
This review will discuss our current understanding of the human gut phageome and gut phage-bacteria interactions. We will also discuss the importance of isolating and characterizing novel phages and associated bacterial hosts from the gut while reviewing current bioinformatic and lab-based methodologies as well as their challenges. The ultimate goal is to overcome these hurdles and fully understand how phages shape the microbiome and their role in human health and disease.
The human gut is a densely populated ecosystem within a complex structure with significant variations in physical conditions, abiotic and biotic factors including pH, oxygen, nutrient and water availability, immunoregulators and bile acids (Mirzaei and Maurice, 2017). This creates a gradient of conditions from the mouth to the large intestine resulting in microbial niche specificity (Pereira and Berry, 2017; Bauer et al., 2018). In addition to longitudinal variation, the GIT varies radially due to features such as the lumen, mucosa, villi and crypts (Donaldson et al., 2016; Zhao et al., 2019). Within this complex ecosystem bacteriophages can partake in intricate dynamics with bacteria and can influence microbiome homeostasis.
Traditionally, phages have been classified as lytic/virulent or lysogenic/temperate based on the method of infection they employ when targeting a permissive host (Hobbs and Abedon, 2016). Both life cycles are initiated by adsorption where binding occurs between the phage receptor binding protein and a bacterial cell surface feature which acts as the corresponding receptor. Following this the phage injects its genetic material, which can be either single or double-stranded DNA or RNA, from its capsid into the host cell. It is at this point that the life cycles diverge. Virulent phages hijack host replication machinery to replicate their genome, assemble phage components and produce progeny. This ultimately ends in lysis of the host cell and release of progeny to commence subsequent rounds of host infection. In contrast, lysogenic phages can integrate their genomes into the host chromosome forming a prophage that replicates in tandem with the bacterial genome. Alternative lifecycles such as pseudolysogeny, carrier state and chronic phage infections are less characterized in comparison to the lytic and lysogenic lifecycles (Figure 1). However, the overall prevalence of each of these lifecycles remains poorly understood in the context of the human gut. To avoid ambiguity, when discussing the phageome we generally refer to phages present within bacterial genomes as temperate, while free phage virions are referred to as virus-like particles (VLPs).
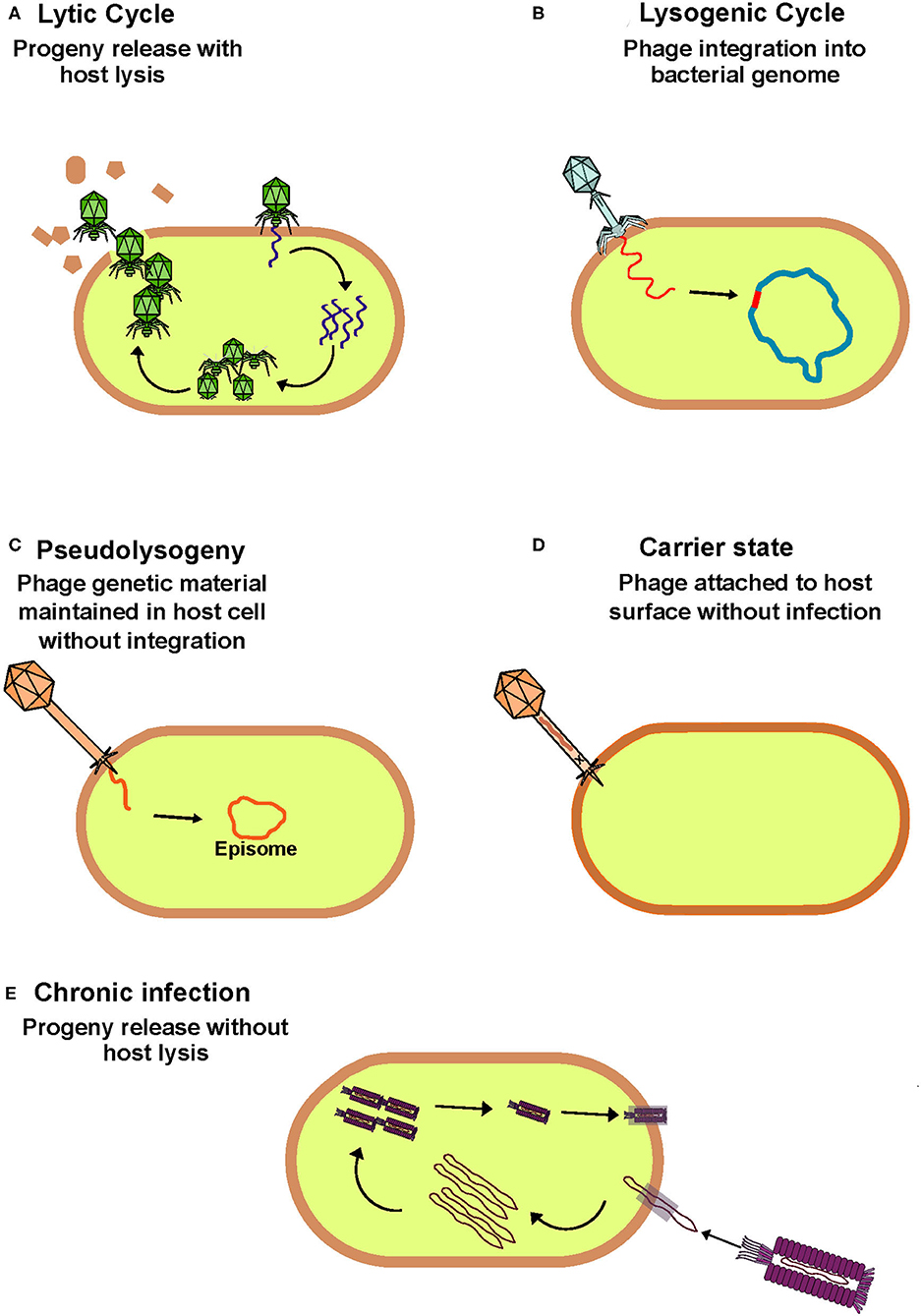
Figure 1. Overview of bacteriophage lifecycles. (A) Lytic phages hijack host cell machinery to replicate, assemble and produce progeny which are released from the cytoplasm on host cell lysis to initiation further rounds of infection. This phage lifecycle is thought to be the most prevalent in the healthy human gut. (B) Lysogenic phages integrate their genome into the bacterial host genome with which they passively replicate until stress signals trigger their induction and switch to the lytic cycle. (C) Phages that follow a pseudolysogenic lifecycle also passively replicate with the host, but their genome remains independent from that of the host and is maintained in the cytoplasm as an episome. (D) When phages are in a carrier state they can remain attached to the surface of a non-permissive host without infection. (E) During a chronic infection phages produce progeny similarly to that of lytic phages but without host cell lysis.
Phage lifecycles have an important role in shaping the bacteriome both directly and indirectly. Phages directly drive the bacteriome composition by predation but also through prophage integration or indirectly by horizontal gene transfer (HGT). HGT can confer bacteria with new functionalities and in turn contributes to evolution and the expansion of bacterial diversity in the gut. Temperate phages mediate this transfer of genetic material either by lysogenic conversion or by generalized, specialized, or lateral transduction (Touchon et al., 2017; McInnes et al., 2020). The acquisition of new genes can be favorable to the bacterial host with benefits including improved colonization of the gut, virulence factors, improved stress tolerance, biofilm formation, motility, or immunity in the case of prophages (Obeng et al., 2016). However, the integration of a prophage or new genes can also be detrimental if essential genes are interrupted. If gut commensals acquire genes conducive to virulence, they can evolve to become pathogens, thus, phages can contribute to the spread of virulence and disease development in the human gut. Indeed, prophages or phage genes have been detected in many pathogens. A classic example of this is Shiga-toxin producing E. coli. Many non-pathogenic E. coli strains reside in the human gut but through the acquisition of phage encoded Shiga toxins, certain strains have evolved into pathogens (Brüssow et al., 2004). Additionally, it is debated that phages carrying antibacterial resistant genes (ARGs) at high abundances, transfer them to bacterial hosts in the gut and fuel the spread of antibiotic resistance. Although, such ARGs have been detected in bacteria it is thought the abundance at which phages carry them is greatly overestimated, largely due to bacterial contamination among viral sequences (Enault et al., 2017; Debroas and Siguret, 2019). It is thought that other mobile genetic elements have a greater role at transferring ARGs among bacteria in the gut than phages (McInnes et al., 2020). The role, if any, that phages play in the dissemination of ARGs in the gut merits further investigation.
The International Committee on Taxonomy of Viruses (ICTV) is responsible for the taxonomic classification of prokaryotic viruses which has been traditionally based on virion morphology (Lefkowitz et al., 2018). The best characterized of the phage families are the Siphoviridae, Podoviridae and Myoviridae that make up the Caudovirales order, all of which are tailed with dsDNA genomes. Members of these phage families have distinct head and tail morphologies: Siphoviridae have long non-contractile tails, Podoviridae have short non-contractile tails and Myoviridae have long contractile tails (Ackermann, 1998). Electron microscopy of human fecal filtrates shows a dominance of these phage morphologies (Hoyles et al., 2014). However, the accuracy of morphology-based taxonomy has been questioned. An interesting argument was put forward by Sutton and Hill regarding the current discrepancies in phage taxonomy, where two phages that shared similar genomic and functional characteristics were classified into different phage families solely based on virion morphology (Sutton and Hill, 2019). This highlights the need for a move toward the use of sequence-based taxonomic classification. Currently, efforts are being made to develop a genome-based taxonomic scheme (Bolduc et al., 2017; Eloe-Fadrosh, 2019; Jang et al., 2019). Indeed, in 2020 ICTV proposed a new 15-rank classification hierarchy of virus taxonomy to accommodate viral genetic diversity (Gorbalenya et al., 2020). This will be important for future virome studies and for the classification of novel phages.
In the human gut, phages are estimated at ~1010 g−1 while the bacteria they infect equate to ~1011 g−1 (Shkoporov and Hill, 2019). It has been long postulated that phages greatly outnumber their bacterial hosts at 10:1 virus-to-microbe ratio (VMR) (Bergh et al., 1989; Wommack and Colwell, 2000; Chibani-Chennoufi et al., 2004). VMR has been associated with influencing lifestyle switches of phages (Howard-Varona et al., 2017). With shifts to a high VMR (high phage to low host densities), it was thought that phages would enter a lysogenic life cycle to ensure persistence in the community. However, more recently it has been proposed that with increased bacterial abundances, phages will transition from lysis to lysogeny allowing them to take advantage of the success of their host in “Piggy-Back-The-Winner” dynamics (Knowles et al., 2016). It is important to note however, that this is largely based on marine and aquatic environments and may not necessarily be representative of the human gut. The VMR in the human gut is significantly lower compared to other ecosystems, estimated at ~0.1:1 (Shkoporov and Hill, 2019).
The gut phageome starts to develop in infancy. In the early stages of microbial and phage colonization of the gut, rapid changes in composition occurs (Sharon et al., 2013). A longitudinal study of twenty healthy infant twins observed a shift from a microbiome with high phage-low bacterial diversity at 0 months to a low phage-high bacterial diversity by the time they reached 24 months old (Lim et al., 2015). In comparison to adults, infants have higher bacteriophage diversity but the infant gut phageome is less stable over time than that of an adult. Despite this, both infants and adults have been observed to have high interindividual variation of the phageome (Lim et al., 2016). Although the phageome starts to develop in infanthood, it is still unclear what factors influence the colonization of phages. Proposed influencing factors include diet, genetics, birth mode, vertical transmission, and environment (Lim et al., 2016). The influence of birth mode on infant gut phageome composition is currently debated, however, more recent findings indicate delivery route has less impact on bacteriophage composition compared to bacterial composition (McCann et al., 2018; Maqsood et al., 2019; Siranosian et al., 2020). Interestingly, one study found that as much as 63% of bacterial communities in the infant gut are inherited from the mother compared to 15% of viruses thus suggesting that vertical transmission has minimal influence on phageome composition (Maqsood et al., 2019). This is consistent with the low level of VLPs detected in infant meconium stool but by 1-month high abundances of VLPs are detectable (Liang et al., 2020). It is suggested that soon after birth the neonate gut becomes colonized with pioneer bacteria and prophage induction from these bacteria is the major contributor to the bacteriophage diversity in the early infant gut (Liang et al., 2020). With time there is a shift from temperate phage dominance and high diversity toward decreased richness and abundance by the second year of life with an increase in phages of the Microviridae family (Lim et al., 2015). It is now thought that environmental and age-dependent factors likely have an important role in shaping the phageome composition in early life (Maqsood et al., 2019). For example, twin infants have more a shared phageome than that of an older non-twin sibling (Reyes et al., 2015). Furthermore, bacteriophage composition varies between infants that are breastfed or formula fed and mode of feeding also influences eukaryotic virus colonization (Liang et al., 2020). It will be important to further expand on the role of diet and environment in shaping the early life phageome as this may have life-long health implications.
The ratio of virulent and temperate phages in the healthy human adult gut has been a question of interest. The main consensus based on both microscopic and sequence-based virome studies is that the human gut is dominated by temperate dsDNA phages of the Caudovirales order and ssDNA phages of the Microviridae family (Reyes et al., 2010; Kim et al., 2011; Minot et al., 2011; Hoyles et al., 2014; Moreno-Gallego et al., 2019). These phages have been described as the core of a healthy human adult phageome with a high occurrence of crAss-like phages (Manrique et al., 2016; Moreno-Gallego et al., 2019). CrAss-like phages form the most abundant phage family in the human gut identified to date with the founder member of the family discovered only in 2014 (Dutilh et al., 2014). These Bacteroidetes-infecting phages showed little to no sequence similarity with phages in pre-2014 viral databases and have the remarkable ability to persist in the gut over extensive periods (Shkoporov et al., 2019). The identified core was found to be highly stable over time which is consistent with reports on crAss-like phages and the slow evolution rates associated with temperate phages compared to virulent phages (Minot et al., 2011; Dutilh et al., 2014; Guerin et al., 2018; Shkoporov et al., 2018a, 2019). Studies have found that 95% of viral genotypes are maintained after 1 year and 80% after 2 years (Reyes et al., 2010; Minot et al., 2013). This was linked to the dominance of temperate phages which have low mutation rates compared to virulent phages, such as Microviridae, which have mutation rates as high as 10−5 substitutions per day (Minot et al., 2013). Changes in the composition and the richness of the temperate core have also been associated with diseases states such as IBD (Norman et al., 2015). However, the existence of a phage core in the human gut that is shared across individuals has been disputed (Gregory et al., 2020).
A key issue with virome studies and verification of the proposed core phageome is that analytical pipelines tend to focus solely on the known annotated component of viral datasets (~1–14% of sequences generated) and exclude viral dark matter which currently constitutes the majority of sequences (Aggarwala et al., 2017). If only a fraction of the reads is examined this leads to an incomplete and potentially biased analysis. More recent studies have attempted to take this into account by implementing stricter criteria and limiting the use of database-dependent methods. A one-year longitudinal study of the healthy human gut virome of ten individuals implemented a database-independent analysis approach and identified a personal persistent virome (PPV) (Shkoporov et al., 2019). This study clarifies multiple discrepancies in virome studies by showing that temperate phages do not dominate the gut in health as previously thought and confirm that although there is a predominant core of phages in the healthy gut, the idea that a defined set of phages is shared among all individuals cannot be supported which is in agreement with other analyses (Gregory et al., 2020). A total of 22 abundant and temporally stable phage clusters were identified; however, none of these were present in all individuals at one time and the presence/abundance of each cluster was individual specific. These 22 clusters were classified as virulent crAss-like phages, other virulent Caudovirales and Microviridae, with only three being identified as temperate. A transiently detected virome (TDV) was also observed but in contrast to the PPV, it was less abundant and less stable but shared to a greater extent across individuals. The TDV is comprised of Siphoviridae and phages such as Inoviridae. Interestingly, CRISPR-spacer analyses found that the PPV was associated with abundant and stable bacterial genera specifically adapted to a healthy gut environment such as Bacteroides, Faecalibacterium, Prevotella, Eubacterium, and Parabacteroides whereas more transient genera such as Escherichia, Akkermansia, Listeria, Clostridium and Streptococcus were associated with the TDV (Shkoporov et al., 2019). In summary, the PPV can be defined as the component of phages in the gut that remain abundant and stable when monitored over 1 year whereas phages that are less abundant and less stable over the same time period are defined as the TDV. These findings also show that the gut is dominated by virulent phages in health as opposed to temperate phages (Reyes et al., 2010; Minot et al., 2011; Moreno-Gallego et al., 2019). If temperate phages are not dominant as previously thought, this begs the question of how virulent phages maintain themselves in the human gut at high levels over extensive periods of time?
With high inter-individual variation across viromes, the identification of viral signals or biomarkers that could differentiate and group individuals based on disease states is challenging. Clooney et al. examined the potential of the virome in distinguishing cohorts through an analysis of healthy and IBD viromes (Norman et al., 2015; Clooney et al., 2019). This was performed using a database-independent method based on the complete virome, i.e., both the known viral sequences and dark matter. Protein-based clustering was performed to identify compositional patterns that potentially separated the two cohorts using vConTACT2, a protein clustering program and the same pipeline implemented by Jang et al. (2019) and Shkoporov et al. (2019). Interestingly, this protocol also revealed a person-specific virulent core of crAss-like phages and Microviridae in health which was absent in IBD patients. IBD viromes were found to have an increased abundance of induced temperate phages and this was reflected by a reduction in bacterial alpha-diversity (Clooney et al., 2019). These findings support the observed expansion of specific phage populations in other IBD virome studies (Norman et al., 2015; Duerkop et al., 2018). Duerkop and colleagues proposed that members of the Spounaviridae subfamily could serve as a biomarker of colitis (Duerkop et al., 2018). Clooney et al. observed an expansion of temperate Myoviridae and Siphoviridae (Clooney et al., 2019). As Spounaviridae resolve into the Myoviridae family, further investigation of this biomarker is merited. In summary, this work shows that loss of, or deviation from, the healthy virulent core to a virome with increased temperate phage induction is indicative of IBD. It would be worthwhile expanding on these findings to identify genus or species-specific changes and apply the same experimental protocol to examine the phageome in other disease states.
The maintenance of a phage in an ecosystem is dependent on its ability to infect a suitable bacterial host. However, bacterial hosts can employ an arsenal of defense mechanisms. This leads to antagonism between the two populations. If selective pressures occur, co-evolution can result in genotypic variants due to mutations at specific sites within both entities, such as in the genes encoding phage tail fibers and bacterial surface receptors. These complex interactions are believed to be an important shaping force of multiple ecosystems. To add another level of complexity there is also evidence that phages interact with the mammalian host, both directly and indirectly, via the immune system (Duerkop and Hooper, 2013; Tetz et al., 2017). For clarity, when discussing phage-host interactions, ‘host' refers solely to the bacterial host unless stated otherwise.
Both bacterial defense and phage counter-defense strategies have been reviewed extensively (Labrie et al., 2010; Samson et al., 2013; Seed, 2015; Rostøl and Marraffini, 2019). Bacteria can employ an array of defense mechanisms when faced with phage predation. These include preventing phage entry, blocking injection of or elimination of nucleic acids, inhibiting the hijacking of replication machinery and programmed cell death. An overview of these anti-phage mechanisms is provided in (Table 1, Figure 2A).
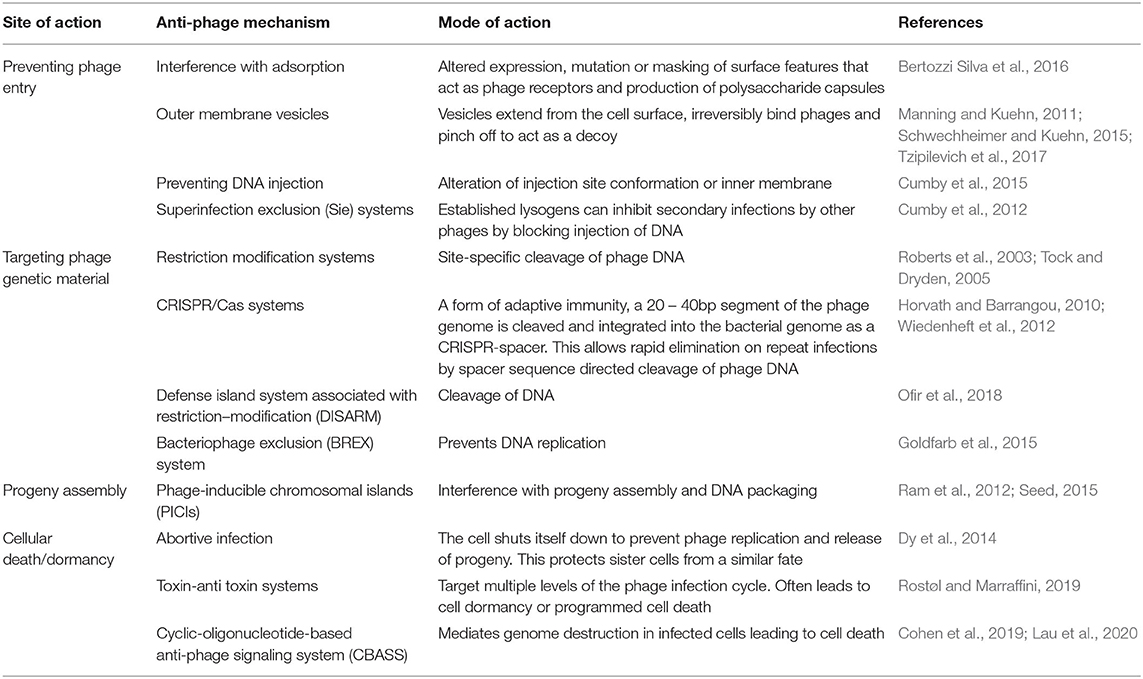
Table 1. A summary of phage defense mechanisms employed by bacteria.
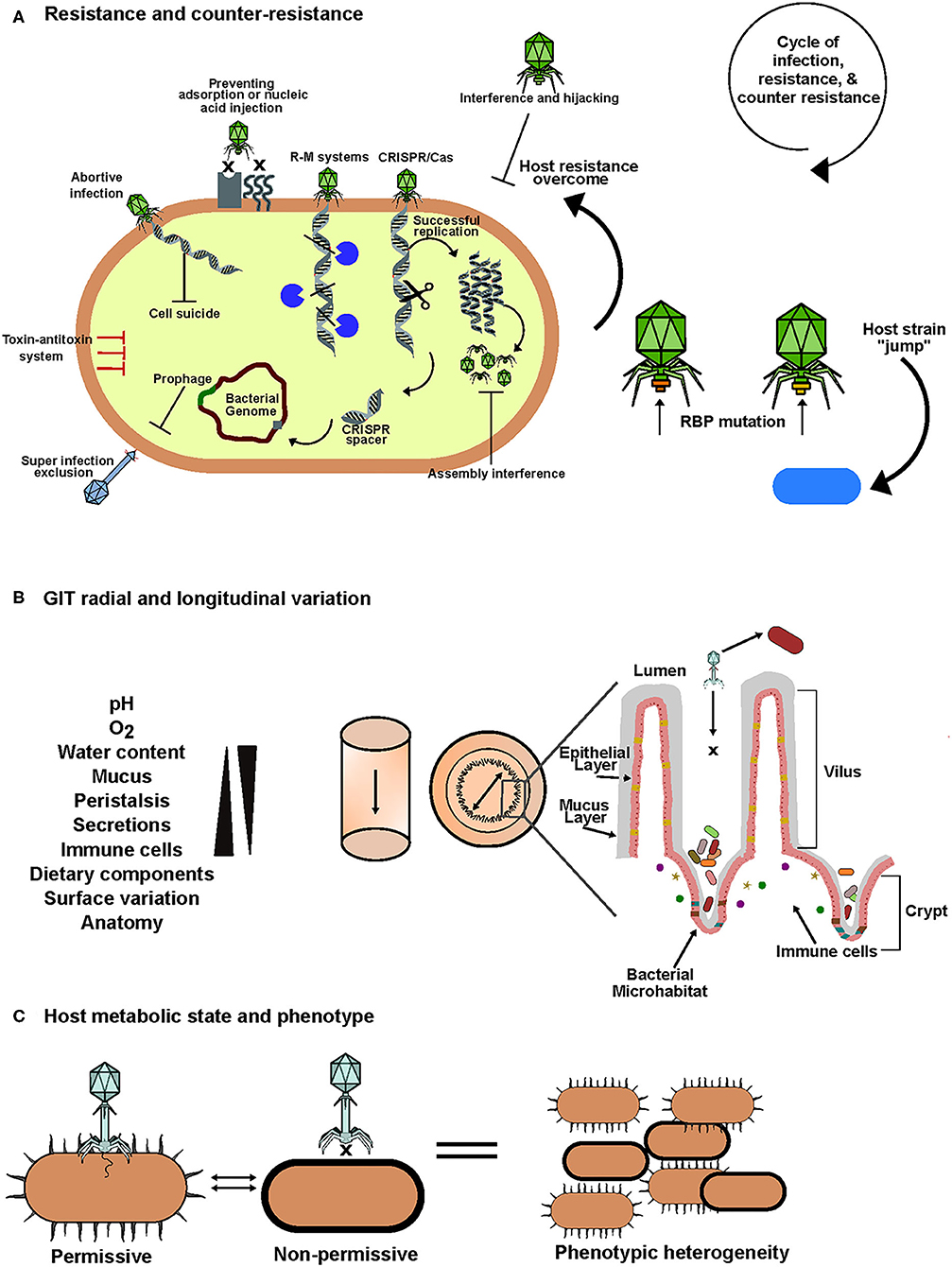
Figure 2. An overview of factors that influence phage-bacteria interactions in the human gut. (A) To overcome phage predation, bacteria possess an arsenal of defense mechanisms that target one or more stages of phage infection cycles. In retaliation phages have evolved an array of counter-defense mechanisms. This results in a cycle of infection, resistance and counter-resistance that leads to evolution and diversity in the human gut. (B) In the gastrointestinal tract there is significant variation in biotic and abiotic factors both longitudinally and radially. This results in sectional variation in bacterial composition and spatial heterogeneity between phages and their hosts. Anatomical features of the gut such as mucus, crypts and villi create bacterial microhabitats which are inaccessible to phages allowing them to escape predation, gradually seed the lumen and maintain homeostasis. (C) Biotic and abiotic factors can also influence the metabolic state of the bacterial host. This may result in transient phenotypic changes mediated by mechanisms such as phase variation allowing adaptation to stress which includes phage infection. These phenotypic changes may inhibit phage infection and can create isogenic populations consisting of phage permissive and non-permissive host variants allowing the co-existence of phage and bacteria.
Despite the efforts of bacteria to ward off phage attack, their predators employ mechanisms to resist. In the case of masked receptors, phages can encode enzymes which degrade capsular polysaccharides (Leiman et al., 2007; Cornelissen et al., 2012). Phages can modify their genomes to avoid restriction modification systems or they can encode anti-CRISPR proteins (Samson et al., 2013; Wiedenheft, 2013). Phages can also mimic host antitoxins to neutralize host produced toxins in host toxin-antitoxin defenses allowing the phage to proceed with propagation (Blower et al., 2012). Interestingly, phage encoded CRISPR-Cas systems have also been detected which allow the phage to hijack host defenses (Seed et al., 2013). When presented with a resistant host with modified receptors, phages can modify their receptor binding protein to continue replication on the same host. For example, lambda phage originally absorbed to its host Escherichia coli B, at cognate receptor LamB but with the introduction of four point mutations into the gene encoding its RBP, the phage was able to bind at a new host receptor, OmpF, while maintaining its ability to bind at the original receptor (Chatterjee and Rothenberg, 2012; Meyer et al., 2012). These bacterial adaptations during resistance and phage counter resistance have given rise to co-evolution and arms-race dynamics. This phage driven diversification of bacteria in the gut can have both positive and negative impacts for the human host. These interactions may select for bacteria with phenotypes conducive to disease development or disease resistance (Scanlan, 2017).
Several ecological models have been developed that describe phage-bacteria interactions in ecosystems. One of the earliest defined ecological models describing these interactions is the classical “kill the winner” model which is derived from the Lotka-Volterra equation (Thingstad, 2000). This model describes the predation of the most abundant bacteria in an ecosystem by their associated phages in a manner that leads to an abrupt reduction in their population. This in turn results in the expansion of another “winner” and a subsequent round of predation leading to fluctuations of the dominant phages and bacteria in an ecosystem (Maslov and Sneppen, 2017). This pattern of predator-prey dynamics has generally not been associated with the GIT, at least not at the genus level (Reyes et al., 2010; Minot et al., 2011; Shkoporov et al., 2019). Furthermore, it does not take lysogenic conversion into account. This is considered in the piggyback-the-winner model which was previously discussed in relation to VMR. Traditionally, this model proposed the lysogenic conversion of phages in a high VMR (high phage: low bacterial densities) to ensure survival until conditions favored reversion to lysis. However, more recently lysogenic conversion was also described in the case of a low VMR (low phage: high bacterial densities) in which phages take advantage of and “piggyback” on host prosperity (Knowles et al., 2016; Silveira and Rohwer, 2016).
Contact rates in the gut can also influence phage-host interactions and perhaps persistence of virulent phages; for example, motility and diffusion can be affected by variations in mucus concentrations. A “continuous time random walk” model was developed supporting this hypothesis (Joiner et al., 2019). Anatomical features in the gut create radial variation and influence the spatial dissemination of phages and their hosts (Figure 2B). An example of such spatial heterogeneity was identified in a study of virulent phage-bacteria coexistence in the gut using a murine model. It was found that the examined phages were in lower abundance in the mucosa while bacterial counts were high and vice versa in the lumen, thus, creating a luminal-mucosal gradient of phages and bacteria (Lourenço et al., 2020). In this example, the mucosa (source) was described as a phage-inaccessible refuge for the bacteria from where they can gradually disseminate into the lumen (sink) and phage predation occurs. These observations suggest that the biogeographical variation of the gut can create source-sink dynamics and spatial distribution that provides certain phage-permissive bacteria with refuges that limit phage predation and thus limits evolutionary selective pressure. As a result, when these bacteria disseminate from their refuge and come in contact with a cognate phage they remain susceptible to infection thus allowing both the bacteria and phage to be maintained. These dynamics can interplay to support the coexistence of virulent phages and susceptible hosts in the gut (Holt, 1985; Lourenço et al., 2020). Interestingly, this is consistent with the idea that populations of intestinal bacteria, particularly commensals, reside in microhabitats such as crypts (Figure 2B). This allows bacteria to form reservoirs and manage the population by re-seeding of the lumen after perturbations (Donaldson et al., 2016). Multiple studies support the idea of spatial refuges for bacteria to avoid or limit phage predation (Schrag and Mittler, 1996; Bruttin et al., 1997; Weiss et al., 2009).
The ability of a phage to infect its host is not solely dependent on resistance and counter resistance. The lifestyle and physiological state of the target host is a significant influencing factor on the success of a phage infection. The environmental conditions of the gut have an important role in this due to the impact they can have on the metabolic state and stress levels in bacteria (Figures 2B,C). These conditions include the physical and anatomical variation along the radial and longitudinal axes of the GIT, mucous, bile acids, pH, oxygen, water levels, nutrient levels, peristalsis and dietary components (Koziolek et al., 2015; Vandeputte et al., 2016; Shkoporov and Hill, 2019; Zhao et al., 2019).
The physiological state of a bacterial host can dictate whether it is phage permissive. This can also generate phenotypic heterogeneity within isogenic populations with some showing phage sensitivity while others are resistant (Bull et al., 2014) (Figure 2C). A phenomenon called phase variation is thought to be an important mediator of transient resistance that results in phenotypic variation in response to environmental conditions. Recently, the significance of phase variation in dictating phage-host interactions in the gut has become of interest (Jiang et al., 2019; Turkington et al., 2019; Porter et al., 2020). It occurs by variable expression of gene loci due to hypermutation or methylation resulting in genes being reversibly switched “on or off” (Bayliss, 2009). This is mediated by recombinases, integrases or invertases that act on invertible regions containing promotors of gene loci often associated with expression of surface features such as Sus-like systems, capsule polysaccharides, S-layer and Ton-dependent transporters which can act as phage receptors, outer membrane vesicles, flagella or restriction modification systems (Zaleski et al., 2005; Coward et al., 2006; Nakayama-Imaohji et al., 2009, 2016; Zitomersky et al., 2011; Porter et al., 2020). These phenotypic switches allow the host to adapt to environmental stresses, which are abundant in the human gut, while minimizing the fitness impact on the total population (Moxon et al., 1994). It has been demonstrated that phase variable invertons are more dominant in the human gut compared to other environments (Jiang et al., 2019). Phenotypic heterogeneity is thought to provide bacteria with a form of herd immunity equivalent to that seen among humans. Non-phage permissive host variants limit the viral load and thus the impact on sensitive variants is reduced preventing a drastic reduction in the overall population (Turkington et al., 2019). The use of herd immunity style dynamics to limit phage predation is further supported by the persistent propagation of virulent phages on a microcolony without its elimination (Eriksen et al., 2018).
Interestingly, phages can hijack host quorum sensing to monitor metabolic conditions and population densities, thus granting them the ability to make informed lytic-lysogenic switch decision (Laganenka et al., 2019). Quorum sensing is a phenomenon employed by bacteria to control the expression of specific genes for processes such as virulence and biofilm formation. This occurs through cell-cell communication mediated by bacterial produced extracellular signaling molecules that allow bacteria to monitor population density and make an informed collective decision on whether expression of specific genes is a fitness cost or beneficial to the overall population (Whitehead et al., 2001; Turovskiy et al., 2007; Ng and Bassler, 2009; Papenfort and Bassler, 2016). Bacteria can use this to assess the risk of phage predation and act accordingly. Quorum sensing homologs have also been identified in phage genomes deposited in NCBI databases. More recently, it was shown that Vibrio cholerae infecting temperate phage VP882 encodes a QS receptor (VqmAPhage) homologous to that of the host allowing it to “listen in” on its target via host produced autoinducer. In turn, the phage can monitor population densities thus allowing an informed lytic/lysogenic lifestyle switch. In high host cell density, lysis occurs resulting in optimal propagation (Silpe and Bassler, 2019). Collectively, the above demonstrates that there are a multitude of factors that influence the interactions between phages and their bacterial hosts. These factors are important for the success of both in the complex gut environment and are summarized in Figure 2.
In addition to phage-bacteria interactions, phages may interact directly or indirectly with the mammalian host, adding another level of complexity when studying the human gut phageome. Ig-like domains have been identified in certain phages which help them adhere to and remain in the outer mucosal layer in the GIT. This led to the proposition of the “bacteriophage adherence to mucus” (BAM) model which describes the idea that such phages protect the mammalian host from bacterial infections (Barr et al., 2013). However, it has also been demonstrated, in vitro, that phages can migrate across the mucus layer, cross the gut epithelial barrier or enter systemic circulation either by transcytosis or by peptide-guided transport and subsequently interact directly with eukaryotic cells or cells of both the innate and adaptive immune system (Duerr et al., 2004; Belleghem et al., 2017; Moustafa et al., 2017; Nguyen et al., 2017; Van Belleghem et al., 2019). This was further supported by in vivo studies in a germ-free murine model that examined the use of endotoxin-free lytic phages in the alleviation of bacterially driven colon cancer. The administered phages, specifically Caudovirales phage with DNA and not empty capsids, interacted directly with the murine immune system. This was indicated by interferon production (virus-specific immunity) which led to the exacerbation of colon inflammation (Gogokhia et al., 2019). Phages have also been detected in cerebrospinal fluid, possibly due to blood-brain barrier permeability, and have been shown to increase intestinal permeability (Tetz and Tetz, 2018). This can impair gut barrier function leading to leaky gut and components such as bacterial extracellular DNA entering circulation which is associated with inflammation and diseases including cancer (Tetz and Tetz, 2019) Taken together, these findings suggest that certain phages could have also potential roles as human pathogens or contribute to disease development. This has been indeed observed for Parkinson's Disease, type 1 diabetes, type 2 diabetes and gut diseases (Ma et al., 2018; Tetz et al., 2018, 2019; Clooney et al., 2019; Gogokhia et al., 2019) A number of extensive articles discuss these interactions in detail (Tetz and Tetz, 2018; Van Belleghem et al., 2019). Findings thus far suggests that phages can directly influence mammalian health through interaction with eukaryotic cells and indirectly by shaping the gut microbiome altering bacteriome composition or via interactions with the immune system; however, we still have little understanding of the mechanistic details and further studies are required to fully understand how phages can impact the mammalian host.
In the past decade there have been significant advances in our understanding of the human gut phageome. Findings to date show that the healthy gut phageome is individual specific and temporally stable comprising a virulent core that is dominated by crAss-like phages, other Caudovirales and Microviridae, but which are not shared among all individuals. However, many important questions remain unanswered and our overall understanding of this elusive component of the gut microbiome remains poor in comparison to our understanding of the bacterial fraction which is largely due to methodological challenges. Efforts are ongoing to overcome shortcomings, to develop and standardize protocols, and improve overall reproducibility and comparability.
Next generation sequencing technologies, bioinformatic tools and metagenomic studies have provided important insights into the human gut phageome. The majority of these studies have used feces as the sample source of phage genetic material due to the practical difficulties of using other GIT sampling sites (Reyes et al., 2010; Minot et al., 2011; Dutilh et al., 2014; Norman et al., 2015; Guerin et al., 2018; Clooney et al., 2019; Devoto et al., 2019; Shkoporov et al., 2019). In brief, the key protocol steps involved in most viral metagenomics studies includes VLP enrichment, nucleic acid extraction, sequencing library preparation, followed by in silico characterization and annotation of generated viral sequences using bioinformatic pipelines. However, due to the lack of universally reproducible methods cross-study comparisons of findings are difficult, and disparities can occur across studies.
The first step in viral metagenomic studies is the removal of non-target contaminants from the biological sample and the enrichment of VLPs to ensure optimal yield and quality of nucleic acids for library preparations (Kleiner et al., 2015). This involves the elimination of prokaryotic and eukaryotic DNA and RNA as well as cellular and dietary debris using physical and chemical process such as homogenisation, filtration, chloroform treatment and enzymatic action (Conceição-Neto et al., 2015). There are multiple phage nucleic acid isolation protocols described and while CsCl gradient centrifugation can yield highly purified samples, this protocol is time consuming, laborious and can introduce bias (Castro-Mejía et al., 2015; Kleiner et al., 2015). It is important to consider that the phageome is comprised of virulent and temperate phages that vary in nucleic acid content (ssDNA, dsDNA, ssRNA, and dsRNA). Developing a protocol to consider all these components is difficult and can affect our overall picture of human gut phage profile. Due to the shared physio-chemical properties among temperate and virulent Caudovirales phages, many techniques have been developed to allow their co-purification. It is important to note that using the incorrect protocol can lead to a biased virome composition (Gregory et al., 2020). The majority of metagenomic studies have focused on enrichment of VLPs followed by extraction of nucleic acids, although many of the protocols promote a biased focus on free-phage and the enrichment of DNA phages (Kleiner et al., 2015).
Protocols may also generate low nucleic acid yields and to overcome this, amplification methods are performed such as multiple-displacement amplification (MDA) (Kim et al., 2011; Marine et al., 2014). However, this leads to the biased amplification of ssDNA viruses such as Microviridae by as much as 10-fold which in turn skews phage community composition. This is thought to be the confounding factor behind why certain viral datasets have an extremely high relative abundances of Microviridae (Kim et al., 2011; Roux et al., 2016; McCann et al., 2018; d'Humières et al., 2019; Garmaeva et al., 2019; Shkoporov and Hill, 2019; Gregory et al., 2020). Many virome studies interpret findings based on relative abundance and thus results may be skewed where MDA has been implemented (Sutton and Hill, 2019). MDA has also been shown to affect phage diversity and method reproducibility (d'Humières et al., 2019). MDA can introduce GC bias and coverage extremes which can have implications for de novo genome assemblies (Chen et al., 2013; Sutton et al., 2019). Having identified the effects of amplification on viral nucleic acids, there is a call to eliminate this step from metagenomic protocols. However, this is challenging due to the variable concentration yields of phage genetic material that can be acquired from samples as a result of differences in the viral load and the strict input requirements for sequencing library preparation (Shkoporov and Hill, 2019). For example, one study found that DNA yields, despite enrichment, could range from 4 to 500 ng of DNA per gram of feces (Shkoporov et al., 2018b). Attempts have been made to improve sequencing library preparations, such as the adaptase-linker amplification method, by excluding the need for MDA with the goal of eliminating downstream issues and providing a more accurate representation of phage composition in a community (Roux et al., 2016).
The elimination of bacterial contamination has also been a hurdle both in terms of viral nucleic acid extraction and in silico analyses. Although it is difficult to completely avoid, contamination levels should be limited to minimize impact on the validity of viral sequences deposited in database (Roux et al., 2013). During the enrichment of VLPs and extraction of viral nucleic acids, the choice of filter pore size is important to avoid contaminants. The use of a larger pore size filter such as 0.8 μm is less biased as it allows larger viruses to be retained but this also leads to high levels of bacterial contamination. A pore size of 0.45 is thought to be optimum (Conceição-Neto et al., 2015; Shkoporov et al., 2018b). The consequence of bacterial contamination among viral sequences can impact on conclusions drawn from datasets. This was highlighted in a study that demonstrated the over-representation of antibiotic resistant genes of bacterial origin among phage genomes (Enault et al., 2017). The issue of contamination was further highlight by Zolfo and colleagues by examination of virome sequences from 35 datasets. They detected an abundance of bacterial, archaeal and fungal contaminants irrespective of VLP enrichment technique (Zolfo et al., 2019). Bioinformatic pipeline development and standardization of detection criteria for bacterial contamination is important in ensuring the quality of viral databases and the de novo detection of phages. For example, ViromeQC has significant potential in detecting such contaminants. This pipeline was developed to allow stringent quality control of viral sequences using 31 universal bacterial genes in addition to the 16S/18S rRNA genes and 23S/28S rRNA genes (Zolfo et al., 2019). In addition to contamination, Shkoporov et al., also considered the effects of sample storage conditions and operator bias in virome analyses and has proposed the use of an exogenous phage standard to spike fecal samples to allow absolute quantification of viral loads across samples (Shkoporov et al., 2018b). The disparity between relative and absolute quantification in microbiome profiling can be significant (Stämmler et al., 2016; Vandeputte et al., 2017). This issue was recently highlight by Shanahan and Hill. They discussed the issue of microbiome misrepresentation due to relative abundance and how it can mask variations between microbiomes that would be highlighted by absolute abundances (Shanahan and Hill, 2019). Thus, the inclusion of a standard to allow absolute quantification of sequences and viral load should form part of future virome studies to give a more accurate representation of composition. Many protocols in phageome studies have been developed with a particular focus on DNA phages. This results in RNA phages being underrepresented in viral databases and highlights the need for the development of protocols specific to the isolation of these phages (Callanan et al., 2018). The potential array of human gut RNA phages was recently highlighted with the in silico discovery of over one thousand near complete RNA phage genomes and over fifteen thousand non-redundant genomes following analysis of 82 publicly available metatranscriptomic datasets, generated from activated sludge and aquatic environments studies, using profile hidden Markov models to detect conserved proteins. Overall this represents a 60-fold increase in identified ssRNA phage genomes (Callanan et al., 2020).
The choice of sequencing platform for virome analysis can influence read length output. Following quality control, reads are assembled into contigs generally by referenced-based or de novo assembly methods (Garmaeva et al., 2019). Short read platforms allow deep sequencing with low error rates and require low DNA concentrations. However, de novo assembly of short reads is arduous due to the modular nature of phage genomes, strain heterogeneity, high incidences of hypervariable and repeat regions and MDA influenced variations in coverage and GC content (Lima-Mendez et al., 2011; Minot et al., 2012, 2013; Chen et al., 2013; Sutton et al., 2019). This leads to fragmented assemblies and hampers downstream in silico analyses (Sutton et al., 2019). It has been suggested that the use of long-read sequencing platforms, such as Oxford Nanopore, which can generate reads that are representative of near complete genomes may overcome the issues associated with short-read sequencing (Somerville et al., 2019; Warwick-Dugdale et al., 2019). The major drawback with this method is that high DNA concentrations are required which, as discussed previously, can be difficult to attain. Therefore, when performing assemblies from short reads, the choice of assembly software and stringent criteria to recruit true viral sequences is essential (Roux et al., 2017; Sutton et al., 2019).
Sequence-based methods have played an important role in providing insights into the human gut phageome. Nevertheless, a pressing issue is the dependency on insufficiently developed viral databases. Most phage sequences in these databases are not annotated due to the lack of homology with known phages. When newly generated sequence reads are aligned to these databases as much as 99% of the reads from a dataset can fail to align to known phage genomes or homologs and thus these reads remain as dark matter (Aggarwala et al., 2017). Therefore, multiple studies have based their findings solely on the identifiable fraction of reads and have excluded the abundant unexplored dark matter. Although these studies have identified cohort variation, it is unknown how the inclusion of viral dark matter would influence and validate the overall findings (Lim et al., 2015; Norman et al., 2015; Monaco et al., 2016; Zuo et al., 2019). To overcome this issue, there has been a move toward the use of database-independent methods, such as open-reference and clustering approaches, to include both the identifiable and unknown fraction of reads in a dataset (Shkoporov et al., 2018b, 2019; Clooney et al., 2019; Moreno-Gallego et al., 2019). Protein-clustering programs, such as vContact2, offer an effective solution and allow the implementation of a whole virome analysis (Bolduc et al., 2017). These work by building a gene-sharing network based on shared protein families across genomes, an approach similar to that employed in the development of the crAss-like phage family taxonomy (Guerin et al., 2018; Yutin et al., 2018; Jang et al., 2019). In addition, this approach overcomes the challenge of detecting cohort variation within datasets due to the high inter-individual variation associated with the human gut phageome (Clooney et al., 2019; Shkoporov et al., 2019; Sutton and Hill, 2019). Also, where annotation is possible, it is important to consider that some phages use alternative genetic codes such as Lak phages and crAss-like phages of specific candidate genera (Guerin et al., 2018; Devoto et al., 2019).
Although the number of phage genomes being generated is increasing rapidly many fail to be taxonomically assigned (Korf et al., 2019). Often the newly deposited phages are de novo assembled without in vitro characterization and therefore do not necessarily slot into a morphology-based taxonomic scheme (Ackermann, 2012). As a result, there is a significant push toward a genome-based taxonomic classification to allow universal and accurate taxonomic assignment of phages that have not yet been cultured in vitro (Meier-Kolthoff and Göker, 2017; Simmonds et al., 2017; Aiewsakun et al., 2018; Eloe-Fadrosh, 2019). This is not without challenges due to the current incompleteness of viral databases, the mosaic structure of phages and the lack of a universal taxonomic marker shared across phages (Lima-Mendez et al., 2011; Shapiro and Putonti, 2018). The application of gene-sharing networks, protein clustering, and whole virome analysis provides possible methodologies to aid the development of such a scheme (Clooney et al., 2019; Jang et al., 2019). This can provide insights into evolution and shared functions across phages (Shkoporov and Hill, 2019). Taken together, this highlights the need for the development, optimisation, and standardization of protocols in many aspects of viral metagenomics and in silico tools. Sequence-based analyses have expanded our insights into the composition of the human phageome which would not be possible in vitro. Despite this, in silico analysis alone can provide little insights into phage-host interactions and biological properties of de novo phages as is possible with in vitro and in vivo methods. Both methodologies in unison will be essential in fully understanding the interactions between phages and the gut microbiome and how these relationships ultimately impact on health.
With recent findings, several questions have arisen regarding the human gut phageome. The observed ability of certain virulent phages to stably co-exist with their host over time remains poorly understood and requires further elucidation (Shkoporov et al., 2018a, 2019). The isolation of dominant gut phages and their hosts, such as the crAss-like phages, which form the virulent core phageome in healthy individuals would provide insights into the mechanisms behind these interactions. CRISPR-spacer analyses have provided some direction for targeting potential hosts (Shkoporov et al., 2019).
Gut bacteria can be difficult to culture as they are often strict anaerobes and the conditions necessary for their growth may be unknown. Metagenomic studies have demonstrated the diversity of bacterial species in the human gut but this is of limited value when an isolate is required in vitro (Gill et al., 2006; Turnbaugh et al., 2007; Huttenhower et al., 2012). Until recently, as many as ~80% of the bacteria identified by these studies were uncultured or unculturable (Eckburg et al., 2005). Nevertheless, significant efforts are being made to culture the “unculturable.” As a result, there has been a push toward the isolation and culturing of bacteria using novel techniques with the complement of genome sequencing. Culturing of gut bacteria is not only necessary to improve our understanding of their role in the microbiome, it is also key for investigating phage-host dynamics and the validation and expansion of in silico findings. Improvements in culturing methods have played an important role in cultivating bacterial species of the human gut. Media developments have aided the isolation of novel obligate anaerobes from human feces. For example, yeast extract, casitone and fatty acid (YCFA) medium has been shown to support the growth of these bacteria to high levels and can be modified with antibiotics, carbohydrates or other components to select for less abundant species or phenotypes of interest (Duncan et al., 2002; Browne et al., 2016; Das et al., 2018; Forster et al., 2019). Culturomics has also proved to be an important tool in expanding the repertoire of bacterial isolates from the human gut (Bilen et al., 2018). This involves coupling high-throughput culture-dependent and culture-independent methods. Samples from which bacteria are to be isolated are tested in multiple optimized culture conditions and rapid identification of isolates is performed using MALDI-TOF MS and 16S rRNA gene sequencing (Lagier et al., 2012, 2016). In the context of the human gut, culturomics has been successfully applied in the isolation of bacteria from feces, small intestine and colonic samples (Lagier et al., 2016). The potential of this method was extensively reviewed by Lagier and colleagues (Lagier et al., 2018). It is also important to consider screening conditions in relation to phenotypic features. For example, the human gut contains a significant population of spore-forming bacterial species which are troublesome to culture (Abecasis et al., 2013; Rajilić-Stojanović and de Vos, 2014; Browne et al., 2016). Browne and colleagues successfully cultured an array of spore-formers from fecal samples using ethanol shock enrichment to distinguish them from vegetative cells (Riley et al., 1987; Browne et al., 2016). This led to the isolation of 137 bacterial species from six healthy individuals, 90 of which were on the Human Microbiome Project's “most wanted” list of species that had yet to be cultured and sequenced (Browne et al., 2016). This emphasizes that even the most stubborn gut residents can be cultured in vitro when the correct conditions are applied; however, identifying these conditions can be difficult.
Fecal samples have been widely used when screening for novel gut phages. Traditional methods of plaque and spot assays are still widely used in this process. This involves screening a phage rich suspension, such as fecal filtrate, from which bacteria and debris have been removed, against a lawn of pure bacterial culture in overlay agar. A successful phage-host pair is indicated by host lysis through spot or plaque formation. These are picked, enriched, and purified on the specific host (Furuse et al., 1978; Kai et al., 1985). Due to the variable abundance of different phages in fecal samples, enrichment is performed to allow phage-host interactions establish and increase titres to detectable levels which is generally indicated by clearing of culture (Salem et al., 2015). This is followed by sequencing of the phage genome. Plaque assays may fail to detect temperate phages unless an induction treatment, such as mitomycin C, is included to induce the phage to a lytic state (Castellazzi et al., 1972). It has also become apparent that not all virulent phages form clear plaques or spots on a suitable host nor do they clear liquid cultures (Porter et al., 2020). This can be due to phenotypic heterogeneity within isogenic populations. Enrichment of phages on intestinal bacteria, either acquired from culture collection or through selective enrichment from feces, followed by shotgun sequencing is a useful approach for screening of novel gut phages. This approach led to the isolation of ΦcrAss001, the first member of the elusive crAss-like phage family to be isolated in pure culture with its host Bacteroides intestinalis (Shkoporov et al., 2018a). To achieve this, 20 healthy fecal donors were recruited, fecal filtrates were prepared, pooled and screened against 53 individually cultured bacterial strains in modified YCFA broth under anaerobic conditions over 3 days, with each filtered lysate used to inoculate the subsequent round of enrichment. The enriched lysates were then sequenced to identify expansion on a specific host (Shkoporov et al., 2018a). Chemostats are also convenient for phage-host enrichment from feces. They allow more controlled conditions than can be provided in vitro and are useful for examining phage-host interactions (Santiago-Rodriguez et al., 2015a).
Culture-independent methodologies have also been applied in the isolation of novel phage-host pairs. Within biological samples there are constantly a percentage of bacterial cells that either have free virions attached to their surface or contain phages internally either replicating or as prophages. Several protocols take advantage of this to match phages to their hosts. Viral tagging (VT) is a useful tool for making this match. A sample of interest is obtained, and virions are randomly tagged with a generic fluorescent marker that binds to nucleic acids. The tagged phages are then applied to potential bacterial hosts. If interactions occur, fluorescence-activated cell sorting (FACS) can isolate and discriminate between tagged phage-host pairs and free virions and the isolated phage can then be sequenced (Deng et al., 2012, 2014). Single bacterial cells can also be isolated from samples using FACS. This allows complete sequencing of the bacterial genome to be performed and in parallel also capture the genomes of phages which were infecting or attached to the cell. This method of identifying phage-host pairs is called single amplified genomes (SAGs) (Swan et al., 2013; Labonté et al., 2015). It is also possible to directly isolate single uncultured viruses from environmental samples using flow cytometry. This allows for single viral genomics (SVGs) which involves sequencing isolated viruses individually which overcomes certain assembly limitations associated with metagenomics and provides insights into strain variation and genetic diversity (Allen et al., 2011; Martinez-Hernandez et al., 2017). In one case, this method allowed the recovery of the genetic information from 5,000 individual viruses from a marine sample (Martínez et al., 2014). SVGs, however, exclude the host and provides little insight into phage-host pairing or interactions. FACs, although very useful in its applications, is also biased toward the isolation of more abundant viruses (Martinez-Hernandez et al., 2017; Lawrence et al., 2019). VT proves to be a particularly powerful tool for examining phage-host interactions. It has been largely applied with known panels of bacteria but has potential in identifying pairing between both unknown bacteria and novel phages from a specific sample. A workflow incorporating single-cell VT in parallel with metagenomics and SAGs has been developed using human fecal samples which led to the identification of 363 novel phage-host pairs (Džunková et al., 2019).
As mentioned previously studies frequently use feces as the sampling source for examining the virome due to difficulties associated with sampling other sites of the gut and acquiring human biopsies. It has been shown that consistency and transit time of feces can influence bacterial composition and in turn can cause discrepancies in studies, thus highlighting the importance of recording this information (Vandeputte et al., 2016). Feces is the most easily accessible and ethical sample source for examining the gut, although, it is important to consider that phage-host interactions vary radially and longitudinally along the GIT (Maura et al., 2012a; Galtier et al., 2017). Indeed, examination of the virome along different sites of the GIT in non-human primates found that the virome of the large intestine and rectum were similar but distinct from the ileum. These findings indicate that feces provide a good representation of the colon specifically rather than the gut as a whole (Zhao et al., 2019). Another example of sampling site variation was observed in the identification of microbial signatures associated with Crohn's disease in children. Such signatures were observed in the mucosa biopsies but went undetected in assessment of stool samples (Gevers et al., 2014). Therefore, focusing solely on stool samples would have had significant implications for the findings of this study. Nonetheless, feces is a useful proxy for studying the gut phageome but animal models allow for biogeographical variation to be accounted for.
The examination of phages at multiple experimental levels (in vitro to ex vivo into in vivo), the importance of incorporating relevant conditions to experimental models and considering the influence of bacterial metabolic state is essential when examining phage-host interactions and are important factors to consider when developing an experimental pipeline (Lourenço et al., 2018). While in vitro studies can be of great importance, they are not necessarily always conducive to realistic conditions. Several studies have demonstrated the usefulness of in vivo models in examining the gut phageome (Maura et al., 2012a,b; Reyes et al., 2013; De Sordi et al., 2017; Galtier et al., 2017; Hsu et al., 2019). For example, De Sordi and colleagues examined phage-host strain jumping both in vitro and in vivo in a conventional murine model, however, the phenomenon was only detected under the latter conditions (De Sordi et al., 2017). This emphasizes the importance of testing under multiple experimental conditions. Murine models or other in vivo models can overcome the issue of acquiring samples that would be ethically difficult to access, such as biopsies, and they also allow spatial heterogeneity in the GIT to be considered (De Sordi et al., 2019). Furthermore, overlap between the murine and human gut phageome has been observed. A study that examined the role of intestinal phages in IBD using a murine model of ulcerative colitis, with healthy mice as a control, mapped sequencing reads to contigs assembled from healthy humans and IBD patient fecal viral reads. An overlap of the murine phageome and the human gut phageome was observed in both health and disease states. These findings suggest that murine models are suitable for studying phage-bacteria interactions in the gut (Duerkop et al., 2018).
There are several other techniques that can be incorporated into human gut phageome studies including transcriptome profiling and metatranscriptomics to give insights into gene expression during individual or community phage-host interactions. Few studies have examined this in the context of the human gut. Transcriptome profiling of phage communities in the oral cavity was carried out to compare gene expression in health vs. periodontal disease (Santiago-Rodriguez et al., 2015b). Similar studies should be performed to examine gene expression in a healthy compared to a diseased gut. Additionally, transcriptomics is useful for examining changes in bacterial host gene expression in the presence of a phage (Porter et al., 2020). It can also determine the effect of an experimental model on bacterial host gene expression (Denou et al., 2007). Consequently, this can aid the development of experimental pipelines and avoid inaccurate conclusions. Bioelectronics could be explored as means of examining phage-bacteria interactions in the gut. It has provided many useful biological and biomedical applications (Strakosas et al., 2015; Pitsalidis et al., 2018). With this, it is possible to mimic bacterial and mammalian membranes, called supported lipid bilayers, using bioelectronic tools and membrane biosensors which can detect specific interactions. For example, this method was used to examine membrane interactions with antibiotics to determine antibiotic targets that were discriminatory between bacterial and mammalian membranes (Su et al., 2019). Similar tools were used to monitor Salmonella typhimurium infection of epithelial cells (Tria et al., 2014). Bioelectronics could have potential applications for the detailed examination of phage-bacteria interactions.
Methodology can have a greater influence on the virome than the examined disease state (Gregory et al., 2020). The progression in metagenomics is more rapid than the isolation and characterization of novel phage-host pairs from the human gut. To overcome this, both bioinformatics and wet lab work can assist each other to expand on findings. A top-down approach that uses in silico findings to guide design of lab-based experiments can allow a more targeted approach. For example, genome sequences of novel phages detected in silico following metagenomic sequencing of biological samples can be used to perform host predictions and develop real-time PCR (qRT-PCR) primers to specifically target these phages. This allows the phages to be monitored in vitro/ex vivo when enriched from the sample of origin on a panel of predicted bacterial hosts thus aiding their isolation in pure culture. When isolation is successful further biological characterisations can be performed while the already available in silico data provides information on genome functions and allows relative phages to be identified. Novel phages that have been identified in silico need to be isolated and characterized in vitro/in vivo to understand the mechanisms they employ when interacting with their host, and the health and disease implications that this may have for the human host. Experimental pipelines that bring the above together will be key to this progress (Figure 3).
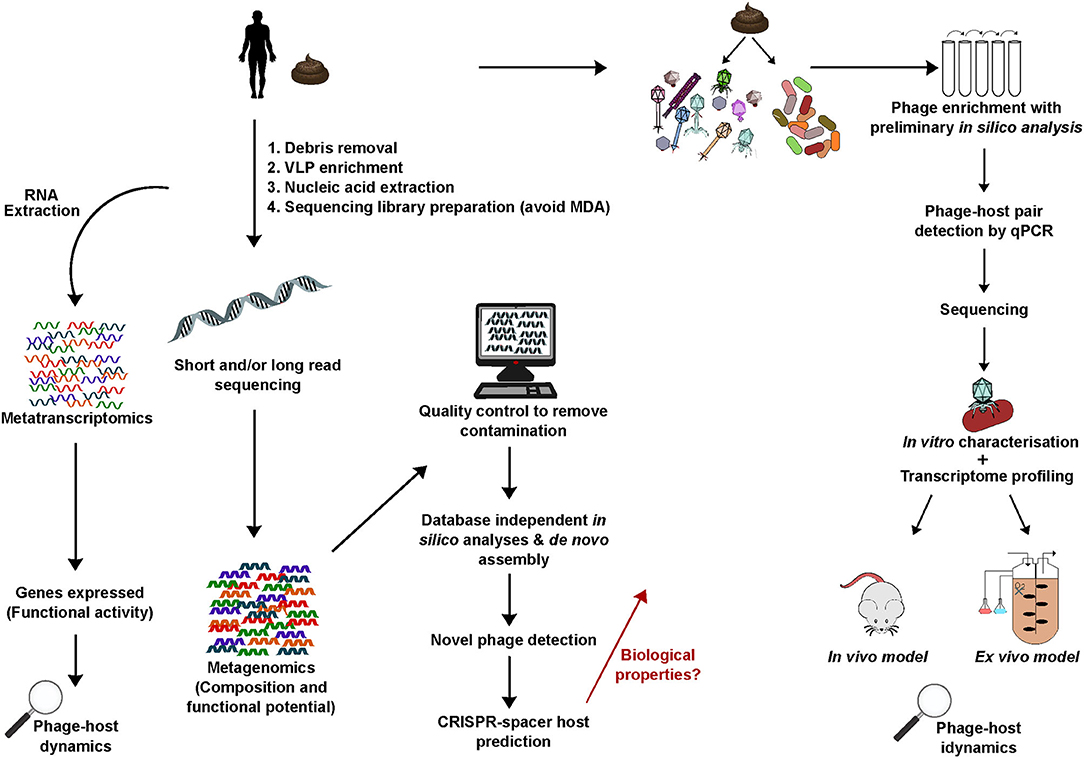
Figure 3. A generic overview of key experimental steps important in studying the human gut phageome: from metagenomics, database-independent whole virome analyses, in silico identification of novel phages to isolation and characterization. Linking bioinformatics and lab-based research provides important insights into phageome composition, aids novel phage isolation and characterization of biological properties including interactions with the bacterial and mammalian host.
CrAssphage provides an important example of novel phage discovery using in silico methods to overcome the limitations of database-dependency. For years following this discovery crAssphages largely only existed in silico due to the challenges faced in achieving isolation on a suitable host. Here, we will discuss the crAssphage timeline from an in silico discovery to an in vitro reality, and highlight the role that this phage family has played in pushing human gut phage research toward looking beyond the known (Figure 4).
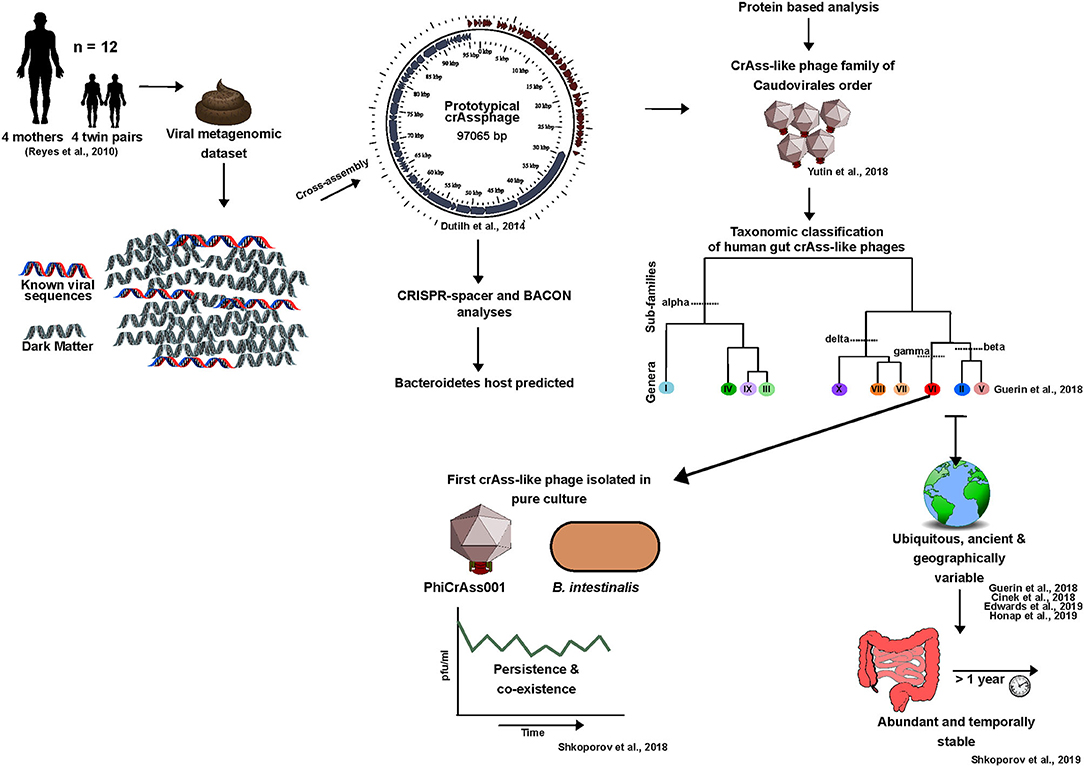
Figure 4. The crAss-like phage family timeline: from in silico discovery to an in vitro reality (2014-2020).
Despite one century of phage research, the most abundant phage in the human gut remained undetected until 2014. The majority of its encoded proteins had no homology with phage proteins in public viral databases (Dutilh et al., 2014). In this sense, crAssphage is an ideal representative of the current status of human gut phage research in that it highlights the potential for novel phage discovery within viral dark matter. In the few years since its discovery, this phage has also played an important role in providing essential insights into the human gut phageome.
For clarity, the initial crAssphage detected in 2014 will be referred to as prototypical-crAssphage (p-crAssphage). P-crAssphage was discovered through mining of a previously published human fecal metagenomic dataset (Reyes et al., 2010). Co-occurring contigs were detected based on depth-profile similarities thus ensuring grouped contigs originated from the same phage genome. Using this information, the p-crAssphage genome was de novo assembled into a complete circular genome ~97 kbp in size using Cross-Assembly (crAss) software, a reference-independent tool from where the phage received its name (Dutilh et al., 2012, 2014). The novel dsDNA phage was further analyzed against public metagenomes sequenced from human feces in Europe, Korea and the USA and was detected in 73% of the datasets examined. This analysis indicated that the phage was present in ~50% of individuals and was found to be six times more abundant in public metagenomes than all other known phages combined (Dutilh et al., 2014). Despite the abundance at which this phage occurs, it remained undetected prior to 2014 due to the database-dependency of bioinformatic pipelines.
Following studies detected a number of p-crAssphage variants (Liang et al., 2016; Manrique et al., 2016; García-Aljaro et al., 2017). These was further explored in 2018 by Yutin and colleagues through a detailed protein sequence-based analysis of p-crAssphage. This led to the proposition of p-crAssphage as the original member of an expansive family of phages, the crAss-like phage family, which includes the IAS-virus (Oude Munnink et al., 2014; Yutin et al., 2018). These phages were predicted to have a podovirus-like morphology, resolve into the Caudovirales order, and were initially predicted to be predominately temperate (Yutin et al., 2018).
In 2018, a taxonomic classification scheme for crAss-like phages of human gut origin was proposed, with four sub-families (Alphacrassvirinae, Betacrassvirinae, Gammacrassvirinae and Deltacrassvirinae) and ten candidate genera. This was developed using 249 crAss-like phages, 244 of which were de novo assembled (Guerin et al., 2018). Members of the same subfamily shared 20–40% orthologous protein similarity and crAss-like phages that clustered into each genus shared >40% protein similarity. Classification was performed using a protein clustering approach that allowed similarity to be identified across the phages despite sharing little to no similarity at the nucleotide level. The developed scheme was further supported by phylogenetic analyses of four conserved genes (capsid, primase, portal protein and terminase) which is consistent with earlier studies (Guerin et al., 2018; Yutin et al., 2018).
Through CRISPR-spacer profiling and bacterial co-abundance analysis, it was predicted that crAss-like phages infect bacteria of the Bacteroides genus or other members of the Bacteroidetes phylum (Dutilh et al., 2014).
In 2018, the first crAssphage-host pair was isolated in pure culture using a human fecal enrichment protocol and provided some insight into the biological properties of these phages (Shkoporov et al., 2018a). The isolated IAS-like crAssphage, ΦcrAss001, of candidate genus VI, sub-family Betacrassvirinae, infects Bacteroides intestinalis thus confirming in silico host predictions. Electron micrographs also confirmed the podovirus-like morphology predicted for these phages. The ΦcrAss001 genome is absent of a lysogeny module but intriguingly the phage can co-exist with its host at high titers in co-culture over 3 weeks without significant impact on host growth (Shkoporov et al., 2018a). This is consistent with the observed persistence and stability of crAss-like phages in metagenomics datasets over time (Guerin et al., 2018; Edwards et al., 2019; Shkoporov et al., 2019). This also supports the stable engraftment of these phages in certain individuals for up to a year following FMT (Draper et al., 2018; Siranosian et al., 2020). More recently, the isolation of two crAss-like phages closely related to ΦcrAss001 which infect Bacteroides thetaiotaomicron have been reported, DAC15 and DAC17 (Hryckowian et al., 2020). Detailed biological characterization of these phages has yet to be performed but will shine further light on the traits shared across this phage family.
Thus far we know that crAss-like phages are generally absent from the neonate gut but are acquired in infanthood at low abundances, however, the influence of birth mode on the transmission of these phages has been debated (McCann et al., 2018; Liang et al., 2020; Siranosian et al., 2020). It was initially thought that crAss-like phages were absent from the infant gut, however, now it is believed that they can be vertically transferred from mother to infant (Siranosian et al., 2020). The early colonization of crAss-like phages and their universal ubiquity is not surprising considering the predominance of Bacteroidetes in the healthy human gut from infancy (Rodríguez et al., 2015). These phages have also been detected in the elderly virome (Stockdale et al., 2018). The crAssphage family is globally distributed, with one or more strain being detected in 77% of individuals with geographic clustering (Cinek et al., 2018; Guerin et al., 2018; Edwards et al., 2019). For example, p-crAssphage (candidate genus I), is largely absent from the gut of hunter-gatherer populations and is more abundant among industrialized populations (Honap et al., 2020). CrAss-like phages have not been linked to factors such as disease, age, gender, or body mass index, but a weak link to diet has been detected as well as potential links to ethnicity and geography (Guerin et al., 2018; Edwards et al., 2019; Honap et al., 2020). Diet may be one factor driving the crAssphage family variation observed between Western and more rural non-industrialized populations. The Western diet generally drives a microbiota dominated in Bacteroides/Clostridia, whereas a non-Western fiber rich diet is associated with high Prevotella/low Bacteroides. Therefore, this diet driven variation in dominant bacterial species in the gut may also influence crAss-like phage composition (Gorvitovskaia et al., 2016; Guerin et al., 2018).
One of the most intriguing characteristics of crAss-like phages is their ability to persist in the human gut (Guerin et al., 2018; Shkoporov et al., 2018a). With these phages forming a major component of the predominantly virulent personal persistent virome, mechanisms other than lysogeny must be at play to maintain these phages in the human gut over time (Shkoporov et al., 2019). This is supported by the absence of lysogeny genes in the ΦcrAss001 genome and crAss-like prophages going undetected in Bacteroidales genomes deposited in databases such as NCBI RefSeq (Shkoporov et al., 2018a). Although, it should be noted that lysogeny-modules have been predicted for some crAss-like phages detected in silico (Yutin et al., 2018). “Royal-family” model dynamics may provide an explanation for the observed stability. If kill-the-winner dynamics occur at a strain or sub-strain level, stability is maintained at the species or genus taxonomic levels over time or it is possible that these phages engage in “piggyback-the-winner” dynamics (Silveira and Rohwer, 2016; Breitbart et al., 2018). The spatial heterogeneity of the gut also provides hosts with phage-inaccessible reservoirs in microhabitats creating source-sink dynamics that reduce selective pressures and allow the coexistence of both (Lourenço et al., 2020). Additionally, the oscillation of bacteria between phage-permissive and non-permissive phenotypes could also have an important role in this persistence (Porter et al., 2020). Overall, it is likely that the persistence of crAss-like phages in the human gut is due to an inter-play of multiple mechanisms.
The isolation of further crAss-like phage representatives will be necessary to identify shared and unique biological properties across this family. The precise mechanisms mediating the stable and persistent colonization of these phages at high levels also needs to be further explored. Ideally, future studies should use in vivo models that simulate gut conditions as best as possible. The goal will be to understand how these phages shape our microbiome and influence health. Although we still have a lot to learn about these enigmatic phages, they have played a significant role in highlighting the potential of viral dark matter and in improving our understanding of the human gut phageome both in terms of composition and interactions (Figure 4).
Phages have many potential applications which could be beneficial to humans in addition to forming an important component of the microbiome. The resurging interest in phages largely coincides with the current and projected severity of the antibiotic resistance crises. It is estimated that the inability to treat bacterial diseases due to multi-drug resistance may lead to 10 million additional deaths globally per annum by 2,050 (Sugden et al., 2016). Phage therapy is a potential alternative or aid to antibiotic treatment, however, hurdles associated with this include undesirable immune responses, host resistance development, identifying suitable phage(s), and regulatory issues (Oechslin, 2018; Brüssow, 2019). Several human diseases, such as IBD, have a bacterial component that worsens disease status due to over-stimulation of the immune system (Gevers et al., 2014). Phage-driven selective elimination of pathogens from the gut to shape microbiome composition toward homeostasis is also being examined. The ability of phages to shape the microbiome is supported by outcomes of FFT/FMT studies (Ott et al., 2017; Draper et al., 2018). Phages also produce endolysins which degrade the cell wall of their associated bacterial host and have therapeutic potential as alternatives to antibiotics (Love et al., 2018). While phage therapy is not within the scope of this review, we will mention a few cases of its application. The current status of phage therapy has been extensively reviewed and assessed (Czaplewski et al., 2016; Cisek et al., 2017; Lin et al., 2017; Lourenço et al., 2018; Brüssow, 2019; Hyman, 2019). Phages have also been proposed as alternatives to preservatives or antibiotics in food production and biocontrol (O'Sullivan et al., 2019; Lewis and Hill, 2020; Rabiey et al., 2020). We have seen the antimicrobial capacity of phages in selectively eradicating infection and alleviating diseases with a microbial component. Recently, phages were successfully administered in eliminating cytolytic E. faecalis in humanized mice, a pathogen responsible for increased disease severity and mortality in patients with alcoholic hepatitis (Duan et al., 2019). Phage-guided nanoparticles have been used in the eradication of pro-tumoral Fusobacterium nucleatum and allowed the site-specific delivery of chemotherapeutic drugs in parallel. This resulted in colorectal cancer tumor reduction in a mouse model with limited impact on healthy tissue (Zheng et al., 2019). This technology was also applied in a piglet model without induction of the immune system or adverse effects (Zheng et al., 2019). However, phage associated immune activation resulting in the exacerbation of symptoms has also be observed (Gogokhia et al., 2019). Remarkably, phage therapy led to the eradication of multi-drug resistant Klebsiella pneumoniae by oral and rectal administration of one lytic phage in a human patient without any adverse effects (Corbellino et al., 2019). Phages can also act in synergy with innate immune cells to eliminated pathogens. This was observed in a murine lung model of drug resistant Pseudomonas aeruginosa in which phages could only eradicate the pneumonia-causing pathogen in synergy with neutrophils (Roach et al., 2017). These are some examples of the potential of phage therapy in selectively eliminating pathogens. Nonetheless, we need to fully decipher phage dynamics with both the bacterial and human host before we apply phages for therapeutic use. Considering the impending outcome of the antibiotic resistance crisis and the importance of microbiome homeostasis we need to expand our knowledge rapidly.
As we begin to understand phage-host dynamics and how these influence the microbiome, specific phages or phage compositions may be identified as biomarkers of disease or health. For example, whole virome analysis of the IBD and healthy virome identified a shift from a lytic core of a few but abundant clusters of Microviridae and crAss-like phages in health to a virome with increased induction of temperate phages such as Myoviridae and Siphoviridae in the disease state (Clooney et al., 2019). Further development of these findings and the identification of compositional patterns in the context of other diseases states may lead to the use of virome biomarkers in the future. Elucidation of the personal persistent virome will serve as an important window into the gut phageome and provide us with insights into ecology, composition and dynamics including the persistence of virulent phages.
One of the critical shortcomings of human gut phage research is the incomplete analysis of metagenomic datasets due to the dependency on a poorly curated viral database. Furthermore, little progress has been made in the isolation and characterization of novel gut phage-host pairs. In recent years, efforts are being made to overcome bottlenecks. This requires the development of universal and easily reproducible methods that limit bias during viral enrichment, nucleic acid extraction, and sequencing library preparation. In silico analyses need to be performed using database-independent methods that allow for a complete virome analysis using benchmarked criteria. The development of a sequence-based taxonomic scheme is needed to facilitate the rapid expansion of phage sequences as a result of high-throughput sequencing technology. The current contradictions among different gut virome studies need to be rectified and clarified. Overall progression in metagenomic analyses of the gut phageome has been more rapid than the isolation of gut phages which is ultimately the key for expanding our scope of the mechanisms mediating phage-host relationships. Recent findings have provided important insights, but they also highlight how little we know about this important and enigmatic component of our gut.
EG and CH drafted and approved this manuscript.
The authors declare that this study received funding from Janssen Biotech. The funder was not involved in the study design, collection, analysis, interpretation of data, the writing of this article or the decision to submit it for publication. This research was conducted with the financial support of Science Foundation Ireland (SFI) under Grant Number SFI/12/RC/2273 a Science Foundation Ireland's Spokes Programme which is co-funded under the European Regional Development Fund under Grant Number SFI/14/SP APC/B3032, and a research grant from Janssen Biotech, Inc.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The authors thank Dr. Stephen Stockdale and Dr. Thomas Sutton for their critical remarks during the preparation of this manuscript.
Abecasis, A. B., Serrano, M., Alves, R., Quintais, L., Pereira-Leal, J. B., and Henriques, A. O. (2013). A genomic signature and the identification of new sporulation genes. J. Bacteriol. 195, 2101–2115. doi: 10.1128/JB.02110-12
Ackermann, H.-W. (1998). “Tailed bacteriophages: the order caudovirales,” in Advances in Virus Research, eds K. Maramorosch, F. A. Murphy, and A. J. Shatkin (Quebec, QC: Academic Press), 135–201.
Ackermann, H.-W. (2012). “Chapter 1 - bacteriophage electron microscopy,” in Advances in Virus Research Bacteriophages, Part A, eds M. Łobocka and W. T. Szybalski (Academic Press), 1–32.
Aggarwala, V., Liang, G., and Bushman, F. D. (2017). Viral communities of the human gut: metagenomic analysis of composition and dynamics. Mob. DNA 8:12. doi: 10.1186/s13100-017-0095-y
Aiewsakun, P., Adriaenssens, E. M., Lavigne, R., Kropinski, A. M., and Simmonds, P. (2018). Evaluation of the genomic diversity of viruses infecting bacteria, archaea and eukaryotes using a common bioinformatic platform: steps towards a unified taxonomy. J. Gen. Virol. 99, 1331–1343. doi: 10.1099/jgv.0.001110
Allen, L. Z., Ishoey, T., Novotny, M. A., McLean, J. S., Lasken, R. S., and Williamson, S. J. (2011). Single virus genomics: a new tool for virus discovery. PLOS ONE 6:e17722. doi: 10.1371/journal.pone.0017722
Baquero, F., and Nombela, C. (2012). The microbiome as a human organ. Clin. Microbiol. Infect. 18, 2–4. doi: 10.1111/j.1469-0691.2012.03916.x
Barr, J. J., Auro, R., Furlan, M., Whiteson, K. L., Erb, M. L., Pogliano, J., et al. (2013). Bacteriophage adhering to mucus provide a non-host-derived immunity. Proc. Natl. Acad. Sci. U.S.A. 110, 10771–10776. doi: 10.1073/pnas.1305923110
Bauer, M. A., Kainz, K., Carmona-Gutierrez, D., and Madeo, F. (2018). Microbial wars: Competition in ecological niches and within the microbiome. Microb. Cell Graz Austria 5, 215–219. doi: 10.15698/mic2018.05.628
Bayliss, C. D. (2009). Determinants of phase variation rate and the fitness implications of differing rates for bacterial pathogens and commensals. FEMS Microbiol. Rev. 33, 504–520. doi: 10.1111/j.1574-6976.2009.00162.x
Belleghem, J. D. V., Clement, F., Merabishvili, M., Lavigne, R., and Vaneechoutte, M. (2017). Pro- and anti-inflammatory responses of peripheral blood mononuclear cells induced by Staphylococcus aureus and Pseudomonas aeruginosa phages. Sci. Rep. 7, 1–13. doi: 10.1038/s41598-017-08336-9
Bergh, O., Børsheim, K. Y., Bratbak, G., and Heldal, M. (1989). High abundance of viruses found in aquatic environments. Nature 340, 467–468. doi: 10.1038/340467a0
Bertozzi Silva, J., Storms, Z., and Sauvageau, D. (2016). Host receptors for bacteriophage adsorption. FEMS Microbiol. Lett. 363. doi: 10.1093/femsle/fnw002
Bilen, M., Dufour, J.-C., Lagier, J.-C., Cadoret, F., Daoud, Z., Dubourg, G., et al. (2018). The contribution of culturomics to the repertoire of isolated human bacterial and archaeal species. Microbiome 6:94. doi: 10.1186/s40168-018-0485-5
Blower, T. R., Evans, T. J., Przybilski, R., Fineran, P. C., and Salmond, G. P. C. (2012). Viral evasion of a bacterial suicide system by RNA–based molecular mimicry enables infectious altruism. PLOS Genet. 8:e1003023. doi: 10.1371/journal.pgen.1003023
Bolduc, B., Jang, H. B., Doulcier, G., You, Z.-Q., Roux, S., and Sullivan, M. B. (2017). vConTACT: an iVirus tool to classify double-stranded DNA viruses that infect Archaea and Bacteria. PeerJ. 5:3243. doi: 10.7717/peerj.3243
Breitbart, M., Bonnain, C., Malki, K., and Sawaya, N. A. (2018). Phage puppet masters of the marine microbial realm. Nat. Microbiol. 3, 754–766. doi: 10.1038/s41564-018-0166-y
Browne, H. P., Forster, S. C., Anonye, B. O., Kumar, N., Neville, B. A., Stares, M. D., et al. (2016). Culturing of ‘unculturable' human microbiota reveals novel taxa and extensive sporulation. Nature 533, 543–546. doi: 10.1038/nature17645
Brüssow, H. (2019). Hurdles for phage therapy to become a reality—an editorial comment. Viruses 11, 557. doi: 10.3390/v11060557
Brüssow, H., Canchaya, C., and Hardt, W.-D. (2004). Phages and the evolution of bacterial pathogens: from genomic rearrangements to lysogenic conversion. Microbiol. Mol. Biol. Rev. MMBR 68, 560–602. doi: 10.1128/MMBR.68.3.560-602.2004
Bruttin, A., Desiere, F., d'Amico, N., Guérin, J. P., Sidoti, J., Huni, B., et al. (1997). Molecular ecology of Streptococcus thermophilus bacteriophage infections in a cheese factory. Appl. Environ. Microbiol. 63, 3144–3150.
Bull, J. J., Vegge, C. S., Schmerer, M., Chaudhry, W. N., and Levin, B. R. (2014). Phenotypic resistance and the dynamics of bacterial escape from phage control. PLoS ONE 9:e94690. doi: 10.1371/journal.pone.0094690
Callanan, J., Stockdale, S. R., Shkoporov, A., Draper, L. A., Ross, R. P., and Hill, C. (2018). RNA phage biology in a metagenomic era. Viruses 10:386. doi: 10.3390/v10070386
Callanan, J., Stockdale, S. R., Shkoporov, A., Draper, L. A., Ross, R. P., and Hill, C. (2020). Expansion of known ssRNA phage genomes: from tens to over a thousand. Sci. Adv. 6:eaay5981. doi: 10.1126/sciadv.aay5981
Cani, P. D. (2018). Human gut microbiome: hopes, threats and promises. Gut 67, 1716–1725. doi: 10.1136/gutjnl-2018-316723
Casén, C., Vebø, H. C., Sekelja, M., Hegge, F. T., Karlsson, M. K., Ciemniejewska, E., et al. (2015). Deviations in human gut microbiota: a novel diagnostic test for determining dysbiosis in patients with IBS or IBD. Aliment. Pharmacol. Ther. 42, 71–83. doi: 10.1111/apt.13236
Castellazzi, M., George, J., and Buttin, G. (1972). Prophage induction and cell division in E. coli. Mol. Gen. Genet. MGG 119, 153–174. doi: 10.1007/BF00269134
Castro-Mejía, J. L., Muhammed, M. K., Kot, W., Neve, H., Franz, C. M. A. P., Hansen, L. H., et al. (2015). Optimizing protocols for extraction of bacteriophages prior to metagenomic analyses of phage communities in the human gut. Microbiome 3:64. doi: 10.1186/s40168-015-0131-4
Chatterjee, S., and Rothenberg, E. (2012). Interaction of bacteriophage λ with its E. coli Receptor, LamB. Viruses 4, 3162–3178. doi: 10.3390/v4113162
Chen, Y.-C., Liu, T., Yu, C.-H., Chiang, T.-Y., and Hwang, C.-C. (2013). Effects of GC bias in next-generation-sequencing data on de novo genome assembly. PLoS ONE 8:e62856. doi: 10.1371/journal.pone.0062856
Chibani-Chennoufi, S., Bruttin, A., Dillmann, M.-L., and Brüssow, H. (2004). Phage-host interaction: an ecological perspective. J. Bacteriol. 186, 3677–3686. doi: 10.1128/JB.186.12.3677-3686.2004
Cinek, O., Mazankova, K., Kramna, L., Odeh, R., Alassaf, A., Ibekwe, M. U., et al. (2018). Quantitative CrAssphage real-time PCR assay derived from data of multiple geographically distant populations. J. Med. Virol. 90, 767–771. doi: 10.1002/jmv.25012
Cisek, A. A., Dabrowska, I., Gregorczyk, K. P., and Wyzewski, Z. (2017). Phage therapy in bacterial infections treatment: one hundred years after the discovery of bacteriophages. Curr. Microbiol. 74, 277–283. doi: 10.1007/s00284-016-1166-x
Clooney, A. G., Sutton, T. D. S., Shkoporov, A. N., Holohan, R. K., Daly, K. M., O'Regan, O., et al. (2019). Whole-virome analysis sheds light on viral dark matter in inflammatory bowel disease. Cell Host Microbe 26, 764–778.e5. doi: 10.1016/j.chom.2019.10.009
Cohen, D., Melamed, S., Millman, A., Shulman, G., Oppenheimer-Shaanan, Y., Kacen, A., et al. (2019). Cyclic GMP–AMP signalling protects bacteria against viral infection. Nature 574, 691–695. doi: 10.1038/s41586-019-1605-5
Conceição-Neto, N., Zeller, M., Lefrère, H., De Bruyn, P., Beller, L., Deboutte, W., et al. (2015). Modular approach to customise sample preparation procedures for viral metagenomics: a reproducible protocol for virome analysis. Sci. Rep. 5:16532. doi: 10.1038/srep16532
Corbellino, M., Kieffer, N., Kutateladze, M., Balarjishvili, N., Leshkasheli, L., Askilashvili, L., et al. (2019). Eradication of a multi-drug resistant, carbapenemase-producing Klebsiella pneumoniae isolate following oral and intra-rectal therapy with a custom-made, lytic bacteriophage preparation. Clin. Infect. Dis. Off. Publ. Infect. Dis. Soc. Am. 70, 1998–2001. doi: 10.1093/cid/ciz782
Cornelissen, A., Ceyssens, P.-J., Krylov, V. N., Noben, J.-P., Volckaert, G., and Lavigne, R. (2012). Identification of EPS-degrading activity within the tail spikes of the novel Pseudomonas putida phage AF. Virology 434, 251–256. doi: 10.1016/j.virol.2012.09.030
Coward, C., Grant, A. J., Swift, C., Philp, J., Towler, R., Heydarian, M., et al. (2006). Phase-variable surface structures are required for infection of campylobacter jejuni by bacteriophages. Appl. Environ. Microbiol. 72, 4638–4647. doi: 10.1128/AEM.00184-06
Cumby, N., Edwards, A. M., Davidson, A. R., and Maxwell, K. L. (2012). The bacteriophage HK97 gp15 moron element encodes a novel superinfection exclusion protein. J. Bacteriol. 194, 5012–5019. doi: 10.1128/JB.00843-12
Cumby, N., Reimer, K., Mengin-Lecreulx, D., Davidson, A. R., and Maxwell, K. L. (2015). The phage tail tape measure protein, an inner membrane protein and a periplasmic chaperone play connected roles in the genome injection process of E. coli phage HK97. Mol. Microbiol. 96, 437–447. doi: 10.1111/mmi.12918
Czaplewski, L., Bax, R., Clokie, M., Dawson, M., Fairhead, H., Fischetti, V. A., et al. (2016). Alternatives to antibiotics—a pipeline portfolio review. Lancet Infect. Dis. 16, 239–251. doi: 10.1016/S1473-3099(15)00466-1
Das, P., Ji, B., Kovatcheva-Datchary, P., Bäckhed, F., and Nielsen, J. (2018). In vitro co-cultures of human gut bacterial species as predicted from co-occurrence network analysis. PLoS ONE 13:e195161. doi: 10.1371/journal.pone.0195161
De Sordi, L., Khanna, V., and Debarbieux, L. (2017). The gut microbiota facilitates drifts in the genetic diversity and infectivity of bacterial viruses. Cell Host Microbe 22, 801–808.e3. doi: 10.1016/j.chom.2017.10.010
De Sordi, L., Lourenço, M., and Debarbieux, L. (2019). The battle within: interactions of bacteriophages and bacteria in the gastrointestinal tract. Cell Host Microbe 25, 210–218. doi: 10.1016/j.chom.2019.01.018
Debroas, D., and Siguret, C. (2019). Viruses as key reservoirs of antibiotic resistance genes in the environment. ISME J. 13, 2856–2867. doi: 10.1038/s41396-019-0478-9
Deng, L., Gregory, A., Yilmaz, S., Poulos, B. T., Hugenholtz, P., and Sullivan, M. B. (2012). Contrasting life strategies of viruses that infect photo- and heterotrophic bacteria, as revealed by viral tagging. mBio 3:e00373-12. doi: 10.1128/mBio.00373-12
Deng, L., Ignacio-Espinoza, J. C., Gregory, A. C., Poulos, B. T., Weitz, J. S., Hugenholtz, P., et al. (2014). Viral tagging reveals discrete populations in Synechococcus viral genome sequence space. Nature 513, 242–245. doi: 10.1038/nature13459
Denou, E., Berger, B., Barretto, C., Panoff, J.-M., Arigoni, F., and Brüssow, H. (2007). Gene expression of commensal lactobacillus johnsonii strain NCC533 during in vitro growth and in the murine gut. J. Bacteriol. 189, 8109–8119. doi: 10.1128/JB.00991-07
Devoto, A. E., Santini, J. M., Olm, M. R., Anantharaman, K., Munk, P., Tung, J., et al. (2019). Megaphages infect Prevotella and variants are widespread in gut microbiomes. Nat. Microbiol. 4, 693. doi: 10.1038/s41564-018-0338-9
d'Humières, C., Touchon, M., Dion, S., Cury, J., Ghozlane, A., Garcia-Garcera, M., et al. (2019). A simple, reproducible and cost-effective procedure to analyse gut phageome: from phage isolation to bioinformatic approach. Sci. Rep. 9, 1–13. doi: 10.1038/s41598-019-47656-w
Donaldson, G. P., Lee, S. M., and Mazmanian, S. K. (2016). Gut biogeography of the bacterial microbiota. Nat. Rev. Microbiol. 14, 20–32. doi: 10.1038/nrmicro3552
Draper, L. A., Ryan, F. J., Smith, M. K., Jalanka, J., Mattila, E., Arkkila, P. A., et al. (2018). Long-term colonisation with donor bacteriophages following successful faecal microbial transplantation. Microbiome 6, 220. doi: 10.1186/s40168-018-0598-x
Duan, Y., Llorente, C., Lang, S., Brandl, K., Chu, H., Jiang, L., et al. (2019). Bacteriophage targeting of gut bacterium attenuates alcoholic liver disease. Nature 575, 505–511. doi: 10.1038/s41586-019-1742-x
Duerkop, B. A., and Hooper, L. V. (2013). Resident viruses and their interactions with the immune system. Nat. Immunol. 14, 654–659. doi: 10.1038/ni.2614
Duerkop, B. A., Kleiner, M., Paez-Espino, D., Zhu, W., Bushnell, B., Hassell, B., et al. (2018). Murine colitis reveals a disease-associated bacteriophage community. Nat. Microbiol. 1:210. doi: 10.1038/s41564-018-0210-y
Duerr, D. M., White, S. J., and Schluesener, H. J. (2004). Identification of peptide sequences that induce the transport of phage across the gastrointestinal mucosal barrier. J. Virol. Methods 116, 177–180. doi: 10.1016/j.jviromet.2003.11.012
Duncan, S. H., Hold, G. L., Harmsen, H. J., Stewart, C. S., and Flint, H. J. (2002). Growth requirements and fermentation products of Fusobacterium prausnitzii, and a proposal to reclassify it as Faecalibacterium prausnitzii gen. nov., comb. nov. Int. J. Syst. Evol. Microbiol. 52, 2141–2146.
Dutilh, B. E., Cassman, N., McNair, K., Sanchez, S. E., Silva, G. G. Z., Boling, L., et al. (2014). A highly abundant bacteriophage discovered in the unknown sequences of human faecal metagenomes. Nat. Commun. 5, 4498. doi: 10.1038/ncomms5498
Dutilh, B. E., Schmieder, R., Nulton, J., Felts, B., Salamon, P., Edwards, R. A., et al. (2012). Reference-independent comparative metagenomics using cross-assembly: crAss. Bioinformatics 28, 3225–3231. doi: 10.1093/bioinformatics/bts613
Dy, R. L., Przybilski, R., Semeijn, K., Salmond, G. P. C., and Fineran, P. C. (2014). A widespread bacteriophage abortive infection system functions through a Type IV toxin–antitoxin mechanism. Nucleic Acids Res. 42, 4590–4605. doi: 10.1093/nar/gkt1419
Džunková, M., Low, S. J., Daly, J. N., Deng, L., Rinke, C., and Hugenholtz, P. (2019). Defining the human gut host–phage network through single-cell viral tagging. Nat. Microbiol. 4, 2192–2203. doi: 10.1038/s41564-019-0526-2
Eckburg, P. B., Bik, E. M., Bernstein, C. N., Purdom, E., Dethlefsen, L., Sargent, M., et al. (2005). Diversity of the human intestinal microbial flora. Science 308, 1635–1638. doi: 10.1126/science.1110591
Edwards, R. A., Vega, A. A., Norman, H. M., Ohaeri, M., Levi, K., Dinsdale, E. A., et al. (2019). Global phylogeography and ancient evolution of the widespread human gut virus crAssphage. Nat. Microbiol. 4, 1727–1736. doi: 10.1038/s41564-019-0494-6
Eloe-Fadrosh, E. A. (2019). Towards a genome-based virus taxonomy. Nat. Microbiol. 4, 1249–1250. doi: 10.1038/s41564-019-0511-9
Enault, F., Briet, A., Bouteille, L., Roux, S., Sullivan, M. B., and Petit, M.-A. (2017). Phages rarely encode antibiotic resistance genes: a cautionary tale for virome analyses. ISME J. 11, 237–247. doi: 10.1038/ismej.2016.90
Eriksen, R. S., Svenningsen, S. L., Sneppen, K., and Mitarai, N. (2018). A growing microcolony can survive and support persistent propagation of virulent phages. Proc. Natl. Acad. Sci. U.S.A. 115, 337–342. doi: 10.1073/pnas.1708954115
Faith, J. J., Guruge, J. L., Charbonneau, M., Subramanian, S., Seedorf, H., Goodman, A. L., et al. (2013). The long-term stability of the human gut microbiota. Science 341. doi: 10.1126/science.1237439
Forster, S. C., Kumar, N., Anonye, B. O., Almeida, A., Viciani, E., Stares, M. D., et al. (2019). A human gut bacterial genome and culture collection for improved metagenomic analyses. Nat. Biotechnol. 37, 186–192. doi: 10.1038/s41587-018-0009-7
Furuse, K., Sakurai, T., Hirashima, A., Katsuki, M., Ando, A., and Watanabe, I. (1978). Distribution of ribonucleic acid coliphages in south and east Asia. Appl. Environ. Microbiol. 35, 995–1002.
Galtier, M., De Sordi, L., Sivignon, A., de Vallée, A., Maura, D., Neut, C., et al. (2017). Bacteriophages targeting adherent invasive escherichia coli strains as a promising new treatment for Crohn's Disease. J. Crohns Colitis 11, 840–847. doi: 10.1093/ecco-jcc/jjw224
García-Aljaro, C., Ballest,é, E., Muniesa, M., and Jofre, J. (2017). Determination of crAssphage in water samples and applicability for tracking human faecal pollution. Microb. Biotechnol. 10, 1775–1780.
Garmaeva, S., Sinha, T., Kurilshikov, A., Fu, J., Wijmenga, C., and Zhernakova, A. (2019). Studying the gut virome in the metagenomic era: challenges and perspectives. BMC Biol. 17:84. doi: 10.1186/s12915-019-0704-y
Gevers, D., Kugathasan, S., Denson, L. A., Vázquez-Baeza, Y., Van Treuren, W., Ren, B., et al. (2014). The treatment-naive microbiome in new-onset Crohn's Disease. Cell Host Microbe 15, 382–392. doi: 10.1016/j.chom.2014.02.005
Gill, S. R., Pop, M., DeBoy, R. T., Eckburg, P. B., Turnbaugh, P. J., Samuel, B. S., et al. (2006). Metagenomic analysis of the human distal gut microbiome. Science 312, 1355–1359. doi: 10.1126/science.1124234
Giongo, A., Gano, K. A., Crabb, D. B., Mukherjee, N., Novelo, L. L., Casella, G., et al. (2011). Toward defining the autoimmune microbiome for type 1 diabetes. ISME J. 5, 82–91. doi: 10.1038/ismej.2010.92
Gkouskou, K. K., Deligianni, C., Tsatsanis, C., and Eliopoulos, A. G. (2014). The gut microbiota in mouse models of inflammatory bowel disease. Front. Cell. Infect. Microbiol. 4:28. doi: 10.3389/fcimb.2014.00028
Gogokhia, L., Buhrke, K., Bell, R., Hoffman, B., Brown, D. G., Hanke-Gogokhia, C., et al. (2019). Expansion of bacteriophages is linked to aggravated intestinal inflammation and colitis. Cell Host Microbe 25, 285–299.e8. doi: 10.1016/j.chom.2019.01.008
Goldfarb, T., Sberro, H., Weinstock, E., Cohen, O., Doron, S., Charpak-Amikam, Y., et al. (2015). BREX is a novel phage resistance system widespread in microbial genomes. EMBO J. 34, 169–183. doi: 10.15252/embj.201489455
Goodrich, J. K., Waters, J. L., Poole, A. C., Sutter, J. L., Koren, O., Blekhman, R., et al. (2014). Human genetics shape the gut microbiome. Cell 159, 789–799. doi: 10.1016/j.cell.2014.09.053
Gorbalenya, A. E., Krupovic, M., Mushegian, A., Kropinski, A. M., Siddell, S. G., Varsani, A., et al. (2020). The new scope of virus taxonomy: partitioning the virosphere into 15 hierarchical ranks. Nat. Microbiol. 5, 668–674. doi: 10.1038/s41564-020-0709-x
Gorvitovskaia, A., Holmes, S. P., and Huse, S. M. (2016). Interpreting Prevotella and Bacteroides as biomarkers of diet and lifestyle. Microbiome 4:15. doi: 10.1186/s40168-016-0160-7
Gregory, A. C., Zablocki, O., Zayed, A. A., Howell, A., Bolduc, B., and Sullivan, M. B. (2020). The gut virome database reveals age-dependent patterns of virome diversity in the human gut. Cell Host Microbe. 28, 1–17. doi: 10.1016/j.chom.2020.08.003
Guerin, E., Shkoporov, A., Stockdale, S. R., Clooney, A. G., Ryan, F. J., Sutton, T. D. S., et al. (2018). Biology and taxonomy of crass-like bacteriophages, the most abundant virus in the human gut. Cell Host Microbe 24, 653–664.e6. doi: 10.1016/j.chom.2018.10.002
Halfvarson, J., Brislawn, C. J., Lamendella, R., Vázquez-Baeza, Y., Walters, W. A., Bramer, L. M., et al. (2017). Dynamics of the human gut microbiome in inflammatory bowel disease. Nat. Microbiol. 2:17004. doi: 10.1038/nmicrobiol.2017.4
Hartstra, A. V., Bouter, K. E. C., Bäckhed, F., and Nieuwdorp, M. (2015). Insights into the role of the microbiome in obesity and type 2 diabetes. Diabetes Care 38, 159–165. doi: 10.2337/dc14-0769
Helmink, B. A., Khan, M. A. W., Hermann, A., Gopalakrishnan, V., and Wargo, J. A. (2019). The microbiome, cancer, and cancer therapy. Nat. Med. 25, 377–388. doi: 10.1038/s41591-019-0377-7
Hobbs, Z., and Abedon, S. T. (2016). Diversity of phage infection types and associated terminology: the problem with ‘Lytic or lysogenic.' FEMS Microbiol. Lett. 363. doi: 10.1093/femsle/fnw047
Holt, R. D. (1985). Population dynamics in two-patch environments: Some anomalous consequences of an optimal habitat distribution. Theor. Popul. Biol. 28, 181–208. doi: 10.1016/0040-5809(85)90027-9
Honap, T. P., Sankaranarayanan, K., Schnorr, S. L., Ozga, A. T., Warinner, C., and C. M. L Jr.. (2020). Biogeographic study of human gut-associated crAssphage suggests impacts from industrialization and recent expansion. PLoS ONE 15:e0226930. doi: 10.1371/journal.pone.0226930
Horvath, P., and Barrangou, R. (2010). CRISPR/Cas, the immune system of bacteria and archaea. Science 327, 167–170. doi: 10.1126/science.1179555
Howard-Varona, C., Hargreaves, K. R., Abedon, S. T., and Sullivan, M. B. (2017). Lysogeny in nature: mechanisms, impact and ecology of temperate phages. ISME J. 11, 1511–1520. doi: 10.1038/ismej.2017.16
Hoyles, L., McCartney, A. L., Neve, H., Gibson, G. R., Sanderson, J. D., Heller, K. J., et al. (2014). Characterization of virus-like particles associated with the human faecal and caecal microbiota. Res. Microbiol. 165, 803–812. doi: 10.1016/j.resmic.2014.10.006
Hryckowian, A. J., Merrill, B. D., Porter, N. T., Treuren, W. V., Nelson, E. J., Garlena, R. A., et al. (2020). Bacteroides thetaiotaomicron-infecting bacteriophage isolates inform sequence-based host range predictions. bioRxiv, doi: 10.1101/2020.03.04.977157
Hsu, B. B., Gibson, T. E., Yeliseyev, V., Liu, Q., Lyon, L., Bry, L., et al. (2019). Dynamic modulation of the gut microbiota and metabolome by bacteriophages in a mouse model. Cell Host Microbe 25, 803–814.e5. doi: 10.1016/j.chom.2019.05.001
Huttenhower, C., Gevers, D., Knight, R., Abubucker, S., Badger, J. H., Chinwalla, A. T., et al. (2012). Structure, function and diversity of the healthy human microbiome. Nature 486, 207–214. doi: 10.1038/nature11234
Hyman, P. (2019). Phages for phage therapy: isolation, characterization, and host range breadth. Pharmaceuticals 12, 35. doi: 10.3390/ph12010035
Jang, H. B., Bolduc, B., Zablocki, O., Kuhn, J. H., Roux, S., Adriaenssens, E. M., et al. (2019). Taxonomic assignment of uncultivated prokaryotic virus genomes is enabled by gene-sharing networks. Nat. Biotechnol. 37, 632–639. doi: 10.1038/s41587-019-0100-8
Jiang, X., Hall, A. B., Arthur, T. D., Plichta, D. R., Covington, C. T., Poyet, M., et al. (2019). Invertible promoters mediate bacterial phase variation, antibiotic resistance, and host adaptation in the gut. Science 363, 181–187. doi: 10.1126/science.aau5238
Joiner, K. L., Baljon, A., Barr, J., Rohwer, F., and Luque, A. (2019). Impact of bacteria motility in the encounter rates with bacteriophage in mucus. Sci. Rep. 9, 1–12. doi: 10.1038/s41598-019-52794-2
Kai, M., Watanabe, S., Furuse, K., and Ozawa, A. (1985). Bacteroides bacteriophages isolated from human feces. Microbiol. Immunol. 29, 895–899. doi: 10.1111/j.1348-0421.1985.tb00891.x
Karlsson, F. H., Ussery, D. W., Nielsen, J., and Nookaew, I. (2011). A closer look at bacteroides: phylogenetic relationship and genomic implications of a life in the human gut. Microb. Ecol. 61, 473–485. doi: 10.1007/s00248-010-9796-1
Kieser, S., Sarker, S. A., Sakwinska, O., Foata, F., Sultana, S., Khan, Z., et al. (2018). Bangladeshi children with acute diarrhoea show faecal microbiomes with increased Streptococcus abundance, irrespective of diarrhoea aetiology. Environ. Microbiol. 20, 2256–2269. doi: 10.1111/1462-2920.14274
Kim, M.-S., Park, E.-J., Roh, S. W., and Bae, J.-W. (2011). Diversity and abundance of single-stranded DNA viruses in human feces. Appl. Environ. Microbiol. 77, 8062–8070. doi: 10.1128/AEM.06331-11
Kleiner, M., Hooper, L. V., and Duerkop, B. A. (2015). Evaluation of methods to purify virus-like particles for metagenomic sequencing of intestinal viromes. BMC Genomics 16:1207. doi: 10.1186/s12864-014-1207-4
Knowles, B., Silveira, C. B., Bailey, B. A., Barott, K., Cantu, V. A., Cobián-Güemes, A. G., et al. (2016). Lytic to temperate switching of viral communities. Nature 531, 466–470. doi: 10.1038/nature17193
Korf, I. H. E., Meier-Kolthoff, J. P., Adriaenssens, E. M., Kropinski, A. M., Nimtz, M., Rohde, M., et al. (2019). Still something to discover: novel insights into escherichia coli phage diversity and taxonomy. Viruses 11, 454. doi: 10.3390/v11050454
Koskella, B., and Meaden, S. (2013). Understanding bacteriophage specificity in natural microbial communities. Viruses 5, 806–823. doi: 10.3390/v5030806
Koziolek, M., Grimm, M., Becker, D., Iordanov, V., Zou, H., Shimizu, J., et al. (2015). Investigation of pH and temperature profiles in the GI tract of fasted human subjects using the intellicap(®) system. J. Pharm. Sci. 104, 2855–2863. doi: 10.1002/jps.24274
Labonté, J. M., Swan, B. K., Poulos, B., Luo, H., Koren, S., Hallam, S. J., et al. (2015). Single-cell genomics-based analysis of virus–host interactions in marine surface bacterioplankton. ISME J. 9, 2386–2399. doi: 10.1038/ismej.2015.48
Labrie, S. J., Samson, J. E., and Moineau, S. (2010). Bacteriophage resistance mechanisms. Nat. Rev. Microbiol. 8, 317–327. doi: 10.1038/nrmicro2315
Laganenka, L., Sander, T., Lagonenko, A., Chen, Y., Link, H., and Sourjik, V. (2019). Quorum sensing and metabolic state of the host control lysogeny-lysis switch of bacteriophage T1. mBio 10:19. doi: 10.1128/mBio.01884-19
Lagier, J.-C., Armougom, F., Million, M., Hugon, P., Pagnier, I., Robert, C., et al. (2012). Microbial culturomics: paradigm shift in the human gut microbiome study. Clin. Microbiol. Infect. 18, 1185–1193. doi: 10.1111/1469-0691.12023
Lagier, J.-C., Dubourg, G., Million, M., Cadoret, F., Bilen, M., Fenollar, F., et al. (2018). Culturing the human microbiota and culturomics. Nat. Rev. Microbiol. 16, 540–550. doi: 10.1038/s41579-018-0041-0
Lagier, J.-C., Khelaifia, S., Alou, M. T., Ndongo, S., Dione, N., Hugon, P., et al. (2016). Culture of previously uncultured members of the human gut microbiota by culturomics. Nat. Microbiol. 1, 1–8. doi: 10.1038/nmicrobiol.2016.203
Lau, R. K., Ye, Q., Birkholz, E. A., Berg, K. R., Patel, L., Mathews, I. T., et al. (2020). Structure and mechanism of a cyclic trinucleotide-activated bacterial endonuclease mediating bacteriophage immunity. Mol. Cell 77, 723–733.e6. doi: 10.1016/j.molcel.2019.12.010
Lawrence, D., Baldridge, M. T., and Handley, S. A. (2019). Phages and human health: more than idle hitchhikers. Viruses 11, 587. doi: 10.3390/v11070587
Lefkowitz, E. J., Dempsey, D. M., Hendrickson, R. C., Orton, R. J., Siddell, S. G., and Smith, D. B. (2018). Virus taxonomy: the database of the International Committee on Taxonomy of Viruses (ICTV). Nucleic Acids Res. 46, D708–D717. doi: 10.1093/nar/gkx932
Leiman, P. G., Battisti, A. J., Bowman, V. D., Stummeyer, K., Mühlenhoff, M., Gerardy-Schahn, R., et al. (2007). The structures of bacteriophages K1E and K1-5 explain processive degradation of polysaccharide capsules and evolution of new host specificities. J. Mol. Biol. 371, 836–849. doi: 10.1016/j.jmb.2007.05.083
Lewis, R., and Hill, C. (2020). Overcoming barriers to phage application in food and feed. Curr. Opin. Biotechnol. 61, 38–44. doi: 10.1016/j.copbio.2019.09.018
Liang, G., Zhao, C., Zhang, H., Mattei, L., Sherrill-Mix, S., Bittinger, K., et al. (2020). The stepwise assembly of the neonatal virome is modulated by breastfeeding. Nature 581, 470–474. doi: 10.1038/s41586-020-2192-1
Liang, Y. Y., Zhang, W., Tong, Y. G., and Chen, S. P. (2016). CrAssphage is not associated with diarrhoea and has high genetic diversity. Epidemiol. Infect. 144, 3549–3553.
Lim, E. S., Wang, D., and Holtz, L. R. (2016). The bacterial microbiome and virome milestones of infant development. Trends Microbiol. 24, 801–810. doi: 10.1016/j.tim.2016.06.001
Lim, E. S., Zhou, Y., Zhao, G., Bauer, I. K., Droit, L., Ndao, I. M., et al. (2015). Early life dynamics of the human gut virome and bacterial microbiome in infants. Nat. Med. 21, 1228–1234. doi: 10.1038/nm.3950
Lima-Mendez, G., Toussaint, A., and Leplae, R. (2011). A modular view of the bacteriophage genomic space: identification of host and lifestyle marker modules. Res. Microbiol. 162, 737–746. doi: 10.1016/j.resmic.2011.06.006
Lin, D. M., Koskella, B., and Lin, H. C. (2017). Phage therapy: an alternative to antibiotics in the age of multi-drug resistance. World J. Gastrointest. Pharmacol. Ther. 8, 162–173. doi: 10.4292/wjgpt.v8.i3.162
Lourenço, M., Chaffringeon, L., Lamy-Besnier, Q., Pédron, T., Campagne, P., Eberl, C., et al. (2020). The spatial heterogeneity of the gut limits predation and fosters coexistence of bacteria and bacteriophages. Cell Host Microbe. 28, 1–12. doi: 10.1016/j.chom.2020.06.002
Lourenço, M., De Sordi, L., and Debarbieux, L. (2018). The diversity of bacterial lifestyles hampers bacteriophage tenacity. Viruses 10:327. doi: 10.3390/v10060327
Love, M. J., Bhandari, D., Dobson, R. C. J., and Billington, C. (2018). Potential for bacteriophage endolysins to supplement or replace antibiotics in food production and clinical care. Antibiotics 7:17. doi: 10.3390/antibiotics7010017
Lozupone, C. A., Stombaugh, J. I., Gordon, J. I., Jansson, J. K., and Knight, R. (2012). Diversity, stability and resilience of the human gut microbiota. Nature 489, 220–230. doi: 10.1038/nature11550
Ma, Y., You, X., Mai, G., Tokuyasu, T., and Liu, C. (2018). A human gut phage catalog correlates the gut phageome with type 2 diabetes. Microbiome 6:24. doi: 10.1186/s40168-018-0410-y
Manning, A. J., and Kuehn, M. J. (2011). Contribution of bacterial outer membrane vesicles to innate bacterial defense. BMC Microbiol. 11:258. doi: 10.1186/1471-2180-11-258
Manrique, P., Bolduc, B., Walk, S. T., van der Oost, J., de Vos, W. M., and Young, M. J. (2016). Healthy human gut phageome. Proc. Natl. Acad. Sci. U.S.A. 113, 10400–10405.
Maqsood, R., Rodgers, R., Rodriguez, C., Handley, S. A., Ndao, I. M., Tarr, P. I., et al. (2019). Discordant transmission of bacteria and viruses from mothers to babies at birth. Microbiome 7, 156. doi: 10.1186/s40168-019-0766-7
Marine, R., McCarren, C., Vorrasane, V., Nasko, D., Crowgey, E., Polson, S. W., et al. (2014). Caught in the middle with multiple displacement amplification: the myth of pooling for avoiding multiple displacement amplification bias in a metagenome. Microbiome 2, 3. doi: 10.1186/2049-2618-2-3
Martínez, J. M., Swan, B. K., and Wilson, W. H. (2014). Marine viruses, a genetic reservoir revealed by targeted viromics. ISME J. 8, 1079–1088. doi: 10.1038/ismej.2013.214
Martinez-Hernandez, F., Fornas, O., Gomez, M. L., Bolduc, B., Peña, M. J., de la, C., et al. (2017). Single-virus genomics reveals hidden cosmopolitan and abundant viruses. Nat. Commun. 8, 1–13. doi: 10.1038/ncomms15892
Maslov, S., and Sneppen, K. (2017). Population cycles and species diversity in dynamic Kill-the-Winner model of microbial ecosystems. Sci. Rep. 7, 39642. doi: 10.1038/srep39642
Maura, D., Galtier, M., Bouguénec, C. L., and Debarbieux, L. (2012a). Virulent bacteriophages can target O104:H4 enteroaggregative Escherichia coli in the mouse intestine. Antimicrob. Agents Chemother. 56, 6235–6242. doi: 10.1128/AAC.00602-12
Maura, D., Morello, E., du Merle, L., Bomme, P., Le Bouguénec, C., and Debarbieux, L. (2012b). Intestinal colonization by enteroaggregative Escherichia coli supports long-term bacteriophage replication in mice. Environ. Microbiol. 14, 1844–1854. doi: 10.1111/j.1462-2920.2011.02644.x
McCann, A., Ryan, F. J., Stockdale, S. R., Dalmasso, M., Blake, T., Ryan, C. A., et al. (2018). Viromes of one year old infants reveal the impact of birth mode on microbiome diversity. PeerJ 6:e4694. doi: 10.7717/peerj.4694
McInnes, R. S., McCallum, G. E., Lamberte, L. E., and van Schaik, W. (2020). Horizontal transfer of antibiotic resistance genes in the human gut microbiome. Curr. Opin. Microbiol. 53, 35–43. doi: 10.1016/j.mib.2020.02.002
Meier-Kolthoff, J. P., and Göker, M. (2017). VICTOR: genome-based phylogeny and classification of prokaryotic viruses. Bioinformatics 33, 3396–3404. doi: 10.1093/bioinformatics/btx440
Meyer, J. R., Dobias, D. T., Weitz, J. S., Barrick, J. E., Quick, R. T., and Lenski, R. E. (2012). Repeatability and contingency in the evolution of a key innovation in phage lambda. Science 335, 428–432. doi: 10.1126/science.1214449
Minot, S., Bryson, A., Chehoud, C., Wu, G. D., Lewis, J. D., and Bushman, F. D. (2013). Rapid evolution of the human gut virome. Proc. Natl. Acad. Sci. U.S.A. 110, 12450–12455. doi: 10.1073/pnas.1300833110
Minot, S., Grunberg, S., Wu, G. D., Lewis, J. D., and Bushman, F. D. (2012). Hypervariable loci in the human gut virome. Proc. Natl. Acad. Sci. U.S.A. 109, 3962–3966. doi: 10.1073/pnas.1119061109
Minot, S., Sinha, R., Chen, J., Li, H., Keilbaugh, S. A., Wu, G. D., et al. (2011). The human gut virome: Inter-individual variation and dynamic response to diet. Genome Res. 21, 1616–1625. doi: 10.1101/gr.122705.111
Mirzaei, M. K., and Maurice, C. F. (2017). Ménage à trois in the human gut: interactions between host, bacteria and phages. Nat. Rev. Microbiol. 15, 397–408. doi: 10.1038/nrmicro.2017.30
Monaco, C. L., Gootenberg, D. B., Zhao, G., Handley, S. A., Ghebremichael, M. S., Lim, E. S., et al. (2016). Altered virome and bacterial microbiome in human immunodeficiency virus-associated acquired immunodeficiency syndrome. Cell Host Microbe 19, 311–322. doi: 10.1016/j.chom.2016.02.011
Moreno-Gallego, J. L., Chou, S.-P., Rienzi, S. C. D., Goodrich, J. K., Spector, T. D., Bell, J. T., et al. (2019). Virome diversity correlates with intestinal microbiome diversity in adult monozygotic twins. Cell Host Microbe 25, 261–272.e5. doi: 10.1016/j.chom.2019.01.019
Moustafa, A., Xie, C., Kirkness, E., Biggs, W., Wong, E., Turpaz, Y., et al. (2017). The blood DNA virome in 8,000 humans. PLOS Pathog. 13:e1006292. doi: 10.1371/journal.ppat.1006292
Moxon, E. R., Rainey, P. B., Nowak, M. A., and Lenski, R. E. (1994). Adaptive evolution of highly mutable loci in pathogenic bacteria. Curr. Biol. 4, 24–33.
Nakayama-Imaohji, H., Hirakawa, H., Ichimura, M., Wakimoto, S., Kuhara, S., Hayashi, T., et al. (2009). Identification of the site-specific DNA invertase responsible for the phase variation of SusC/SusD family outer membrane proteins in bacteroides fragilis. J. Bacteriol. 191, 6003–6011. doi: 10.1128/JB.00687-09
Nakayama-Imaohji, H., Hirota, K., Yamasaki, H., Yoneda, S., Nariya, H., Suzuki, M., et al. (2016). DNA inversion regulates outer membrane vesicle production in bacteroides fragilis. PLoS ONE 11:e0148887. doi: 10.1371/journal.pone.0148887
Ng, W.-L., and Bassler, B. L. (2009). Bacterial quorum-sensing network architectures. Annu. Rev. Genet. 43, 197–222. doi: 10.1146/annurev-genet-102108-134304
Nguyen, S., Baker, K., Padman, B. S., Patwa, R., Dunstan, R. A., Weston, T. A., et al. (2017). Bacteriophage transcytosis provides a mechanism to cross epithelial cell layers. mBio 8, e01874–e01817. doi: 10.1128/mBio.01874-17
Norman, J. M., Handley, S. A., Baldridge, M. T., Droit, L., Liu, C. Y., Keller, B. C., et al. (2015). Disease-specific alterations in the enteric virome in inflammatory bowel disease. Cell 160, 447–460. doi: 10.1016/j.cell.2015.01.002
Obeng, N., Pratama, A. A., and van Elsas, J. D. (2016). The significance of mutualistic phages for bacterial ecology and evolution. Trends Microbiol. 24, 440–449. doi: 10.1016/j.tim.2015.12.009
Oechslin, F. (2018). Resistance development to bacteriophages occurring during bacteriophage therapy. Viruses 10, 351. doi: 10.3390/v10070351
Ofir, G., Melamed, S., Sberro, H., Mukamel, Z., Silverman, S., Yaakov, G., et al. (2018). DISARM is a widespread bacterial defence system with broad anti-phage activities. Nat. Microbiol. 3, 90–98. doi: 10.1038/s41564-017-0051-0
O'Hara, A. M., and Shanahan, F. (2006). The gut flora as a forgotten organ. EMBO Rep. 7, 688–693. doi: 10.1038/sj.embor.7400731
O'Sullivan, L., Bolton, D., McAuliffe, O., and Coffey, A. (2019). Bacteriophages in food applications: from foe to friend. Annu. Rev. Food Sci. Technol. 10, 151–172. doi: 10.1146/annurev-food-032818-121747
Ott, S. J., Waetzig, G. H., Rehman, A., Moltzau-Anderson, J., Bharti, R., Grasis, J. A., et al. (2017). Efficacy of sterile fecal filtrate transfer for treating patients with clostridium difficile infection. Gastroenterology 152, 799–811.e7. doi: 10.1053/j.gastro.2016.11.010
Oude Munnink, B. B., Canuti, M., Deijs, M., de Vries, M., Jebbink, M. F., Rebers, S., et al. (2014). Unexplained diarrhoea in HIV-1 infected individuals. BMC Infect. Dis. 14:22. doi: 10.1186/1471-2334-14-22
Papenfort, K., and Bassler, B. L. (2016). Quorum sensing signal–response systems in Gram-negative bacteria. Nat. Rev. Microbiol. 14, 576–588. doi: 10.1038/nrmicro.2016.89
Pereira, F. C., and Berry, D. (2017). Microbial nutrient niches in the gut. Environ. Microbiol. 19, 1366–1378. doi: 10.1111/1462-2920.13659
Pitsalidis, C., Pappa, A.-M., Porel, M., Artim, C. M., Faria, G. C., Duong, D. D., et al. (2018). Biomimetic electronic devices for measuring bacterial membrane disruption. Adv. Mater. 30:1803130. doi: 10.1002/adma.201803130
Porter, N. T., Hryckowian, A. J., Merrill, B. D., Fuentes, J. J., Gardner, J. O., Glowacki, R. W. P., et al. (2020). Phase-variable capsular polysaccharides and lipoproteins modify bacteriophage susceptibility in Bacteroides thetaiotaomicron. Nat. Microbiol. 5, 1170–1181. doi: 10.1038/s41564-020-0746-5
Rabiey, M., Roy, S. R., Holtappels, D., Franceschetti, L., Quilty, B. J., Creeth, R., et al. (2020). Phage biocontrol to combat Pseudomonas syringae pathogens causing disease in cherry. Microb. Biotechnol. 13, 1428–1445. doi: 10.1111/1751-7915.13585
Rajilić-Stojanović, M., and de Vos, W. M. (2014). The first 1000 cultured species of the human gastrointestinal microbiota. FEMS Microbiol. Rev. 38, 996–1047. doi: 10.1111/1574-6976.12075
Ram, G., Chen, J., Kumar, K., Ross, H. F., Ubeda, C., Damle, P. K., et al. (2012). Staphylococcal pathogenicity island interference with helper phage reproduction is a paradigm of molecular parasitism. Proc. Natl. Acad. Sci. U.S.A. 109, 16300–16305. doi: 10.1073/pnas.1204615109
Reyes, A., Blanton, L. V., Cao, S., Zhao, G., Manary, M., Trehan, I., et al. (2015). Gut DNA viromes of Malawian twins discordant for severe acute malnutrition. Proc. Natl. Acad. Sci. U.S.A. 112, 11941–11946. doi: 10.1073/pnas.1514285112
Reyes, A., Haynes, M., Hanson, N., Angly, F. E., Heath, A. C., Rohwer, F., et al. (2010). Viruses in the faecal microbiota of monozygotic twins and their mothers. Nature 466, 334–338. doi: 10.1038/nature09199
Reyes, A., Wu, M., McNulty, N. P., Rohwer, F. L., and Gordon, J. I. (2013). Gnotobiotic mouse model of phage-bacterial host dynamics in the human gut. Proc. Natl. Acad. Sci. U.S.A. 110, 20236–20241. doi: 10.1073/pnas.1319470110
Riley, T. V., Brazier, J. S., Hassan, H., Williams, K., and Phillips, K. D. (1987). Comparison of alcohol shock enrichment and selective enrichment for the isolation of Clostridium difficile. Epidemiol. Infect. 99, 355–359. doi: 10.1017/S0950268800067832
Roach, D. R., Leung, C. Y., Henry, M., Morello, E., Singh, D., Santo, J. P. D., et al. (2017). Synergy between the host immune system and bacteriophage is essential for successful phage therapy against an acute respiratory pathogen. Cell Host Microbe 22, 38-47.e4. doi: 10.1016/j.chom.2017.06.018
Roberts, R. J., Belfort, M., Bestor, T., Bhagwat, A. S., Bickle, T. A., Bitinaite, J., et al. (2003). A nomenclature for restriction enzymes, DNA methyltransferases, homing endonucleases and their genes. Nucleic Acids Res. 31, 1805–1812. doi: 10.1093/nar/gkg274
Rodríguez, J. M., Murphy, K., Stanton, C., Ross, R. P., Kober, O. I., Juge, N., et al. (2015). The composition of the gut microbiota throughout life, with an emphasis on early life. Microb. Ecol. Health Dis. 26, 26050. doi: 10.3402/mehd.v26.26050
Rostøl, J. T., and Marraffini, L. (2019). (Ph)ighting phages: how bacteria resist their parasites. Cell Host Microbe 25, 184–194. doi: 10.1016/j.chom.2019.01.009
Roux, S., Emerson, J. B., Eloe-Fadrosh, E. A., and Sullivan, M. B. (2017). Benchmarking viromics: an in silico evaluation of metagenome-enabled estimates of viral community composition and diversity. PeerJ. 5:e3817. doi: 10.7717/peerj.3817
Roux, S., Krupovic, M., Debroas, D., Forterre, P., and Enault, F. (2013). Assessment of viral community functional potential from viral metagenomes may be hampered by contamination with cellular sequences. Open Biol. 3:130160. doi: 10.1098/rsob.130160
Roux, S., Solonenko, N. E., Dang, V. T., Poulos, B. T., Schwenck, S. M., Goldsmith, D. B., et al. (2016). Towards quantitative viromics for both double-stranded and single-stranded DNA viruses. PeerJ. 4:e2777. doi: 10.7717/peerj.2777
Salem, M., Virtanen, S., Korkeala, H., and Skurnik, M. (2015). Isolation and characterization of Yersinia-specific bacteriophages from pig stools in Finland. J. Appl. Microbiol. 118, 599–608. doi: 10.1111/jam.12722
Samson, J. E., Magadán, A. H., Sabri, M., and Moineau, S. (2013). Revenge of the phages: defeating bacterial defences. Nat. Rev. Microbiol. 11, 675–687. doi: 10.1038/nrmicro3096
Santiago-Rodriguez, T. M., Ly, M., Daigneault, M. C., Brown, I. H. L., McDonald, J. A. K., Bonilla, N., et al. (2015a). Chemostat culture systems support diverse bacteriophage communities from human feces. Microbiome 3, 58. doi: 10.1186/s40168-015-0124-3
Santiago-Rodriguez, T. M., Naidu, M., Abeles, S. R., Boehm, T. K., Ly, M., and Pride, D. T. (2015b). Transcriptome analysis of bacteriophage communities in periodontal health and disease. BMC Genomics 16:549. doi: 10.1186/s12864-015-1781-0
Scanlan, P. D. (2017). Bacteria–bacteriophage coevolution in the human gut: implications for microbial diversity and functionality. Trends Microbiol. 25, 614–623. doi: 10.1016/j.tim.2017.02.012
Schrag, S. J., and Mittler, J. E. (1996). Host-parasite coexistence: the role of spatial refuges in stabilizing bacteria-phage interactions. Am. Nat. 148, 348–377.
Schwechheimer, C., and Kuehn, M. J. (2015). Outer-membrane vesicles from Gram-negative bacteria: biogenesis and functions. Nat. Rev. Microbiol. 13, 605–619. doi: 10.1038/nrmicro3525
Seed, K. D. (2015). Battling phages: how bacteria defend against viral attack. PLOS Pathog. 11:e1004847. doi: 10.1371/journal.ppat.1004847
Seed, K. D., Lazinski, D. W., Calderwood, S. B., and Camilli, A. (2013). A bacteriophage encodes its own CRISPR/Cas adaptive response to evade host innate immunity. Nature 494, 489–491. doi: 10.1038/nature11927
Sender, R., Fuchs, S., and Milo, R. (2016). Revised estimates for the number of human and bacteria cells in the body. PLOS Biol. 14:e1002533. doi: 10.1371/journal.pbio.1002533
Shanahan, F., and Hill, C. (2019). Language, numeracy and logic in microbiome science. Nat. Rev. Gastroenterol. Hepatol. 16, 387–388. doi: 10.1038/s41575-019-0163-5
Shapiro, J. W., and Putonti, C. (2018). Gene co-occurrence networks reflect bacteriophage ecology and evolution. mBio 9:e01870-17. doi: 10.1128/mBio.01870-17
Sharon, I., Morowitz, M. J., Thomas, B. C., Costello, E. K., Relman, D. A., and Banfield, J. F. (2013). Time series community genomics analysis reveals rapid shifts in bacterial species, strains, and phage during infant gut colonization. Genome Res. 23, 111–120. doi: 10.1101/gr.142315.112
Shkoporov, A. N., Clooney, A. G., Sutton, T. D. S., Ryan, F. J., Daly, K. M., Nolan, J. A., et al. (2019). The human gut virome is highly diverse, stable, and individual specific. Cell Host Microbe 26, 527–541.e5. doi: 10.1016/j.chom.2019.09.009
Shkoporov, A. N., and Hill, C. (2019). Bacteriophages of the human gut: the “known unknown” of the microbiome. Cell Host Microbe 25, 195–209. doi: 10.1016/j.chom.2019.01.017
Shkoporov, A. N., Khokhlova, E. V., Fitzgerald, C. B., Stockdale, S. R., Draper, L. A., Ross, R. P., et al. (2018a). ΦCrAss001 represents the most abundant bacteriophage family in the human gut and infects Bacteroides intestinalis. Nat. Commun. 9:225. doi: 10.1038/s41467-018-07225-7
Shkoporov, A. N., Ryan, F. J., Draper, L. A., Forde, A., Stockdale, S. R., Daly, K. M., et al. (2018b). Reproducible protocols for metagenomic analysis of human faecal phageomes. Microbiome 6:68. doi: 10.1186/s40168-018-0446-z
Silpe, J. E., and Bassler, B. L. (2019). A host-produced quorum-sensing autoinducer controls a phage lysis-lysogeny decision. Cell 176, 268–280.e13. doi: 10.1016/j.cell.2018.10.059
Silveira, C. B., and Rohwer, F. L. (2016). Piggyback-the-Winner in host-associated microbial communities. Npj Biofilms Microbiomes 2:16010. doi: 10.1038/npjbiofilms.2016.10
Simmonds, P., Adams, M. J., Benk,o, M., Breitbart, M., Brister, J. R., Carstens, E. B., et al. (2017). Consensus statement: virus taxonomy in the age of metagenomics. Nat. Rev. Microbiol. 15, 161–168. doi: 10.1038/nrmicro.2016.177
Siranosian, B. A., Tamburini, F. B., Sherlock, G., and Bhatt, A. S. (2020). Acquisition, transmission and strain diversity of human gut-colonizing crAss-like phages. Nat. Commun. 11, 1–11. doi: 10.1038/s41467-019-14103-3
Somerville, V., Lutz, S., Schmid, M., Frei, D., Moser, A., Irmler, S., et al. (2019). Long-read based de novo assembly of low-complexity metagenome samples results in finished genomes and reveals insights into strain diversity and an active phage system. BMC Microbiol. 19:143. doi: 10.1186/s12866-019-1500-0
Stämmler, F., Gläsner, J., Hiergeist, A., Holler, E., Weber, D., Oefner, P. J., et al. (2016). Adjusting microbiome profiles for differences in microbial load by spike-in bacteria. Microbiome 4, 28. doi: 10.1186/s40168-016-0175-0
Stockdale, S. R., Ryan, F. J., McCann, A., Dalmasso, M., Ross, P. R., and Hill, C. (2018). Viral dark matter in the gut virome of elderly humans. Preprint. doi: 10.20944/preprints201807.0128.v1
Strakosas, X., Bongo, M., and Owens, R. M. (2015). The organic electrochemical transistor for biological applications. J. Appl. Polym. Sci. 132. doi: 10.1002/app.41735
Su, H., Liu, H.-Y., Pappa, A.-M., Hidalgo, T. C., Cavassin, P., Inal, S., et al. (2019). Facile generation of biomimetic-supported lipid bilayers on conducting polymer surfaces for membrane biosensing. ACS Appl. Mater. Interfaces. 11, 43799–43810. doi: 10.1021/acsami.9b10303
Sugden, R., Kelly, R., and Davies, S. (2016). Combatting antimicrobial resistance globally. Nat. Microbiol. 1, 1–2. doi: 10.1038/nmicrobiol.2016.187
Sutton, T. D. S., Clooney, A. G., Ryan, F. J., Ross, R. P., and Hill, C. (2019). Choice of assembly software has a critical impact on virome characterisation. Microbiome 7, 12. doi: 10.1186/s40168-019-0626-5
Sutton, T. D. S., and Hill, C. (2019). Gut bacteriophage: current understanding and challenges. Front. Endocrinol. 10:784. doi: 10.3389/fendo.2019.00784
Swan, B. K., Tupper, B., Sczyrba, A., Lauro, F. M., Martinez-Garcia, M., González, J. M., et al. (2013). Prevalent genome streamlining and latitudinal divergence of planktonic bacteria in the surface ocean. Proc. Natl. Acad. Sci. U.S.A. 110, 11463–11468. doi: 10.1073/pnas.1304246110
Tetz, G., Brown, S. M., Hao, Y., and Tetz, V. (2018). Parkinson's disease and bacteriophages as its overlooked contributors. Sci. Rep. 8, 1–11. doi: 10.1038/s41598-018-29173-4
Tetz, G., Brown, S. M., Hao, Y., and Tetz, V. (2019). Type 1 diabetes: an association between autoimmunity, the dynamics of gut amyloid-producing E. coli and their phages. Sci. Rep. 9:9685. doi: 10.1038/s41598-019-46087-x
Tetz, G., and Tetz, V. (2018). Bacteriophages as new human viral pathogens. Microorganisms 6:54. doi: 10.3390/microorganisms6020054
Tetz, G. V., Ruggles, K. V., Zhou, H., Heguy, A., Tsirigos, A., and Tetz, V. (2017). Bacteriophages as potential new mammalian pathogens. Sci. Rep. 7, 7043. doi: 10.1038/s41598-017-07278-6
Tetz, V., and Tetz, G. (2019). Bacterial DNA induces the formation of heat-resistant disease-associated proteins in human plasma. Sci. Rep. 9, 1–10. doi: 10.1038/s41598-019-54618-9
Thingstad, T. F. (2000). Elements of a theory for the mechanisms controlling abundance, diversity, and biogeochemical role of lytic bacterial viruses in aquatic systems. Limnol. Oceanogr. 45, 1320–1328. doi: 10.4319/lo.2000.45.6.1320
Tock, M. R., and Dryden, D. T. F. (2005). The biology of restriction and anti-restriction. Curr. Opin. Microbiol. 8, 466–472. doi: 10.1016/j.mib.2005.06.003
Touchon, M., Moura de Sousa, J. A., and Rocha, E. P. (2017). Embracing the enemy: the diversification of microbial gene repertoires by phage-mediated horizontal gene transfer. Curr. Opin. Microbiol. 38, 66–73. doi: 10.1016/j.mib.2017.04.010
Tria, S. A., Ramuz, M., Huerta, M., Leleux, P., Rivnay, J., Jimison, L. H., et al. (2014). Dynamic monitoring of salmonella typhimurium infection of polarized epithelia using organic transistors. Adv. Healthc. Mater. 3, 1053–1060. doi: 10.1002/adhm.201300632
Turkington, C. J. R., Morozov, A., Clokie, M. R. J., and Bayliss, C. D. (2019). Phage-resistant phase-variant sub-populations mediate herd immunity against bacteriophage invasion of bacterial meta-populations. Front. Microbiol. 10:473. doi: 10.3389/fmicb.2019.01473
Turnbaugh, P. J., Ley, R. E., Hamady, M., Fraser-Liggett, C. M., Knight, R., and Gordon, J. I. (2007). The human microbiome project. Nature 449, 804–810. doi: 10.1038/nature06244
Turovskiy, Y., Kashtanov, D., Paskhover, B., and Chikindas, M. L. (2007). Quorum sensing: fact, fiction, and everything in between. Adv. Appl. Microbiol. 62, 191–234. doi: 10.1016/S0065-2164(07)62007-3
Tzipilevich, E., Habusha, M., and Ben-Yehuda, S. (2017). Acquisition of phage sensitivity by bacteria through exchange of phage receptors. Cell 168, 186–199.e12. doi: 10.1016/j.cell.2016.12.003
Ursell, L. K., Metcalf, J. L., Parfrey, L. W., and Knight, R. (2012). Defining the human microbiome. Nutr. Rev. 70, S38–S44. doi: 10.1111/j.1753-4887.2012.00493.x
Van Belleghem, J. D., Dabrowska, K., Vaneechoutte, M., Barr, J. J., and Bollyky, P. L. (2019). Interactions between bacteriophage, bacteria, and the mammalian immune system. Viruses 11:10. doi: 10.3390/v11010010
Vandeputte, D., Falony, G., Vieira-Silva, S., Tito, R. Y., Joossens, M., and Raes, J. (2016). Stool consistency is strongly associated with gut microbiota richness and composition, enterotypes and bacterial growth rates. Gut 65, 57–62. doi: 10.1136/gutjnl-2015-309618
Vandeputte, D., Kathagen, G., D'hoe, K., Vieira-Silva, S., Valles-Colomer, M., Sabino, J., et al. (2017). Quantitative microbiome profiling links gut community variation to microbial load. Nature 551, 507–511. doi: 10.1038/nature24460
Vogt, N. M., Kerby, R. L., Dill-McFarland, K. A., Harding, S. J., Merluzzi, A. P., Johnson, S. C., et al. (2017). Gut microbiome alterations in Alzheimer's disease. Sci. Rep. 7, 13537. doi: 10.1038/s41598-017-13601-y
Warwick-Dugdale, J., Solonenko, N., Moore, K., Chittick, L., Gregory, A. C., Allen, M. J., et al. (2019). Long-read viral metagenomics captures abundant and microdiverse viral populations and their niche-defining genomic islands. PeerJ 7, e6800. doi: 10.7717/peerj.6800
Weiss, M., Denou, E., Bruttin, A., Serra-Moreno, R., Dillmann, M.-L., and Brüssow, H. (2009). In vivo replication of T4 and T7 bacteriophages in germ-free mice colonized with Escherichia coli. Virology 393, 16–23. doi: 10.1016/j.virol.2009.07.020
Whitehead, N. A., Barnard, A. M. L., Slater, H., Simpson, N. J. L., and Salmond, G. P. C. (2001). Quorum-sensing in Gram-negative bacteria. FEMS Microbiol. Rev. 25, 365–404. doi: 10.1111/j.1574-6976.2001.tb00583.x
Wiedenheft, B. (2013). In defense of phage: viral suppressors of CRISPR-mediated adaptive immunity in bacteria. RNA Biol. 10, 886–890. doi: 10.4161/rna.23591
Wiedenheft, B., Sternberg, S. H., and Doudna, J. A. (2012). RNA-guided genetic silencing systems in bacteria and archaea. Nature 482, 331–338. doi: 10.1038/nature10886
Wommack, K. E., and Colwell, R. R. (2000). Virioplankton: viruses in aquatic ecosystems. Microbiol. Mol. Biol. Rev. 64, 69–114. doi: 10.1128/mmbr.64.1.69-114.2000
Yutin, N., Makarova, K. S., Gussow, A. B., Krupovic, M., Segall, A., Edwards, R. A., et al. (2018). Discovery of an expansive bacteriophage family that includes the most abundant viruses from the human gut. Nat. Microbiol. 3, 38–46. doi: 10.1038/s41564-017-0053-y
Zaleski, P., Wojciechowski, M., and Piekarowicz, A. (2005). The role of Dam methylation in phase variation of Haemophilus influenzae genes involved in defence against phage infection. Microbiol. Read. Engl. 151, 3361–3369. doi: 10.1099/mic.0.28184-0
Zhao, G., Droit, L., Gilbert, M. H., Schiro, F. R., Didier, P. J., Si, X., et al. (2019). Virome biogeography in the lower gastrointestinal tract of rhesus macaques with chronic diarrhea. Virology 527, 77–88. doi: 10.1016/j.virol.2018.10.001
Zhao, G., Vatanen, T., Droit, L., Park, A., Kostic, A. D., Poon, T. W., et al. (2017). Intestinal virome changes precede autoimmunity in type I diabetes-susceptible children. Proc. Natl. Acad. Sci. U.S.A. 114, E6166–E6175. doi: 10.1073/pnas.1706359114
Zheng, D.-W., Dong, X., Pan, P., Chen, K.-W., Fan, J.-X., Cheng, S.-X., et al. (2019). Phage-guided modulation of the gut microbiota of mouse models of colorectal cancer augments their responses to chemotherapy. Nat. Biomed. Eng. 3, 717–728. doi: 10.1038/s41551-019-0423-2
Zitomersky, N. L., Coyne, M. J., and Comstock, L. E. (2011). Longitudinal analysis of the prevalence, maintenance, and IgA response to species of the order Bacteroidales in the human gut. Infect. Immun. 79, 2012–2020. doi: 10.1128/IAI.01348-10
Zolfo, M., Pinto, F., Asnicar, F., Manghi, P., Tett, A., Bushman, F. D., et al. (2019). Detecting contamination in viromes using ViromeQC. Nat. Biotechnol., 1–5. doi: 10.1038/s41587-019-0334-5
Keywords: bacteriophage, phage-bacteria interactions, gut microbiota, microbiome, virome
Citation: Guerin E and Hill C (2020) Shining Light on Human Gut Bacteriophages. Front. Cell. Infect. Microbiol. 10:481. doi: 10.3389/fcimb.2020.00481
Received: 17 June 2020; Accepted: 04 August 2020;
Published: 10 September 2020.
Edited by:
Eleanor Jameson, University of Warwick, United KingdomReviewed by:
Sabrina Ingrid Green, Baylor College of Medicine, United StatesCopyright © 2020 Guerin and Hill. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Colin Hill, Yy5oaWxsQHVjYy5pZQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.