 Ying Fan1,2
Ying Fan1,2 Sumana Sanyal
Sumana Sanyal Roberto Bruzzone
Roberto Bruzzone- 1HKU-Pasteur Research Pole, LKS Faculty of Medicine, School of Public Health, The University of Hong Kong, Hong Kong, Hong Kong
- 2MRC Protein Phosphorylation and Ubiquitylation Unit, School of Life Sciences, University of Dundee, Dundee, United Kingdom
- 3LKS Faculty of Medicine, School of Biomedical Sciences, The University of Hong Kong, Hong Kong, Hong Kong
- 4Department of Cell Biology and Infection, Institut Pasteur, Paris, France
Interactions between the host and viruses during the course of their co-evolution have not only shaped cellular function and the immune system, but also the counter measures employed by viruses. Relatively small genomes and high replication rates allow viruses to accumulate mutations and continuously present the host with new challenges. It is therefore, no surprise that they either escape detection or modulate host physiology, often by redirecting normal cellular pathways to their own advantage. Viruses utilize a diverse array of strategies and molecular targets to subvert host cellular processes, while evading detection. These include cell-cycle regulation, major histocompatibility complex-restricted antigen presentation, intracellular protein transport, apoptosis, cytokine-mediated signaling, and humoral immune responses. Moreover, viruses routinely manipulate the host cell cycle to create a favorable environment for replication, largely by deregulating cell cycle checkpoints. This review focuses on our current understanding of the molecular aspects of cell cycle regulation that are often targeted by viruses. Further study of their interactions should provide fundamental insights into cell cycle regulation and improve our ability to exploit these viruses.
Introduction
The study of host-pathogen interaction and the search for host factors that are crucial for virus life cycle is critical to the understanding of viral pathogenesis, and may shed light on the development of therapeutic approaches. Viruses are obligate intracellular parasites that constantly evolve strategies to subvert their host cellular environment. As survival of viruses depend on the ability to replicate in living organisms, it is not surprising that they are able to arrest or promote cell cycle progression, depending on the purpose, to their advantage. Viruses that are associated with malignancies, such as human T-cell leukemia virus type I (HTLV-1), Epstein-Barr virus (EBV), human papilloma virus (HPV), and Kaposi's sarcoma-associated herpesvirus (KSHV), have long been known to antagonize cell cycle checkpoints for neoplastic transformation of infected cells (Dyson et al., 1989; Moore and Chang, 1998; Knight and Robertson, 2004; Iwanaga et al., 2008). Thus, Simian virus 40 (SV40) and HPV promote entry of host cell cycle into the S phase as viral DNA replication depends on the host DNA replication machinery (Lehman et al., 2000; Banerjee et al., 2011). In contrast, human immunodeficiency virus type 1 (HIV-1) infection of T lymphocytes results in a cell cycle arrest in G2, suggesting that HIV-1 may disrupt T-cell-mediated immune response by preventing its clonal expansion (Zimmerman et al., 2006). The avian coronavirus infectious bronchitis virus (IBV) also promotes favorable conditions for viral protein synthesis and, hence, progeny virus production, by inducing cell cycle G2/M phase arrest of virus-infected cells (Dove et al., 2006).
The investigation of mechanisms viruses employ to hijack host cell cycle is, therefore, of significance not only for insights into the intracellular viral lifecycle, but also for development of strategies to counteract viral infection. Viruses associated with human cancer have been the focus of scores of studies that have led to the identification of cellular networks regulating the cell cycle, including the discovery of oncogenes and tumor suppressor genes (Bishop, 1991; Weinberg, 1991). While discussion of their manipulation of the cell cycle is inevitable, oncogenic viruses will not be the focus of this article and the reader is referred to a number of excellent reviews on this topic (Weinberg, 1997; zur Hausen, 2001; Chang et al., 2017). Rather, we summarize and discuss here current concepts of the mechanistic features that non-oncogenic viruses adopt to modify host cell cycle machinery.
Overview of the Cell Cycle
Cell cycle, or cell-division cycle, is a highly regulated process during which a cell undergoes duplication and division leading to the generation of two daughter cells. The eukaryotic cell cycle is generally divided into four stages: gap 1 phase (G1), synthesis phase (S), gap 2 phase (G2), and mitotic phase (M). In between M and G1, a cell may enter a quiescent state called gap 0 phase (G0), during which cells are neither dividing nor preparing to divide.
G1 is the phase during which metabolic changes take place to prepare the cell for division. Events that take place in G1 include growth in cell size as well as mRNA and protein synthesis. Initiation of G1 phase requires growth factor stimulation and a continuous supply of growth factor until the cell cycle passes through the restriction point (Pardee, 1974) in G1, after which cells are committed to DNA synthesis to complete the cell cycle and become extracellular growth factor-independent throughout the remainder of the cell cycle (Zetterberg et al., 1995). The retinoblastoma (Rb) protein is the guardian of restriction point (Bartek et al., 1996). When conditions are propitious for cell division without warning signals such as DNA damage or metabolic disturbances, Rb undergoes phosphorylation and become functionally inactivated, permitting the cell to proceed into late G1 (Weinberg, 1995). DNA synthesis takes place in S phase, generating exactly two identical sister chromosomes. G2 phase is a period of rapid cell growth and protein synthesis during which cells get ready for mitosis. DNA damage and replication checkpoint exists in G2 to monitor if chromosome replication is successfully completed and whether DNA damage occurs during the process (Hartwell and Weinert, 1989). This checkpoint determines the fate of a cell, either to enter mitosis or to undergo growth arrest for DNA repair. Mitosis is the process during which eukaryotic sister chromatid get separated to generate two nuclei (Pines and Rieder, 2001). A metaphase checkpoint detects whether all chromosomes are properly attached to mitotic spindles (spindle checkpoint), to ensure equal separation of sister chromatid (Gorbsky, 1995). Cell division is completed by cytokinesis, which segregates the nuclei, cytoplasm, organelles, and cell membrane into two genetically identical daughter cells (Straight and Field, 2000; Glotzer, 2001). Thus, cell cycle is a highly regulated process where multiple checkpoints determine whether to continue or abort a division.
The initiation of specific events during cell cycle, such as preparation for cell division, DNA replication, nuclear membrane breakdown, spindle formation and chromosome segregation, is triggered by a series of protein complexes that are activated in a concerted fashion. The protein complexes, consisting of cyclin and cyclin dependent kinase (CDK), constitute the cell cycle engine, which is driven in a precise sequence with different cyclin-CDK partners activated at specific points of the cell cycle. Mammalian cells accommodate multiple cell cycle-regulatory cyclin and CDKs throughout the cell cycle machinery probably to fine-tune the flexibility of cell cycle control (Nigg, 1995).
After mammalian cells are released from the quiescent state, the first cyclin-CDK holoenzyme known to be activated is composed of a D-type cyclin and either CDK4 or CDK6 depending on the cell type (Matsushime et al., 1994). Cyclin D functions as growth factor sensor, the expression of which is stimulated by growth factor and is independent of the state of the cell cycle (Sherr, 1993). Knowledge of the role of D-type cyclin in G1 progression was obtained from the observation that microinjection of anti-cyclin D1 antibodies into fibroblasts that were in G1 phase prevent them from entering into S phase (Baldin et al., 1993). On the contrary, overexpression of cyclin D1 shortens the duration of G1 phase in mouse fibroblasts, demonstrating cyclin D1 is a rate limiting factor for G1 progression (Quelle et al., 1993). Assembly of cyclin D-CDK4/6 complexes facilitates the phosphorylation of CDK4/6 by a CDK-activating kinase (CAK), which is required for the phosphorylation and enzymatic activation of CDKs (Kato et al., 1994; Kaldis et al., 1998). CAK is composed of three subunits, CDK7, cyclin H and MAT1 (ménage à trois) (Devault et al., 1995). Specifically, phosphorylation of CDKs at Thr161/160/172 by CAK is essential for stabilization of CDK/cyclin complexes by inducing a conformational change that increases the flexibility of the T-loops of all CDKs tested and enables access of the cyclin substrate (Matsuoka et al., 1994; Brown et al., 1999; Morris et al., 2002).
Activated cyclin D-CDK4/6 complexes phosphorylate Rb, which gets further phosphorylated by cyclin E-CDK2 complex at the mid-to-late G1 phase. Hyperphosphorylation inactivates Rb and leads to the release of transcription factor E2F, which activates the transcription of other cell cycle promoting genes, from the blocking effect imposed by hypophosphorylated Rb (Ewen et al., 1993; Weinberg, 1995). From this time on, cell cycle becomes independent of growth factor stimulation for continued cell cycle progression.
Further progression into S phase is driven by cyclin E-CDK2, and thereafter cyclin A-CDK2 promotes the completion of S phase. The precise role cyclin-CDK complexes play in the control of DNA replication is poorly defined. It appears that cyclin E-CDK2 is involved in centrosome duplication (Lacey et al., 1999), and CDK2 is necessary for DNA replication in a cell-free system (Krude et al., 1997). Following DNA synthesis, cells enter into G2 phase and CDK1 is believed to catalyze events in G2/M phase through sequential association with cyclin A and cyclin B. It is reported that the complexing of both cyclin A-CDK2 in late S phase and cyclin A-CDK1 in G2/M phase inactivate anaphase promoting complex/cyclosome (APC/C) to ensure accumulation of key mitotic regulators (Mitra et al., 2006).
Maturation/M phase-promoting factor (MPF) refers to the universal inducer of entry into M phase in eukaryotic cell. Cyclin B-CDK1 is one major component of the MPF complex (Hunt, 1989; Nurse, 1990). Purified cyclin B-CDK1 can induce meiotic G2/M transition upon injection into immature oocytes (Lohka et al., 1988). Proteins that regulate chromosomal condensation, formation of mitotic spindles, and fragmentation of the Golgi apparatus become the substrates of CDK1 during M phase, underscoring the importance of cyclin B-CDK1 complex (Lowe et al., 1998).
Study with CDK or cyclin knockout mice challenged the traditional orderly scheme of cell cycle progression described above. Mice survive in the absence of individual interface CDKs (Rane et al., 1999; Tsutsui et al., 1999; Ye et al., 2001; Berthet et al., 2003; Malumbres et al., 2004), and similar results have been obtained on ablation of cyclin D and cyclin E (Geng et al., 2003; Kozar et al., 2004). It has been shown that CDK4 and CDK6 partner only D-type cyclins, whereas both CDK1 and CDK2 show promiscuity in their choice of cyclin partners and exhibit binding ability with cyclin A, B, D, and E (Aleem et al., 2005; Petri et al., 2007; Santamaría et al., 2007). Thus, a minimal threshold model of cell cycle control has emerged, which suggests that pairing of cyclin A to either CDK1 or CDK2 is sufficient to trigger G1/S transition, whereas cyclin B-CDK1 activity seems to be required for mitosis.
Cell Cycle Subversion in Viral Infection
Viruses keep evolving strategies to subvert the cellular environment of the host for replication and survival. For example, some viral infections induce cell cycle arrest in lymphocytes to inhibit the clonal expansion of anti-viral T or B lymphocytes as a way of immune evasion, whereas carcinogenic viruses are known to antagonize cell cycle checkpoints for neoplastic transformation of infected cells. Thus, elucidating mechanisms of virus-induced deregulation of host cell cycle bears both fundamental and translational relevance.
Subversion by Protein-Protein Interaction (Table 1, Figure 1)
Normal cellular function including signal transduction and metabolic processes is often mediated through protein-protein interactions, and are routinely targeted by viruses. The host cell cycle is no exception, as illustrated below.
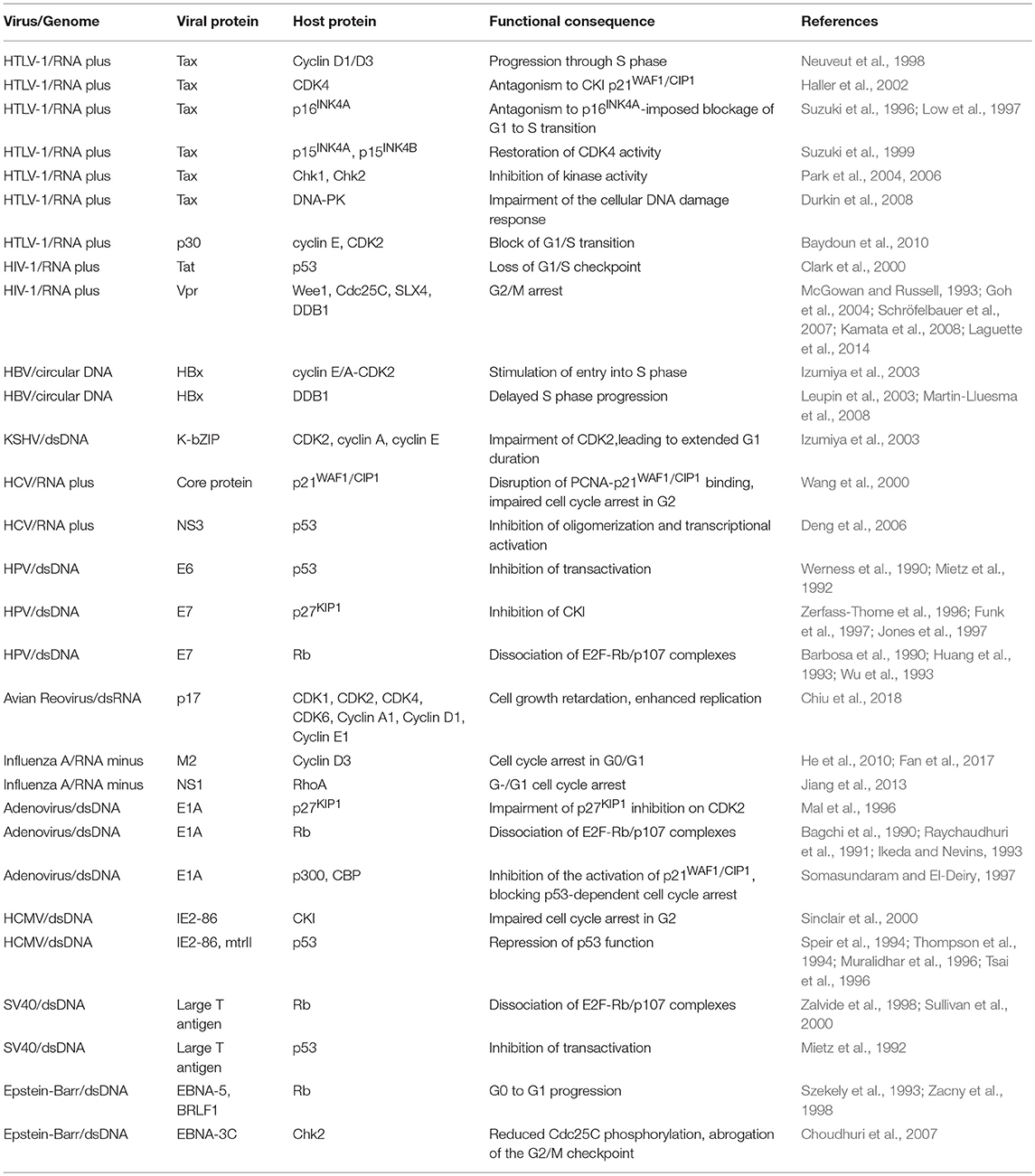
Table 1. Cell cycle subversion by protein-protein interaction.
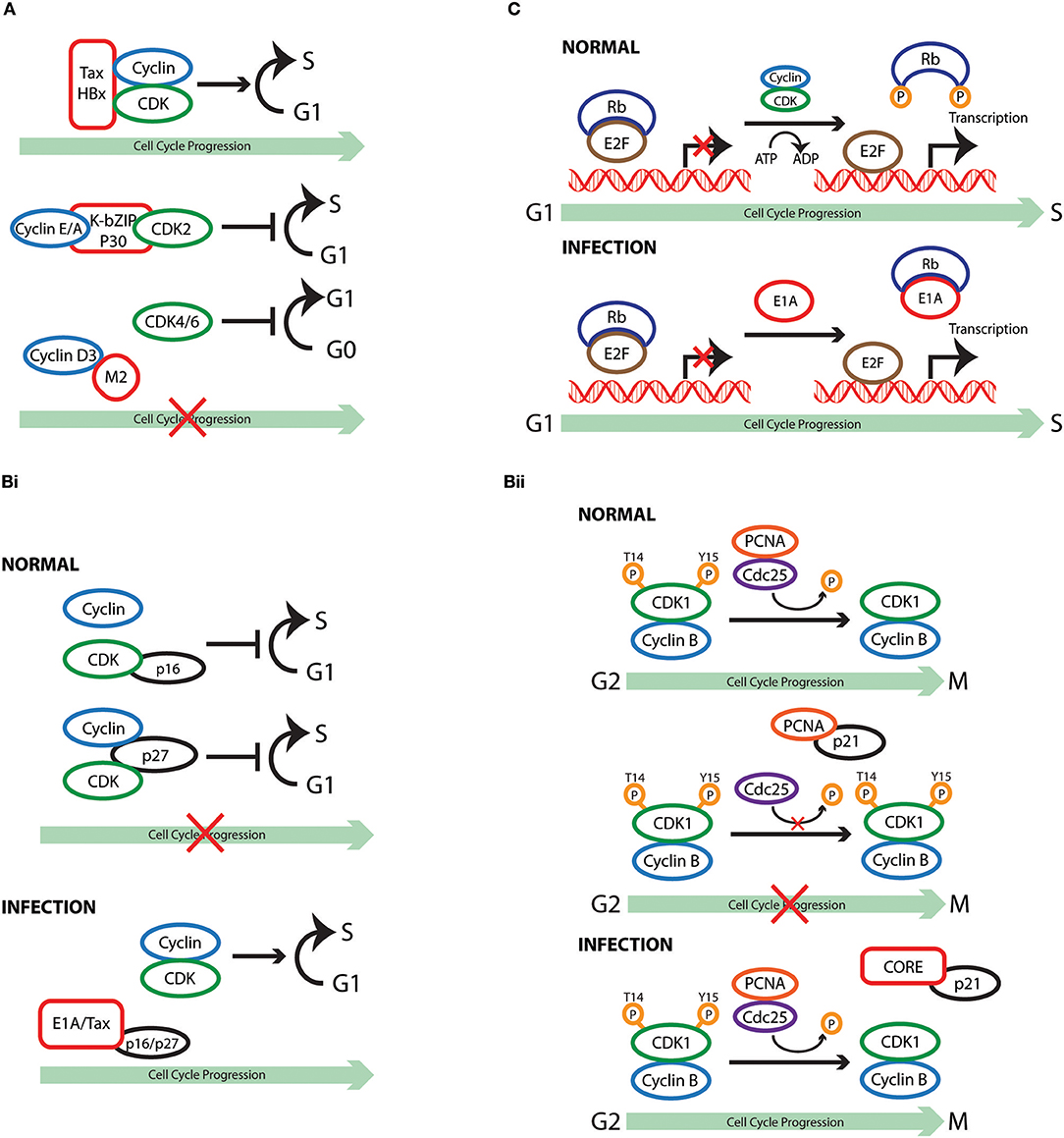
Figure 1. Cell cycle subversion by protein-protein interaction. (A) Binding of Tax and HBx proteins to cyclin and/or CDK promotes cell cycle progression either by enhancing kinase activity and/or weakening the inhibitory effect of CKI (Benn and Schneider, 1995; Neuveut et al., 1998; Haller et al., 2002). Direct binding of either K-bZIP protein of KSHV or p30 of HTLV-1 to both CDK2 and cyclin E leads to extended G1 duration or block of G1/S transition (Izumiya et al., 2003; Baydoun et al., 2010). Similarly, direct interaction of M2 of influenza A virus with cyclin D3 arrested cell cycle in G0/G1 phase (Fan et al., 2017). (Bi) Binding of E1A to p27KIP1 blocks its inhibition on CDK2 kinase activity, overcoming cell cycle arrest in late G1 phase (Mal et al., 1996). In addition to its association with cyclins, HTLV-1 Tax is also able to interact with p16INK4A and relieve p16INK4A-imposed blockage of G1 to S transition (Suzuki et al., 1996; Low et al., 1997). (Bii) p21WAF1/CIP1 can block the interaction between Cdc25C and proliferating cell nuclear antigen (PCNA) by competing with Cdc25C for PCNA binding. This observation points to a role of p21WAF1/CIP1 in G2 cell cycle arrest upon DNA damage (Ando et al., 2001). Competition between core protein of hepatitis C virus and PCNA for the association with p21WAF1/CIP1 may disrupt PCNA-p21WAF1/CIP1 binding, leading to impaired cell cycle arrest in G2 and DNA repair in response to damage signals (Wang et al., 2000). (C) E1A protein of adenovirus functions to dissociate E2F-Rb/p107 complexes owing to the interaction of its two conserved regions with Rb (Bagchi et al., 1990; Raychaudhuri et al., 1991). The release of E2F, therefore, transcriptionally activates various downstream target genes that are required for proliferation and DNA synthesis, including c-myc and cyclin E (Roussel et al., 1994; Ohtani et al., 1995).
Cyclin or CDK
Cyclin and CDK are components that drive the cell cycle clock by forming an active protein complex that could be directly targeted by viruses to modulate cell cycle progression. Binding of the oncoprotein Tax of HTLV-1 to cyclin D1/D3 was observed in transfected cells by co-immunoprecipitation (Neuveut et al., 1998). This interaction is implicated in the ability of Tax to promote progression through S phase, as cells expressing a cyclin D3deficient in Tax-binding exhibit much lesser thymidine incorporation (Neuveut et al., 1998). Interestingly, Haller et al. later demonstrated that Tax could also stimulate CDK4 activity through direct physical interaction via its N-terminal region (Haller et al., 2002). A binding-deficient Tax variant failed to stimulate CDK4-cyclin D complex formation and lost its ability to antagonize the inhibitory effect of cyclin-dependent kinase inhibitor (CKI) p21WAF1/CIP1, underlying the importance of protein interaction in Tax-mediated cell cycle regulation (Haller et al., 2002). Chronic hepatitis B virus (HBV) infection is associated with the development of hepatocellular carcinoma, and the X protein of HBV (HBx) was found to stimulate S phase entry of growth-arrested cells (Benn and Schneider, 1995). Co-immunoprecipitation in HBx-expressing cells demonstrated that this viral protein binds to cyclin E/A-CDK2 complexes. Mutant HBx, which displayed disrupted interaction with cyclin-CDK complexes, failed to increase CDK2 activity and, similarly to Tax, lost its effectiveness in counteracting the action ofp27KIP1 (Mukherji et al., 2007).
The examples of Tax and Hobs illustrate how binding of viral protein to cyclin or CDK promotes cell cycle progression either by enhancing kinase activity and/or weakening the inhibitory effect of CKI. The interaction of viral protein with cyclin or CDK could also results in hampered cell division. A direct binding of the K-bZIP protein of KSHV to CDK2, cyclin A, or cyclin E has been demonstrated using purified proteins (Izumiya et al., 2003). These interactions were found to impair the kinase activity of CDK2, leading to extended G1 duration, which is accompanied by increased production of the negative regulators p21WAF1/CIP1 and p27KIP1 (Izumiya et al., 2003). Interestingly p30, another HTLV-1 expressing protein, blocks G1/S transition in contrast to the neoplastic effect of Tax. CyclinE-CDK2 complex formation is hindered in p30 expressing cells due to the interaction of p30 with both cyclin E and CDK2, leading to reduced phosphorylation of the Rb tumor suppressor protein (Baydoun et al., 2010). Infection of influenza A virus (IAV) has been reported to induce host cell cycle G0/G1 phase accumulation (He et al., 2010; Fan et al., 2017). A yeast two-hybrid screen using the cytoplasmic tail of matrix protein 2 (M2) from the highly pathogenic H5N1 strain revealed a high-score interaction with cyclin D3. The M2 ion channel protein is a multifunctional protein with a highly conserved sequence among IAV isolates that approaches 95% identity in some regions (Le and Leluk, 2011; Pielak and Chou, 2011). The physical interaction between M2 and cyclin D3 was confirmed in virus-infected cells, was accompanied by cyclin D3 relocalization and degradation, and resulted in host cell cycle arrest in the G0/G1 phase (Fan et al., 2017). The interaction of viral protein with host cell cycle machinery is not always favorable for viral life cycle. Using a combination of small interfering RNA (siRNA) mediated genetic analysis it has become clear that cyclin D3 restricts IAV production independent of its role in cell cycle. The biogenesis of viral particles involves proper packaging and assembly of viral proteins and RNA to form intact virions at discrete budding sites. M2 is a multifunctional protein: its cytosolic tail interacts with M1, promoting the recruitment of viral internal proteins and viral RNA to the plasma membrane for efficient virus assembly (Chen et al., 2008). The inhibitory effect of cyclin D3 on IAV infection is due to its interaction with viral M2 protein, as measured by competitive co-immunoprecipitation assays, indicating that cyclin D3-M2 interaction either masks the domain on M2 mediating binding to M1, or has a higher affinity than that between M1 and M2. Consequently, limited M1-M2 binding during the budding process of IAV infection results in less progeny virions being efficiently packaged and released from host cells (Fan et al., 2017). These observations suggest a novel function of cyclin D3 that is beyond its classical function in cell cycle regulation. Hence, in this armed-race between cyclin D3 and M2 during IAV infection, the virus evolved strategies to counteract the inhibition imposed by cyclinD3, resulting in hampered cell cycle progression. Most recently, Chiu et al. identified a negatively charged pocket, formed by several conserved acidic residues, which is required for the binding of p17 protein of avian reovirus (ARV) to CDK1 (Chiu et al., 2018). Through GST-pull down assay and in vitro kinase assay, the authors showed that p17 competes with cyclin B1 for CDK1 binding and this direct interaction of p17 with CDK1 inhibits the kinase activity of the latter, thus preventing cells from entering mitosis and enhancing virus replication (Chiu et al., 2016, 2018). Unlike its exquisite binding to CDK1 but not cyclin B1, p17 could also suppress CDK2 and CKD4 kinase activities by direct binding to CDKs, partner cyclins, and CDK/cyclin complexes (Chiu et al., 2018).
CKI
Cyclin-dependent kinase inhibitors are a family of proteins that, acting through separate pathways, determine in cooperation with cyclins and CDKs the decision of the cell to progress through the cell cycle. The adenovirus early region 1A (E1A) protein displays the ability to bind multiple host factors for the manipulation of cell cycle progression. Binding of E1A to p27KIP1 blocks its inhibitiononCDK2 kinase activity, overcoming transforming growth factor (TGF)-β-induced cell cycle arrest in late G1 phase (Mal et al., 1996). This provided the first evidence that a viral oncoprotein could manipulate the cell cycle by counteracting an inhibitor of CDKs. In addition to its association with cyclins, HTLV-1 Tax is also able to interact with p16INK4A and relieve p16INK4A-imposed blockage of G1 to S transition, demonstrated by the finding that binding-deficient Tax could not protect CDK4 from the inhibitory effect of p16INK4A (Suzuki et al., 1996; Low et al., 1997). Suzuki et al. found that the co-precipitation of p16INK4A by CDK4 was drastically reduced in the presence of Tax, providing direct proof that Tax activates CDK4 by disabling the formation of p16INK4A-CDK4 complex (Suzuki et al., 1996). In a similar manner, Tax restores CDK4 activity through interaction with p15INK4B, further contributing to the immortalization of T cells (Suzuki et al., 1999). Interestingly, while Tax directly antagonizes p16INK4A and p15INK4B through protein-protein interaction, it utilizes a different mechanism to repress p18INK4C transcription and further promote cell transformation (Suzuki et al., 1999).
It was reported that p21WAF1/CIP1 could block the interaction between cell division cycle 25C (Cdc25C), a phosphatase of CDK1 that activates the CDK1/cyclin B1 complex (Strausfeld et al., 1991), and proliferating cell nuclear antigen (PCNA) by competing with Cdc25C for PCNA binding. This observation points to a role of p21WAF1/CIP1 in G2 cell cycle arrest upon DNA damage (Ando et al., 2001). Formation of a complex between the core protein of hepatitis C virus (HCV), which plays an important role in the development of hepatocellular carcinomas, and p21WAF1/CIP1 was mapped to the C-terminus of this CKI, a region in the close vicinity of the PCNA binding site of p21WAF1/CIP1 (Wang et al., 2000). In vitro experiments revealed that a competition exists between core protein and PCNA for the association with p21WAF1/CIP1 (Wang et al., 2000). It is plausible to speculate that, in the case of HCV infection, expression of core protein may disrupt PCNA-p21WAF1/CIP1 binding, leading to impaired cell cycle arrest in G2 and DNA repair in response to damage signals. Other examples of viral protein-mediated inhibition of CKIs include the E7 oncoprotein of HPV-16 and the immediate-early (IE) protein IE2-86 of human cytomegalovirus (HCMV) (Zerfass-Thome et al., 1996; Funk et al., 1997; Jones et al., 1997; Sinclair et al., 2000).
Rb/Pocket Proteins or E2Fs
Growth factors are necessary to drive the cell cycle machinery to the restriction point, beyond which commitment to cell cycle progression occurs and the cell enters S phase without requiring extracellular mitogenic signals. Retinoblastoma (Rb) protein, which serves as the guardian of restriction point, represents a non-redundant checkpoint that can be targeted by viruses to modulate host cell cycle under growth limiting conditions (Blagosklonny and Pardee, 2002). For example, the adenovirus E1A not only associates with CKI, but also functions to dissociate E2F-Rb/p107 complexes owing to the interaction of its two conserved regions (CRs) with Rb (Bagchi et al., 1990; Raychaudhuri et al., 1991). It was proposed that the LXCXE motif-containing CR2 of E1A mediates its initial binding to Rb, allowing CR1 to block the sequences on Rb that are involved in E2F recognition by means of physical interaction (Ikeda and Nevins, 1993). The release of E2F, therefore, transcriptionally activates various downstream target genes that are required for proliferation and DNA synthesis, including c-myc and cyclin E (Roussel et al., 1994; Ohtani et al., 1995). Similarly, HPV E7 possesses two homology regions of CR1 and CR2, which contribute to the transformation properties of E7 through disruption of E2F-Rb complexes (Barbosa et al., 1990; Huang et al., 1993; Wu et al., 1993). The association of the SV40 large T-antigen (L-Tag), which is expressed early during infection and is essential for viral replication, with Rb, p107, and p130 is attributed to the viral LXCXE residues, which are required but not sufficient in L-Tag-mediated transformation (DeCaprio et al., 1988; Chen and Paucha, 1990; Thompson et al., 1990; Stubdal et al., 1996). It was reported that the N-terminus of L-Tag, which shares sequence homology with the J domain of heat shock protein 40 (HSP40), is also required to disrupt the complex formation between the three “pocket” proteins of the Rb family with E2F (Zalvide et al., 1998; Sullivan et al., 2000).
The nuclear antigen 5 (EBNA-5), also known as nuclear antigen leader protein (EBNA-LP), of Epstein-Barr virus (EBV) cooperates with its nuclear antigen 2 (EBNA-2) to prompt G0 to G1 progression during the immortalization of virus-infected cells (Sinclair et al., 1994). Unlike the E1A of adenovirus, E7 of HPV, and L-Tag of SV40, EBNA-5 localizes with and binds to Rb without possessing a LXCXE motif. Although study revealed that the carboxy-terminal 45 amino acids of EBNA-5 are crucial for B cell transformation (Jiang et al., 1991; Mannick et al., 1991; Szekely et al., 1993), the Rb binding region on EBNA-5 was mapped to a 66-amino acid-long peptide that locates on the N-terminal half of this viral protein, which is also the binding site for p53 (Szekely et al., 1993). It was found that p53 competes with Rb for EBNA-5 binding as it inhibits EBNA-5-Rb complex formation in vitro in a dose-dependent manner. However, inhibition of p53-EBNA-5 interaction by Rb was not observed in a reciprocal experiment (Szekely et al., 1993). The fact that the transformation-capable region on EBNA-5 is distinct from its Rb/p53-binding motif suggests interaction with Rb/p53 is not related to its ability to induce B cell transformation. Indeed, Inman et al. showed that expression of EBNA-5 could neither relieve Rb-mediated repression of E2F1 transactivation, nor prevent p53-induced transactivation (Inman and Farrell, 1995). However, it is possible that the association of EBNA-5 to Rb affects other aspects of its function, such as its control of c-Abl tyrosine kinase, to aid in viral infection. In addition, Zancy et al. reported an interaction of EBV immediate-early lytic gene product BRLF1 with Rb in virus-infected cells (Zacny et al., 1998). Although the interaction motif on Rb for BRLF1 binding is outside of its pocket region and BRLF1 does not interact with E2F, a correlation was still observed between the kinetics of BRLF1-Rb binding and the displacement of E2F1 from Rb, suggesting the potential of BRLF1 in initiating cell cycle progression (Zacny et al., 1998).
Chk
Checkpoint kinase (Chk) 1 and 2 are tumor repressor proteins that are activated upon replication defect and/or DNA damage (Bartek and Lukas, 2003; Haoudi et al., 2003; Park et al., 2004, 2006). Being a multifunctional viral protein, Tax of HTLV-1 is also known to interact with Chk. Thus, expression of Tax inhibits the kinase activity of Chk1, whereas the effect of Tax on the kinase activity of Chk2 is controversial. Park et al. showed that Cdc25A degradation and p53 phosphorylation (Ser20), the downstream events of Chk1 activation, were attenuated by Tax expression in a dose-dependent manner, suggesting that Tax inactivates Chk1 (Hirao et al., 2000; Shieh et al., 2000; Xiao et al., 2003; Park et al., 2004). Two domains on Tax were identified to be important for both binding to and inhibition of Chk2, drawing a causal relationship between physical protein interaction and the functional inactivation of Chk family of tumor repressors (Park et al., 2006). By contrast, Haoudi et al. described an induction of the steady-state level of Chk2, but not Chk1, by Tax with increased Cdc25C degradation and p53 phosphorylation (Ser15) (Haoudi et al., 2003). Despite the fact that Chk2 was revealed to be activated by Tax, the overall response of Chk2 to DNA damage was impaired through inhibiting the release of Chk2 from chromatin, which is normally observed upon exposure to ionizing radiation (Li and Stern, 2005a; Gupta et al., 2007). This discrepancy in the regulatory role of Tax on Chk2 activity may be explained by the differences in the assay systems adopted by these studies. Alternatively, it may reflect the dual action of Tax during different phases of the same event, as Tax could first induce the accumulation of Cdc25C, which undergoes degradation at a later stage (Haoudi et al., 2003). However, regardless of whether Tax activates or inhibits the activity of Chk2, the cellular DNA damage response mediated by Chk2 is dampened. In addition, an association of Tax with the DNA-dependent protein kinase (DNA-PK), an early mediator of cell cycle arrest via the activation of Chk2, has also been reported (Li and Stern, 2005b; Durkin et al., 2008). Durkin et al. observed increased DNA-PK phosphorylation, an initiating event required for the activation of the kinase activity of DNA-PK (Li and Stern, 2005b), in Tax-expressing cells (Durkin et al., 2008). Tax-induced constitutive signaling of the DNA-PK pathway, however, impairs cellular response to new damage (Durkin et al., 2008). Thus, through saturating DNA-PK-mediated damage repair response, Tax inactivates Chk2. Furthermore, the delay and impairment of the cellular DNA damage response imposed by Tax appears to be regulated by molecular sequestration of DNA-PK by Tax through physical interaction (Durkin et al., 2008). The nuclear antigen 3C (EBNA-3C) of Bevies is another viral protein that interacts with Chk2, resulting in reduced Cdc25C phosphorylation at Ser216. This Chk2-dependent step causes sequestration of Cdc25C in the cytoplasm, which prevents removal of the inhibitory phosphates from CDKs, leading to EBNA3C-mediated abrogation of the G2/M checkpoint and transformation in lymphoblastoid cell lines (Choudhuri et al., 2007).
P53
The tumor suppressor, p53, was first identified as a host protein that stably interacts with the L-Tag of SV40 (Lane and Crawford, 1979; Linzer and Levine, 1979; Tan et al., 1986). The use of chloramphenicol (CAT) assay demonstrated that L-Tag inhibits the transactivation function of p53, which is mediated through physical interaction as L-Tag mutants deficient in p53 binding fail to prevent downstream activation of gene expression (Mietz et al., 1992). Similarly, complex formation between HPV E6, limited only to the oncogenic HPV types (Werness et al., 1990; Mietz et al., 1992), and p53 leads to a drastic reduction (more than 80%) in the level of p53-mediated transactivation in CAT assay system. HCMV IE2-86 is able to repress p53 function via physical interaction and this inhibitory effect requires two functional domains on IE2-86 (Speir et al., 1994; Tsai et al., 1996). Tsai et al. showed that the N-terminus of IE2-86 was necessary for p53-binding while the C-terminal domain was needed to inhibit p53-mediated transactivation, suggesting that IE2-86 is recruited by protein interaction to target p53 and exert its inhibitory effect (Tsai et al., 1996). Another HCMV protein, which alters p53-orchestrated cellular regulatory mechanisms, leading to tumorigenic transformation in rodent cells, is the morphological transforming region II (mtrII) (Thompson et al., 1994; Muralidhar et al., 1996). It was reported that the first 49 amino acids of mtrII mediate its binding to p53 and are sufficient to repress p53-activated transcription (Muralidhar et al., 1996).
Binding of the nonstructural protein 3 (NS3) of HCV to p53 has been located to the C-terminus, a region that also contains the oligomerization domains required for the latter to form the bioactive homo-tetramer (Levine, 1997; Ishido and Hotta, 1998). It has been proposed that binding of NS3 to p53 interferes with the formation of p53 tetramer, which is most effective in transactivating its target genes, leading to impaired p53-dependent transcriptional activation (Deng et al., 2006). Interestingly, other than binding to p53 itself, adenovirus E1A targets p53-mediated transcription through interacting with the transcriptional coactivators p300 and CREB-binding protein (CBP) of p53 (Somasundaram and El-Deiry, 1997). As a result, E1A inhibits the activation of p21WAF1/CIP1, abolishing p53-dependent cell cycle arrest. An interaction between the transactivator of transcription (Tat) of HIV-1 and p53 was correlated to the development of acquired immune deficiency syndrome (AIDS)-related malignancies. Thus, Clark et al. found that the presence of Tat almost completely abolished p53-mediated activation, leading to reduced levels of p21CIP1/WAF1 and the loss of G1/S checkpoint (Clark et al., 2000). However, it remains to be addressed whether a causal relationship exists between Tat-p53 interaction and the repression of p53 function, which will require the use of p53 binding-deficient Tat.
Other Factors
Interactions of viral component with other cell-cycle regulators have been reported for several viruses. For example, Jiang et al. have shown that expression of influenza A virus nonstructural protein 1 (NS1) downregulates the protein level and inhibits the function of the Ras homolog gene family member A (RhoA), a small GTPase that, besides its primary involvement with formation of actin stress fibers and cytoskeletal remodeling, mediates cell cycle G1/S transition (Jiang et al., 2013). The inhibition of RhoA activity by NS1 was reported to be achieved through physical interaction between the two proteins.
Wee1, a protein kinase which inactivates CDK1 by phosphorylating the latter on Tyr15, can interact with HIV-1 viral protein R (Vpr) in cell culture, resulting in enhanced kinase activity of Wee1 (McGowan and Russell, 1993; Kamata et al., 2008). Although this interaction is required,it is not sufficient for the induction of cell cycle arrest as Vpr mutants, which are unable to induce cell cycle arrest, maintain their binding and activation to Wee1, suggesting the existence of additional factors in Vpr-mediated activation of the G2 checkpoint (Kamata et al., 2008). It is interesting to note that Wee1 blocks G2toM transition by phosphorylating CDK1, whereas Cdc25 triggers this transition and entry into mitosis by dephosphorylating the same residues on CDK1 (Millar and Russell, 1992). In addition to its binding with Wee1, Vpr was also shown to associate with Cdc25C. This binding was mapped to a site that lies near but is distinct from the catalytic domain of Cdc25C (Goh et al., 2004). In accordance with its activation of Wee1, Vpr inhibits the activity of Cdc25C, most likely through its ability to bind Cdc25C (Goh et al., 2004). Thus, Vpr targets CDK1 to mediate G2/M arrest acting on both Wee1 and Cdc25C.
Furthermore, several groups recently demonstrated a direct interaction of Vpr withSLX4, a scaffold for the formation of a heterodimeric structure-specific endonuclease consisted of MUS81 and EME1 (Fekairi et al., 2009; Muñoz et al., 2009; Svendsen et al., 2009; Kim et al., 2013; Laguette et al., 2014). Thus, binding of Vpr to SLX4 activates the MUS81-EME1 endonuclease, leading to replication stress. The observation that G2/M arrest-deficient Vpr mutants fail to interact with SLX4, and that silencing of SLX4 reduces Vpr-dependent cell cycle perturbation highlight the significance of Vpr-SLX4 association in Vpr-induced G2/M arrest (Laguette et al., 2014). In accordance, the use of a panel of simian immunodeficiency virus (SIV) Vprs that differ in their ability to promote cell cycle arrest demonstrated the correlation between Vpr-SLX4binding and the G2/M arrest effect of Vpr (Berger et al., 2015).
DNA damage-binding (DDB) protein, which constitutesDDB1 and DDB2 subunits, translocates to the nucleus to mediate DNA repair in response to DNA damage signal (Wittschieben and Wood, 2003; Iovine et al., 2011). The formation of a complex comprising HIV-1 Vpr, DDB1-Cullin 4A-associated factor 1 (DCAF1), and DDB1 has been implicated as the upstream event in Vpr-triggered G2/M arrest (Le Rouzic et al., 2007; Schröfelbauer et al., 2007). It is known that Vpr hampers the nuclear localization of DDB1, and interferes with the binding of DDB1 to damaged DNA as detected by an electrophoretic mobility shift assay (EMSA) (Schröfelbauer et al., 2007). Moreover, the association of Vpr with DDB1 disrupts complex formation between DDB1 and DDB2, which is a prerequisite for DNA repair (Schröfelbauer et al., 2007). The significance of Vpr-DDB1 interaction in Vpr-mediated cytostatic effect was revealed through the use of DDB1 binding-deficient Vpr mutant (Schröfelbauer et al., 2007). It is worth mentioning that one of the multiple functions of DDB1 is to act as a component of an ubiquitin-E3 ligase complex that targets cell cycle regulatory proteins such as p27KIP1 for proteasomal degradation (Bondar et al., 2006). It will be of interests to investigate whether the association of Vpr with DDB1 could antagonize its role in the hydrolysis of p27KIP1 and other cell cycle effectors, thus contributing to cell cycle arrest. Indirect support for this hypothesis stems from experiments with transient expression of HBV HBx, which induces accumulation of lagging chromosomes and other mitotic aberrations, leading to the activation of DNA replication checkpoint with a delayed S phase progression (Martin-Lluesma et al., 2008). The use of an HBx mutant that is deficient for DDB1 binding suggests that this perturbation of cell cycle progression is attributable to the interaction of HBx with DDB1 (Leupin et al., 2003; Martin-Lluesma et al., 2008).
Subversion by Protein Phosphorylation (Table 2 and Figure 2)
Phosphorylation and dephosphorylation govern the function of many cell cycle regulators, thus acting as switches to ensure its normal progression. For instance, the kinase activity of CDK4 cannot be merely regulated by its association with D-type cyclin. CDK4 phosphorylation onThr172 by CAK is also required to catalyze the process (Kato et al., 1994). As another example, the inhibitory effect of Rb on transcription factor E2F1is suppressed through CDK4/6-mediated hyperphosphorylation of Rb (Sidle et al., 1996). Not surprisingly, viruses have evolved mechanisms to modulate phosphorylation and dephosphorylation events as additional means to subvert host cell division cycle for their own benefit.
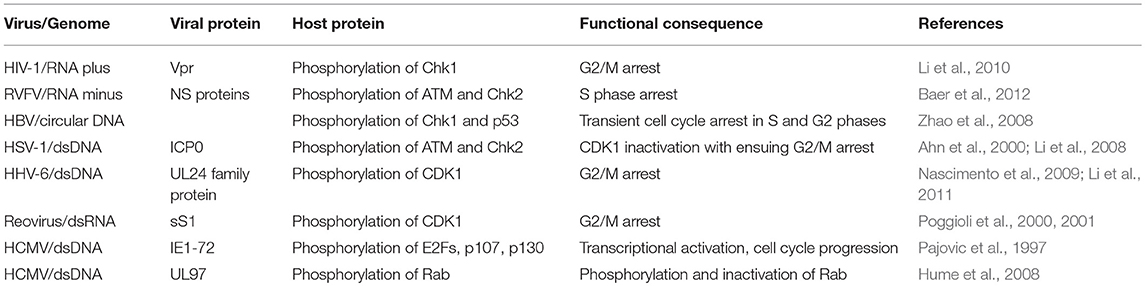
Table 2. Cell cycle subversion by protein phosphorylation.
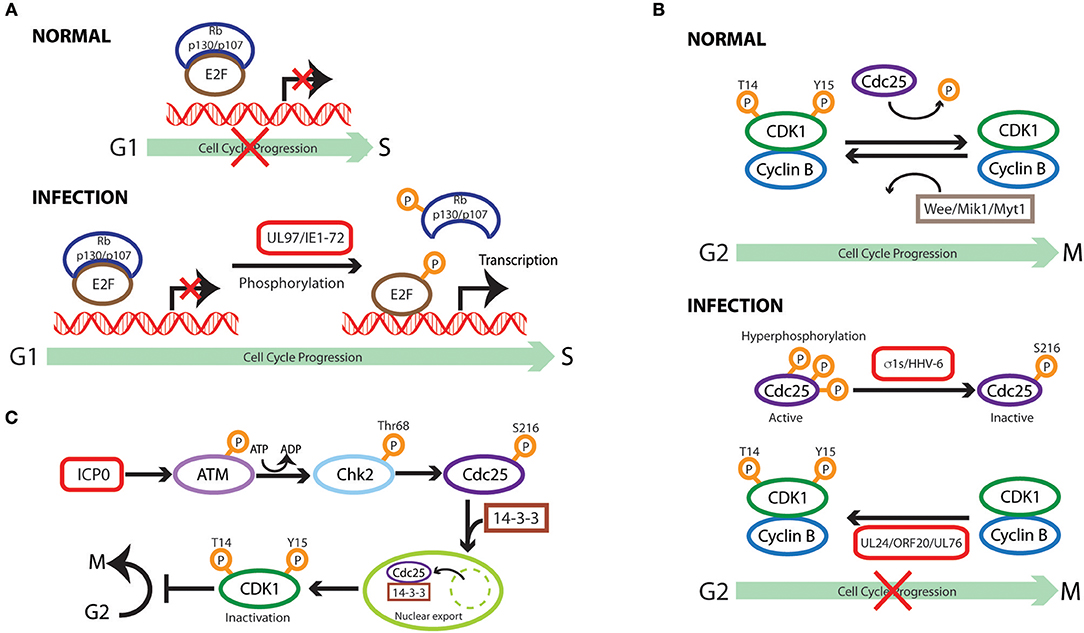
Figure 2. Cell cycle subversion by protein phosphorylation. (A) Direct phosphorylation of Rb by HCMV UL97 protein dissociates E2F-Rb/p107 complexes releasing E2F to activate transcription and cell cycle progression (Hume et al., 2008). HCMV IE1-72 is a viral protein kinase able to directly and selectively phosphorylate transcription factors of the E2F family (E2F1,−2, and−3), leading to E2F-dependent transcriptional activation, thus regulating cell cycle events (Pajovic et al., 1997). (B) Expression of viral S1 gene-encoded σ1s nonstructural protein during reovirus infection leads to G2/M arrest in host cells via σ1s-mediated CDK1 hyperphosphorylation (Poggioli et al., 2000, 2001). Similarly, human herpesvirus 6 (HHV-6)-induces G2/M arrest by increasing the inactive Ser216-phosphorylated form Cdc25C phosphatase that accumulates in the cytoplasm where it has no access to its CDK1 substrate (Li et al., 2011). Members of herpesvirus share the conserved UL24 family of proteins (i.e., UL24, ORF20, and UL76), whose expression induces CDK1 phosphorylation at Tyr15 inhibitory site with ensuing cell cycle G2/M arrest (Nascimento et al., 2009). (C) HSV-1 immediate-early gene product ICP0 triggers a series of phosphorylation events resulting in cytoplasmic sequestration of Cdc25C, which maintains high levels of CDK1 inhibitory phosphorylation, leading to CDK1 inactivation with ensuing G2/M arrest (Li et al., 2008).
Viral Kinases
Direct phosphorylation of Rb was reported for HCMV UL97 protein as the first example of a virus-encoded functional ortholog of CDK (Hume et al., 2008). As expected, expression of mutant UL97 in cells, which are intrinsically deficient in the ability to phosphorylate Rb, induces Rb phosphorylation on the inactivating residues. Moreover, UL97 rescues the proliferation of a yeast mutant which lacks CDK activity, suggesting UL97 functions similar to CDK. Sequence comparison between CDK and UL97 revealed that this viral protein does not require cyclin binding for kinase activity and, therefore, is resistant to the inhibition of p21WAF1/CIP1, features which shield UL97 from the normal cellular control mechanisms that could attenuate CDK activity (Hume et al., 2008).
HCMV IE1-72 is a viral protein kinase, which is able to directly and selectively phosphorylate transcription factors of the E2F family (E2F1,−2, and−3) and pocket proteins (p107 and p130) (Pajovic et al., 1997). Its kinase activity is required for IE1-72-mediatedformation and/or dissociation of inhibitory complexes that lead to E2F-dependent transcriptional activation, thus regulating cell cycle events (Pajovic et al., 1997). It is interesting to note that the kinase activity of IE1-72 resides in the portion encoded by exon 4, which is not the same region necessary for its binding to p107 (encoded by exon 3) as mentioned above (Johnson et al., 1999). It is possible that this interaction functions as the first step to bridge IE1-72 to its target for later phosphorylation, or that IE1-72 can independently exert its role in cell cycle regulation through either physical interaction or its kinase activity.
CDK1/Cdc25
Reovirus infection and the sole expression of viral S1 gene-encoded σ1s nonstructural protein could lead to G2/M arrest in host celosia σ1s-mediated CDK1 hyperphosphorylation as revealed with the use of σ1s-mutant virus (Poggioli et al., 2000, 2001). It was reported that the inhibitory phosphorylation of CDK1 by reovirus decreased the hyperphosphorylated form (active form) of Cdc25C, the CDK1-specific phosphatase (Poggioli et al., 2001), hence preventing CDK1 dephosphorylation, an absolute requirement for the onset of mitosis (Jin et al., 1996). Similarly, human herpesvirus 6 (HHV-6)-induced G2/M arrest correlates with enhanced phosphorylation of CDK1 at the Tyr15 inhibitory site. This is achieved through both elevating the level of the negative regulator Wee1 and inhibiting the activity of Cdc25C phosphatase by increasing the inactive Ser216-phosphorylated form that accumulates in the cytoplasm where it has no access to its CDK1 substrate (Li et al., 2011). Members of herpesvirus share the conserved UL24 family of proteins, namely UL24 in herpes simplex virus type 1 (HSV-1), ORF20 in KSHV, and UL76 in HCMV. Expression of these UL24 proteins led to CDK1 phosphorylation at Tyr15, the dephosphorylation of which is required for the initiation of mitosis (Jin et al., 1996), with ensuing cell cycle G2/M arrest (Nascimento et al., 2009). Similar observations were made with murine gammaherpesvirus 68 (MHV68) infection (Nascimento and Parkhouse, 2007).
The expression of HSV-1 immediate-early gene product ICP0 in HEK 293T cells activates Chk2, but not Chk1, through Thr68 phosphorylation (Li et al., 2008). This activation relies on ICP0-dependent phosphorylation of ataxia telangiectasia mutated (ATM), an upstream effector kinase of Chk2 (Ahn et al., 2000), as revealed in ATM-deficient cell lines (Li et al., 2008). Activated Chk2 in turn phosphorylates Cdc25 on Ser216, which promotes binding of the molecule to 14-3-3 proteins, resulting in cytoplasmic sequestration of Cdc25C, as discussed above, which maintains high levels of CDK1 inhibitory phosphorylation (Matsuoka et al., 1998; Graves et al., 2001). The requirement for the kinase activity of Chk2 in ICP0-induced inhibitory-phosphorylation of Cdc25C was demonstrated by reconstituting Chk2-deficient cells with kinase-defective mutant form of Chk2 (Li et al., 2008). Thus, through serial phosphorylation events, ICP0 leads to CDK1 inactivation with ensuing G2/M arrest. Similarly, ATM and Chk2 phosphorylation are required for Rift Valley fever virus (RVFV)-induced S phase arrest, a phenomenon attributed to the activity of viral nonstructural proteins (Baer et al., 2012).
Infection of a hepatocyte cell line with HBV causes phosphorylation of Chk1 and p53at Ser345 and Ser15respectively, which is accompanied by a transient cell cycle arrest in S and G2 phases (Zhao et al., 2008). Although more remains to be done to elucidate the signaling pathway concerning these phosphorylation events, it is known that Chk1 phosphorylation on Ser345 is essential for the activation of checkpoint arrest in G2/M phase (Lopez-Girona et al., 2001). Similarly, Chk1 phosphorylation at Ser345 is required in G2/M arrest induced by HIV-1 Vpr as re-introduction of a phosphorylation-resistant Chk1 mutant fails to restore Vpr-induced G2/M arrest in Chk1-depleted cells (Li et al., 2010). Thus, by phosphorylating Chk1, Vpr facilitates proteasome-mediated degradation of Cdc25C and, to a lesser extent, Cdc25B, contributing to G2/M arrest (Lopez-Girona et al., 1999; Graves et al., 2001; Bulavin et al., 2003).
CDK2
CDKs remain catalytically inactive until cyclin-bound CDKs undergo phosphorylation at Thr161/160/172 by CAK (Gould et al., 1991; Norbury et al., 1991; Desai et al., 1992; Gu et al., 1992; Krek and Nigg, 1992; Solomon et al., 1992). The current model of CDK/cyclin activation posits two steps: (i) formation of an intermediate complex composed by the PSTAIRE helix of CDK2 and helices 3 and 5 of the cyclin; (ii) isomerization of the CDK/cyclin complex to expose the T-loop of CDK, which is then accessible to phosphorylation by CAK to form the substrate binding site (Morris et al., 2002). CDK2/CDK4 phosphorylation at their respective Thr sites by CAK subunits, for example CDK7/cyclin H (Fisher and Morgan, 1994), is essential for both the stabilization of CDK/cyclin complexes and could increase the flexibility of the CDK2 T-loop (Matsuoka et al., 1994; Morris et al., 2002). Overexpression of the ARV p17 protein inhibited CDK2 phosphorylation at T160, which was dependent on CDK7/cyclin H disassociation due to increased p53/cyclin H interaction (Chiu et al., 2018). Thus, by diminishing the CAK activity, namely CDK7/cyclin H association, p17 prevents CDK2 phosphorylation and therefore inhibits its kinase activity. Although the mechanism of how p17 diminishes CAK activity in a p53-dependent manner remains to be elucidated, it is another example illustrating how viruses regulate the cell cycle machinery (Chiu et al., 2018).
Subversion by Protein Degradation (Table 3 and Figure 3)
Proteolysis of cell cycle regulators at defined stages plays a crucial role in cell cycle progression. While purified recombinant IE2-86 of HCMV is able to block the inhibitory effect of the cyclin-dependent kinase inhibitor p21WAF1/CIP1 through physical interaction as previously mentioned, HCMV infection can also target p21WAF1/CIP1 todegradation. Thus, a disparity between p21WAF1/CIP1 RNA and protein levels was observed after HCMV infection, implying possible degradation of p21WAF1/CIP1 in virus-infected cells (Chen et al., 2001). Infection could induce sustained increase in calpain activity and the use of specific inhibitors demonstrated that calpain, and not proteasome activity is required for HCMV-mediated p21WAF1/CIP1 reduction, although the molecular mechanism by which p21WAF1/CIP1 is targeted by calpain is not clear (Chen et al., 2001).
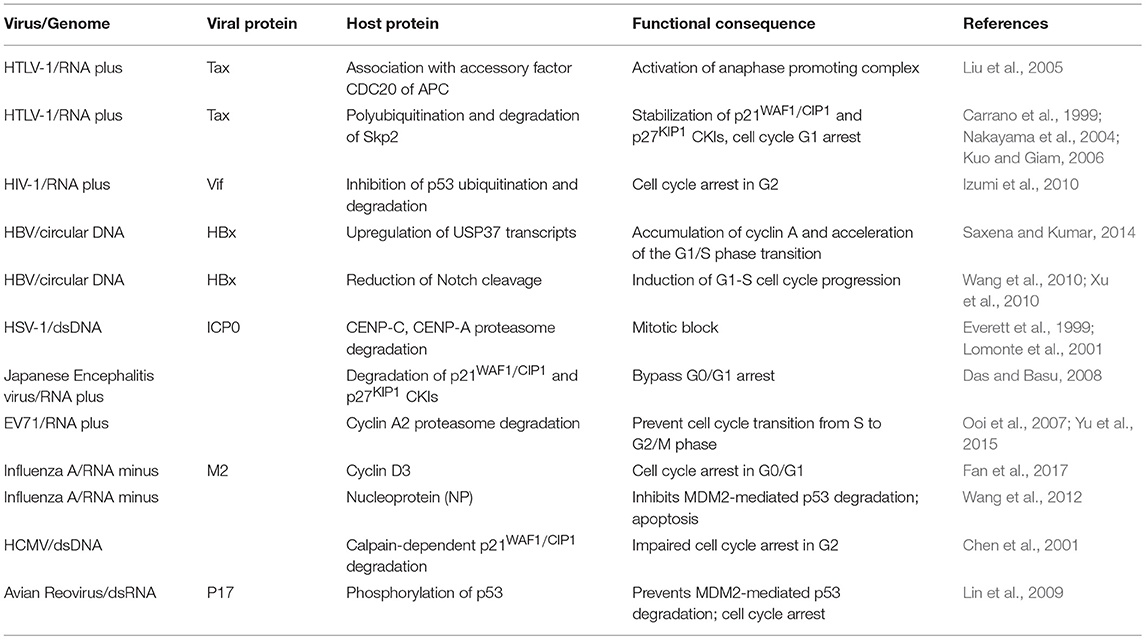
Table 3. Cell cycle subversion by protein degradation.
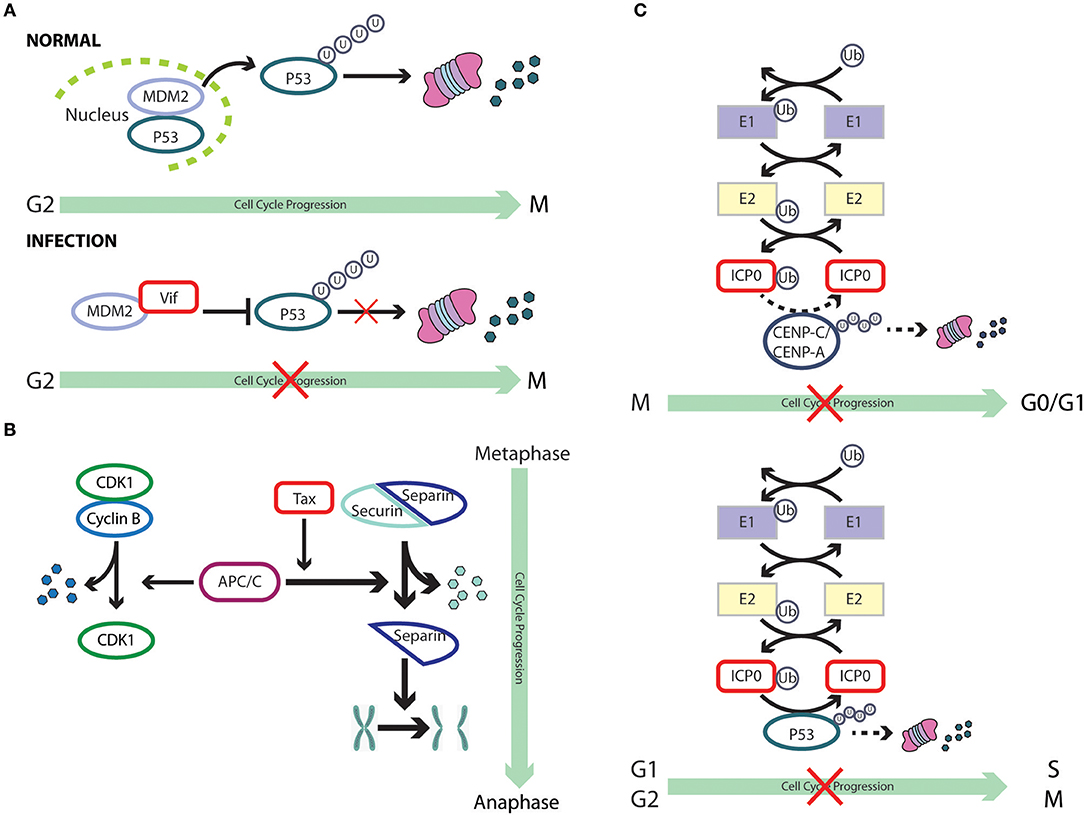
Figure 3. Cell cycle subversion by protein degradation. (A) Expression of (Vif) protein of HIV-1 blocks MDM2-mediated proteasomal degradation of p53, by binding to both MDM2 and p53, leading to cell cycle arrest in G2 (Izumi et al., 2010). (B) The degradation of securin by the anaphase promoting complex/cyclosome (APC/C) is essential for the completion of mitosis, leading to correct transmission of chromosomes from mother to daughter cells. The oncoprotein Tax of HTLV-1 is able to promote securin and cyclin B1 degradation, leading to chromosome instability (Liu et al., 2003). (C) ICP0 of HSV-1 possesses an ubiquitin E3 ligase activity (Boutell et al., 2002; Hagglund et al., 2002), which results in rapid loss of the centromeric protein CENP-C and CENP-A in a proteasome-dependent manner and mitotic block (Everett et al., 1999; Lomonte et al., 2001). However, it remains unclear whether these two centromere components are direct substrates of the ICP0 E3 ligase (dashed line). Likewise, ICP0 can directly ubiquitinate p53 (Boutell and Everett, 2003), although p53 levels are not reduced during HSV-1 infection (Hobbs and DeLuca, 1999) and, hence, the role of p53 in ICP0-induced perturbation of cell cycle progression remains unclear (dashed line).
The interaction between cyclin D3 and M2 during Influenza A infection provides another example of the involvement of the proteasomal degradation pathway. Although the interaction was found to inhibit virus production, this mode of restriction eventually surrenders to viral infection as cyclin D3 becomes trapped in the cytoplasm by M2 and eventually undergoes proteasomal degradation upon prolonged infection (Fan et al., 2017). Indeed, mislocalization and subsequent downregulation of restriction factors is one of the recurring mechanisms viruses have adopted to overcome host defenses (Ma et al., 2012). In the case of IAV, two possible mechanisms account for the relocalization of cyclin D3 to the cytoplasm: (i) M2 interacts with newly synthesized cyclin D3 and prevents it from translocating into the nucleus, or (ii) cyclin D3 is retained by M2 in the cytoplasm after its nuclear export. It will be interesting to explore these mechanisms in greater detail in future studies.
HIV-1 viral infectivity factor (Vif) binds to the central domain of mouse double-minute 2 protein (MDM2), the primary E3 ubiquitin ligase for p53 (Momand et al., 2000; Izumi et al., 2009). Izumi et al. showed that expression of Vif could block MDM2-mediated proteasomal degradation of p53, leading to cell cycle arrest in G2 (Izumi et al., 2010). More interestingly, the same study reported that Vif could directly interact with p53, independent of its association with MDM2, leading to a partial blockage of p53-MDM2 binding and, hence, impairment of MDM2-mediated p53 degradation. This strategy appears to be very effective and economical, as it has been adopted by other viruses. For example, the IAV nucleoprotein (NP) binds to p53 with ensuing protein stabilization as a result of compromised MDM2-mediated p53 degradation (Wang et al., 2012). ARV infection promotes p53 phosphorylation at multiple sites, an event which alleviates p53 inhibition by MDM2 (Shieh et al., 1997), and activates the transcriptional function of p53 (Lin et al., 2009). It was later discovered that p17 protein of ARV could robustly enhance MDM2 binding to ribosomal proteins, thereby blocking MDM2-mediated p53 ubiquitination and degradation (Zhang and Lu, 2009; Huang et al., 2017). Thus, through multiple mechanisms, ARV impairs the ability of MDM2 to target p53 for degradation. There is evidence that both PTEN and p21WAF1/CIP1 are required for p53-mediated cell cycle arrest (el-Deiry et al., 1993; Stambolic et al., 2001), which is dependent on a complex signaling cascade that brings about Akt-induced phosphorylation of MDM2, antagonized by PTEN, and the ensuing translocation of MDM2 to the nucleus to downregulate p53 (Mayo and Donner, 2002). Thus, the interplay between viruses and tumor suppressor proteins is likely to target multiple networks that ultimately affect the cell cycle.
The anaphase promoting complex/cyclosome (APC/C) was characterized as a securin- and cyclin B-specific ubiquitin ligase, functioning mainly to trigger metaphase to anaphase transition by tagging these proteins for degradation (Irniger et al., 1995; King et al., 1995; Sudakin et al., 1995; Thornton and Toczyski, 2003; Peters, 2006). Securin sequesters separin, the “sister-separating” protease, preventing the separation and segregation of sister chromatids (Nasmyth et al., 2000; Yanagida, 2000). Thus, the degradation of securin by APC/C is essential for the completion of mitosis, leading to correct transmission of chromosomes from mother to daughter cells (Zou et al., 1999). The oncoprotein Tax of HTLV-1 is able to promote securin and cyclin B1 degradation, a process which requires the function of the accessory factor CDC20 of APC, leading to chromosome instability (Liu et al., 2003). Although the mechanism by which Tax activates APC is still unclear, an association of Tax with APCCDC20 was reported (Liu et al., 2005). Interestingly, premature activation of APC by Tax could also result in a surge in the levels of p21WAF1/CIP1 and p27KIP1, leading to G1 arrest that resembles cellular senescence (Kuo and Giam, 2006). This was found to be a result of polyubiquitination and degradation of Skp2, the substrate recognition subunit of the SCFSkp2 E3 ubiquitin ligase that regulates the destruction of p21WAF1/CIP1 and p27KIP1, by Tax-activated APC (Carrano et al., 1999; Nakayama et al., 2004; Kuo and Giam, 2006). Thus, by targeting the E3 ligase of p21WAF1/CIP1 and p27KIP1 for degradation, Tax stabilizes these two CKIs. On the contrary, the association of chicken anemia virus protein Apoptin with APC1 inhibits the activity of theAPC/C ubiquitin ligase, leading to the stabilization of cyclin B1 and other APC/C substrates, with ensuing cell cycle G2/M arrest and apoptosis (Teodoro et al., 2004).
The deubiquitinase (DUB) USP37 has been found to stabilize cyclin A by removing the polyubiquitin from the latter and, hence, accelerates entry into S phase (Huang et al., 2011). HBx of HBV could upregulate USP37 transcripts in both hepatic and non-hepatic cells and also prevents USP37 from proteasomal degradation. HBx acts as a chaperone of USP37 and shuttles it out of the nucleus, where the ubiquitin E3 ligase CDC20 homolog 1 (CDH1) and b-TrCP associate with USP37 (Zhou et al., 2003; von Mikecz, 2006; Saxena and Kumar, 2014). Thus, by stabilizing the expression of USP37, HBx promotes the accumulation of cyclin A to modulate cell cycle progression. Furthermore, HBx hinders the proteolytic cleavage of Notch1, which plays a critical role in proliferation (Artavanis-Tsakonas et al., 1999; Wang et al., 2010; Xu et al., 2010). This is achieved through HBx-mediated transcriptional downregulation of presenilin 1, a catalytic subunit of the large protease complex γ-secretase which mediates the cleavage of Notch 1 (Artavanis-Tsakonas et al., 1999; Kopan and Ilagan, 2009; Xu et al., 2010).
In addition to hijacking the host ubiquitination machinery, viruses could also encode their own degradation enzymes. The zinc-binding RING finger domain of ICP0 of HSV-1 possesses a ubiquitin E3 ligase activity (Boutell et al., 2002; Hagglund et al., 2002). Sole expression of ICP0 is able to induce rapid loss of the centromeric protein CENP-C and CENP-A in a proteasome-dependent manner, resulting in mitotic block (Everett et al., 1999; Lomonte et al., 2001). However, it remains unclear whether these two centromere components are direct substrates of the ICP0 E3 ligase. The use of purified p53 in the presence of ICP0, together with a full complement of the ubiquitin-conjugating enzymes in test tubes, revealed a direct ubiquitination of p53 by ICP0 (Boutell and Everett, 2003); p53 levels, howver, were not reduced during HSV-1 infection and, hence, the role of p53 in ICP0-induced perturbation of cell cycle progression and accumulation in G1/S and G2/M needs further investigation (Hobbs and DeLuca, 1999). Still, it provides a novel mechanism by which a virus-encoded protein functions as an E3 ligase to catalyze the degradation of cell cycle checkpoint proteins.
Acute Japanese encephalitis virus (JEV) infection leads to cell cycle G0/G1 arrest with elevated p21WAF1/CIP1 and p27KIP1 levels, and reduced production of cyclin D (Das and Basu, 2008). However, in the case of persistent JEV infection, expression of these CKIs and GSK-3β, which mediates the degradation of cyclin D, is suppressed (Diehl et al., 1998; Kim et al., 2015). Thus, by extending the stability of cyclin D, persistent JEV infection bypasses cell cycle arrest, striking a balance between the persistence of lytic RNA virus and host survival. Enterovirus 71 (EV71), the etiological agent of hand, foot, and mouth disease, was shown to prevent cell cycle transition from S to G2/M phase (Ooi et al., 2007; Yu et al., 2015). Although the molecular mechanisms is still unclear, it was observed that cyclin A2 was targeted for proteasome-mediated degradation during infection (Yu et al., 2015).
Subversion by Protein Redistribution (Table 4 and Figure 4)
While it is well established that several biochemical processes serve to orchestrate cell cycle control, including protein-protein interaction and dissociation, phosphorylation/ dephosphorylation, and the synthesis/ destruction of cell cycle regulators at specific time point, the importance of subcellular localization of CDK-cyclins complexes and their regulators is receiving increasing attention for proper cell-cycle coordination. Hence, viruses have also evolved mechanisms to deregulate cell cycle progression by targeting the localization of cell cycle regulators.

Table 4. Cell cycle subversion by protein redistribution.
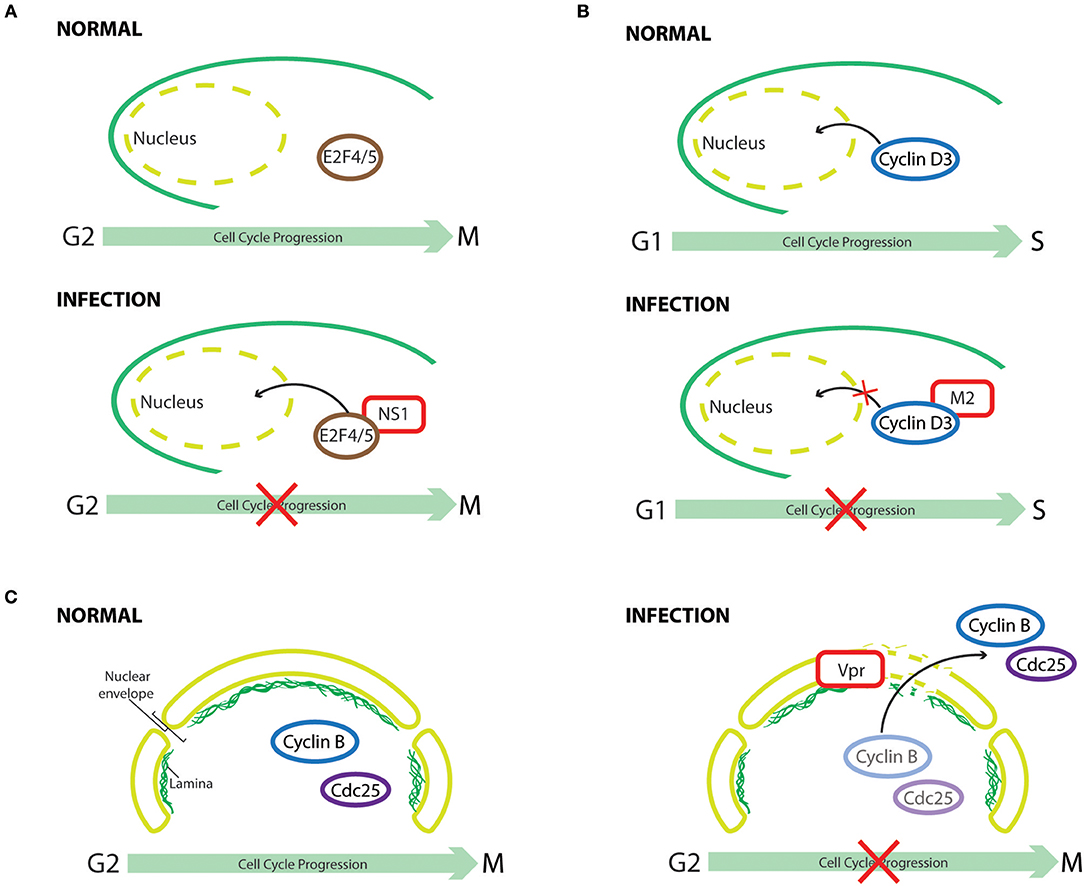
Figure 4. Cell cycle subversion by protein redistribution. (A) Nonstructural protein 1 (NS1) of human parvovirus B19 (B19V) leads to G2 arrest, which is mediated by upregulating nuclear localization of repressive E2F4 and E2F5 transcription factors (Wan et al., 2010). (B) Cyclin D3 needs to be imported into the nucleus to assist G0/G1 cell cycle progression (Mahony et al., 1998). Influenza A virus infection perturbs cell cycle progression by redistributing cyclin D3 from the nucleus to the cytoplasm, triggering its proteasomal degradation (Fan et al., 2017). (C) Expression of HIV-1 Vpr induces localized-disruption in the lamin architecture, resulting in G2/M arrest that is dependent on cytoplasmic compartmentalization of Cdc25C, cyclin B1, and Wee1 (Peng et al., 1997; Laronga et al., 2000; de Noronha et al., 2001; Kino et al., 2005).
One early example of virus-induced translocation of cell cycle regulators is exemplified by HCMV infection, which can mitogenically stimulate growth-arrested cells to enter the cell cycle by promoting nuclear uptake of CDK2 (Bresnahan et al., 1997). This suggests that HCMV possesses mechanisms to regulate the subcellular distribution of CDK2, thus facilitating the activation of cyclin E-CDK2 complex and, consequently, modulating cell cycle progression. It is possible that CDK2 could be transported into the nucleus in a “piggyback” manner, interacting with a viral protein that shuttles between nucleus and cytoplasm, or that viral infection activates the intrinsic CDK2 import machinery. Indeed, infection of primary erythroid progenitor cells by human parvovirus B19 (B19V) leads to G2 arrest, which is dependent on viral nonstructural protein 1 (NS1), as its sole expression could recapitulate the same phenomenon (Wan et al., 2010). The cell cycle perturbation effect results from NS1-mediated downregulation of activating E2Fs transcription factors (E2F1 to E2F3a) and upregulation of repressive E2Fs (E2F4 to E2F8). Interestingly, co-immunoprecipitation experiments in NS1 expressing cells identified NS1-E2F4 and NS1-E2F5 complexes, which could enhance the nuclear import of these transcription-repressive factors. Normal cell cycle progression was observed in cells infected with a mutant B19V, which encodes a nuclear-localization-deficient NS1, demonstrating the importance of NS1-mediated redistribution of E2F4 and E2F5 in causing cell cycle perturbation (Wan et al., 2010).
For cyclin D3 to exert its effect in cell cycle G0/G1 progression, it needs to be transported from the cytoplasm to the nucleus (Mahony et al., 1998). It was demonstrated through confocoal imaging and subcellular fractionation assay that, in influenza A virus infected cells, cyclin D3 was significantly redistributed from the nucleus to the cytoplasm, followed by its proteasomal degradation (Fan et al., 2017). Hence, through multiple mechanisms, influenza A virus perturbs cell cycle progression for enhanced progeny virus production. Similarly, a significant increase in the cytoplasmic level of CDK1 was observed in ARV-infected or p17-expressing cells (Chiu et al., 2018). Thus, p17 of ARV not only perturbs cell cycle progression into mitosis through direct inhibition of CDK1 kinase activity, but also disrupts CDK1/cyclin B1 complex formation in the nucleus by sequestering CDK1 in the cytoplasm. Although total p53 levels were elevated after infection, a reduction of p53 level in nuclear extracts of HCMV-infected human umbilical vein endothelial cells was observed, resulting in an anti-apoptotic phenotype of HCMV-infected cells (Kovacs et al., 1996). The reduced nuclear-translocation of p53 by HCMV, as suggested by immunofluoresence, provides a plausible explanation for the oncogenic potential of HCMV (Kovacs et al., 1996). Wang et al. later reported that the cytoplasmic sequestration of p53 by HCMV occurs by blocking the nuclear localization signal (NLS) of p53 (Wang et al., 2001). Interestingly, in addition to perturbing the nuclear-import of p53, HCMV could also inhibit the function of nuclear p53 through sequestering it into discrete foci, viz., the replication centers, within the nuclei of HCMV-infected cells (Fortunato and Spector, 1998). Changes in the subcellular localization of p53 were also observed in two different cell lines that expressed the HCV NS5A protein (Majumder et al., 2001). Endogenous p53 is a nuclear protein, whereas it was retained at the perinuclear membrane following NS5A expression (Shaulsky et al., 1991). A physical association of NS5A with p53 was demonstrated both in vitro and in vivo; however, it is not the cause for this sequestration as NS5A binding-deficient p53 was still observed to localize on the perinuclear membrane. In accordance, hTAFII32, a transcriptional coactivator of p53, was also partially sequestered by NS5A in the cytoplasm, thus suggesting an indirect mechanism (Lu and Levine, 1995; Lan et al., 2002). Although p53 is known as a tumor suppressor gene, increasing evidence suggest that nasopharyngeal carcinoma (NPC) is associated with overexpression and accumulation of p53 (Gulley et al., 1998; Yip et al., 2006). It is interesting to find that the late membrane protein 1 (LMP1) of EBV not only upregulates the expression and phosphorylation of p53 and survivin, which possesses a p53-binding element in its promoter region and facilitates G1/S progression by interacting with CDK4 in the nucleus (Suzuki et al., 2000; Hoffman et al., 2002; Mirza et al., 2002; Guo et al., 2012), but also increases the nuclear localization of both proteins (Guo et al., 2012).
Expression of HIV-1 Vpr could induce localized-disruption in the lamin architecture, which is known to interfere with DNA synthesis and changes in the intracellular trafficking of cell cycle regulators (Moir et al., 2000; de Noronha et al., 2001). This structural defect of lamin leads to the formation of nuclear envelope herniations as revealed by immunofluorescence, resulting in changes in the subcellular compartmentalization of Cdc25C, cyclin B1, and Wee1 (de Noronha et al., 2001). Thus, interaction of HIV-1 Vpr with the 14-3-3σ scaffolding protein, a G2/M transition regulator that inactivates Cdc25C by changing its subcellular localization and/or stability (Fu et al., 2000; Muslin and Xing, 2000), has been implicated in Vpr-induced cell cycle perturbation (Peng et al., 1997; Lopez-Girona et al., 1999; Laronga et al., 2000; Kino et al., 2005). Vpr could mediate protein assembly among 14-3-3σ, Cdc25C, CDK1, and cyclin B1, leading to G2/M arrest by either sequestering Cdc25 in the cytoplasm or impairing the normal association of 14-3-3σ with centrosomal proteins (Kino et al., 2005; Bolton et al., 2008). An association of Vpr with MAPKAP kinase-2 (MK2), a possible regulatory kinase of Cdc25, was reported by Huard et al. (2008). Although the relevance of this interaction in Vpr-mediated cell cycle arrest needs further investigation, it appears that MK2 is required for Vpr-induced Cdc25 phosphorylation and nuclear export (López-Avilés et al., 2005; Manke et al., 2005; Huard et al., 2008). The accessory protein viral infectivity factor (Vif) of HIV-1 also inhibits cell cycle progression at the G2 phase by interfering with the nuclear translocation of CDK1 and cyclin B1, a finding corroborated by infection experiments with Vif-deficient HIV-1 virus that point to a role of this viral protein in mediating CDK1 cytoplasmic-retention (Sakai et al., 2011).
HHV-6A and HHV-6B provide other examples of mislocalization coupled to cell cycle arrest, as their infection reduces E2F1-Rb complex formation and leads to accumulation of E2F1 both in the cytoplasmic and nuclear fractions, as opposed to its strict nuclear localization in normal cells (Mlechkovich and Frenkel, 2007). Although the relevance of E2F1 translocation in HHV-6-induced cell cycle arrest is not completely understood, it illustrates that viruses could also target E2F family of protein in this context (Sharon et al., 2014).
Subversion by Virus-Encoded Homologs of Cell Cycle Regulators (Table 5 and Figure 5)
Herpesvirus saimiri (HVS), a T-lymphotropic gamma-herpesvirus that establishes persistent conditions in primate host species, encodes the first reported viral-cyclin (v-cyclin) (Nicholas et al., 1992; Fickenscher and Fleckenstein, 2001). Similarly, amino acid and DNA sequence analyses of KSHV, which has been strongly implicated as the etiologic factor in the development of Kaposi's sarcoma and primary effusion lymphomas (PEL) (Chang et al., 1994; Cesarman et al., 1995), identified a “cyclin-box” showing similarity to human cyclin D2 (Chang et al., 1996; Li et al., 1997). In addition, murine gammaherpesvirus 68 (MHV68), another member of the gammaherpesviruses, shares with HVS and KSHV similarities in the sequence of an open reading frame that is predicted to encode a D-type cyclin (Virgin et al., 1997). Taken together, these findings argue for a conserved role of these v-cyclins in gammaherpesvirus biology. Indeed, both HVS and KSHV v-cyclins show strong association with CDK6, leading to the phosphorylation of Rb and histone H1 (Jung et al., 1994; Li et al., 1997). Surprisingly, despite the fact that v-cyclin of MHV68 displays only around 20% similarity to cyclin A/E, it associates with CDK2 rather than CDK6 (Card et al., 2000). Herpesvirus ateles (AtHV-2) causes lethal lymphomas in various new world primates and is closely related to HVS. The light DNA segment of AtHV-2 possesses an open reading frame which shows significant homology to D-type cyclins although its function was not studied in molecular terms (Albrecht, 2000).
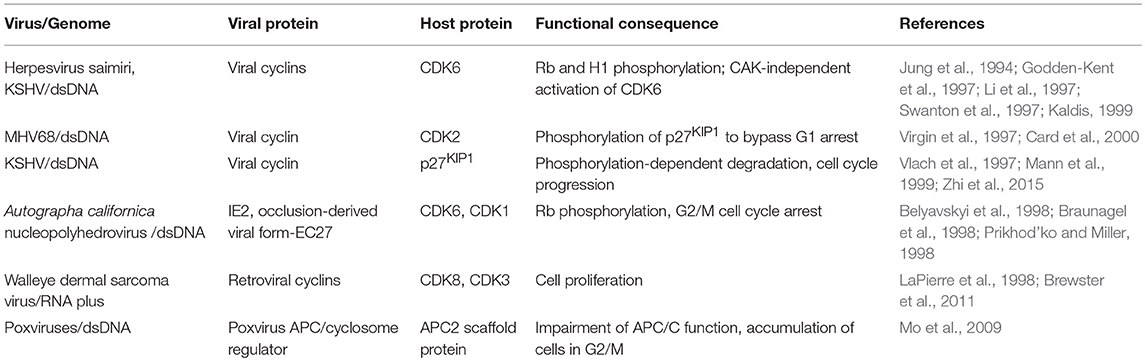
Table 5. Cell cycle subversion by virus-encoded homologs of cell cycle regulators.
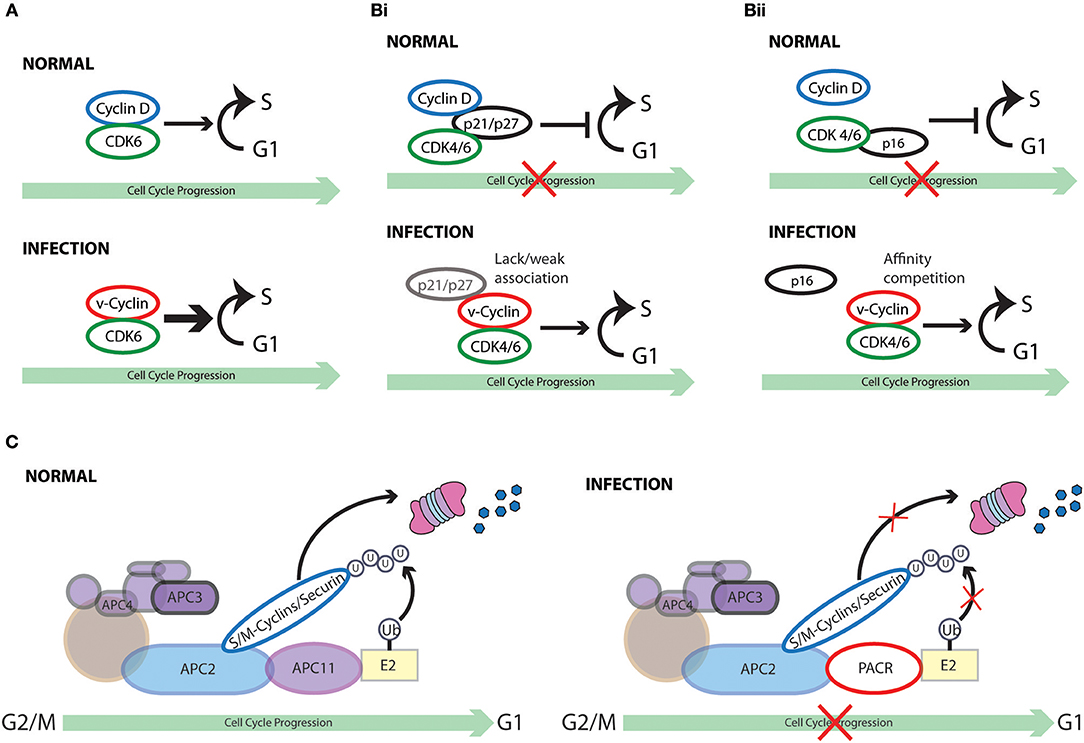
Figure 5. Cell cycle subversion by virus-encoded homologs of cell cycle regulators. (A) Several gammaherpesviruses encode viral-cyclins (v-cyclin) (Nicholas et al., 1992; Chang et al., 1994, 1996; Cesarman et al., 1995; Li et al., 1997; Virgin et al., 1997; Fickenscher and Fleckenstein, 2001) that display strong association with CDK6, leading to the phosphorylation of Rb and histone H1. (Bi) V-cyclin-CDK complexes are significantly more resistant to the inhibition imposed by p21WAF1/CIP1, p27KIP1, and p16INK4A than cyclin D1-CDK complexes in vitro (Swanton et al., 1997). Failure or weak association of p21WAF1/CIP1 and p27KIP1 with v-cyclin likely defines the inefficiency of these CKI to inhibit them (Swanton et al., 1997; Schulze-Gahmen et al., 1999; Card et al., 2000). (Bii) Higher affinity of v-cyclin for CDK6 than p16INK4A may prevent its displacement by p16INK4A. (C) Both crocodile and squirrel poxviruses encode the APC/cyclosome regulator (PACR), which shares sequence similarities to the APC subunit 11 (APC11). PACR mimics the binding ability of APC11 to APC2 but not the ubiquitin ligase activity of APC11. Thus, poxvirus hinders normal cell cycle progression by encoding an inactive homolog of APC11 (Mo et al., 2009).
Interestingly, unlike D-type cyclins that have broad association with different CDKs, the v-cyclins of HVS and KSHV bind almost exclusively to CDK6 (Godden-Kent et al., 1997). Of note, KSHV-cyclin can activate CDK6 independent of phosphorylation by CAK in vitro and in vivo (Kaldis, 1999). Moreover, the kinase activity of v-cyclin-CDK6 appears to be much stronger than cyclin D1-CDK6, targeting also histone H1, which is a preferred substrate of CDK2 rather than CDK6 (Alexandrow and Hamlin, 2005). This finding suggests a transformation potential of v-cyclin through “hyperactivation” of CDK6 (Jung et al., 1994; Swanton et al., 1997). In fact, ectopic expression of KSHV v-cyclin abolishes the cell cycle arrest effect imposed by Rb, which is related to Rb hyperphosphorylation and functional inactivation (Chang et al., 1996). In support of these finding, MHV v-cyclin-transgenic mice display a significant increase in thymocyte number with promoted cell cycle progression and also develop high-grade lymphoblastic lymphoma (van Dyk et al., 1999).
Furthermore, the v-cyclin-CDK complexes are significantly more resistant to the inhibition imposed by p21WAF1/CIP1, p27KIP1, and p16INK4A than cyclin D1-CDK complexes in vitro (Swanton et al., 1997). Lack or weak association of p21WAF1/CIP1 and p27KIP1 with v-cyclin likely defines the inefficiency of these CKI to inhibit them (Swanton et al., 1997; Schulze-Gahmen et al., 1999; Card et al., 2000). It is currently unclear how v-cyclins antagonize the inhibitory effect of p16INK4A since this CKI acts by inhibiting the catalytic subunit of CDK4 and CDK6 (Sherr and Roberts, 1999). One hypothesis is that higher affinity of v-cyclin for CDK6 than p16INK4A may prevent its displacement by p16INK4A. Deletion of the p16INK4A locus was identified in KSHV associated PEL primary samples, suggesting that loss of p16INK4A may be another mechanism of KSHV-induced transformation (Platt et al., 2002). In addition, by targeting p27KIP1 to phosphorylation-dependent degradation, the v-cyclin of KSHV could prevent the G1 arrest induced by the viral latency protein vFLIP, which is co-translated from the same viral mRNA as v-cyclin (Vlach et al., 1997; Mann et al., 1999; Zhi et al., 2015). This was also demonstrated by the inability of KSHV v-cyclin to escape p27KIP1 mediated cell cycle arrest when p27KIP1 is mutated to a form that cannot be phosphorylated. Moreover, p27KIP1 phosphorylation on Ser10 by KSHV v-cyclin-CDK6 complex enhances its cytoplasmic dislocation, which serves as another means to facilitate cell cycle progression (Boehm et al., 2002; Ishida et al., 2002; Sarek et al., 2006). Similarly, CDK2 associated with v-cyclin of MHV68 exhibits substantial resistance to both p21WAF1/CIP1 and p27KIP1, while inducing phosphorylation of p27KIP1 to overcome p27KIP1-mediated G1 arrest (Yarmishyn et al., 2008). However, v-cyclin of MHV68 is only able to subvert p27KIP1-imposed G1 arrest, but not that imposed by p21WAF1/CIP1 (Yarmishyn et al., 2008).
Autographa californica nucleopolyhedrovirus (AcMNPV) infects pest insect and causes cycle arrest at G2/M phase in host cells with continuing viral DNA replication (Braunagel et al., 1998). Sole expression of the IE2 protein of AcMNPV results in accumulation of enlarged cells with DNA content greater than 4N (Prikhod'ko and Miller, 1998). More interestingly, the structural protein occlusion-derived viral form (ODV)-EC27 is endowed with cyclin-like activity, making it the second viral-encoded cyclin homolog reported (Belyavskyi et al., 1998). Similar to the v-cyclins of herpesviruses, association of ODV-EC27 with CDK6 leads to Rb phosphorylation (Huard et al., 2008). Additionally, ODV-EC27 was found to interact with CDK1, exerting cyclin B-like activity.
Walleye dermal sarcoma virus (WDSV) and walleye epidermal hyperplasia virus (WEHV) are retroviruses that encode human D-type cyclin homologs. A standard yeast complementation assay showed that the retroviral-cyclins (rv-cyclins) of WDSV, but not that of WEHV, induce cell cycle progression in G1-cyclin-deficient yeast (LaPierre et al., 1998). In addition to sharing sequence similarity with D-type cyclins, WDSV rv-cyclin was shown to be a structural ortholog of cyclin C (Brewster et al., 2011), which was identified owing to its ability to rescue cyclin G1 function in yeast (Huard et al., 2008). Cyclin C has been shown to promote G0/G1 transition through phosphorylating Rb in combination with CDK3 (Lew et al., 1991; Ren and Rollins, 2004) and was later recognized as part of the Mediator complex, which links gene-specific activators to the general RNA Pol II transcription machinery, in complex with CDK8 (Tassan et al., 1995; Conaway and Conaway, 2011; Malumbres, 2014). As expected, WDSV rv-cyclin has been shown to interact with either CDK8, component of the Mediator complex (Rovnak and Quackenbush, 2002; Poss et al., 2013), or CDK3, leading to enhanced host gene expression or cell proliferation without serum stimulation, respectively (Brewster et al., 2011). Other viruses have been shown to interact with subunits of the Mediator complex, but they are doing so independently of viral cyclins. For example, varicella-zoster virus (VZV), an alpha-herpes virus that causes varicella and zoster, targets the MED25 subunit of Mediator through its major transactivator protein, immediate early gene 62 (IE62) (Yang et al., 2008), whereas herpes simplex virus utilizes VP16, which controls the transcription of immediate early genes, to activate transcription through a specific MED25-associated mediator deficient in CDK8 (Uhlmann et al., 2007). Interestingly, it was later found that CDK8 could positively regulate VP16-dependent transcriptional regulation, while CDK11 and CDK19 exhibit opposing roles (Furumoto et al., 2007; Tsutsui et al., 2008, 2011).
Sequence analysis of available poxvirus genome has revealed that both crocodile and squirrel poxviruses encode a RING-H2 protein, the poxvirus APC/cyclosome regulator (PACR),which shares sequence similarities to the APC subunit 11 (APC11). Together with the scaffold protein APC2, APC11 forms the catalytic core of APC/C, which regulates the progression through mitosis by targeting several anaphase inhibitory proteins for proteolysis and the maintenance of G1 phase (Tang et al., 2001; Hsu et al., 2002; Song et al., 2004; Wei et al., 2004; Wehman et al., 2007; Mo et al., 2009). Interestingly, PACR mimics the binding ability of APC11 to APC2 but not the ubiquitin ligase activity of APC11. Expression of PACR impairs APC/C function, accompanied by the accumulation of cells in G2/M phase. The cell cycle perturbation effect of PACR requires its N-terminal region, which is also the region responsible for association with APC2 (Mo et al., 2009). Thus, poxvirus hinders normal cell cycle progression by encoding an inactive homolog of APC11.
Subversion by Other Mechanisms (Table 6)
HIV-1 Vpr-induced G2 arrest depends on the activation of ATR, the ataxia telangiectasia mutated (ATM)- and Rad3-related protein, which phosphorylates Chk1 and, in turn, leads to both phosphorylation-induced degradation of Cdc25A and Cdc25C cytosolic sequestration (Peng et al., 1997; Sanchez et al., 1997; Zhao and Piwnica-Worms, 2001; Zhao et al., 2002; Roshal et al., 2003). It has been proposed that the activation of ATR-dependent DNA damage G2/M checkpoint by Vpr involves the direct binding of this viral protein to chromatin, as mutants which are unable to bind chromatin fail to activate ATR (Lai et al., 2005), although the underlying mechanism remains controversial. Lai et al. reported that Vpr does not induce DNA double-strand breaks (DSBs), but increases the association of replication protein A (RPA) with chromatin, an upstream event required for ATR activation (Cortez et al., 2001; Zou and Elledge, 2003; Lai et al., 2005). However, using the same pulsed-field gel electrophoresis assay, Tachiwana et al. later showed that Vpr expression could induce chromosomal DSBs (Tachiwana et al., 2006). Differences in experimental settings may underpin this discrepancy. Lai et al. relied on Vpr-transfected cells for the PFGE analysis, whereas isolated nuclei that were treated with purified Vpr were used in the other study. The NS1 of the autonomous parvovirus minute virus of mice (MVMp) is another example of viral protein that affects the integrity of chromatin. MVMp infection leads to cell cycle arrest in S phase as a result of NS1-induced nicks in the cellular chromatin (Op De Beeck et al., 1995; Op De Beeck and Caillet-Fauquet, 1997).
Table 6. Cell cycle subversion by other mechanisms.
Infection of adeno-associated virus (AAV) leads to accumulation of cells with 4N DNA content in p53-competent cells, whereas it induces apoptosis in p53-deficient cells (Raj et al., 2001). Interestingly, virus replication or viral protein expression is not required for AAV-mediated cell cycle arrest as ultraviolet-treated AAV maintain their ability to perturb cell cycle, pointing to the role of AAV genome in these effects (Raj et al., 2001). In support of this hypothesis, empty AAV particles, which lack viral genome, fail to affect cell growth (Raj et al., 2001). It was proposed that the special structure of AAV genome, a single-stranded DNA with hairpin loops at both ends, could trigger host DNA damage response and mediate cell cycle arrest in a p53/p21WAF1/CIP1-dependent manner (Raj et al., 2001). Indeed, using an oligonucleotide corresponding to the AAV hairpin structure but not the AAV coding sequence, the same group successfully replicated the phenotypes induced by AAV infection (Raj et al., 2001). Likewise, expression of the individual proteins of Bocavirus minute virus of canines (MVC) has no effect on cell cycle progression (Chen et al., 2010). However, inoculation of UV-inactivated virus or a nonreplicative mutant virus phenocopied the G2/M arrest induced by MVC infection, suggesting that viral genome counts for the effect (Chen et al., 2010). Indeed, the use of a series of MVC mutant constructs indicated the terminal repeats of its genome, which form a strong secondary structure, are essential in the G2/M arrest feature of MVC (Sun et al., 2009; Chen et al., 2010).
Other than viral proteins and genome, virus-encoded microRNAs (miRNAs) have been also implicated to play a role in regulating host cell cycle. For example, KSHV miRNA K1 specifically represses the expression of p21WAF1/CIP1 via the 3′-UTR of this CKI, antagonizing p21WAF1/CIP1-mediated cell cycle arrest (Gottwein and Cullen, 2010). The significance of viral miRNA in favoring cell cycle progression was studied by introducing mutations into the precursor miRNA transcripts of EBV, which prevents the subsequent production of mature viral miRNAs thus antagonizes both early B-cell proliferation and miRNA protection from spontaneous apoptosis (Seto et al., 2010).
Measles virus (MV) was reported to subvert dendritic cells (DCs) and induce immune suppression. Dubois et al. found that infection of DCs by MV led to their functional maturation, allowing the migration of DCs from mucosal surfaces to draining lymph nodes to prime naive T cells (Dubois et al., 2001). Interestingly, instead of activating T cells, MV-infected DCs block T-cell expansion through cell-to-cell contact. The use of anti-MV HA or fusion protein (F) antibodies revealed that these two viral proteins, which are expressed on virus-infected DCs, are responsible for the suppression of T-cell proliferation (Dubois et al., 2001). Thus, through surface expression of HA and F on virus-infected DCs, MV takes a free ride on DCs to reach draining lymph nodes and subverts the normal function of DCs to inhibit T-cell proliferation. This infection-independent action in T cells results in accumulation of cells in G0/G1, owing to the inhibition of PI3K signaling in response to T-cell receptor binding (Griffin, 2010).
Zika virus (ZIKV), a flavivirus that is transmitted to humans primarily by Aedes mosquitoes, has been shown to cause congenital microcephaly when it infects women early in pregnancy (de Araújo et al., 2018; Hoen et al., 2018). This has led to a flurry of studies in different experimental models that have established the ZIKV induces massive abnormalities of cell division, including the presence of supernumerary centrosomes, that lead to virally-induced caspase 3-mediated apoptotic cell death and cell cycle abnormalities of human neural progenitor cells (Faizan et al., 2016; Souza et al., 2016; Gabriel et al., 2017), although how it interferes with the cell cycle has not been precisely defined. Moreover, transcriptional profiling of infected human neural progenitor cells has revealed a dysregulation of cell cycle- and differentiation-related pathways in neural progenitor cells (Dang et al., 2016; Li et al., 2016; Tang et al., 2016). Interestingly, it has been observed that an antiproliferactive and pro-apoptotic p53-related response markedly different from p53-induced transcriptional changes upon infection by HCMV, which can also cause microcephaly, is activated in ZIKA-infected human neural progenitor cells (Ghouzzi et al., 2016). More recently, by investigating the effect of individual ZIKV proteins in vitro, it was found that expression of the membrane-anchored capsid anaC, structural envelope E, membrane M proteins, and nonstructural protein 4A (NS4A) could trigger G2/M cell cycle arrest and apoptosis (Li et al., 2017; Liu et al., 2018). The NS4A induces growth restriction via the target of rapamycin (TOR) cellular stress pathway (Li et al., 2017), whereas envelope-triggered apoptosis is associated with upregulation of both p53 and p21Cip1/Waf1 and downregulation of cyclin B1 (Liu et al., 2018). Interestingly, ZIKA membrane protein (M) induce arrest of the cell cycle in G2/M, whereas the pre-membrane protein (prM) results in G1 accumulation (Li et al., 2017), revealing that posttranslational processing modifies its interference with cell cycle progression.
Conclusions
The interplay between the life cycle of viruses and the series of events leading to cell division has been actively investigated since the discovery that certain tumors were associated to viral infection. Although estimates of the incidence of cancers caused by viruses vary, both DNA and RNA viruses have been clearly linked to the development of human cancers (Liao, 2006; Mesri et al., 2014). However, most viruses have evolved a more complicated relationship with the cell cycle contenting themselves with surreptitious tampering for more effective replication and productive infection, without leading to cell transformation and tumorigenesis. The most obvious mechanism is the direct interaction of a viral gene product with a host protein to disarm its function, which has been adopted by many viruses as a common strategy to exploit host cell cycle. It is commonly held that, by interacting with cell cycle regulators and delaying cell division, these viruses can subvert the biosynthetic apparatus, re-directing it from the production of host proteins needed for cell cycle progression to suit their replication needs. Viruses stop at nothing and use any means necessary to survive in a hostile environment by inducing posttranslational modifications and dislocating host restriction factors to modulate their function and/or degradation. Co-evolution and adaptation of virus-host interactions is best described as a zero-sum biological arms race. It is clear that viruses could utilize different nodes of the cell cycle machinery to hamper cell division to escape the dependency on the normal progression of cellular proliferation. The mechanistic study of these interactions has revealed unexpected, non-canonical roles of cellular proteins. Further work should bring about a more refined understanding that will shed new light on the panoply of assets available to viruses to subvert the process of cell division, while at the same time providing insight into the function of host proteins that can offer unexpected handles to counteract viral infection.
Author Contributions
All authors listed have made a substantial, direct and intellectual contribution to the work, and approved it for publication.
Funding
Work in the authors' lab was supported by grants from the Research Fund for the Control of Infectious (RFCID 11101332), Area of Excellence Scheme of the University Grants Committee (AoE/M-12/06), the Research Grants Council of the Hong Kong Special Administrative Region, PR China (T11-705/14N and 17113915), and BNP Paribas CIB. YF was the recipient of a Hong Kong PhD Fellowship from the Hong Kong Research Grants Council/HKU Foundation Postgraduate Fellowship. SS is supported in part by the Croucher Foundation.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Ahn, J. Y., Schwarz, J. K., Piwnica-Worms, H., and Canman, C. E. (2000). Threonine 68 phosphorylation by ataxia telangiectasia mutated is required for efficient activation of Chk2 in response to ionizing radiation. Cancer Res 60, 5934–5936.
Albrecht, J. C. (2000). Primary structure of the Herpesvirus ateles genome. J. Virol. 74, 1033–1037. doi: 10.1128/JVI.74.2.1033-1037.2000
Aleem, E., Kiyokawa, H., and Kaldis, P. (2005). Cdc2-cyclin E complexes regulate the G1/S phase transition. Nat. Cell Biol. 7, 831–893. doi: 10.1038/ncb1284
Alexandrow, M. G., and Hamlin, J. L. (2005). Chromatin decondensation in S-phase involves recruitment of Cdk2 by Cdc45 and histone H1 phosphorylation. J. Cell Biol. 168, 875–886. doi: 10.1083/jcb.200409055
Ando, T., Kawabe, T., Ohara, H., Ducommun, B., Itoh, M., and Okamoto, T. (2001). Involvement of the interaction between p21 and proliferating cell nuclear antigen for the maintenance of G2/M arrest after DNA damage. J. Biol. Chem. 276, 42971–42977. doi: 10.1074/jbc.M106460200
Artavanis-Tsakonas, S., Rand, M. D., and Lake, R. J. (1999). Notch signaling: cell fate control and signal integration in development. Science 284, 770–776. doi: 10.1126/science.284.5415.770
Baer, A., Austin, D., Narayanan, A., Popova, T., Kainulainen, M., Bailey, C., et al. (2012). Induction of DNA damage signaling upon Rift Valley fever virus infection results in cell cycle arrest and increased viral replication. J. Biol. Chem. 287, 7399–7410. doi: 10.1074/jbc.M111.296608
Bagchi, S., Raychaudhuri, P., and Nevins, J. R. (1990). Adenovirus E1A proteins can dissociate heteromeric complexes involving the E2F transcription factor: a novel mechanism for E1A trans-activation. Cell 62, 659–669. doi: 10.1016/0092-8674(90)90112-R
Baldin, V., Lucas, J., Marcote, M. J., Pagano, M., and Draetta, G. (1993). Cyclin D1 is a nuclear protein required for cell cycle progression in G1. Genes Dev. 7, 812–821. doi: 10.1101/gad.7.5.812
Banerjee, N. S., Wang, H. K., Broker, T. R., and Chow, L. T. (2011). Human papillomavirus (HPV) E7 induces prolonged G2 following S phase reentry in differentiated human keratinocytes. J. Biol. Chem. 286, 15473–15482. doi: 10.1074/jbc.M110.197574
Barbosa, M. S., Edmonds, C., Fisher, C., Schiller, J. T., Lowy, D. R., and Vousden, K. H. (1990). The region of the HPV E7 oncoprotein homologous to adenovirus E1a and Sv40 large T antigen contains separate domains for Rb binding and casein kinase II phosphorylation. EMBO J. 9, 153–160. doi: 10.1002/j.1460-2075.1990.tb08091.x
Bartek, J., Bartkova, J., and Lukas, J. (1996). The retinoblastoma protein pathway and the restriction point. Curr. Opin. Cell Biol. 8, 805–814. doi: 10.1016/S0955-0674(96)80081-0
Bartek, J., and Lukas, J. (2003). Chk1 and Chk2 kinases in checkpoint control and cancer. Cancer Cell 3, 421–429. doi: 10.1016/S1535-6108(03)00110-7
Baydoun, H. H., Pancewicz, J., Bai, X., and Nicot, C. (2010). HTLV-I p30 inhibits multiple S phase entry checkpoints, decreases cyclin E-CDK2 interactions and delays cell cycle progression. Mol. Cancer 9:302. doi: 10.1186/1476-4598-9-302
Belyavskyi, M., Braunagel, S. C., and Summers, M. D. (1998). The structural protein ODV-EC27 of Autographa californica nucleopolyhedrovirus is a multifunctional viral cyclin. Proc. Natl. Acad. Sci. U.S.A. 95, 11205–11210. doi: 10.1073/pnas.95.19.11205
Benn, J., and Schneider, R. J. (1995). Hepatitis B virus HBx protein deregulates cell cycle checkpoint controls. Proc. Natl. Acad. Sci. U.S.A. 92, 11215–11219. doi: 10.1073/pnas.92.24.11215
Berger, G., Lawrence, M., Hue, S., and Neil, S. J. (2015). G2/M cell cycle arrest correlates with primate lentiviral Vpr interaction with the SLX4 complex. J. Virol. 89, 230–240. doi: 10.1128/JVI.02307-14
Berthet, C., Aleem, E., Coppola, V., Tessarollo, L., and Kaldis, P. (2003). Cdk2 knockout mice are viable. Curr. Biol. 13, 1775–1785. doi: 10.1016/j.cub.2003.09.024
Bishop, J. M. (1991). Molecular themes in oncogenesis. Cell 64, 235–248. doi: 10.1016/0092-8674(91)90636-D
Blagosklonny, M. V., and Pardee, A. B. (2002). The restriction point of the cell cycle. Cell Cycle 1, 103–110. doi: 10.4161/cc.1.2.108
Boehm, M., Yoshimoto, T., Crook, M. F., Nallamshetty, S., True, A., Nabel, G. J., et al. (2002). A growth factor-dependent nuclear kinase phosphorylates p27(Kip1) and regulates cell cycle progression. EMBO J. 21, 3390–3401. doi: 10.1093/emboj/cdf343
Bolton, D. L., Barnitz, R. A., Sakai, K., and Lenardo, M. J. (2008). 14-3-3 theta binding to cell cycle regulatory factors is enhanced by HIV-1 Vpr. Biol. Direct 3:17. doi: 10.1186/1745-6150-3-17
Bondar, T., Kalinina, A., Khair, L., Kopanja, D., Nag, A., Bagchi, S., et al. (2006). Cul4A and DDB1 associate with Skp2 to target p27Kip1 for proteolysis involving the COP9 signalosome. Mol. Cell. Biol. 26, 2531–2539. doi: 10.1128/MCB.26.7.2531-2539.2006
Boutell, C., and Everett, R. D. (2003). The herpes simplex virus type 1 (HSV-1) regulatory protein ICP0 interacts with and Ubiquitinates p53. J. Biol. Chem. 278, 36596–36602. doi: 10.1074/jbc.M300776200
Boutell, C., Sadis, S., and Everett, R. D. (2002). Herpes simplex virus type 1 immediate-early protein ICP0 and is isolated RING finger domain act as ubiquitin E3 ligases in vitro. J. Virol. 76, 841–850. doi: 10.1128/JVI.76.2.841-850.2002
Braunagel, S. C., Parr, R., Belyavskyi, M., and Summers, M. D. (1998). Autographa californica nucleopolyhedrovirus infection results in Sf9 cell cycle arrest at G2/M phase. Virology 244, 195–211. doi: 10.1006/viro.1998.9097
Bresnahan, W. A., Thompson, E. A., and Albrecht, T. (1997). Human cytomegalovirus infection results in altered Cdk2 subcellular localization. J. Gen. Virol. 78, 1993–1997. doi: 10.1099/0022-1317-78-8-1993
Brewster, C. D., Birkenheuer, C. H., Vogt, M. B., Quackenbush, S. L., and Rovnak, J. (2011). The retroviral cyclin of walleye dermal sarcoma virus binds cyclin-dependent kinases 3 and 8. Virology 409, 299–307. doi: 10.1016/j.virol.2010.10.022
Brown, N. R., Noble, M. E., Lawrie, A. M., Morris, M. C., Tunnah, P., Divita, G., et al. (1999). Effects of phosphorylation of threonine 160 on cyclin-dependent kinase 2 structure and activity. J. Biol. Chem. 274, 8746–8756. doi: 10.1074/jbc.274.13.8746
Bulavin, D. V., Higashimoto, Y., Demidenko, Z. N., Meek, S., Graves, P., Phillips, C., et al. (2003). Dual phosphorylation controls Cdc25 phosphatases and mitotic entry. Nat. Cell Biol. 5, 545–551. doi: 10.1038/ncb994
Card, G. L., Knowles, P., Laman, H., Jones, N., and McDonald, N. Q. (2000). Crystal structure of a gamma-herpesvirus cyclin-cdk complex. EMBO J. 19, 2877–2888. doi: 10.1093/emboj/19.12.2877
Carrano, A. C., Eytan, E., Hershko, A., and Pagano, M. (1999). SKP2 is required for ubiquitin-mediated degradation of the CDK inhibitor p27. Nat. Cell Biol. 1, 193–199. doi: 10.1038/12013
Cesarman, E., Chang, Y., Moore, P. S., Said, J. W., and Knowles, D. M. (1995). Kaposi's sarcoma-associated herpesvirus-like DNA-sequences in AIDS-related body-cavity-based lymphomas. N. Engl. J. Med. 332, 1186–1191. doi: 10.1056/NEJM199505043321802
Chang, Y., Cesarman, E., Pessin, M. S., Lee, F., Culpepper, J., Knowles, D. M., et al. (1994). Identification of herpesvirus-like DNA-sequences in AIDS-associated Kaposi's-sarcoma. Science 266, 1865–1869. doi: 10.1126/science.7997879
Chang, Y., Moore, P. S., Talbot, S. J., Boshoff, C. H., Zarkowska, T., Godden, K., et al. (1996). Cyclin encoded by KS herpesvirus. Nature 382:410. doi: 10.1038/382410a0
Chang, Y., Moore, P. S., and Weiss, R. A. (2017). Human oncogenic viruses: nature and discovery. Philos. Trans. R. Soc. Lond. B. Biol. Sci. 372:20160264. doi: 10.1098/rstb.2016.0264
Chen, A. Y., Luo, Y., Cheng, F., Sun, Y., and Qiu, J. (2010). Bocavirus infection induces mitochondrion-mediated apoptosis and cell cycle arrest at G(2)/M phase. J. Virol. 84, 5615–5626. doi: 10.1128/JVI.02094-09
Chen, B. J., Leser, G. P., Jackson, D., and Lamb, R. A. (2008). The influenza virus M2 protein cytoplasmic tail interacts with the M1 protein and influences virus assembly at the site of virus budding. J. Virol. 82, 10059–10070. doi: 10.1128/JVI.01184-08
Chen, S., and Paucha, E. (1990). Identification of a region of simian virus 40 large T antigen required for cell transformation. J. Virol. 64, 3350–3357.
Chen, Z., Knutson, E., Kurosky, A., and Albrecht, T. (2001). Degradation of p21cip1 in cells productively infected with human cytomegalovirus. J. Virol. 75, 3613–3625. doi: 10.1128/JVI.75.8.3613-3625.2001
Chiu, H. C., Huang, W. R., Liao, T. L., Chi, P. I., Nielsen, B. L., Liu, J. H., et al. (2018). Mechanistic insights into avian reovirus p17-modulated suppression of cell-cycle CDK/cyclin complexes and enhancement of p53 and cyclin H interaction. J. Biol. Chem. 293, 12542–12562. doi: 10.1074/jbc.RA118.002341
Chiu, H. C., Huang, W. R., Liao, T. L., Wu, H. Y., Munir, M., Shih, W. L., et al. (2016). Suppression of vimentin phosphorylation by the avian reovirus p17 through inhibition of CDK1 and Plk1 impacting the G2/M phase of the cell cycle. PLoS ONE 11:e0162356. doi: 10.1371/journal.pone.0162356
Choudhuri, T., Verma, S. C., Lan, K., Murakami, M., and Robertson, E. S. (2007). The ATM/ATR signaling effector Chk2 is targeted by Epstein-Barr virus nuclear antigen 3C to release the G(2)/M cell cycle block. J. Virol. 81, 6718–6730. doi: 10.1128/JVI.00053-07
Clark, E., Santiago, F., Deng, L. W., Chong, S. Y., de la Fuente, C., Wang, L., et al. (2000). Loss of G(1)/S checkpoint in human immunodeficiency virus type 1-infected cells is associated with a lack of cyclin-dependent kinase inhibitor p21/Waf1. J. Virol. 74, 5040–5052. doi: 10.1128/JVI.74.11.5040-5052.2000
Conaway, R. C., and Conaway, J. W. (2011). Function and regulation of the Mediator complex. Curr. Opin. Genet. Dev. 21, 225–230. doi: 10.1016/j.gde.2011.01.013
Cortez, D., Guntuku, S., Qin, J., and Elledge, S. J. (2001). ATR and ATRIP: partners in checkpoint signaling. Science 294, 1713–1716. doi: 10.1126/science.1065521
Dang, J., Tiwari, S. K., Lichinchi, G., Qin, Y., Patil, V. S., Eroshkin, A. M., et al. (2016). Zika virus depletes neural progenitors in human cerebral organoids through activation of the innate immune receptor TLR3. Cell Stem Cell 19, 258–265. doi: 10.1016/j.stem.2016.04.014
Das, S., and Basu, A. (2008). Japanese encephalitis virus infects neural progenitor cells and decreases their proliferation. J. Neurochem. 106, 1624–1636. doi: 10.1111/j.1471-4159.2008.05511.x
de Araújo, T. V. B., Ximenes, R. A. A., Miranda-Filho, D. B., Souza, W. V., Montarroyos, U. R., de Melo, A. P. L., et al. (2018). Association between microcephaly, Zika virus infection, and other risk factors in Brazil: final report of a case-control study. Lancet Infect. Dis. 18, 328–336. doi: 10.1016/S1473-3099(17)30727-2
de Noronha, C. M., Sherman, M. P., Lin, H. W., Cavrois, M. V., Moir, R. D., Goldman, R. D., et al. (2001). Dynamic disruptions in nuclear envelope architecture and integrity induced by HIV-1 Vpr. Science 294, 1105–1108. doi: 10.1126/science.1063957
DeCaprio, J. A., Ludlow, J. W., Figge, J., Shew, J. Y., Huang, C. M., Lee, W. H., et al. (1988). SV40 large tumor antigen forms a specific complex with the product of the retinoblastoma susceptibility gene. Cell 54, 275–283. doi: 10.1016/0092-8674(88)90559-4
Deng, L., Nagano-Fujii, M., Tanaka, M., Nomura-Takigawa, Y., Ikeda, M., Kato, N., et al. (2006). NS3 protein of Hepatitis C virus associates with the tumour suppressor p53 and inhibits its function in an NS3 sequence-dependent manner. J. Gen. Virol. 87, 1703–1713. doi: 10.1099/vir.0.81735-0
Desai, D., Gu, Y., and Morgan, D. O. (1992). Activation of human cyclin-dependent kinases in vitro. Mol. Biol. Cell 3, 571–582. doi: 10.1091/mbc.3.5.571
Devault, A., Martinez, A. M., Fesquet, D., Labbe, J. C., Morin, N., Tassan, J. P., et al. (1995). MAT1 ('menage a trois') a new RING finger protein subunit stabilizing cyclin H-cdk7 complexes in starfish and Xenopus, CAK. EMBO J. 14, 5027–5036. doi: 10.1002/j.1460-2075.1995.tb00185.x
Diehl, J. A., Cheng, M., Roussel, M. F., and Sherr, C. J. (1998). Glycogen synthase kinase-3beta regulates cyclin D1 proteolysis and subcellular localization. Genes Dev. 12, 3499–3511. doi: 10.1101/gad.12.22.3499
Dove, B., Brooks, G., Bicknell, K., Wurm, T., and Hiscox, J. A. (2006). Cell cycle perturbations induced by infection with the coronavirus infectious bronchitis virus and their effect on virus replication. J. Virol. 80, 4147–4156. doi: 10.1128/JVI.80.8.4147-4156.2006
Dubois, B., Lamy, P. J., Chemin, K., Lachaux, A., and Kaiserlian, D. (2001). Measles virus exploits dendritic cells to suppress CD4(+) T-cell proliferation via expression of surface viral glycoproteins independently of T-cell trans-infection. Cell. Immunol. 214, 173–183. doi: 10.1006/cimm.2001.1898
Durkin, S. S., Guo, X., Fryrear, K. A., Mihaylova, V. T., Gupta, S. K., Belgnaoui, S. M., et al. (2008). HTLV-1 Tax oncoprotein subverts the cellular DNA damage response via binding to DNA-dependent protein kinase. J. Biol. Chem. 283, 36311–36320. doi: 10.1074/jbc.M804931200
Dyson, N., Howley, P. M., Munger, K., and Harlow, E. (1989). The human papilloma virus-16 E7 oncoprotein is able to bind to the retinoblastoma gene product. Science 243, 934–937. doi: 10.1126/science.2537532
el-Deiry, W. S., Tokino, T., Velculescu, V. E., Levy, D. B., Parsons, R., Trent, J. M., et al. (1993). WAF1, a potential mediator of p53 tumor suppression. Cell 75, 817–825. doi: 10.1016/0092-8674(93)90500-P
Everett, R. D., Earnshaw, W. C., Findlay, J., and Lomonte, P. (1999). Specific destruction of kinetochore protein CENP-C and disruption of cell division by herpes simplex virus immediate-early protein Vmw110. EMBO J. 18, 1526–1538. doi: 10.1093/emboj/18.6.1526
Ewen, M. E., Sluss, H. K., Sherr, C. J., Matsushime, H., Kato, J., and Livingston, D. M. (1993). Functional interactions of the retinoblastoma protein with mammalian D-type cyclins. Cell 73, 487–497. doi: 10.1016/0092-8674(93)90136-E
Faizan, M. I., Abdullah, M., Ali, S., Naqvi, I. H., Ahmed, A., and Parveen, S. (2016). Zika virus-induced microcephaly and its possible molecular mechanism. Intervirology 59, 152–158. doi: 10.1159/000452950
Fan, Y., Mok, C. K., Chan, M. C., Zhang, Y., Nal, B., Kien, F., et al. (2017). Cell cycle-independent role of cyclin D3 in host restriction of influenza virus infection. J. Biol. Chem. 292, 5070–5088. doi: 10.1074/jbc.M117.776112
Fekairi, S., Scaglione, S., Chahwan, C., Taylor, E. R., Tissier, A., Coulon, S., et al. (2009). Human SLX4 is a Holliday junction resolvase subunit that binds multiple DNA repair/recombination endonucleases. Cell 138, 78–89. doi: 10.1016/j.cell.2009.06.029
Fickenscher, H., and Fleckenstein, B. (2001). Herpesvirus saimiri. Philos. Trans. R. Soc. Lond. B. Biol. Sci. 356, 545–567. doi: 10.1098/rstb.2000.0780
Fisher, R. P., and Morgan, D. O. (1994). A novel cyclin associates with MO15/CDK7 to form the CDK-activating kinase. Cell 78, 713–724. doi: 10.1016/0092-8674(94)90535-5
Fortunato, E. A., and Spector, D. H. (1998). p53 and RPA are sequestered in viral replication centers in the nuclei of cells infected with human cytomegalovirus. J. Virol. 72, 2033–2039.
Fu, H., Subramanian, R. R., and Masters, S. C. (2000). 14-3-3 proteins: structure, function, and regulation. Annu. Rev. Pharmacol. Toxicol. 40, 617–647. doi: 10.1146/annurev.pharmtox.40.1.617
Funk, J. O., Waga, S., Harry, J. B., Espling, E., Stillman, B., and Galloway, D. A. (1997). Inhibition of CDK activity and PCNA-dependent DNA replication by p21 is blocked by interaction with the HPV-16 E7 oncoprotein. Genes Dev. 11, 2090–2100. doi: 10.1101/gad.11.16.2090
Furumoto, T., Tanaka, A., Ito, M., Malik, S., Hirose, Y., Hanaoka, F., et al. (2007). A kinase subunit of the human mediator complex, CDK8, positively regulates transcriptional activation. Genes Cells 12, 119–132. doi: 10.1111/j.1365-2443.2007.01036.x
Gabriel, E., Ramani, A., Karow, U., Gottardo, M., Natarajan, K., Gooi, L. M., et al. (2017). Recent Zika virus isolates induce premature differentiation of neural progenitors in human brain organoids. Cell Stem Cell 20, 397–406. doi: 10.1016/j.stem.2016.12.005
Geng, Y., Yu, Q. Y., Sicinska, E., Das, M., Schneider, J. E., Bhattacharya, S., et al. (2003). Cyclin E ablation in the mouse. Cell 114, 431–443. doi: 10.1016/S0092-8674(03)00645-7
Ghouzzi, V. E., Bianchi, F. T., Molineris, I., Mounce, B. C., Berto, G. E., Rak, M., et al. (2016). ZIKA virus elicits P53 activation and genotoxic stress in human neural progenitors similar to mutations involved in severe forms of genetic microcephaly. Cell Death Dis. 7:e2440. doi: 10.1038/cddis.2016.266
Glotzer, M. (2001). Animal cell cytokinesis. Annu. Rev. Cell Dev. Biol. 17, 351–386. doi: 10.1146/annurev.cellbio.17.1.351
Godden-Kent, D., Talbot, S. J., Boshoff, C., Chang, Y., Moore, P., Weiss, R. A., et al. (1997). The cyclin encoded by Kaposi's sarcoma-associated herpesvirus stimulates cdk6 to phosphorylate the retinoblastoma protein and histone H1. J. Virol. 71, 4193–4198.
Goh, W. C., Manel, N., and Emerman, M. (2004). The human immunodeficiency virus Vpr protein binds Cdc25C: implications for G2 arrest. Virology 318, 337–349. doi: 10.1016/j.virol.2003.10.007
Gorbsky, G. J. (1995). Kinetochores, microtubules and the metaphase checkpoint. Trends Cell Biol. 5, 143–148. doi: 10.1016/S0962-8924(00)88968-0
Gottwein, E., and Cullen, B. R. (2010). A human herpesvirus microRNA inhibits p21 expression and attenuates p21-mediated cell cycle arrest. J. Virol. 84, 5229–5237. doi: 10.1128/JVI.00202-10
Gould, K. L., Moreno, S., Owen, D. J., Sazer, S., and Nurse, P. (1991). Phosphorylation at Thr167 is required for Schizosaccharomyces pombe p34cdc2 function. EMBO J. 10, 3297–3309. doi: 10.1002/j.1460-2075.1991.tb04894.x
Graves, P. R., Lovly, C. M., Uy, G. L., and Piwnica-Worms, H. (2001). Localization of human Cdc25C is regulated both by nuclear export and 14-3-3 protein binding. Oncogene 20, 1839–1851. doi: 10.1038/sj.onc.1204259
Griffin, D. E. (2010). Measles virus-induced suppression of immune responses. Immunol. Rev. 236, 176–189. doi: 10.1111/j.1600-065X.2010.00925.x
Gu, Y., Rosenblatt, J., and Morgan, D. O. (1992). Cell cycle regulation of CDK2 activity by phosphorylation of Thr160 and Tyr15. EMBO J. 11, 3995–4005. doi: 10.1002/j.1460-2075.1992.tb05493.x
Gulley, M. L., Burton, M. P., Allred, D. C., Nicholls, J. M., Amin, M. B., Ro, J. Y., et al. (1998). Epstein-Barr virus infection is associated with p53 accumulation in nasopharyngeal carcinoma. Hum. Pathol. 29, 252–259. doi: 10.1016/S0046-8177(98)90044-2
Guo, L., Tang, M., Yang, L., Xiao, L., Bode, A. M., Li, L., et al. (2012). Epstein-Barr virus oncoprotein LMP1 mediates survivin upregulation by p53 contributing to G1/S cell cycle progression in nasopharyngeal carcinoma. Int. J. Mol. Med. 29, 574–580. doi: 10.3892/ijmm.2012.889
Gupta, S. K., Guo, X., Durkin, S. S., Fryrear, K. F., Ward, M. D., and Semmes, O. J. (2007). Human T-cell leukemia virus type 1 Tax oncoprotein prevents DNA damage-induced chromatin egress of hyperphosphorylated Chk2. J. Biol. Chem. 282, 29431–29440. doi: 10.1074/jbc.M704110200
Hagglund, R., Van Sant, C., Lopez, P., and Roizman, B. (2002). Herpes simplex virus 1-infected cell protein 0 contains two E3 ubiquitin ligase sites specific for different E2 ubiquitin-conjugating enzymes. Proc. Natl. Acad. Sci. U.S.A. 99, 631–636. doi: 10.1073/pnas.022531599
Haller, K., Wu, Y. L., Derow, E., Schmitt, I., Jeang, K. T., and Grassmann, R. (2002). Physical interaction of human T-cell leukemia virus type 1 Tax with cyclin-dependent kinase 4 stimulates the phosphorylation of retinoblastoma protein. Mol. Cell. Biol. 22, 3327–3338. doi: 10.1128/MCB.22.10.3327-3338.2002
Haoudi, A., Daniels, R. C., Wong, E., Kupfer, G., and Semmes, O. J. (2003). Human T-cell leukemia virus-I tax oncoprotein functionally targets a subnuclear complex involved in cellular DNA damage-response. J. Biol. Chem. 278, 37736–37744. doi: 10.1074/jbc.M301649200
Hartwell, L. H., and Weinert, T. A. (1989). Checkpoints: controls that ensure the order of cell cycle events. Science 246, 629–634. doi: 10.1126/science.2683079
He, Y. A., Xu, K., Keiner, B., Zhou, J. F., Czudai, V., Li, T. X., et al. (2010). Influenza A virus replication induces cell cycle arrest in G(0)/G(1) phase. J. Virol. 84, 12832–12840. doi: 10.1128/JVI.01216-10
Hirao, A., Kong, Y. Y., Matsuoka, S., Wakeham, A., Ruland, J., Yoshida, H., et al. (2000). DNA damage-induced activation of p53 by the checkpoint kinase Chk2. Science 287, 1824–1827. doi: 10.1126/science.287.5459.1824
Hobbs, W. E. II., and DeLuca, N. A. (1999). Perturbation of cell cycle progression and cellular gene expression as a function of herpes simplex virus ICP0. J. Virol. 73, 8245–8255.
Hoen, B., Schaub, B., Funk, A. L., Ardillon, V., Boullard, M., Cabie, A., et al. (2018). Pregnancy outcomes after ZIKV infection in French Territories in the Americas. N. Engl. J. Med. 378, 985–994. doi: 10.1056/NEJMoa1709481
Hoffman, W. H., Biade, S., Zilfou, J. T., Chen, J., and Murphy, M. (2002). Transcriptional repression of the anti-apoptotic survivin gene by wild type p53. J. Biol. Chem. 277, 3247–3257. doi: 10.1074/jbc.M106643200
Hsu, J. Y., Reimann, J. D. R., Sorensen, C. S., Lukas, J., and Jackson, P. K. (2002). E2F-dependent accumulation of hEmi1 regulates S phase entry by inhibiting APC(Cdh1). Nat. Cell Biol. 4, 358–366. doi: 10.1038/ncb785
Huang, P. S., Patrick, D. R., Edwards, G., Goodhart, P. J., Huber, H. E., Miles, L., et al. (1993). Protein domains governing interactions between E2F, the retinoblastoma gene product, and human papillomavirus type 16 E7 protein. Mol. Cell. Biol. 13, 953–960. doi: 10.1128/MCB.13.2.953
Huang, W. R., Chi, P. I., Chiu, H. C., Hsu, J. L., Nielsen, B. L., Liao, T. L., et al. (2017). Avian reovirus p17 and sigmaA act cooperatively to downregulate Akt by suppressing mTORC2 and CDK2/cyclin A2 and upregulating proteasome PSMB6. Sci. Rep. 7:5226. doi: 10.1038/s41598-017-05510-x
Huang, W. R., Chiu, H. C., Liao, T. L., Chuang, K. P., Shih, W. L., and Liu, H. J. (2015). Avian reovirus protein p17 functions as a nucleoporin Tpr suppressor leading to activation of p53, p21 and PTEN and inactivation of PI3K/AKT/mTOR and ERK signaling pathways. PLoS One . 10:e0138627. doi: 10.1371/journal.pone.0138627
Huang, X. D., Summers, M. K., Pham, V., Lill, J. R., Liu, J. F., Lee, G., et al. (2011). Deubiquitinase USP37 is activated by CDK2 to antagonize APC(CDH1) and promote S phase entry. Mol. Cell 42, 511–523. doi: 10.1016/j.molcel.2011.03.027
Huard, S., Elder, R. T., Liang, D., Li, G., and Zhao, R. Y. (2008). Human immunodeficiency virus type 1 vpr induces cell cycle G(2) arrest through Srk1/MK2-mediated phosphorylation of Cdc25. J. Virol. 82, 2904–2917. doi: 10.1128/JVI.01098-07
Hume, A. J., Finkel, J. S., Kamil, J. P., Coen, D. M., Culbertson, M. R., and Kalejta, R. F. (2008). Phosphorylation of retinoblastoma protein by viral protein with cyclin-dependent kinase function. Science 320, 797–799. doi: 10.1126/science.1152095
Hunt, T. (1989). Maturation promoting factor, cyclin and the control of M-phase. Curr. Opin. Cell Biol. 1, 268–274. doi: 10.1016/0955-0674(89)90099-9
Ikeda, M. A., and Nevins, J. R. (1993). Identification of distinct roles for separate E1A domains in disruption of E2F complexes. Mol. Cell. Biol. 13, 7029–7035. doi: 10.1128/MCB.13.11.7029
Inman, G. J., and Farrell, P. J. (1995). Epstein-Barr virus EBNA-LP and transcription regulation properties of pRB, p107 and p53 in transfection assays. J. Gen. Virol. 76, 2141–2149. doi: 10.1099/0022-1317-76-9-2141
Iovine, B., Iannella, M. L., and Bevilacqua, M. A. (2011). Damage-specific DNA binding protein 1 (DDB1): a protein with a wide range of functions. Int. J. Biochem. Cell Biol. 43, 1664–1667. doi: 10.1016/j.biocel.2011.09.001
Irniger, S., Piatti, S., Michaelis, C., and Nasmyth, K. (1995). Genes involved in sister chromatid separation are needed for B-type cyclin proteolysis in budding yeast. Cell 81, 269–278. doi: 10.1016/0092-8674(95)90337-2
Ishida, N., Hara, T., Kamura, T., Yoshida, M., Nakayama, K., and Nakayama, K. I. (2002). Phosphorylation of p27Kip1 on serine 10 is required for its binding to CRM1 and nuclear export. J. Biol. Chem. 277, 14355–14358. doi: 10.1074/jbc.C100762200
Ishido, S., and Hotta, H. (1998). Complex formation of the nonstructural protein 3 of hepatitis C virus with the p53 tumor suppressor. FEBS Lett. 438, 258–262. doi: 10.1016/S0014-5793(98)01312-X
Iwanaga, R., Ozono, E., Fujisawa, J., Ikeda, M. A., Okamura, N., Huang, Y., et al. (2008). Activation of the cyclin D2 and cdk6 genes through NF-kappaB is critical for cell-cycle progression induced by HTLV-I Tax. Oncogene 27, 5635–5642. doi: 10.1038/onc.2008.174
Izumi, T., Io, K., Matsui, M., Shirakawa, K., Shinohara, M., Nagai, Y., et al. (2010). HIV-1 viral infectivity factor interacts with TP53 to induce G2 cell cycle arrest and positively regulate viral replication. Proc. Natl. Acad. Sci. U.S.A. 107, 20798–20803. doi: 10.1073/pnas.1008076107
Izumi, T., Takaori-Kondo, A., Shirakawa, K., Higashitsuji, H., Itoh, K., Io, K., et al. (2009). MDM2 is a novel E3 ligase for HIV-1 Vif. Retrovirology 6:1. doi: 10.1186/1742-4690-6-1
Izumiya, Y., Lin, S. F., Ellison, T. J., Levy, A. M., Mayeur, G. L., Izumiya, C., et al. (2003). Cell cycle regulation by Kaposi's sarcoma-associated herpesvirus K-bZIP: Direct interaction with cyclin-CDK2 and induction of G(1) growth arrest. J. Virol. 77, 9652–9661. doi: 10.1128/JVI.77.17.9652-9661.2003
Jiang, W., Wang, Q., Chen, S., Gao, S., Song, L., Liu, P., et al. (2013). Influenza A virus NS1 induces G0/G1 cell cycle arrest by inhibiting the expression and activity of RhoA protein. J. Virol. 87, 3039–3052. doi: 10.1128/JVI.03176-12
Jiang, W. Q., Szekely, L., Wendel-Hansen, V., Ringertz, N., Klein, G., and Rosen, A. (1991). Co-localization of the retinoblastoma protein and the Epstein-Barr virus-encoded nuclear antigen EBNA-5. Exp. Cell Res. 197, 314–318. doi: 10.1016/0014-4827(91)90438-Z
Jin, P., Gu, Y., and Morgan, D. O. (1996). Role of inhibitory CDC2 phosphorylation in radiation-induced G2 arrest in human cells. J. Cell Biol. 134, 963–970. doi: 10.1083/jcb.134.4.963
Johnson, R. A., Yurochko, A. D., Poma, E. E., Zhu, L., and Huang, E. S. (1999). Domain mapping of the human cytomegalovirus IE1-72 and cellular p107 protein-protein interaction and the possible functional consequences. J. Gen. Virol. 80, 1293–1303. doi: 10.1099/0022-1317-80-5-1293
Jones, D. L., Alani, R. M., and Munger, K. (1997). The human papillomavirus E7 oncoprotein can uncouple cellular differentiation and proliferation in human keratinocytes by abrogating p21Cip1-mediated inhibition of cdk2. Genes Dev. 11, 2101–2111. doi: 10.1101/gad.11.16.2101
Jung, J. U., Stager, M., and Desrosiers, R. C. (1994). Virus-encoded cyclin. Mol. Cell. Biol. 14, 7235–7244. doi: 10.1128/MCB.14.11.7235
Kaldis, P. (1999). The cdk-activating kinase (CAK): from yeast to mammals. Cell. Mol. Life Sci. 55, 284–296. doi: 10.1007/s000180050290
Kaldis, P., Russo, A. A., Chou, H. S., Pavletich, N. P., and Solomon, M. J. (1998). Human and yeast cdk-activating kinases (CAKs) display distinct substrate specificities. Mol. Biol. Cell 9, 2545–2560. doi: 10.1091/mbc.9.9.2545
Kamata, M., Watanabe, N., Nagaoka, Y., and Chen, I. S. (2008). Human immunodeficiency virus type 1 Vpr binds to the N lobe of the Wee1 kinase domain and enhances kinase activity for CDC2. J. Virol. 82, 5672–5682. doi: 10.1128/JVI.01330-07
Kato, J. Y., Matsuoka, M., Strom, D. K., and Sherr, C. J. (1994). Regulation of cyclin D-dependent kinase 4 (cdk4) by cdk4-activating kinase. Mol. Cell. Biol. 14, 2713–2721. doi: 10.1128/MCB.14.4.2713
Kim, J. Y., Park, S. Y., Lyoo, H. R., Koo, E. S., Kim, M. S., and Jeong, Y. S. (2015). Extended stability of cyclin D1 contributes to limited cell cycle arrest at G1-phase in BHK-21 cells with Japanese encephalitis virus persistent infection. J. Microbiol. 53, 77–83. doi: 10.1007/s12275-015-4661-z
Kim, Y., Spitz, G. S., Veturi, U., Lach, F. P., Auerbach, A. D., and Smogorzewska, A. (2013). Regulation of multiple DNA repair pathways by the Fanconi anemia protein SLX4. Blood 121, 54–63. doi: 10.1182/blood-2012-07-441212
King, R. W., Peters, J. M., Tugendreich, S., Rolfe, M., Hieter, P., and Kirschner, M. W. (1995). A 20S complex containing CDC27 and CDC16 catalyzes the mitosis-specific conjugation of ubiquitin to cyclin B. Cell 81, 279–288. doi: 10.1016/0092-8674(95)90338-0
Kino, T., Gragerov, A., Valentin, A., Tsopanomihalou, M., Ilyina-Gragerova, G., Erwin-Cohen, R., et al. (2005). Vpr protein of human immunodeficiency virus type 1 binds to 14-3-3 proteins and facilitates complex formation with Cdc25C: implications for cell cycle arrest. J. Virol. 79, 2780–2787. doi: 10.1128/JVI.79.5.2780-2787.2005
Knight, J. S., and Robertson, E. S. (2004). Epstein-Barr virus nuclear antigen 3C regulates cyclin A/p27 complexes and enhances cyclin A-dependent kinase activity. J. Virol. 78, 1981–1991. doi: 10.1128/JVI.78.4.1981-1991.2004
Kopan, R., and Ilagan, M. X. (2009). The canonical Notch signaling pathway: unfolding the activation mechanism. Cell 137, 216–233. doi: 10.1016/j.cell.2009.03.045
Kovacs, A., Weber, M. L., Burns, L. J., Jacob, H. S., and Vercellotti, G. M. (1996). Cytoplasmic sequestration of p53 in cytomegalovirus-infected human endothelial cells. Am. J. Pathol. 149, 1531–1539.
Kozar, K., Ciemerych, M. A., Rebel, V. I., Shigematsu, H., Zagozdzon, A., Sicinska, E., et al. (2004). Mouse development and cell proliferation in the absence of D-cyclins. Cell 118, 477–491. doi: 10.1016/j.cell.2004.07.025
Krek, W., and Nigg, E. A. (1992). Cell cycle regulation of vertebrate p34cdc2 activity: identification of Thr161 as an essential in vivo phosphorylation site. New Biol. 4, 323–329.
Krude, T., Jackman, M., Pines, J., and Laskey, R. A. (1997). Cyclin/Cdk-dependent initiation of DNA replication in a human cell-free system. Cell 88, 109–119. doi: 10.1016/S0092-8674(00)81863-2
Kuo, Y. L., and Giam, C. Z. (2006). Activation of the anaphase promoting complex by HTLV-1 tax leads to senescence. EMBO J. 25, 1741–1752. doi: 10.1038/sj.emboj.7601054
Lacey, K. R., Jackson, P. K., and Stearns, T. (1999). Cyclin-dependent kinase control of centrosome duplication. Proc. Natl. Acad. Sci. U.S.A. 96, 2817–2822. doi: 10.1073/pnas.96.6.2817
Laguette, N., Bregnard, C., Hue, P., Basbous, J., Yatim, A., Larroque, M., et al. (2014). Premature activation of the SLX4 complex by Vpr promotes G2/M arrest and escape from innate immune sensing. Cell 156, 134–145. doi: 10.1016/j.cell.2013.12.011
Lai, M., Zimmerman, E. S., Planelles, V., and Chen, J. (2005). Activation of the ATR pathway by human immunodeficiency virus type 1 Vpr involves its direct binding to chromatin in vivo. J. Virol. 79, 15443–15451. doi: 10.1128/JVI.79.24.15443-15451.2005
Lan, K. H., Sheu, M. L., Hwang, S. J., Yen, S. H., Chen, S. Y., Wu, J. C., et al. (2002). HCV NS5A interacts with p53 and inhibits p53-mediated apoptosis. Oncogene 21, 4801–4811. doi: 10.1038/sj.onc.1205589
Lane, D. P., and Crawford, L. V. (1979). T antigen is bound to a host protein in SV40-transformed cells. Nature 278, 261–263. doi: 10.1038/278261a0
LaPierre, L. A., Casey, J. W., and Holzschu, D. L. (1998). Walleye retroviruses associated with skin tumors and hyperplasias encode cyclin D homologs. J. Virol. 72, 8765–8771.
Laronga, C., Yang, H. Y., Neal, C., and Lee, M. H. (2000). Association of the cyclin-dependent kinases and 14-3-3 sigma negatively regulates cell cycle progression. J. Biol. Chem. 275, 23106–23112. doi: 10.1074/jbc.M905616199
Le Rouzic, E., Belaidouni, N., Estrabaud, E., Morel, M., Rain, J.-C., Transy, C., et al. (2007). HIV1 Vpr arrests the cell cycle by recruiting DCAF1/VprBP, a receptor of the Cul4-DDB1 ubiquitin ligase. Cell Cycle 6, 182–188. doi: 10.4161/cc.6.2.3732
Le, L., and Leluk, J. (2011). Study on phylogenetic relationships, variability, and correlated mutations in M2 proteins of influenza virus A. PLoS ONE 6:e22970. doi: 10.1371/journal.pone.0022970
Lehman, J. M., Laffin, J., and Friedrich, T. D. (2000). Simian virus 40 induces multiple S phases with the majority of viral DNA replication in the G2 and second S phase in CV-1 cells. Exp. Cell Res. 258, 215–222. doi: 10.1006/excr.2000.4927
Leupin, O., Bontron, S., and Strubin, M. (2003). Hepatitis B virus X protein and simian virus 5 V protein exhibit similar UV-DDB1 binding properties to mediate distinct activities. J. Virol. 77, 6274–6283. doi: 10.1128/JVI.77.11.6274-6283.2003
Levine, A. J. (1997). p53, the cellular gatekeeper for growth and division. Cell 88, 323–331. doi: 10.1016/S0092-8674(00)81871-1
Lew, D. J., Dulic, V., and Reed, S. I. (1991). Isolation of three novel human cyclins by rescue of G1 cyclin (Cln) function in yeast. Cell 66, 1197–1206. doi: 10.1016/0092-8674(91)90042-W
Li, C., Xu, D., Ye, Q., Hong, S., Jiang, Y., Liu, X., et al. (2016). Zika virus disrupts neural progenitor development and leads to microcephaly in mice. Cell Stem Cell 19, 120–126. doi: 10.1016/j.stem.2016.04.017
Li, G., Park, H. U., Liang, D., and Zhao, R. Y. (2010). Cell cycle G2/M arrest through an S phase-dependent mechanism by HIV-1 viral protein R. Retrovirology 7:59. doi: 10.1186/1742-4690-7-59
Li, G., Poulsen, M., Fenyvuesvolgyi, C., Yashiroda, Y., Yoshida, M., Simard, J. M., et al. (2017). Characterization of cytopathic factors through genome-wide analysis of the Zika viral proteins in fission yeast. Proc. Natl. Acad. Sci. U.S.A. 114, E376–E385. doi: 10.1073/pnas.1619735114
Li, H., Baskaran, R., Krisky, D. M., Bein, K., Grandi, P., Cohen, J. B., et al. (2008). Chk2 is required for HSV-1 ICP0-mediated G2/M arrest and enhancement of virus growth. Virology 375, 13–23. doi: 10.1016/j.virol.2008.01.038
Li, J., and Stern, D. F. (2005a). DNA damage regulates Chk2 association with chromatin. J. Biol. Chem. 280, 37948–37956. doi: 10.1074/jbc.M509299200
Li, J., and Stern, D. F. (2005b). Regulation of CHK2 by DNA-dependent protein kinase. J. Biol. Chem. 280, 12041–12050. doi: 10.1074/jbc.M412445200
Li, L., Gu, B., Zhou, F., Chi, J., Wang, F., Peng, G., et al. (2011). Human herpesvirus 6 suppresses T cell proliferation through induction of cell cycle arrest in infected cells in the G2/M phase. J. Virol. 85, 6774–6783. doi: 10.1128/JVI.02577-10
Li, M., Lee, H., Yoon, D. W., Albrecht, J. C., Fleckenstein, B., Neipel, F., et al. (1997). Kaposi's sarcoma-associated herpesvirus encodes a functional cyclin. J. Virol. 71, 1984–1991.
Lin, P. Y., Lee, J. W., Liao, M. H., Hsu, H. Y., Chiu, S. J., Liu, H. J., et al. (2009). Modulation of p53 by mitogen-activated protein kinase pathways and protein kinase C delta during avian reovirus S1133-induced apoptosis. Virology 385, 323–334. doi: 10.1016/j.virol.2008.12.028
Linzer, D. I., and Levine, A. J. (1979). Characterization of a 54K dalton cellular SV40 tumor antigen present in SV40-transformed cells and uninfected embryonal carcinoma cells. Cell 17, 43–52. doi: 10.1016/0092-8674(79)90293-9
Liu, B., Hong, S., Tang, Z., Yu, H., and Giam, C. Z. (2005). HTLV-I Tax directly binds the Cdc20-associated anaphase-promoting complex and activates it ahead of schedule. Proc. Natl. Acad. Sci. U.S.A. 102, 63–68. doi: 10.1073/pnas.0406424101
Liu, B., Liang, M. H., Kuo, Y. L., Liao, W., Boros, I., Kleinberger, T., et al. (2003). Human T-lymphotropic virus type 1 oncoprotein tax promotes unscheduled degradation of Pds1p/securin and Clb2p/cyclin B1 and causes chromosomal instability. Mol. Cell. Biol. 23, 5269–5281. doi: 10.1128/MCB.23.15.5269-5281.2003
Liu, J., Li, Q., Li, X., Qiu, Z., Li, A., Liang, W., et al. (2018). Zika Virus Envelope Protein induces G2/M cell cycle arrest and apoptosis via an intrinsic cell death signaling pathway in neuroendocrine PC12 cells. Int. J. Biol. Sci. 14, 1099–1108. doi: 10.7150/ijbs.26400
Lohka, M. J., Hayes, M. K., and Maller, J. L. (1988). Purification of maturation-promoting factor, an intracellular regulator of early mitotic events. Proc. Natl. Acad. Sci. U.S.A. 85, 3009–3013. doi: 10.1073/pnas.85.9.3009
Lomonte, P., Sullivan, K. F., and Everett, R. D. (2001). Degradation of nucleosome-associated centromeric histone H3-like protein CENP-A induced by herpes simplex virus type 1 protein ICP0. J. Biol. Chem. 276, 5829–5835. doi: 10.1074/jbc.M008547200
López-Avilés, S., Grande, M., Gonzalez, M., Helgesen, A. L., Alemany, V., Sanchez-Piris, M., et al. (2005). Inactivation of the Cdc25 phosphatase by the stress-activated Srk1 kinase in fission yeast. Mol. Cell 17, 49–59. doi: 10.1016/j.molcel.2004.11.043
Lopez-Girona, A., Furnari, B., Mondesert, O., and Russell, P. (1999). Nuclear localization of Cdc25 is regulated by DNA damage and a 14-3-3 protein. Nature 397, 172–175. doi: 10.1038/16488
Lopez-Girona, A., Tanaka, K., Chen, X. B., Baber, B. A., McGowan, C. H., and Russell, P. (2001). Serine-345 is required for Rad3-dependent phosphorylation and function of checkpoint kinase Chk1 in fission yeast. Proc. Natl. Acad. Sci. U.S.A. 98, 11289–11294. doi: 10.1073/pnas.191557598
Low, K. G., Dorner, L. F., Fernando, D. B., Grossman, J., Jeang, K. T., and Comb, M. J. (1997). Human T-cell leukemia virus type 1 Tax releases cell cycle arrest induced by p16INK4a. J. Virol. 71, 1956–1962.
Lowe, M., Rabouille, C., Nakamura, N., Watson, R., Jackman, M., Jamsa, E., et al. (1998). Cdc2 kinase directly phosphorylates the cis-Golgi matrix protein GM130 and is required for Golgi fragmentation in mitosis. Cell 94, 783–793. doi: 10.1016/S0092-8674(00)81737-7
Lu, H., and Levine, A. J. (1995). Human TAFII31 protein is a transcriptional coactivator of the p53 protein. Proc. Natl. Acad. Sci. U.S.A. 92, 5154–5158. doi: 10.1073/pnas.92.11.5154
Ma, H., Kien, F., Maniere, M., Zhang, Y., Lagarde, N., Tse, K. S., et al. (2012). Human annexin A6 interacts with influenza A virus M2 protein and negatively modulates infection. J. Virol. 86, 1789–1801. doi: 10.1128/JVI.06003-11
Mahony, D., Parry, D. A., and Lees, E. (1998). Active cdk6 complexes are predominantly nuclear and represent only a minority of the cdk6 in T cells. Oncogene 16, 603–611. doi: 10.1038/sj.onc.1201570
Majumder, M., Ghosh, A. K., Steele, R., Ray, R., and Ray, R. B. (2001). Hepatitis C virus NS5A physically associates with p53 and regulates p21/waf1 gene expression in a p53-dependent manner. J. Virol. 75, 1401–1407. doi: 10.1128/JVI.75.3.1401-1407.2001
Mal, A., Poon, R. Y., Howe, P. H., Toyoshima, H., Hunter, T., and Harter, M. L. (1996). Inactivation of p27Kip1 by the viral E1A oncoprotein in TGFbeta-treated cells. Nature 380, 262–265. doi: 10.1038/380262a0
Malumbres, M., Sotillo, R., Santamaria, D., Galan, J., Cerezo, A., Ortega, S., et al. (2004). Mammalian cells cycle without the D-type cyclin-dependent kinases Cdk4 and Cdk6. Cell 118, 493–504. doi: 10.1016/j.cell.2004.08.002
Manke, I. A., Nguyen, A., Lim, D., Stewart, M. Q., Elia, A. E., and Yaffe, M. B. (2005). MAPKAP kinase-2 is a cell cycle checkpoint kinase that regulates the G2/M transition and S phase progression in response to UV irradiation. Mol. Cell 17, 37–48. doi: 10.1016/j.molcel.2004.11.021
Mann, D. J., Child, E. S., Swanton, C., Laman, H., and Jones, N. (1999). Modulation of p27(Kip1) levels by the cyclin encoded by Kaposi's sarcoma-associated herpesvirus. EMBO J. 18, 654–663. doi: 10.1093/emboj/18.3.654
Mannick, J. B., Cohen, J. I., Birkenbach, M., Marchini, A., and Kieff, E. (1991). The Epstein-Barr virus nuclear protein encoded by the leader of the EBNA RNAs is important in B-lymphocyte transformation. J. Virol. 65, 6826–6837.
Martin-Lluesma, S., Schaeffer, C., Robert, E. I., van Brengel, P. C., Leupin, O., Hantz, O., et al. (2008). Hepatitis B virus X protein affects S phase progression leading to chromosome segregation defects by binding to damaged DNA binding protein 1. Hepatology 48, 1467–1476. doi: 10.1002/hep.22542
Matsuoka, M., Kato, J. Y., Fisher, R. P., Morgan, D. O., and Sherr, C. J. (1994). Activation of cyclin-dependent kinase 4 (cdk4) by mouse MO15-associated kinase. Mol. Cell. Biol. 14, 7265–7275. doi: 10.1128/MCB.14.11.7265
Matsuoka, S., Huang, M., and Elledge, S. J. (1998). Linkage of ATM to cell cycle regulation by the Chk2 protein kinase. Science 282, 1893–1897. doi: 10.1126/science.282.5395.1893
Matsushime, H., Quelle, D. E., Shurtleff, S. A., Shibuya, M., Sherr, C. J., and Kato, J. Y. (1994). D-type cyclin-dependent kinase activity in mammalian cells. Mol. Cell. Biol. 14, 2066–2076. doi: 10.1128/MCB.14.3.2066
Mayo, L. D., and Donner, D. B. (2002). The PTEN, Mdm2, p53 tumor suppressor-oncoprotein network. Trends Biochem. Sci. 27, 462–467. doi: 10.1016/S0968-0004(02)02166-7
McGowan, C. H., and Russell, P. (1993). Human Wee1 kinase inhibits cell division by phosphorylating p34cdc2 exclusively on Tyr15. EMBO J. 12, 75–85. doi: 10.1002/j.1460-2075.1993.tb05633.x
Mesri, E. A., Feitelson, M. A., and Munger, K. (2014). Human viral oncogenesis: a cancer hallmarks analysis. Cell Host Microbe 15, 266–282. doi: 10.1016/j.chom.2014.02.011
Mietz, J. A., Unger, T., Huibregtse, J. M., and Howley, P. M. (1992). The transcriptional transactivation function of wild-type-p53 is inhibited by SV40 large T-antigen and by Hpv-16 E6-oncoprotein. EMBO J. 11, 5013–5020. doi: 10.1002/j.1460-2075.1992.tb05608.x
Millar, J. B., and Russell, P. (1992). The cdc25 M-phase inducer: an unconventional protein phosphatase. Cell 68, 407–410. doi: 10.1016/0092-8674(92)90177-E
Mirza, A., McGuirk, M., Hockenberry, T. N., Wu, Q., Ashar, H., Black, S., et al. (2002). Human survivin is negatively regulated by wild-type p53 and participates in p53-dependent apoptotic pathway. Oncogene 21, 2613–2622. doi: 10.1038/sj.onc.1205353
Mitra, J., Enders, G. H., Azizkhan-Clifford, J., and Lengel, K. L. (2006). Dual regulation of the anaphase promoting complex in human cells by cyclin A-Cdk2 and cyclin A-Cdk1 complexes. Cell Cycle 5, 661–666. doi: 10.4161/cc.5.6.2604
Mlechkovich, G., and Frenkel, N. (2007). Human herpesvirus 6A (HHV-6A) and HHV-6B alter E2F1/Rb pathways and E2F1 localization and cause cell cycle arrest in infected T cells. J. Virol. 81, 13499–13508. doi: 10.1128/JVI.01496-07
Mo, M., Fleming, S. B., and Mercer, A. A. (2009). Cell cycle deregulation by a poxvirus partial mimic of anaphase-promoting complex subunit 11. Proc. Natl. Acad. Sci. U.S.A. 106, 19527–19532. doi: 10.1073/pnas.0905893106
Moir, R. D., Spann, T. P., Herrmann, H., and Goldman, R. D. (2000). Disruption of nuclear lamin organization blocks the elongation phase of DNA replication. J. Cell Biol. 149, 1179–1192. doi: 10.1083/jcb.149.6.1179
Momand, J., Wu, H. H., and Dasgupta, G. (2000). MDM2–master regulator of the p53 tumor suppressor protein. Gene 242, 15–29. doi: 10.1016/S0378-1119(99)00487-4
Moore, P. S., and Chang, Y. (1998). Antiviral activity of tumor-suppressor pathways: clues from molecular piracy by KSHV. Trends Genet. 14, 144–150. doi: 10.1016/S0168-9525(98)01408-5
Morris, M. C., Gondeau, C., Tainer, J. A., and Divita, G. (2002). Kinetic mechanism of activation of the Cdk2/cyclin A complex. Key role of the C-lobe of the Cdk. J. Biol. Chem. 277, 23847–23853. doi: 10.1074/jbc.M107890200
Mukherji, A., Janbandhu, V. C., and Kumar, V. (2007). HBx-dependent cell cycle deregulation involves interaction with cyclin E/A-cdk2 complex and destabilization of p27Kip1. Biochem. J. 401, 247–256. doi: 10.1042/BJ20061091
Muñoz, I. M., Hain, K., Declais, A. C., Gardiner, M., Toh, G. W., Sanchez-Pulido, L., et al. (2009). Coordination of structure-specific nucleases by human SLX4/BTBD12 is required for DNA repair. Mol. Cell 35, 116–127. doi: 10.1016/j.molcel.2009.06.020
Muralidhar, S., Doniger, J., Mendelson, E., Araujo, J. C., Kashanchi, F., Azumi, N., et al. (1996). Human cytomegalovirus mtrII oncoprotein binds to p53 and down-regulates p53-activated transcription. J. Virol. 70, 8691–8700.
Muslin, A. J., and Xing, H. (2000). 14-3-3 proteins: regulation of subcellular localization by molecular interference. Cell. Signal. 12, 703–709. doi: 10.1016/S0898-6568(00)00131-5
Nakayama, K., Nagahama, H., Minamishima, Y. A., Miyake, S., Ishida, N., Hatakeyama, S., et al. (2004). Skp2-mediated degradation of p27 regulates progression into mitosis. Dev. Cell 6, 661–672. doi: 10.1016/S1534-5807(04)00131-5
Nascimento, R., Dias, J. D., and Parkhouse, R. M. (2009). The conserved UL24 family of human alpha, beta and gamma herpesviruses induces cell cycle arrest and inactivation of the cyclinB/cdc2 complex. Arch. Virol. 154, 1143–1149. doi: 10.1007/s00705-009-0420-y
Nascimento, R., and Parkhouse, R. M. E. (2007). Murine gammaherpesvirus 68 ORF20 induces cell-cycle arrest in G(2) by inhibiting the Cdc2-cyclin B complex. J. Gen. Virol. 88, 1446–1453. doi: 10.1099/vir.0.82589-0
Nasmyth, K., Peters, J. M., and Uhlmann, F. (2000). Splitting the chromosome: cutting the ties that bind sister chromatids. Science 288, 1379–1385. doi: 10.1126/science.288.5470.1379
Neuveut, C., Low, K. G., Maldarelli, F., Schmitt, I., Majone, F., Grassmann, R., et al. (1998). Human T-cell leukemia virus type 1 Tax and cell cycle progression: role of cyclin D-cdk and p110Rb. Mol. Cell. Biol. 18, 3620–3632. doi: 10.1128/MCB.18.6.3620
Nicholas, J., Cameron, K. R., and Honess, R. W. (1992). Herpesvirus saimiri encodes homologues of G protein-coupled receptors and cyclins. Nature 355, 362–365. doi: 10.1038/355362a0
Nigg, E. A. (1995). Cyclin-dependent protein kinases: key regulators of the eukaryotic cell cycle. Bioessays 17, 471–480. doi: 10.1002/bies.950170603
Norbury, C., Blow, J., and Nurse, P. (1991). Regulatory phosphorylation of the p34cdc2 protein kinase in vertebrates. EMBO J. 10, 3321–3329. doi: 10.1002/j.1460-2075.1991.tb04896.x
Nurse, P. (1990). Universal control mechanism regulating onset of M-phase. Nature 344, 503–508. doi: 10.1038/344503a0
Ohtani, K., DeGregori, J., and Nevins, J. R. (1995). Regulation of the cyclin E gene by transcription factor E2F1. Proc. Natl. Acad. Sci. U.S.A. 92, 12146–12150. doi: 10.1073/pnas.92.26.12146
Ooi, M. H., Solomon, T., Podin, Y., Mohan, A., Akin, W., Yusuf, M. A., et al. (2007). Evaluation of different clinical sample types in diagnosis of human enterovirus 71-associated hand-foot-and-mouth disease. J. Clin. Microbiol. 45, 1858–1866. doi: 10.1128/JCM.01394-06
Op De Beeck, A., Anouja, F., Mousset, S., Rommelaere, J., and Caillet-Fauquet, P. (1995). The nonstructural proteins of the autonomous parvovirus minute virus of mice interfere with the cell cycle, inducing accumulation in G2. Cell Growth Differ. 6, 781–787.
Op De Beeck, A., and Caillet-Fauquet, P. (1997). The NS1 protein of the autonomous parvovirus minute virus of mice blocks cellular DNA replication: a consequence of lesions to the chromatin? J. Virol. 71, 5323–5329.
Pajovic, S., Wong, E. L., Black, A. R., and Azizkhan, J. C. (1997). Identification of a viral kinase that phosphorylates specific E2Fs and pocket proteins. Mol. Cell. Biol. 17, 6459–6464. doi: 10.1128/MCB.17.11.6459
Pardee, A. B. (1974). A restriction point for control of normal animal cell proliferation. Proc. Natl. Acad. Sci. U.S.A. 71, 1286–1290. doi: 10.1073/pnas.71.4.1286
Park, H. U., Jeong, J. H., Chung, J. H., and Brady, J. N. (2004). Human T-cell leukemia virus type 1 Tax interacts with Chk1 and attenuates DNA-damage induced G2 arrest mediated by Chk1. Oncogene 23, 4966–4974. doi: 10.1038/sj.onc.1207644
Park, H. U., Jeong, S. J., Jeong, J. H., Chung, J. H., and Brady, J. N. (2006). Human T-cell leukemia virus type 1 Tax attenuates gamma-irradiation-induced apoptosis through physical interaction with Chk2. Oncogene 25, 438–447. doi: 10.1038/sj.onc.1209059
Peng, C. Y., Graves, P. R., Thoma, R. S., Wu, Z., Shaw, A. S., and Piwnica-Worms, H. (1997). Mitotic and G2 checkpoint control: regulation of 14-3-3 protein binding by phosphorylation of Cdc25C on serine-216. Science 277, 1501–1505. doi: 10.1126/science.277.5331.1501
Peters, J. M. (2006). The anaphase promoting complex/cyclosome: a machine designed to destroy. Nat. Rev. Mol. Cell Biol. 7, 644–656. doi: 10.1038/nrm1988
Petri, E. T., Errico, A., Escobedo, L., Hunt, T., and Basavappa, R. (2007). The crystal structure of human cyclin B. Cell Cycle 6, 1342–1349. doi: 10.4161/cc.6.11.4297
Pielak, R. M., and Chou, J. J. (2011). Influenza M2 proton channels. Biochim. Biophys. Acta 1808, 522–529. doi: 10.1016/j.bbamem.2010.04.015
Pines, J., and Rieder, C. L. (2001). Re-staging mitosis: a contemporary view of mitotic progression. Nat. Cell Biol. 3:E3–E6. doi: 10.1038/35050676
Platt, G., Carbone, A., and Mittnacht, S. (2002). p16INK4a loss and sensitivity in KSHV associated primary effusion lymphoma. Oncogene 21, 1823–1831. doi: 10.1038/sj.onc.1205360
Poggioli, G. J., Dermody, T. S., and Tyler, K. L. (2001). Reovirus-induced sigma1s-dependent G(2)/M phase cell cycle arrest is associated with inhibition of p34(cdc2). J. Virol. 75, 7429–7434. doi: 10.1128/JVI.75.16.7429-7434.2001
Poggioli, G. J., Keefer, C., Connolly, J. L., Dermody, T. S., and Tyler, K. L. (2000). Reovirus-induced G(2)/M cell cycle arrest requires sigma 1s and occurs in the absence of apoptosis. J. Virol. 74, 9562–9570. doi: 10.1128/JVI.74.20.9562-9570.2000
Poss, Z. C., Ebmeier, C. C., and Taatjes, D. J. (2013). The Mediator complex and transcription regulation. Crit. Rev. Biochem. Mol. Biol. 48, 575–608. doi: 10.3109/10409238.2013.840259
Prikhod'ko, EA., and Miller, L. K. (1998). Role of baculovirus IE2 and its RING finger in cell cycle arrest. J. Virol. 72, 684–692.
Quelle, D. E., Ashmun, R. A., Shurtleff, S. A., Kato, J. Y., Bar-Sagi, D., Roussel, M. F., et al. (1993). Overexpression of mouse D-type cyclins accelerates G1 phase in rodent fibroblasts. Genes Dev. 7, 1559–1571. doi: 10.1101/gad.7.8.1559
Raj, K., Ogston, P., and Beard, P. (2001). Virus-mediated killing of cells that lack p53 activity. Nature 412, 914–917. doi: 10.1038/35091082
Rane, S. G., Dubus, P., Mettus, R. V., Galbreath, E. J., Boden, G., Reddy, E. P., et al. (1999). Loss of Cdk4 expression causes insulin-deficient diabetes and Cdk4 activation results in beta-islet cell hyperplasia. Nat. Genet. 22, 44–52. doi: 10.1038/8751
Raychaudhuri, P., Bagchi, S., Devoto, S. H., Kraus, V. B., Moran, E., and Nevins, J. R. (1991). Domains of the adenovirus E1A protein required for oncogenic activity are also required for dissociation of E2F transcription factor complexes. Genes Dev. 5, 1200–1211. doi: 10.1101/gad.5.7.1200
Ren, S., and Rollins, B. J. (2004). Cyclin C/cdk3 promotes Rb-dependent G0 exit. Cell 117, 239–251. doi: 10.1016/S0092-8674(04)00300-9
Roshal, M., Kim, B., Zhu, Y., Nghiem, P., and Planelles, V. (2003). Activation of the ATR-mediated DNA damage response by the HIV-1 viral protein R. J. Biol. Chem. 278, 25879–25886. doi: 10.1074/jbc.M303948200
Roussel, M. F., Davis, J. N., Cleveland, J. L., Ghysdael, J., and Hiebert, S. W. (1994). Dual control of myc expression through a single DNA binding site targeted by ets family proteins and E2F-1. Oncogene 9, 405–415.
Rovnak, J., and Quackenbush, S. L. (2002). Walleye dermal sarcoma virus cyclin interacts with components of the mediator complex and the RNA polymerase II holoenzyme. J. Virol. 76, 8031–8039. doi: 10.1128/JVI.76.16.8031-8039.2002
Sakai, K., Barnitz, R. A., Chaigne-Delalande, B., Bidere, N., and Lenardo, M. J. (2011). Human Immunodeficiency Virus Type 1 Vif causes dysfunction of Cdk1 and CyclinB1: implications for cell cycle arrest. Virol. J. 8:219. doi: 10.1186/1743-422X-8-219
Sanchez, Y., Wong, C., Thoma, R. S., Richman, R., Wu, Z., Piwnica-Worms, H., et al. (1997). Conservation of the Chk1 checkpoint pathway in mammals: linkage of DNA damage to Cdk regulation through Cdc25. Science 277, 1497–1501. doi: 10.1126/science.277.5331.1497
Santamaría, D., Barriere, C., Cerqueira, A., Hunt, S., Tardy, C., Newton, K., et al. (2007). Cdk1 is sufficient to drive the mammalian cell cycle. Nature 448, 811–818. doi: 10.1038/nature06046
Sarek, G., Jarviluoma, A., and Ojala, P. M. (2006). KSHV viral cyclin inactivates p27KIP1 through Ser10 and Thr187 phosphorylation in proliferating primary effusion lymphomas. Blood 107, 725–732. doi: 10.1182/blood-2005-06-2534
Saxena, N., and Kumar, V. (2014). The HBx oncoprotein of hepatitis B virus deregulates the cell cycle by promoting the intracellular accumulation and re-compartmentalization of the cellular deubiquitinase USP37. PLoS ONE 9:e111256. doi: 10.1371/journal.pone.0111256
Schröfelbauer, B., Hakata, Y., and Landau, N. R. (2007). HIV-1 Vpr function is mediated by interaction with the damage-specific DNA-binding protein DDB1. Proc. Natl. Acad. Sci. U.S.A. 104, 4130–4135. doi: 10.1073/pnas.0610167104
Schulze-Gahmen, U., Jung, J. U., and Kim, S. H. (1999). Crystal structure of a viral cyclin, a positive regulator of cyclin-dependent kinase 6. Structure 7, 245–254. doi: 10.1016/S0969-2126(99)80035-5
Seto, E., Moosmann, A., Groemminger, S., Walz, N., Grundhoff, A., and Hammerschmidt, W. (2010). Micro RNAs of Epstein-Barr virus promote cell cycle progression and prevent apoptosis of primary human B cells. PLoS Pathog. 6:e1001063. doi: 10.1371/journal.ppat.1001063
Sharon, E., Volchek, L., and Frenkel, N. (2014). Human herpesvirus 6 (HHV-6) alters E2F1/Rb pathways and utilizes the E2F1 transcription factor to express viral genes. Proc. Natl. Acad. Sci. U.S.A. 111, 451–456. doi: 10.1073/pnas.1308854110
Shaulsky, G., Goldfinger, N., Tosky, M. S., Levine, A. J., and Rotter, V. (1991). Nuclear-localization is essential for the activity of P53 protein. Oncogene 6, 2055–2065.
Sherr, C. J., and Roberts, J. M. (1999). CDK inhibitors: positive and negative regulators of G1-phase progression. Genes Dev. 13, 1501–1512. doi: 10.1101/gad.13.12.1501
Shieh, S. Y., Ahn, J., Tamai, K., Taya, Y., and Prives, C. (2000). The human homologs of checkpoint kinases Chk1 and Cds1 (Chk2) phosphorylate p53 at multiple DNA damage-inducible sites. Genes Dev. 14, 289–300.
Shieh, S. Y., Ikeda, M., Taya, Y., and Prives, C. (1997). DNA damage-induced phosphorylation of p53 alleviates inhibition by MDM2. Cell 91, 325–334. doi: 10.1016/S0092-8674(00)80416-X
Sidle, A., Palaty, C., Dirks, P., Wiggan, O., Kiess, M., Gill, R. M., et al. (1996). Activity of the retinoblastoma family proteins, pRB, p107, and p130, during cellular proliferation and differentiation. Crit. Rev. Biochem. Mol. Biol. 31, 237–271. doi: 10.3109/10409239609106585
Sinclair, A. J., Palmero, I., Peters, G., and Farrell, P. J. (1994). EBNA-2 and EBNA-LP cooperate to cause G0 to G1 transition during immortalization of resting human B lymphocytes by Epstein-Barr virus. EMBO J. 13, 3321–3328. doi: 10.1002/j.1460-2075.1994.tb06634.x
Sinclair, J., Baillie, J., Bryant, L., and Caswell, R. (2000). Human cytomegalovirus mediates cell cycle progression through G(1) into early S phase in terminally differentiated cells. J. Gen. Virol. 81, 1553–1565. doi: 10.1099/0022-1317-81-6-1553
Solomon, M. J., Lee, T., and Kirschner, M. W. (1992). Role of phosphorylation in p34cdc2 activation: identification of an activating kinase. Mol. Biol. Cell 3, 13–27. doi: 10.1091/mbc.3.1.13
Somasundaram, K., and El-Deiry, W. S. (1997). Inhibition of p53-mediated transactivation and cell cycle arrest by E1A through its p300/CBP-interacting region. Oncogene 14, 1047–1057. doi: 10.1038/sj.onc.1201002
Song, M. S., Song, S. J., Ayad, N. G., Chang, J. S., Lee, J. H., Hong, H. K., et al. (2004). The tumour suppressor RASSF1A regulates mitosis by inhibiting the APC-Cdc20 complex. Nat. Cell Biol. 6, 129–137. doi: 10.1038/ncb1091
Souza, B. S., Sampaio, G. L., Pereira, C. S., Campos, G. S., Sardi, S. I., Freitas, L. A., et al. (2016). Zika virus infection induces mitosis abnormalities and apoptotic cell death of human neural progenitor cells. Sci. Rep. 6:39775. doi: 10.1038/srep39775
Speir, E., Modali, R., Huang, E. S., Leon, M. B., Shawl, F., Finkel, T., et al. (1994). Potential role of human cytomegalovirus and p53 interaction in coronary restenosis. Science 265, 391–394. doi: 10.1126/science.8023160
Stambolic, V., MacPherson, D., Sas, D., Lin, Y., Snow, B., Jang, Y., et al. (2001). Regulation of PTEN transcription by p53. Mol. Cell 8, 317–325. doi: 10.1016/S1097-2765(01)00323-9
Straight, A. F., and Field, C. M. (2000). Microtubules, membranes and cytokinesis. Curr. Biol. 10, R760–R770. doi: 10.1016/S0960-9822(00)00746-6
Strausfeld, U., Labbe, J. C., Fesquet, D., Cavadore, J. C., Picard, A., Sadhu, K., et al. (1991). Dephosphorylation and activation of a p34cdc2/cyclin B complex in vitro by human CDC25 protein. Nature 351, 242–245. doi: 10.1038/351242a0
Stubdal, H., Zalvide, J., and DeCaprio, J. A. (1996). Simian virus 40 large T antigen alters the phosphorylation state of the RB-related proteins p130 and p107. J. Virol. 70, 2781–2788.
Sudakin, V., Ganoth, D., Dahan, A., Heller, H., Hershko, J., Luca, F. C., et al. (1995). The cyclosome, a large complex containing cyclin-selective ubiquitin ligase activity, targets cyclins for destruction at the end of mitosis. Mol. Biol. Cell 6, 185–197. doi: 10.1091/mbc.6.2.185
Sullivan, C. S., Cantalupo, P., and Pipas, J. M. (2000). The molecular chaperone activity of simian virus 40 large T antigen is required to disrupt Rb-E2F family complexes by an ATP-dependent mechanism. Mol. Cell. Biol. 20, 6233–6243. doi: 10.1128/MCB.20.17.6233-6243.2000
Sun, Y., Chen, A. Y., Cheng, F., Guan, W., Johnson, F. B., and Qiu, J. (2009). Molecular characterization of infectious clones of the minute virus of canines reveals unique features of bocaviruses. J. Virol. 83, 3956–3967. doi: 10.1128/JVI.02569-08
Suzuki, A., Hayashida, M., Ito, T., Kawano, H., Nakano, T., Miura, M., et al. (2000). Survivin initiates cell cycle entry by the competitive interaction with Cdk4/p16(INK4a) and Cdk2/cyclin E complex activation. Oncogene 19, 3225–3234. doi: 10.1038/sj.onc.1203665
Suzuki, T., Kitao, S., Matsushime, H., and Yoshida, M. (1996). HTLV-1 Tax protein interacts with cyclin-dependent kinase inhibitor p16INK4A and counteracts its inhibitory activity towards CDK4. EMBO J. 15, 1607–1614. doi: 10.1002/j.1460-2075.1996.tb00505.x
Suzuki, T., Narita, T., Uchida-Toita, M., and Yoshida, M. (1999). Down-regulation of the INK4 family of cyclin-dependent kinase inhibitors by tax protein of HTLV-1 through two distinct mechanisms. Virology 259, 384–391. doi: 10.1006/viro.1999.9760
Svendsen, J. M., Smogorzewska, A., Sowa, M. E., O'Connell, B. C., Gygi, S. P., Elledge, S. J., et al. (2009). Mammalian BTBD12/SLX4 assembles a Holliday junction resolvase and is required for DNA repair. Cell 138, 63–77. doi: 10.1016/j.cell.2009.06.030
Swanton, C., Mann, D. J., Fleckenstein, B., Neipel, F., Peters, G., and Jones, N. (1997). Herpes viral cyclin/Cdk6 complexes evade inhibition by CDK inhibitor proteins. Nature 390, 184–187. doi: 10.1038/36606
Szekely, L., Selivanova, G., Magnusson, K. P., Klein, G., and Wiman, K. G. (1993). EBNA-5, an Epstein-Barr virus-encoded nuclear antigen, binds to the retinoblastoma and p53 proteins. Proc. Natl. Acad. Sci. U.S.A. 90, 5455–5459. doi: 10.1073/pnas.90.12.5455
Tachiwana, H., Shimura, M., Nakai-Murakami, C., Tokunaga, K., Takizawa, Y., Sata, T., et al. (2006). HIV-1 Vpr induces DNA double-strand breaks. Cancer Res. 66, 627–631. doi: 10.1158/0008-5472.CAN-05-3144
Tan, T. H., Wallis, J., and Levine, A. J. (1986). Identification of the p53 protein domain involved in formation of the simian virus-40 large T-antigen p53 protein complex. J. Virol. 59, 574–583.
Tang, H., Hammack, C., Ogden, S. C., Wen, Z., Qian, X., Li, Y., et al. (2016). Zika virus infects human cortical neural progenitors and attenuates their growth. Cell Stem Cell 18, 587–590. doi: 10.1016/j.stem.2016.02.016
Tang, Z., Li, B., Bharadwaj, R., Zhu, H., Ozkan, E., Hakala, K., et al. (2001). APC2 Cullin protein and APC11 RING protein comprise the minimal ubiquitin ligase module of the anaphase-promoting complex. Mol. Biol. Cell 12, 3839–3851. doi: 10.1091/mbc.12.12.3839
Tassan, J. P., Jaquenoud, M., Leopold, P., Schultz, S. J., and Nigg, E. A. (1995). Identification of human cyclin-dependent kinase 8, a putative protein kinase partner for cyclin C. Proc. Natl. Acad. Sci. U.S.A. 92, 8871–8875. doi: 10.1073/pnas.92.19.8871
Teodoro, J. G., Heilman, D. W., Parker, A. E., and Green, M. R. (2004). The viral protein Apoptin associates with the anaphase-promoting complex to induce G2/M arrest and apoptosis in the absence of p53. Genes Dev. 18, 1952–1957. doi: 10.1101/gad.1198404
Thompson, D. L., Kalderon, D., Smith, A. E., and Tevethia, M. J. (1990). Dissociation of Rb-binding and anchorage-independent growth from immortalization and tumorigenicity using SV40 mutants producing N-terminally truncated large T antigens. Virology 178, 15–34. doi: 10.1016/0042-6822(90)90375-2
Thompson, J., Doniger, J., and Rosenthal, L. J. (1994). A 79 amino acid oncogene is responsible for human cytomegalovirus mtrII induced malignant transformation. Arch. Virol. 136, 161–172. doi: 10.1007/BF01538825
Thornton, B. R., and Toczyski, D. P. (2003). Securin and B-cyclin/CDK are the only essential targets of the APC. Nat. Cell Biol. 5, 1090–1094. doi: 10.1038/ncb1066
Tsai, H. L., Kou, G. H., Chen, S. C., Wu, C. W., and Lin, Y. S. (1996). Human cytomegalovirus immediate-early protein IE2 tethers a transcriptional repression domain to p53. J. Biol. Chem. 271, 3534–3540. doi: 10.1074/jbc.271.7.3534
Tsutsui, T., Fukasawa, R., Tanaka, A., Hirose, Y., and Ohkuma, Y. (2011). Identification of target genes for the CDK subunits of the Mediator complex. Genes Cells 16, 1208–1218. doi: 10.1111/j.1365-2443.2011.01565.x
Tsutsui, T., Hesabi, B., Moons, D. S., Pandolfi, P. P., Hansel, K. S., Koff, A., et al. (1999). Targeted disruption of CDK4 delays cell cycle entry with enhanced p27(Kip1) activity. Mol. Cell. Biol. 19, 7011–7019. doi: 10.1128/MCB.19.10.7011
Tsutsui, T., Umemura, H., Tanaka, A., Mizuki, F., Hirose, Y., and Ohkuma, Y. (2008). Human mediator kinase subunit CDK11 plays a negative role in viral activator VP16-dependent transcriptional regulation. Genes Cells 13, 817–826. doi: 10.1111/j.1365-2443.2008.01208.x
Uhlmann, T., Boeing, S., Lehmbacher, M., and Meisterernst, M. (2007). The VP16 activation domain establishes an active mediator lacking CDK8 in vivo. J. Biol. Chem. 282, 2163–2173. doi: 10.1074/jbc.M608451200
van Dyk, L. F., Hess, J. L., Katz, J. D., Jacoby, M., Speck, S. H., and Virgin, H. I. (1999). The murine gammaherpesvirus 68 v-cyclin gene is an oncogene that promotes cell cycle progression in primary lymphocytes. J. Virol. 73, 5110–5122.
Virgin, H. W. T., Latreille, P., Wamsley, P., Hallsworth, K., Weck, K. E., Dal Canto, A. J., et al. (1997). Complete sequence and genomic analysis of murine gammaherpesvirus 68. J. Virol. 71, 5894–5904.
Vlach, J., Hennecke, S., and Amati, B. (1997). Phosphorylation-dependent degradation of the cyclin-dependent kinase inhibitor p27. EMBO J. 16, 5334–5344. doi: 10.1093/emboj/16.17.5334
von Mikecz, A. (2006). The nuclear ubiquitin-proteasome system. J. Cell Sci. 119, 1977–1984. doi: 10.1242/jcs.03008
Wan, Z., Zhi, N., Wong, S., Keyvanfar, K., Liu, D., Raghavachari, N., et al. (2010). Human parvovirus B19 causes cell cycle arrest of human erythroid progenitors via deregulation of the E2F family of transcription factors. J. Clin. Invest. 120, 3530–3544. doi: 10.1172/JCI41805
Wang, F., Yoshida, I., Takamatsu, M., Ishido, S., Fujita, T., Oka, K., et al. (2000). Complex formation between hepatitis C virus core protein and p21Waf1/Cip1/Sdi1. Biochem. Biophys. Res. Commun. 273, 479–484. doi: 10.1006/bbrc.2000.2970
Wang, F., Zhou, H., Xia, X., Sun, Q., Wang, Y., and Cheng, B. (2010). Activated Notch signaling is required for hepatitis B virus X protein to promote proliferation and survival of human hepatic cells. Cancer Lett. 298, 64–73. doi: 10.1016/j.canlet.2010.06.003
Wang, J., Belcher, J. D., Marker, P. H., Wilcken, D. E., Vercellotti, G. M., and Wang, X. L. (2001). Cytomegalovirus inhibits p53 nuclear localization signal function. J. Mol. Med. 78, 642–647. doi: 10.1007/s001090000157
Wang, X., Deng, X., Yan, W., Zhu, Z., Shen, Y., Qiu, Y., et al. (2012). Stabilization of p53 in influenza A virus-infected cells is associated with compromised MDM2-mediated ubiquitination of p53. J. Biol. Chem. 287, 18366–18375. doi: 10.1074/jbc.M111.335422
Wehman, A. M., Staub, W., and Baier, H. (2007). The anaphase-promoting complex is required in both dividing and quiescent cells during zebrafish development. Dev. Biol. 303, 144–156. doi: 10.1016/j.ydbio.2006.10.043
Wei, W., Ayad, N. G., Wan, Y., Zhang, G. J., Kirschner, M. W., and Kaelin, W. G. (2004). Degradation of the SCF component Skp2 in cell-cycle phase G1 by the anaphase-promoting complex. Nature 428, 194–198. doi: 10.1038/nature02381
Weinberg, R. A. (1991). Tumor suppressor genes. Science 254, 1138–1146. doi: 10.1126/science.1659741
Weinberg, R. A. (1995). The retinoblastoma protein and cell cycle control. Cell 81, 323–330. doi: 10.1016/0092-8674(95)90385-2
Weinberg, R. A. (1997). The cat and mouse games that genes, viruses, and cells play. Cell 88, 573–575. doi: 10.1016/S0092-8674(00)81897-8
Werness, B. A., Levine, A. J., and Howley, P. M. (1990). Association of human papillomavirus types 16 and 18 E6 proteins with p53. Science 248, 76–79. doi: 10.1126/science.2157286
Wittschieben, B. O., and Wood, R. D. (2003). DDB complexities. DNA Repair (Amst). 2, 1065–1069. doi: 10.1016/S1568-7864(03)00113-7
Wu, E. W., Clemens, K. E., Heck, D. V., and Munger, K. (1993). The human papillomavirus E7 oncoprotein and the cellular transcription factor E2F bind to separate sites on the retinoblastoma tumor suppressor protein. J. Virol. 67, 2402–2407.
Xiao, Z., Chen, Z., Gunasekera, A. H., Sowin, T. J., Rosenberg, S. H., Fesik, S., et al. (2003). Chk1 mediates S and G2 arrests through Cdc25A degradation in response to DNA-damaging agents. J. Biol. Chem. 278, 21767–21773. doi: 10.1074/jbc.M300229200
Xu, J., Yun, X., Jiang, J., Wei, Y., Wu, Y., Zhang, W., et al. (2010). Hepatitis B virus X protein blunts senescence-like growth arrest of human hepatocellular carcinoma by reducing Notch1 cleavage. Hepatology 52, 142–154. doi: 10.1002/hep.23613
Yanagida, M. (2000). Cell cycle mechanisms of sister chromatid separation; Roles of Cut1/separin and Cut2/securin. Genes Cells 5, 1–8. doi: 10.1046/j.1365-2443.2000.00306.x
Yang, M., Hay, J., and Ruyechan, W. T. (2008). Varicella-zoster virus IE62 protein utilizes the human mediator complex in promoter activation. J. Virol. 82, 12154–12163. doi: 10.1128/JVI.01693-08
Yarmishyn, A., Child, E. S., Elphick, L. M., and Mann, D. J. (2008). Differential regulation of the cyclin-dependent kinase inhibitors p21(Cip1) and p27(Kip1) by phosphorylation directed by the cyclin encoded by Murine Herpesvirus 68. Exp. Cell Res. 314, 204–212. doi: 10.1016/j.yexcr.2007.09.016
Ye, X., Zhu, C. H., and Harper, J. W. (2001). A premature-termination mutation in the Mus musculus cyclin-dependent kinase 3 gene. Proc. Natl. Acad. Sci. U.S.A. 98, 1682–1686. doi: 10.1073/pnas.98.4.1682
Yip, K. W., Shi, W., Pintilie, M., Martin, J. D., Mocanu, J. D., Wong, D., et al. (2006). Prognostic significance of the Epstein-Barr virus, p53, Bcl-2, and survivin in nasopharyngeal cancer. Clin. Cancer Res. 12, 5726–5732. doi: 10.1158/1078-0432.CCR-06-0571
Yu, J., Zhang, L., Ren, P., Zhong, T., Li, Z., Wang, Z., et al. (2015). Enterovirus 71 mediates cell cycle arrest in S phase through non-structural protein 3D. Cell Cycle 14, 425–436. doi: 10.4161/15384101.2014.980631
Zacny, V. L., Wilson, J., and Pagano, J. S. (1998). The Epstein-Barr virus immediate-early gene product, BRLF1, interacts with the retinoblastoma protein during the viral lytic cycle. J. Virol. 72, 8043–8051.
Zalvide, J., Stubdal, H., and DeCaprio, J. A. (1998). The J domain of simian virus 40 large T antigen is required to functionally inactivate RB family proteins. Mol. Cell. Biol. 18, 1408–1415. doi: 10.1128/MCB.18.3.1408
Zerfass-Thome, K., Zwerschke, W., Mannhardt, B., Tindle, R., Botz, J. W., and Jansen-Durr, P. (1996). Inactivation of the cdk inhibitor p27KIP1 by the human papillomavirus type 16 E7 oncoprotein. Oncogene 13, 2323–2330.
Zetterberg, A., Larsson, O., and Wiman, K. G. (1995). What is the restriction point. Curr. Opin. Cell Biol. 7, 835–842. doi: 10.1016/0955-0674(95)80067-0
Zhang, Y., and Lu, H. (2009). Signaling to p53: ribosomal proteins find their way. Cancer Cell 16, 369–377. doi: 10.1016/j.ccr.2009.09.024
Zhao, F., Hou, N. B., Yang, X. L., He, X., Liu, Y., Zhang, Y. H., et al. (2008). Ataxia telangiectasia-mutated-Rad3-related DNA damage checkpoint signaling pathway triggered by hepatitis B virus infection. World J. Gastroenterol. 14, 6163–6170. doi: 10.3748/wjg.14.6163
Zhao, H., and Piwnica-Worms, H. (2001). ATR-mediated checkpoint pathways regulate phosphorylation and activation of human Chk1. Mol. Cell. Biol. 21, 4129–4139. doi: 10.1128/MCB.21.13.4129-4139.2001
Zhao, H., Watkins, J. L., and Piwnica-Worms, H. (2002). Disruption of the checkpoint kinase 1/cell division cycle 25A pathway abrogates ionizing radiation-induced S and G2 checkpoints. Proc. Natl. Acad. Sci. U.S.A. 99, 14795–14800. doi: 10.1073/pnas.182557299
Zhi, H., Zahoor, M. A., Shudofsky, A. M., and Giam, C. Z. (2015). KSHV vCyclin counters the senescence/G1 arrest response triggered by NF-kappaB hyperactivation. Oncogene 34, 496–505. doi: 10.1038/onc.2013.567
Zhou, Y., Ching, Y. P., Ng, R. W. M., and Jin, D. Y. (2003). Differential expression, localization and activity of two alternatively spliced isoforms of human APC regulator CDH1. Biochem. J. 374, 349–358. doi: 10.1042/bj20030600
Zimmerman, E. S., Sherman, M. P., Blackett, J. L., Neidleman, J. A., Kreis, C., Mundt, P., et al. (2006). Human immunodeficiency virus type 1 Vpr induces DNA replication stress in vitro and in vivo. J. Virol. 80, 10407–10418. doi: 10.1128/JVI.01212-06
Zou, H., McGarry, T. J., Bernal, T., and Kirschner, M. W. (1999). Identification of a vertebrate sister-chromatid separation inhibitor involved in transformation and tumorigenesis. Science 285, 418–422. doi: 10.1126/science.285.5426.418
Zou, L., and Elledge, S. J. (2003). Sensing DNA damage through ATRIP recognition of RPA-ssDNA complexes. Science 300, 1542–1548. doi: 10.1126/science.1083430
Keywords: viruses, cell cycle, checkpoint, infection, life cycle, host-pathogen interactions, phosphorylation, degradation
Citation: Fan Y, Sanyal S and Bruzzone R (2018) Breaking Bad: How Viruses Subvert the Cell Cycle. Front. Cell. Infect. Microbiol. 8:396. doi: 10.3389/fcimb.2018.00396
Received: 11 June 2018; Accepted: 22 October 2018;
Published: 19 November 2018.
Edited by:
Jianming Qiu, University of Kansas Medical Center, United StatesReviewed by:
Hung Jen Liu, National Chung Hsing University, TaiwanTian Wang, The University of Texas Medical Branch at Galveston, United States
Zhengwen Liu, First Affiliated Hospital of Xi'an Jiaotong University, China
Copyright © 2018 Fan, Sanyal and Bruzzone. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Sumana Sanyal, c2FueWFsQGhrdS5oaw==
Roberto Bruzzone, YnJ1enpvbmVAaGt1Lmhr