 Qing-Ming Qin1,2†
Qing-Ming Qin1,2† Jianwu Pei3†
Jianwu Pei3† Gabriel Gomez3
Gabriel Gomez3 Allison Rice-Ficht4
Allison Rice-Ficht4 Thomas A. Ficht3*
Thomas A. Ficht3* Paul de Figueiredo2,3,5*
Paul de Figueiredo2,3,5*- 1College of Plant Sciences, Key Laboratory of Zoonosis Research, Ministry of Education, Jilin University, Changchun, China
- 2Department of Microbial Pathogenesis and Immunology, Texas A&M Health Science Center, Bryan TX, United States
- 3Department of Veterinary Pathobiology, Texas A&M University, College Station, TX, United States
- 4Department of Molecular and Cellular Medicine, Texas A&M Health Science Center, Bryan, TX, United States
- 5Norman Borlaug Center, Texas A&M University, College Station, TX, United States
Acinetobacter baumannii is an important causative agent of nosocomial infections worldwide. The pathogen also readily acquires resistance to antibiotics, and pan-resistant strains have been reported. A. baumannii is widely regarded as an extracellular bacterial pathogen. However, accumulating evidence demonstrates that the pathogen can invade, survive or persist in infected mammalian cells. Unfortunately, the molecular mechanisms controlling these processes remain poorly understood. Here, we show that Drosophila S2 cells provide several attractive advantages as a model system for investigating the intracellular lifestyle of the pathogen, including susceptibility to bacterial intracellular replication and limited infection-induced host cell death. We also show that the Drosophila system can be used to rapidly identify host factors, including MAP kinase proteins, which confer susceptibility to intracellular parasitism. Finally, analysis of the Drosophila system suggested that host proteins that regulate organelle biogenesis and membrane trafficking contribute to regulating the intracellular lifestyle of the pathogen. Taken together, these findings establish a novel model system for elucidating interactions between A. baumannii and host cells, define new factors that regulate bacterial invasion or intracellular persistence, and identify subcellular compartments in host cells that interact with the pathogen.
Introduction
Acinetobacter baumannii is a clinically important pathogen that can survive on hospital equipment and can cause serious nosocomial infections. The organism also causes serious wound infections in injured combat soldiers and in victims of traumatic injury. In addition, the bacterium can readily acquire multidrug, extensive-drug and even pan-drug resistance phenotypes. These attributes render A. baumannii a potential global threat to health-care settings (Mortensen and Skaar, 2013; Harding et al., 2018). A. baumannii is widely regarded as an extracellular bacterial pathogen; however, accumulating evidence indicates that the pathogen can invade and persist within an iron-starved compartment of mammalian cells (Mortensen and Skaar, 2013; Harding et al., 2018). In the past two decades, progress has been made in identifying and characterizing host factors that regulate the intracellular lifestyle of diverse pathogens, including A. baumannii (Choi et al., 2008; Smani et al., 2012; Rumbo et al., 2014; Wang et al., 2016; Parra-Millan et al., 2018; An et al., 2019). However, this aspect of the infection process remains poorly understood.
The evolutionarily divergent Drosophila S2 cell, a macrophage-like cell line, model system has been exploited as an alternative host system for studying mammalian host-pathogen interactions since it recapitulates conserved aspects of innate immunity (e.g., the detection or recognition of microbial infection and the activation of inflammatory and antimicrobial innate immune responses by Toll-like receptors in mammals and insects) (Kim and Kim, 2005; Criscitiello and de Figueiredo, 2013; Pandey et al., 2014). We previously demonstrated that mammalian orthologs of hits, e.g., IRE1a, a key unfolded protein response (UPR) sensor of endoplasmic reticulum (ER) stress, and autophagy related proteins, identified in RNAi (RNA interference) screens of the Drosophila S2 cells for host factors mediating pathogen infection are important for bacterial and fungal infection of mammalian cells, thereby validating the utility and convenience of this insect cell model for host-pathogen interaction studies (Qin et al., 2008, 2011; Pandey et al., 2018). The combination of the Drosophila S2 cell system and RNAi technology for depletion of host gene expression has also provided unprecedented opportunities for rapid functional elucidation of host factors. Here, we show that Drosophila S2 cells provide a convenient system for interrogating interactions between A. baumannii and host cells. We demonstrate the utility of this system by showing its use for identifying a role for host MAP kinase proteins in conferring susceptibility to intracellular parasitism. Ultimately, these findings may facilitate the development of novel host-directed anti-infectives for combatting the bacterium.
Results
A. baumannii Infection Induces Host Cell Death in Alveolar Macrophages but Not Lung Epithelial Cells
The lung is an important site of A. baumannii infection. We therefore used gentamicin protection assays to determine whether host cells susceptible to A. baumannii intracellular parasitism. We used several cell lines for our experiments, including alveolar macrophage cells (MH-S, an SV40 transformed alveolar macrophage line), lung epithelial cells (TC-1, a tumor line derived from primary murine lung epithelial cells, or pulmonary tumor cells (MLE-12, SV40 large T antigen transformed line). We found that the pathogen successfully invaded mouse MH-S, TC-1, MLE-12, and Drosophila S2 cells (Figure 1A). However, the levels of bacterial invasion was significantly lower in these cells compared to J774 cells, a murine macrophage-like line that has been previously shown to be susceptible to invasion by A. baumannii (Asplund et al., 2013; May et al., 2019). To confirm our findings, we used gentamicin protection assays to analyze intracellular persistence or replication of the pathogen during a time course of infection. Colony forming unit (CFU) analysis revealed that the pathogen successfully replicated and/or persisted for at least 48 h in alveolar macrophage MH-S, lung epithelial TC-1 or MLE-12 cells (Figures 1B–C). Bacterial replication was further confirmed by confocal immunofluorescence assay (Figure 1D). Previous studies showed that A. baumannii invasion of mammalian cells can ultimately induce host cell death (Choi et al., 2005; Kim et al., 2008; Smani et al., 2011; Tamang et al., 2011). To determine whether the bacterium similarly killed alveolar macrophage MH-S and lung epithelial TC-1 or MLE-12 cells, we measured the release of LDH, an indicator of host cell death, following infection (Pei and Ficht, 2004; Pei et al., 2008). We found that invasive bacterial strains Ab17978 or Ab19606 did indeed induce the death of alveolar MH-S macrophages at 48 h post infection (h.p.i.) in an MOI (multiplicity of infection)-dependent fashion (Figure 1E; Figure S1). The data also show that A. baumannii invades/persists and replicates in lung epithelial (TC-1) cells, and ultimately kills alveolar macrophage cells.
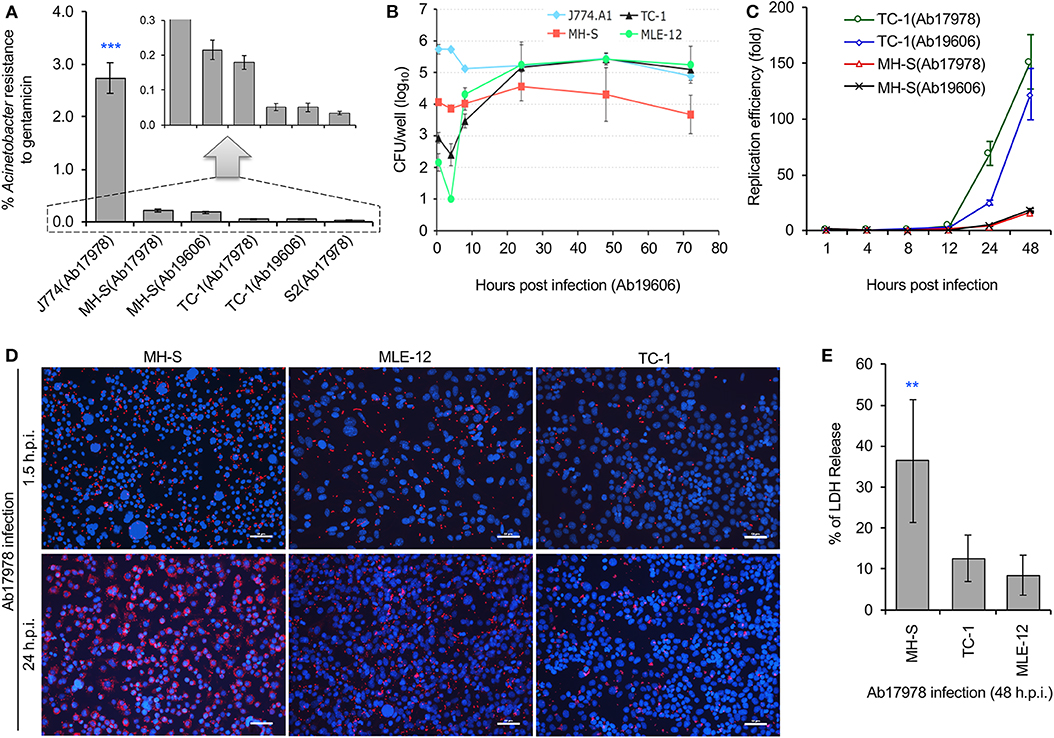
Figure 1. Acinetobacter baumannii invades and persists or replicates within mouse alveolar cells during a time course (72 h) of infections. Mouse macrophage J774 cells, alveolar MH-S macrophages, lung epithelial tumor cells (TC-1 and MLE-12), or Drosophila S2 cells were infected with A. baumannii strains 19606 (Ab19606) or 17978 (Ab17978), at the indicated time points post infection, the infected cells were lysed and subjected to gentamicin protection assay or fixed for confocal immunofluorescence image assay. (A) Successful invasion of A. baumannii into various host cells. S2: Drosophila melanogaster S2 cells. Inset: scale enlargement for the graph within the dash lines. (B) Dynamics of intracellular Ab19606 replication or persistence in the indicated mouse cell lines. (C) Intracellular replication efficiency of A. baumannii in mouse lung epithelial (TC-1) or alveolar MH-S macrophages. (D) Representative images showing intracellular replication or persistence of A. baumannii (Ab17978) in the indicated mammalian cells at 24 h post infection (h.p.i.). Intracellular bacteria and host cell nuclei were stained in red and blue, respectively. Bars: 50 μm. (E) A. baumannii (Ab17978) infection induces varying levels of cell death in alveolar MH-S macrophages and lung epithelial (TC-1 or MLE-12) cells at 48 h.p.i. Data represent the means ± standard deviation (SD) from three independent experiments with triplicate wells examined for each treatment. **, ***: significance at p < 0.01 and 0.001, respectively.
A. baumannii Infects Drosophila S2 Cells
A. baumannii enters membrane bounded vacuoles of epithelial cells in a process that depends upon the activities of microfilaments, microtubules, clathrin and β-arrestins (Choi et al., 2008; Smani et al., 2012). We sought to identify additional host mechanisms that support the intracellular survival or persistence of A. baumannii. However, bacterial invasion of mammalian cells induces cell death (Figure 1E; Figure S1) (Choi et al., 2005, 2008; Smani et al., 2012), an outcome which had the potential of confounding efforts to identify novel host mechanisms. We therefore tested whether Drosophila S2 cells, a model system previously shown to be useful for elucidating host-pathogen interactions (Cherry, 2008; Qin et al., 2008, 2011; Pandey et al., 2014), displayed resistance to bacteria-induced killing. We found that A. baumannii efficiently invaded S2 cells in an MOI-dependent fashion (Figure 1A; Figure S2A) and S2 cells were permissive to intracellular replication of A. baumannii (Figures S2B,C). Moreover, S2 cells were also resistant to pathogen-induced killing (Figure S2D). Similar to previous findings from mammalian host cell systems (Parra-Millan et al., 2018), A. baumanni resided in lysosomal marker LAMP-1 or cathepsin D-positive compartments at later time points post-infection (24 h.p.i.) (Figure S3). Interestingly, intracellular bacteria were also located in ER marker calreticulin-positive vacuoles (Figure S3). These data demonstrated that Drosophila S2 cells support A. baumannii infection. We therefore performed several experiments to determine the suitability of Drosophila S2 cells for identifying host factors that control the intracellular survival or persistence of A. baumannii.
Drosophila S2 Cells Can Be Used as an Alternative Host to Rapidly Identify Host Factors That Control the Invasion, Intracellular Survival, or Persistence of A. baumannii
To identify host functions that confer susceptibility to A. baumannii internalization, survival or intracellular replication, we pre-treated S2 cells with compounds that disrupt host cell functions, and then measured levels of bacterial internalization or intracellular replication. We found that pre-treatment with brefeldin A (BFA), a molecule that disrupts ER to Golgi anterograde transport and the morphology and function of the trans-Golgi network and endosomes (de Figueiredo et al., 1998, 2000), altered the internalization, but not the replication, of the pathogen (Figures 2A,B). Pre-treatment of host cells with myriocin, which depletes host cells of sphingolipids, did not alter either of these processes (Figures 2A,B). However, host cells pre-treated with cytochalasin D, a compound that depolymerizes host cell actin, or wortmannin (WM), a class III phosphatidylinositol 3-kinase (PI3K) inhibitor, or SP600125, an inhibitor of the MAP kinase JNK, limited the internalization and replication of the pathogen (Figures 2A,B; Figure S4). Interestingly, host cells pre-treated with bafilomycin A (BAF), a commonly used compound that inhibits autophagy by targeting lysosomes, limited the internalization but increased intracellular replication efficiency of the pathogen (Figures 2A,B; Figure S4). Importantly, these compounds did not disturb bacterial grow or cause cytotoxicity on host cells (Qin et al., 2008, 2011; Pandey et al., 2017, 2018), thereby suggesting that the observed reductions in pathogen internalization or intracellular replication did not emerge as a consequence of the drug acting directly upon the pathogen or host cells. Taken together, the data indicate that autophagosome biogenesis and accumulation, as well as JNK activity, conferred susceptibility to A. baumannii infection of Drosophila S2 cells.
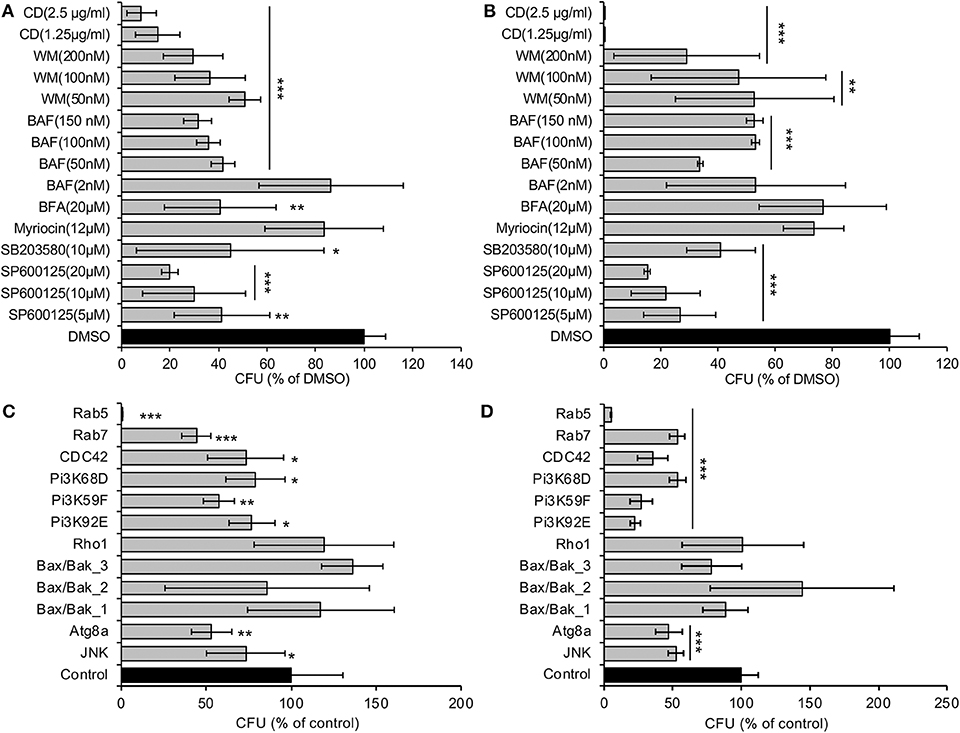
Figure 2. A. baumannii invasion and intracellular replication in Drosophila S2 cells pretreated with pharmacological compounds or treated with dsRNAs to deplete host proteins. (A,B) Drug-treated or untreated S2 cells were infected with Ab17978. At 1.5 (A) or 48 (B) h.p.i., the infected host cells were lysed for CFU analysis as described in the Materials and Methods. (C,D) S2 cells were depleted of the indicated target genes using RNAi approaches and then infected with Ab17978 for 1.5 (C) or 48 (D) h. The infected S2 cells were then lysed for CFU analysis. Data represent the means ± SD from four independent experiments with triplicate wells examined for each treatment. *, **, ***: significance at p < 0.05, 0.01 and 0.001, respectively.
To confirm and extend findings uncovered in our pharmacological analysis, we used RNAi technology to knockdown the expression of selected host target genes. S2 cells that had been treated with dsRNAs that targeted Rab5 and Rab7, which control the biogenesis of early and late endosomes, respectively, displayed resistance to the internalization and intracellular replication or persistence of the pathogen (Figures 2C,D). Similarly, depletion of proteins encoding components of the PI3K complex, Atg8 (a master regulator of autophagosome elongation), or JNK, displayed resistance to bacterial infection (Figures 2C,D). Importantly, trypan blue experiments demonstrated that cells in which target proteins had been knocked down displayed viability that was similar to controls treated with scrambled dsRNAs (Qin et al., 2008, 2011; Pandey et al., 2017, 2018). Thus, the differences in the numbers of recovered intracellular bacteria did not arise as a consequence of differences in the viability of host cells treated with dsRNAs. These data implicated the activities of several key proteins (including JNK) in controlling the subcellular trafficking, survival or replication of the pathogen in Drosophila S2 cells.
Host Factors Identified in Drosophila S2 Cell Infection Models Function Similarly in Mammalian Host Cell Infection Models
We used mammalian macrophages or epithelial cells to validate findings from experiments using Drosophila S2 cells. First, results from Western blot experiments showed that JNK or p38 phosphorylation accompanied A. baumannii invasion of J774 and MH-S macrophages (Figure 3A). Second, we showed that pre-treatment of lung epithelial TC-1 cells with compounds that inactivate JNK or PI3K signaling (SP60012 or wortmannin, respectively) reduced the internalization of A. baumannii into host cells (Figures 3B,C). Pre-treatment of host cells with myriocin did not alter the invasion of the pathogen (Figure 3D), as was observed in fly cells. However, depolymerization of host actin by pre-treating host cells with cytochalasin D did impair A. baumannii invasion of host cells (Figure 3D). Importantly, trypan blue exclusion experiments revealed that the drug treatments did not induce host cell death during bacterial invasion, thereby suggesting that the reductions in bacterial internalization observed in the drug-treated cells resulted from the loss of the activities of the protein drug targets.
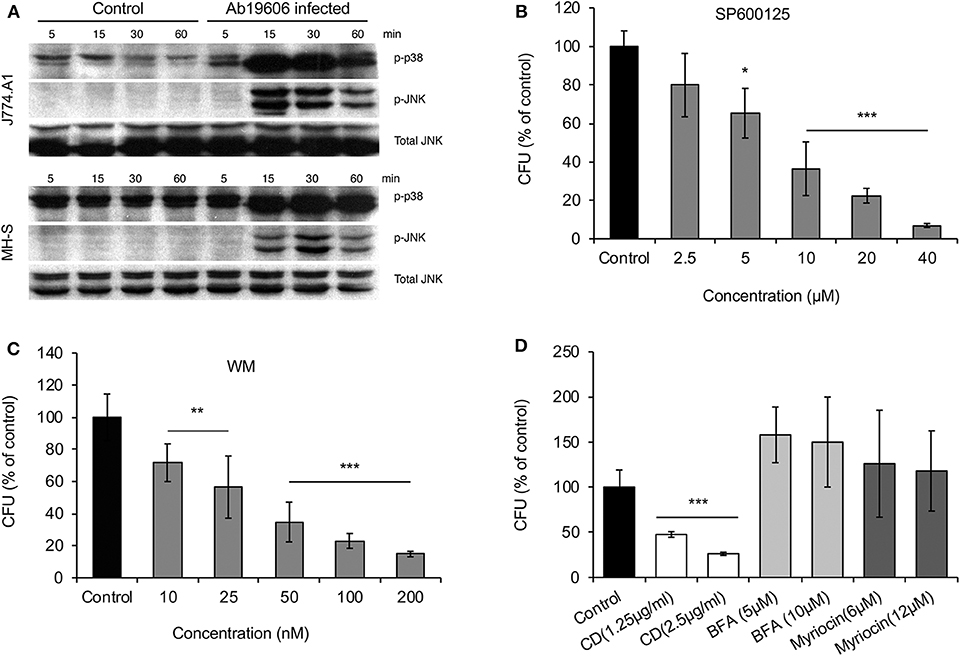
Figure 3. Host factors regulate A. baumannii invasion of mammalian cells. Mouse J774 and MH-S macrophages or lung epithelial TC-1 cells were infected with A. baumannii at an MOI of 50, at the indicated time points post infection, the infected host cells or uninfected controls were lysed for Western blotting analysis or CFU analysis. (A) A. baumannii (Ab19609) infection stimulates p38 and JNK phosphorylation in infected mouse J774 or MH-S macrophages. (B–D) Effects of pre-treating mouse lung epithelial TC-1 cells with pharmacological compounds SP600125 (B), wortmannin (WM) (C) or cytochalasin D (CD), brefeldin A (BFA), and myriocin (D) on Ab17978 invasion of host cells (1.5 h.p.i.). Data represent the means ± SD from four independent experiments with triplicate wells examined for each treatment. *, **, ***: significance at p < 0.05, 0.01 and 0.001, respectively.
Finally, our fly cell findings indicated that proteins regulating organelle biogenesis and membrane trafficking contribute to controlling the intracellular lifestyle of the pathogen. To further explore this possibility, we used immunofluorescence microscopy to monitor the subcellular trafficking of A. baumannii in mammalian cells. We found that A. baumannii colocalized with EEA1 (early endosomal antigen 1)-containing membranes at early time points post-infection (≤1 h.p.i); interestingly, the compartment was also positive for the autophagosomal marker monodansylcadaverin (MDC) (Biederbick et al., 1995) (Figures 4A–C), suggesting that the internalized bacteria are located in autophagic vacuoles. At later time points (4–24 h.p.i), a subpopulation of the bacteria trafficked to a compartment that was decorated with the ER marker calreticulin (Figures 4D,G). At the same time, a large subpopulation trafficked to a compartment that contained lysosomal marker LAMP-1 or cathepsin D (Figures 4E–G). These data indicated that a subpopulation of the pathogen avoided interactions with degradative lysosomal compartments and established a niche in an ER-like vacuole that were decorated with ER proteins. Taken together, findings from confocal immunofluorescence microscopy experiments performed with mammalian cells were consistent with insights garnered from experiments using the Drosophila S2 cell system.
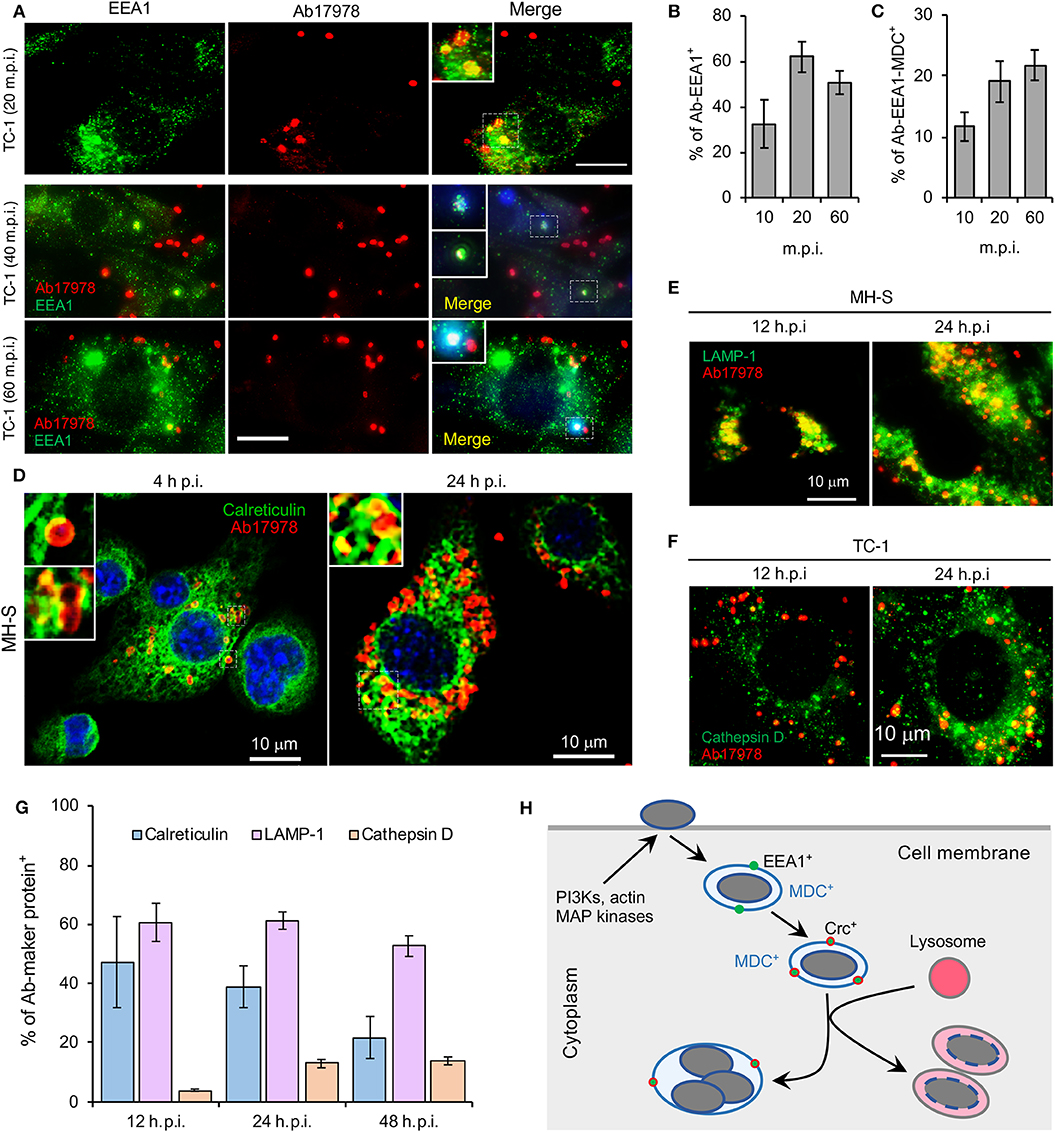
Figure 4. Intracellular trafficking and replication and/or persistence of A. baumannii in mammalian cells. Mouse macrophages (MH-S) and lung epithelial cells (TC-1) were infected with A. baumannii (Ab17978) at an MOI of 20. At the indicated time points post infection, the cells were fixed with 3.7% formaldehyde and stained with the indicated antibodies and processed confocal immunofluorescence microscopy assay. (A) Analysis of A. baumannii localization in host cells strained with the early endosomal marker EEA1 (early-endosomal autoantigen 1) and/or autophagosomal marker monodansylcadaverine (MDC, blue) after internalization. m.p.i.: minutes post infection. Bars: 10 μm. (B,C) Quantification of A. baumannii colocalization with EEA1 (B) and with both EEA1 and MDC (C) at the indicated time points post-infection. (D) A. baumannii infected alveolar MH-S macrophages (D) were fixed and stained antibodies that recognize calreticulin, a host cell ER (endoplasmic reticulum) marker. (E,F) Localization of intracellular bacteria in host cells stained with antibodies that recognize the lysosome marker LAMP1 (E) or cathepsin D (F) at 12 and 24 h.p.i. (G) Quantification of A. baumannii colocalization with calreticulin, LAMP1 or cathepsin D at the indicated time points post infection. (H) A proposal model of A. baumannii intracellular trafficking and persistence or replication. Representative confocal images showing the co-localization of A. baumannii (red) with the indicated host protein markers (green) inside MH-S macrophages or lung epithelial TC-1 cells. Quantitative data represent the means ± SD from four independent experiments with triplicate wells examined for each treatment.
Discussion
The genetically tractable Drosophila S2 cell system possesses innate immune functions that are similar to mammalian cell systems. This attribute, together with efficient RNAi-mediated protein depletion and the availability of genome-scale RNAi libraries, has made the Drosophila S2 cell system a powerful platform for rapidly identifying host factors in large-scale screening campaigns (Qin et al., 2008, 2011). Drosophila S2 cells are also more resistant to A. baumannii-induced host cell death than their mammalian counterparts. The observed resistance facilitates analysis of host cell functions. Finally, the most important criterion for judging the utility of an alternative non-mammalian host-pathogen interaction model system is whether it can shed light on interactions that take place in cells from target organisms (e.g., mammalian cells). In this report, we demonstrate that host factors identified in the Drosophila cell system, including MAP kinases and proteins that regulate organelle biogenesis and membrane trafficking, contribute to regulating the internalization, intracellular trafficking and replication or persistence of bacterial pathogens in mammalian cells. Therefore, these findings not only demonstrate that the Drosophila S2 cell-A. baumannii interaction system recapitulates critical aspects of bacterial infection of mammalian cells, but also demonstrate the utility of the system.
A. baumannii is not predominately an intracellular pathogen. However, the bacterium can invade and persist in mammalian cells, including human lung, laryngeal, and cervical epithelial cells, and thereby promote invasive disease (Mortensen and Skaar, 2013; Harding et al., 2018). Following entry into host cells, the bacterium resides in a membrane-bound vacuole (Choi et al., 2008). Clathrin and β-arrestins are also engaged during the uptake into human lung epithelial cells (Smani et al., 2012). Similar to these findings, we found that upon internalization, A. baumannii resides in vacuoles decorated with the early endosome marker EEA1, or with both EEA1 and the autophagosomal marker MDC. During intracellular trafficking and persistence, some bacteria are observed in calreticulin-positive compartments. However, most intracellular bacteria locate in LAMP-1 or cathepsin D-positive vacuoles. Interestingly, intracellular persistence of A. baumannii was observed while the bacterium colocalized with lysosome, which is consistent with a previous report (Parra-Millan et al., 2018). The destabilization of lysosomal membranes (resulting in the release of cathepsin D outside of bacterium-containing lysosomes and the loss of acidic conditions inside lysosomes) may account for the intracellular persistence of the bacterium (Parra-Millan et al., 2018).
A. baumannii infection induces host cell autophagy, an evolutionary conserved self-eating process that plays critical roles in maintaining cellular homeostasis and host cell defense against microbial infections. Outer membrane protein A (OmpA) is a virulence factor of A. baumannii associated with the bacterial survival and pathogenicity (Sato et al., 2017). The bacterial proteins Omp33-36 can induce incomplete autophagy and assist intracellular replication of A. baumannii (Rumbo et al., 2014). Different pathways are activated during A. baumannii induced autophagy, including the AMPK/ERK/mTOR signaling pathway (Wang et al., 2016), the MAP/JNK signaling pathway (An et al., 2019) and the transcription factor EB (TFEB) pathway (Parra-Millan et al., 2018). Moreover, activation of TFEB could induce the autophagosome-lysosome system and destroys the acidic environment of the lysosome. These changes facilitate A. baumannii invasion, intracellular trafficking and persistence (Parra-Millan et al., 2018). We also found that infection increases the phosphorylation levels of host cell JNK and p38 (Figure 3A), molecules that control (in part) autophagosome biogenesis (Sui et al., 2014). Similar to previous findings (Parra-Millan et al., 2018), both Drosophila S2 and mammalian host cells treated with bafilomycin, an inhibitor of organelle (e.g., endosomes and lysosomes) acidification and fusion between autophagosomes and lysosomes (Rumbo et al., 2014), wortmannin, or SP600125, reduced A. baumannii invasion. In the presence of bafilomycin, replication efficiency of A. baummanii increases, which is consistent with the observation of A. baumannii replication within autophagosomes (Rumbo et al., 2014). Taken together, these finding suggest that the intracellular lifestyle of A. baumannii is controlled by factors that regulate host cell autophagosome and autophagolysosome biogenesis (Figure 4H).
To persist or replicate intracellularly, A. baumannii needs to traffic to a niche that is permissive for these activities. However, the intracellular lifestyle of A. baumannii may be a transient event in some cells. Moreover, the contribution of the intracellular lifestyle of A. baumannii to pathogenesis requires further clarification. Nevertheless, it is notable that A. baumannii displays better survival and/or persistence in Drosophila S2 cells than in mammalian cells. Therefore, host factors identified in the Drosophila S2-A. baumannii interaction system needs to be further evaluated in mammalian hosts. In this report, host factors involved in autophagosome and autophagolysosome biogenesis were found to support A. baumannii intracellular trafficking, survival and replication in mammalian and Drosophila cells. These findings further support idea that the Drosophila cell system can serve as a useful model system for elucidating host-A. baumannii interactions.
The mechanisms of A. baumannii intracellular persistence are poorly understood. Host TFEB is required for bacterial invasion, intracellular trafficking and persistence inside human lung epithelial A549 cells. After infection by the bacterium, host TFEB is activated which results in activation of lysosomal biogenesis and autophagy that could promote the death of host cells. Therefore, TFEB is associated with host defense against bacterial infection. Moreover, HLH-30, the TFEB ortholog of Caenorhabditis elegans, plays an important role in promoting survival of the nematode during infection by the bacterium (Parra-Millan et al., 2018). These findings demonstrate that the functions of host factors are conserved in the pathogenesis of A. baumannii in phylogenetically divergent organisms. Our data also demonstrate that host factors that mediate A. baumannii invasion, intracellular trafficking and persistence are functionally conserved in Drosophila S2 and mammalian host cells. Therefore, the Drosophila S2 cell-A. baumannii interaction system may facilitate identifying and elucidating host factors that regulate the bacterium infection.
In summary, our findings establish a novel model system for defining new host factors that regulate A. baumannii invasion, intracellular trafficking and replication or persistence. By reporting host factor activities that, when disrupted, suppress intracellular survival of the pathogen, this work opens up new research avenues for defining potential therapeutics that target host cell functions. Therefore, this work will ultimately contribute to the fight against this harmful bacterial agent.
Materials and Methods
Cell Cultures and dsRNA-Mediated Knockdown of Target Genes
Mouse alveolar macrophage MH-S (ATCC CRL-2019), mouse lung epithelial cell TC-1 (ATCC, CRL-2785) and MLE12 (ATCC CRL-2110) were routinely cultured at 37°C in a 5% CO2 atmosphere in Dulbecco's Modified Eagle's Medium (DMEM) supplemented with 10% fetal bovine serum (FBS). Drosophila S2 cell cultivation and dsRNA-mediated knockdown of target genes in Drosophila S2 were performed as previously described (Qin et al., 2008; Pandey et al., 2018). Cells were seeded in 24-well plates and cultured overnight prior to infection. For antibiotic protection assays, 2.5 × 105 cells were seeded in each well; for fluorescence microscopy assays (see below), 1.0 × 105 cells were seeded on 12-mm glass coverslips with a thickness of 0.13 mm (Fisherbrand®) placed on the bottom of 24-well plates.
Acinetobacter Infection and Gentamicin Protection Assay
Acinetobacter baumannii strains ATCC17978 (Ab17978) and ATCC19606 (Ab19606) were used in this study. Bacteria were grown in LB broth or on LB agar plates. To prepare bacterial inoculums, 5 ml of LB broth was inoculated with a single colony from a freshly grown LB plate. Cultures were then grown at 37°C overnight with shaking. Bacteria were than washed with 1 × PBS (pH 7.4) and re-suspended in PBS. Host cells were infected with A. baumannii at an MOI of 50. Briefly, infected cells were incubated at 37°C (28°C for Drosophila S2 cells, unless otherwise stated) after centrifugation for 5 min at 200×g. Thirty minutes post-infection, culture media were removed and infected cells were rinsed twice with PBS (pH7.4). Fresh media containing 50 μg/ml gentamicin were added to kill extracellular bacteria. Infected cells were continuously incubated in this antibiotic at 37°C. At various times post-infection, infected cells were lysed with 0.5% Tween 20 in sterile water, and the cell lysates were diluted in peptone saline [1% (wt/vol) Bacto peptone and 0.85% (w/v) NaCl]. Appropriate dilutions were spotted on LB plates and incubated at 37°C overnight to determine colony counts.
Viability and Membrane Permeability Assays of Infected Host Cells
Cells cultured in 24-well plate were infected with A. baumannii as described above. The morphology of infected cells was observed and recorded using a Nikon (Eclipse Ti) laser confocal microscope. To quantitate cytotoxicity, cells cultured in 24-well plates were infected with A. baumannii in triplicate wells as described above. The culture supernatants were collected at various time points p.i., and the lactate dehydrogenase (LDH) release was determined by a CytoTox 96 non-radioactive cytotoxicity assay (Promega, Madison, Wis.) as described previously (Pei and Ficht, 2004; Pei et al., 2008). Cytopathic cell death is expressed as a percentage of maximum LDH release, i.e., 100 × (optical density at 490 nm [OD490] of infected cells – OD490 of uninfected cells)/(OD490 of lysed uninfected cells – OD490 of uninfected cells). A typan blue dye (0.2%) exclusion assay (Qin et al., 2008) was also employed to quantify the cytopathic effect or evaluate membrane permeability on mammalian and Drosophila S2 cells caused by A. baumannii infection or by drug or dsRNA treatment. Host cells in which dsRNA or drug treatment did not induce significant differences in viability and membrane permeability were used in subsequent experiments.
Monodansylcadaverine (MDC) Staining
MDC staining was performed to evaluate the abundance of autophagic vacuoles in A. baumannii infected cells as previously described (Biederbick et al., 1995). Briefly, a 50 mM stock solution of MDC was prepared in dimethyl sulfoxide (DMSO). Cells were stained with MDC at a final concentration of 50 μM for 1 hr at room temperature, washed with phosphate-buffered saline for 3 × 10 min, and then examined by fluorescence microscopy.
Immunofluorescence Microscopy
To determine where A. baumannii replication occurred, host cells were infected with A. baumannii and fixed with 3.7% formaldehyde fixed at 24 h.p.i. The fixed cells were washed and processed for immunofluorescence microscopy (Qin et al., 2008, 2011; Pandey et al., 2017, 2018). The primary antibodies used were as follows: mouse polyclonal anti-Acinetobacter, made by a commercial company, anti-EEA1, anti-LAMP1, anti-calreticulin, anti-cathepsin D (Santa Cruze, CA, USA). Samples were stained with Alexa Fluor 488-conjugated donkey anti goat IgG or Alexa Fluor 488-conjugated chicken anti-rabbit IgG and Alexa Fluor 594-conjugated donkey anti mouse (Molecular Probes). Cover slips were then mounted in Vectashield® mounting media (Vector Laboratories, Inc., CA, USA) and visualized with the Nikon confocal microscope. Image acquisition and processing were performed as previously described (Qin et al., 2008, 2011; Pandey et al., 2017, 2018).
Drug Treatments
Drosophila S2 and/or murine cells were coincubated with assorted pharmacological compounds, including cytochalasin D (CD, actin polymerization inhibitor), wortmannin [WM, phosphoinositide 3-kinase (PI3K) inhibitor)], bafilomycin A (BAF), brefeldin A (BFA) Myriocin, SB203580 (MAP kinase p38 inhibitor) and SP600125 1 hr before, and during, infection with the indicated A. baumannii strains. Uptake of the microorganisms by the treated and untreated cells and intracellular replication and/or persistence of A. baumannii population were determined using the gentamicin protection assay as described above.
Western Blot
To determine MAP kinase activation, cells cultured in 24-well plates were infected with A. baumannii at an MOI of 50. Uninfected cells were used as control. At 5, 15, 30 and 60 min post infection (m.p.i.), the infected and uninfected control cells were lysed and subjected to Western blotting analysis as previously described (Qin et al., 2011; Pandey et al., 2017, 2018). Primary antibodies used in the analysis were: anti-p38, anti-SAPK/JNK, anti-p-SAPK/JNK (Cell signaling). Dilution of primary antibodies and secondary antibody (HRP anti-IgG, Sigma-Aldrich, USA) was 1:1,000 and 1:1,000~5,000, respectively.
Statistical Analysis
All quantitative data were derived from results obtained from at least three independent experiments with triplicate treatments tested. The data of controls were normalized as 1 or 100% to easily compare results from different independent experiments. The significance of the data was assessed using the Student's t-test to assess statistical significance between two experimental data sets or a one-way ANOVA test to evaluate the statistical differences of multiple comparisons of the data sets.
Data Availability Statement
All datasets generated for this study are included in the article/Supplementary Material.
Author Contributions
PF, Q-MQ, JP, AR-F, and TF conceived and designed the experiments. Q-MQ, JP, and GG performed the experiments. Q-MQ, JP, PF, and TF analyzed the data. PF, AR-F, and TF contributed reagents, materials, and analysis tools. Q-MQ and PF wrote the paper.
Funding
PF was supported by funding from the Texas AM Clinical Science Translational Research Institute Pilot Grant CSTR2016-1, DARPA (HR001118A0025-FoF-FP-006), NIH (R21AI139738-01A1, 1 R01AI141607-01A1, 1R21GM132705-01), the National Science Foundation (DBI 1532188, NSF0854684) and the Bill Melinda Gates Foundation. The project depicted was in part sponsored by the Defense Advanced Research Projects Agency (Agreement HR001118A0025-FoF-FP-006). This research was also supported by NIH grant awards NIH 1R01 AI48496-01A1 and NIH 1U54AI057156-0100 to TF; grant awards from the National Natural Science Foundation of China (# 81371773) to Q-MQ. The content of the information does not necessarily reflect the position or the policy of the Government, and no official endorsement should be inferred.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fcimb.2020.00240/full#supplementary-material
References
An, Z., Huang, X., Zheng, C., and Ding, W. (2019). Acinetobacter baumannii outer membrane protein A induces HeLa cell autophagy via MAPK/JNK signaling pathway. Int. J. Med. Microbiol. 309, 97–107. doi: 10.1016/j.ijmm.2018.12.004
Asplund, M. B., Coelho, C., Cordero, R. J., and Martinez, L. R. (2013). Alcohol impairs J774. 16 macrophage-like cell antimicrobial functions in Acinetobacter baumannii infection. Virulence 4, 467–472. doi: 10.4161/viru.25641
Biederbick, A., Kern, H. F., and Elsässer, H. (1995). Monodansylcadaverine (MDC) is a specific in vivo marker for autophagic vacuoles. Eur. J. Cell Biol 66, 3–14.
Cherry, S. (2008). Genomic RNAi screening in Drosophila S2 cells: what have we learned about host-pathogen interactions? Curr. Opin. Microbiol. 11, 262–270. doi: 10.1016/j.mib.2008.05.007
Choi, C. H., Lee, E. Y., Lee, Y. C., Park, T. I., Kim, H. J., Hyun, S. H., et al. (2005). Outer membrane protein 38 of Acinetobacter baumannii localizes to the mitochondria and induces apoptosis of epithelial cells. Cell. Microbiol. 7, 1127–1138. doi: 10.1111/j.1462-5822.2005.00538.x
Choi, C. H., Lee, J. S., Lee, Y. C., Park, T. I., and Lee, J. C. (2008). Acinetobacter baumannii invades epithelial cells and outer membrane protein A mediates interactions with epithelial cells. BMC Microbiol. 8:216. doi: 10.1186/1471-2180-8-216
Criscitiello, M. F., and de Figueiredo, P. (2013). Fifty shades of immune defense. PLoS Pathogens 9:e1003110. doi: 10.1371/journal.ppat.1003110
de Figueiredo, P., Drecktrah, D., Katzenellenbogen, J. A., Strang, M., and Brown, W. J. (1998). Evidence that phospholipase A2 activity is required for Golgi complex and trans Golgi network membrane tubulation. Proc. Natl. Acad. Sci. U.S.A. 95, 8642–8647. doi: 10.1073/pnas.95.15.8642
de Figueiredo, P., Drecktrah, D., Polizotto, R. S., Cole, N. B., Lippincott-Schwartz, J., and Brown, W. J. (2000). Phospholipase A2 antagonists inhibit constitutive retrograde membrane traffic to the endoplasmic reticulum. Traffic 1, 504–511. doi: 10.1034/j.1600-0854.2000.010608.x
Harding, C. M., Hennon, S. W., and Feldman, M. F. (2018). Uncovering the mechanisms of Acinetobacter baumannii virulence. Nat. Rev. Microbiol. 16:91. doi: 10.1038/nrmicro.2017.148
Kim, S. A., Yoo, S. M., Hyun, S. H., Choi, C. H., Yang, S. Y., Kim, H. J., et al. (2008). Global gene expression patterns and induction of innate immune response in human laryngeal epithelial cells in response to Acinetobacter baumannii outer membrane protein A. FEMS Immunol. Med. Microbiol. 54, 45–52. doi: 10.1111/j.1574-695X.2008.00446.x
Kim, T., and Kim, Y. (2005). Overview of innate immunity in Drosophila. J. Biochem. Mol. Biol. 38:121. doi: 10.5483/BMBRep.2005.38.2.121
May, H. C., Yu, J.-J., Shrihari, S., Seshu, J., Klose, K. E., Cap, A. P., et al. (2019). Thioredoxin modulates cell surface hydrophobicity in Acinetobacter baumannii. Front. Microbiol. 10:2849. doi: 10.3389/fmicb.2019.02849
Mortensen, B. L., and Skaar, E. P. (2013). The contribution of nutrient metal acquisition and metabolism to Acinetobacter baumannii survival within the host. Front. Cell. Infect. Microbiol. 3:95. doi: 10.3389/fcimb.2013.00095
Pandey, A., Ding, S. L., Ficht, T. A., and de Figueiredo, P. (2014). “siRNA screens using drosophila cells to identify host factors required for infection,” in Host-Bacteria Interactions, eds A. C. Vergunst and D. O'Callaghan (Springer), 229–244. doi: 10.1007/978-1-4939-1261-2_13
Pandey, A., Ding, S. L., Qin, Q.-M., Gupta, R., Gomez, G., Lin, F., et al. (2017). Global reprogramming of host kinase signaling in response to fungal infection. Cell Host Microbe 21, 637−649. e636. doi: 10.1016/j.chom.2017.04.008
Pandey, A., Lin, F., Cabello, A. L., da Costa, L. F., Feng, X., Feng, H.-Q., et al. (2018). Activation of host IRE1α-dependent signaling axis contributes the intracellular parasitism of Brucella melitensis. Front. Cell. Infect. Microbiol. 8:103. doi: 10.3389/fcimb.2018.00103
Parra-Millan, R., Guerrero-Gomez, D., Ayerbe-Algaba, R., Pachon-Ibanez, M. E., Miranda-Vizuete, A., Pachon, J., et al. (2018). Intracellular trafficking and persistence of Acinetobacter baumannii requires transcription factor EB. mSphere 3:e00106–18. doi: 10.1128/mSphere.00106-18
Pei, J., and Ficht, T. A. (2004). Brucella abortus rough mutants are cytopathic for macrophages in culture. Infect. Immun. 72, 440–450. doi: 10.1128/IAI.72.1.440-450.2004
Pei, J., Wu, Q., Kahl-McDonagh, M., and Ficht, T. A. (2008). Cytotoxicity in macrophages infected with rough Brucella mutants is type IV secretion system dependent. Infect. Immun. 76, 30–37. doi: 10.1128/IAI.00379-07
Qin, Q. M., Luo, J., Lin, X., Pei, J., Li, L., Ficht, T. A., et al. (2011). Functional analysis of host factors that mediate the intracellular lifestyle of Cryptococcus neoformans. PLoS Pathog 7:e1002078. doi: 10.1371/journal.ppat.1002078
Qin, Q. M., Pei, J., Ancona, V., Shaw, B. D., Ficht, T. A., and de Figueiredo, P. (2008). RNAi screen of endoplasmic reticulum-associated host factors reveals a role for IRE1alpha in supporting Brucella replication. PLoS Pathog. 4:e1000110. doi: 10.1371/journal.ppat.1000110
Rumbo, C., Tomás, M., Moreira, E. F., Soares, N. C., Carvajal, M., Santillana, E., et al. (2014). The Acinetobacter baumannii Omp33-36 porin is a virulence factor that induces apoptosis and modulates autophagy in human cells. Infect. Immun. 82, 4666–4680. doi: 10.1128/IAI.02034-14
Sato, Y., Unno, Y., Kawakami, S., Ubagai, T., and Ono, Y. (2017). Virulence characteristics of Acinetobacter baumannii clinical isolates vary with the expression levels of omps. J. Med. Microbiol. 66, 203–212. doi: 10.1099/jmm.0.000394
Smani, Y., Docobo-Perez, F., Lopez-Rojas, R., Dominguez-Herrera, J., Ibanez-Martinez, J., and Pachon, J. (2012). Platelet-activating factor receptor initiates contact of Acinetobacter baumannii expressing phosphorylcholine with host cells. J. Biol. Chem. 287, 26901–26910. doi: 10.1074/jbc.M112.344556
Smani, Y., Dominguez-Herrera, J., and Pachon, J. (2011). Rifampin protects human lung epithelial cells against cytotoxicity induced by clinical multi and pandrug-resistant Acinetobacter baumannii. J. Infect. Dis. 203, 1110–1119. doi: 10.1093/infdis/jiq159
Sui, X., Kong, N., Ye, L., Han, W., Zhou, J., Zhang, Q., et al. (2014). p38 and JNK MAPK pathways control the balance of apoptosis and autophagy in response to chemotherapeutic agents. Cancer Lett. 344, 174–179. doi: 10.1016/j.canlet.2013.11.019
Tamang, M. D., Kim, S., Kim, S. M., Kong, H. H., and Kim, J. (2011). Interaction of Acinetobacter baumannii 19606 and 1656-2 with Acanthamoeba castellanii. J. Microbiol. 49, 841–846. doi: 10.1007/s12275-011-1063-8
Keywords: Acinetobacter baumannii, Drosophila S2 cells, invasion, persistence, host factors
Citation: Qin Q-M, Pei J, Gomez G, Rice-Ficht A, Ficht TA and de Figueiredo P (2020) A Tractable Drosophila Cell System Enables Rapid Identification of Acinetobacter baumannii Host Factors. Front. Cell. Infect. Microbiol. 10:240. doi: 10.3389/fcimb.2020.00240
Received: 15 March 2020; Accepted: 27 April 2020;
Published: 26 May 2020.
Edited by:
D. Scott Merrell, Uniformed Services University, United StatesReviewed by:
Phil Rather, Emory University, United StatesWangxue Chen, National Research Council Canada (NRC-CNRC), Canada
Copyright © 2020 Qin, Pei, Gomez, Rice-Ficht, Ficht and de Figueiredo. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Thomas A. Ficht, dGZpY2h0QGN2bS50YW11LmVkdQ==; Paul de Figueiredo, cGpkZWZpZ3VlaXJlZG9AdGFtdS5lZHU=
†These authors have contributed equally to this work