 Jocelyn R. Hauser1
Jocelyn R. Hauser1 Rama R. Atitkar1
Rama R. Atitkar1 Courtney D. Petro1
Courtney D. Petro1 Rebecca L. Lindsey2
Rebecca L. Lindsey2 Nancy Strockbine2
Nancy Strockbine2 Alison D. O'Brien1
Alison D. O'Brien1 Angela R. Melton-Celsa1*
Angela R. Melton-Celsa1*- 1Department of Microbiology and Immunology, Uniformed Services University of the Health Sciences, Bethesda, MD, United States
- 2Centers for Disease Control and Prevention, Atlanta, GA, United States
In this study we compared nine Shiga toxin (Stx)-producing Escherichia coli O157:H7 patient isolates for Stx levels, stx-phage insertion site(s), and pathogenicity in a streptomycin (Str)-treated mouse model. The strains encoded stx2a, stx1a and stx2a, or stx2a and stx2c. All of the strains elaborated 105-106 cytotoxic doses 50% (CD50) into the supernatant after growth in vitro as measured on Vero cells, and showed variable levels of increased toxin production after growth with sub-inhibitory levels of ciprofloxacin (Cip). The stx2a+stx2c+ isolates were 90–100% lethal for Str-treated BALB/c mice, though one isolate, JH2013, had a delayed time-to-death. The stx2a+ isolate was avirulent. Both an stx2a and a recA deletion mutant of one of the stx2a+stx2c+ strains, JH2010, exhibited at least a three-log decrease in cytotoxicity in vitro and both were avirulent in the mice. Stool from Str-treated mice infected with the highly virulent isolates were 10- to 100-fold more cytotoxic than feces from mice infected with the clinical isolate, JH2012, that made only Stx2a. Taken together these findings demonstrate that the stx2a-phage from JH2010 induces to higher levels in vivo than does the phage from JH2012. The stx1a+stx2a+ clinical isolates were avirulent and neutralization of Stx1 in stool from mice infected with those strains indicated that the toxin produced in vivo was primarily Stx1a. Treatment of mice infected with Stx1a+Stx2a+ isolates with Cip resulted in an increase in Stx2a production in vivo and lethality in the mice. Our data suggest that high levels of Stx2a in stool are predictive of virulence in mice.
Introduction
Shiga toxin (Stx)-producing E. coli (STEC) O157:H7 is a foodborne pathogen estimated to cause more than 265,000 episodes of diarrheal illnesses each year in the United States, with more than 3,600 hospitalizations and 30 deaths (Scallan et al., 2011). The estimated infectious dose for O157:H7 is from 10 to 700 organisms (Tuttle et al., 1999; Hara-Kudo and Takatori, 2011). Spontaneous resolution of the infection usually occurs in about 85% of patients. However, 3–20% of people with confirmed E. coli O157 infection develop hemolytic uremic syndrome (HUS), a sequela characterized by acute kidney failure, thrombocytopenia, and hemolytic anemia (Tarr et al., 2005). Children are disproportionately affected as over 90% of STEC-associated HUS cases occur among children under the age of 5 (Friedrich et al., 2002; Spinale et al., 2013). In addition, the use of antibiotic therapy in patients infected with STEC is linked to an increased risk for the HUS (Freedman et al., 2016). The long-term sequelae of STEC-related HUS in both children and adults can include renal, neurological, pulmonary and cardiac complications (Tarr et al., 2005). The only recommended treatment for STEC-related HUS involves supportive therapy, such as intravenous volume expansion, which has been shown to improve long-term renal outcome (Ake et al., 2005).
The symptoms and sequelae of STEC infection are the result of tissue damage caused by Stx. Stx is a potent AB5 toxin that binds to cells that express the toxin receptor, globotriaosylceramide (GB3) and inhibits protein synthesis in the target cell (see review Melton-Celsa, 2014). There are two antigenically distinct prototype Stxs: Stx1a and Stx2a. There are also subtype variations within each toxin type. Stx subtypes associated with human clinical disease include Stx1a, Stx2a, Stx2c and Stx2d. The Stx operon is encoded on a lysogenic lamdoid-like bacteriophage integrated within the bacterial genome. Phage and host cell factors influence toxin production and release. Although there is a basal level of Stx produced, likely due to spontaneous induction of the stx-phage(s) within a strain, induction of the stx-phage is linked to the bacterium's stress response. When the bacterial cell is grown in the presence of the antibiotic ciprofloxacin (Cip), it undergoes the stress response due to DNA damage and the phage lytic cycle is induced. The latter process is likely the explanation for the increased risk for the development of the HUS after treatment of STEC patients with antibiotics (Freedman et al., 2016).
Although there is a new study that links several single nucleotide polymorphisms in the human genome with an increased susceptibility to post-STEC HUS (Kallianpur et al., 2018), no well-defined patient-specific factor(s) has been found that can be used to predict whether a patient will develop O157:H7-associated post-diarrheal HUS. However, the risk of developing HUS is increased for children under 10 years age and in patients of any age who develop leukocytosis, experience treatment delays, or receive antibiotics and/or anti-motility agents (Tarr et al., 2005). Although the presence of the locus of enterocyte effacement (LEE) in an STEC isolate, the production of specific Stx subtypes, and E. coli O157:H7 clade classification have been associated with STEC that cause severe human disease (Manning et al., 2008; Neupane et al., 2011; Amigo et al., 2015; Bunger et al., 2015), no other pathogen-specific factors have been identified that can be used to predict the severity of STEC related disease. The focus of this study was to determine whether factors, such as stx-subtype, Stx protein levels, stx-phage insertion sites, and virulence of O157:H7 clinical isolates in a mouse model correlated with disease presentation in patients. Although we were unable to identify specific bacterial factors that corresponded to patient clinical outcome data, we did find that that pathogenesis in a streptomycin (Str)-treated mouse model correlated with elevated induction of the stx2a-phage in vivo.
Results
Stx Subtypes, stx-Phage Insertion Site, and Virulence Gene Profiles of Clinical O157:H7 Strains Do Not Correlate With Clinical Presentation
The Stx subtypes, stx-phage insertion sites, and virulence genes profiles of nine clinical O157:H7 isolates from patients that had non-bloody diarrhea (NBD, n = 3), bloody diarrhea (BD, n = 3), or bloody diarrhea that progressed to HUS (n = 3) are summarized in Table 1. The toxin genotypes of the O157:H7 isolates were stx1a and stx2a (n = 3), stx2a (n = 1), or stx2a and stx2c (n = 5). Our finding that five of the strains were stx2a+stx2c+ was not surprising to us because many O157:H7 isolates in the USA and Finland have that genotype (Eklund et al., 2002; Tarr et al., 2019), including the strain from the 2006 spinach outbreak in the USA (Uhlich et al., 2008). Of the nine known loci in which stx phages insert into the host bacterial chromosome (Bonanno et al., 2015), we observed that for strains lysogenized by both stx1a- and stx2a-phages, the stx1a phage occupied yehV, and the stx2-phage occupied wrbA. In the stx2astx2c isolates, the stx2c-phage was in sbcB and the stx2a-phage was in argW. The finding that the stx2a phage was in argW rather than in wrbA has been shown for 40–77% of human O157:H7 isolates (Shaikh and Tarr, 2003; Shringi et al., 2012). Only one strain, JH2012, was lysogenized by a single stx-phage, and that stx2a-phage was also inserted in argW. Although the stx2a+stx2c+ strains did not have stx1, yehV was occupied by an stx1-like defective phage, a finding that has been described for some other STEC (Shaikh and Tarr, 2003).
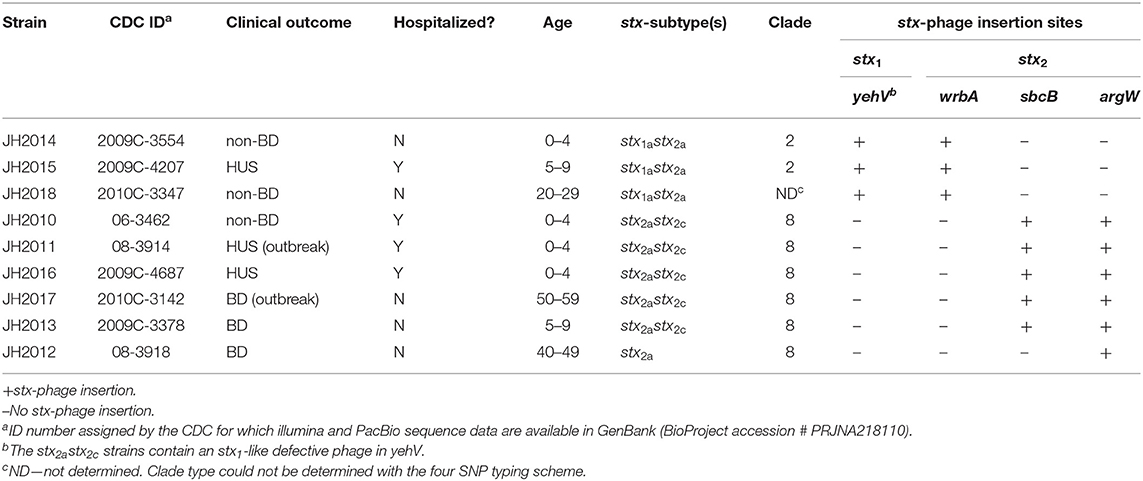
Table 1. Summary of clinical outcomes, stx-subtyping, clades, and phage insertion sites for nine O157:H7 clinical isolates.
We used the four gene typing system described by Riordan et al. (2008) and found that the stx2a+stx2c+ and stx2a+ strains belonged to clade 8. The stx1a+stx2a+ strains (JH2014, JH2015) were identified as clade 2, another O157 clade associated with severe human disease (Manning et al., 2008). JH2018 could not be classified with the four gene typing system. With an E. coli Multilocus Sequence Typing (MLST) scheme (Alikhan et al., 2018), all nine isolates belong to sequence type 11 and possessed the same virulence gene profiles, including genes related to intimin production (eae, tir, tccP), enterohemolysin (ehxA), increased serum survival (iss), non-LEE effectors (nleA, nleB, nleC), Type III secretion proteins (espA, espB, espF, espD, espJ, espP), toxin B (toxB), and catalase (katP). Because the virulence gene profile other than stx type was the same among the isolates, we could not correlate any of the non-stx genes with disease outcome in the patients.
The O157:H7 Strains Are Cytotoxic on Vero Cells and Produce Variable Levels of Stx1 and Stx2 in vitro After Cip Induction
We assessed cytotoxicity of the nine clinical O157:H7 strains and a Str resistant (Strr) derivative of EDL933 [a ground beef isolate from the 1983 hemorrhagic colitis outbreak in Michigan (O'Brien et al., 1983)], on Vero cells after the strains were grown in LB or LB supplemented with the stx-phage inducer, ciprofloxacin (Cip). The minimum inhibitory concentration for the nine clinical isolates ranged between 20 and 40 ng/ml; therefore, a sub-inhibitory concentration of Cip (5 ng/ml) was used for stx induction. After overnight growth in LB with 5 ng/ml Cip there was no statistical difference in CFU count among strains (data not shown). The cell-associated (Figure 1A) or supernatant (Figure 1B) fractions from all the isolates were cytotoxic on Vero cells after growth with or without Cip. The 50% cytotoxic dose (CD50) was calculated as the amount of toxin required to kill 50% of the cells in a well. The stx1a stx2a strains (JH2014, JH2015, and JH2018) exhibited a CD50 of ~105.5−6 with and without Cip for both the cell-associated and supernatant fractions. We separated the cell-associated and supernatant fractions, because in some studies Stx1a is more cell-associated and Stx2 is more consistently found in the supernatant (Shimizu et al., 2009). However, since we measured total toxicity, we cannot say how much of each toxin was present in a particular fraction from this assay. We observed a statistical difference in the CD50 in the cell-associated fraction between the Cip and no Cip samples for stx1astx2a strains JH2014 and JH2015 (Figure 1). Cytotoxicity was not induced for strain JH2018, or, alternatively, it is possible that one of the stx-phages is inducible, but that measuring the total toxicity does not allow us to detect such an increase if the level of toxin produced from the one phage is higher than the induced levels from the second phage. We confirmed that the stx1a+ stx2a+ strains produced both Stx1a and Stx2a by immuno dot blot (data not shown). All strains that produced only Stx2 (we will use Stx2 to mean total Stx2 for strains that may make more than one Stx2 subtype) showed increased cytotoxicity in both the cell-associated and supernatant fractions after growth with Cip, except for JH2013 in the supernatant fraction (Figures 1A,B). Overall, however, we were unable to correlate the in vitro cytotoxicity or inducibility of the strains with disease outcome in the patients.
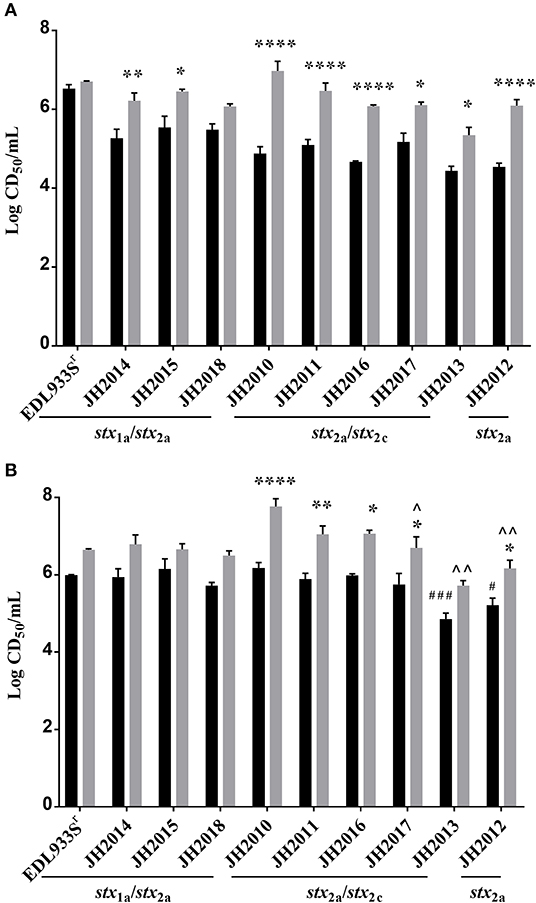
Figure 1. Cytotoxicity of cell-associated or supernatant fractions from cultures of E. coli O157:H7 clinical isolates grown in LB with or without Cip. (A) Cell-associated and (B) supernatant fractions are shown. Results are the mean log CD50/ml ± one standard error for at least 5 independent cultures. Two-way ANOVA with Tukey's posttest was used for multiple comparisons. Comparison between no Cip and Cip samples for each individual strain: *P < 0.05; **P < 0.01; ****P < 0.0001. Comparison of cytotoxicity levels in the cell-associated or supernatant no Cip fractions among the strains that make only Stx2: #P < 0.05; ###P < 0.001 (as compared to JH2010 no Cip). Cytotoxicity levels in the cell-associated and Cip supernatant fractions compared to JH2010; ∧P < 0.05. ∧∧P < 0.0001.
The Most Virulent O157:H7 Clinical Isolates had the Highest Stx2 Levels in Mouse Stool
To determine the virulence potential of the clinical strains in the Str-treated mouse model, mice were gavaged with ~109−10 CFU and followed for 14 days. Of the nine clinical strains, the stx2a+stx2c+ strains JH2010, JH2011, and JH2016 were lethal in 100% of mice by day 7 post-infection (Figure 2A). Of the mice infected with JH2013 or JH2017 just 10% survived the infection, though there was a significant delay in the death of the mice infected with JH2013 (Figure 2A). The stx1astx2a strains JH2014 and JH2015 and the stx2a-only strain, JH2012 were avirulent. Strain JH2018 (stx1astx2a) killed 1/9 mice. There were no differences in mouse colonization levels by the strains on day 1 or day 3 post-infection as measured by the number of O157:H7 shed into the feces (data not shown). Although all three of the stx1a+ stx2a+ strains were avirulent or slightly virulent, stool from mice infected with these strains exhibited cytotoxicity on Vero cells with a CD50 between ~103.5 and 105.5 (Figure 2B). Fecal toxicity data are shown for JH2015 below. Stool from mice infected with the most virulent strains that produced only Stx2 was ~10- to 100-fold more cytotoxic than from avirulent strain JH2012 on days 1 and 3 post-infection and 10-fold more cytotoxic than from the strain with delayed virulence, JH2013, on day 1 post-infection (Figure 2C). We hypothesize from this latter finding that Stx2 from the highly virulent strains is more easily induced in the mouse. Because Stx2 appears to be more easily induced from the virulent strains in vivo, we tested lower inoculum doses (102 or 101 CFU) for one strain, JH2010, in the Str-treated mice. We found that although the time-to-death was delayed for the lower doses of bacteria, all of the mice infected with JH2010 succumbed to infection by day 7 post-infection, Figure 2D.
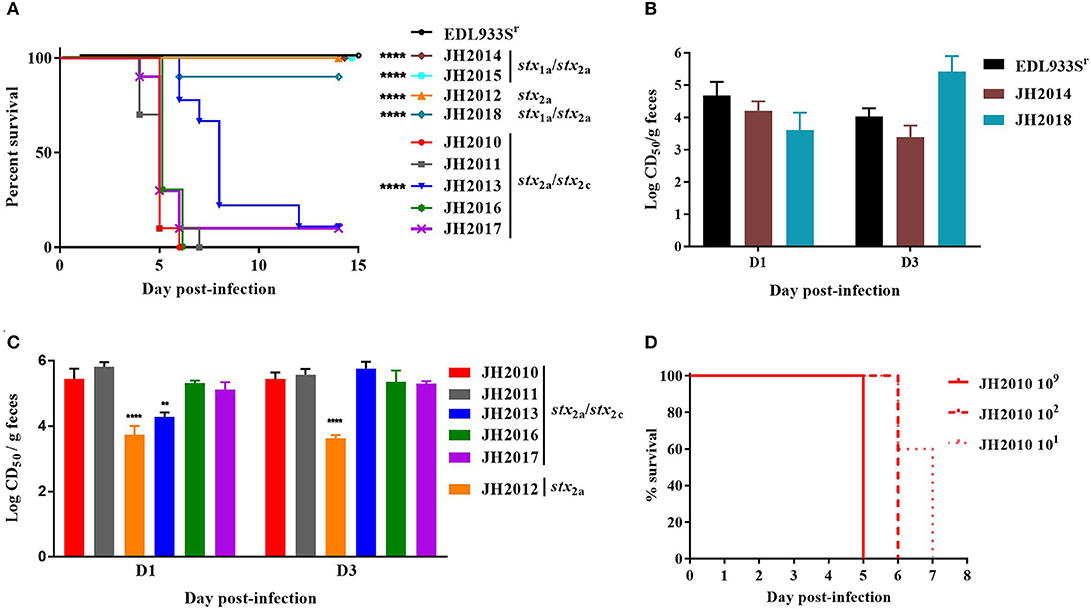
Figure 2. Virulence of clinical O157:H7 strains and cytotoxicity from stool of infected BALB/c mice. (A) Survival of Str-treated mice infected with clinical O157:H7 isolates (n = 9 or 10 mice per isolate) or control strain EDL933Sr (n = 10). Survival of mice infected with JH2010 was different than survival after infection with JH2013 and the stx1astx2a isolates, P < 0.0001, indicated by **** on graph. (B) Cytotoxicity (mean ± one standard error) of stool supernatant from individual mice (n = 5 or 10 mice) infected with either stx1astx2a strain EDL933Sr or JH2014, or JH2018. (C) Cytotoxicity from individual mice (n = 4 or 5) infected with stx2a or stx2astx2c strains. Error bars represent standard deviation of the mean cytotoxicity. Limit of detection was 102 CD50/g feces. **P < 0.01; ****P < 0.0001. (D) Survival of Str-treated mice infected with different doses of JH2010 (n = 5 mice per dose).
Differences in Cytotoxicity and Virulence of JH2010 Were Not Due to Differences in Colonization and Inoculum Dose
We next did a more extensive comparison of colonization levels over time for JH2010 and the less virulent stx2a+stx2c+ strain JH2013. We found that JH2010 colonized similarly or less well than JH2013, as measured by the number of bacteria shed into the feces (Figure 3A). However, the toxicity in stool from mice fed JH2010 was statistically higher on days 1–4 post-infection (Figure 3B). We were unable to compare the toxicity levels in stool on day 5 because too many of the mice had died in the JH2010 group. Taken together, these results suggest that elevated toxin production by JH2010 in vivo contributes to the greater virulence of this strain as compared to JH2013.
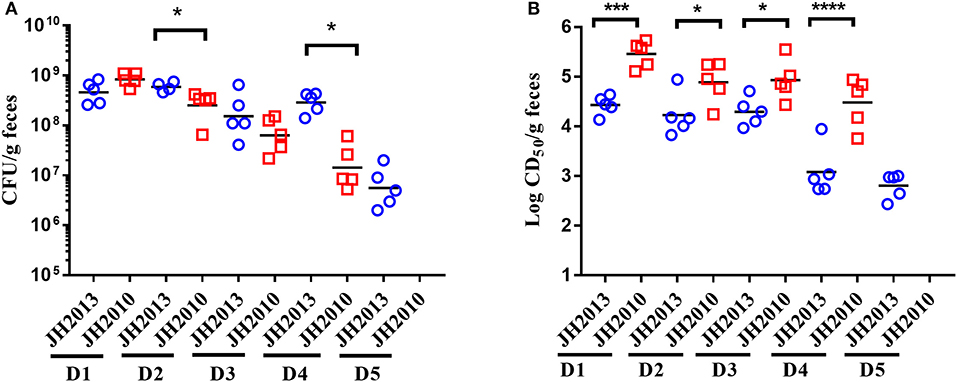
Figure 3. Comparison of colonization and fecal cytotoxicity of two stx2a+stx2c+ O157:H7. (A) Colonization: stool was collected every day for 5 days (D1–D5) post-infection and plated for colonization (n = 5 mice per group) from mice fed 109 CFU JH2013 (circles) or JH2010 (squares). Each symbol represents one animal. The line represents the geometric mean. The limit of detection was 102 CFU/g feces. CFU for each day were compared by unpaired t-test. *P < 0.05. (B) Cytotoxicity of stool collected from infected mice. Each symbol represents one animal and the line indicates the mean log CD50/ml. Cytotoxicity values from the stool samples were compared by unpaired t-test. ***P = 0.0005; ****P < 0.0001. Stool could not be collected from mice infected with JH2010 on D5 so colonization and cytotoxicity levels were not determined.
In vivo Cytotoxicity of Stx1a+ Stx2a+ Strain JH2015 in the Absence of Cip Is Due Mostly to Stx1a
The Stx1a+ Stx2a+ strains JH2015, JH2014, and JH2018 exhibited high levels of colonization (data not shown) and cytotoxicity in vivo, similar to that of virulent strain JH2010 however, these strains were avirulent in mice. We hypothesized that the high level of cytotoxicity from stool of mice infected with the avirulent strains was due primarily to Stx1a. To determine the contribution of Stx1a to the cytotoxicity produced by avirulent strain JH2015 in vivo, we used an anti-Stx1 antibody to neutralize Stx1a in stool supernatants from feces collected from JH2015-infected mice. Incubation of stool from JH2015-infected mice with anti-Stx1 (100 μg/ml) decreased cytotoxicity of the stool 100-fold, a finding that demonstrated that JH2015 produced primarily Stx1a in vivo (Figure 4A). We hypothesize that JH2015 is avirulent in the mice due to the relatively low level of Stx2a produced during infection as indicated by the amount of cytotoxicity that remains after the Stx1a is neutralized (~102 CD50/g stool). To determine whether administration of Cip to JH2015-infected, Str-treated mice would increase Stx2 production in vivo and induce virulence as was previously observed by Zhang et al. for the Stx2-only producing O157:H7 strain 1:361 (Zhang et al., 2000), we gave Cip (5 μg/mouse) or phosphate-buffered saline (PBS) daily to JH2015-infected mice, beginning on day 2 post-infection until the end of the study. Of the Cip- treated mice, 9 out of 10 succumbed to infection, while all infected mice treated with PBS survived the infection (Figure 4B). Stool supernatants from JH2015-infected Cip-treated mice were 100- to 1,000-fold more cytotoxic on Vero cells than similar samples from mice not treated with Cip (Figure 4C compared to Figure 4A, for example, 105.9 compared to 103.0 CD50/g feces on day 3). However, incubation of the stool supernatant from Cip-treated mice with anti-Stx1 did not reduce the toxicity of the samples (Figure 4C); both anti-Stx1 and anti-Stx2 were required for a 1.5-log reduction in toxicity, not shown (The stool samples from the PBS-treated mice in this latter experiment acted similarly to those from untreated mice: the toxicity was neutralized at least 100-fold with anti-Stx1, not shown). These data suggest that treating JH2015-infected mice with Cip increased Stx2a induction and that enhanced level of Stx2 allowed JH2015 to become virulent in mice.
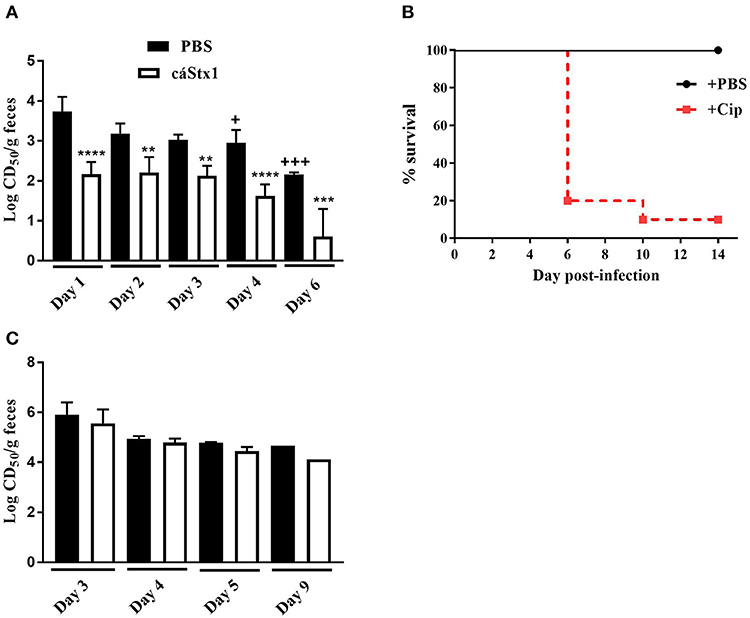
Figure 4. Neutralization of toxin from and survival of Str- or Str- and Cip-treated, JH2015-infected mice. (A) Cytotoxicity of stool samples from mice infected with stx1astx2a strain JH2015 that were incubated with PBS (black bars) or anti-Stx1 antibody, cαStx1 (white bars) (n = 5 mice). Results are the mean log CD50/g feces ± standard error. One-way ANOVA with Tukey's multiple comparisons test. **P < 0.004; ***P = 0.0006; ****P < 0.0001 (as compared to the sample incubated with PBS). +P < 0.05; +++P = 0.0002 (compared to the cytotoxicity of stool from day 1 incubated with PBS). (B) Survival of Str-treated BALB/c mice infected with JH2015 administered PBS (black line) or 0.1 mL Cip (5 μg/ml; dashed red line) starting on day 2 post-infection. (C) Lack of neutralization of Stx1a in stool supernatants from JH2015-infected mice treated parenterally with Cip (5 μg/mouse) beginning on day 2 and administered daily for the remainder of the study. Black bars represent cytotoxicity of fecal supernatants incubated with PBS; white bars represent stool supernatant treated with cαStx1. Results are the mean log CD50/g feces ± standard error.
Deletion of Stx2a in Highly Virulent Strains Attenuates Virulence
JH2010 is lysogenized by an stx2a and an stx2c phage. To determine whether one or both of the stxs plays a role in the virulence of the strain, we generated stx2a and stx2c deletion mutants in the JH2010 background. Supernatant and cell-associated fractions from the stx2c mutant grown with or without Cip were as cytotoxic in vitro as the parental strain (Figure 5A). In contrast, deletion of stx2a caused a >1,000-fold decrease in cytotoxicity when the mutant was grown with or without Cip (Figure 5A). These latter results suggest that a low level of Stx2c is produced from stx2c but that expression is not induced with Cip in vitro (Figure 5A). In vivo, stool supernatant from feces of JH2010Δstx2c-infected mice displayed similar levels of cytotoxicity as the parental strain on Vero cells (Figure 5B) and was equally virulent in mice (Figure 5C). In contrast, stool supernatants from feces of JH2010Δstx2a-infected mice were not cytotoxic (Figure 5B) and the stx2a mutant was avirulent in mice (Figure 5C). These data indicate that Stx2a is responsible for the virulence of JH2010 in Str-treated mice. We observed similar results for another stx2a+stx2c+ strain, JH2011 (Supplemental Figure 1). Taken together our results show that Stx2a is responsible for the virulence of both JH2010 and JH2011.
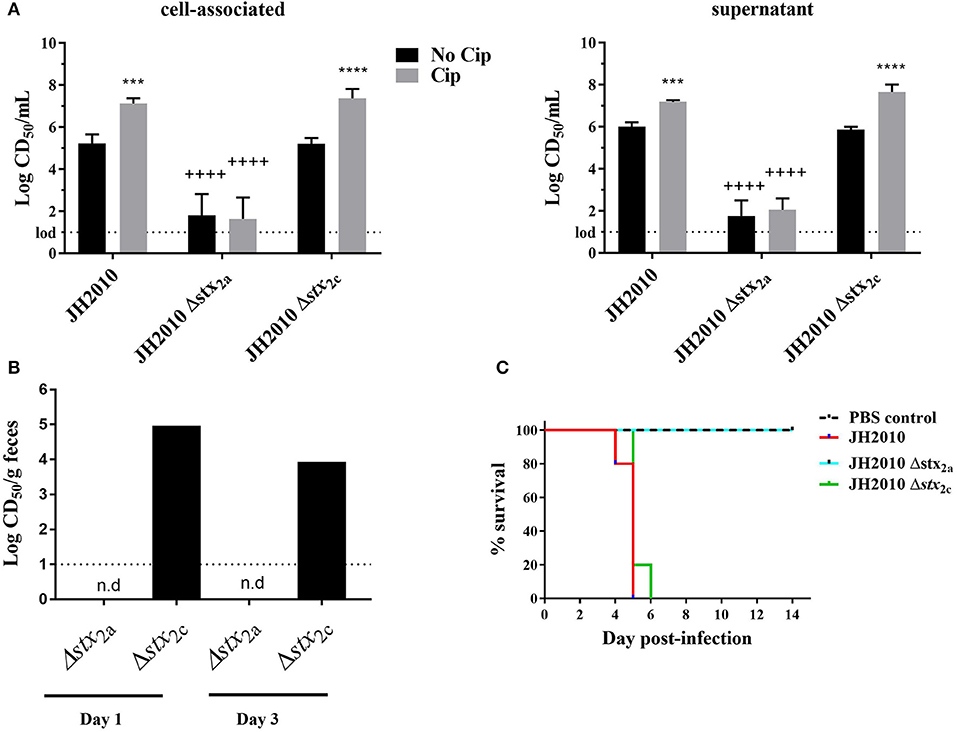
Figure 5. In vitro and in vivo cytotoxicity and survival of JH2010 stx2a and stx2c mutants. (A) Cytotoxicity produced in vitro by the mutants grown in LB or LB with Cip (5 ng/mL). Results are the mean log CD50/ml ± standard error for at least five independent cultures. Two-way ANOVA with Tukey's posttest were used for multiple comparisons. ***P < 0.001; ****P < 0.0001 (as compared to the no Cip control). ++++P < 0.0001 (as compared to the parent strain in the same experimental condition). (B) In vivo cytotoxicity was determined from pooled stool supernatant from feces collected from each mouse post-infection. No cytotoxicity was detected in stx2a mutants (n.d). Bar represents cytotoxicity of pooled feces from five mice. (C) Virulence of stx2 mutants in Str-treated BALB/c mice. JH2010 (n = 15 mice), mutant strains (n = 5 mice per strain).
RecA-Dependent Induction of stx2 Phage Is Required for Virulence of JH2010 in vivo
To determine whether virulence of JH2010 is due to RecA-dependent induction of the stx2 phages, we generated a recA deletion mutant. On agar plates, colonies of the recA deletion mutant appeared smaller compared to the parental strain; however, the mutant did not exhibit a growth defect when grown in LB broth (data not shown). The recA mutant also displayed increased sensitivity to Cip with a minimum inhibitory concentration (MIC) of 2.5 ng/ml compared to the parent (40 ng/ml). Complementation of recA expressed from its native promoter restored both colony morphology and the MIC for Cip. In vitro, deletion of recA reduced cytotoxicity on Vero cells of both the cell-associated and supernatant fractions ~100- fold when grown in LB or LB-Cip (0.6 ng/ml; Figures 6A,B). The recA mutant strain colonized Str-treated mice to similar levels as the parent strain (data not shown); however, the mutant was avirulent (Figure 6C). Str-treated mice fed the complemented mutant strain succumbed to infection by day 5 (Figure 6C). Cytotoxicity of stools collected from mice infected with the ΔrecA mutant were 10- to 100-fold lower compared to the parent (Figure 6D). These data suggest RecA-dependent induction of stx2 phage in JH2010 occurs in vivo and is necessary for the virulence of the strain.
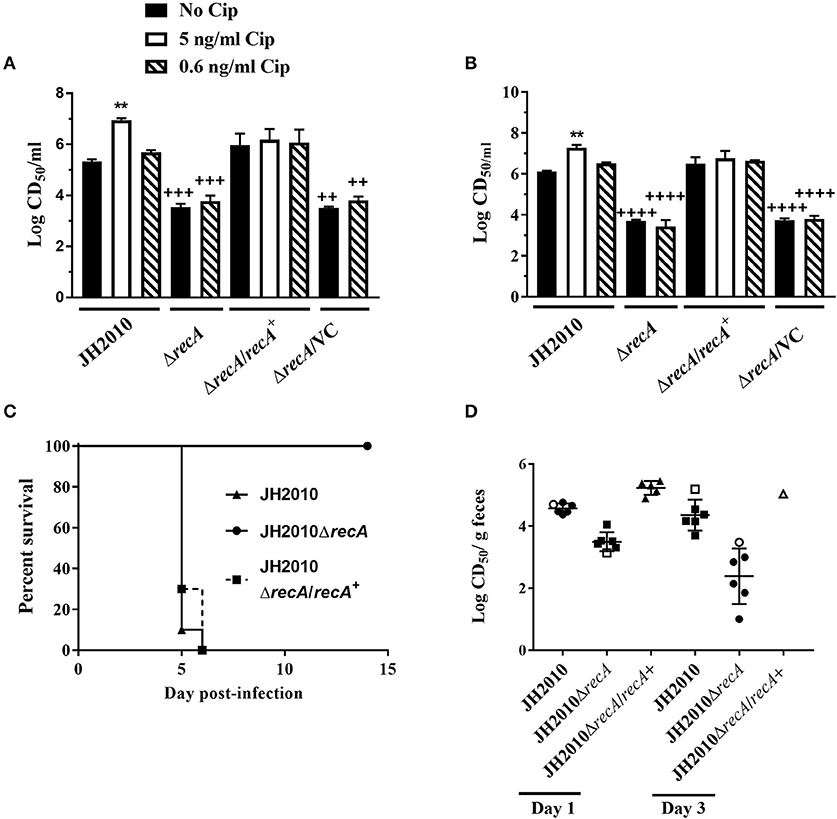
Figure 6. RecA-dependent induction of Stx in JH2010. Cytotoxicity of JH2010, JH2010ΔrecA, JH2010ΔrecA/recA+, and JH2010ΔrecA/VC in vitro of cell-associated (A) and the supernatant (B) fractions. Results are the mean log CD50/ml ±standard error for at least five independent cultures. One-way ANOVA with Tukey's posttest were used for multiple comparisons. **P < 0.01 (as compared to the no Cip control). ++P < 0.01; +++P < 0.001; ++++P < 0.0001 (as compared to the parent strain in the same experimental condition). (C) Survival of mice infected with JH2010, JH2010ΔrecA, or JH2010ΔrecA/recA+. (D) Cytotoxicity of stool supernatant from feces of mice infected with JH2010, the recA mutant, or the complemented strain. Each solid symbol represents one animal, open symbols represent pooled samples from a cage of five mice. The line is the mean log CD50/g feces from the stool supernatant. One-way ANOVA with Tukey's posttest were used for multiple comparisons. *P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001 (as compared to the no Cip control).
Discussion
In this study, we found that four STEC strains from a collection of nine O157:H7 isolates were highly virulent in Str-treated mice. One of those strains, JH2010, killed 100% of Str-treated infected mice even at an inoculum of 101 CFU, and lethality was dependent on Stx2 and RecA. Although JH2010 possesses the genes for both stx2a and stx2c, toxicity could not be detected in stool from mice infected with JH2010Δstx2a, and that mutant was avirulent. Taken together these findings indicate that Stx2a is entirely responsible for the high virulence of JH2010. The toxicity of JH2010 and the other highly virulent strains identified in this study was 10- to 100-fold higher in the feces from infected mice as compared to avirulent strain JH2012. We hypothesize that the higher levels of toxin in the stool from mice infected with the highly virulent O157:H7 isolates is an indication that the stx2a-phage from those strains is readily induced in vivo. A study to compare the induction of stx2a-phage in the stool from mice infected with JH2010 or JH2012 would allow us to address this latter hypothesis.
Our finding that the virulence of JH2010 is dependent on RecA is consistent with a previous study that showed RecA is required for the virulence of STEC isolates EDL933 (Stx1a+ Stx2a+) and 86-24 (Stx2a+) in an intravenous infection model and lung toxicity assay (Fuchs et al., 1999). Furthermore, the stx2a-phage from EDL933 is induced in germ-free mice, a finding that also suggests a requirement for RecA (Tyler et al., 2013) even in the absence of antibiotic treatment to induce the phage. Our findings further indicate that some STEC exhibit enhanced induction of stx2a-encoding bacteriophage in the mouse intestine. Production of Stx2a in strains JH2010 and JH2011 (not shown) was predominately RecA-dependent. Deletion of recA rendered both strains avirulent and decreased cytotoxicity recovered from stool of infected mice. In these strains, there is RecA-independent expression and release into the supernatant of Stx2 in vitro and in vivo. These latter findings demonstrate that there is a non-RecA-mediated way to produce and release Stx2a, a result that contrasts to findings for O157:H7 strain 86-24 in which there was only a low level of Stx2 in the cell-associated fraction in the 86-24 RecA mutant and no Stx2 in the supernatant after growth in LB (Fuchs et al., 1999). However, the level of Stx2a produced in the absence of RecA is not sufficient for lethality in Str-treated mice. We hypothesize that the RecA-independent expression may be due to increased sensitivity to oxidative stress or other factors that lead to expression of Stx2 or induction of the stx2-phage in a RecA-independent manner, as found after growth of cultures in the presence of ethylenediaminetetraacetate (EDTA) (Imamovic and Muniesa, 2012).
Although Stx2a levels were induced from JH2012 after growth with Cip in vitro, and Stx2a was detected in the stool of Str-treated, JH2012-infected mice, JH2012 was avirulent. While it is possible that JH2012 produces a factor that reduces virulence in Str-treated mice, we deem it more likely that mutations exist within the stx2a-phage or the host bacterial genome of JH2012 that prevent complete cell lysis or toxin release from the cell that would be needed for this strain to exhibit mouse pathogenicity. Indeed, another laboratory demonstrated that both phage and bacterial sequences influence toxin production and release in vitro (Yin et al., 2015). Moreover, differences in Stx2 production even among clade 8 strains has previously been reported (Neupane et al., 2011). We compared the sequence upstream of stx2a from JH2012 with those of JH2011, JH2016, and JH2017 (not shown) and found the same SNPs in the q region upstream of stx2a in JH2012 that have been identified in clade 8 strain EC508 as described by Neupane et al. (2011). However, because the same q region SNPs were found in other clade 8 STEC, and those strains express different levels of Stx2 in vitro (Neupane et al., 2011), the q region SNPs alone cannot explain the differences in Stx2a levels we observed in vivo for JH2012 as compared to the stx2astx2c O157:H7 clade 8 strains in this study. However, despite the differences in induction after growth with cip between strain JH2012 and the more virulent strains, we hypothesize that Cip-treatment of mice infected with JH2012 would result in death of the animals because the levels of Stx2a produced in vitro by JH2012 after induction are similar to those from JH2013. The q regions for the virulent strains JH2011, JH2016, and JH1017 are identical to those of the majority of clade 8 strains as described (Neupane et al., 2011).
We observed that the clinical Stx1a+ Stx2a+ strains were avirulent in mice. Neutralization of Stx1a from stool homogenates of mice infected with those Stx1a+ Stx2a+ strains revealed that Stx1a is the predominant toxin produced by these strains in vivo. The relatively low levels of Stx2a made by these strains in vivo was surprising to us but could be related to the insertion site for the stx2a-phage in these strains (wrbA) or possibly to the presence of the stx1a-phage. It has been demonstrated that the presence of two stx2 phages within a single bacterial K12 host lowers the level of toxin produced compared to singly lysogenized strains (Serra-Moreno et al., 2008). Nevertheless, mice infected with Stx1a+ Stx2a+ strain JH2015 and then treated with Cip released high levels of Stx2a into their stool and succumbed to the infection. We attribute the enhanced virulence of the Stx1a+ Stx2a+ strain JH2015 in Cip-treated infected mice to increased production of Stx2a because neutralization of the toxicity in the stool from infected and Cip-treated mice required both Stx1 and Stx2 antibody, a fact that indicates that the Cip treatment greatly enhanced the Stx2a levels in vivo as compared to PBS-treated mice, a phenomenon previously observed in an Stx2-only O157:H7 strain (Zhang et al., 2000). We do not know whether the lack of virulence of the Stx1a+Stx2a+ isolates in the mouse model is due to the relatively low level of Stx2a production in the absence of Cip treatment or if the presence of Stx1a is protective in these strains. Our laboratory previously demonstrated in co-intoxication studies that Stx1a reduces Stx2a mediated toxicity in vivo particularly when more Stx1a than Stx2a is administered (Russo et al., 2016). We also recently found that the presence of Stx1a reduces the morbidity in Str-treated mice after infection with an STEC strain that makes both Stx1a and Stx2a (Petro et al., 2019).
In our subset of strains, we found that Stx2c did not contribute to virulence, because deletion of stx2a in all of the stx2a stx2c isolates resulted in at least a 1,000-fold reduction of toxicity in vitro and complete attenuation in mice. The finding that Stx2c is produced at low levels in some isolates coincide with previous observations (Strauch et al., 2004; Ogura et al., 2015). However, there are other strains that do produce enough Stx2c to be moderately pathogenic in Str-treated mice (Lindgren et al., 1994). We were able to induce Stx2c in vivo in the stx2a phage-cured strain of JH2010 after Cip-treatment of the mice (data not shown). All the strains lysogenized by an stx2c-phage shared 100% sequence identity to stx2c bacteriophage 2851, which is the phage considered to be the progenitor phage for this subtype (Strauch et al., 2004). We also note that stx2a in JH2012 occupies the argW site, a site usually occupied by stx2a in strains that are also lysogenized by an stx2c-phage in the sbcB site. One possibility for this unexpected finding is that JH2012 lost an stx2c phage.
In this study we initially evaluated factors, such as stx subtypes, stx-phage insertion sites, and virulence in Str-treated mice for nine clinical isolates to determine whether these factors could be correlated with severity of human disease. We were unable to make such a correlation, perhaps due to the relatively low number of strains in our study. It might be possible to correlate the stx subtypes and stx-phage insertion sites of O157:H7 strains to the clinical disease manifestation if a large collection of complete whole genome sequences became available for isolates with corresponding clinical data.
However, we did find that the level of increase in cytotoxicity from the strains in response to growth in Cip, or after infection in Str-treated mice varied among the isolates. Furthermore, we demonstrated that the best predictor of virulence in the mice was high levels of Stx2a in the stool. In contrast, one study in humans found that Stx in stool at day 4 or later illness manifestation was inversely correlated with disease (Cornick et al., 2002); however, those authors also suggested that toxin levels were likely high early in infection and then was absorbed. We found that lower levels of Stx2a in stool led to delayed virulence or avirulence in mice. It may be that at lower levels of Stx2a, host factors or other bacterial factors play a role in virulence, and such variables likely also have roles in human disease. However, our results demonstrate that the level of in vivo of induction of the stx2-phage contributes to the severity of disease in mice.
Materials and Methods
Bacterial Strains, Plasmids, and Culture Growth Conditions
All strains and plasmids used in this study are described in Table 2. E. coli O157:H7 strains isolated from patients who developed HUS (n = 3), bloody diarrhea (n = 3) or non-bloody diarrhea (n = 3) came from the CDC Escherichia and Shigella Reference Unit (Centers for Disease Control and Prevention, Atlanta, Ga) and were demonstrated by that group to be agglutinated by both anti-O157 and anti-H7 serum. Isolates were grown at 37°C on Luria-Bertani (LB) agar, Sorbitol-MacConkey (SMAC) agar, or in LB broth with aeration. Spontaneous Str-resistant (Strr) strains were isolated and used for all in vitro and animal studies. For antibiotics, unless stated otherwise, the following concentrations were used: ampicillin (Ap; 100 μg/ml); chloramphenicol (Cm; 30 μg/ml), ciprofloxacin (Cip; 5 ng/ml), Str (50 μg/ml), and tetracycline (Tc; 10 μg/ml).
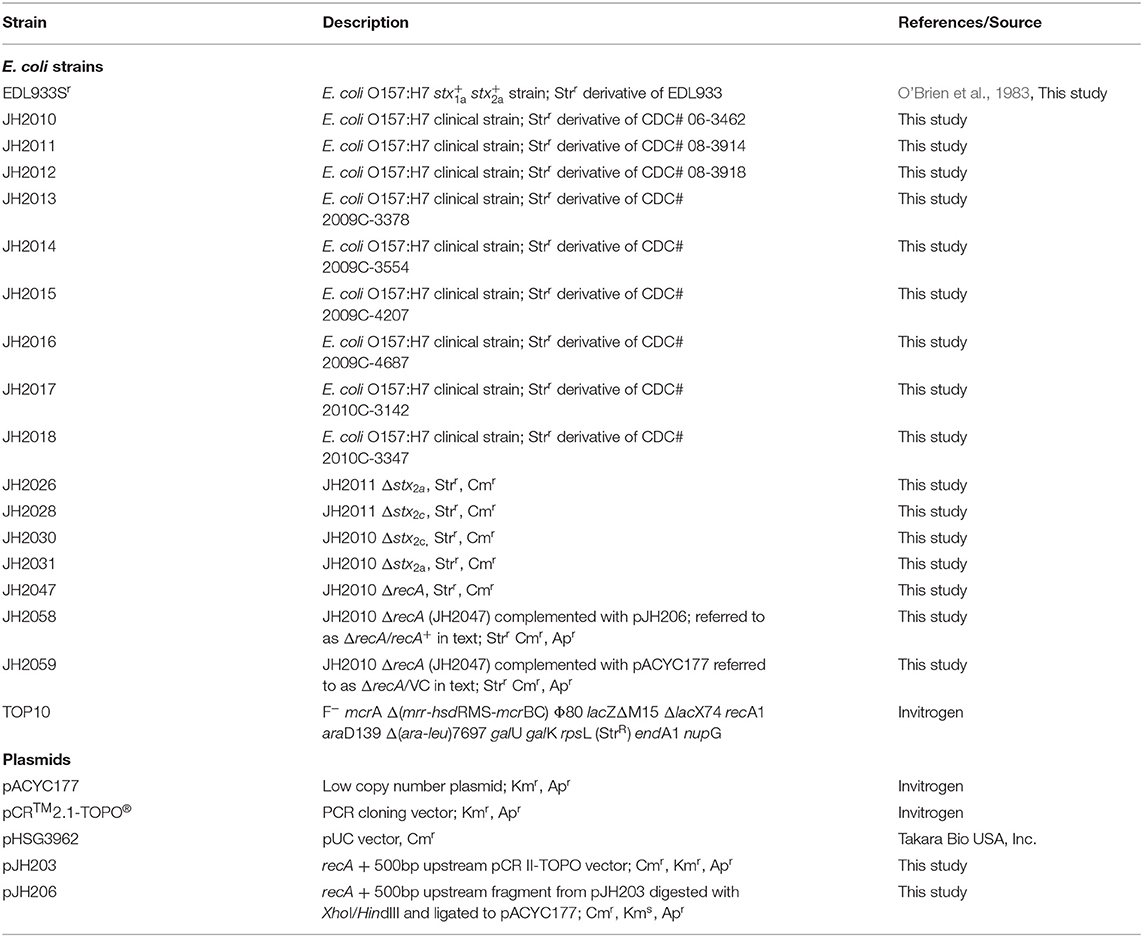
Table 2. Bacterial strains and plasmids used in this study.
stx Subtypes, Phage Insertions Sites, and Clade Determination
DNA was extracted with the Wizard® Genomic DNA Purification kit (Promega) according to manufacturer's protocol. PCR was used to determine the stx subtypes (Scheutz et al., 2012) and stx-phage chromosomal insertion sites (Scheutz et al., 2012; Bonanno et al., 2015) as described in those references. MG1655 was used as the negative control strain used for the stx-phage insertion sites. The positive control strains used for stx-phage insertion site determination were as follows: EDL933 (wrbA, yehV), EC4045 (sbcB, argW), and B2F1 (yecE). The phage insertion sites were confirmed by analysis of the whole genome sequence. Strains were assigned to clades based on known polymorphic SNPs previously described (Riordan et al., 2008).
Sequencing
Pacific BioSciences sequencing was completed as previously described (Lindsey et al., 2017). Briefly, E. coli genomic DNA was extracted according to the manufacturer's protocol (Archive Pure, 5 Prime, Gaithersburg, MD). The DNA was sheared to 20 kb fragments using needle shearing and Blue Pippin was used for size selection. DNA fragments were used to generate large SMRTbell™ libraries using the standard library protocols of the Pacific Biosciences DNA Template Preparation Kit (Menlo Park, CA). One SMRTcell was used to sequence each isolate. Finished libraries were bound to proprietary P6v2 polymerase and sequenced on a PacBio RSII sequencer using C4 chemistry for 360 min movies. Sequence reads were filtered and assembled de novo with the PacBio Hierarchical Genome Assembly Process version 3 and polished using Quiver (Chin et al., 2013). The same DNA extract for all isolates except for 08-3914 and 2009C-4687 were sequenced with an illumina MiSeq following manufacturers protocols (Illumina, USA). The PacBio sequences (except for 08-3914 and 2009C-4687) were Illumina corrected with unicycler_polish that uses Pilon (Walker et al., 2014; Wick et al., 2017).
Genomic analysis was done with CLC Genomics Workbench 9.5.3 (https://www.qiagenbioinformatics.com). Even though whole genome sequence was generated for each of the isolates, for strains 06-3462 (progenitor to JH2010) and 2009C-3378 (progenitor to JH2013), the stx2-phage sequences were not fully resolved. Virulence gene profiles of clinical strains were determined using Virulence finder (https://cge.cbs.dtu.dk/services/VirulenceFinder). MLST typing was assigned by uploading sequences for seven housekeeping genes (adk, fumC, gyrB, icd, mdh, purA, and recA) to The Enterobase MLST database at the University of Warwick [https://enterobase.warwick.ac.uk/species/ecoli/allele_st_search (Alikhan et al., 2018)].
Stx Mutations
Transformation of O157 Strains
The λ Red-mediated gene replacement protocol previously described was used for mutagenesis of O157:H7 strains (Murphy and Campellone, 2003). The Red + Gam-producing plasmid, pTP223, which confers Tc resistance, was electroporated into each isolate (2.5 V; 200 Ω) and plated on LB-Tc.
stx2 mutagenesis: For deletion of stx2, cat, which confers resistance to Cm, was amplified by PCR from pHSG3962 (linearized using HindIII/BamHI) with primers Stx2aCMF (5′- ATGAAGTGTATATTATTTAAATGGGTACTGTGCCTGTTACTGGGTTTTTCG
CACGTAAGAGGTTCCAACTTTCACCATAATG-3′) and Stx2aCMR (5′-TTCAGCAAATCCGGAGCCTGATTCACAGGTACTGGATTTGATTGT
GACAGtTACGCCCCGCCCTGCCACTCATC-3′) that contain 5′ and 3′ overhangs homologous to the first and last 50 base pairs of stx2. Strains with pTP223 that were grown overnight in LB-Tc were diluted 1/50 in LB-Tc with 10 mM IPTG and grown to OD600 ~0.2. The culture was placed on ice for 10 min, then washed and resuspended in 3-(N-Morpholino) propanesulfonic acid (MOPS) with 20% glycerol and made electrocompetent as mentioned above. Five or 10 μl of PCR product were electroporated into competent cells and incubated at 37°C with aeration in Super Optimal broth with Catabolite repression (S.O.C) media (Invitrogen) for 2 h or overnight. Transformants were selected for Cmr and screened for Tc-sensitivity to ensure that the recombineering plasmid was lost. PCR and southern blot were used to confirm deletion of stx2a or stx2c.
recA mutagenesis: recA in O157:H7 strain JH2010 was replaced with cat. cat with 5′ and 3′ overhangs homologous to the first and last 50 base pairs of recA was amplified by PCR from pHSG3962 with primers recAF3 (5′-TTAAAAATCTTCGTTAGTTTCTGCTACGCCTTCGCTATCATCT
ACAGAGAAATTACGCCCCGCCCTGCCACTCATC-3′) and recAR4 (5′-atgGCTATCGACGAAAACAAACAGAAAGCGTTGGCGGCAGCA
CTGGGCCAGCACGTAAGAGGTTCCAACTTTCACCATAATG-3′). The PCR product was electroporated into competent cells and Cmr transformants were selected and screened for Tc-sensitivity to ensure that the recombineering plasmid was lost. Two independent mutants were generated. Mutants were confirmed by PCR and Southern blot (not shown). recA mutants were complemented with recA expressed from its native promoter cloned into the low copy plasmid, pACYC177. The construct was generated by PCR amplification of recA including 500 bp upstream of the recA start site, which includes a hypothetical protein with primers recA_upF (5′-ATGGCTATCGACGAAAACAAAC-3′) and recAR5 (5′-GTATCAAACAAGACGATTAAAAATCTTCG-3′). The PCR fragment was cloned into pCR™2.1-TOPO® vector, digested using HindIII and XbaI, and then ligated to pACYC177. The resulting plasmid, pJH206, was transformed into the JH2010 ΔrecA mutant, and ApR colonies were selected. As a control, parent strains were transformed with an empty pACYC177 vector (denoted as ΔrecA/VC in the figure legends). For all assays, complement and vector control (VC) strains were grown in Ap-containing media.
Vero Cell Assay
Overnight cultures were diluted 1/500 into LB or LB ± Cip (5 ng/ml) and grown overnight. Cells were pelleted by centrifugation at 15,871 × g for 5 min. The supernatants were filter-sterilized with a 0.22 μm filter. The cell pellets were resuspended in an equal volume of water and subjected to three freeze-thaw cycles. The cell-associated fraction and supernatants were serial-diluted in Eagle's Minimal Essential Medium and 100 μl of each dilution was overlaid onto Vero cells that were seeded in microtiter plates 24 h previously. The Vero cell plates were incubated for 48 h at 37°C in 5% CO2, then the media was removed and the cells were then fixed in 10% formalin solution and stained with 0.13% crystal violet. The optical density was measured at 630 nm on BioTek (Winooski, VT, USA) EL800 spectrophotometric plate reader. The 50% cytotoxic dose (CD50) represented the amount of toxin required to kill 50% of the cells in a well.
Mouse Studies
All mouse studies were conducted in accordance with the recommendations of the Guide for the Care and Use of Laboratory Animals and were approved by the Institutional Animal Care and Use Committee of the Uniformed Services University. Male BALB/c mice (weight, 14 g) from Charles River Labs (Wilmington, MA) were used for all mouse experiments. Mice were fed drinking water containing Str (5 g/L) and fasted for ~18 h prior to infection. Mice were infected by oral gavage with 100 μl of ~1010−11 CFU bacteria or PBS. Food was returned after infection and Str water was continued for the remainder of the experiment.
Stool samples were suspended in PBS 1:10 (weight/volume) and homogenized. To determine colonization, supernatants from homogenized stool were serially diluted, plated on SMAC-Str, and incubated overnight at 37°C. Colonies were enumerated to determine the number of CFU/g feces. The limit of detection was 102 CFU. To determine cytotoxicity in vivo, stool homogenates were centrifuged for 10 min at 15,871 × g, the supernatants were collected, serially diluted and a Vero cell assay was done as described above. Mice were monitored for morbidity and mortality for 14 days post-infection. Infected mice were euthanized if they displayed two of the following morbidity symptoms: 25% weight loss, lethargy, ruffled fur, difficulty breathing, and/or difficulty moving.
Neutralization of Stx1 in Stool Samples
To determine the amount of Stx1a present in fecal samples, stool supernatants (as described above) from infected mice were diluted 1/5 in EMEM then mixed 1:1 with EMEM containing cαStx1 (100 μg/ml), a mouse-human chimeric monoclonal antibody against Stx1 (Edwards et al., 1998), then incubated for at 37°C in 5% CO2 for 2 h. Finally, 100 μl of toxin-antibody mixture was overlaid onto Vero cells and the cytotoxicity assay was completed as above.
Statistical Analyses
All statistical analyses were done with GraphPad Prism v7.03 for Windows software (GraphPad Software, San Diego, CA).
Accession Number(s)
The sequences for the nine clinical isolates used in this study are included in BioProject accession # PRJNA218110.
Data Availability Statement
The whole genome sequences for the nine O157:H7 isolates from the CDC described in this study are available through GenBank as part of BioProject PRJNA218110.
Ethics Statement
All mouse studies were conducted in accordance with the recommendations of the Guide for the Care and Use of Laboratory Animals and were approved by the Institutional Animal Care and Use Committee of the Uniformed Services University.
Author Contributions
JH, NS, AO'B, and AM-C conceived this study. JH, CP, and RA conducted the experiments. RL and NS were responsible for sequencing. JH and AM-C wrote the manuscript with the support of all authors.
Funding
This work was supported by National Institutes of Health grant R37 AI020148 to AO'B.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The opinions or assertions contained herein are the private ones of the authors and are not to be construed as official or reflecting the views of the Department of Defense, the Uniformed Services University of the Health Sciences, or the National Institutes of Health. We thank Dr. Cara Olsen for facilitation of statistical analyses.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fcimb.2020.00062/full#supplementary-material
References
Ake, J. A., Jelacic, S., Ciol, M. A., Watkins, S. L., Murray, K. F., Christie, D. L., et al. (2005). Relative nephroprotection during Escherichia coli O157:H7 infections: association with intravenous volume expansion. Pediatrics 115, e673–680. doi: 10.1542/peds.2004-2236
Alikhan, N. F., Zhou, Z., Sergeant, M. J., and Achtman, M. (2018). A genomic overview of the population structure of Salmonella. PLoS Genet. 14:e1007261. doi: 10.1371/journal.pgen.1007261
Amigo, N., Mercado, E., Bentancor, A., Singh, P., Vilte, D., Gerhardt, E., et al. (2015). Clade 8 and Clade 6 strains of Escherichia coli O157:H7 from cattle in Argentina have hypervirulent-like phenotypes. PLoS One 10:e0127710. doi: 10.1371/journal.pone.0127710
Bonanno, L., Loukiadis, E., Mariani-Kurkdjian, P., Oswald, E., Garnier, L., Michel, V., et al. (2015). Diversity of Shiga toxin-producing Escherichia coli (STEC) O26:H11 strains examined via stx subtypes and insertion sites of stx and espk bacteriophages. Appl. Environ. Microbiol. 81, 3712–3721. doi: 10.1128/AEM.00077-15
Bunger, J. C., Melton-Celsa, A. R., Maynard, E. L., and O'brien, A. D. (2015). Reduced toxicity of Shiga toxin (Stx) type 2c in mice compared to Stx2d is associated with instability of Stx2c holotoxin. Toxins (Basel) 7, 2306–2320. doi: 10.3390/toxins7062306
Chin, C. S., Alexander, D. H., Marks, P., Klammer, A. A., Drake, J., Heiner, C., et al. (2013). Nonhybrid, finished microbial genome assemblies from long-read SMRT sequencing data. Nat. Methods 10, 563–569. doi: 10.1038/nmeth.2474
Cornick, N. A., Jelacic, S., Ciol, M. A., and Tarr, P. I. (2002). Escherichia coli O157:H7 infections: discordance between filterable fecal Shiga toxin and disease outcome. J. Infect. Dis. 186, 57–63. doi: 10.1086/341295
Edwards, A. C., Melton-Celsa, A. R., Arbuthnott, K., Stinson, J. R., Schmitt, C. K., Wong, H. C., et al. (1998). “Vero cell neutralization and mouse protective efficacy of humanized monoclonal antibodies against Escherichia coli toxins stx1 and stx2,” in Escherichia Coli O157:H7 and Other Shiga Toxin-Producing E. Coli Strains, eds J. B. Kaper and A. D. O'Brien (Washington, DC: ASM Press), 388–392.
Eklund, M., Leino, K., and Siitonen, A. (2002). Clinical Escherichia coli strains carrying stx genes: stx variants and stx-positive virulence profiles. J. Clin. Microbiol. 40, 4585–4593. doi: 10.1128/JCM.40.12.4585-4593.2002
Freedman, S. B., Xie, J., Neufeld, M. S., Hamilton, W. L., Hartling, L., Tarr, P. I., et al. (2016). Shiga toxin-producing Escherichia coli infection, antibiotics, and risk of developing hemolytic uremic syndrome: a meta-analysis. Clin. Infect. Dis. 62, 1251–1258. doi: 10.1093/cid/ciw099
Friedrich, A. W., Bielaszewska, M., Zhang, W. L., Pulz, M., Kuczius, T., Ammon, A., et al. (2002). Escherichia coli harboring Shiga toxin 2 gene variants: frequency and association with clinical symptoms. J. Infect. Dis. 185, 74–84. doi: 10.1086/338115
Fuchs, S., Muhldorfer, I., Donohue-Rolfe, A., Kerenyi, M., Emody, L., Alexiev, R., et al. (1999). Influence of RecA on in vivo virulence and Shiga toxin 2 production in Escherichia coli pathogens. Microb. Pathog. 27, 13–23. doi: 10.1006/mpat.1999.0279
Hara-Kudo, Y., and Takatori, K. (2011). Contamination level and ingestion dose of foodborne pathogens associated with infections. Epidemiol. Infect. 139, 1505–1510. doi: 10.1017/S095026881000292X
Imamovic, L., and Muniesa, M. (2012). Characterizing RecA-independent induction of Shiga toxin 2-encoding phages by EDTA treatment. PLoS ONE 7:e32393. doi: 10.1371/journal.pone.0032393
Kallianpur, A. R., Bradford, Y., Mody, R. K., Garman, K. N., Comstock, N., Lathrop, S. L., et al. (2018). Genetic susceptibility to postdiarrheal hemolytic-uremic syndrome after Shiga toxin-producing Escherichia coli infection: a centers for disease control and prevention FoodNet study. J. Infect. Dis. 217, 1000–1010. doi: 10.1093/infdis/jix633
Lindgren, S. W., Samuel, J. E., Schmitt, C. K., and O'Brien, A. D. (1994). The specific activities of Shiga-like toxin type II (SLT-II) and SLT-II-related toxins of enterohemorrhagic Escherichia coli differ when measured by vero cell cytotoxicity but not by mouse lethality. Infect. Immun. 62, 623–631. doi: 10.1128/IAI.62.2.623-631.1994
Lindsey, R. L., Batra, D., Rowe, L., Vladimir, N.L., Juieng, P., Garcia-Toledo, L., et al. (2017). High-quality draft genome sequences for four drug-resistant or outbreak-associated Shigella sonnei strains generated with PacBio sequencing and whole-genome maps. Genome Announc. 5:e00906-17. doi: 10.1128/genomeA.00906-17
Manning, S. D., Motiwala, A. S., Springman, A. C., Qi, W., Lacher, D. W., Ouellette, L. M., et al. (2008). Variation in virulence among clades of Escherichia coli O157:H7 associated with disease outbreaks. Proc. Natl. Acad. Sci. U.S.A. 105, 4868–4873. doi: 10.1073/pnas.0710834105
Melton-Celsa, A. R. (2014). Shiga toxin (Stx) classification, structure, and function. Microbiol. Spectr. 2. doi: 10.1128/microbiolspec.EHEC-0024-2013
Murphy, K. C., and Campellone, K. G. (2003). Lambda red-mediated recombinogenic engineering of enterohemorrhagic and enteropathogenic. E. coli. BMC Mol. Biol. 4:11. doi: 10.1186/1471-2199-4-11
Neupane, M., Abu-Ali, G. S., Mitra, A., Lacher, D. W., Manning, S. D., and Riordan, J. T. (2011). Shiga toxin 2 overexpression in Escherichia coli O157:H7 strains associated with severe human disease. Microb. Pathog. 51, 466–470. doi: 10.1016/j.micpath.2011.07.009
O'Brien, A. O., Lively, T. A., Chen, M. E., Rothman, S. W., and Formal, S. B. (1983). Escherichia coli O157:H7 strains associated with haemorrhagic colitis in the United States produce a Shigella dysenteriae 1 (SHIGA) like cytotoxin. Lancet 1:702. doi: 10.1016/S0140-6736(83)91987-6
Ogura, Y., Mondal, S. I., Islam, M. R., Mako, T., Arisawa, K., Katsura, K., et al. (2015). The Shiga toxin 2 production level in enterohemorrhagic Escherichia coli O157:H7 is correlated with the subtypes of toxin-encoding phage. Sci. Rep. 5:16663. doi: 10.1038/srep16663
Petro, C. D., Trojnar, E., Sinclair, J., Liu, Z. M., Smith, M., O'brien, A. D., et al. (2019). Shiga toxin type 1a (Stx1a) reduces the toxicity of the more potent Stx2a in vivo and in vitro. Infect. Immun. 87:e00787–18. doi: 10.1128/IAI.00787-18
Riordan, J. T., Viswanath, S. B., Manning, S. D., and Whittam, T. S. (2008). Genetic differentiation of Escherichia coli O157:H7 clades associated with human disease by real-time PCR. J. Clin. Microbiol. 46, 2070–2073. doi: 10.1128/JCM.00203-08
Russo, L. M., Melton-Celsa, A. R., and O'brien, A. D. (2016). Shiga Toxin (Stx) type 1a reduces the oral toxicity of Stx type 2a. J. Infect. Dis. 213, 1271–1279. doi: 10.1093/infdis/jiv557
Scallan, E., Hoekstra, R. M., Angulo, F. J., Tauxe, R. V., Widdowson, M. A., Roy, S. L., et al. (2011). Foodborne illness acquired in the United States—major pathogens. Emerg. Infect. Dis. 17, 7–15. doi: 10.3201/eid1701.P11101
Scheutz, F., Teel, L. D., Beutin, L., Pierard, D., Buvens, G., Karch, H., et al. (2012). Multicenter evaluation of a sequence-based protocol for subtyping Shiga toxins and standardizing Stx nomenclature. J. Clin. Microbiol. 50, 2951–2963. doi: 10.1128/JCM.00860-12
Serra-Moreno, R., Jofre, J., and Muniesa, M. (2008). The CI repressors of Shiga toxin-converting prophages are involved in coinfection of Escherichia coli strains, which causes a down regulation in the production of Shiga toxin 2. J. Bacteriol. 190, 4722–4735. doi: 10.1128/JB.00069-08
Shaikh, N., and Tarr, P. I. (2003). Escherichia coli O157:H7 Shiga toxin-encoding bacteriophages: integrations, excisions, truncations, and evolutionary implications. J. Bacteriol. 185, 3596–3605. doi: 10.1128/JB.185.12.3596-3605.2003
Shimizu, T., Ohta, Y., and Noda, M. (2009). Shiga toxin 2 is specifically released from bacterial cells by two different mechanisms. Infect. Immun. 77, 2813–2823. doi: 10.1128/IAI.00060-09
Shringi, S., Schmidt, C., Katherine, K., Brayton, K. A., Hancock, D. D., and Besser, T. E. (2012). Carriage of stx2a differentiates clinical and bovine-biased strains of Escherichia coli O157. PLoS ONE 7:e51572. doi: 10.1371/journal.pone.0051572
Spinale, J. M., Ruebner, R. L., Copelovitch, L., and Kaplan, B. S. (2013). Long-term outcomes of Shiga toxin hemolytic uremic syndrome. Pediatr. Nephrol. 28, 2097–2105. doi: 10.1007/s00467-012-2383-6
Strauch, E., Schaudinn, C., and Beutin, L. (2004). First-time isolation and characterization of a bacteriophage encoding the Shiga toxin 2c variant, which is globally spread in strains of Escherichia coli O157. Infect. Immun. 72, 7030–7039. doi: 10.1128/IAI.72.12.7030-7039.2004
Tarr, G. A. M., Stokowski, T., Shringi, S., Tarr, P. I., Freedman, S. B., et al. (2019). Contribution and interaction of Shiga toxin genes to Escherichia coli O157:H7 virulence. Toxins 11:607. doi: 10.3390/toxins11100607
Tarr, P. I., Gordon, C. A., and Chandler, W. L. (2005). Shiga-toxin-producing Escherichia coli and haemolytic uraemic syndrome. Lancet 365, 1073–1086. doi: 10.1016/S0140-6736(05)71144-2
Tuttle, J., Gomez, T., Doyle, M. P., Wells, J. G., Zhao, T., Tauxe, R. V., et al. (1999). Lessons from a large outbreak of Escherichia coli O157:H7 infections: insights into the infectious dose and method of widespread contamination of hamburger patties. Epidemiol. Infect. 122, 185–192. doi: 10.1017/s0950268898001976
Tyler, J. S., Beeri, K., Reynolds, J. L., Alteri, C. J., Skinner, K. G., Friedman, J. H., et al. (2013). Prophage induction is enhanced and required for renal disease and lethality in an EHEC mouse model. PLoS Pathog. 9:e1003236. doi: 10.1371/journal.ppat.1003236
Uhlich, G. A., Sinclair, J. R., Warren, N. G., Chmielecki, W. A., and Fratamico, P. (2008). Characterization of Shiga toxin-producing Escherichia coli isolates associated with two multistate food-borne outbreaks that occurred in 2006. Appl. Environ. Microbiol. 74, 1268–1272. doi: 10.1128/AEM.01618-07
Walker, B. J., Abeel, T., Shea, T., Priest, M., Abouelliel, A., Sakthikumar, S., et al. (2014). Pilon: an integrated tool for comprehensive microbial variant detection and genome assembly improvement. PLoS ONE 9:e112963. doi: 10.1371/journal.pone.0112963
Wick, R. R., Judd, L. M., Gorrie, C. L., and Holt, K. E. (2017). Unicycler: resolving bacterial genome assemblies from short and long sequencing reads. PLoS Comput. Biol. 13:e1005595. doi: 10.1371/journal.pcbi.1005595
Yin, S., Rusconi, B., Sanjar, F., Goswami, K., Xiaoli, L., Eppinger, M., et al. (2015). Escherichia coli O157:H7 strains harbor at least three distinct sequence types of Shiga toxin 2a-converting phages. BMC Genomics 16:733. doi: 10.1186/s12864-015-1934-1
Keywords: Escherichia coli, O157:H7, Shiga toxin, Stx2, phage, recA, mouse model
Citation: Hauser JR, Atitkar RR, Petro CD, Lindsey RL, Strockbine N, O'Brien AD and Melton-Celsa AR (2020) The Virulence of Escherichia coli O157:H7 Isolates in Mice Depends on Shiga Toxin Type 2a (Stx2a)-Induction and High Levels of Stx2a in Stool. Front. Cell. Infect. Microbiol. 10:62. doi: 10.3389/fcimb.2020.00062
Received: 22 October 2019; Accepted: 07 February 2020;
Published: 26 February 2020.
Edited by:
Vernon L. Tesh, Texas A&M University, United StatesReviewed by:
Cheleste Thorpe, Tufts University School of Medicine, United StatesLisa Harrison Plemons, United States Food and Drug Administration, United States
Copyright © 2020 Hauser, Atitkar, Petro, Lindsey, Strockbine, O'Brien and Melton-Celsa. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Angela R. Melton-Celsa, YW5nZWxhLm1lbHRvbi1jZWxzYUB1c3Vocy5lZHU=