 R. K. Subbarao Malireddi
R. K. Subbarao Malireddi Sannula Kesavardhana
Sannula Kesavardhana Thirumala-Devi Kanneganti
Thirumala-Devi Kanneganti
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Cell. Infect. Microbiol., 26 November 2019
Sec. Microbes and Innate Immunity
Volume 9 - 2019 | https://doi.org/10.3389/fcimb.2019.00406
Cell death is central to development, organismal homeostasis, and immune responses. The cell death field has experienced tremendous progress by delineating the molecular programs specific to each of the apoptotic and inflammatory cell death pathways. Moreover, the discovery of the inflammasomes and pyroptosis and necroptosis pathway regulators have provided the genetic basis for the programmed inflammatory cell death pathways. Earlier research highlighted the unique regulation of each of these pathways, but emerging studies discovered co-regulation and crosstalk between these seemingly different cell death complexes. The progress in this area has led to an idea that master regulators play central roles in orchestrating multiple cell death pathways. Here, we provide a brief review of the master regulators, the innate immune sensor ZBP1 and the essential cell survival kinase TAK1, that play vital roles in the regulation of RIPK1/RIPK3–FADD–caspase-8 cell death complex assembly and its versatility in executing Pyroptosis, Apoptosis, and Necroptosis, which we dubbed here as PAN-optosis. Furthermore, we discuss the implications and therapeutic potential of targeting these master regulators in health and disease.
ZBP1 and TAK1 regulate PAN-optosis.
Cell death is fundamentally important for organismal development, homeostasis, and host response to infection. Several cell death pathways have been identified with specific genetically encoded requirements; these have collectively been named programmed cell death pathways (Bergsbaken et al., 2009; Lamkanfi and Dixit, 2010; Pasparakis and Vandenabeele, 2015; Man and Kanneganti, 2016). Apoptosis is the most well-studied form of cell death and was originally considered immunologically silent with major roles in the homeostatic development of multicellular organisms (Kerr et al., 1972; Green et al., 2009). Unlike apoptosis, pyroptosis and necroptosis are considered immunologically active due to their association with membrane lysis and extracellular release of damage-associated molecular patterns (DAMPs), resulting in amplification of inflammation. Receptor-interacting serine/threonine-protein kinase 1 (RIPK1) and RIPK3 play key roles in phosphorylation and activation of the protein mixed lineage kinase domain-like pseudokinase (MLKL) that forms pores in the membrane to execute necroptosis (Feng et al., 2007; Degterev et al., 2008; Cho et al., 2009; He et al., 2009; Zhang et al., 2009; Sun et al., 2012; Zhao et al., 2012; Green, 2019). It is now established that pyroptosis is executed by gasdermin D (GSDMD)-mediated membrane lysis as a result of cleavage by inflammatory caspases to release the functionally active N-terminal fragment (Kayagaki et al., 2015; Shi et al., 2015; Man and Kanneganti, 2016; Van Opdenbosch and Lamkanfi, 2019). The last decade has provided a wealth of information about the molecular details of the inflammasome and inflammatory caspase activation processes that promote pyroptosis and the release of the inflammatory cytokines interleukin (IL)-1 and IL-18; these findings are discussed in detail elsewhere (Kayagaki et al., 2011, 2013; Hagar et al., 2013; Man and Kanneganti, 2016; Van Opdenbosch and Lamkanfi, 2019). As our understanding of these cell death pathways increases, it is becoming clear that in addition to the unique regulation of each of these pathways, there is also significant co-regulation and crosstalk. In this review, we will discuss this co-regulation and the master regulators that play central roles in orchestrating these cell death pathways.
Cell death plays key roles in infection and immunity, as programmed cell death is often part of the host anti-microbial strategy (Man and Kanneganti, 2016; Jorgensen et al., 2017; Van Opdenbosch and Lamkanfi, 2019). The innate immune system recognizes microbial infections through its pattern recognition receptors (PRRs), which can induce a robust pro-inflammatory immune response and cell death (Lamkanfi and Dixit, 2010; Jorgensen et al., 2017). However, membrane-bound PRRs, such as Toll-like receptors (TLRs) and cytokine receptors such as tumor necrosis factor (TNF) receptor 1 (TNFR1), often show a bias toward induction of the inflammatory immune response through the NF-κB and MAPK pathways, despite having all the principal components required for the induction of cell death (Kawai and Akira, 2007; Li et al., 2010; Thapa et al., 2011; Kaiser et al., 2013; Peltzer et al., 2016; Ting and Bertrand, 2016). As a consequence, several microbes have evolved strategies to target these inflammatory mechanisms. In response, hosts have evolved sophisticated feedback mechanisms which are wired to sense the perturbations in these key nodes of their inflammatory signaling pathways (e.g., the NF-κB and MAPK pathways) and initiate the assembly of multifaceted cell death complexes to drive PAN-optosis to promote inflammation, immune responses, and protection against infection (Mukherjee et al., 2006; Paquette et al., 2012; Philip et al., 2014; Weng et al., 2014; Orning et al., 2018; Sarhan et al., 2018). Pathogen- or pharmacologically mediated obstruction of survival signaling acts as a key danger signal to trigger the assembly of PAN-optotic cell death complexes (Figures 1, 2). Alternatively, monogenic master sensors and regulators have evolved to integrate the complex signals coming from multiple ligands and stimuli associated with live microbial infections. These molecules act as critical sensors of specific microbial infections and as central hubs to trigger both multifaceted cell death in the form of PAN-optosis and inflammatory immune responses. Here we focus on two such key master regulators, Z-DNA binding protein 1 (ZBP1, also called DNA-dependent activator of IFN regulatory factor, DAI, and DLM1) and transforming growth factor beta-activated kinase 1 (TAK1, also called mitogen-activated protein kinase kinase kinase 7, MAP3K7).
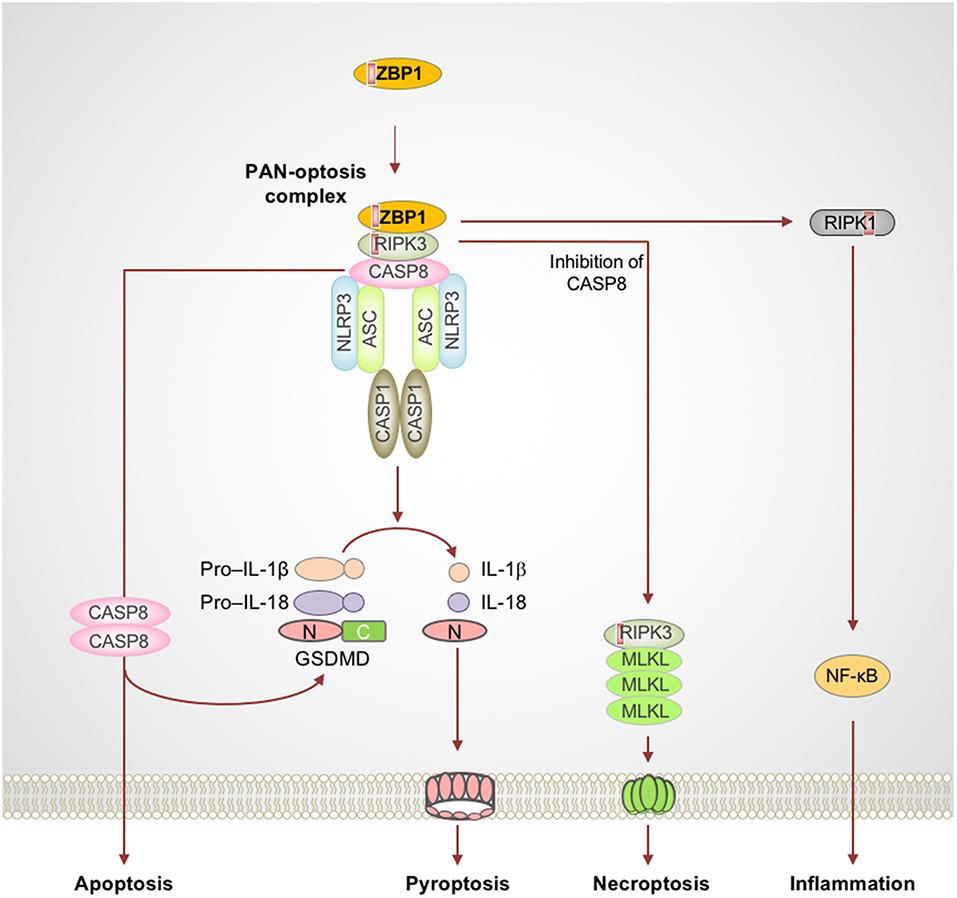
Figure 1. Activation of ZBP1 triggers assembly of signaling complexes to engage PAN-optosis. Z-DNA binding protein 1 (ZBP1) is an innate immune receptor that senses nucleic acids and activates PAN-optosis and inflammation. ZBP1 activation leads to its interaction with receptor-interacting serine/threonine-protein kinase 3 (RIPK3) and recruitment of caspase-8 (CASP8) to form cell death signaling scaffolds. This ZBP1-RIPK3-CASP8 complex engages nucleotide-binding oligomerization domain-like receptor family pyrin domain-containing 3 (NLRP3) inflammasome-dependent pyroptosis, CASP8-mediated apoptosis, and RIPK3-mixed lineage kinase domain-like pseudokinase (MLKL)–driven necroptosis. ZBP1 also induces RIPK1-driven NF-κB activation and inflammation in response to influenza infection. Red boxes within proteins represent the RIP homotypic interaction motif (RHIM) domain. ASC, apoptosis-associated speck-like protein containing a caspase recruitment domain; C, C-terminus; CASP1, caspase-1; GSDMD, gasdermin D; N, N-terminus.
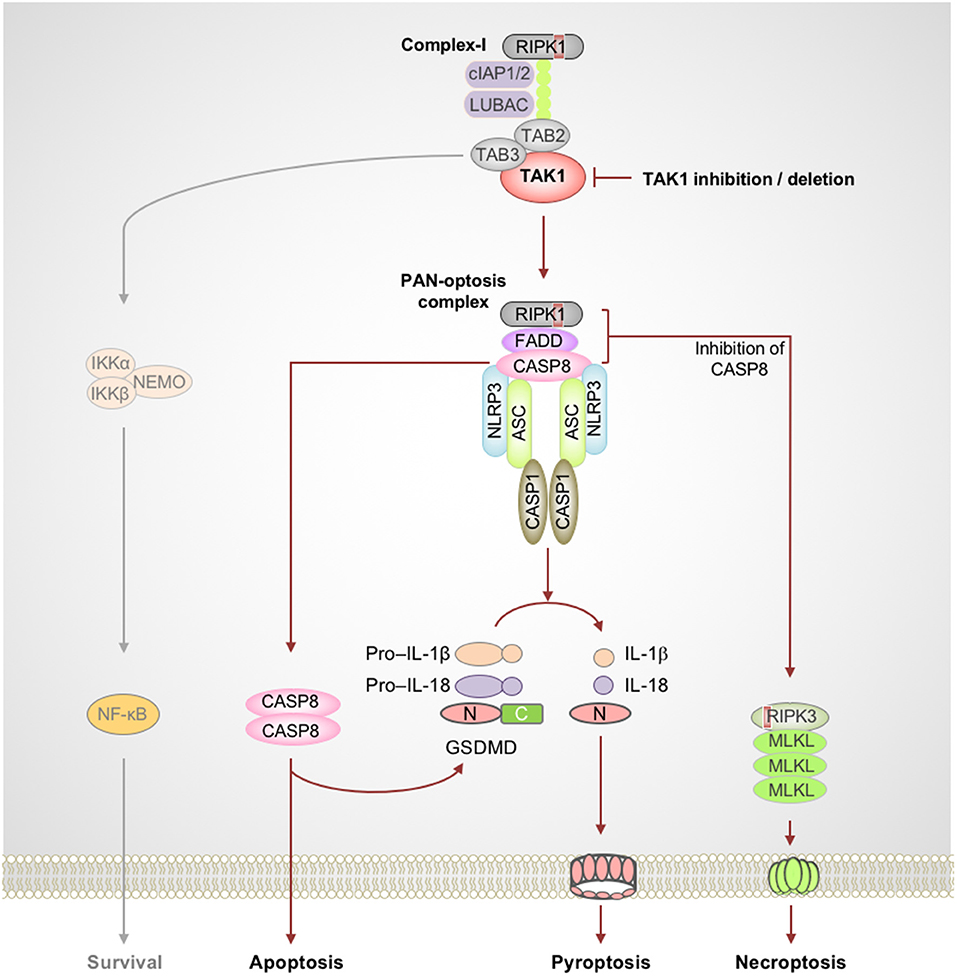
Figure 2. TAK1 acts as a master switch for PAN-optosis quiescence. Genetic deletion or microbial or pharmacological inactivation of transforming growth factor beta-activated kinase 1 (TAK1) function triggers receptor-interacting serine/threonine-protein kinase 1 (RIPK1)-dependent assembly of PAN-optotic cell death complexes. RIPK1 in association with FS7-associated cell surface antigen (Fas)-associated death domain (FADD) and caspase-8 (CASP8) triggers nucleotide-binding oligomerization domain-like receptor family pyrin domain-containing 3 (NLRP3) inflammasome-dependent and CASP8-mediated cleavage of gasdermin D (GSDMD) and the execution of pyroptosis. This complex also engages CASP8-mediated apoptosis, and inhibition of CASP8 activity promotes RIPK3-mixed lineage kinase domain-like pseudokinase (MLKL)–dependent necroptosis. Red boxes within proteins represent the RIP homotypic interaction motif (RHIM) domain. ASC, apoptosis-associated speck-like protein containing a caspase recruitment domain; C, C-terminus; CASP1, caspase-1; cIAP, cellular inhibitor of apoptosis protein; LUBAC, linear ubiquitin chain assembly complex; N, N-terminus; NEMO, NF-κB essential modulator; TAB, TAK1 binding protein.
Influenza A virus (IAV) is a single-stranded RNA virus which infects millions worldwide and poses a major threat to public health. Early reports provided the first evidence of a crucial role for nucleotide-binding oligomerization domain (NOD)-like receptor (NLR) family pyrin domain-containing 3 (NLRP3) in sensing IAV infection and dsRNAs to assemble the inflammasome complex via the adapter protein apoptosis-associated speck-like protein containing a caspase recruitment domain (ASC) and drive caspase-1–mediated inflammation (Kanneganti et al., 2006a,b). Furthermore, IAV-induced activation of the NLRP3 inflammasome controls viral spread and protects against lung damage, promoting host-protective immune responses (Allen et al., 2009; Thomas et al., 2009). While IAV RNA recognition is known to induce inflammation and cell death, the identity of innate sensors that mediate programmed inflammatory cell death and inflammation were not known for many years. ZBP1 was recently identified as an innate sensor of IAV infection (Kuriakose et al., 2016; Thapa et al., 2016; Kesavardhana et al., 2017). Upon sensing IAV infection, ZBP1 activation leads to NLRP3 inflammasome activation and PAN-optosis (Kuriakose et al., 2016; Kesavardhana et al., 2017). ZBP1 also critically regulates the IAV-induced pro-inflammatory cytokine response (Kuriakose et al., 2016; Kesavardhana et al., 2017).
ZBP1 is one of the RIP homotypic interaction motif (RHIM)-containing proteins and is known to activate necroptosis via RIPK3, another RHIM-containing protein (Rebsamen et al., 2009; Upton et al., 2012; Kuriakose and Kanneganti, 2018). ZBP1 is unique among cell death proteins since it contains a RHIM domain to mediate cell death and also a Zα domain which binds Z-nucleic acids (Rebsamen et al., 2009; Kuriakose and Kanneganti, 2018). Thus, ZBP1 not only engages cell death signaling but is also capable of sensing aberrant nucleic acids and instructing the assembly of cell death signaling scaffolds. Recent studies suggest that ZBP1 directly senses IAV infection and facilitates RIPK3 and caspase-8 activation (Kuriakose et al., 2016; Kesavardhana et al., 2017). ZBP1, in association with RIPK3 and caspase-8, promotes assembly of the signaling complexes that mediate PAN-optosis (Kuriakose et al., 2016; Kesavardhana et al., 2017; Kuriakose and Kanneganti, 2018) (Figure 1). Deletion of ZBP1 completely abolishes IAV-induced NLRP3 inflammasome activation and the release of leaderless cytokines. ZBP1-mediated PAN-optosis after IAV infection is dependent on the RIPK3–caspase-8 complex but independent of RIPK1. However, ZBP1 is essential for the RIPK1-dependent pro-inflammatory cytokine response during IAV infection, although ZBP1 and RIPK1 do not directly interact in endogenous conditions. In addition, ZBP1 mediates necroptosis and inflammation when the RIPK1 RHIM domain is mutated and causes perinatal lethality in Ripk1RHIM/RHIM mice (Lin et al., 2016; Newton et al., 2016). ZBP1 also promotes the activation of RIPK3 in the absence of RIPK1-RHIM function. These findings establish that, in physiological settings, RIPK1-RHIM function counteracts ZBP1-mediated cell death and inflammation for organismal homeostasis. Based on these findings, it is possible that a putative signaling complex might control RIPK1-mediated inhibition of ZBP1 function, independent of the canonical ripoptosome complex which contains RIPK1 as a core scaffolding protein.
ZBP1 activation requires upstream activation of other nucleic acid sensors. Recent studies suggest that recognition of IAV RNA by the RIG-I receptor initiates type I IFN/IFNAR signaling to license ZBP1 upregulation and activation (Kesavardhana et al., 2017). These studies found that ZBP1 senses the viral ribonucleoprotein complexes (vRNPs) of IAV that are generated during replication to activate cell death pathways. How ZBP1 is activated in the absence of RIPK1-RHIM function and what the endogenous nucleic acid ligands that activate ZBP1 are still remain as unexplored questions in this field.
The TNF family of cytokine receptors plays important roles in inflammation and cell death and shapes the nature of host innate and adaptive immune responses. Often, the default outcome of triggering these cell surface receptors is cell survival and cytokine production. In immune cells, the TNF-TNFR1 complex proximal to the membrane enables the formation of the RIPK1-containing complex-I which promotes prosurvival and inflammatory responses (Peltzer et al., 2016; Ting and Bertrand, 2016). The inactivation of essential complex-I components such as TAK1 results in the assembly of a cytosolic ripoptosome-like cell death complex with the potential to drive multifaceted cell death, PAN-optosis. The inhibitory phosphorylation of RIPK1 by TAK1 is critical to restrict its activation and blocks the spontaneous activation of PAN-optosis. The PAN-optotic death complex containing RIPK1, FS7-associated cell surface antigen (Fas)-associated death domain (FADD), and caspase-8 acts as the core and is assembled through their death domain (DD) interactions. This complex promotes FADD–caspase-8–dependent apoptosis through the activation of effector caspase-3 and -7 and promotes necroptosis by RIPK3-mediated phosphorylation of MLKL (Figure 2). Recent studies demonstrated that this ripoptosome-like complex also plays a key role in activation of the NLRP3 inflammasome and pyroptosis when TAK1 is inhibited (Malireddi et al., 2018, in press; Orning et al., 2018; Sarhan et al., 2018).
The essential role of TAK1 in innate immunity against microbes resulted in the evolution of microbial inhibitors that block its function to promote immune evasion (Mukherjee et al., 2006; Paquette et al., 2012; Philip et al., 2014; Weng et al., 2014; Malireddi et al., 2018; Orning et al., 2018; Sarhan et al., 2018). For example, the intracellular bacteria Yersinia has evolved to produce the toxin YopJ that inactivates TAK1 (Mukherjee et al., 2006; Paquette et al., 2012). However, feedback mechanisms in the host sense the perturbation of TAK1 signaling as an intracellular pathogenic insult and trigger RIPK1 kinase activity-dependent PAN-optosis (Malireddi et al., 2018). Furthermore, recent studies focused on pharmacological- or pathogen-based inhibition of TAK1 have revealed a direct role for caspase-8, which is activated in the RIPK1–FADD–caspase-8 complex, in driving GSDMD cleavage and pyroptosis, independent of caspase-1 (Orning et al., 2018; Sarhan et al., 2018). Additionally, it was shown that microbial priming bypasses the requirement for RIPK1 kinase activity to drive PAN-optosis when TAK1 is inactivated (Malireddi et al., in press). Together, these findings are particularly important since they demonstrate the versatility of this cell death complex and provide the proof-of-concept for the existence of the PAN-optotic complex. Moreover, mice deficient in components of the PAN-optotic complex were shown to be susceptible to Yersinia infection (Philip et al., 2014; Weng et al., 2014), demonstrating the functional importance of this cell death complex.
Recent research focused on inflammasomes and inflammatory caspases led to the discovery of extensive crosstalk between the apoptotic and inflammatory cell death pathways. Studies focused on the RHIM domain-containing proteins (RIPK1 and RIPK3) and their crosstalk with FADD–caspase-8–mediated pathways improved our understanding of the apoptotic and necroptotic pathways and their intricate regulatory mechanisms (Holler et al., 2000; He et al., 2009; Zhang et al., 2009; Gunther et al., 2011; Kaiser et al., 2011; Oberst et al., 2011; Peter, 2011; Welz et al., 2011; Wrighton, 2011). In parallel, several studies of innate immune receptors showed unique and overlapping roles in assembling a diverse array of caspase-activating inflammasomes and activating pyroptotic cell death (Kesavardhana and Kanneganti, 2017; Karki and Kanneganti, 2019). Of these, the NLRP3 inflammasome has emerged as an extremely versatile sensor of stress that responds to a wide range of microbial and damage-promoting cytotoxic insults (Kesavardhana and Kanneganti, 2017; Karki and Kanneganti, 2019). Generation and characterization of NLRP3-deficient mice provided the first concrete and genetic evidence for its importance in sensing bacterial and viral components and in the induction of inflammatory caspase-1 activation and maturation of pro–IL-1β and pro–IL-18 (Kanneganti et al., 2006a,b; Mariathasan et al., 2006; Sutterwala et al., 2006). More recently, studies have revealed an unexpected amount of crosstalk between pyroptosis and apoptosis and necroptosis. While early studies indicated that caspase-8 could contribute to inflammatory functions by driving maturation of inflammatory IL-1 cytokines (Maelfait et al., 2008; Bossaller et al., 2012; Gringhuis et al., 2012; Vince et al., 2012; Man et al., 2014), Gurung et al. provided the first definitive evidence for direct crosstalk between the apoptotic and pyroptotic components. This study demonstrated that FADD and caspase-8 are crucial for canonical and non-canonical inflammasome activation and inflammatory cell death (Gurung et al., 2014). Moreover, ASC, an important adapter for inflammasome assembly, was also shown to be recruited to the caspase-8–containing cell death complex (Van Opdenbosch et al., 2017; Lee et al., 2018). More recent studies focused on the innate immune sensor ZBP1 and the essential kinase TAK1 have further strengthened our understanding of the versatility of the RIPK1/RIPK3–FADD–caspase-8 cell death complex and provided a strong foundation for the emerging concept of PAN-optosis (Kuriakose et al., 2016; Malireddi et al., 2018; Orning et al., 2018; Sarhan et al., 2018) (Figures 1, 2). Furthermore, defects in TAK1-associated cell survival signaling molecules also trigger similar multifaceted cell death pathways and promote inflammatory immune responses (Gerlach et al., 2011; Vince et al., 2012; Dondelinger et al., 2013, 2015; Lawlor et al., 2015; Moriwaki et al., 2015; Peltzer et al., 2018; Zhang et al., 2019). This newly proposed concept of PAN-optosis should encourage further future attempts to expand our understanding of the co-regulation of multifaceted cell death complexes during cell death and inflammation and the relevance of this co-regulation to health and disease.
The studies focused on caspases, cell death, and inflammation have demonstrated that there is a constant competition between the host and microbes to exploit mechanisms of cell death and inflammation to optimize their own survival, and the outcome is not always beneficial to the host (Lamkanfi and Dixit, 2010; Malireddi and Kanneganti, 2013). Loss- or gain-of-function mutations in the key components of different cell death pathways result in dysregulated immune responses to infection or promote the development of debilitating inflammatory diseases. For example, liberation of constraints on ZBP1 function leads to autoinflammation and perinatal lethality in mice, indicating manifestations of PAN-optosis in pathophysiology and organismal development (Lin et al., 2016; Newton et al., 2016). Consistent with its role in driving robust cell death and inflammation, inhibition of TAK1 can lead to unwanted activation of PAN-optosis, predisposing an individual to the development of inflammatory diseases. In support of this concept, genetic deletion of TAK1 in mice was shown to result in spontaneous cell death and embryonic lethality (Sato et al., 2005; Shim et al., 2005). Moreover, mutations resulting in reduced TAK1 function in humans and mice lead to loss of immune homeostasis and myeloid-proliferation syndromes (Ajibade et al., 2012; Eftychi et al., 2012; Lamothe et al., 2012). Interestingly, an aging-associated reduction of TAK1 expression was shown to cooperate with other genetic risk factors to promote the RIPK1-dependent onset of neuroinflammation and the development of amyotrophic lateral sclerosis (ALS) in humans (Xu et al., 2018). Furthermore, loss of TAK1 in myeloid cells in mice results in hypersusceptibility to LPS shock and enhanced neutrophilic proliferation, supporting a crucial role for TAK1-mediated inhibition of PAN-optosis in preventing inflammation and maintaining immune homeostasis (Ajibade et al., 2012; Eftychi et al., 2012; Malireddi et al., 2018; Sanjo et al., 2019). These studies support that ZBP1 and TAK1 act as master regulators of multiple cell death pathways and are essential to control cellular homeostasis and PAN-optosis and to prevent inflammatory pathophysiology.
Similarly, genetic deficiency of the NF-κB and MAPK components suggests a role for the necrosome components RIPK1 and RIPK3 in promoting NLRP3 inflammasome activation, triggering multiple forms of cell death and the development of inflammatory diseases (Ikeda et al., 2011; Matmati et al., 2011; Tokunaga et al., 2011; Vince et al., 2012; Kumari et al., 2014; Rickard et al., 2014; Vande Walle et al., 2014; Gurung et al., 2015; Xu et al., 2018; Peltzer and Walczak, 2019; Polykratis et al., 2019; Yuan et al., 2019). Similarly, disease in Pstpip2cmo mice, which have a missense mutation in the proline-serine-threonine phosphatase-interacting protein 2 (Pstpip2) gene that results in debilitating inflammatory arthritis similar to that of patients with chronic multifocal osteomyelitis (cmo), is rescued when caspase-8 is inactivated in combination with either caspase-1 or the NLRP3 inflammasome (Ferguson et al., 2006; Grosse et al., 2006; Lukens et al., 2014; Gurung et al., 2016). These observations indicate that the inflammasome, caspase-1, and caspase-8 have redundant roles and potentially become activated in a multifaceted PAN-optotic complex to promote the inflammatory disease in Pstpip2cmo mice. Together, these findings support the convergent evolution and shared mechanism of PAN-optosis with potential implications in the development of inflammatory disease.
Recent work has resulted in tremendous progress in our understanding of cell death pathways with the discovery of novel regulators and the crosstalk between pyroptosis, apoptosis, and necroptosis. These studies also revealed the existence of master regulators such as ZBP1 and TAK1 that act as key signaling nodes for cell death. Recent progress in this area has also revealed an intimate connection between the RIPK1/RIPK3–FADD–caspase-8 signaling complex and the execution of PAN-optosis. It is clear that the PAN-optotic death complexes activated by the master regulators are multifaceted in nature with the potential to execute diverse forms of caspase-mediated cell death and promote necroptosis when caspases are inhibited. Together, these findings lay the foundation for the concept of master regulators controlling multifaceted cell death, PAN-optosis. Future studies should reveal additional mechanisms of PAN-optosis to better understand its evolutionary relevance and significance in health and disease. Further understanding of the master regulators of PAN-optosis may also allow us to develop superior therapeutic approaches to target cancer, infection, and inflammatory diseases.
RM, SK, and T–DK drafted and edited the review.
Research in the Kanneganti lab is supported by the National Institutes of Health grants CA163507, AR056296, AI124346, and AI101935 and by the American Lebanese Syrian Associated Charities. The funding agencies had no role in the analysis, decision to publish, or preparation of the manuscript.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The authors acknowledge many investigators in the field whose primary data could not be cited in this review because of space limitations. We would like to thank the members of the Kanneganti lab for their comments and suggestions and Rebecca Tweedell, Ph.D., for scientific editing.
Ajibade, A. A., Wang, Q., Cui, J., Zou, J., Xia, X., Wang, M., et al. (2012). TAK1 negatively regulates NF-kB and p38 MAP kinase activation in Gr-1+CD11b+ neutrophils. Immunity 36, 43–54. doi: 10.1016/j.immuni.2011.12.010
Allen, I. C., Scull, M. A., Moore, C. B., Holl, E. K., McElvania-TeKippe, E., Taxman, D. J., et al. (2009). The NLRP3 inflammasome mediates in vivo innate immunity to influenza a virus through recognition of viral RNA. Immunity 30, 556–565. doi: 10.1016/j.immuni.2009.02.005
Bergsbaken, T., Fink, S. L., and Cookson, B. T. (2009). Pyroptosis: host cell death and inflammation. Nat. Rev. Microbiol. 7, 99–109. doi: 10.1038/nrmicro2070
Bossaller, L., Chiang, P. I., Schmidt-Lauber, C., Ganesan, S., Kaiser, W. J., Rathinam, V. A., et al. (2012). Cutting edge: FAS (CD95) mediates noncanonical IL-1β and IL-18 maturation via caspase-8 in an RIP3-independent manner. J. Immunol. 189, 5508–5512. doi: 10.4049/jimmunol.1202121
Cho, Y. S., Challa, S., Moquin, D., Genga, R., Ray, T. D., Guildford, M., et al. (2009). Phosphorylation-driven assembly of the RIP1-RIP3 complex regulates programmed necrosis and virus-induced inflammation. Cell 137, 1112–1123. doi: 10.1016/j.cell.2009.05.037
Degterev, A., Hitomi, J., Germscheid, M. I. L., Ch'en Korkina, O., Teng, X., Abbott, D., et al. (2008). Identification of RIP1 kinase as a specific cellular target of necrostatins. Nat. Chem. Biol. 4, 313–321. doi: 10.1038/nchembio.83
Dondelinger, Y., Aguileta, M. A., Goossens, V., Dubuisson, C., Grootjans, S., Dejardin, E., et al. (2013). RIPK3 contributes to TNFR1-mediated RIPK1 kinase-dependent apoptosis in conditions of cIAP1/2 depletion or TAK1 kinase inhibition. Cell Death Differ. 20, 1381–1392. doi: 10.1038/cdd.2013.94
Dondelinger, Y., Jouan-Lanhouet, S., Divert, T., Theatre, E., Bertin, J., Gough, P. J., et al. (2015). NF-κB-independent roISle of IKKα/IKKβ in preventing RIPK1 kinase-dependent apoptotic and necroptotic cell death during TNF signaling. Mol. Cell 60, 63–76. doi: 10.1016/j.molcel.2015.07.032
Eftychi, C., Karagianni, N., Alexiou, M., Apostolaki, M., and Kollias, G. (2012). Myeloid TAKI [corrected] acts as a negative regulator of the LPS response and mediates resistance to endotoxemia. PLoS ONE 7:e31550. doi: 10.1371/annotation/ea1a4c80-8dfd-496a-a273-d74c1fd6e069
Feng, S., Yang, Y., Mei, Y., Ma, L., Zhu, D. E., Hoti, N., et al. (2007). Cleavage of RIP3 inactivates its caspase-independent apoptosis pathway by removal of kinase domain. Cell Signal. 19, 2056–2067. doi: 10.1016/j.cellsig.2007.05.016
Ferguson, P. J., Bing, X., Vasef, M. A., Ochoa, L. A., Mahgoub, A., Waldschmidt, T. J., et al. (2006). A missense mutation in pstpip2 is associated with the murine autoinflammatory disorder chronic multifocal osteomyelitis. Bone 38, 41–47. doi: 10.1016/j.bone.2005.07.009
Gerlach, B., Cordier, S. M., Schmukle, A. C., Emmerich, C. H., Rieser, E., Haas, T. L., et al. (2011). Linear ubiquitination prevents inflammation and regulates immune signalling. Nature 471, 591–596. doi: 10.1038/nature09816
Green, D. R. (2019). The coming decade of cell death research: five riddles. Cell 177, 1094–1107. doi: 10.1016/j.cell.2019.04.024
Green, D. R., Ferguson, T., Zitvogel, L., and Kroemer, G. (2009). Immunogenic and tolerogenic cell death. Nat. Rev. Immunol. 9, 353–363. doi: 10.1038/nri2545
Gringhuis, S. I., Kaptein, T. M., Wevers, B. A., Theelen, B. M., van der Vlist, M., Boekhout, T., and Geijtenbeek, T. B. (2012). Dectin-1 is an extracellular pathogen sensor for the induction and processing of IL-1β via a noncanonical caspase-8 inflammasome. Nat. Immunol. 13, 246–254. doi: 10.1038/ni.2222
Grosse, J., Chitu, V., Marquardt, A., Hanke, P., Schmittwolf, C., Zeitlmann, L., et al. (2006). Mutation of mouse Mayp/Pstpip2 causes a macrophage autoinflammatory disease. Blood 107, 3350–3358. doi: 10.1182/blood-2005-09-3556
Gunther, C., Martini, E., Wittkopf, N., Amann, K., Weigmann, B., Neumann, H., et al. (2011). Caspase-8 regulates TNF-α-induced epithelial necroptosis and terminal ileitis. Nature 477, 335–339. doi: 10.1038/nature10400
Gurung, P., Anand, P. K., Malireddi, R. K., Vande Walle, L., Van Opdenbosch, N., Dillon, C. P., et al. (2014). FADD and caspase-8 mediate priming and activation of the canonical and noncanonical Nlrp3 inflammasomes. J. Immunol. 192, 1835–1846. doi: 10.4049/jimmunol.1302839
Gurung, P., Burton, A., and Kanneganti, T. D. (2016). NLRP3 inflammasome plays a redundant role with caspase 8 to promote IL-1β-mediated osteomyelitis. Proc. Natl. Acad. Sci. U.S.A. 113, 4452–4457. doi: 10.1073/pnas.1601636113
Gurung, P., Lamkanfi, M., and Kanneganti, T. D. (2015). Cutting edge: SHARPIN is required for optimal NLRP3 inflammasome activation. J. Immunol. 194, 2064–2067. doi: 10.4049/jimmunol.1402951
Hagar, J. A., Powell, D. A., Aachoui, Y., Ernst, R. K., and Miao, E. A. (2013). Cytoplasmic LPS activates caspase-11: implications in TLR4-independent endotoxic shock. Science 341, 1250–1253. doi: 10.1126/science.1240988
He, S., Wang, L., Miao, L., Wang, T., Du, F., Zhao, L., et al. (2009). Receptor interacting protein kinase-3 determines cellular necrotic response to TNF-alpha. Cell 137, 1100–1111. doi: 10.1016/j.cell.2009.05.021
Holler, N., Zaru, R., Micheau, O., Thome, M., Attinger, A., Valitutti, S., et al. (2000). Fas triggers an alternative, caspase-8-independent cell death pathway using the kinase RIP as effector molecule. Nat. Immunol. 1, 489–495. doi: 10.1038/82732
Ikeda, F., Deribe, Y. L., Skanland, S. S., Stieglitz, B., Grabbe, C., Franz-Wachtel, M., et al. (2011). SHARPIN forms a linear ubiquitin ligase complex regulating NF-κB activity and apoptosis. Nature 471, 637–641. doi: 10.1038/nature09814
Jorgensen, I., Rayamajhi, M., and Miao, E. A. (2017). Programmed cell death as a defence against infection. Nat. Rev. Immunol. 17, 151–164. doi: 10.1038/nri.2016.147
Kaiser, W. J., Sridharan, H., Huang, C., Mandal, P., Upton, J. W., Gough, P. J., et al. (2013). Toll-like receptor 3-mediated necrosis via TRIF, RIP3, and MLKL. J. Biol. Chem. 288, 31268–31279. doi: 10.1074/jbc.M113.462341
Kaiser, W. J., Upton, J. W., Long, A. B., Livingston-Rosanoff, D., Daley-Bauer, L. P., Hakem, R., et al. (2011). RIP3 mediates the embryonic lethality of caspase-8-deficient mice. Nature 471, 368–372. doi: 10.1038/nature09857
Kanneganti, T. D., Body-Malapel, M., Amer, A., Park, J. H., Whitfield, J., Franchi, L., et al. (2006a). Critical role for0 Cryopyrin/Nalp3 in activation of caspase-1 in response to viral infection and double-stranded RNA. J. Biol. Chem. 281, 36560–36568. doi: 10.1074/jbc.M607594200
Kanneganti, T. D., Ozoren, N., Body-Malapel, M., Amer, A., Park, J. H., Franchi, L., et al. (2006b). Bacterial RNA and small antiviral compounds activate caspase-1 through cryopyrin/Nalp3. Nature 440, 233–236. doi: 10.1038/nature04517
Karki, R., and Kanneganti, T. D. (2019). Diverging inflammasome signals in tumorigenesis and potential targeting. Nat. Rev. Cancer 19, 197–214. doi: 10.1038/s41568-019-0123-y
Kawai, T., and Akira, S. (2007). Signaling to NF-κB by Toll-like receptors. Trends Mol. Med. 13, 460–469. doi: 10.1016/j.molmed.2007.09.002
Kayagaki, N., Stowe, I. B., Lee, B. L., O'Rourke, K., Anderson, K., Warming, S., et al. (2015). Caspase-11 cleaves gasdermin D for non-canonical inflammasome signalling. Nature 526, 666–671. doi: 10.1038/nature15541
Kayagaki, N., Warming, S., Lamkanfi, M., Vande Walle, L., Louie, S., Dong, J., et al. (2011). Non-canonical inflammasome activation targets caspase-11. Nature 479, 117–121. doi: 10.1038/nature10558
Kayagaki, N., Wong, M. T., Stowe, I. B., Ramani, S. R., Gonzalez, L. C., Akashi-Takamura, S., et al. (2013). Noncanonical inflammasome activation by intracellular LPS independent of TLR4. Science 341, 1246–1249. doi: 10.1126/science.1240248
Kerr, J. F., Wyllie, A. H., and Currie, A. R. (1972). Apoptosis: a basic biological phenomenon with wide-ranging implications in tissue kinetics. Br. J. Cancer 26, 239–257. doi: 10.1038/bjc.1972.33
Kesavardhana, S., and Kanneganti, T. D. (2017). Mechanisms governing inflammasome activation, assembly and pyroptosis induction. Int. Immunol. 29, 201–210. doi: 10.1093/intimm/dxx018
Kesavardhana, S., Kuriakose, T., Guy, C. S., Samir, P., Malireddi, R. K. S., Mishra, A., et al. (2017). ZBP1/DAI ubiquitination and sensing of influenza vRNPs activate programmed cell death. J. Exp. Med. 214, 2217–2229. doi: 10.1084/jem.20170550
Kumari, S., Redouane, Y., Lopez-Mosqueda, J., Shiraishi, R., Romanowska, M., Lutzmayer, S., et al. (2014). Sharpin prevents skin inflammation by inhibiting TNFR1-induced keratinocyte apoptosis. Elife 2:3. doi: 10.7554/eLife.03422.019
Kuriakose, T., and Kanneganti, T. D. (2018). ZBP1: innate sensor regulating cell death and inflammation. Trends Immunol. 39, 123–134. doi: 10.1016/j.it.2017.11.002
Kuriakose, T., Man, S. M., Malireddi, R. K., Karki, R., Kesavardhana, S., Place, D. E., et al. (2016). ZBP1/DAI is an innate sensor of influenza virus triggering the NLRP3 inflammasome and programmed cell death pathways. Sci. Immunol. 1:aag2045. doi: 10.1126/sciimmunol.aag2045
Lamkanfi, M., and Dixit, V. M. (2010). Manipulation of host cell death pathways during microbial infections. Cell Host Microbe. 8, 44–54. doi: 10.1016/j.chom.2010.06.007
Lamothe, B., Lai, Y., Hur, L., Orozco, N. M., Wang, J., Campos, A. D., et al. (2012). Deletion of TAK1 in the myeloid lineage results in the spontaneous development of myelomonocytic leukemia in mice. PLoS ONE 7:e51228. doi: 10.1371/journal.pone.0051228
Lawlor, K. E., Khan, N., Mildenhall, A., Gerlic, M., Croker, B. A., D'Cruz, A. A., et al. (2015). RIPK3 promotes cell death and NLRP3 inflammasome activation in the absence of MLKL. Nat. Commun. 6:6282. doi: 10.1038/ncomms7282
Lee, B. L., Mirrashidi, K. M., Stowe, I. B., Kummerfeld, S. K., Watanabe, C., Haley, B., et al. (2018). ASC- and caspase-8-dependent apoptotic pathway diverges from the NLRC4 inflammasome in macrophages. Sci. Rep. 8:3788. doi: 10.1038/s41598-018-21998-3
Li, X., Jiang, S., and Tapping, R. I. (2010). Toll-like receptor signaling in cell proliferation and survival. Cytokine 49, 1–9. doi: 10.1016/j.cyto.2009.08.010
Lin, J., Kumari, S., Kim, C., Van, T. M., Wachsmuth, L., Polykratis, A., et al. (2016). RIPK1 counteracts ZBP1-mediated necroptosis to inhibit inflammation. Nature 540, 124–128. doi: 10.1038/nature20558
Lukens, J. R., Gurung, P., Vogel, P., Johnson, G. R., Carter, R. A., McGoldrick, D. J., et al. (2014). Dietary modulation of the microbiome affects autoinflammatory disease. Nature 516, 246–249. doi: 10.1038/nature13788
Maelfait, J., Vercammen, E., Janssens, S., Schotte, P., Haegman, M., Magez, S., et al. (2008). Stimulation of Toll-like receptor 3 and 4 induces interleukin-1beta maturation by caspase-8. J. Exp. Med. 205, 1967–1973. doi: 10.1084/jem.20071632
Malireddi, R. K., and Kanneganti, T. D. (2013). Role of type I interferons in inflammasome activation, cell death, and disease during microbial infection. Front. Cell. Infect. Microbiol. 3:77. doi: 10.3389/fcimb.2013.00077
Malireddi, R. K. S., Gurung, P., Kesavardhana, S., Samir, P., Burton, A., Mummareddy, H., et al. (in press). Innate immune priming in the absence of TAK1 drives RIPK1 kinase activity-independent pyroptosis, apoptosis, necroptosis, and inflammatory disease. J. Exp. Med.
Malireddi, R. K. S., Gurung, P., Mavuluri, J., Dasari, T. K., Klco, J. M., Chi, H., et al. (2018). TAK1 restricts spontaneous NLRP3 activation and cell death to control myeloid proliferation. J. Exp. Med. 215, 1023–1034. doi: 10.1084/jem.20171922
Man, S. M., Hopkins, L. J., Nugent, E., Cox, S., Gluck, I. M., Tourlomousis, P., et al. (2014). Inflammasome activation causes dual recruitment of NLRC4 and NLRP3 to the same macromolecular complex. Proc. Natl. Acad. Sci. U.S.A. 111, 7403–7408. doi: 10.1073/pnas.1402911111
Man, S. M., and Kanneganti, T. D. (2016). Converging roles of caspases in inflammasome activation, cell death and innate immunity. Nat. Rev. Immunol. 16, 7–21. doi: 10.1038/nri.2015.7
Mariathasan, S., Weiss, D. S., Newton, K., McBride, J., O'Rourke, K., Roose-Girma, M., et al. (2006). Cryopyrin activates the inflammasome in response to toxins and ATP. Nature 440, 228–232. doi: 10.1038/nature04515
Matmati, M., Jacques, P., Maelfait, J., Verheugen, E., Kool, M., Sze, M., et al. (2011). A20 (TNFAIP3) deficiency in myeloid cells triggers erosive polyarthritis resembling rheumatoid arthritis. Nat. Genet. 43, 908–912. doi: 10.1038/ng.874
Moriwaki, K., Bertin, J., Gough, P. J., and Chan, F. K. (2015). A RIPK3-caspase 8 complex mediates atypical pro-IL-1β processing. J. Immunol. 194, 1938–1944. doi: 10.4049/jimmunol.1402167
Mukherjee, S., Keitany, G., Li, Y., Wang, Y., Ball, H. L., Goldsmith, E. J., et al. (2006). Yersinia YopJ acetylates and inhibits kinase activation by blocking phosphorylation. Science 312, 1211–1214. doi: 10.1126/science.1126867
Newton, K., Wickliffe, K. E., Maltzman, A., Dugger, D. L., Strasser, A., Pham, V. C., et al. (2016). RIPK1 inhibits ZBP1-driven necroptosis during development. Nature 540, 129–133. doi: 10.1038/nature20559
Oberst, A., Dillon, C. P., Weinlich, R., McCormick, L. L., Fitzgerald, P., Pop, C., et al. (2011). Catalytic activity of the caspase-8-FLIP(L) complex inhibits RIPK3-dependent necrosis. Nature 471, 363–367. doi: 10.1038/nature09852
Orning, P., Weng, D., Starheim, K., Ratner, D., Best, Z., Lee, B., et al. (2018). Pathogen blockade of TAK1 triggers caspase-8-dependent cleavage of gasdermin D and cell death. Science 362, 1064–1069. doi: 10.1126/science.aau2818
Paquette, N., Conlon, J., Sweet, C., Rus, F., Wilson, L., Pereira, A., et al. (2012). Serine/threonine acetylation of TGFbeta-activated kinase (TAK1) by Yersinia pestis YopJ inhibits innate immune signaling. Proc. Natl. Acad. Sci. U.S.A. 109, 12710–12715. doi: 10.1073/pnas.1008203109
Pasparakis, M., and Vandenabeele, P. (2015). Necroptosis and its role in inflammation. Nature 517, 311–320. doi: 10.1038/nature14191
Peltzer, N., Darding, M., Montinaro, A., Draber, P., Draberova, H., Kupka, S., et al. (2018). LUBAC is essential for embryogenesis by preventing cell death and enabling haematopoiesis. Nature 557, 112–117. doi: 10.1038/s41586-018-0064-8
Peltzer, N., Darding, M., and Walczak, H. (2016). Holding RIPK1 on the ubiquitin leash in TNFR1 signaling. Trends Cell Biol. 26, 445–461. doi: 10.1016/j.tcb.2016.01.006
Peltzer, N., and Walczak, H. (2019). Cell death and inflammation - a vital but dangerous liaison. Trends Immunol. 40, 387–402. doi: 10.1016/j.it.2019.03.006
Peter, M. E. (2011). Programmed cell death: apoptosis meets necrosis. Nature 471, 310–312. doi: 10.1038/471310a
Philip, N. H., Dillon, C. P., Snyder, A. G., Fitzgerald, P., Wynosky-Dolfi, M. A., Zwack, E. E., et al. (2014). Caspase-8 mediates caspase-1 processing and innate immune defense in response to bacterial blockade of NF-kB and MAPK signaling. Proc. Natl. Acad. Sci. U.S.A. 111, 7385–7390. doi: 10.1073/pnas.1403252111
Polykratis, A., Martens, A., Eren, R. O., Shirasaki, Y., Yamagishi, M., Yamaguchi, Y., et al. (2019). A20 prevents inflammasome-dependent arthritis by inhibiting macrophage necroptosis through its ZnF7 ubiquitin-binding domain. Nat. Cell Biol. 21, 731–742. doi: 10.1038/s41556-019-0324-3
Rebsamen, M., Heinz, L. X., Meylan, E., Michallet, M. C., Schroder, K., Hofmann, K., et al. (2009). DAI/ZBP1 recruits RIP1 and RIP3 through RIP homotypic interaction motifs to activate NF-kappaB. EMBO Rep. 10, 916–922. doi: 10.1038/embor.2009.109
Rickard, J. A., Anderton, H., Etemadi, N., Nachbur, U., Darding, M., Peltzer, N., et al. (2014). TNFR1-dependent cell death drives inflammation in sharpin-deficient mice. Elife 2:3. doi: 10.7554/eLife.03464.016
Sanjo, H., Nakayama, J., Yoshizawa, T., Fehling, H. J., Akira, S., and Taki, S. (2019). Cutting edge: TAK1 safeguards macrophages against proinflammatory cell death. J. Immunol. 203, 783–788. doi: 10.4049/jimmunol.1900202
Sarhan, J., Liu, B. C., Muendlein, H. I., Li, P., Nilson, R., Tang, A. Y., et al. (2018). Caspase-8 induces cleavage of gasdermin D to elicit pyroptosis during Yersinia infection. Proc. Natl. Acad. Sci. U.S.A. 115, E10888–E10897. doi: 10.1073/pnas.1809548115
Sato, S., Sanjo, H., Takeda, K., Ninomiya-Tsuji, J., Yamamoto, M., Kawai, T., et al. (2005). Essential function for the kinase TAK1 in innate and adaptive immune responses. Nat. Immunol. 6, 1087–1095. doi: 10.1038/ni1255
Shi, J., Zhao, Y., Wang, K., Shi, X., Wang, Y., Huang, H., et al. (2015). Cleavage of GSDMD by inflammatory caspases determines pyroptotic cell death. Nature 526, 660–665. doi: 10.1038/nature15514
Shim, J. H., Xiao, C., Paschal, A. E., Bailey, S. T., Rao, P., Hayden, M. S., et al. (2005). TAK1, but not TAB1 or TAB2, plays an essential role in multiple signaling pathways in vivo. Genes Dev. 19, 2668–2681. doi: 10.1101/gad.1360605
Sun, L., Wang, H., Wang, Z., He, S., Chen, S., Liao, D., et al. (2012). Mixed lineage kinase domain-like protein mediates necrosis signaling downstream of RIP3 kinase. Cell 148, 213–227. doi: 10.1016/j.cell.2011.11.031
Sutterwala, F. S., Ogura, Y., Szczepanik, M., Lara-Tejero, M., Lichtenberger, G. S., Grant, E. P., et al. (2006). Critical role for NALP3/CIAS1/Cryopyrin in innate and adaptive immunity through its regulation of caspase-1. Immunity 24, 317–327. doi: 10.1016/j.immuni.2006.02.004
Thapa, R. J., Basagoudanavar, S. H., Nogusa, S., Irrinki, K., Mallilankaraman, K., Slifker, M. J., et al. (2011). NF-kappaB protects cells from gamma interferon-induced RIP1-dependent necroptosis. Mol. Cell. Biol. 31, 2934–2946. doi: 10.1128/MCB.05445-11
Thapa, R. J., Ingram, J. P., Ragan, K. B., Nogusa, S., Boyd, D. F., Benitez, A. A., et al. (2016). DAI senses influenza a virus genomic RNA and activates RIPK3-dependent cell death. Cell Host Microbe 20, 674–681. doi: 10.1016/j.chom.2016.09.014
Thomas, P. G., Dash, P., Aldridge, J. R Jr., Ellebedy, A. H., Reynolds, C., Funk, A. J., et al. (2009). The intracellular sensor NLRP3 mediates key innate and healing responses to influenza a virus via the regulation of caspase-1. Immunity 30, 566–575. doi: 10.1016/j.immuni.2009.02.006
Ting, A. T., and Bertrand, M. J. M. (2016). More to life than NF-κB in TNFR1 signaling. Trends Immunol. 37, 535–545. doi: 10.1016/j.it.2016.06.002
Tokunaga, F., Nakagawa, T., Nakahara, M., Saeki, Y., Taniguchi, M., Sakata, S., et al. (2011). SHARPIN is a component of the NF-κB-activating linear ubiquitin chain assembly complex. Nature 471, 633–636. doi: 10.1038/nature09815
Upton, J. W., Kaiser, W. J., and Mocarski, E. S. (2012). DAI/ZBP1/DLM-1 complexes with RIP3 to mediate virus-induced programmed necrosis that is targeted by murine cytomegalovirus vIRA. Cell Host Microbe 11, 290–297. doi: 10.1016/j.chom.2012.01.016
Van Opdenbosch, N., and Lamkanfi, M. (2019). Caspases in cell death, inflammation, and disease. Immunity 50, 1352–1364. doi: 10.1016/j.immuni.2019.05.020
Van Opdenbosch, N., Van Gorp, H., Verdonckt, M., Saavedra, P. H. V., de Vasconcelos, N. M., Gonçalves, A., et al. (2017). Caspase-1 engagement and TLR-induced c-flip expression suppress ASC/Caspase-8-dependent apoptosis by inflammasome sensors NLRP1b and NLRC4. Cell Rep. 21, 3427–3444. doi: 10.1016/j.celrep.2017.11.088
Vande Walle, L., Van Opdenbosch, N., Jacques, P., Fossoul, A., Verheugen, E., Vogel, P., et al. (2014). Negative regulation of the NLRP3 inflammasome by A20 protects against arthritis. Nature 512, 69–73. doi: 10.1038/nature13322
Vince, J. E., Wong, W. W., Gentle, I., Lawlor, K. E., Allam, R., O'Reilly, L., et al. (2012). Inhibitor of apoptosis proteins limit RIP3 kinase-dependent interleukin-1 activation. Immunity 36, 215–227. doi: 10.1016/j.immuni.2012.01.012
Welz, P. S., Wullaert, A., Vlantis, K., Kondylis, V., Fernandez-Majada, V., Ermolaeva, M., et al. (2011). FADD prevents RIP3-mediated epithelial cell necrosis and chronic intestinal inflammation. Nature 477, 330–334. doi: 10.1038/nature10273
Weng, D., Marty-Roix, R., Ganesan, S., Proulx, M. K., Vladimer, G. I., Kaiser, W. J., et al. (2014). Caspase-8 and RIP kinases regulate bacteria-induced innate immune responses and cell death. Proc. Natl. Acad. Sci. U.S.A. 111, 7391–7396. doi: 10.1073/pnas.1403477111
Wrighton, K. H. (2011). Cell death: a killer puts a stop on necroptosis. Nat. Rev. Mol. Cell Biol. 12, 279. doi: 10.1038/nrm3101
Xu, D., Jin, T., Zhu, H., Chen, H., Ofengeim, D., Zou, C., et al. (2018). TBK1 Suppresses RIPK1-driven apoptosis and inflammation during development and in aging. Cell 174, 1477–1491.e19. doi: 10.1016/j.cell.2018.07.041
Yuan, J., Amin, P., and Ofengeim, D. (2019). Necroptosis and RIPK1-mediated neuroinflammation in CNS diseases. Nat. Rev. Neurosci. 20, 19–33. doi: 10.1038/s41583-018-0093-1
Zhang, D. W., Shao, J., Lin, J., Zhang, N., Lu, B. J., Lin, S. C., et al. (2009). RIP3, an energy metabolism regulator that switches TNF-induced cell death from apoptosis to necrosis. Science 325, 332–336. doi: 10.1126/science.1172308
Zhang, J., Webster, J. D., Dugger, D. L., Goncharov, T., Roose-Girma, M., Hung, J., et al. (2019). Ubiquitin ligases cIAP1 and cIAP2 limit cell death to prevent inflammation. Cell Rep. 27, 2679–2689.e3. doi: 10.1016/j.celrep.2019.04.111
Keywords: caspase-1, gasdermin D, MLKL, inflammasome, infection, innate immunity, inflammation, TLR priming
Citation: Malireddi RKS, Kesavardhana S and Kanneganti T-D (2019) ZBP1 and TAK1: Master Regulators of NLRP3 Inflammasome/Pyroptosis, Apoptosis, and Necroptosis (PAN-optosis). Front. Cell. Infect. Microbiol. 9:406. doi: 10.3389/fcimb.2019.00406
Received: 29 October 2019; Accepted: 12 November 2019;
Published: 26 November 2019.
Edited by:
Yousef Abu Kwaik, University of Louisville, United StatesReviewed by:
Dmitry M. Shayakhmetov, Emory University, United StatesCopyright © 2019 Malireddi, Kesavardhana and Kanneganti. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Thirumala-Devi Kanneganti, dGhpcnVtYWxhLWRldmkua2FubmVnYW50aUBzdGp1ZGUub3Jn
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.